
Comparison of Long-term Ambulatory Function in Patients with Duchenne Muscular Dystrophy Treated with Eteplirsen and Matched Natural History Controls
Abstract
Background:
Duchenne muscular dystrophy (DMD) is a rare, X-linked, fatal, degenerative neuromuscular disease caused by DMD gene mutations. A relationship between exon skipping and dystrophin production in exon 51-amenable patients treated with eteplirsen (EXONDYS 51®) is established. Once-weekly eteplirsen significantly increased dystrophin, with slower decline in ambulatory function compared to baseline. Long-term treatment with eteplirsen leads to accumulation of dystrophin over time and observed functional benefits in patients with DMD.
Objective:
Compare long-term ambulatory function in eteplirsen-treated patients versus controls.
Methods:
Study 201/202 included 12 eteplirsen-treated patients assessed twice/year for ambulatory function over 4 years. Ambulatory evaluations (6-minute walk test [6MWT], loss of ambulation, and North Star Ambulatory Assessment [NSAA]) were compared with matched controls from Italian Telethon and Leuven registries.
Results:
At Years 3 and 4, eteplirsen-treated patients demonstrated markedly greater mean 6MWT than controls (difference in change from baseline of 132 m [95%CI (29, 235), p = 0.015] at Year 3 and 159 m [95%CI (66, 253), p = 0.002] at Year 4). At Year 4, a significantly greater proportion of eteplirsen-treated patients were still ambulant versus controls (10/12 vs 3/11; p = 0.020). At Year 3, eteplirsen-treated patients demonstrated milder NSAA decline versus controls (difference in change from baseline of 2.6, 95%CI [-6, 11]), however, the difference was not statistically significant; Year 4 control NSAA data were not available.
Conclusion:
In this retrospective matched control study, eteplirsen treatment resulted in attenuation of ambulatory decline over a 4-year observation period, supporting long-term benefit in patients with DMD.
INTRODUCTION
Duchenne muscular dystrophy (DMD) is a rare, X-linked, fatal, degenerative neuromuscular disease that occurs in approximately 1 in every 3,500 to 5,000 males born worldwide [1–4]. In the US, the prevalence of DMD is approximately 9,000 to 12,000 based on the global estimate of DMD prevalence [4–8]. This irreversible, progressive disease is caused by mutations in the dystrophin gene (DMD) [9, 10]. Mutations that are due to deletions in the dystrophin gene result in an out-of-frame messenger ribonucleic acid (mRNA) that prevents translation of functional dystrophin protein [11, 12]. Dystrophin is a component of the dystrophin-associated protein complex (DAPC) that links the intracellular cytoskeleton to the extracellular matrix, and its molecular function is critical for maintenance of structural integrity of muscle fibers and protection against damage. Thus, absence of this protein leads to progressive deterioration of muscle fibers and loss of function [13]. Muscle damage is evident in patients with DMD upon histologic examination, with an increase in inflammation and fibrosis in the first years of life. However, because of the rapid motor development associated with this early stage of life, the underlying progressive and irreversible muscle damage and resulting muscle weakness are often unnoticed until later. Although early signs include delayed milestones and presence of the Gowers’ sign when rising from the floor, functional assessments of patients with DMD in the first decade of life generally have demonstrated maturational improvements in ambulatory function until age 7 years; pulmonary function remains close to age-predicted normative values until the age of 10 years, with progression correlated to loss of ambulation [14–19].
Declining ambulatory function in patients with DMD is reflected by reductions in numerous assessments, including North Star Ambulatory Assessment (NSAA) total score and 6-minute walk test (6MWT). Loss of ambulation is a notable milestone and indicator of disease progression and severity [20–22], and it has been used to predict other milestones such as cardiopulmonary health in the clinical course and medical care of patients with DMD [20]. In particular, the age at loss of ambulation is closely linked to milestones of pulmonary decline such as FVC < 1L or FVC%p < 30%, both of which are associated with increased mortality and increased cost of care [22].
Eteplirsen (EXONDYS 51®; Sarepta Therapeutics, Inc., Cambridge, MA) is an antisense phospho-rodiamidate morpholino oligomer (PMO) approved by the US Food and Drug Administration for treatment of patients with DMD having a confirmed mutation amenable to the skipping of exon 51 [23–26], comprising approximately 13%of patients with DMD [27]. Eteplirsen binds to exon 51 of dystrophin pre-mRNA to induce skipping of exon 51 in order to restore the mRNA reading frame and enable translation of internally truncated, yet functional, dystrophin protein [23, 24]. Once-weekly eteplirsen has been shown to significantly increase dystrophin expression and slow the decline of ambulatory and pulmonary function compared with natural history controls [23–25, 28]. This report compares ambulatory function data from eteplirsen-treated patients from the 201/202 study with observational patient data from natural history cohorts [23, 29–31] to further understand the impact of eteplirsen on long-term disease trajectory.
MATERIALS AND METHODS
Patients
Twelve boys between the ages of 7 and 13 years, with DMD gene mutations amenable to skipping exon 51, were enrolled in the study. All patients were on stable glucocorticoids for at least 24 weeks prior to baseline and throughout the duration of the study. Two-thirds (8/12) of eteplirsen-treated patients received deflazacort (0.9 mg/kg) versus prednisone (0.75 mg/kg). The mean age at steroid initiation of the eteplirsen-treated patients was 5.2 years (N = 12; standard deviation [SD], 1.07 years). All 12 patients were ambulatory at baseline with a 6MWT between 256 and 416 meters. The study protocol was approved by an institutional review board and conducted in accordance with the principles of the Declaration of Helsinki and Good Clinical Practice guidelines. Prior to enrolment, written informed consent (and assent as appropriate for age) was obtained from each patient or parent/caregiver, respectively.
Study design
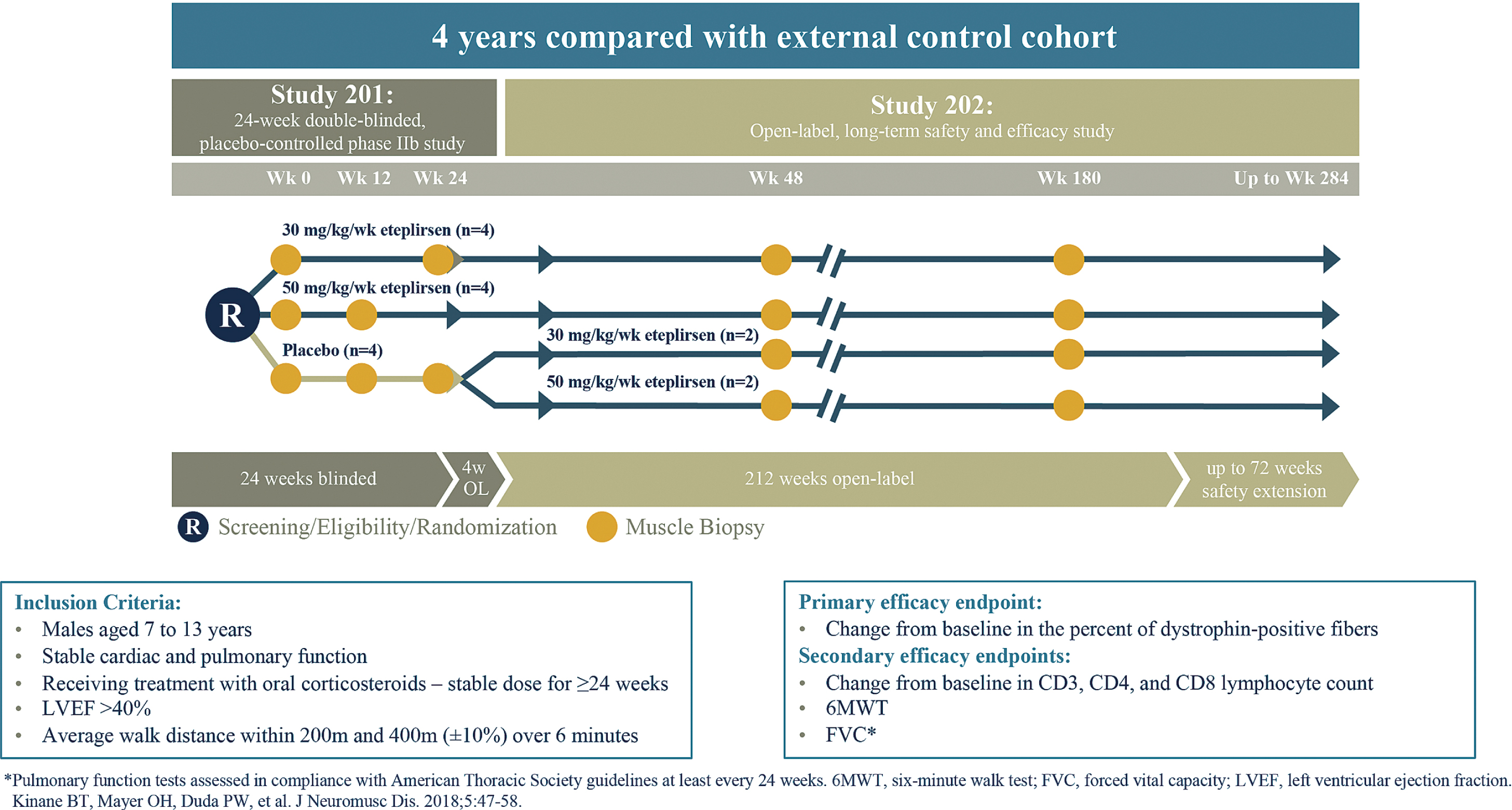
Study 201/202 (NCT01396239 and NCT01540409) has been described previously [23–25] and is shown in Fig. 1. Briefly, patients (N = 12) were assessed at time points permitting long-term analysis of the relationship between treatment duration, dystrophin production, and clinical efficacy. Patients were randomized to eteplirsen 30 mg/kg (n = 4), eteplirsen 50 mg/kg (n = 4), or placebo (n = 4) treatment groups for 24 weeks. Study 201 included a 24-week placebo-controlled period, followed by an open-label eteplirsen treatment period; efficacy data from the 4 patients randomized to receive placebo and subsequent eteplirsen treatment were pooled with the efficacy data from the 8 patients originally randomized to eteplirsen. Muscle biopsies were obtained for all patients at baseline and at a second time point (half of the study patients at Week 12 and the other half at Week 24). At completion of 24 weeks, patients randomized to receive placebo and subsequent eteplirsen treatment (n = 4) were switched to open-label treatment with either 30 or 50 mg/kg eteplirsen. All patients had a third biopsy at Week 48. Eleven of the 12 patients consented to a fourth biopsy at Week 180; optimized, validated methods for dystrophin measurement were used in analysis of the Week 180 biopsies. Clinical outcomes of 6MWT (primary endpoint), loss of ambulation, and NSAA were collected through Year 4 for patients enrolled in Study 201/202.
Fig. 1
Study design for eteplirsen studies 201/202. Twelve patients with Duchenne muscular dystrophy were randomly assigned to 1 of 3 cohorts receiving weekly intravenous (IV) infusions in a 24-week, double-blind, placebo-controlled study (Study 201): eteplirsen 30 mg/kg, eteplirsen 50 mg/kg, or placebo. At Week 25, eteplirsen-treated patients continued the same weekly dose open-label, and placebo patients were randomized to open-label treatment with eteplirsen 30 mg/kg or 50 mg/kg weekly IV (Study 202).
Evaluations
For Study 201/202 and natural history control patients derived from the Italian Telethon and Leuven registries (primary external controls), ambulatory evaluations included: 6MWT [32–35], loss of ambulation (defined as 0 meters on the 6MWT), and NSAA [31, 36–39]. Lead physical therapists representing multiple regions were engaged in a collaborative training on administration of these ambulatory assessments.
Natural history control groups for ambulatory assessments
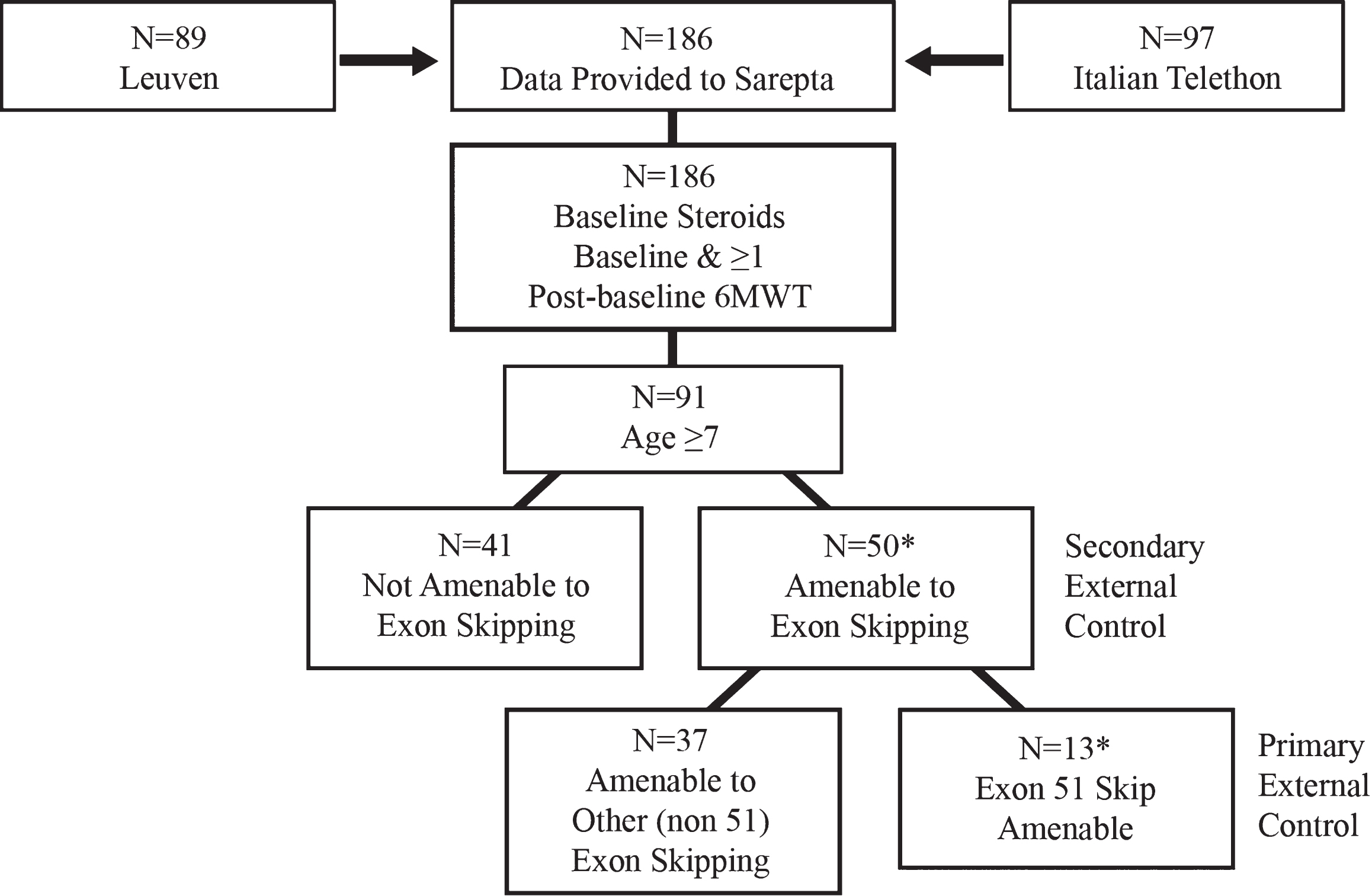
The natural history controls were derived from well-characterized and established patient-level natural history studies from DMD centers of excellence participating in the Italian Telethon and Leuven registries (referred to as primary external controls throughout this manuscript) [23, 24, 40] (Fig. 2). In brief, of the 186 ambulatory patients selected from the 2 registries, 116 patients were receiving steroids and had≥1 post-baseline 6MWT measurement; and 91 were≥7 years of age. Following application of the selection criteria to the combined dataset, a primary external control group amenable to exon 51 skipping (N = 11) was identified (Fig. 2). For the primary external controls, 63.6%(7/11) patients received deflazacort (0.9 mg/kg) versus prednisone (0.75 mg/kg). The mean age at steroid initiation of the primary external controls was 6.8 years (N = 11; SD, 2.08 years). All primary external control patients were ambulatory at baseline with a 6MWT between 200 and 458 meters.
Fig. 2
Primary external controls for ambulatory assessment were derived from patient-level natural history data from Duchenne muscular dystrophy centers of excellence participating in the Italian Telethon and Leuven registries.* *2 patients were excluded from the analysis for the following reasons: •Patient OBG 16 withdrew consent. •Though included in earlier reports, it was later determined that Patient OBG 20 was enrolled in a drisapersen trial in Year 2 and lost ambulation while on drisapersen.
Statistical analyses
The eteplirsen intent-to-treat population (N = 12) was compared to natural history controls for the on-treatment time period. This was done by excluding the first 24 weeks of data for the 4 patients initially on placebo for 24 weeks. After this adjustment, Week 24 was considered the baseline (Week 0) assessment for these patients and all subsequent time points for the purpose of data analyses were counted from this new readjusted baseline for the 4 patients originally on placebo.
Study 201/202 included patients dosed at either 30 mg/kg or 50 mg/kg and data for both dosing levels were pooled. The ambulatory assessments of 6MWT, NSAA, and loss of ambulation were assessed at baseline and at 1-year (defined as 48 weeks) intervals over the course of 4 years for 6MWT and loss of ambulation, and 3 years for NSAA (NSAA analysis was limited to 3 years based on availability of NSAA data from the Italian telethon registry). If a patient was unable to perform the 6MWT, they were considered to have loss of ambulation and were assigned a score of 0 for all subsequent assessments, and a 0 value was included in the calculation of mean 6MWT at each time point.
Although the study protocol specified 6MWT assessments on 2 consecutive days for some time points, we used only Day 1 values for the purposes of this analysis. This approach was more consistent with methodology employed in the natural history cohorts, where patients were only assessed on 1 day, once per visit. Baseline visit was defined as the first visit for the natural history patients and was considered the first recorded visit within the registry; 1 year was considered to be 52 weeks. Derivation of natural history control cohorts has been described extensively [23, 40]. Sensitivity analyses were performed to assess potential bias arising from any imbalances in baseline characteristics (eg, 6MWT, age, height, weight, glucocorticoid schedule, age of steroid initiation) and any difference in assessment schedule.
Mean 6MWT data for eteplirsen-treated patients and primary external controls were plotted at baseline (Year 0) and every year thereafter. Standard errors were also plotted. For the purposes of this analysis, a study year was counted as 48 weeks for eteplirsen-treated patients, and 52 weeks for primary external controls. Means and standard errors were plotted over time for both groups. The groups were compared using the analysis of covariance (ANCOVA) model, with treatment group as a fixed-effect term and baseline 6MWT as a covariate; adjustment was calculated for change from baseline for each group and for difference of change from baseline. No adjustments were made for multiple comparisons. The approach to analysis of NSAA was consistent with that of the 6MWT, with the exception that NSAA data were not available from the Leuven registry so this resulted in only 8 available patients for comparison, with data provided through Year 3. 95%confidence intervals for change from baseline at Year 3 in NSAA total score were generated using Welch’s unequal variances t-test.
RESULTS
Baseline characteristics
Baseline characteristics in the eteplirsen group and the primary external control group used for the ambulatory measures are shown in Table 1. All patients in both groups were ambulatory at baseline. The mean age of the eteplirsen-treated patients was 9.4 years (SD, 1.18 years) compared with 9.6 years (SD, 1.52 years) in the primary external controls. Baseline 6MWT for the eteplirsen group was 363.2 m (SD, 42.19 m) compared with 361.3 m (SD, 69.93 m) for the primary external control group. Baseline NSAA score for the eteplirsen group was 24.9 (SD, 4.93) compared with 22.8 (SD, 6.54) in the primary external control group. At baseline, 92%of patients in the eteplirsen-treated group and 64%of patients in the primary external control group received continuous (vs intermittent) dosing of glucocorticoids. Patients across both groups were comparable in terms of their deletion mutations, heights, and weights.
Table 1
Summary of baseline characteristics: Study 201/202 and primary external controls
| Parameter | Combined 201/202 Studies (N = 12) | Primary external controls (N = 11) |
| Age (years) | ||
| n | 12 | 11 |
| Mean (SD) | 9.4 (1.18) | 9.6 (1.52) |
| Median (Min, Max) | 9.7 (7, 11) | 9.0 (7, 12) |
| Baseline 6MWT (m) | ||
| n | 12 | 11 |
| Mean (SD) | 363.2 (42.19) | 361.3 (69.93) |
| Median (Min, Max) | 370.0 (256, 416) | 373.0 (200, 458) |
| NSAA score | ||
| n | 12 | 8a |
| Mean (SD) | 24.9 (4.93) | 22.8 (6.54) |
| Median (Min, Max) | 25.5 (17, 31) | 24.0 (10, 31) |
| Able to rise from floor independently, n (%) | 11 (92%) | 8 (80%)b |
| Rise time (seconds) | ||
| n | 12 | 10c |
| Mean (SD) | 8.2 (7.57) | 10.0 (10.74) |
| Median (Min, Max) | 5.5 (3, 30) | 5.3 (2, 30) |
| Deletion mutations | 45–50, 48–50, 49–50, 50, 52 | 45–50, 48–50, 49–50, 50, 52 |
| Height (cm) | ||
| n | 12 | 11 |
| Mean (SD) | 123.9 (8.37) | 129.7 (8.25) |
| Median (Min, Max) | 119.0 (117, 138) | 131.5 (107, 136) |
| Weight (kg) | ||
| n | 12 | 11 |
| Mean (SD) | 32.4 (6.75) | 34.2 (10.22) |
| Median (Min, Max) | 34.6 (24, 41) | 31.0 (17, 48) |
6MWT, 6-minute walk test; NSAA, North Star Ambulatory Assessment. aSince only the Italian Telethon registry provided NSAA data, the primary external control consisted of 8 patients. bAt Baseline (on Day 1), 1 patient was unable to rise; however, he was able to rise at the Week 4 and subsequent assessment time points. cFor patients unable to rise independently, a rise time of 30 seconds was used. Data are missing for 1 patient; data were not imputed as 30 seconds for this patient.
Ambulatory function
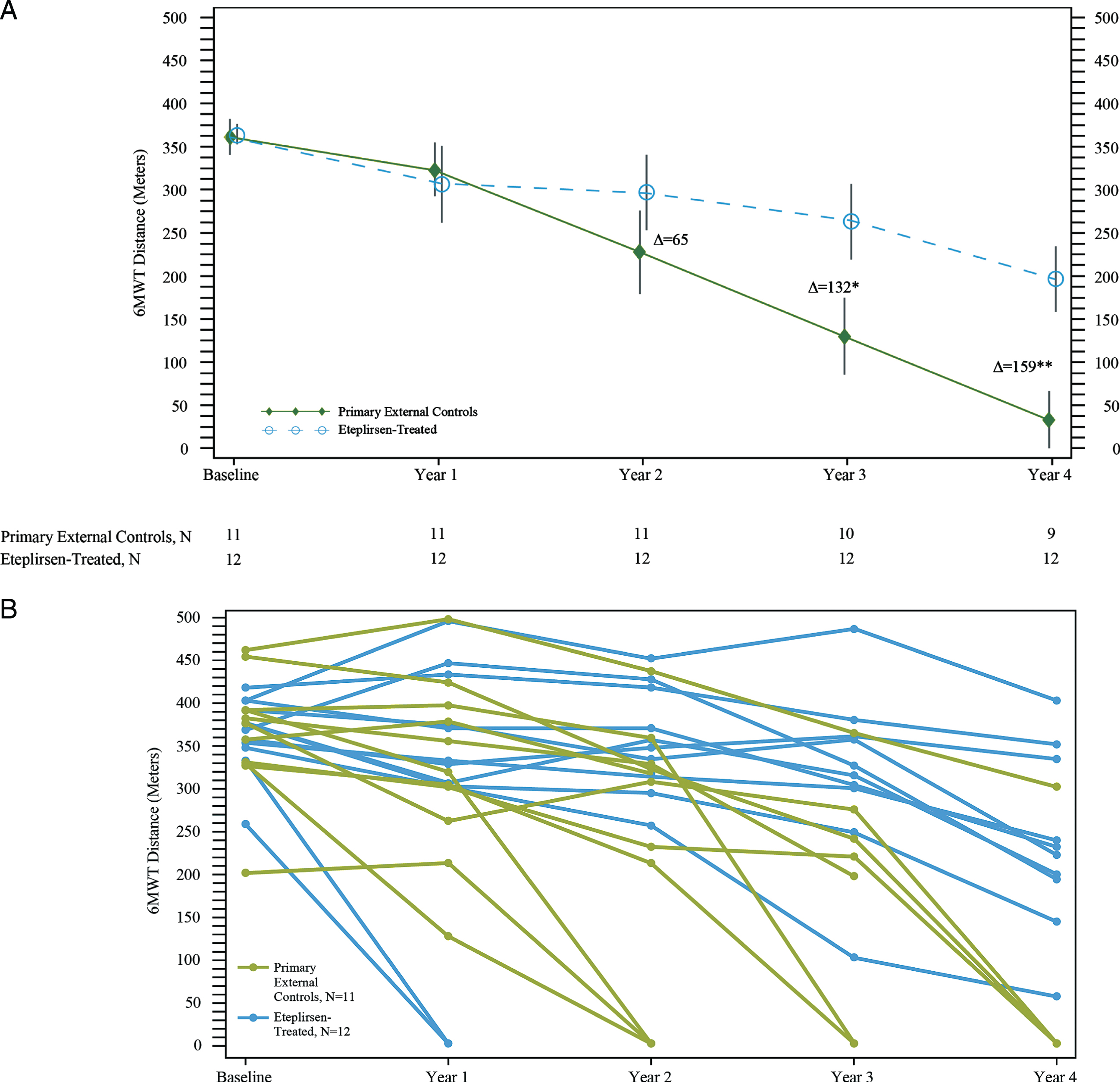
Eteplirsen-treated patients experienced less decline across all measures of ambulatory function compared with primary external controls. Eteplirsen-treated and primary external control groups had similar baseline 6MWT, and their disease progression trajectories overlapped through Year 1 (Fig. 3A, 3B). After Year 1, mean walking distance stabilized for eteplirsen-treated patients but declined substantially for the primary external controls. Eteplirsen-treated patients experienced less decline with a notable difference observed at Year 2 compared with primary external controls (295.9 m vs. 227.5 m, respectively). In Years 3 and 4, eteplirsen-treated patients demonstrated significantly greater mean walking distance of 263.1 m and 196.3 m, respectively (difference between the adjusted change from baseline values between eteplirsen-treated patients and control patients = 132 m [95%CI (29, 235), p = 0.015] and 159 m [95%CI (66, 253), p = 0.002], respectively). Individual data series for 6MWT over time are shown in Fig. 3B. All sensitivity analyses, which were performed to assess potential bias arising from any imbalances in baseline characteristics and any difference in assessment schedule, continued to demonstrate a clinically meaningful difference of more than 100 meters on the 6MWT favoring eteplirsen-treated patients, and maintained statistical significance (p < 0.05) (Supplement).
Fig. 3
6-minute walk test (6MWT) in eteplirsen-treated patients and primary external controls over 4 years. (A) Mean 6MWT results; *p = 0.0145; **p = 0.0022. N represents the number of patients at the time point specified. One external control patient did not have data at Year 3; 2 patients did not have data at Year 4. Δ represents the adjusted difference between change in baseline values of eteplirsen-treated patients and untreated primary external controls from the ANCOVA model. (B) Individual 6MWT results.
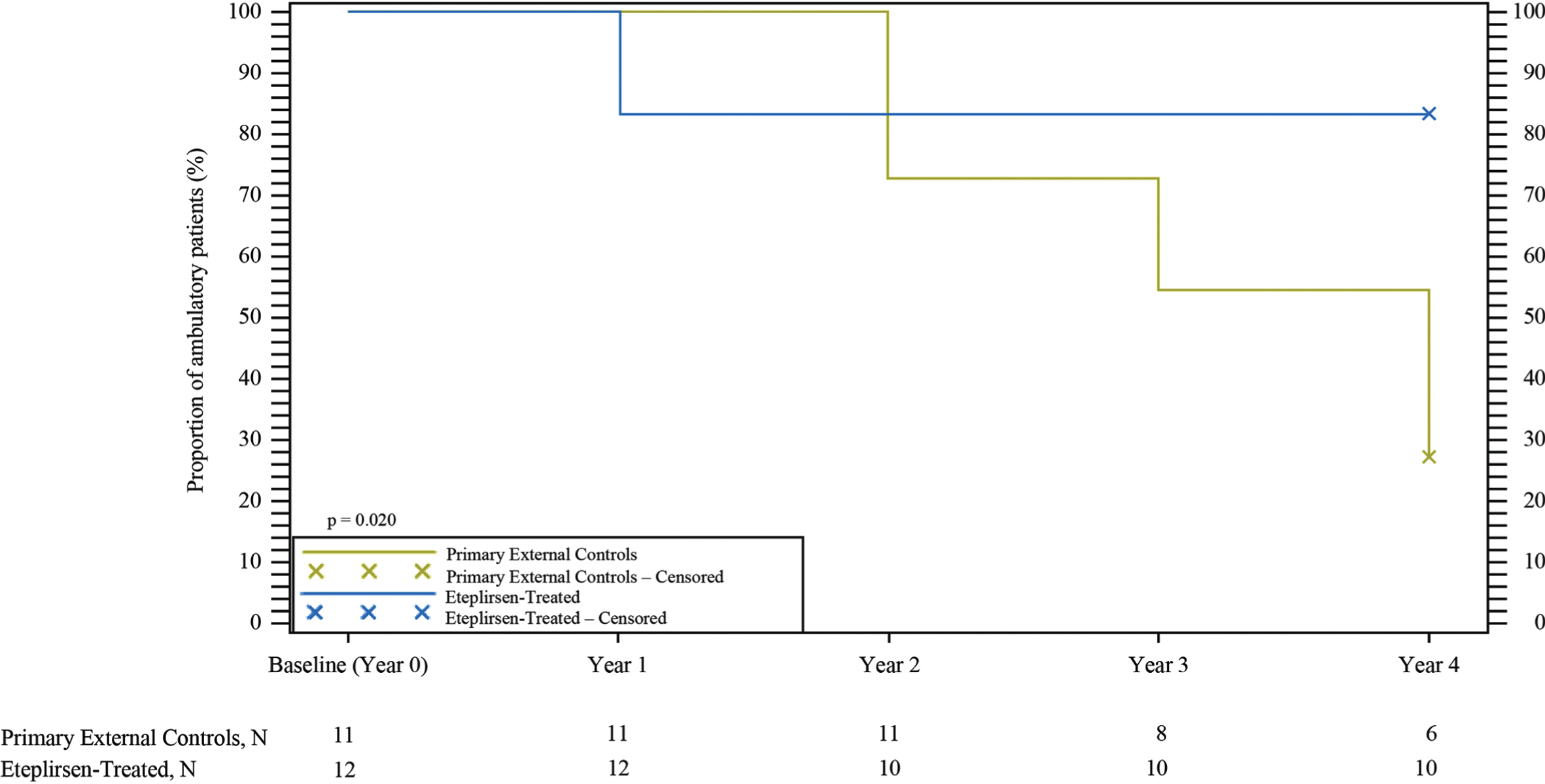
Eteplirsen-treated patients showed a preservation of ambulation compared with primary external controls (Fig. 4). At Year 1, 17%of eteplirsen-treated patients had lost ambulation. Despite this, no additional eteplirsen-treated patients lost ambulation through Year 4. Conversely, the proportion of primary external controls who lost ambulation steadily increased over time. At 4-year follow-up, significantly more primary external controls were not ambulant compared with eteplirsen-treated patients (73%[8/11] vs. 17%[2/12], p = 0.020). Eight of 11 patients in the primary external control group lost ambulation, with the median age for loss of ambulation equal to 13 years. In contrast, at the time of the last 6MWT (Week 216), 10 of 12 eteplirsen-treated patients were ambulant, hence the median time to loss of ambulation was not yet reached. The median age for eteplirsen-treated boys at Week 216 of eteplirsen treatment was 13.8 years. Moreover, 4 of 12 (33.3%) eteplirsen-treated patients were older than 14 years of age, and all were still ambulant.
Fig. 4
Kaplan-Meier analysis of loss of ambulation. Eteplirsen-treated patients showed a preservation of ambulation over 4 years compared with primary external controls.
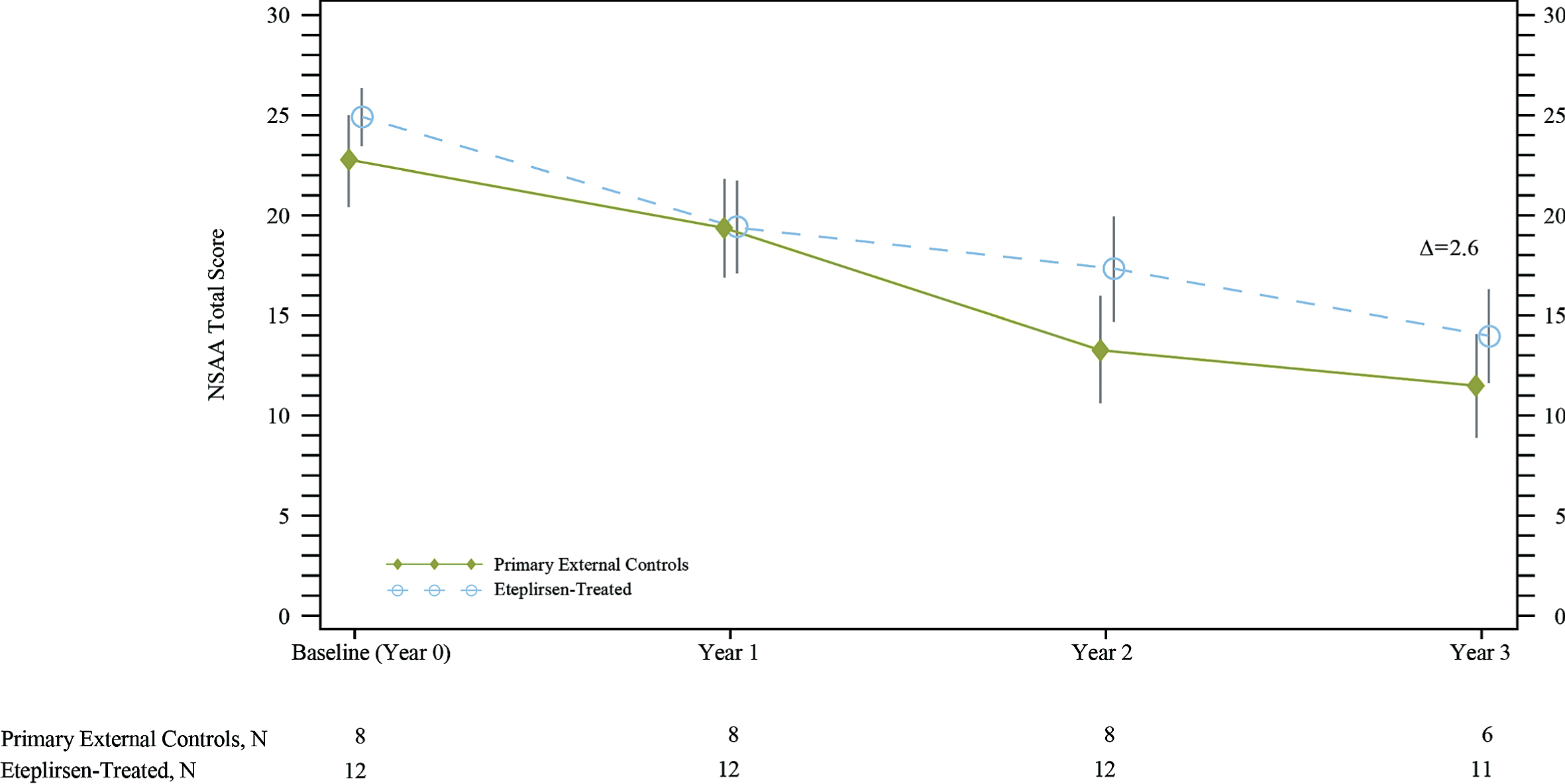
Over the first year, eteplirsen-treated patients and primary external controls demonstrated a decline in NSAA total score (Fig. 5). As with the 6MWT, however, after 1 year, the decline in NSAA total score for the eteplirsen-treated patients appears slower over time compared with primary external controls. Over 3 years, eteplirsen-treated patients experienced less decline in NSAA total score, with a change from baseline of –11.3 vs. –13.8 for the primary external controls (difference = 2.6, 95%CI [–6, 11]); this difference was not statistically significant.
Fig. 5
North Star Ambulatory Assessment (NSAA) total score showed that eteplirsen-treated patients experienced less decline compared with primary external controls over 3 years. Δ represents the unadjusted difference in mean change from baseline in eteplirsen-treated patients versus external controls. NSAA data for primary external controls were available from the Italian Telethon registry patients only (8 of the 12 natural history patients used for ambulatory comparisons). In the eteplirsen-treated group, 1 patient did not have NSAA measured at Year 3.
DISCUSSION
In Study 201/202, over the course of 4 years, eteplirsen-treated patients outperformed patients from well-matched, natural history databases for ambulatory outcomes.
The divergence from natural history seen in eteplirsen-treated patients in Study 201/202 is demonstrated by 5-fold fewer eteplirsen-treated patients who lost ambulation by Year 4 compared with primary external controls. The median age for loss of ambulation for the controls of 13 years is consistent with the published natural history data for ambulatory decline in exon 51-amenable patients with DMD [41]. The loss of ambulation data are consistent with other ambulatory assessments in the eteplirsen-treated patients. After Year 1, the observed deviation in 6MWT trajectory for eteplirsen-treated patients from primary external controls may be due to dystrophin accumulation, as supported by a large, US-based, multi-center study of efficacy and safety in exon 51-amenable patients with DMD [42]. The magnitude of difference seen in the 6MWT after Year 2 far exceeds the minimal clinically important difference of 30 meters for patients with DMD established in studies observing patients with DMD for 1 year [35, 43]. Loss of ambulation is a major milestone of disease progression in DMD as this function is irreversible and prognostic for further irreversible losses of function. The 6MWT results account for this important milestone of disease progression by assigning a value of zero in the case of lost ambulation. All patients in both groups were ambulatory at baseline, the groups were well-matched for known prognostic factors, and any loss of ambulation-driven differences in 6MWT between groups over time highlight the important role of eteplirsen in delaying irreversible disease progression. Future analyses with larger sample sizes will allow additional evaluation of 6MWT in patients who did not lose ambulation. The NSAA total score for the eteplirsen-treated patients demonstrated a trend toward a slower decline compared with primary external controls after 1 year of treatment.
The sustained long-term slowing of ambulatory decline observed with eteplirsen-treated patients is promising. Of note, differences between eteplirsen-treated patients and natural history patients emerged after at least 1 year of treatment, and this striking consistency between treated and untreated groups during the first year, in addition to the closely matched baseline parameters, suggests the differences between the eteplirsen-treated patients and natural history patients after Year 1 are due to eteplirsen treatment.
Dystrophin production has been shown in eteplirsen-treated patients, with higher levels of dystrophin seen after 180 weeks on treatment in Study 201/202 compared to untreated patients with DMD [40]. Further, previous reports have shown dystrophin levels increase over time with increased duration of eteplirsen treatment [24]. These findings, combined with the data reported here, suggest continued treatment with eteplirsen results in the accumulation of dystrophin over time, and this ultimately translates to clinical divergence of eteplirsen-treated patients from natural history. Further study is needed to confirm this relationship, as there are some limitations to the present work.
Study 201/202 was a long-term, open-label extension study over the course of 4 years. While there was an initial 24-week placebo period, the subsequent portion of the study did not have a placebo cohort as a comparator due to challenges associated with running long-term, multi-year, placebo-controlled studies in an irreversible, progressive and fatal rare disease like DMD. Due to the lack of long-term placebo controls, the data from the treated patients were compared to well-matched natural history cohorts, but the comparisons were post-hoc. Additional limitations include restrictions in statistical comparison associated with small number sample size and non-randomized design.
There are 2 imbalances in the comparison of steroid treatment between the primary external control group and eteplirsen-treated patients. First, the mean age at the time of steroid initiation for the primary external controls was 1.6 years older than in the eteplirsen-treated group (6.8 vs. 5.2 years, respectively). Second, at baseline, a higher percentage of patients in the eteplirsen-treated group received glucocorticoids daily (continuous regimen): 92%in the eteplirsen-treated group compared with 64%in primary external control group. Sensitivity analyses for the 6MWT using covariates of the age of steroid initiation and steroid regimen (intermittent vs continuous) demonstrate minimal impact on the primary endpoint of 6MWT. Despite possible imbalances, all sensitivity analyses indicate a statistically significant (p < 0.05) and clinically meaningful difference of more than 100 meters on the 6MWT, favoring eteplirsen-treated patients. When interpreting the mean 6MWT data, we recognize that due to loss of ambulation, 8 patients with 6MWT data in the primary external control group included at Year 4 are assigned a value of zero, which lowers the mean. However, these zeros are not null values, but instead represent the valid, actual effort and ability of patients on the day of their 6MWT. Figure 3B shows individual loss of ambulation data (primary external controls, 73%[8/11] vs. eteplirsen-treated, 17%[2/12], p = 0.020) as well as each 6MWT score for patients still able to perform the test over time.
The present work demonstrates consistency be-tween ambulatory measures over 4 years of etep-lirsen treatment. During the initial year of treatment, eteplirsen-treated patients performed no different from natural history controls, while significant and meaningful differences emerged between treated and untreated patients over time. Treatment with eteplirsen slowed the decline in ambulatory compared to natural history controls, indicating that eteplirsen alters the long-term disease trajectory for patients with DMD. The long-term clinical benefit of eteplirsen treatment in ambulatory outcomes shows potential for the production of low levels of novel dystrophin protein in patients amenable to exon 51 skipping to be a significant advance in clinical amelioration of this progressive and life-limiting disease.
COLLABORATIVE STUDY GROUP AUTHORS
Eteplirsen Study Group investigators
Study 201: Jerry R. Mendell (Nationwide Children’s Hospital).
Study 202: Jerry R. Mendell (Nationwide Children’s Hospital); Kevin O’Brien (Miller Children’s Hospital); Ludwig Matthew Frank (Children’s Hospital of the King’s Daughter); Barry Byrne (University of Florida); Erika S. Helgerson (Osceola Medical Center); Amy Harper (Levine Children’s Hospital at the Carolinas Medical Center); Peter Heydemann (Rush University); Hoda Abdel-Hamid (Children’s Hospital of Pittsburgh –UPMC); Robert Dracker (Summerwood Pediatrics/Infusa-care); Anne Connolly (Washington University School of Medicine); Linda Specht (Dartmouth –Hitchcock Medical Center); Fawn Leigh (Massachusetts General Hospital).
CONFLICTS OF INTEREST
This study was sponsored by Sarepta Therapeutics, Inc. N. Khan, N. Sha, and H. Eliopolous were employees of Sarepta Therapeutics, Inc. at the time of this study and during manuscript preparation, and may own stock/option in the company. L. Alfano, J. R. Mendell, C. McDonald, E. Mercuri, and L. P. Lowes serve as remunerated consultants to Sarepta Therapeutics, Inc. N. Goemans has nothing to disclose.
Employees of Sarepta Therapeutics, Inc. contributed to the study design, data analysis, and critical review of the manuscript. The decision to submit resided solely with the authors.
ACKNOWLEDGMENTS
This study was sponsored by Sarepta Therapeutics, Inc. Medical writing and editorial support were provided by Stephen Collins, PhD, of Peloton Advantage, LLC (Parsippany, NJ), and Purvi Kobawala Smith, MS, MPH, of Health & Wellness Partners, LLC (Upper Saddle River, NJ), funded by Sarepta Therapeutics, Inc. The authors thank Aji Nair, PhD, and Aaron Novack, PhD, of Sarepta Therapeutics, Inc. for their guidance.
SUPPLEMENTARY MATERIAL
[1] The supplementary material is available in the electronic version of this article: https://dx.doi.org/10.3233/JND-200548.
REFERENCES
[1] | Aartsma-Rus A , Straub V , Hemmings R , Haas M , Schlosser-Weber G , Stoyanova-Beninska V , et al. Development of exon skipping therapies for Duchenne muscular dystrophy: A critical review and a perspective on the outstanding issues. Nucleic Acid Ther. (2017) ;27: (5):251–9. |
[2] | Emery AE Population frequencies of inherited neuromuscular disease –a world survey. Neuromuscul Disord. (1991) ;1: (1):19–29. |
[3] | Mendell JR , Lloyd-Puryear M Report of MDA muscle disease symposium on newborn screening for newborn muscular dystrophy. Muscle Nerve. (2013) ;48: (1)21–6. |
[4] | Mendell JR , Shilling C , Leslie ND , Flanigan KM , al-Dahhak R , Gastier-Foster J , et al. Evidence-based path to newborn screening for Duchenne muscular dystrophy. Ann Neurol. (2012) ;71: (3):304–13. |
[5] | Centers for Disease Control and Prevention. Prevalence of Duchenne/Becker muscular dystrophy among males aged 5-24 years- four states, 2007. MMWR Morb Mortal Wkly Rep. (2009) ;58: (40):1119–22. |
[6] | Dooley J , Gordon KE , Dodds L , MacSween J Duchenne muscular dystrophy: A 3-year population-based incidence study. Clin Pediatr (Phila). (2010) ;49: (2):177–9. |
[7] | Jeppesen J , Green A , Steffensen BF , Rahbek J The Duchenne muscular dystrophy population in Denmark, 1977-2001: Prevalence, incidence and survival in relation to the introduction of ventilator use. Neuromuscul Disord. (2003) ;13: (10):804–12. |
[8] | Norwood FLM , Harling C , Chinnery PF , Eagle M , Bushby K , Straub V Prevalence of genetic muscle disease in Northern England: In-depth analysis of a muscle clinic population. Brain. (2009) ;132: (11):3175–86. |
[9] | Bushby K , Finkel R , Birnkrant DJ , Case LE , Clemens PR , Cripe L , et al. Diagnosis and management of Duchenne muscular dystrophy, part Diagnosis, and pharmacological and psychosocial management. Lancet Neurol.. (2010) ;9: (1):77–93. |
[10] | Blake DJ , Weir A , Newey SE , Davies KE Function and genetics of dystrophin and dystrophin-related proteins in muscle. Physiol Rev. (2002) ;82: (2):291–329. |
[11] | Mah JK Current and emerging treatment strategies for Duchenne muscular dystrophy. Neuropsychiatr Dis Treat. (2016) ;12: : 1795–807. |
[12] | Kole R , Krieg AM Exon skipping therapy for Duchenne muscular dystrophy. Adv Drug Deliv Rev. (2015) ;87: : 104–7. |
[13] | Aartsma-Rus A , Ginjaar IB , Bushby K The importance of genetic diagnosis for Duchenne muscular dystrophy. J Med Genet. (2016) ;53: (3):145–51. |
[14] | Ricotti V , Ridout DA , Pane M , Main M , Mayhew A , Mercuri E , et al. The NorthStar Ambulatory Assessment in Duchenne muscular dystrophy: Considerations for the design of clinical trials. J Neurol Neurosurg Psychiatr. (2016) ;87: (2):149–55. |
[15] | Ricotti V , Ridout DA , Scott E , Quinlivan R , Robb SA , Manzur AY , et al. Long-term benefits and adverse effects of intermittent versus daily glucocorticoids in boys with Duchenne muscular dystrophy. J Neurol Neurosurg Psychiatry. (2013) ;84: (6):698–705. |
[16] | Mazzone ES , Pane M , Sormani MP , Scalise R , Berardinelli A , Messina S , et al. 24 month longitudinal data in ambulant boys with Duchenne muscular dystrophy. PLoS One. (2013) ;8: (1):e52512. |
[17] | Mayer OH , Henricson EK , McDonald CM , Buyse GM Advances in pulmonary care in Duchenne muscular dystrophy. US Neurol. (2017) ;13: (1):35–41. |
[18] | Meier T , Rummey C , Leinonen M , Spagnolo P , Mayer OH , Buyse GM ; DELOS Study Group. Characterization of pulmonary function in 10-18 year old patients with Duchenne muscular dystrophy. Neuromuscul Disord. (2017) ;27: (4):307–14. |
[19] | McDonald CM , Gordish-Dressman H , Henricson EK , Duong T , Joyce NC , Jhawar S , et al. Longitudinal pulmonary function testing outcome measures in Duchenne muscular dystrophy: Long-term natural history with and without glucocorticoids. Neuromuscul Disord.. (2018) ;28: (11):897–909. |
[20] | Finder JD , Birnkrant D , Carl J , Farber HJ , Goza D , Iannacone ST , et al. Respiratory care of the patient with Duchenne muscular dystrophy: ATS consensus statement. Am J Respir Crit Care Med. (2004) ;170: (4):456–65. |
[21] | Phillips MF , Quinlivan RC , Edwards RH , Calverley PM Changes in spirometry over time as a prognostic marker in patients with Duchenne muscular dystrophy. Am J Respir Crit Care Med. (2001) ;164: (12):2191–4. |
[22] | Birnkrant DJ , Panitch HB , Benditt JO , Boitano LJ , Carter ER , Cwik VA , et al. American College of Chest Physicians consensus statement on the respiratory and related management of patients with Duchenne muscular dystrophy undergoing anesthesia or sedation. Chest.. (2007) ;132: (6):1977–86. |
[23] | Mendell JR , Goemans N , Lowes LP , Alfano LN , Berry K , Shao J , et al. Longitudinal effect of eteplirsen versus historical control on ambulation in Duchenne muscular dystrophy. Ann Neurol. (2016) ;79: : 257–71. |
[24] | Mendell JR , Rodino-Klapac LR , Sahenk Z , Roush K , Bird L , Lowes LP , et al. Eteplirsen for the treatment of Duchenne muscular dystrophy. Ann Neurol. (2013) ;74: (5):637–47. |
[25] | Kinane TB , Mayer OH , Duda PW , Lowes LP , Moody SL , Mendell JR . Long-term pulmonary function in Duchenne muscular dystrophy: Comparison of eteplirsen-treated patients to natural history. J Neuromuscul Dis. (2018) ;5: (1):47–58. |
[26] | US Food & Drug Administration. September 19, 2016. FDA grants accelerated approval to first drug for Duchenne muscular dystrophy. Available from: https://www.fda.gov/newsevents/newsroom/pressannouncements/ucm521263.htm. Available from:. Accessed March 15, 2020. |
[27] | Aartsma-Rus A , Fokkema I , Verschuuren J , Ginjaar I , van Deutekom J , van Ommen GJ , den Dunnen JT . Theoretic applicability of antisense-mediated exon skipping for Duchenne muscular dystrophy mutations. Hum Mutat. (2009) ;30: (3):293–9. |
[28] | Khan N , Eliopoulos H , Han L , Kinane TB , Lowes LP , Mendell JR , et al. Eteplirsen treatment attenuates respiratory decline in ambulatory and non-ambulatory patients with Duchenne muscular dystrophy. J Neuromuscul Dis. (2019) ;6: (2):213–25. |
[29] | Pane M , Mazzone ES , Sivo S , Sormani MP , Messina S , D’Amico A , et al. Long term natural history data in ambulant boys with Duchenne muscular dystrophy: 36-month changes. PLoS One. (2014) ;9: (10):e108205. |
[30] | Goemans N , van den Hauwe M , Wilson R , van Impe A , Klingels K , Buyse G . Ambulatory capacity and disease progression as measured by the 6-minute-walk-distance in Duchenne muscular dystrophy subjects on daily corticosteroids. Neuromuscul Disord. (2013) ;23: (8):618–23. |
[31] | Mazzone ES , Messina S , Vasco G , Main M , Eagle M , D’Amico A , et al. Reliability of the North Star Ambulatory Assessment in a multicentric setting. Neuromusc Disord. (2009) ;19: (7):458–61. |
[32] | McDonald CM , Henricson EK , Han JJ , Abresch RT , Nicorici A , Elfring GL , et al. The 6-minute walk test as a new outcome measure in Duchenne muscular dystrophy. Muscle Nerve. (2009) ;41: (4):500–10. |
[33] | McDonald CM , Henricson EK , Han JJ , Abresch RT , Nicorici A , Atkinson L , et al. The 6-minute walk test in Duchenne/Becker muscular dystrophy: Longitudinal observations. Muscle Nerve. (2010) ;42: (6):966–74. |
[34] | McDonald CM , Henricson EK , Abresch RT , Florence J , Eagle M , Gappmaier E , et al. The 6-minute walk test and other clinical endpoints in Duchenne muscular dystrophy: Reliability, concurrent validity, and minimal clinically important differences from a multicenter study. Muscle Nerve. (2013) ;48: (3):357–68. |
[35] | McDonald CM , Henricson EK , Abresch RT , Florence JM , Eagle M , Gappmaier E , et al. The 6-minute walk test and other endpoints in Duchenne muscular dystrophy: Longitudinal natural history observations over 48 weeks from a multicenter study. Muscle Nerve. (2013) ;48: (3):343–56. |
[36] | Scott E , Mawson SJ . Measurement in Duchenne muscular dystrophy: considerations in the development of a neuromuscular assessment tool. Dev Med Child Neurol. (2006) ;48: (6):540–44. |
[37] | Mazzone E , Martinelli D , Berardinelli A , Messina S , D’Amico A , Vasco G , et al. North Star Ambulatory Assessment, 6-minute walk test and timed items in ambulant boys with Duchenne muscular dystrophy. Neuromuscul Disord. (2010) ;20: (11):712–16. |
[38] | Mayhew A , Cano S , Scott E , Eagle M , Bushby K , Muntoni F , et al. Moving towards meaningful measurement: Rasch analysis of the North Star Ambulatory Assessment in Duchenne muscular dystrophy. Dev Med Child Neurol. (2011) ;53: (6):535–42. |
[39] | Scott E , Eagle M , Mayhew A , Freeman J , Main M , Sheehan J , et al. Development of a functional assessment scale for ambulatory boys with Duchenne muscular dystrophy. Physiother Res Int. (2012) ;17: (2):101–9. |
[40] | Charleston JS , Schnell FJ , Dworzak J , Donoghue C , Lewis S , Chen L , et al. Eteplirsen treatment for Duchenne muscular dystrophy: Exon skipping and dystrophin production. Neurology. (2018) ;90: (24):e2146–54. |
[41] | Bello L , Morgenroth LP , Gordish-Dressman H , Hoffman EP , McDonald CM , Cirak S , et al. DMD genotypes and loss of ambulation in the CINRG Duchenne natural history study. Neurology. (2016) ;87: (4):401–9. |
[42] | McDonald C , Shieh P , Abdel-Hamid HZ , Connolly AM , Khan N , Koenig E , et al. Open-label evaluation of eteplirsen in patients with Duchenne uscular dystrophy amenable to exon 51 skipping: PROMOVI trial. Poster presented at: 2020 Muscular Dystrophy Association (MDA) Clinical and Scientific Conference; Virtual Meeting. |
[43] | Henricson E , Abresch R , Han JJ , Nicorici A , Goude Keller E , de Bie E , et al. The 6-minute walk test and person-reported outcomes in boys with Duchenne muscular dystrophy and typically developing controls: Longitudinal comparisons and clinically-meaningful changes over one year. PLoS Curr. 2013;5. |


