
Disorders of sterol biosynthesis
Abstract
As a major component of the cell membrane of most eukaryotic cells, cholesterol assumes crucial roles in myelination of the central and peripheral nervous systems; in the synthesis of bile acids, steroids, and vitamin D; in sterol signaling; and in embryonic development. Inborn errors of cholesterol biosynthesis comprise a heterogeneous group of disorders characterized by developmental delays, increased tissue levels of specific sterol intermediates, and complex malformations involving many organ systems. Here we present the basics of cholesterol biosynthesis and review the clinical phenotype, biochemistry, molecular pathogenesis, and management of the 8 known errors of sterol metabolism in humans: Antley-Bixler syndrome, Greenberg dysplasia, X-linked dominant congenital hemidysplasia with ichthyosiform erythroderma and limb defects (CHILD syndrome), X-linked dominant chondrodysplasia punctacta (Conradi–Hünermann syndrome; CDPX2), lathosterolosis, Smith-Lemli-Opitz syndrome, desmosterolosis, and methyl sterol oxidase (MSOM1/SC4MOL) deficiency.
1Sterols: Background
A eukaryotic cell is a highly complex structure with a true nucleus (hence, the term eukaryote), mitochondria, chromosomes, a cytoskeleton, and membrane-enclosed organelles, including, in more complex organisms, lysosomes, peroxisomes, and mitochondria.
Whereas the cell membrane of archaea contains branched isoprene chains in ether linkage to L-glycerol, the membranes of eukarya (and some bacteria) use unbranched fatty acids in ester linkages to D-glycerol in addition to sterols, and sphingolipids (Fig. 1). Interestingly, the evolution of eukaryotes and sterol synthesis coincided with—and possibly because of—the great oxygenation between 2.2 and 2.4 BYA.
Porifera (the least complex of the metazoans, “true animals”) appear to have had the capacity for sterol synthesis during the Cryogenian period, i.e. before 635 MYA. This inference is derived from the discovery of “abundant fossil” sterols in the South Oman Salt Basin, namely 24-isopropyl cholestanes derived from C30 (30-carbon) sterols produced by sponges (the “porifera”) [1], the only metazoans known to methylate the sterol side chain, a requirement for the formation of the 24-isopropyl cholestanes (Fig. 2). And, given that 1) the choanocytes (collar cells) of sponges are morphologically strikingly similar to choanoflagellates (neither containing chloroplasts), and 2) that colonial (plasmodial) choanoflagellates, such as Diaphanoeca, resemble free-swimming collar chambers of sponges [2], it is not surprising that both groups of organisms have a robust sterol metabolism, suggesting that the choanoflagellates and the fungi shared a common ancestor. Recently, Kodner et al. [3] showed that the unicellular choanoflagellate Monosiga brevicollis [3] has a C7-sterol reductase but no C22 sterol reductase, and therefore is unable to make genuine cholesterol (Fig. 3a, b). Thus, sterols were already present in the unicellular “half-sibs” of the porifera (morphologically least complex of the eumetazoans), in the fungi and plants able to synthesize ergosterol via C7 (Δ7-sterol reductase), and, ultimately, in the animals using that same phylogenetically ancient C7 sterol reductase, “DHCR7,”to convert 7-dehydrocholesterol to cholesterol. (Fig. 4a-c) Autosomal recessive inactivating mutations of DHCR7 in humans lead to Smith-Lemli-Opitz (RSH) syndrome, better known as Smith-Lemli-Opitz syndrome (SLOS), and, in thale cress (Arabidopsis thaliana), to the dwarf 5 mutant, which is unable to convert ergosterol (ergosta-5,7,22-trien-3β-ol) into brassicasterol [4] (Fig. 5). Choe et al. [4] show convincingly the homology-by-descent from a common ancestor of DHCR7 in humans, rat, chicken, yeast and A. thaliana. Indeed, this close genetic relatedness of proteins with DHCR7 activity together with the determination of the Arabidopsis DHCR7 cDNA sequence allowed Fitzky et al. [5], Wassif et al. [6] and Waterham et al. [7] to clone the human DHCR7 gene.
1.1Lipid rafts
The plasma membranes of eukaryotic cells are remarkably “heterogeneous” in their constituents and function; heterogeneous in quotes since that term, in genetics, refers to different causes of the same phenomenon, as illustrated by the identification of aneuploidy, teratogens, and single gene mutations as causes of histologically identical forms of holoprosencephaly (more on HPE below). Studies of cell membranes and their biophysical properties have a direct bearing on phylogeny of the more complex forms of life on earth. Analysis of the genomes of a dozen or so cyanobacteriae showed that none contain genes for sterol synthesis [8]. In contrast, plants synthesize sitosterol, animals cholesterol and fungi ergosterol as their dominant sterols (Fig. 6). The synthesis of all such sterols begins with the linear molecule squalene, which undergoes epoxidation and then cyclization by oxidosqualene cylase to form the plant sterol precursor, cycloartenol, and, in both fungi and animals, the corresponding sterol, lanosterol. After final conversion of 7-dehydrocholesterol (7DHC) to cholesterol, this tetracyclic alcohol assumes its crucial roles in CNS myelination; the synthesis of bile acids, steroids, and vitamin D; sterol signaling (Fig. 7a, b); and the structure and function of cytoplasmic organelles and plasma membranes. Mouritsen and Zuckermann [9] note that the “striking gradient” in cholesterol concentration from mitochondria (3 wt%), to endoplasmic reticulum (6 wt%) to Golgi (8 wt%) plus the low cholesterol concentration in mitochondria support Margulis’s [10] endosymbiosis hypothesis of mitochondria being phagocytosed ancient prokaryotes, which then evolved to their current respiratory processes for themselves and their host cells.
About half of lipid mass of plasma membranes consists of non-sterol lipids, including phosphatidylcholine, phosphatidylethanolamine, phosphatidyl serine and sphingomyelin and half of approximately an equimolar amount of cholesterol [11]. These constituents are organized asymmetrically into a lipid bilayer, with an outer leaflet comprised mainly of phosphatidylcholine and sphingomyelin and an inner leaflet of phosphatidylethanolamine, phosphatidylserine, and phosphatidylinositol. Cholesterol by itself is unable to organize into membranes, but, instead, is dispersed among the other lipids of bilayer membrane with its polar 3ß-hydroxyl group in close proximity to the anionic headgroups of the phospholipids (Fig. 8)
Important structures of biological phospholipid bilayer membranes, Lipid rafts, subserving both cell-cell signaling and endocytosis, consist of non-random clusters of cholesterol, sphingomyelin and glycolipids having a different melting point, greater solidity, and a more highly organized structure than the rest of the plasma membrane and subserving both cell-cell signaling and endocytosis. The physico-chemical properties of cell membranes prevent free diffusion across the lipid bilayer of charged molecules, such as salt ions, amino acids, large polar molecules (e.g., glucose) but allow passage into cells of other small molecules, such as gases and hydrophobic compounds (e.g. benzene), and some small polar molecules, such as ethanol and water. Carrier proteins span the cell membrane and allow passage of specific molecules into the cytoplasm through conformational changes, whereas channel proteins form open pores through the plasma membrane allowing influx and efflux depending on the size and charge of the molecules. Other important functions of plasma membranes include clathrin-dependent endocytosis, pinocytosis and clathrin-independent endocytosis through small caveolae (50–80 nm in diameter) organized by caveolin proteins. These proteins form a complex with cholesterol in lipid rafts inserted into the cell membrane and interact with other caveolins to form caveolae, which are involved in the uptake of specific molecules, including high density lipoproteins (HDLs) [11]. Caveolae are sufficiently well-defined structures to be seen in the plasma membrane by electron microscopy.
Given this essential role of cholesterol in determining the structure and function of cell membranes, it is surprising that human fetuses homozygous for null mutations in the terminal enzyme of cholesterol biosynthesis, DHCR7, can survive to term.
1.2Morphogenetic implications
The report in 1993 that individuals with SLOS have low serum cholesterol levels and up to 1000-fold increased levels of 7-dehydrocholesterol (7DHC) not only led to the identification of the genetic cause of SLOS, a deficiency of the terminal enzyme in the cholesterol biosynthetic pathway,7-dehydrocholesterol reductase (DHCR7), but also brought into focus the role of cholesterol metabolism in morphogenesis, as first studied in the relationship of cholesterol metabolism to the causes of holoprosencephaly (HPE) [12]. The full development of these studies showed that HPE could be caused both by genetic defects in the paracrine signaling protein, Sonic Hedgehog (SHH), and by changes in cholesterol metabolism that impair downstream SHH signaling (Fig. 9). Since then, it has been estimated that 4 to 5% of SLOS fetuses have HPE, usually, when tested, those with especially low tissue levels of cholesterol. Null mutations of Shh in the mouse can lead to severe HPE and defective induction of the floorplate of the entire neural tube [25, 26]. Similar inhibitory effects of low cholesterol levels on the actions of the SHH-related proteins, Desert Hedgehog (DHH) and Indian Hedgehog (IHH) probably also obtain and explain some of the extracranial malformation of SLOS. Inhibitors of cholesterol metabolism acting on DHCR7 (AY-9944) and 24-dehydrocholesterol reductase (triparanol) given to pregnant rats also cause lowered cholesterol levels and HPE in the fetuses and can be prevented by cholesterol feeding of the dams during the critical periods of cerebral lobar development [13]. Certain plant-derived toxins, such as cyclopamine and jervine from the corn-lily (Veratrum californicum), also cause HPE and inhibit SHH signaling, but probably not by depressing cholesterol synthesis, as once thought. Thus, 30 years after clinical beginnings in Madison [28] and the first teratological studies in 1964 by Roux and his coworkers in Paris [29], these two strands of investigation finally flowed together in the recognition that humans and other animals developed HPE because of defective SHH/Shh function.
Fig. 10 illustrates the 8 known errors of sterol metabolism in humans.
2Antley-Bixler syndrome
Antley-Bixler syndrome (ABS) is a rare heterogeneous syndrome characterized by a wide spectrum of congenital anomalies. The cardinal features of ABS include craniosynostosis, radiohumeral synostosis, multiple joint contractures, midface hypoplasia, proptosis, choanal atresia/stenosis, and visceral abnormalities, with craniosynostosis and radiohumeral synostosis being considered for many years the minimum diagnostic criteria for ABS [14–20]. Abnormal genitalia and/or complex steroid abnormalities are also found in a subset of ABS patients.
The first description of ABS was, apparently, by Lacheretz et al. [21] who reported a 10-year-old boy born to consanguineous parents, who had coronal and sagittal synostosis, bilateral radio-humeral synostosis, fused trapezium and trapezoid, club feet, complete absence of palmar flexion creases, absent gonion, malformed auricles, ectopic testes resistant to gonadotropin treatment, conductive hearing loss, and moderate intellectual disability. The eponym of the syndrome, however, derives from Ray Antley and David Bixler [14], who described a term female infant with striking features, including trapezoidocephaly, midface hypoplasia, low-set ears with prominent downfolded upper helices, mild micrognathia, bilateral choanal stenosis, multiple synostoses, bilateral hip dislocation, cutaneous syndactyly of second and third toes, short 4th metatarsals, tarsal and carpal fusions, overtubulation of tibiae and fibulae, and bilateral femoral fractures. Autopsy at 8 months after a respiratory infection showed patent foramen ovale and right atrial dilatation. The authors did not comment on the child’s genitalia. In a subsequent publication, DeLozier and co-workers described an additional patient with craniosynostosis, radiohumeral synostosis, femoral bowing, midface hypoplasia and multiple minor anomalies of the limbs [22]. In that report, the authors also described that testing of Antley and Bixler’s initial patient showed no evidence for a defect of steroid metabolism as the cause of the patient’s virilized external genitalia. Neither did the patient of DeLozier and co-workers [22]. Although both of these original cases were sporadic, the later report of Schinzel et al. [23] provided the first hints of heterogeneity and familial occurrence of ABS.
Currently, ABS with and without genital anomalies and/or disordered steroidogenesis are considered to be two separate genetic entities. ABS without genital ambiguity and/or impaired steroidogenesis (ABS2; OMIM 207410) is caused by dominant mutations in the fibroblast growth factor receptor 2 gene (FGFR2), located at 10q26 [24]. FGFR2 contains 16 exons that encode the transmembrane protein FGFR2, a tyrosine kinase receptor that plays an important role in craniofacial development. FGFR2 contains an extracellular signal peptide, three extracellular immunoglobulin-like domains (Ig1, Ig2 and Ig3), an extracellular acid box domain, a transmembrane segment and a cytoplasmic tyrosine kinase domain. Alternative splicing involving exons encoding the latter portion of the Ig3 domain results in two FGFR2 isoforms: KGFR, which expresses an epithelial cell lineage receptor, FGFR2IIIb, that preferentially plays a role in skin development, and BEK, a product of the mesenchymal cell lineage receptor, FGFR2IIIc, which is preferentially expressed during osteogenesis. [42] Most of the dominant missense mutations reported in ABS occur in the Ig3 domain of FGFR2, encoded by exons 8 and 10, mutations that have also been associated with Crouzon and Pfeiffer type 2 craniosynostosis syndromes [25, 26]. A viable Fgfr2IIIc knockout mouse manifests synostosis of skull base sutures, craniosynostosis and dwarfism [27], while gene targeted fgfr2 knockouts are embryonic lethals and show trophoblast defects, abnormal placental formation, and defective limb bud development [28]. FGFR2 mutations have not been associated with steroid abnormalities or genital anomalies in humans or mice. The laboratory diagnosis of ABS without genital anomalies and/or disordered steroidogenesis is made by mutation analysis of FGFR2.
Based on the observation that lymphoblasts from ABS patients with ambiguous genitalia and/or disordered steroid metabolism (ABS1; OMIM 201750) show functional deficits in the sterol enzyme, lanosterol demethylase (CYP51A1), this form of ABS was shown to be caused by genetic deficiency of the CYP51A1-associated P450 cytochrome oxidoreductase (POR) gene [29], located at 7q11.23. POR, a flavoprotein with 680 amino acids localized to the endoplasmic reticulum (ER), catalyzes the transfer of electrons from reduced NADPH to all microsomal P450 enzymes, including the steroidogenic enzymes CYP17A1 (17-alpha-hydroxylase and 17, 20-lyase activities), CYP21A2 (21-hydroxylase activity), and CYP19A1 (aromatase activity) and lanosterol 14α-demethylase (CYP51A1), a key enzyme in the cholesterol biosynthetic pathway [30]. Because POR regulates multiple P450 enzymes, deficient activity of POR causes abnormalities in both sterol and steroid metabolism. Abnormal lanosterol 14α-demethylase activity is thought to be responsible for the skeletal alterations identified in POR-mutated ABS patients, and inhibition of CYP51A1 has been implicated as the cause of the ABS-like skeletal phenotype associated with exposure to high doses of fluconazole during early pregnancy [30–34]. Nonetheless, the mechanism by which POR deficiency causes the skeletal anomalies remains unproven. Impaired steroidogenesis due to POR deficiency causes both underdevelopment of the genitalia in males due to deficient activity of 17,20 lyase (CYP17A1) and virilization in females due to 21α-hydroxylase or aromatase (CYP19A1) deficiency. It is noteworthy that male undervirilization is a less consistent feature than female virilization, due to the major alternative androgen biosynthesis pathway, “the backdoor pathway,” responsible for production of dihydrotestosterone (DHT) in the human testis [35]. In this pathway, 17α-hydroxyprogesterone or progesterone is converted to 5α-pregnane-3α,17α-diol-20-one via 5α- and 3α-reductases and sequentially converted to androsterone by CYP17A, which is then transformed to DHT by 3α-hydroxysteroid dehydrogenase and 17β-hydroxysteroid dehydrogenase. This contrasts the principal pathway, in which DHEA, androstenedione and testosterone serve as intermediates in the synthesis of DHT [reviewed in [36]]. Other consequences of abnormal fetal steroid metabolism in POR-deficiency include reversible gestational maternal virilization and low/undetectable serum unconjugated estriol (uE3) [37, 38]; primary amenorrhea and enlarged cystic ovaries in some POR-deficient females; and failure of pubertal development and fertility in ABS1 patients from both sexes [39]. POR mutations may also explain a case of ABS1 in a newborn girl with virilized genitalia and disordered steroidogenesis, whose mother had a pregnancy luteoma and virilization [40].
Thus far, more than 60 pathogenic mutations of POR have been reported, including missense and frameshift mutations, indels and splice-site variations [41]. Of these, two missense mutations, R457H and A287P, are common, accounting for 50% and 60% of mutant alleles among individuals of Japanese and European ancestry, respectively [42]. Phenotype correlation exists for various POR mutations. For instance, R457H has been associated with fewer severe skeletal abnormalities and less frequent adrenal crises, while the A284P mutation causes more severe skeletal anomalies and a greater disturbance of steroidogenesis [43]. The wide phenotypic spectrum of POR-deficiency might be explained by mutation-related structural changes in the POR protein that variably affect protein-protein interactions between POR and its client enzymes. This genetic variability in POR could represent another aspect of the selective pressure to maintain a relatively high level of individual variability in p450-linked xenobiotic metabolism [44].
The mechanism underlying the abnormal morphogenesis of ABS1 is still under debate, in part because of the multiple P450 enzymes potentially affected by insufficient POR activity, including pathways in retinoic acid, cholesterol, and arachidonic acid metabolism. In mice, Por knockout leads to embryonic lethality and severe maldevelopment of the neural tube, heart, eye, and limbs [45, 46], while liver-specific knockout of Por in mice leads to a significant decrease of hepatic drug metabolizing ability, with no morphological or reproductive differences between the wild-type and liver-specific Por knockout mice [47, 48].
A biochemical diagnosis of POR-deficiency ABS is most easily made by identifying a characteristically abnormal pattern of urinary steroid metabolites, including increased levels of corticosterone, deoxycorticosterone, 21-deoxycortisol, and 17 alpha-hydroxyprogesterone and decreased levels of cortisol, dehydroepiandrosterone (DHEA), DHEA sulfate and androstenedione In plasma, the level of cortisol tends to be decreased and that of ACTH increased. Under conditions of severe cholesterol depletion, cultured lymphoblasts and fibroblasts from ABS1 patients will show increased levels of lanosterol and dihydrolanosterol, reflecting impaired function of POR-dependent CYP51A1, but this is a cumbersome and not entirely specific method to use for diagnosis. Today, mutation analysis of POR is the most common method for diagnosing ABS1.
Differentiating between the two forms of ABS is relevant, because the natural history, clinical manifestations and treatment for the two diagnoses differ substantially. For example, patients with POR deficiency can die suddenly and are at risk for developing adrenal insufficiency or an Addisonian crisis during surgery or illness if not provided with adequate steroid hormone supplementation. Other important considerations include the management of a malformed airway and orthopedic treatment of congenital deformities and fractures in ABS1.
Figures 11–14 shows clinical and radiographic features of a patient with ABS.
3Greenberg dysplasia
Hydrops-ectopic calcification-“moth-eaten” skeletal dysplasia (HEM skeletal dysplasia, Greenberg dysplasia, OMIM 215140) was first described as a lethal autosomal recessive syndrome characterized by hydrops fetalis, intrauterine growth retardation, severe tetrabrachymelia, postaxial polydactyly, narrow thorax, and chondro-osseous dystrophy [42, 49–55]. Radiographs demonstrated a mottled, “moth-eaten” appearance of bones with multiple ectopic ossification centers. Other radiologic findings include deficient ossification of skull, depressed nasal bridge, midface hypoplasia, prominent supraorbital ridges, micrognathia, elongated clavicles, brachyplatyspondyly, hypoplasia of vertebral bodies, and abnormal development of vertebral pedicles and laminae [49, 51, 52]. Histologic examination of cartilage and bones shows disorganization of cartilaginous architecture with lack of chondrocyte columns, abrupt bone-cartilage transition due to deficient growth plate formation, zones of endochondral ossification, multifocal deposits of calcification on cartilage and bone matrix, and, most distinctively, infiltration of cartilage and bone by mesenchymal tissue overgrowth. The internal organs usually lack congenital anomalies, although marked extramedullary hematopoiesis secondary to marrow fibrosis and absent/deficient hematopoietic activity is also a characteristic of the disorder[49, 52, 53].
Greenberg dysplasia is caused by mutations in LBR, a gene encoding a protein first identified as a nuclear Lamin B receptor but later found to have dehydrosterol-14-redutase (DHCR14) activity, an essential step in cholesterol biosynthesis [56]. The explanation for the apparent dual function of the 615 amino acid LBR protein is revealed in the fine analysis and its structure [57]. Of the 13 LBR exons, exons 1–4 encode the Lamin B receptor amino-terminus domain of the protein, whereas exons 5–13 encode a polypeptide with high homology to TM7SF2, a microsomal protein with DHCR14 activity that is heavily expressed in liver and adrenal, two tissues with high rates of cholesterol synthesis [58, 59]. The existence of an especially large intron connecting these two segments of LBR is evidence that LBR is a fusion protein. The nucleoplasmic, amino-terminal domain of LBR interacts with nuclear components, such as chromatin, lamins, double-stranded DNA and histones and, among other functions, is involved in the morphological and functional development of granulocytes [59, 60], as reflected in the high expression of LBR in myeloid and erythroid bone marrow lineages [55, 56, 61–64]. In Greenberg dysplasia, there is sufficient reduction in total 14-sterol reductase activity that small amounts of the cholesterol precursor, cholesta-8,14-dien-3β-ol [55, 56, 65], can accumulate in cartilaginous tissues and cultured skin fibroblasts. Because of the only slightly increased levels of cholesta-8,14-dien-3β-ol and even smaller amounts of cholesta-8,14,24-trien-3β-ol, several authors once speculated that the partial loss of DHCR14 activity had little if any role in the pathophysiology of Greenberg dysplasia [66].
Thus far, 4 pathogenic LBR mutations causing Greenberg dysplasia have been reported [54–56]. Waterham et al. [56] described a homozygous 7-base-pair substitution (c.1599–1605 TCTTCTA⟶CTAGAAG) in exon 13 of LBR in an affected 18-week fetus of consanguineous Turkish parents. Sterol studies showed increased levels of cholesta-8,14-dien-3β-ol in cultured skin fibroblasts. Functional complementation studies in Greenberg cells with control LBR cDNA confirmed the role of deficient LBR in the pathogenesis of Greenberg. Subsequently, Konstantinidou et al. [54] described three affected fetuses of a consanguineous Greek couple. LBR mutation analysis showed a missense mutation in exon 13, c.1639A>G (p.N547D), which was homozygous in tissues of the third fetus and heterozygous in the parents. Recently, Clayton et al. [55] studied three fetuses with Greenberg dysplasia and found a homozygous frameshift mutation, c.1492delT (p.Y468TfsX475) in a 16-week of gestation male fetus (fetus A), product of consanguineous parents; a compound heterozygous mutation, c.32delTGGT (p.V11EfsX24, maternal), and c.1748G>A (R583Q, paternal), in a 14-week female fetus [fetus B, previously described by Offiah and co-workers [67]]; and a homozygous missense mutation, c.1639A>G (N547D), in a fetus of consanguineous parents (fetus C). Sterol studies, available only for fetus B, showed increased tissue levels of cholesta-8,14-dien-3β-ol.
Because the levels of cholesta-8(9),14-dien-3β-ol and cholesta-8(9),14,24-dien-3β-ol in fetal tissues are relatively low and measurement in cultured fibroblasts is both difficult and could yield falsely negative results, sequencing of LBR in fetal tissues or cultured cells is the preferred method of diagnosis of Greenberg dysplasia.
LBR mRNA is expressed in human fibroblasts, lymphoid cells, osteoblast-like cells and osteoclasts [55]. Homozygous deficiency of LBR is lethal in humans but not necessarily in mice The Lbr deficient mouse (ichthyosis, ic/ic) shows alopecia, ichthyosis, hydrocephalus, deficient growth, variable syndactyly, misshapen granulocyte nuclei and increased perinatal mortality. Hypotheses to explain the survival of some Lbr mutant mice include 1) greater enzymatic redundancy of Lbr and Dhcr14 in the murine cholesterol biosynthetic pathway [68, 69] and 2) transplacental transfer of cholesterol in mice, which is quantitatively much greater in mice than in humans. Human LBR is homologous to Erg24 of fungi, which also has both C14 sterol reductase and Lamin B receptor activities. Erg24 yeast mutants have a lethal outcome or weak viability, depending on their genetic backgrounds [70]. In contrast, LBR in Drosophila [71] lacks C14 sterol reductase activity.
In addition to LBR, identified as a protein of the nuclear envelope, humans also have 3β-hydroxysteroid-Δ14-reductase activity expressed in the endoplasmatic reticulum protein, DHCR14, encoded by the transmembrane 7 superfamily, member 2 (TM7SF2, DHRC14) gene [72]. No mutations in TM7SF2 have been detected in patients with Greenberg dysplasia, nor have any human conditions yet been attributed to a deficiency of TM7SF2/DHCR14. To what degree the loss of LBR’s 14-sterol reductase activity contributes to the abnormal development of Greenberg fetuses has been controversial because of the relatively minor amounts of 14-unsaturated sterols found in the most severely affected tissue, i.e., bone. The widely disparate levels of mRNA expression for these two proteins in different tissues—LBR high in bone marrow-derived cell lineages and fetal liver and DHCR14 highest in liver and adrenal—and the lack of adequate biochemical analysis of human Greenberg tissues has limited our understanding of the individual roles of LBR and DHCR14 in human fetal development. Furthermore, studies of LBR and DHCR14-knockout mice have not been able to answer questions about the significance of abnormal sterol metabolism in human LBR-deficiency, possibly because studies indicate that 3 out of 4 murine 14-sterol reductase alleles must be inactivated before 14-unsaturated sterols accumulate in tissues [66].
No treatment for Greenberg dysplasia is available to date.
In addition to Greenberg dysplasia, certain mutations of LBR in the heterozygous state cause Pelger-Huët anomaly (PHA [OMIM 169400]) [73], an autosomal dominant hematological disorder characterized by granulocytes with hypolobulated nuclei and abnormal chromatin structure. PHA homozygotes exhibit a phenotype distinct from Greenberg dysplasia featuring neutrophils with misshapen nuclei, variable skeletal anomalies unlike those of Greenberg dysplasia, and developmental delays [74, 75]. Most cases of homozygous PHA have been diagnosed by demonstrating PHA in both parents combined with finding the more severe neutrophil abnormality in the propositus. However, one patient was shown to be homozygous for an intronic deletion, IVS12–5–10del, leading to a splice variant cDNA lacking exon 13, although a trace amount of apparently normal LBR protein in the homozygote was found by Western blot. Despite the physical and genomic separation of the functional units of LBR, Lamin B binding and sterol reductase, the known pathological mutations for Greenberg dysplasia vs. PHA show no preference for exons encoding their segments of the protein. Rather, most mutations associated with either isolated PHA or Greenberg dysplasia are in the 14-sterol reductase-related exons or introns between exons 9 and 13. Moreover, whereas all 5 known LBR mutations associated with Greenberg dysplasia are either missense or deletion-frameshift mutations, 6 of 12 mutations found in PHA alone are splice site variants. In view of the small amount of residual normal-sized protein found in cells of the IVS12–5–10del exon-skipping homozygote [93], LBR missense and splice site mutations associated with persistence of small amounts of 14-sterol reductase activity in chondrocytes might prevent the more severe skeletal deformities of Greenberg dysplasia, since chondrocytes have a limited ability to import systemic cholesterol synthesized via TM7SF2/DHCR14 [76]. In this manner, chondrocytes in the severe but remitting chondrodystrophy associated with compound heterozygosity for LBR mutations R76X and N547S could have just enough 14-sterol reductase activity to prevent classic Greenberg dysplasia. With the gradual age-dependent reduction in growth velocity, there would be a proportional decrease in the relative deficit in cholesterol synthesis, eventually allowing more normal chondrocyte growth and maturation. A change in the affected patient’s bone disease in the future during the progress of puberty should provide additional insight into the pathogenesis of the boy’s chondrodystrophy. Of interest with regard to the different pathogenesis of PHA vs. Greenberg dysplasia are the studies of Clayton and colleagues [75], who show biochemically and histologically the separate effects of disruption of the Lamin B binding and sterol reductase portions of the LBR fusion protein. They offer strong evidence that Greenberg chondrodystrophy results from the loss of the sterol reductase activity of LBR, not its nuclear protein binding functions.
4Congenital hemidysplasia with ichthyosiform erythroderma and limb defects (CHILD syndrome)
X-linked dominant, congenital hemidysplasia with ichthyosiform erythroderma and limb defects (CHILD syndrome, OMIM 308050) [77] is a rare disorder of cholesterol biosynthesis caused by mutations of the gene NSDHL, which encodes the NAD(P)H steroid dehydrogenase-like protein, sterol-4α-carboxylate 3β-hydroxysteroid dehydrogenase, located at Xq28. NSDHL is an integral membrane protein located in the endoplasmic reticulum (ER) and at the surface of lipid droplets. It has an N-terminal NADH cofactor binding site, a central Tyr–X3–Lys active site motif common to all known 3β-hydroxysteroid dehydrogenases, and a C-terminal amino acid sequence “KKXX,” which acts as a consensus signal sequence for retrograde transport of ER membrane proteins from the Golgi [reviewed in [78]]. In concert with C4-sterol methyl oxidase (SC4MOL) and a 3-ketosteroid reductase (17β-HSD), NSDHL catalyzes the sequential removal of two C4-methyl groups from 14-dehydrolanosterol in the post-squalene cholesterol biosynthetic pathway [79]. Pathogenic NSDHL mutations causing CHILD syndrome are distributed over all 8 exons of the gene and include missense mutations, nonsense mutations, large deletions, frameshift insertions, and splicing mutations [80–87]. Missense mutations affecting highly conserved amino acids, particularly c.314C>T (p.A105V), c.613G>A (p.G205S), c.1046A>G (p.Y349C), and c.370G>A (p.G124S) are most frequently identified, [85]. Most NSDHL mutations lead to near-complete or total loss of functional NSDHL protein, which, with rare exceptions, is not manifest in plasma of affected females as increased levels of 4-methyl and 4,4-dimethyl sterol intermediates, as it is cultured fibroblasts and lymphobasts [79, 88]. The usual absence of sterol abnormalities in plasma is presumed to be due to the expression of the normal NSDHL allele in cells of affected heterozygous females via either skewed Lyonization in liver (which governs plasma sterol levels) or simple systemic biochemical compensation. Among manifesting CHILD syndrome females, there appears to be no phenotype-genotype correlation, although hypomorphic NSDHL mutations causing a different syndrome in hemizygous males are known. Most cases of CHILD syndrome reported to date have been sporadic, although rare familial cases with mother to daughter transmission have been described [85, 89].
A biochemical diagnosis of CHILD syndrome can be made by sterol analysis of fibroblasts or lymphoblasts grown in lipid-depleted culture medium to activate fully endogenous cholesterol synthesis. This yields, in lymphoblasts and in fibroblasts from both affected and unaffected skin, a diagnostic pattern of metabolites by GC/MS. These include increased levels of 4-methyl- 4,4-dimethyl-, and, pathognomonically, 4-carboxysterol intermediates. However, direct sterol analysis of flakes or scrapings from the characteristic ichthyosiform skin lesions of CHILD syndrome is simpler and rapid and always yields a diagnostic pattern of sterol intermediates in NSDHL mutation heterozygotes. Such skin testing can be used to guide mutation testing when assessing the cause of an atypical skin lesion or an isolated skin lesion in the absence of skeletal features of heterozygous NSDHL deficiency. However, because of the limited availability of biochemical testing for NSDHL deficiency in skin or cultured cells, mutation analysis and deletion/duplication testing of NSDHL [reviewed in [90]] is currently the favored method for diagnosis.
The phenotype of NSDHL deficiency in heterozygous females includes radiographic stippling of the epiphyses (typically asymmetric in distribution), unilateral or occasionally bilateral ichthyosiform nevus, ipsilateral limb defects, and less common and variable visceral malformations that usually are ipsilateral to the skin and skeletal anomalies [77, 80, 83, 91]. The ichthyosiform nevus of CHILD syndrome is often large; sharply delimited at the midline; characterized by wax-like scaling on top of a warty, erythematous base; and most commonly affecting the right side of the body [77], although some skin lesions occur on the contralateral side, and, in rare individuals, bilaterally [92]. The skin lesions are frequently present at birth and often persist throughout life, but they can also recede or appear in previously unaffected skin, especially after surgery or injury to the skin of the affected side. The lesions often involve the skin folds (ptychotropism) [93], tend to spare the face, and generally do not follow the lines of Blaschko, as would be expected for an X-linked dominant skin disease [94]. Histologic examination of areas of the ichthyosiform nevus shows papillomatosis, hyperkeratosis (with ortho- and parakeratosis), acanthosis, subcorneal accumulation of neutrophils, and, in some cases, accumulation of foamy histiocytes in the dermal papillae (verruciform xanthoma) [77, 95, 96]. Abnormal lamellar granules have been identified by electron microscopy in the granular layer cells of the epidermis of CHILD patients, suggesting metabolite-induced toxicity [95–97]. The clinical presentation in affected females can be variable, preumably due to differential Lyonization of the normal and mutant NSDHL alleles. However, because the distribution of the skin and skeletal lesions is unlike that of most other X-linked genodermatoses, and the biochemical abnormality can be expressed in fibroblasts from the normal side of the body (R. I. Kelley, unpublished), the timing and mechanism of the asymmetries in CHILD syndrome remains to be explained. Although NSDHL mutations typically result in very early lethality in males, rare surviving males reported with CHILD syndrome have had phenotypic findings of affected females [98]. Survival among 46, XY NSDHL-deficient males has been attributed to an early postzygotic (somatic) mutation in NSDHL or a hypomorphic NSDHL mutation, a phenomenon that has been documented in the related and more common X-linked genodermatosis, Conradi–Hünermann syndrome (chondrodysplasia punctacta; CDPX2; OMIM 302960), caused by mutations in EBP. A constellation of clinical features similar to CHILD syndrome, including right-sided hemidysplasia, occurs in rare female heterozygotes with X-linked dominant CDPX2. [65, 99], The almost exclusively unilateral distribution of abnormalities in CHILD syndrome versus the nearly universal bilateral pathology of CDPX2, both otherwise classic X-linked dominant disorders with high male in utero lethality, remains to be explained The pathogenesis of the anomalies of CHILD syndrome remains unknown. Whereas CHILD syndrome truncation limb defects similar to those of the Shh KO mouse probably are caused by focal areas severe cholesterol deficiency in cartilaginous limb anlage Lyonized in favor of the mutant NSDHL allele, the proliferative skin lesions of CHILD syndrome cannot be explained by impaired SHH function. However, two C4-dimethyl sterol intermediates that accumulate behind the NSDHL enzymatic block are the growth-stimulating follicular and testicular meiosis activating sterols. Therefore, some authors have proposed that one or both of these steroid-like C29 sterols accumulate locally in fibroblasts and stimulate the hyperproliferative growth of the overlying epidermis. Indeed, the observation that a new lesion can erupt at the site of surgery, progressively expand outward argues that fibroblasts stimulated to grow and, thereby, to activate cholesterol biosynthesis produce locally high levels of the meiosis activating sterols, which induce the hyperproliferative ichthyosiform epidermal skin lesions of CHILD syndrome. Interestingly, the murine equivalents of human NSDHL deficiency, the X-linked male-lethals bare patches (Bpa) and striated (Str) mutations in Nsdhl [79], have phenotypes more like heterozygous females with EBP-deficiency CDPX2 than CHILD syndrome. Indeed, both mouse models were initially studied as possible murine equivalents of CDPX2. However, sterol analysis of plasma and fibroblasts of heterozygous female Bpa mice show a sterol profile essentially identical to that of human CHILD syndrome fibroblasts with normal levels of the characteristic sterols of CDPX2, 8(9)-cholestenol and 8-dehydrocholesterol. Consistent with Str’s appearing to be a hypomorphic form of Bpa limited to having mosaic coat striations following the lines of Blaschko, the sterol profile of Str females is the same as, but less severe than, the Bpa sterol profile.
In view of the possible role of C4,4’-dimethylsterols in inducing the proliferative ichythyosiform nevus of CHILD syndrome, Paller et al. [100] and Liu et al. [101] showed that, respectively, topical lovastatin+cholesterol and topical ketoconazole alone cause almost complete resolution of the skin lesions in patients with CHILD syndrome caused by heterozygosity for pathogenic mutations in NSDHL [100, 101]. Both treatments are expected to lower the levels of C4,4’-dimethylsterols in skin by upregulating the expression of residual NSDHL activity, in the case of lovastatin treatment, and, in the case of ketoconazole, by partial inhibition of the action of lanosterol demethylase, thereby directly reducing the levels of the C4,4’-dimethylsterols. Because many NSDHL mutations are predicted to leave little or no residual NSDHL activity, ketoconazole would be the preferred treatment for most patients with NSDHL-related skin lesions.
Although human NSDHL deficiency for many years was assumed to be male-lethal in utero, hypomorphic NSDHL mutations have been found to cause CK syndrome (CKS, OMIM 300831), a recently described X-linked recessive intellectual disability disorder characterized by seizures, microcephaly, thin body habitus, and cortical malformations [102]. CKS males and their mothers exhibit sterol abnormalities in cultured cells similar to, but milder than, those reported for allelic CHILD syndrome. Despite their enzymatic deficiency in cholesterol synthesis, CKS patients tend to have normal plasma cholesterol and steroid hormone levels and normal lipoprotein profiles. Thus, the diversity of congenital anomalies in NSDHL deficiency ranges from limb loss secondary to locally severe cholesterol deficiency in mutant allele-expressing limb buds, to hyperplastic ichthyosiform skin lesions, and to cerebral dysgenesis from an as yet unidentified pathophysiology [103].
5X-Linked dominant chondrodysplasia punctata (Conradi–Hünermann syndrome; CDPX2; EBP mutations)
X-linked dominant, chondrodysplasia punctacta (Conradi–Hünermann syndrome; CDPX2, OMIM 302960) is a rare disorder of cholesterol biosynthesis caused by mutations of the gene, EBP, encoding the emopamil binding protein [104, 105] and located at Xp11.22-p11.23 [106]. EBP contains 5 exons, the last four of which encode a protein initially defined by its ability to bind the phenylalkylamine Ca+2 antagonist, emopamil. EBP is an integral membrane protein, located mainly in the endoplasmic reticulum and having four predicted transmembrane domains and a high content of aromatic amino acids [107]. Amino acids within transmembrane domains 2–4 and from the cytoplasmatic linker domain are essential for enzymatic activity of EBP in cholesterol biosynthesis [108]. The protein acts as a 3β-hydroxysteroid-Δ8, Δ7-isomerase converting cholest-8(9)-en-3β-ol (8(9)-cholestenol) to cholest-7-en-3β-ol (lathosterol) as the penultimate step in post-squalene cholesterol biosynthesis [109] (Fig. 10). Thus far, over 70 pathogenic EBP mutations causing CDPX2 have been reported with no clear genotype–phenotype correlation [110]. Most mutations are single nucleotide substitutions, although a relatively high proportion of EBP mutations associated with typical CDPX2 are stop mutations or other mutations predicted to abolish all or most enzyme activity. The mutations are distributed in all exons of the gene, but mostly affect exons 2 and 4 [104, 111, 112]. However, because the GC-enriched exon 5 is difficult to amplify, mutations in the exon 5 could be underdetected. Deficient activity of 3β-hydroxysteroid Δ8, Δ7 isomerase leads to an accumulation of 8(9)-cholestenol and 8-dehydrocholesterol, the latter compound most likely arising from the 8(9)-cholestenol being a substrate for lathosterol 5-desaturase (SC5D).
The laboratory diagnosis of CDPX2 is made by sterol analysis of plasma and/or tissue by GC/MS to demonstrate the increased levels of 8(9)-cholestenol and 8-dehydrocholesterol, and by mutation analysis of EBP. Prenatal diagnosis by sterol analysis is available, although direct mutation analysis of amniotic cells is now sometimes performed as a confirmatory test following sterol analysis.
The phenotype of heterozygous EBP deficiency, CDPX2, includes radiographic stippling of the epiphyses (typically asymmetric in distribution), a depressed nasal bridge, scoliosis from vertebral defects, short stature, congenital cataracts, and patchy skin changes, often with mosaic patterning observing the lines of Blaschko, including ichthyosis, follicular atrophoderma, pigmentary dysplasia, and cicatricial alopecia [104, 111, 113–115]. Histologic examination of areas of ichthyosis in infancy shows acanthosis, hyperkeratosis, and parakeratosis, with calcium in the stratum corneum and follicular plugging. The follicular plugging is a distinctive histopathologic feature of CDPX2 not seen in other ichthyoses [116]. Abnormal lamellar granules have been identified by electron microscopy in the granular layer cells of the epidermis of a newborn girl with CDPX2 [117]. The marked clinical variability of involvement of affected heterozygous females is presumed to be due to Lyonization.
The distinctive restriction of most pathology of CDPX2 and CHILD syndrome heterozygotes to the skeleton and epidermis is more easily explained than the pathophysiology of their very different skin lesions. Because liver has a remarkable capacity to increase cholesterol synthesis by coordinated upregulation of the entire cholesterol biosynthetic pathway, even liver Lyonized such that only 10 or 20% of hepatocytes express the normal EBP or NSDHL allele should still be able to export enough LDL cholesterol to supply the needs of most cells and tissues in the growing fetus, as long as cells expressing the abnormal allele are able to transport cholesterol. The focal death of chondrocytes causing the calcific stippling is most likely explained by the inability of mutant NSDHL or EBP allele-expressing chondrocytes to compensate for their loss of cholesterol-synthesizing capacity by importing LDL cholesterol through an increasingly thick surrounding cartilage matrix. Thus, initially expanding local populations of chondrocytes evolving from single cells expressing the mutant EBP or NSDHL allele eventually die and the mutant lineages are lost, leaving a reduced number of viable chondrocytes and, as a result, bony hypoplasia. However, as noted above, total hemidysplasia, amelia, and, more rarely, dysgenesis of solid tissues are unique phenomena, that, although also probably related to early Lyonization, are more difficult to explain mechanistically.
Most EBP mutations causing typical CDPX2 result in early in utero lethality in males. Rare surviving 46,XY males with a CDPX2 biochemical profile usually have apparently hypomorphic EBP mutations, which typically have no or minimal physical effects in heterozygote mothers, most of whom have at most only mild increased levels of 8(9)-cholestenol and 8-dehydrocholesterol. In addition, 47,XXY males and 46,XY males with apparent post-zygotic EBP mutations have been described. As expected, such mosaic heterozygous EBP males clinically resemble CDPX2 females [138–141].
Recently, Furtado et al. [118, 119] reported a novel hemizygous, non-conservative EBP mutation (c.141G>T, p.W47C) in two unrelated males who died within the first months of life and displayed a unique and reproducible pattern of physical features differing from those of CDPX2. The distinctive phenotype is characterized by Dandy-Walker variant; hydrocephalus; dysplasia of the corpus callosum; cataracts; cardiovascular, craniofacial, and skeletal anomalies; 2,3-toe syndactyly; collodion skin; cryptorchidism; and no epiphyseal stippling and occurred in multiple branches and generations in one of the kindreds as an X-linked, male-lethal disorder with no clinical manifestations in obligate carrier females [118]. The biochemical phenotype was similar to that of CDPX2, with markedly increased levels of 8(9)-cholestenol and 8-dehydrocholesterol.
A similar constellation of clinical and laboratory features was described by Kelley et al. [120] in four previously unreported affected males with increased levels of 8(9)-cholestenol and 8-dehydrocholesterol. Although EBP sequence analysis of these patients was not reported at the time, mutations in EBP were subsequently identified in 3 of 4 of the affected males (R. I. Kelley, unpublished). These severe findings contrast the milder but not dissimilar phenotype of the first reported hemizygous EBP male [121], who carried the missense mutation, c.53C>T; p.L18P.
Recently, Bode and colleagues reported an almost 3-year-old boy with craniofacial anomalies, tetralogy of Fallot, pterygial contractures of the elbows, short and broad fingers, bilateral single palmar creases, cutaneous syndactyly of the second and third toes, cryptorchidism, parchment-like ichthyosiform skin and markedly increased plasma level of 8(9)-cholestenol and 8-dehydrocholesterol. Mutation analysis disclosed a novel, non-mosaic c.356T>G, p.V119G, EBP mutation [122].
The different phenotypes associated with heterozygous and hemizygous human EBP mutations is reproduced in the mouse Ebp mutant, “Tattered” (Td), and resemble in many ways the “bare patches” (Bpa) and “Striated” (Str) mice, who carry mutations in the other X-enzyme of sterol biosynthesis, Nsdhl. Whereas female Tattered heterozygotes develop dermal defects delimited by the lines of Blaschko, similar to CDPX2, hemizygous Tattered males have a phenotype of small size, edema, craniofacial anomalies, rhizomelia, and retarded ossification and die in utero [79, 105, 123].
The pathogenesis of classical, asymmetric CDPX2 in females and the more severe, symmetric cerebral and systemic abnormalities in EBP mutation hemizygotes doubtless reflects the different roles of Lyonization and toxic effects of increased levels of one or both diagnostic sterols, 8(9)-cholestenol and 8-dehydrocholesterol. However, that even much higher levels 8-dehydrocholesterol occur in SLOS, which shares almost no features with hemizgous EBP deficiency, argues that 8(9)-cholestenol is the principal damaging metabolite in EBP deficiency syndromes. The phenotype of Ebp female Tattered mouse is similar to that of heterozygous females with CDPX2 and another X-linked mouse mutation, “bare patches” (Bpa), due to Nsdhl deficiency [101]. Consistent with their enzymatic defect in the sterol-Δ8, Δ7-isomerase, female heterozygote Td mice also have increased levels of 8(9)-cholestenol and 8-dehydrocholesterol, similar in amounts to those found in human CDPX2 patients.. The Td mutation causes late prenatal death in hemizygous males with a phenotype of modest growth retardation, generalized edema, craniofacial anomalies, severe rhizomelic chondrodystrophy, and delayed ossification [79, 105, 123].
No treatment for this non-progressive disorder is available to date apart from standard orthopedic and dermatologic management of the associated anomalies. However, the recent success in treating the skin lesions of NSDHL-related CHILD syndrome [100, 101] should also obtain with EBP skin lesions, which sometimes can be quite severe and persist in an active form beyond the first month of life.
6Lathosterolosis
Lathosterolosis (OMIM 607330) is a rare autosomal recessive inborn error of cholesterol biosynthesis caused by mutations of the sterol-C5-desaturase-like (SC5DL, SC5D) gene, located at 11p23.3 [124, 125]. SC5D contains 5 exons that encode the enzyme, 3β-hydroxysteroid-Δ5-desaturase (sterol-Δ5-desaturase, SC5D) [126]. The SC5D gene is homologous to the ERG3 (C-5 desaturase) gene of fungi, the third terminal enzyme of fungal ergosterol synthesis, which introduces a C5 double bond into the B ring of ergosterol [124, 127]. In humans, SC5D catalyzes the conversion of cholest-7-en-3β-ol (lathosterol) into 7-dehydrocholesterol (7DHC) in the post-squalene cholesterol biosynthetic pathway (Fig. 1). SC5D is located in the endoplasmatic reticulum and is predicted to be a 299-amino-acid protein with an iron binding domain, four transmembrane domains, and an oxido-reductase domain containing an NAD/NADP binding motif. The 204-amino acid middle portion of the enzyme shows 48%, 46%, and 22% amino acid identity to the corresponding regions of the enzyme in Candida glabrata, Saccharomyces cerevisae and Arabidopsis thaliana, respectively [124]. SC5D mRNA expression occurs in most human tissues and shows notably high levels of expression in liver, testis, prostate, spinal cord, and peripheral nerve [126].
The clinical phenotype of lathosterolosis is characterized by multiple congenital anomalies and signs of lysosomal storage. The diagnosis is made by demonstrating increased levels of lathosterol in plasma, tissue, or cultured cells, and by mutation analysis of SC5D.
Thus far, only three lathosterolosis patients have been reported [128–130]. The first, in 2002, was a 2-year-old girl with multiple congenital malformations, liver disease, developmental delays and minor facial anomalies resembling those of SLOS [128]. She presented at birth with microcephaly, receding forehead, micrognathia, anteverted nares, highly arched palate, postaxial polydactyly of the left foot, and polysyndactyly. A liver biopsy demonstrated intrahepatic cholestasis with irregular bile ducts and cholangiolitis. The patient also had a bilobed gall bladder, a cleft 8th thoracic vertebra, sensorineural hearing loss, and normal brain magnetic resonance imaging (MRI) apart from micrencephaly. Plasma sterol profiling (GC/MS) demonstrated an increased lathosterol but normal cholesterol level. SC5D deficiency was confirmed by studies of cholesterol biosynthesis and direct enzymatic assay in fibroblasts. Sequence analysis of SC5D showed compound heterozygosity for mutations c.88G>A (maternal, p.R29Q) and c.632G>A (paternal, p.G211D). The heterozygous parents had slightly increased lathosterol levels in plasma and cultured cells. At age 6 years, the patient had bilateral lens opacities that progressed to a unilateral cataract. By age 7, she had developed severe liver fibrosis, portal hypertension, cholestasis, and generalized osteoporosis. Examination of fibroblasts by electron microscopy identified concentric lamellar vacuoles within lysosomes, similar to those seen in Niemann-Pick type C disease and, to a lesser degree, SLOS. Retrospective study of the first stillborn female sib of this patient, who died at 21 weeks of gestation, showed type II Arnold Chiari malformation, microcephaly, tetramelic hexadactyly, talipes equinovarus and lumbosacral meningocele. Histologic examination of liver showed normal portal tracts and marked extramedullary hematopoiesis. Sequence analysis of SC5D in the fetus demonstrated the same compound heterozygous mutations, c.88G>A (p.R29Q) and c.632G>A (p.G211D), found in the sister [129].
The second patient, reported by Krakowiak et al. [130], had multiple congenital malformations, intracellular accumulation of mucopolysaccharides and lipids, and minor facial anomalies resembling those seen in SLOS and died at age 18 months. Sterol analysis of stored tissue showed a markedly increased level of lathosterol with an otherwise normal tissue sterol profile. Sequence analysis of SC5D identified homozygosity for a missense mutation, c.137A>C (p.Y46S), carried by both parents [130]. A third patient with lathosterolosis was reported in 2013 by Ho et al., who described an almost 2-year-old girl who had many features of SLOS, but who on plasma sterol analysis was found to have a lathosterol level of 81.6μmol/L and normal levels of 7DHC and 8DHC. Her phenotype included microcephaly, bitemporal narrowing, broad nasal tip without anteverted nares, micrognathia, postaxial hexadactyly of both feet, 2-3 toe syndactyly, hypotonia, and moderate global developmental delays. Mutation analysis showed compound heterozygosity for mutations in SC5D: c.442A>G (p.K148E) and c.630C>A (p.D210E). After 4 weeks treatment with simvastatin (max 1.0 mg/kg/d), the patient’s level of lathosterol normalized [131].
The pathogenesis of lathosterolosis in humans is still uncertain. Sc5d knockout mice are stillborn and have severe craniofacial defects, including cleft palate and micrognathia, and limb patterning defects such as postaxial polydactyly [130]. The close correspondence of these defects to those of SLOS and the similar facial features of both syndromes suggests that many of the embryonic defects of lathosterolosis are caused by cholesterol deficiency. Supporting this notion is that the plasma level of lathosterol in the three patients is higher than the corresponding abnormal sterol levels in sterol disorders other than SLOS, arguing that the tissue level of cholesterol during the rapid cell growth of embryogenesis is low enough to impair Hedgehog protein signaling. Other metabolic features of the disorder, such as lysosomal lipid storage, could be responsible for the progressive cholestatic liver disease, which is not seen in SLOS unless the cholesterol level is much lower than that found in the 3 lathosterolosis patients. However, comparative proteomic analysis of control and Sc5d–/– embryonic mouse brains showed that multiple pathways, including mevalonate metabolism, glycolysis, antioxidant system, apoptosis, protein biosynthesis, intracellular trafficking, and cytoskeleton systems are affected in Sc5d–/– brain tissue, offering many other pathophysiologies to consider [132].
In addition to statin treatment to lower the level of lathosterol by SREBP-mediated upregulation of SC5D transcription, orthotopic liver transplantation at age 7 years for end-stage liver disease in the first reported patient with lathosterolosis [133, 151] led to normalization of the plasma cholesterol level and, at 5-year follow-up, arrest of mental deterioration with return of many lost physical abilities.
7Smith-Lemli-Opitz Syndrome
Smith-Lemli-Opitz syndrome (SLOS); originally described as “RSH syndrome;”) OMIM 270400, 268670, and 602858) is a multiple congenital anomalies/intellectual disabilities (MCA/ID) syndrome first described in 1964 [134]. SLOS was initially considered an autosomal or possibly X-linked recessive disorder but not a possible metabolic disorder, because, in 1964, Garrodian inborn errors of metabolism were not thought to cause or be involved in the pathogenesis of MCA or MCA/ID syndromes. Diagnostic considerations then were focused on chromosomal aneuploidy while plasma and urine screening for inborn errors of amino acid and organic acid metabolism was more or less limited to non-dysmorphic patients with intellectual and other developmental disabilities, following the metabolic disease paradigm of phenylketonuria. The low cholesterol levels in SLOS children were successfully ignored for 30 years, as was pioneering French research on developmental defects resembling those of SLOS caused by of inhibitors of cholesterol synthesis administered to pregnant rats [12]. The breakthrough came in 1993 when several investigators [135–137], prompted by the finding of low urinary bile acid levels in a patient with SLOS, discovered to general amazement that the SLOS was, after all, a “simple” Garrodian inborn error of metabolism, namely, the failure of 7-dehydrocholesterol (7DHC) to be converted to cholesterol, due to autosomal recessive deficiency of the enzyme, 7DHC reductase (DHCR7) [5–7]
Excellent reviews of SLOS and its abnormal sterol metabolism include (but are not limited to): Porter, Herman [42]; Miller [138]; Irons [139]; Yu, Patel [140]; Porter [141]; Herman [142]; Porter [143], Kelley, Herman [65]; Herman [144]. Hammerschmidt et al. [145] provide an authoritative and illuminating analysis of hedgehog functions in Drosophila and vertebrates, perhaps complemented best by such a developmental biology text as Gilbert (10th ed, 2014).
7.1Clinical manifestations
Because cholesterol is necessary for the formation of all cell membranes, it should be no surprise that many SLOS homozygotes/embryos do not survive early gestation, and that the clinical picture, with few exceptions, of longer-surviving fetuses can vary from cyclopia (holoprosencephaly, HPE) to fetal akinesia with renal dysgenesis. Pediatric pathologists must be exquisitely sensitive to the uncommon but almost always unexpected occurrence of such SLOS homozygotes (think: 2/3 toe syndactyly if nothing else) among stillbirths and neonatal deaths with or without other anomalies, not only to make a diagnosis, but also, more importantly, to counsel parents about recurrence risk. The carrier state is sufficiently common (q.v. Nowaczyk et al., [146], Opitz et al., [147] up to almost 4%) in some populations that, by chance and without consanguinity, affected children can be born in different branches of the same family. It is now probable that other reports of phenotypically similar and more severe disorders, especially with fetal or perinatal death [148–150] are examples of SLOS. The extraordinary stability over time of cholesterol and cholesterol-derived saturated steranes in ancient stratigraphic deposits allows diagnosis of SLOS in preserved museum specimens as old as 130 years [151] and tracing the origins of early multicellular or colonial life in >635-million-year old geologic deposits.
In general, SLOS fetuses and infants have a disproportionately small head; small anterior fontanel; a characteristic combination of minor facial anomalies, including micrognathia with highly arched (or cleft) palate/uvula; a rugose anterior palate mucosa; congenital or early postnatal cataracts; short “swan-neck” thumbs; polydactyly – mostly postaxial; and partial or more extensive cutaneous syndactyly of toes 2 and 3 (initially Y-shaped, then more tuning-fork shaped). Less common limb anomalies include congenital contractures, oligodactyly and ectrodactyly in various combinations. A short hamartomatous-tipped tongue and fingerprints with all whorls are especially important clues to the diagnosis of SLOS.
SLOS infants frequently are unusually irritable, and cry excessively and have failure to thrive, projectile vomiting, resting hypotonia with waking hypertonia, and hyperactive deep tendon reflexes. Less frequent problems include pyloric stenosis, intestinal malrotation, and Hirschsprung “disease.” All but the most mildly affected infants with SLOS have feeding disorders ranging from oral hypersensitivity to severe intestinal dysmotility with volume-dependent feeding intolerance. As a result, most SLOS infants are finicky eaters and are constipated from birth. Males often have underdeveloped external genitalia or genital malformations ranging from mild or severe hypospadias and cryptorchidism to the occasional male fetus with female external genitalia. More severe genito-urinary malformations include uni- or bilateral renal agenesis and renal hypoplasia, with or without hypertension.
Cognitive impairment can be severe to profound, but the more mildly affected children, now more easily identified by biochemical screening, are able to attend public school with special education assistance. Characteristic behavior disturbances include irritability, tactile defensiveness, hyperactivity, aggressiveness, self-injurious behaviors, sleep disturbances, autistic-like behaviors and a peculiar propensity for sudden retropulsion, “opistokinesis,” regardless of risks for self-injury consequences. Although cholesterol does not cross the blood-brain barrier, treatment of SLOS patients at any age often leads improved behavior, including reduced irritability, better sleeping, and reduced autistic behaviors, sometimes within a day or two of starting treatment. Because the adrenal cortex preferentially uses circulating LDL cholesterol to synthesize steroids, which can readily cross the blood-brain barrier, the rapid improvement in behavior with cholesterol treatment likely is caused by increased adrenal production of pregnenolone, the precursor of all steroid hormones, or other steroids. Neuroradiological findings include, but are not limited to, hypoplasia/dysgenesis/agenesis of the corpus callosum, gyral and cerebellar abnormalities, and, rarely, an HPE defect ranging from cyclopia [152] to semi-lobar prosencephaly.
The pathogenesis of the common and often severe and damaging photosensitivity in SLOS has been much debated [14, 153–159], but evidence suggests that it results from the interaction of UVA light with 7DHC or, more likely, one of its metabolites. The evidence in favor of a metabolite of 7DHC, such as 7-peroxy-DHC, rather than 7DHC itself is that 7DHC has a low extinction coefficient for UVA-1 (340–400 nm) light, which is the cause of SLOS photosensitivity. Unfortunately, because sunscreens sold in the US other than zinc oxide absorb minimally in the UVA-1 range, the only practical outdoor protection is wearing UV-filtering sunglasses and clothing with minimal light penetration. “Roast” RSH infants/children after a day at the lake are very unhappy campers indeed.
A “mildly” affected girl with characteristic facial appearance is illustrated in Fig. 15.
7.2Cardiovascular system
Perhaps the most characteristic malformation in SLOS is TAPVR (total anomalous pulmonary venous return), but more common malformations include atrial septal defect, ventricular septal defect, and atrioventricular (AV) canal. One of the most characteristic combinations of anomalies a (pediatric) pathologist will see in an SLOS fetus is non- or hypolobation of lungs with an AV canal. Whether to proceed to surgical correction (Norwood procedure) in an infant with proven SLOS and hypoplastic left heart can be a most difficult decision for parents, who might ultimately have to choose to withdraw life support.
7.3Adrenal function
Bianconi et al. [160] recently studied adrenal function in 35 individuals with SLOS and found normal results of ACTH stimulation in 28 of 29 tested, and subsequently normal ovine corticotropin testing in the one exception. They found adequate glucocorticoid responses and concluded that stress steroid coverage may not be warranted in mildly to moderately affected SLOS children. However, this can be different in severely affected SLOS infants. For example, Andersson et al. [161] studied 3 such infants (2 of whom died neonatally), all with very low cholesterol levels, who had abnormal serum electrolytes and inadequate cortisol responses at times of stress. In addition, Chemaitilly et al. [162] reported an SLOS infant with salt wasting on day 4, and the severely affected patient of Nowaczyk et al. [163] developed signs of moderate mineralocorticoid deficiency despite a comparatively high serum cholesterol level of 62 mg/dL on day 25 of life. He responded quickly to steroid replacement but subsequently died at age 3.5 months from progressive cholestatic liver disease.
7.4Prenatal diagnosis
With better understanding of the prenatal development and physiology of SLOS, affected fetuses are now often diagnosed by sonographically identified malformations and by maternal second trimester serum screening, from which can be calculated accurate risk estimates for SLOS in the fetus. When the risk determined by maternal serum screening is high, typically 2% or more, prenatal diagnosis can be accurately made by sterol analysis of amniotic fluid or by measurement of 7- and 8-dehydrosteroids in maternal urine after 16 weeks gestation. Although mutation testing of DHCR7 in amniotic fluid cells—directly or after culturing—is possible, molecular analysis takes longer than biochemical testing and has a history of rare technical failures. [42, 164–171].
7.5Postnatal diagnosis
A prenatally or postnatally suspected diagnosis of SLOS is most easily and swiftly confirmed or rejected by quantification of 7-dehydro- (7DHC) and 8-dehydro- (8DHC) cholesterol in plasma [172]. When SLOS is suspected in a prenatal or perinatal death, measuring the 7DHC/cholesterol ratio in any tissue can render a secure diagnosis in most cases. There are two conditions, however, when the results of biochemical testing can yield ambiguous results. First, patients with behavioral disorders treated with medications classified as “sigma inhibitors,” such as risperidone and haloperidol, often have mildly increased plasma levels of both 7DHC and 8DHC indistinguishable from mild SLOS, because such drugs inhibit DHCR7. Second, because the degree of biochemical abnormality in SLOS depends, in part, on the rate of growth and the need for de novo cholesterol synthesis, which is highest in utero and during the first 18 months of life, some mildly affected SLOS individuals—those who often escape perinatal clinical diagnosis—can have normal plasma sterols when tested at a later age. Although in such situations a biochemical diagnosis historically has been made by sterol analysis in cultured fibroblasts or lymphoblasts, today mutation testing of DHCR7 is easier for diagnosis of SLOS in patients whose biochemical testing is inconclusive. In general, DHCR7 mutation analysis is now also performed to confirm positive biochemical diagnoses and to provide information needed for genetic counseling.
7.6Cause and genetics
Autosomal recessive SLOS appears to be non-heterogeneous with only one locus known at 11q12-q13. In 1998, Moebius et al. successfully mapped and cloned the gene [173], DHCR7, after which the mutant nature of DHCR7 in affected SLOS homozygotes was determined, in part through comparative analysis of the homologous gene structure of Arabidopsis thaliana and other organisms [5–7]. DHCR7 converts 7DHC to cholesterol, has a mass of 55 kDa, and is bound to the endoplasmic reticulum, with an estimated 9 transmembrane domains. Fig. 16 (from Yu and Patel) [140] shows the distribution of the then-known 121 mutations, among 685 alleles tested, in DHCR7 (see also Correa-Cerro, Porter [174], who analyzed 105 different mutant DHCR7 alleles). In order of frequency, the mutant alleles include: splice-site, missense, and nonsense mutations distributed throughout the coding region (exons 3-9) of the gene. Yu and Patel [140] list 105 missense, 5 nonsense, 3 splice-site and 8 nucleotide deletions or insertions, with missense mutations accounting for the great majority of individual mutations (over 87%). The commonest mutations are: IVS8-1G>C (over one-quarter of alleles), T93M, W151X, V326L, and R404C, all of which are null or almost-null mutations. Yu et al. [175] found at least one IVS8-1G>C allele in over 60% of American Caucasian SLOS propositi, but none among Finns, Chinese (Han), Japanese, or Sierra Leoneans. In their analysis of DHCR7 mutations in 59 European SLOS patients, Witsch-Baumgartner et al. [176] noted a west-to-east cline of IVS8-1G>C (highest in England) and a reverse east-to-west cline for W151X and V326L.They also determined by haplotype analysis that the IVS8-1G>C and W151X alleles are the oldest of the common DHCR7 mutations in Europe. An observed deficiency of IVS8-1G>C homozygotes [by at least 50%, Kelley and Herman, Table 2 [65]] can, to our mind, reasonably be understood as representing embryonic lethality, a notion now supported by the epidemiologic studies of Jezela-Stanek et al. [177]. Although no heterozygote advantage explaining the high prevalence of DHCR7 mutations in Europe has been proven, evidence argues that heterozygotes carrying common severe mutations, most of whom have mildly increased levels of 7DHC, the precursor of vitamin D, produce higher levels of vitamin D under the reduced sunlight conditions of Northern Europe [178], a selective advantage for late Neolithic farmers, e.g. the Funnelbeaker culture, who repopulated northern Europe and had more limited natural sources of vitamin D than earlier hunter-gatherer societies.
7.7Teratology
Charles Roux of Paris and his collaborators, continuing the wonderful tradition of t é ratologie proper begun some 150 years earlier by the great French naturalist (and Napoléon’s naturalist in Egypt) Étienne Geoffroy Saint-Hilaire (q.v. Dareste, 1877) have made many contributions of fundamental biological importance to an understanding of the role of cholesterol in animal development [179–186], continued in the US by Batta and Salen [187], Honda et al. [188] and Lanoue et al. [189]. The teratogenic agents, triparanol, AY9944 and BM15766 used to inhibit post-squalene cholesterol synthesis in pregnant rats or mice, cause many defects of early, primary morphogenesis, including several types of limb defects, microcephaly, holoprosencephaly (HPE), pituitary agenesis and genital defects, similar to the defects produced by Sulik and her collaborators in mice by combined genetic- and drug-induced cholesterol deficiency [189, 190].
7.8Animal models (i.e. transgenic and knockout mice)
In 2004, collaborators from the NIH and Johns Hopkins Hospital reported creating a hypomorphic mouse (DHCR7T93M), which as homozygotes or compound heterozygotes with a null allele appear normal, are viable and fertile, but have mild dilation of the third and lateral ventricles. The T93M/null mice also have 2-3 toe syndactyly [174]. Subsequently, the NIH SLOS mouse [191] was created by homologous recombination inserting the neomycin phosphotransferase gene (PGKneo) and deleting exons 3, 4 and part of 5. In these essentially DHCR7–/– mice, there was a marked reduction of cholesterol in serum and tissues and a striking increase in 7DHC levels. Homozygous pups showed intra-uterine growth retardation, craniofacial anomalies, cleft palate in 8%, apparent congenital hypotonia with paucity of spontaneous movements, inadequate suck, persistence of nasal plugs, and complete absence of nasal openings in 9%, causing death on day 1 of life. Brain weight was reduced in proportion to body size, and the cerebral cortex showed a substantially impaired response to exogenous glutamate.
The “UNC SLOS mouse” was created at the University of North Carolina, Chapel Hill [192] by deleting from Dhcr7 exon 8 and its flanking splice acceptor, i.e. the sequence for amino acids 318-471 coding for about one-third of the protein, thereby creating the equivalent of the human IVS8-1G>C DHCR7 null mutation. Results were very similar to those with the NIH mouse in that the homozygous mutant pups were smaller, had respiratory difficulties and did not suck. They died on day 1 having frequent cleft palate and compact and immature lungs with sparse, unconnected airspaces. The sterol abnormalities in Dhcr7 –/– mice and humans are similar. Cholesterol levels were low, but the mRNA levels for HMG-CoA reductase, the LDL receptor, and SREBP2 were normal. Yet, protein levels and activities of HMG-CoA reductase were markedly reduced, apparently reflecting a proteolytic effect of 7DHC or a derived metabolite on HMG-CoA reductase [192].
7.9Carrier and homozygote frequency
In 2001, Davies and Metherall at the University of Utah began work to estimate as accurately as possible the prevalence of the commonest (IVS8-1G>C) mutation in our population. It was found in 1.13% of normal individuals and, if IVS8-1G>C represented some 29% of all DHCR7 mutations, the estimated carrier frequency of all DHCR7 mutations (3.896%) would predict a homozygote incidence at birth of slightly less than 1/2500. Battaile et al. [193, 194], also analyzing the IVS8-1G>C mutation in 1503 anonymous blood samples (random newborn screening blood cards), found 16 such carriers. Further studies by Nowaczyk and Waye [195], Witsch-Baumgartner et al. [176] and Yu et al (2001) have established a 4% carrier (2pq) frequency in mostly Caucasian populations (= 2% gene frequency). If the recent estimates of homozygotes in newborn infants of 1/101,000 is correct then prenatal mortality must be over 97% [196], a proportion supported by the recent studies of Jezela-Stanek et al. [177].
7.10Treatment
Recently, Porter and Steiner [197] stated that [cholesterol] “has not yet been proven to be effective in SLO” [syndrome]. Yet, unless something more effective becomes available, current practice remains dietary supplementation of cholesterol practice in the form of egg yolk, pharmacist-prepared cholesterol suspensions, or commercially manufactured cholesterol powders. Because cholesterol supplementation rarely raises the serum total and LDL cholesterol levels above normal, there would seem to be little risk of accelerated atherosclerosis from cholesterol supplementation. The occasional child with SLOS who is found, either before or after cholesterol supplementation, to have a high cholesterol level usually has a parent with a classic hyperlipoproteinemia disorder.
8Desmosterolosis
Desmosterolosis (OMIM 602398) is a rare autosomal recessive inborn error of cholesterol biosynthesis caused by deficient activity of 3β-hydroxysteroid-Δ24-reductase (desmosterol-Δ24-reductase, DHCR24), a flavin adenine dinucleotide (FAD)-dependent oxidoreductase that catalyzes the reduction of the 24-unsaturated bond of desmosterol to yield cholesterol in the last step of the canonical Bloch post-squalene cholesterol biosynthetic pathway In contrast, in the Kandutch-Russell pathway (Fig. 10), reduction of the 24-unsaturated bond by DHCR24 occurs at the 30-carbon level of the pathway. DHCR24 is located mainly in the endoplasmatic reticulum and also has a role protecting cells against oxidative stress-induced apoptosis [198–200], although the mechanism by which the DHCR24 protein exerts this action is unknown. DHCR24 is predicted to be a 516-amino acid protein with a leader sequence, 4 possible transmembrane domains, and an oxidoreductase domain, including an FAD binding motif. The protein shows 97%, 49%, and 4% amino acid identity with its orthologs in mouse, Caenorhabditis elegans, and Arabidopsis thaliana, respectively, but no sequence similarity with other plant or yeast sterol reductases [201]. When there is insufficient activity of DHCR24, there is a pathognomonic accumulation of desmosterol detectable in plasma and tissues by GC/MS. The gene encoding 3β-hydroxysteroid-Δ24-reductase, DHCR24, contains 9 exons and is located at 1p32.3 [201]. DHCR24 was initially demonstrated to have reduced expression in the inferior temporal lobes of patients with Alzheimer disease, and was therefore designated seladin1 (selective Alzheimer disease indicator 1) [202]. DHCR24 mRNA is almost ubiquitously expressed in adult and fetal tissues, with highest levels of expression in adult adrenal cortex, liver, and spinal cord [201].
Thus far, only five patients with desmosterolosis have been reported [203–205]. Their clinical findings are characterized by varied central nervous system, craniofacial, and skeletal anomalies sufficiently diverse among the several patients to make generalizing about the embryology of desmosterolosis difficult. The first patient, reported by FitzPatrick et al. in [203], a female with multiple lethal congenital anomalies, died within an hour of birth at 34 weeks gestation. Autopsy showed macrocephaly, facial anomalies similar to Potter facies, thick alveolar ridges, gingival nodules, cleft palate, total anomalous pulmonary venous return, hypoplastic kidneys, ambiguous genitalia, rhizomelia, and generalized osteosclerosis resembling Raine syndrome. Neuropathological examination demonstrated an immature gyral pattern, hypoplastic corpus callosum, dilatation of the ventricles, white matter changes suggestive of hypoxia, and focal hypoplasia of the cerebellar cortex. Insufficiently vascularized villi were seen in the placental parenchyma. Plasma sterol analysis showed increased desmosterol/cholesterol ratios of 6-, 107-, and 11-fold above control values for brain, liver and kidney, respectively. DHCR24 sequence analysis demonstrated two apparently pathogenic missense mutations, c.1412A>C (p.Y471S, maternal) and c.881A>C (p.N294T, paternal) The second patient, reported by Andersson et al. [204], was a 3-1/2–year-old boy with severe congenital microcephaly, multiple facial anomalies unlike those of other desmosterolosis patients, agenesis of corpus callosum, focal cutis aplasia (cranial), submucous cleft palate, arthrogryposis, and severe developmental delays. Although the plasma cholesterol level was normal, the desmosterol level was increased 120-fold above controls, and both parents had approximately twice normal desmosterol levels. The child was homozygous for a missense mutation, c.571G>A (p.E191K), in DHCR24 carried by both parents. The third patient, reported by Schaaf et al. [205], was a girl with IUGR, relative macrocephaly, hypoplastic corpus callosum, non-communicating hydrocephalus, incomplete opercularization, diaphragmatic eventration, and rhizomelia. Her plasma cholesterol level was slightly above upper limit of normal for age, whereas the level of desmosterol was increased 300-fold above controls. DHCR24 enzyme activity was significantly decreased, and sequence analysis of DHCR24 demonstrated compound heterozygosity for mutations c.281G>A (maternal, p.R94H) and c.1438G>A (paternal, p.E480K). Karyotype was normal in all three patients; and in all cases, analysis of the parents’ plasma sterols revealed mildly increased levels of desmosterol.
Recently, Dias et al. [206] described two sisters homozygous for a c.571G>A (p.E191K) DHCR24 mutation, who presented for evaluation of intellectual disability and “dysmorphic features” at ages 10 and 14 years. They had short stature, hirsutism, craniofacial dysmorphism, optic atrophy, nystagmus, spasticity, deep tendon hyperreflexia, camptodactyly of fingers and toes, and muscle wasting, particularly of the thenar and hypothenar muscles. Cranial MRI showed prominent, irregular ventricles, thin corpus callosum, and Chiari I malformation in both sisters.
The underlying mechanisms involved in the pathogenesis of desmosterolosis remain uncertain and could be attributed, at least in part, to prenatal deficiency of cholesterol, especially in the CNS and cartilage, or toxic effects of high levels of desmosterol. Cholesterol and its derivatives are important for cell membrane biology and signaling. During later embryogenesis and fetal life, in situ synthesis of cholesterol could be the main or only way to support the increased demand for cholesterol in tissues undergo rapid prenatal growth and differentiation [205]. As in other sterol disorder with quantitatively reduced cholesterol synthesis in fetal life, brain and cartilage in DHCR24 deficiency show the greatest teratological effects, possibly because of the inability of these tissues to import hepatic LDL cholesterol, as can most other tissues. Although the functional role of desmosterol in cells and why its level should be increased in brain during fetal life is unknown, desmosterol increases membrane fluidity by about one-third [207], which could alter intercellular interactions and embryonic cell migration during morphogenesis. Desmosterol is a prominent sterol constituent of the human central nervous system during the embryonic period and in later fetal life [208], and it is also present at high concentration in mammalian testes [209] and spermatozoa [210]. Nevertheless, a teratogenic action of desmosterol in humans has been not been established. In rats, the DHCR24 inhibitors triparanol [12] and U18666A [211] have teratogenic effects, especially during the early development of the brain. Dhcr24 knockout mice have variable viability ranging from survival to adulthood with infertility and reduction of subcutaneous and mesenteric fat, decrease of myelination of the central nervous system, and hypotonia, but no major malformations [212] to mutants exhibiting a lethal restrictive dermopathy without congenital malformations [213]. Desmosterol accumulation in Dhcr24 knockout mice stimulates LXR (liver X receptor) – targeted genes [214], which may be relevant for lipid homeostasis in cells and tissues with high desmosterol content.
No effective treatment for desmosterolosis has been proven, although, in theory, treatment with high-dose riboflavin might have an effect with mutations affecting the FAD binding site, and, as in other sterol disorders, statin treatment should be able to upregulate residual DHCR24 activity. Use of simvastatin, which crosses the blood-brain barrier, would be a way to determine what aspects of the associated developmental delays are ongoing effects of high CNS desmosterol levels.
9Methyl sterol oxidase (sc4 mol) deficiency
Recently, the first patient with methyl sterol oxidase (MSOM1/SC4MOL) deficiency was described [215]. Her condition was caused by two different mutations of the sterol-C4-methyl oxidase-like gene (SC4MOL) and manifested not as multiple malformations with conspicuous skeletal involvement as in several other sterol defects, but rather as a syndrome with:
– Postnatal onset of an ichthyosiform erythroderma-like severe psoriasis.
– Congenital cataracts.
– Mild developmental delay with normal OFC (53.5 = 50th centile).
– Relative failure to thrive (BW, BL, and BOFC not given); length between 9-39 months was at the 3rd centile; height at 13 years (140 cm, – 2.5 SD); weight was 28.6 Kg (<3rd, 50th for 9.5 years); bone age was delayed; but skeletal morphology was normal.
– Skin histology showed psoriasiform changes with oil red O-positive, foamy, lipid-laden fibroblasts in the dermis (CD68-negative, hence, not macrophages, as in CHILD syndrome or verruciform xanthoma). The skin had been normal until age 2 when the dermopathy appeared around the umbilicus; and by age 6 had extended to cover the entire body except for palms.
The serum lipid profile showed (result, normal range): cholesterol: 85 mg/dL [140–176]; HDL: 28 mg/dL [35–75]; LDL: 49 mg/dL [70–160], but normal triglyceride and VLDL levels.
Plasma sterol analysis showed 20–500 fold elevation of 4α-monomethyl and 4,4’-dimethylsterols, compatible with deficient activity of methylsterol oxidase (SC4MOL), an enzyme previously identified in yeast (erg25) and plants. Sequencing of SC4MOL in the patient identified the mutations c.519T>A (p.H173Q, paternal) and c.731A>G (p.Y244C, maternal), both mutations being in codons mapping to the highly conserved metal-binding domains of the enzyme. Cholesterol supplementation for 3 months lowered the patient’s methylsterol level maximally by about 20%, with concomitant rise in cholesterol level to the lower limit of normal but no effect on the skin disease. However, later treatment with simvastatin led to almost complete resolution of the skin pathology [216].
Immunologically, the patient and her carrier father had activated CD16+ granulocyte levels 30 and 20 fold, respectively, above control levels. Typical of psoriasis, the patient also had an upregulation of TLR-2 granulocytes concomitant with a marked down regulation of the granulocyte-specific CD16b isoform. Analysis of cytokines also showed increased levels of pro-inflammatory chemokines, including GM-CSF, known to trigger psoriasis, possibly due to the defect in cholesterol synthesis. However, because the heterozygote father, but not the mother, manifested quantitatively similar immunological abnormalities as his affected daughter, the relatedness of the immunological abnormalities to SC4MOL deficiency remains to be clarified. A second patient with SC4MOL deficiency described by the same authors had congenital microcephaly and cataracts and developmental delays but no skin disease by age 2 years.
He et al. point out that in humans SC4MOL is expressed ubiquitously but has highest mRNA levels in liver, brain, adrenal, lymphoblasts, skin, testes and blood vessels. Because SC4MOL/MSO is present in plants, animals and yeast (all eukaryotes), it must have been present in their common ancestor but not in LUCA (the last universal common ancestor of the archaea, bacteria and eukaryotes). Also, among the sterol precursors accumulating in defects of sterol metabolism, 4,4dimethylsterols appear to be unique in stimulating hyperproliferative skin disease with barrier dysfunction and, possibly, immune dysfunction. Methylsterols are known to be potent ligands for liver X receptors (LXRs) and, therefore, in this case, could affect lipid transport and skin cytokine response to bacterial infections.
References
[1] | Love G.D. , Grosjean E. , Stalvies C. , Fike D.A. , Grotzinger J.P. , Bradley A.S. et al., Fossil steroids record the appearance of Demospongiae during the Cryogenian period, Nature 457: (7230) ((2009) ), 718–721. |
[2] | Nielsen C. , Animal Evolution 3rd ed: Oxford University Press; (2012) . |
[3] | Kodner R.B. , Summons R.E. , Pearson A. , King N. and Knoll A.H. , Sterols in a unicellular relative of the metazoans, Proceedings of the National Academy of Sciences of the United States of America 105: (29) ((2008) ), 9897–9902. |
[4] | Choe S. , Tanaka A. , Noguchi T. , Fujioka S. , Takatsuto S. , Ross A.S. et al., Lesions in the sterol Delta(7) reductase gene of Arabidopsis cause dwarfism due to a block in brassinosteroid biosynthesis, Plant J 21: (5) ((2000) ), 431–443. |
[5] | Fitzky B.U. , Witsch-Baumgartner M. , Erdel M. , Lee J.N. , Paik Y.K. , Glossmann H. et al., Mutations in the Delta7-sterol reductase gene in patients with the Smith-Lemli-Opitz syndrome, Proceedings of the National Academy of Sciences of the United States of America 95: (14) ((1998) ), 8181–8186. |
[6] | Wassif C.A. , Maslen C. , Kachilele-Linjewile S. , Lin D. , Linck L.M. , Connor W.E. et al., Mutations in the human sterol Delta(7)-reductase gene at 11q12-13 cause Smith-Lemli-Opitz syndrome, American Journal of Human Genetics 63: (1) ((1998) ), 55–62. |
[7] | Waterham H.R. , Wijburg F.A. , Hennekam R.C.M. , Vreken P. , Poll-The B.T. , Dorland L. et al., Smith-Lemli-Opitz syndrome is caused by mutations in the 7-dehydrocholesterol reductase gene, American Journal of Human Genetics 63: (2) ((1998) ), 329–338. |
[8] | Summons R.E. , Bradley A.S. , Jahnke L.L. and Waldbauer J.R. , Steroids, triterpenoids and molecular oxygen, Philosophical Transactions of the Royal Society of London Series B, Biological sciences 361: (1470) ((2006) ), 951–968. |
[9] | Mouritsen O.G. and Zuckermann M.J. , What’s so special about cholesterol? Lipids 39: (11) ((2004) ), 1101–1113. |
[10] | Margulis L. , Origins of species: Acquired genomes and individuality, Bio Systems 31: (2-3) ((1993) ), 121–125. |
[11] | Cooper G.M. and Hausman R.E. eds. The Cell, A Molecular Approach: Sinauer; (2009) . |
[12] | Roux C. , [Teratogenic Action of Triparanol in Animals], Archives Francaises de Pediatrie 21: ((1964) ), 451–464. |
[13] | Haas D. and Muenke M. , Abnormal sterol metabolism in holoprosencephaly, American Journal of Medical Genetics Part C, Seminars in Medical Genetics 154C: (1) ((2010) ), 102–108. |
[14] | Antley R. and Bixler D. , Trapezoidocephaly, midfacial hypoplasia and cartilage abnormalities with multiple synostoses and skeletal fractures, Birth Defects Original Article Series 11: (2) ((1975) ), 397–401. |
[15] | Bottero L. , Cinalli G. , Labrune P. , Lajeunie E. and Renier D. , Antley-Bixler syndrome, Description of two new cases and a review of the literature, Child’s Nervous System: ChNS: Official Journal of the International Society for Pediatric Neurosurgery 13: (5) ((1997) ), 275–280; discussion 81. |
[16] | Crisponi G. , Porcu C. and Piu M.E. , Antley-Bixler syndrome: Case report and review of the literature, Clinical Dysmorphology 6: (1) ((1997) ), 61–68. |
[17] | Robinson L.K. , Powers N.G. , Dunklee P. , Sherman S. and Jones K.L. , The Antley-Bixler syndrome, The Journal of Pediatrics 101: (2) ((1982) ), 201–205. |
[18] | Rumball K.M. , Pang E. and Letts R.M. , Musculoskeletal manifestations of the Antley-Bixler syndrome, Journal of Pediatric Orthopedics Part B 8: (2) ((1999) ), 139–143. |
[19] | Lee H.J. , Cho D.Y. , Tsai F.J. and Shen W.C. , Antley-Bixler syndrome, description of two new cases and review of the literature, Pediatric Neurosurgery 34: (1) ((2001) ), 33–39. |
[20] | McGlaughlin K.L. , Witherow H. , Dunaway D.J. , David D.J. and Anderson P.J. , Spectrum of Antley-Bixler syndrome, The Journal of Craniofacial Surgery 21: (5) ((2010) ), 1560–1564. |
[21] | Lacheretz M. , Walbaum R. and Tourgis C. , L’acrocephalosynankie: A propos d’une observation avec synostoses multiples, Pediatrie 39: ((1974) ), 169–177. |
[22] | DeLozier C.D. , Antley R.M. , Williams R. , Green N. , Heller R.M. , Bixler D. et al., The syndrome of multisynostotic osteodysgenesis with long-bone fractures, American Journal of Medical Genetics 7: (3) ((1980) ), 391–403. |
[23] | Schinzel A. , Savoldelli G. , Briner J. , Sigg P. and Massini C. , Antley-Bixler syndrome in sisters: A term newborn and a prenatally diagnosed fetus, American Journal of Medical Genetics 14: (1) ((1983) ), 139–147. |
[24] | Chun K. , Siegel-Bartelt J. , Chitayat D. , Phillips J. and Ray P.N. , FGFR2 mutation associated with clinical manifestations consistent with Antley-Bixler syndrome, American Journal of Medical Genetics 77: (3) ((1998) ), 219–224. |
[25] | Schaefer F. , Anderson C. , Can B. and Say B. , Novel mutation in the FGFR2 gene at the same codon as the Crouzon syndrome mutations in a severe Pfeiffer syndrome type 2 case, American Journal of Medical Genetics 75: (3) ((1998) ), 252–255. |
[26] | Gripp K.W. , Stolle C.A. , McDonald-McGinn D.M. , Markowitz R.I. , Bartlett S.P. , Katowitz J.A. et al., Phenotype of the fibroblast growth factor receptor 2 Ser351Cys mutation: Pfeiffer syndrome type III, American Journal of Medical Genetics 78: (4) ((1998) ), 356–360. |
[27] | De Moerlooze L. , Spencer-Dene B., Revest J.M., Hajihosseini M., Rosewell I. and Dickson C., An important role for the IIIb isoform of fibroblast growth factor receptor 2 (FGFR2) in mesenchymal-epithelial signalling during mouse organogenesis, Development 127: (3) ((2000) ), 483–492. |
[28] | Xu X.L. , Weinstein M. , Li C.L. , Naski M. , Cohen R.I. , Ornitz D.M. et al., Fibroblast growth factor receptor 2 (FGFR2)-mediated reciprocal regulation loop between FGF8 and FGF10 is essential for limb induction, Development 125: (4) ((1998) ), 753–765. |
[29] | Fluck C.E. , Tajima T. , Pandey A.V. , Arlt W. , Okuhara K. , Verge C.F. et al., Mutant P450 oxidoreductase causes disordered steroidogenesis with and without Antley-Bixler syndrome, Nat Genet 36: (3) ((2004) ), 228–230. |
[30] | Fluck C.E. and Pandey A.V. , Clinical and biochemical consequences of p450 oxidoreductase deficiency, Endocrine Development 20: ((2011) ), 63–79. |
[31] | Lee B.E. , Feinberg M. , Abraham J.J. and Murthy A.R. , Congenital malformations in an infant born to a woman treated with fluconazole, The Pediatric Infectious Disease Journal 11: (12) ((1992) ), 1062–1064. |
[32] | Pursley T.J. , Blomquist I.K. , Abraham J. , Andersen H.F. and Bartley J.A. , Fluconazole-induced congenital anomalies in three infants, Clinical Infectious Diseases: An Official Publication of the Infectious Diseases Society of America 22: (2) ((1996) ), 336–340. |
[33] | Aleck K.A. and Bartley D.L. , Multiple malformation syndrome following fluconazole use in pregnancy: Report of an additional patient, American Journal of Medical genetics 72: (3) ((1997) ), 253–256. |
[34] | Kelley R.I. , Kratz L.E. , Glaser R.L. , Netzloff M.L. , Wolf L.M. and Jabs E.W. , Abnormal sterol metabolism in a patient with Antley-Bixler syndrome and ambiguous genitalia, American Journal of Medical Genetics 110: (2) ((2002) ), 95–102. |
[35] | Wilson J.D. , Auchus R.J. , Leihy M.W. , Guryev O.L. , Estabrook R.W. , Osborn S.M. et al., 5alpha-androstane-3alpha,17beta-diol is formed in tammar wallaby pouch young testes by a pathway involving 5alpha-pregnane-3alpha,17alpha-diol-20-one as a key intermediate, Endocrinology 144: (2) ((2003) ), 575–580. |
[36] | Fukami M. , Homma K. , Hasegawa T. and Ogata T. , Backdoor pathway for dihydrotestosterone biosynthesis: Implications for normal and abnormal human sex development, Developmental Dynamics: An Official Publication of the American Association of Anatomists 242: (4) ((2013) ), 320–329. |
[37] | Cragun D.L. , Trumpy S.K. , Shackleton C.H. , Kelley R.I. , Leslie N.D. , Mulrooney N.P. et al., Undetectable maternal serum uE3 and postnatal abnormal sterol and steroid metabolism in Antley-Bixler syndrome, American Journal of Medical Genetics Part A 129A: (1) ((2004) ), 1–7. |
[38] | Shackleton C. , Marcos J. , Arlt W. and Hauffa B.P. , Prenatal diagnosis of P450 oxidoreductase deficiency (ORD): A disorder causing low pregnancy estriol, maternal and fetal virilization, and the Antley-Bixler syndrome phenotype, American Journal of Medical Genetics Part A 129A: (2) ((2004) ), 105–112. |
[39] | Fukami M. , Horikawa R. , Nagai T. , Tanaka T. , Naiki Y. , Sato N. et al., Cytochrome P450 oxidoreductase gene mutations and Antley-Bixler syndrome with abnormal genitalia and/or impaired steroidogenesis: Molecular and clinical studies in 10 patients, The Journal of Clinical Endocrinology and Metabolism 90: (1) ((2005) ), 414–426. |
[40] | Roth C. , Hinney B. , Peter M. , Steinberger D. and Lakomek M. , Features of Antley-Bixler syndrome in an infant born to a mother with pregnancy luteoma, European Journal of Pediatrics 159: (3) ((2000) ), 189–192. |
[41] | The Human Gene Mutation Database. [cited 7/20/2015]. Available from: http://www.hgmd.cf.ac.uk/ac/index.php |
[42] | Porter F.D. and Herman G.E. , Malformation syndromes caused by disorders of cholesterol synthesis, Journal of Lipid Research 52: (1) ((2011) ), 6–34. |
[43] | Fukami M. , Nishimura G. , Homma K. , Nagai T. , Hanaki K. , Uematsu A. et al., Cytochrome P450 oxidoreductase deficiency: Identification and characterization of biallelic mutations and genotype-phenotype correlations in 35 Japanese patients, The Journal of Clinical Endocrinology and Metabolism 94: (5) ((2009) ), 1723–1731. |
[44] | Zanger U.M. and Schwab M. , Cytochrome P450 enzymes in drug metabolism: Regulation of gene expression, enzyme activities, and impact of genetic variation, Pharmacology & Therapeutics 138: (1) ((2013) ), 103–141. |
[45] | Shen A.L. , O’Leary K.A. and Kasper C.B. , Association of multiple developmental defects and embryonic lethality with loss of microsomal NADPH-cytochrome P450 oxidoreductase, The Journal of Biological Chemistry 277: (8) ((2002) ), 6536–6541. |
[46] | Otto D.M. , Henderson C.J. , Carrie D. , Davey M. , Gundersen T.E. , Blomhoff R. et al., Identification of novel roles of the cytochrome p450 system in early embryogenesis: Effects on vasculogenesis and retinoic Acid homeostasis, Molecular and Cellular Biology 23: (17) ((2003) ), 6103–6116. |
[47] | Gu J. , Weng Y. , Zhang Q.Y. , Cui H. , Behr M. , Wu L. et al., Liver-specific deletion of the NADPH-cytochrome P450 reductase gene: Impact on plasma cholesterol homeostasis and the function and regulation of microsomal cytochrome P450 and heme oxygenase, The Journal of Biological Chemistry 278: (28) ((2003) ), 25895–25901. |
[48] | Henderson C.J. , Otto D.M. , Carrie D. , Magnuson M.A. , McLaren A.W. , Rosewell I. et al., Inactivation of the hepatic cytochrome P450 system by conditional deletion of hepatic cytochrome P450 reductase, The Journal of Biological Chemistry 278: (15) ((2003) ), 13480–13486. |
[49] | Greenberg C.R. , Rimoin D.L. , Gruber H.E. , DeSa D.J. , Reed M. and Lachman R.S. , A new autosomal recessive lethal chondrodystrophy with congenital hydrops, American Journal of Medical Genetics 29: (3) ((1988) ), 623–632. |
[50] | Spranger J. and Maroteaux P. , The lethal osteochondrodysplasias, Advances in Human Genetics 19: ((1990) ), 1–103, 331–332. |
[51] | Chitayat D. , Gruber H. , Mullen B.J. , Pauzner D. , Costa T. , Lachman R. et al., Hydrops-Ectopic Calcification - Moth-Eaten Skeletal Dysplasia (Greenberg Dysplasia) - Prenatal-Diagnosis and Further Delineation of a Rare Genetic Disorder, American Journal of Medical Genetics 47: (2) ((1993) ), 272–277. |
[52] | Horn L.C. , Faber R. , Meiner A. , Piskazeck U. and Spranger J. , Greenberg dysplasia: First reported case with additional non-skeletal malformations and without consanguinity, Prenatal Diag 20: (12) ((2000) ), 1008–1011. |
[53] | Trajkovski Z. , Vrcakovski M. , Saveski J. and Gucev Z.S. , Greenberg dysplasia (hydrops-ectopic calcification-moth-eaten skeletal dysplasia): Prenatal ultrasound diagnosis and review of literature, American Journal of Medical Genetics 111: (4) ((2002) ), 415–419. |
[54] | Konstantinidou A. , Karadimas C. , Waterham H.R. , Superti-Furga A. , Kaminopetros P. , Grigoriadou M. et al., Pathologic, radiographic and molecular findings in three fetuses diagnosed with HEM/Greenberg skeletal dysplasia, Prenat Diagn 28: (4) ((2008) ), 309–312. |
[55] | Clayton P. , Fischer B. , Mann A. , Mansour S. , Rossier E. , Veen M. et al., Mutations causing Greenberg dysplasia but not Pelger anomaly uncouple enzymatic from structural functions of a nuclear membrane protein, Nucleus-Austin 1: (4) ((2010) ), 354–366. |
[56] | Waterham H.R. , Koster J. , Mooyer P. and van Noort G. , Kelley R.I., Wilcox W.R., et al., Autosomal recessive HEM/greenberg skeletal dysplasia is caused by 3 beta-hydroxysterol Delta(14)-reductase deficiency due to mutations in the lamin B receptor gene, American Journal of Human Genetics 72: (4) ((2003) ), 1013–1017. |
[57] | Wydner K.L. , McNeil J.A. , Lin F. , Worman H.J. and Lawrence J.B. , Chromosomal assignment of human nuclear envelope protein genes LMNA, LMNB1, and LBR by fluorescence in situ hybridization, Genomics 32: (3) ((1996) ), 474–478. |
[58] | Worman H.J. , Evans C.D. and Blobel G. , The lamin B receptor of the nuclear envelope inner membrane: A polytopic protein with eight potential transmembrane domains, The Journal of Cell Biology 111: (4) ((1990) ), 1535–1542. |
[59] | Ye Q. and Worman H.J. , Primary structure analysis and lamin B and DNA binding of human LBR, an integral protein of the nuclear envelope inner membrane, The Journal of Biological Chemistry 269: (15) ((1994) ), 11306–11311. |
[60] | Ye Q. and Worman H.J. , Interaction between an integral protein of the nuclear envelope inner membrane and human chromodomain proteins homologous to Drosophila HP1, Journal of Biological Chemistry 271: (25) ((1996) ), 14653–14656. |
[61] | Canonica L. , Fiecchi A. , Kienle M.G. , Scala A. , Galli G. , Paoletti E.G. et al., The fate of the 15-beta hydrogen of lanosterol in cholesterol biosynthesis, Journal of the American Chemical Society 90: (13) ((1968) ), 3597–3598. |
[62] | Fiecchi A. , Canonica L. , Scala A. , Cattabeni F. , Paoletti E.G. and Paoletti R. , 4,4-dimethyl-5-alpha-cholesta-8,14-dien-3-beta-ol. A new precursor of cholesterol in mammalian tissues, Life Sciences 8: (12) ((1969) ), 629–634. |
[63] | Silve S. , Dupuy P.H. , Ferrara P. and Loison G. , Human lamin B receptor exhibits sterol C14-reductase activity in Saccharomyces cerevisiae, Biochimica et Biophysica Acta 1392: (2-3) ((1998) ), 233–244. |
[64] | Prakash A. , Sengupta S. , Aparna K. and Kasbekar D.P. , The erg-3 (sterol delta14,15-reductase) gene of Neurospora crassa: Generation of null mutants by repeat-induced point mutation and complementation by proteins chimeric for human lamin B receptor sequences, Microbiology 145: (Pt 6) ((1999) ), 1443–1451. |
[65] | Kelley R.I. and Herman G.E. , Inborn errors of sterol biosynthesis, Annual Review of Genomics and Human Genetics 2: ((2001) ), 299–341. |
[66] | Wassif C.A. , Brownson K.E. , Sterner A.L. , Forlino A. , Zerfas P.M. , Wilson W.K. et al., HEM dysplasia and ichthyosis are likely laminopathies and not due to 3beta-hydroxysterol Delta14-reductase deficiency, Hum Mol Genet 16: (10) ((2007) ), 1176–1187. |
[67] | Offiah A.C. , Mansour S. , Jeffrey I. , Nash R. , Whittock N. , Pyper R. et al., Greenberg dysplasia (HEM) and lethal X linked dominant Conradi-Hunermann chondrodysplasia punctata (CDPX2): Presentation of two cases with overlapping phenotype, Journal of Medical Genetics 40: (12) ((2003) ), e129. |
[68] | Shultz L.D. , Lyons B.L. , Burzenski L.M. , Gott B. , Samuels R. , Schweitzer P.A. et al., Mutations at the mouse ichthyosis locus are within the lamin B receptor gene: A single gene model for human Pelger-Huet anomaly, Hum Mol Genet 12: (1) ((2003) ), 61–69. |
[69] | Shah Alam Bhuiyan M. , Eckstein J., Barbuch R. and Bard M., Synthetically lethal interactions involving loss of the yeast ERG24: The sterol C-14 reductase gene, Lipids 42: (1) ((2007) ), 69–76. |
[70] | Wagner N. , Weber D. , Seitz S. and Krohne G. , The lamin B receptor of Drosophila melanogaster, Journal of Cell Science 117: (Pt 10) ((2004) ), 2015–2028. |
[71] | Holmer L. , Pezhman A. and Worman H.J. , The human lamin B receptor/sterol reductase multigene family, Genomics 54: (3) ((1998) ), 469–476. |
[72] | Lemmens I.H. , Kas K. , Merregaert J. and Van de Ven W.J. , Identification and molecular characterization of TM7SF2 in the FAUNA gene cluster on human chromosome 11q13, Genomics 49: (3) ((1998) ), 437–442. |
[73] | Hoffmann K. , Dreger C.K. , Olins A.L. , Olins D.E. , Shultz L.D. , Lucke B. et al., Mutations in the gene encoding the lamin B receptor produce an altered nuclear morphology in granulocytes (Pelger-Huet anomaly), Nat Genet 31: (4) ((2002) ), 410–414. |
[74] | Oosterwijk J.C. , Mansour S. , van Noort G. , Waterham H.R., Hall C.M. and Hennekam R.C.M., Congenital abnormalities reported in Pelger-Huet homozygosity as compared to Greenberg/HEM dysplasia: Highly variable expression of allelic phenotypes, Journal of Medical Genetics 40: (12) ((2003) ), 937–941. |
[75] | Speeckaert M.M. , Verhelst C. , Koch A. , Speeckaert R. and Lacquet F. , Pelger-Huet anomaly: A critical review of the literature, Acta Haematologica 121: (4) ((2009) ), 202–206. |
[76] | Villalvilla A. , Gomez R. , Largo R. and Herrero-Beaumont G. , Lipid transport and metabolism in healthy and osteoarthritic cartilage, International Journal of Molecular Sciences 14: (10) ((2013) ), 20793–20808. |
[77] | Happle R. , Koch H. and Lenz W. , The CHILD syndrome. Congenital hemidysplasia with ichthyosiform erythroderma and limb defects, European Journal of Pediatrics 134: (1) ((1980) ), 27–33. |
[78] | Caldas H. and Herman G.E. , NSDHL, an enzyme involved in cholesterol biosynthesis, traffics through the Golgi and accumulates on ER membranes and on the surface of lipid droplets, Hum Mol Genet 12: (22) ((2003) ), 2981–2991. |
[79] | Liu X.Y. , Dangel A.W. , Kelley R.I. , Zhao W. , Denny P. , Botcherby M. et al., The gene mutated in bare patches and striated mice encodes a novel 3beta-hydroxysteroid dehydrogenase, Nat Genet 22: (2) ((1999) ), 182–187. |
[80] | Konig A. , Happle R. , Bornholdt D. , Engel H. and Grzeschik K.H. , Mutations in the NSDHL gene, encoding a 3beta-hydroxysteroid dehydrogenase, cause CHILD syndrome, American Journal of Medical Genetics 90: (4) ((2000) ), 339–346. |
[81] | Saito M. and Ishiko A. , A novel silent mutation in the NSDHL gene causing CHILD syndrome as a result of aberrant splicing, Brit J Dermatol 159: (5) ((2008) ), 1204–1206. |
[82] | Schmidt-Sidor B. , Obersztyn E. , Szymanska K. , Wychowski J. , Mierzewska H. , Wierzba-Bobrowicz T. et al., Brain and cerebellar hemidysplasia in a case with ipsilateral body dysplasia and suspicion of CHILD syndrome, Folia Neuropathologica / Association of Polish Neuropathologists and Medical Research Centre, Polish Academy of Sciences 46: (3) ((2008) ), 232–237. |
[83] | Avgerinou G.P. , Asvesti A.P. , Katsambas A.D. , Nikolaou V.A. , Christofidou E.C. , Grzeschik K.H. et al., CHILD syndrome: The NSDHL gene and its role in CHILD syndrome, a rare hereditary disorder, Journal of the European Academy of Dermatology and Venereology: JEADV 24: (6) ((2010) ), 733–736. |
[84] | Yang Z. , Hartmann B. , Xu Z. , Ma L. , Happle R. , Schlipf N. , et al. Large Deletions in the NSDHL Gene in Two Patients with CHILD Syndrome. Acta dermato-venereologica. (2015) . |
[85] | Bornholdt D. , Konig A. , Happle R. , Leveleki L. , Bittar M. , Danarti R. et al., Mutational spectrum of NSDHL in CHILD syndrome, Journal of Medical Genetics 42: (2) ((2005) ), e17. |
[86] | Kim C.A. , Konig A. , Bertola D.R. , Albano L.M. , Gattas G.J. , Bornholdt D. et al., CHILD syndrome caused by a deletion of exons 6-8 of the NSDHL gene, Dermatology 211: (2) ((2005) ), 155–158. |
[87] | Raychaudhury T. , George R. , Mandal K. , Srivastava V.M. , Thomas M. , Bornholdt D. et al., A novel X-chromosomal microdeletion encompassing congenital hemidysplasia with ichthyosiform erythroderma and limb defects, Pediatric Dermatology 30: (2) ((2013) ), 250–252. |
[88] | Hummel M. , Cunningham D. , Mullett C.J. , Kelley R.I. and Herman G.E. , Left-sided CHILD syndrome caused by a nonsense mutation in the NSDHL gene, American Journal of Medical Genetics Part A 122A: (3) ((2003) ), 246–251. |
[89] | Bittar M. , Happle R. , Grzeschik K.H. , Leveleki L. , Hertl M. , Bornholdt D. et al., CHILD syndrome in 3 generations: The importance of mild or minimal skin lesions, Archives of Dermatology 142: (3) ((2006) ), 348–351. |
[90] | NSDHL-Related Disorders [Internet]. University of Washington, Seattle. 2013. Available from: http://www.ncbi.nlm.nih.gov/books/NBK51754/. |
[91] | Happle R. , Mittag H. and Kuster W. , The CHILD nevus: A distinct skin disorder, Dermatology 191: (3) ((1995) ), 210–216. |
[92] | Konig A. , Happle R. , Fink-Puches R. , Soyer H.P. , Bornholdt D. , Engel H. et al., A novel missense mutation of NSDHL in an unusual case of CHILD syndrome showing bilateral, almost symmetric involvement, J Am Acad Dermatol 46: (4) ((2002) ), 594–596. |
[93] | Happle R. , Ptychotropism as a Cutaneous Feature of the Child Syndrome, J Am Acad Dermatol 23: (4) ((1990) ), 763–766. |
[94] | Happle R. , Mosaicism in human skin. Understanding the patterns and mechanisms, Archives of Dermatology 129: (11) ((1993) ), 1460–1470. |
[95] | Hebert A.A. , Esterly N.B. , Holbrook K.A. and Hall J.C. , The CHILD syndrome. Histologic and ultrastructural studies, Archives of Dermatology 123: (4) ((1987) ), 503–509. |
[96] | Hashimoto K. , Prada S. , Lopez A.P. , Hoyos J.G. and Escobar M. , CHILD syndrome with linear eruptions, hypopigmented bands, and verruciform xanthoma, Pediatric Dermatology 15: (5) ((1998) ), 360–366. |
[97] | Ishibashi M. , Matsuda F. , Oka H. and Ishiko A. , Abnormal lamellar granules in a case of CHILD syndrome, Journal of Cutaneous Pathology 33: (6) ((2006) ), 447–453. |
[98] | Happle R. , Effendy I. , Megahed M. , Orlow S.J. and Kuster W. , CHILD syndrome in a boy, American Journal of Medical Genetics 62: (2) ((1996) ), 192–194. |
[99] | Grange D.K. , Kratz L.E. , Braverman N.E. and Kelley R.I. , CHILD syndrome caused by deficiency of 3beta-hydroxysteroid-delta8, delta7-isomerase, American Journal of Medical Genetics 90: (4) ((2000) ), 328–335. |
[100] | Paller A.S. , van Steensel M.A. , Rodriguez-Martin M., Sorrell J., Heath C., Crumrine D., et al., Pathogenesis-based therapy reverses cutaneous abnormalities in an inherited disorder of distal cholesterol metabolism, The Journal of Investigative Dermatology 131: (11) ((2011) ), 2242–2248. |
[101] | Liu T. , Qian G. , Wang X.X. and Zhang Y.G. , CHILD syndrome: Effective treatment of ichthyosiform naevus with oral and topical ketoconazole, Acta Dermato-Venereologica 95: (1) ((2015) ), 91–92. |
[102] | du Souich C. , Chou A., Yin J., Oh T., Nelson T.N., Hurlburt J., et al., Characterization of a new X-linked mental retardation syndrome with microcephaly, cortical malformation, and thin habitus, American Journal of Medical Genetics Part A 149A: (11) ((2009) ), 2469–2478. |
[103] | McLarren K.W. , Severson T.M. and du Souich C. , Stockton D.W., Kratz L.E., Cunningham D., et al., Hypomorphic temperature-sensitive alleles of NSDHL cause CK syndrome, American Journal of Human Genetics 87: (6) ((2010) ), 905–914. |
[104] | Braverman N. , Lin P. , Moebius F.F. , Obie C. , Moser A. , Glossmann H. et al., Mutations in the gene encoding 3 beta-hydroxysteroid-delta 8, delta 7-isomerase cause X-linked dominant Conradi-Hunermann syndrome, Nat Genet 22: (3) ((1999) ), 291–294. |
[105] | Derry J.M. , Gormally E. , Means G.D. , Zhao W. , Meindl A. , Kelley R.I. et al., Mutations in a delta 8-delta 7 sterol isomerase in the tattered mouse and X-linked dominant chondrodysplasia punctata. [email protected], Nat Genet 22: (3) ((1999) ), 286–290. |
[106] | Schindelhauer D. , Hellebrand H. , Grimm L. , Bader I. , Meitinger T. , Wehnert M. et al., Long-range map of a 3.5-Mb region in Xp11.23-22 with a sequence-ready map from a 1.1-Mb gene-rich interval, Genome Research 6: (11) ((1996) ), 1056–1069. |
[107] | Hanner M. , Moebius F.F. , Weber F. , Grabner M. , Striessnig J. and Glossmann H. , Phenylalkylamine Ca2+ antagonist binding protein, Molecular cloning, tissue distribution, and heterologous expression. The Journal of Biological Chemistry, 270: (13) ((1995) ), 7551–7557. |
[108] | Moebius F.F. , Soellner K.E. , Fiechtner B. , Huck C.W. , Bonn G. and Glossmann H. , Histidine77, glutamic acid81, glutamic acid123, threonine126, asparagine194, and tryptophan197 of the human emopamil binding protein are required for in vivo sterol delta 8-delta 7 isomerization, Biochemistry 38: (3) ((1999) ), 1119–1127. |
[109] | Silve S. , Dupuy P.H. , LabitLebouteiller C. , Kaghad M. , Chalon P. , Rahier A. et al., Emopamil-binding protein, a mammalian protein that binds a series of structurally diverse neuroprotective agents, exhibits Delta 8-Delta 7 sterol isomerase activity in yeast, Journal of Biological Chemistry 271: (37) ((1996) ), 22434–22440. |
[110] | [cited 7/20/2015]. Available from: http://www.hgmd.cf.ac.uk/ac/index.php. |
[111] | Has C. , Bruckner-Tuderman L. , Muller D. , Floeth M. , Folkers E. and Donnai D. , et al., The Conradi-Hunermann-Happle syndrome (CDPX2) and emopamil binding protein: Novel mutations, and somatic and gonadal mosaicism, Hum Mol Genet 9: (13) ((2000) ), 1951–1955. |
[112] | Feldmeyer L. , Mevorah B. , Grzeschik K.H. , Huber M. and Hohl D. , Clinical variation in X-linked dominant chondrodysplasia punctata (X-linked dominant ichthyosis), The British Journal of Dermatology 154: (4) ((2006) ), 766–769. |
[113] | Happle R. , X-linked dominant chondrodysplasia punctata. Review of literature and report of a case, Human Genetics 53: (1) ((1979) ), 65–73. |
[114] | Manzke H. , Christophers E. and Wiedemann H.R. , Dominant sex-linked inherited chondrodysplasia punctata: A distinct type of chondrodysplasia punctata, Clinical Genetics 17: (2) ((1980) ), 97–107. |
[115] | Ikegawa S. , Ohashi H. , Ogata T. , Honda A. , Tsukahara M. , Kubo T. et al., Novel and recurrent EBP mutations in X-linked dominant chondrodysplasia punctata, American Journal of Medical Genetics 94: (4) ((2000) ), 300–305. |
[116] | Hoang M.P. , Carder K.R. , Pandya A.G. and Bennett M.J. , Ichthyosis and keratotic follicular plugs containing dystrophic calcification in newborns: Distinctive histopathologic features of x-linked dominant chondrodysplasia punctata (Conradi-Hunermann-Happle syndrome), The American Journal of Dermatopathology 26: (1) ((2004) ), 53–58. |
[117] | Akiyama M. , Sakai K. , Hayasaka K. , Tabata N. , Yamada M. , Ujiie H. et al., Conradi-Hunermann-Happle syndrome with abnormal lamellar granule contents, The British Journal of Dermatology 160: (6) ((2009) ), 1335–1337. |
[118] | Furtado L.V. , Botto L. , Carey J.C. , Stevenson D.A. , Viskochil D.H. and Male editors. , infant with ectodermal and biochemical phenotype of CDPX2 without chondrodysplasia punctata, Proc Greenwood Genet Center 27: ((2007) ), 124. |
[119] | Furtado L.V. , Bayrak-Toydemir P. , Hulinsky B. , Damjanovich K. , Carey J.C. and Rope A.F. , A novel X-linked multiple congenital anomaly syndrome associated with an EBP mutation, American Journal of Medical Genetics Part A 152A: (11) ((2010) ), 2838–2844. |
[120] | Kelley R.I. , Maegawa G.H. , Leite J.C. , Kratz L. and The editors. , male with Conradi–Hunermann syndrome (CDPX2): A distinct phenotype, Proc Greenwood Genet Center 24: (3) ((2005) ), 95. |
[121] | Milunsky J.M. , Maher T.A. and Metzenberg A.B. , Molecular, biochemical, and phenotypic analysis of a hemizygous male with a severe atypical phenotype for X-linked dominant Conradi-Hunermann-Happle syndrome and a mutation in EBP, American Journal of Medical Genetics Part A 116A: (3) ((2003) ), 249–254. |
[122] | Bode H. , Galm C. , Hummler H. , Teller C. , Haas D. and Gencik M. , Non-lethal non-mosaic male with Conradi-Hunermann syndrome caused by a novel EBP c.356T>G mutation, American Journal of Medical Genetics Part A 161A: (9) ((2013) ), 2385–2388. |
[123] | Uwechue I.C. , Cooper B.F. , Goble C. , Hacker T. , Blair H.J. , Burke D.T. et al., The mouse X-linked developmental mutant, tattered, lies between DXMit55 and Xkh and is associated with hyperkeratinization, Genomics 37: (2) ((1996) ), 238–241. |
[124] | Matsushima M. , Inazawa J. , Takahashi E. , Suzumori K. and Nakamura Y. , Molecular cloning and mapping of a human cDNA (SC5DL) encoding a protein homologous to fungal sterol-C5-desaturase, Cytogenetics and Cell Genetics 74: (4) ((1996) ), 252–254. |
[125] | Nishi S. , Nishino H. and Ishibashi T. , cDNA cloning of the mammalian sterol C5-desaturase and the expression in yeast mutant, Biochimica et Biophysica Acta 1490: (1-2) ((2000) ), 106–108. |
[126] | Sugawara T. , Fujimoto Y. and Ishibashi T. , Molecular cloning and structural analysis of human sterol C5 desaturase, Biochimica et Biophysica Acta 1533: (3) ((2001) ), 277–284. |
[127] | Arthington B.A. , Bennett L.G. , Skatrud P.L. , Guynn C.J. , Barbuch R.J. , Ulbright C.E. et al., Cloning, disruption and sequence of the gene encoding yeast C-5 sterol desaturase, Gene 102: (1) ((1991) ), 39–44. |
[128] | Brunetti-Pierri N. , Corso G. , Rossi M. , Ferrari P. , Balli F. , Rivasi F. et al., Lathosterolosis, a novel multiple-malformation/mental retardation syndrome due to deficiency of 3 beta-hydroxysteroid-Delta(5)-desaturase, American Journal of Human Genetics 71: (4) ((2002) ), 952–958. |
[129] | Rossi M. , D’Armiento M. , Parisi I. , Ferrari P. , Hall C.M. , Cervasio M. et al., Clinical phenotype of lathosterolosis, American Journal of Medical Genetics Part A 143A: (20) ((2007) ), 2371–2381. |
[130] | Krakowiak P.A. , Wassif C.A. , Kratz L. , Cozma D. , Kovarova M. , Harris G. et al., Lathosterolosis: An inborn error of human and murine cholesterol synthesis due to lathosterol 5-desaturase deficiency, Hum Mol Genet 12: (13) ((2003) ), 1631–1641. |
[131] | Ho A.C. , Fung C.W. , Siu T.S. , Ma O.C. , Lam C.W. , Tam S. et al., Lathosterolosis: A disorder of cholesterol biosynthesis resembling smith-lemli-opitz syndrome, JIMD reports 12: ((2014) ), 129–134. |
[132] | Jiang X.S. , Backlund P.S. , Wassif C.A. , Yergey A.L. and Porter F.D. , Quantitative proteomics analysis of inborn errors of cholesterol synthesis: Identification of altered metabolic pathways in DHCR7 and SC5D deficiency, Molecular & Cellular Proteomics: MCP 9: (7) ((2010) ), 1461–1475. |
[133] | Calvo P.L. , Brunati A. , Spada M. , Romagnoli R. , Corso G. , Parenti G. et al., Liver transplantation in defects of cholesterol biosynthesis: The case of lathosterolosis, American Journal of Transplantation: Official Journal of the American Society of Transplantation and the American Society of Transplant Surgeons 14: (4) ((2014) ), 960–965. |
[134] | Smith D.W. , Lemli L. and Opitz J.M. , A Newly Recognized Syndrome of Multiple Congenital Anomalies, The Journal of Pediatrics 64: ((1964) ), 210–217. |
[135] | Irons M. , Elias E.R. , Salen G. , Tint G.S. and Batta A.K. , Defective cholesterol biosynthesis in Smith-Lemli-Opitz syndrome, Lancet 341: (8857) ((1993) ), 1414. |
[136] | Tint G.S. , Irons M. , Elias E.R. , Batta A.K. , Frieden R. , Chen T.S. et al., Defective cholesterol biosynthesis associated with the Smith-Lemli-Opitz syndrome, The New England Journal of Medicine 330: (2) ((1994) ), 107–113. |
[137] | Natowicz M.R. and Evans J.E. , Abnormal bile acids in the Smith-Lemli-Opitz syndrome, American Journal of Medical Genetics 50: (4) ((1994) ), 364–367. |
[138] | Miller W.L. , P450 oxidoreductase. In: Estein CJ, Erickson RP, Wynshaw-Boris A, editors. Inborn Errors of Development 2nd ed: Oxford University Press; (2008) ; pp. 1531–1539. |
[139] | Irons M. , DHCR7 and the Smith-Lemli-Opitz (RSH) syndrome and Cyclopamine Teratogenesis. In: Estein CJ, Erickson RP, Wynshaw-Boris A, editors. Inborn Errors of Development 2nd ed: Oxford University Press; (2008) ; pp. 1531–1539. |
[140] | Yu H. and Patel S.B. , Recent insights into the Smith-Lemli-Opitz syndrome, Clinical Genetics 68: (5) ((2005) ), 383–391. |
[141] | Porter F.D. , Human malformation syndromes due to inborn errors of cholesterol synthesis, Curr Opin Pediatr 15: (6) ((2003) ), 607–613. |
[142] | Herman G.E. , Disorders of cholesterol biosynthesis: Prototypic metabolic malformation syndromes, Hum Mol Genet 12 Spec No 1: ((2003) ), R75–R88. |
[143] | Porter F.D. , Malformation syndromes due to inborn errors of cholesterol synthesis, J Clin Invest 110: (6) ((2002) ), 715–724. |
[144] | Herman G.E. , X-linked dominant disorders of cholesterol biosynthesis in man and mouse, Bba-Mol Cell Biol L 1529: (1-3) ((2000) ), 357–373. |
[145] | Hammerschmidt M. , Brook A. and McMahon A.P. , The world according to hedgehog, Trends in Genetics: TIG 13: (1) ((1997) ), 14–21. |
[146] | Nowaczyk M.J. , Whelan D.T. and Hill R.E. , Smith-Lemli-Opitz syndrome: Phenotypic extreme with minimal clinical findings, American Journal of Medical Genetics 78: (5) ((1998) ), 419–423. |
[147] | Opitz J.M. , Smith-Lemli-Opitz syndrome: Clinical and biochemical findings in Brazilian patients. Scalco et al. Genetics and molecular biology (this issue), Genet Mol Biol 29: (3) ((2006) ), 437–438. |
[148] | Rutledge J.C. , Friedman J.M. , Harrod M.J. , Currarino G. , Wright C.G. , Pinckney L. et al., A “new” lethal multiple congenital anomaly syndrome: Joint contractures, cerebellar hypoplasia, renal hypoplasia, urogenital anomalies, tongue cysts, shortness of limbs, eye abnormalities, defects of the heart, gallbladder agenesis, and ear malformations, American Journal of Medical Genetics 19: (2) ((1984) ), 255–264. |
[149] | Curry C.J. , Carey J.C. , Holland J.S. , Chopra D. , Fineman R. , Golabi M. et al., Smith-Lemli-Opitz syndrome-type II: Multiple congenital anomalies with male pseudohermaphroditism and frequent early lethality, American Journal of Medical Genetics 26: (1) ((1987) ), 45–57. |
[150] | Donnai D. , Young I.D. , Owen W.G. , Clark S.A. , Miller P.F. and Knox W.F. , The lethal multiple congenital anomaly syndrome of polydactyly, sex reversal, renal hypoplasia, and unilobular lungs, Journal of Medical Genetics 23: (1) ((1986) ), 64–71. |
[151] | Oostra R.J. , Baljet B. , Schutgens R.B. and Hennekam R.C. , Smith-Lemli-Opitz syndrome diagnosed in a 130-year-old anatomical specimen, American Journal of Medical Genetics 68: (3) ((1997) ), 257–259. |
[152] | Weaver D.D. , Solomon B.D. , Akin-Samson K. , Kelley R.I. and Muenke M. , Cyclopia (synophthalmia) in Smith-Lemli-Opitz syndrome: First reported case and consideration of mechanism, American Journal of Medical Genetics Part C, Seminars in Medical Genetics 154C: (1) ((2010) ), 142–145. |
[153] | Ryan A.K. , Bartlett K. , Clayton P. , Eaton S. , Mills L. , Donnai D. et al., Smith-Lemli-Opitz syndrome: A variable clinical and biochemical phenotype, Journal of Medical Genetics 35: (7) ((1998) ), 558–565. |
[154] | Charman C.R. , Ryan A. , Tyrrell R.M. , Pearse A.D. , Arlett C.F. , Kurwa H.A. et al., Photosensitivity associated with the Smith-Lemli-Opitz syndrome, The British Journal of Dermatology 138: (5) ((1998) ), 885–888. |
[155] | Elias E.R. , Irons M.B. , Hurley A.D. , Tint G.S. and Salen G. , Clinical effects of cholesterol supplementation in six patients with the Smith-Lemli-Opitz syndrome (SLOS), American Journal of Medical Genetics 68: (3) ((1997) ), 305–310. |
[156] | Azurdia R.M. , Anstey A.V. and Rhodes L.E. , Cholesterol supplementation objectively reduces photosensitivity in the Smith-Lemli-Opitz syndrome, The British Journal of Dermatology 144: (1) ((2001) ), 143–145. |
[157] | Anstey A.V. , Ryan A. , Rhodes L.E. , Charman C.R. , Arlett C.F. , Tyrrell R.M. et al., Characterization of photosensitivity in the Smith-Lemli-Opitz syndrome: A new congenital photosensitivity syndrome, The British Journal of Dermatology 141: (3) ((1999) ), 406–414. |
[158] | Chignell C.F. , Kukielczak B.M. , Sik R.H. , Bilski P.J. and He Y.Y. , Ultraviolet A sensitivity in Smith-Lemli-Opitz syndrome: Possible involvement of cholesta-5,7,9(11)-trien-3 beta-ol, Free Radical Biology & Medicine 41: (2) ((2006) ), 339–346. |
[159] | Valencia A. , Rajadurai A. , Carle A.B. and Kochevar I.E. , 7-Dehydrocholesterol enhances ultraviolet A-induced oxidative stress in keratinocytes: Roles of NADPH oxidase, mitochondria, and lipid rafts, Free Radical Biology & Medicine 41: (11) ((2006) ), 1704–1718. |
[160] | Bianconi S.E. , Conley S.K. , Keil M.F. , Sinaii N. , Rother K.I. , Porter F.D. et al., Adrenal function in Smith-Lemli-Opitz syndrome, American Journal of Medical Genetics Part A 155A: (11) ((2011) ), 2732–2738. |
[161] | Andersson H.C. , Frentz J. , Martinez J.E. , Tuck-Muller C.M. and Bellizaire J. , Adrenal insufficiency in Smith-Lemli-Opitz syndrome, American Journal of Medical Genetics 82: (5) ((1999) ), 382–384. |
[162] | Chemaitilly W. , Goldenberg A. , Baujat G. , Thibaud E. , Cormier-Daire V. and Abadie V. , Adrenal insufficiency and abnormal genitalia in a 46XX female with Smith-Lemli-Opitz syndrome, Horm Res 59: (5) ((2003) ), 254–256. |
[163] | Nowaczyk M.J. , Siu V.M. , Krakowiak P.A. and Porter F.D. , Adrenal insufficiency and hypertension in a newborn infant with Smith-Lemli-Opitz syndrome, American Journal of Medical Genetics 103: (3) ((2001) ), 223–225. |
[164] | Dallaire L. , Mitchell G. , Giguere R. , Lefebvre F. , Melancon S.B. and Lambert M. , Prenatal-diagnosis of smith-lemli-opitz syndrome is possible by measurement of 7-dehydrocholesterol in amniotic-fluid, Prenatal Diag 15: (9) ((1995) ), 855–858. |
[165] | Kratz L.E. and Kelley R.I. , Prenatal diagnosis of the RSH/Smith-Lemli-Opitz syndrome, American Journal of Medical Genetics 82: (5) ((1999) ), 376–381. |
[166] | Shackleton C.H. , Marcos J. , Palomaki G.E. , Craig W.Y. , Kelley R.I. , Kratz L.E. et al., Dehydrosteroid measurements in maternal urine or serum for the prenatal diagnosis of Smith-Lemli-Opitz syndrome (SLOS), American Journal of Medical Genetics Part A 143A: (18) ((2007) ), 2129–2136. |
[167] | Shackleton C.H. , Roitman E. and Kelley R. , Neonatal urinary steroids in Smith-Lemli-Opitz syndrome associated with 7-dehydrocholesterol reductase deficiency, Steroids 64: (7) ((1999) ), 481–490. |
[168] | Shackleton C.H. , Roitman E. , Kratz L.E. and Kelley R.I. , Midgestational maternal urine steroid markers of fetal Smith-Lemli-Opitz (SLO) syndrome (7-dehydrocholesterol 7-reductase deficiency), Steroids 64: (7) ((1999) ), 446–452. |
[169] | Loeffler J. , Utermann G. and Witsch-Baurngartner M. , Molecular prenatal diagnosis of Smith-Lemli-Opitz syndrome is reliable and efficient, Prenatal Diag 22: (9) ((2002) ), 827–830. |
[170] | Schoen E. , Norem C. , O’Keefe J. , Krieger R. , Walton D. and To T.T. , Maternal serum unconjugated estriol as a predictor for Smith-Lemli-Opitz syndrome and other fetal conditions, Obstet Gynecol 102: (1) ((2003) ), 167–172. |
[171] | Rossiter J.P. , Hofman K.J. and Kelley R.I. , Smith-Lemli-Opitz syndrome: Prenatal diagnosis by quantification of cholesterol precursors in amniotic fluid, American Journal of Medical Genetics 56: (3) ((1995) ), 272–275. |
[172] | Kelley R.I. , Diagnosis of Smith-Lemli-Opitz syndrome by gas chromatography/mass spectrometry of 7-dehydrocholesterol in plasma, amniotic fluid and cultured skin fibroblasts, Clinica Chimica acta; International Journal of Clinical Chemistry 236: (1) ((1995) ), 45–58. |
[173] | Moebius F.F. , Fitzky B.U. , Lee J.N. , Paik Y.K. and Glossmann H. , Molecular cloning and expression of the human delta7-sterol reductase, Proceedings of the National Academy of Sciences of the United States of America 95: (4) ((1998) ), 1899–1902. |
[174] | Correa-Cerro L.S. and Porter F.D. , 3beta-hydroxysterol Delta7-reductase and the Smith-Lemli-Opitz syndrome, Molecular Genetics and Metabolism 84: (2) ((2005) ), 112–126. |
[175] | Yu H.W. , Tint G.S. , Salen G. and Patel S.B. , Detection of a common mutation in the RSH or Smith-Lemli-Opitz syndrome by a PCR-RFLP assay: IVS8-1G ->C is found in over sixty percent of US propositi, American Journal of Medical Genetics 90: (4) ((2000) ), 347–50. |
[176] | Witsch-Baumgartner M. , Ciara E. , Loffler J. , Menzel H.J. , Seedorf U. , Burn J. et al., Frequency gradients of DHCR7 mutations in patients with Smith-Lemli-Opitz syndrome in Europe: Evidence for different origins of common mutations, European Journal of Human Genetics: EJHG 9: (1) ((2001) ), 45–50. |
[177] | Jezela-Stanek A. , Ciara E. , Malunowicz E. , Chrzanowska K. , Latos-Bielenska A. and Krajewska-Walasek M. , et al. Differences between predicted and established diagnoses of Smith-Lemli-Opitz syndrome in the Polish population: Underdiagnosis or loss of affected fetuses? Journal of Inherited Metabolic Disease 33: ((2010) ), S241–S248. |
[178] | Berry D. and Hypponen E. , Determinants of vitamin D status: Focus on genetic variations, Current Opinion in Nephrology and Hypertension 20: (4) ((2011) ), 331–276. |
[179] | Roux C. and Aubry M. , [Teratogenic action in the rat of an inhibitor of cholesterol synthesis, AY 9944], Comptes Rendus des Seances de la Societe de Biologie et de Ses Filiales 160: (7) ((1966) ), 1353–1357. |
[180] | Roux C. , Horvath C. and Dupuis R. , Teratogenic action and embryo lethality of AY 9944R. Prevention by a hypercholesterolemia-provoking diet, Teratology 19: (1) ((1979) ), 35–38. |
[181] | Roux C. , Dupuis R. , Horvath C. and Talbot J.N. , Teratogenic effect of an inhibitor of cholesterol synthesis (AY 9944) in rats: Correlation with maternal cholesterolemia, The Journal of Nutrition 110: (11) ((1980) ), 2310–2312. |
[182] | Roux C. , Wolf C. , Mulliez N. , Gaoua W. , Cormier V. , Chevy F. et al., Role of cholesterol in embryonic development, The American Journal of Clinical Nutrition 71: (5 Suppl) ((2000) ), 1270S–1279S. |
[183] | Barbu V. , Roux C. , Dupuis R. , Gardette J. and Maziere J.C. , Teratogenic effect of AY 9944 in rats: Importance of the day of administration and maternal plasma cholesterol level, Proceedings of the Society for Experimental Biology and Medicine Society for Experimental Biology and Medicine 176: (1) ((1984) ), 54–59. |
[184] | Barbu V. , Roux C. , Lambert D. , Dupuis R. , Gardette J. , Maziere J.C. et al., Cholesterol prevents the teratogenic action of AY 9944: Importance of the timing of cholesterol supplementation to rats, The Journal of Nutrition 118: (6) ((1988) ), 774–779. |
[185] | Repetto M. , Maziere J.C. , Citadelle D. , Dupuis R. , Meier M. , Biade S. et al., Teratogenic effect of the cholesterol synthesis inhibitor AY 9944 on rat embryos in vitro, Teratology 42: (6) ((1990) ), 611–618. |
[186] | Wolf C. , Chevy F. , Pham J. , Kolf-Clauw M. , Citadelle D. , Mulliez N. et al., Changes in serum sterols of rats treated with 7-dehydrocholesterol-delta 7-reductase inhibitors: Comparison to levels in humans with Smith-Lemli-Opitz syndrome, Journal of Lipid Research 37: (6) ((1996) ), 1325–1333. |
[187] | Batta A.K. and Salen G. , Abnormal cholesterol biosynthesis produced by AY 9944 in the rat leads to skeletal deformities similar to the Smith-Lemli-Opitz syndrome, The Journal of Laboratory and Clinical Medicine 131: (3) ((1998) ), 192–193. |
[188] | Honda M. , Tint G.S. , Honda A. , Batta A.K. , Chen T.S. , Shefer S. et al., Measurement of 3 beta-hydroxysteroid Delta(7)-reductase activity in cultured skin fibroblasts utilizing ergosterol as a substrate: A new method for the diagnosis of the Smith-Lemli-Opitz syndrome, Journal of Lipid Research 37: (11) ((1996) ), 2433–2438. |
[189] | Lanoue L. , Dehart D.B. , Hinsdale M.E. , Maeda N. , Tint G.S. and Sulik K.K. , Limb, genital, CNS, and facial malformations result from gene/environment-induced cholesterol deficiency: Further evidence for a link to sonic hedgehog, American Journal of Medical Genetics 73: (1) ((1997) ), 24–31. |
[190] | Dehart D.B. , Lanoue L. , Tint G.S. and Sulik K.K. , Pathogenesis of malformations in a rodent model for Smith-Lemli-Opitz syndrome, American Journal of Medical Genetics 68: (3) ((1997) ), 328–337. |
[191] | Wassif C.A. , Zhu P. , Kratz L. , Krakowiak P.A. , Battaile K.P. , Weight F.F. et al., Biochemical, phenotypic and neurophysiological characterization of a genetic mouse model of RSH/Smith–Lemli–Opitz syndrome, Hum Mol Genet 10: (6) ((2001) ), 555–564. |
[192] | Fitzky B.U. , Moebius F.F. , Asaoka H. , Waage-Baudet H. , Xu L. , Xu G. et al., 7-Dehydrocholesterol-dependent proteolysis of HMG-CoA reductase suppresses sterol biosynthesis in a mouse model of Smith-Lemli-Opitz/RSH syndrome, J Clin Invest 108: (6) ((2001) ), 905–915. |
[193] | Battaile K.P. and Steiner R.D. , Smith-Lemli-Opitz syndrome: The first malformation syndrome associated with defective cholesterol synthesis, Molecular Genetics and Metabolism 71: (1-2) ((2000) ), 154–162. |
[194] | Battaile K.P. , Battaile B.C. , Merkens L.S. , Maslen C.L. and Steiner R.D. , Carrier frequency of the common mutation IVS8-1G>C in DHCR7 and estimate of the expected incidence of Smith-Lemli-Opitz syndrome, Molecular Genetics and Metabolism 72: (1) ((2001) ), 67–71. |
[195] | Nowaczyk M.J. and Waye J.S. , The Smith-Lemli-Opitz syndrome: A novel metabolic way of understanding developmental biology, embryogenesis, and dysmorphology, Clinical Genetics 59: (6) ((2001) ), 375–386. |
[196] | Craig W.Y. , Haddow J.E. , Palomaki G.E. , Kelley R.I. , Kratz L.E. , Shackleton C.H. et al., Identifying Smith-Lemli-Opitz syndrome in conjunction with prenatal screening for Down syndrome, Prenat Diagn 26: (9) ((2006) ), 842–849. |
[197] | Porter F.D. and Steiner R.D. , Research updates: Cholesterol supplementation in Smith-Lemli-Opitz syndrome, The Exchange (http://wwwsmithlemliopitzorg) 18: (1) ((2012) ), 16–18. |
[198] | Lu X. , Kambe F. , Cao X. , Kozaki Y. , Kaji T. , Ishii T. et al., 3 beta-hydroxysteroid-Delta 24 reductase is a hydrogen peroxide scavenger, protecting cells from oxidative stress-induced apoptosis, Endocrinology 149: (7) ((2008) ), 3267–3273. |
[199] | Kuehnle K. , Crameri A. , Kalin R.E. , Luciani P. , Benvenuti S. , Peri A. et al., Prosurvival effect of DHCR24/Seladin-1 in acute and chronic responses to oxidative stress, Molecular and Cellular Biology 28: (2) ((2008) ), 539–550. |
[200] | Wu C. , Miloslavskaya I. , Demontis S. , Maestro R. and Galaktionov K. , Regulation of cellular response to oncogenic and oxidative stress by Seladin-1, Nature 432: (7017) ((2004) ), 640–645. |
[201] | Waterham H.R. , Koster J. , Romeijn G.J. , Hennekam R.C. , Vreken P. , Andersson H.C. et al., Mutations in the 3beta-hydroxysterol Delta24-reductase gene cause desmosterolosis, an autosomal recessive disorder of cholesterol biosynthesis, American Journal of Human Genetics 69: (4) ((2001) ), 685–694. |
[202] | Greeve I. , Hermans-Borgmeyer I. , Brellinger C. , Kasper D. , Gomez-Isla T. , Behl C. et al., The human DIMINUTO/DWARF1 homolog seladin-1 confers resistance to Alzheimer’s disease-associated neurodegeneration and oxidative stress, The Journal of Neuroscience: The Official Journal of the Society for Neuroscience 20: (19) ((2000) ), 7345–7352. |
[203] | FitzPatrick D.R. , Keeling J.W. , Evans M.J. , Kan A.E. , Bell J.E. , Porteous M.E. et al., Clinical phenotype of desmosterolosis, American Journal of Medical Genetics 75: (2) ((1998) ), 145–152. |
[204] | Andersson H.C. , Kratz L. and Kelley R. , Desmosterolosis presenting with multiple congenital anomalies and profound developmental delay, American Journal of Medical Genetics 113: (4) ((2002) ), 315–319. |
[205] | Schaaf C.P. , Koster J. , Katsonis P. , Kratz L. , Shchelochkov O.A. , Scaglia F. et al., Desmosterolosis-phenotypic and molecular characterization of a third case and review of the literature, American Journal of Medical Genetics Part A 155A: (7) ((2011) ), 1597–1604. |
[206] | Dias C. , Rupps R. , Millar B. , Choi K. , Marra M. , Demos M. et al., Desmosterolosis: An illustration of diagnostic ambiguity of cholesterol synthesis disorders, Orphanet Journal of Rare Diseases 9: ((2014) ), 94. |
[207] | Ashraf J. , Feix J.B. and Butterfield D.A. , Membrane fluidity and myotonia: Effects of cholesterol and desmosterol on erythrocyte membrane fluidity in rats with 20,25-diazacholesterol-induced myotonia and on phospholipid liposomes, Bioscience Reports 4: (2) ((1984) ), 115–120. |
[208] | Dennick R.G. , Dean P.D. and Abramovich D.A. , Desmosterol levels in human foetal brain–a reassessment, Journal of Neurochemistry 20: (4) ((1973) ), 1293–1294. |
[209] | Connor W.E. , Lin D.S. and Neuringer M. , Biochemical markers for puberty in the monkey testis: Desmosterol and docosahexaenoic acid, The Journal of Clinical Endocrinology and Metabolism 82: (6) ((1997) ), 1911–1916. |
[210] | Awano M. , Kawaguchi A. , Morisaki M. and Mohri H. , Identification of cholesta-7,24-dien-3 beta-ol and desmosterol in hamster cauda epididymal spermatozoa, Lipids 24: (7) ((1989) ), 662–664. |
[211] | Fliesler S.J. , Richards M.J. , Miller C.Y. , Peachey N.S. and Cenedella R.J. , Retinal structure and function in an animal model that replicates the biochemical hallmarks of desmosterolosis, Neurochem Res 25: (5) ((2000) ), 685–694. |
[212] | Wechsler A. , Brafman A. , Shafir M. , Heverin M. , Gottlieb H. , Damari G. et al., Generation of viable cholesterol-free mice, Science 302: (5653) ((2003) ), 2087. |
[213] | Mirza R. , Hayasaka S. , Takagishi Y. , Kambe F. , Ohmori S. , Maki K. et al., DHCR24 gene knockout mice demonstrate lethal dermopathy with differentiation and maturation defects in the epidermis, The Journal of Investigative Dermatology 126: (3) ((2006) ), 638–647. |
[214] | Rodriguez-Acebes S. and de la Cueva P. , Fernandez-Hernando C., Ferruelo A.J., Lasuncion M.A., Rawson R.B., et al., Desmosterol can replace cholesterol in sustaining cell proliferation and regulating the SREBP pathway in a sterol-Delta(24)-reductase-deficient cell line, Biochem J 420: ((2009) ), 305–315. |
[215] | He M. , Kratz L.E. , Michel J.J. , Vallejo A.N. , Ferris L. , Kelley R.I. et al., Mutations in the human SC4MOL gene encoding a methyl sterol oxidase cause psoriasiform dermatitis, microcephaly, and developmental delay, J Clin Invest 121: (3) ((2011) ), 976–984. |
[216] | He M. , Smith L.D. , Chang R. , Li X. and Vockley J. , The role of sterol-C4-methyl oxidase in epidermal biology, Biochimica et Biophysica Acta 1841: (3) ((2014) ), 331–335. |
Figures and Tables
Fig.1
Cell membrane composition of archaeans, bacteria and eukaryotes. Adapted from http://www.ucmp.berkeley.edu/archaea/archaeamm.html and http://wikipedia.org/wiki/Archaea.
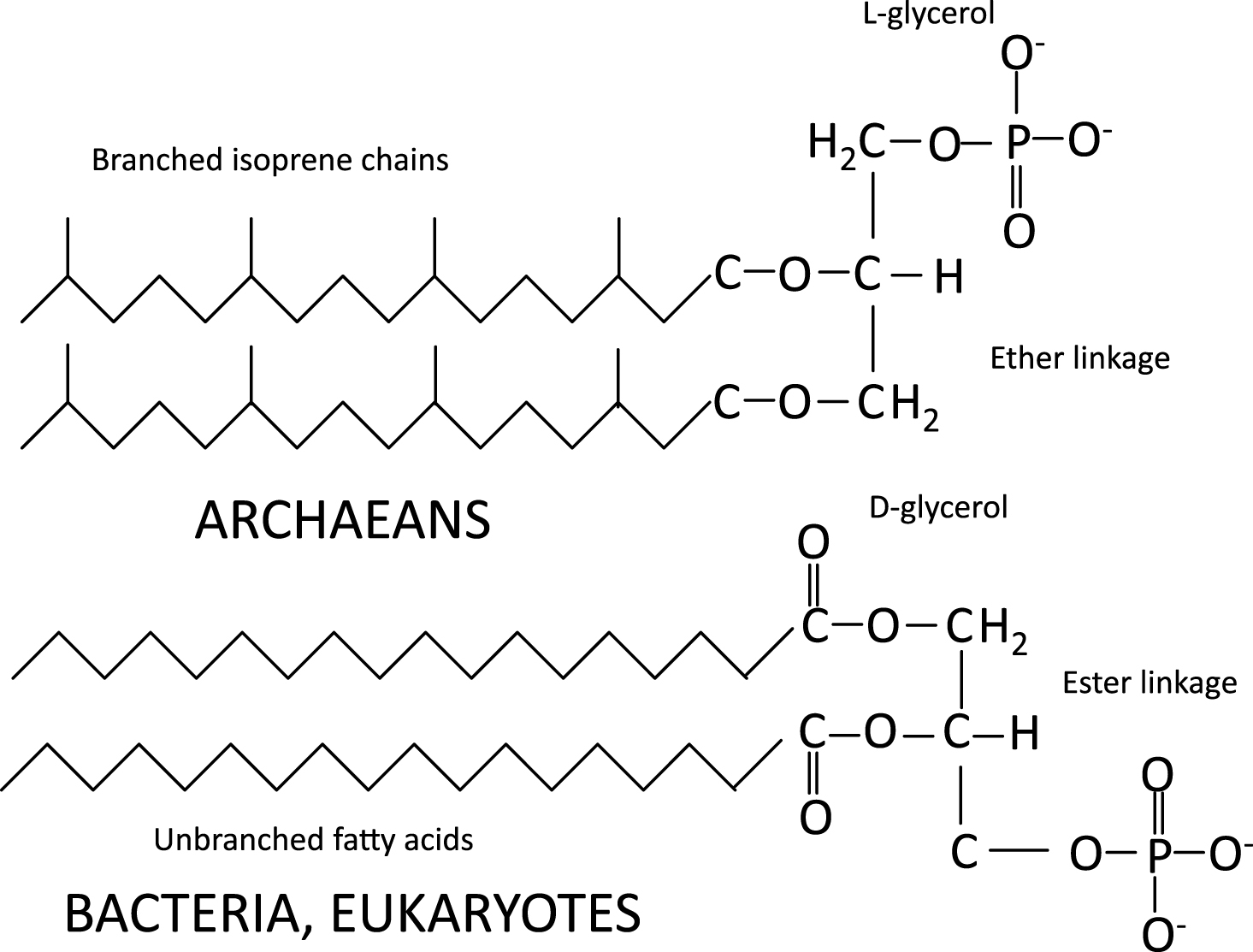
Fig.2
C30-desmethylsteranes. Note: C-24 isopropyl side chain, no double bonds and loss of 3ß-hydroxyl moiety. Adapted from Love et al. [1]. Molecule derived from demosponges over 635 million years ago (MYA).
![C30-desmethylsteranes. Note: C-24 isopropyl side chain, no double bonds and loss of 3ß-hydroxyl moiety. Adapted from Love et al. [1]. Molecule derived from demosponges over 635 million years ago (MYA).](https://content.iospress.com:443/media/trd/2016/1-2/trd-1-2-trd004/trd-1-trd004-g002.jpg)
Fig.3
a: Compound b in Fig. 1 of Kodner et al. [3]: C27 :4 Cholesta 5,7,9(11)-22-tetraene-3ßol. b: Compound c in Fig. 1 of Kodner et al. [3]: C27 :3 Cholesta, 5,7,22triene-3ßol, two molecules synthesized by the phylogenetically ancient choanoflagellate Monosiga brevicollis. Ergosterol (see Fig. 4) is the second most common sterol in choanoflagellates; hence, M. brevicollis must have a C-24 sterol-methyl transferase.
![a: Compound b in Fig. 1 of Kodner et al. [3]: C27 :4 Cholesta 5,7,9(11)-22-tetraene-3ßol. b: Compound c in Fig. 1 of Kodner et al. [3]: C27 :3 Cholesta, 5,7,22triene-3ßol, two molecules synthesized by the phylogenetically ancient choanoflagellate Monosiga brevicollis. Ergosterol (see Fig. 4) is the second most common sterol in choanoflagellates; hence, M. brevicollis must have a C-24 sterol-methyl transferase.](https://content.iospress.com:443/media/trd/2016/1-2/trd-1-2-trd004/trd-1-trd004-g003.jpg)
Fig.4
a: Plants synthesize sitosterol (hence, human sitosterolemia). b: Animals: cholesterol; c: Fungi and choanoflagellates: ergosterol, hence, probably had a common ancestor. Modified from Desmond and Gribaldo, 2009.
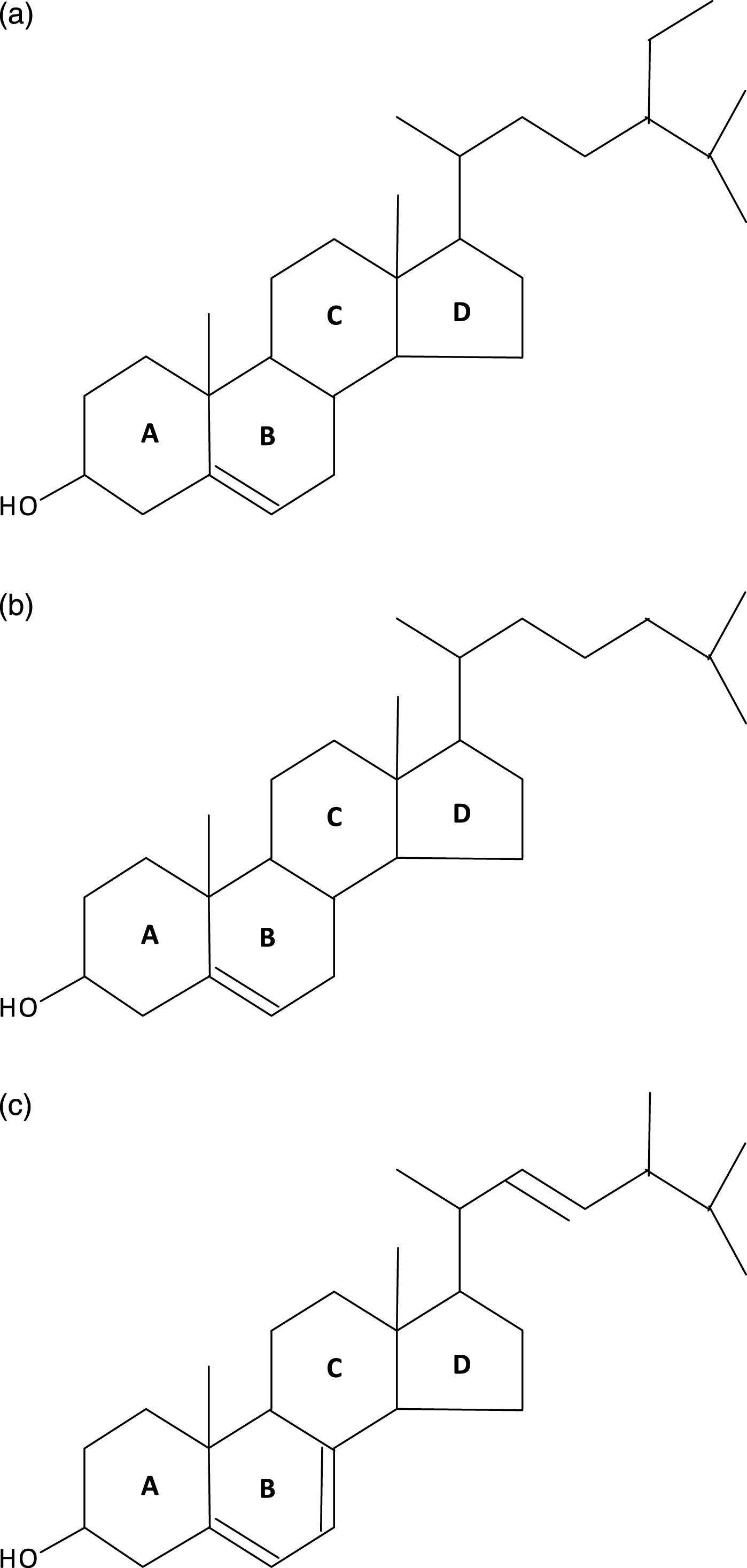
Fig.5
a: Humans; inability to convert 7-dehydrocholesterol to cholesterol due to mutant 7-dehydrocholesterol reductase causes RSH (so-called Smith-Lemli-Opitz, SLO) syndrome in the homozygous survivors of the severe embryonic and fetal selection against them. b: Plants (Arabidopsis thaliana) corresponding deficiency of this highly conserved enzyme with inability to convert ergosterol into brassicasterol causes the dwarf 5 mutations of A. thaliana.
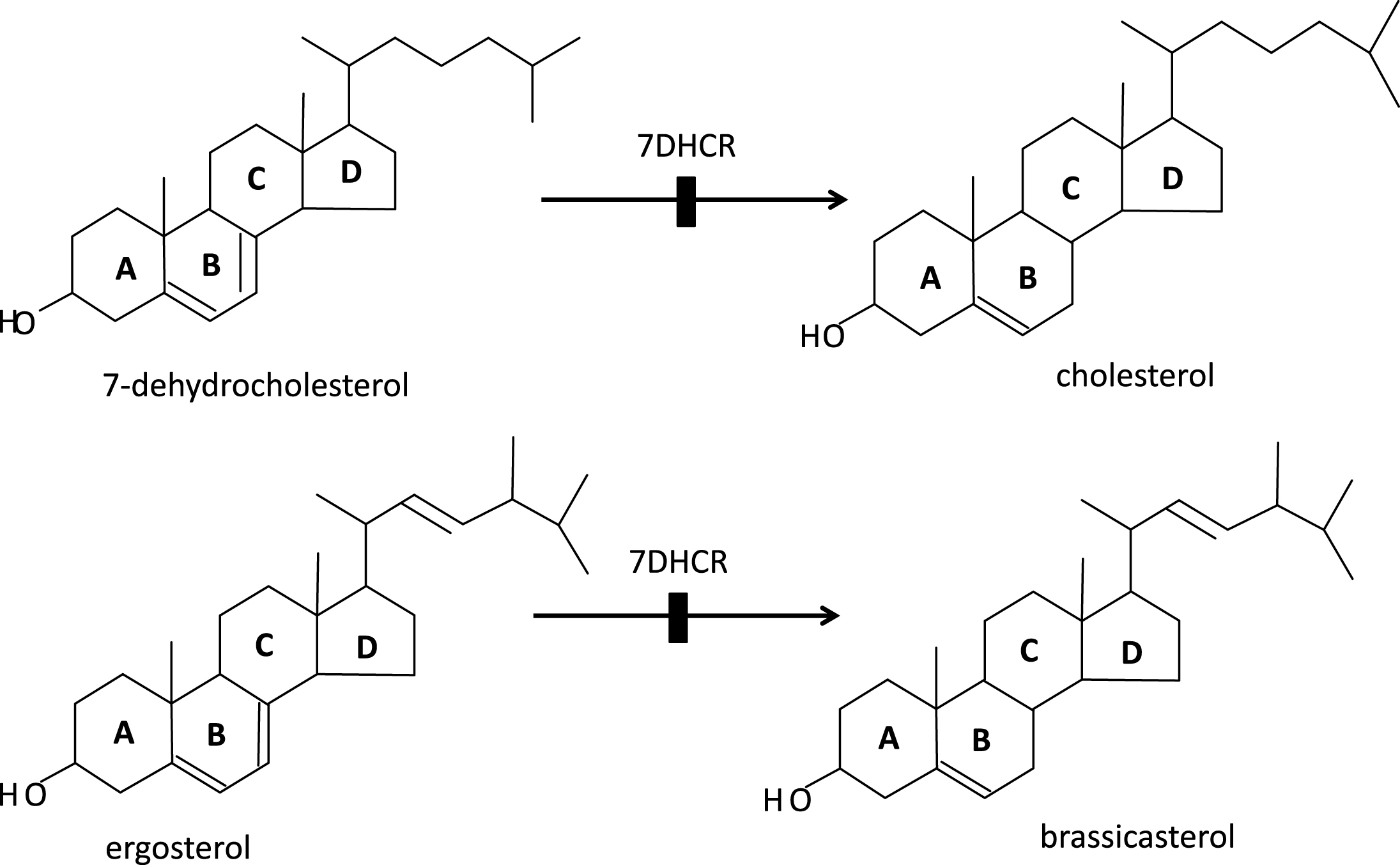
Fig.6
Ultimate synthesis of cholesterol in animals from squalene and of sitosterol in plants from squalene. Greatly modified from Desmond and Gribaldo, 2009.
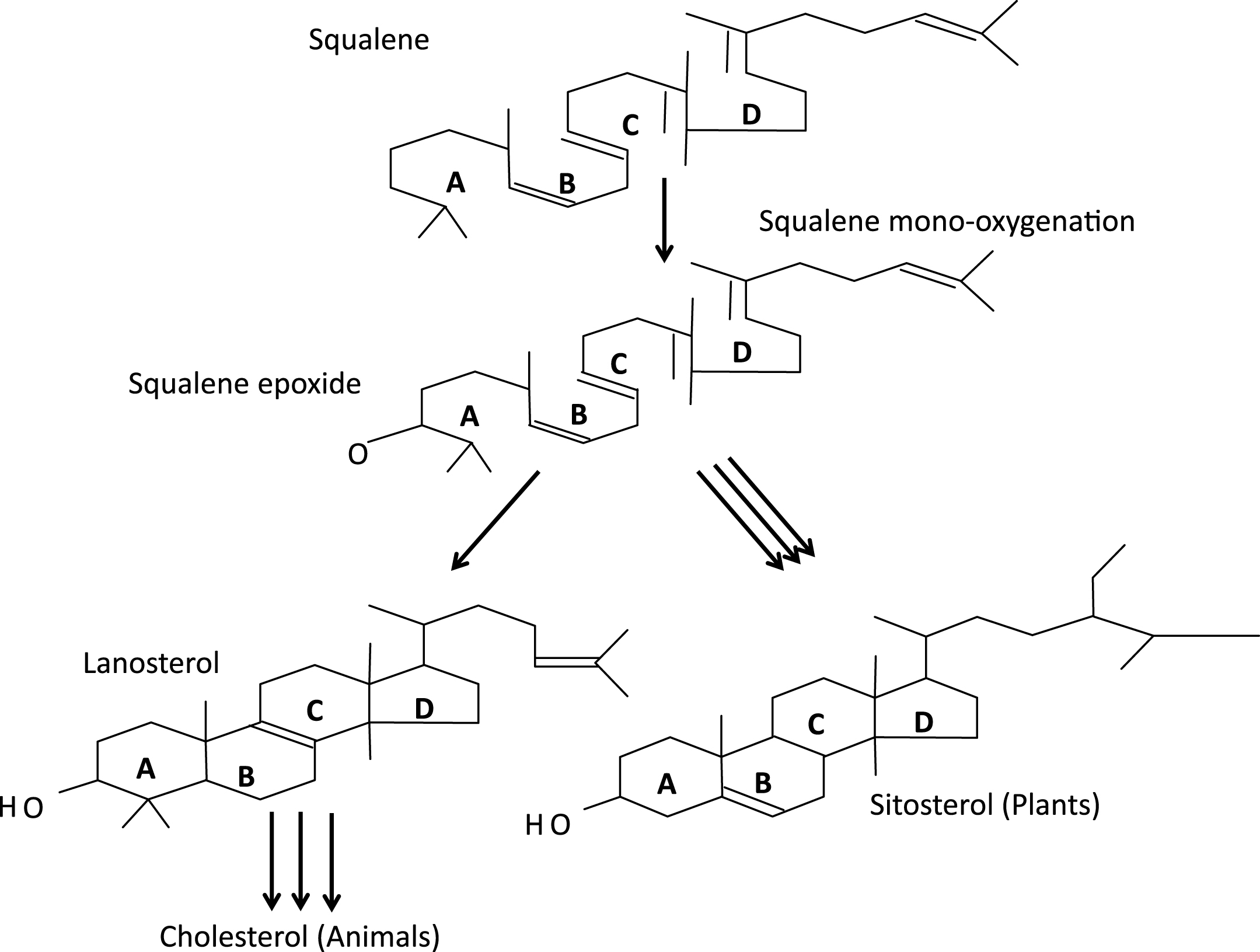
Fig.7
a. Gross outline of cholesterol biosynthesis in humans – note negative feedback by cholesterol on HMG-CoA reductase, the rate-regulating step of cholesterol synthesis. b. Cholesterol biosynthesis – implications for other metabolic functions and requirements.
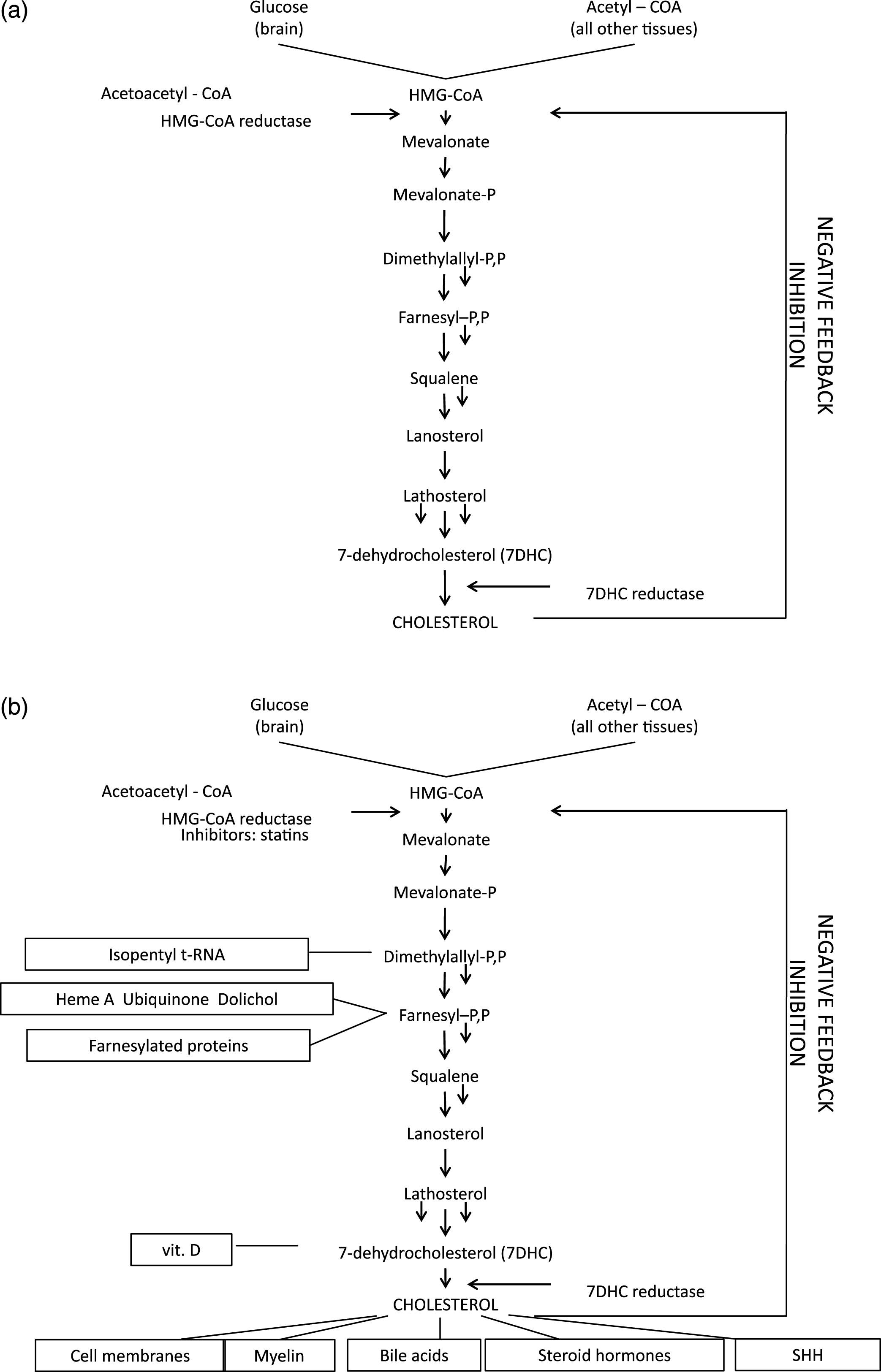
Fig.8
Cholesterol in the structure of the cell membrane; from Cooper, Hausman with permission.
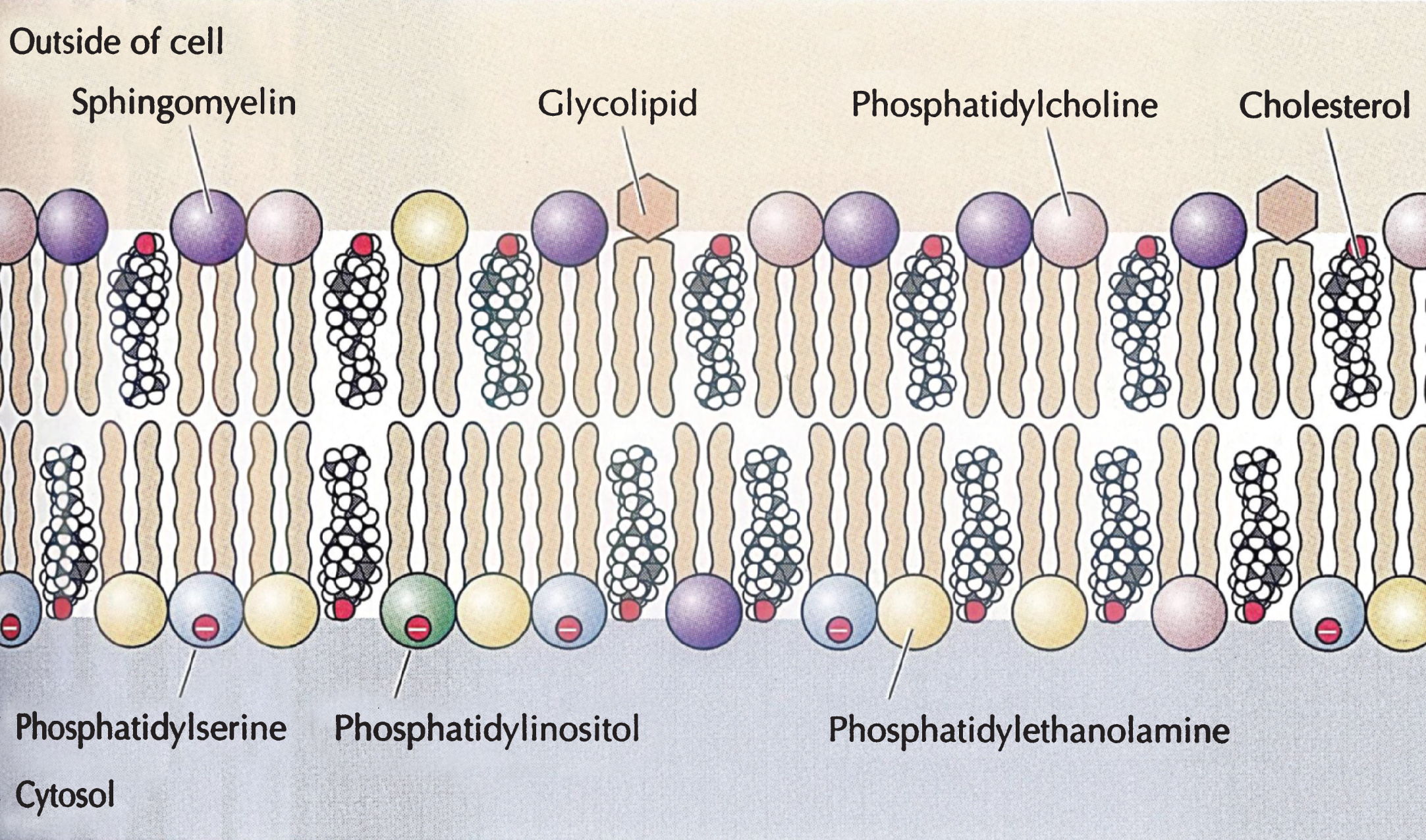
Fig.9
Cholesterol function in the synthesis of the sonic hedgehog molecule. The gene product of SHH is a paracrine, i.e secreted molecule, produced early during embryogenesis by autocatalytic cleavage into a SHH-C cholesterol transferase domain and a SHH-N signaling domain bound covalently to cholesterol before activation as morphogen which requires also palmitoylation to become an effective ligand.
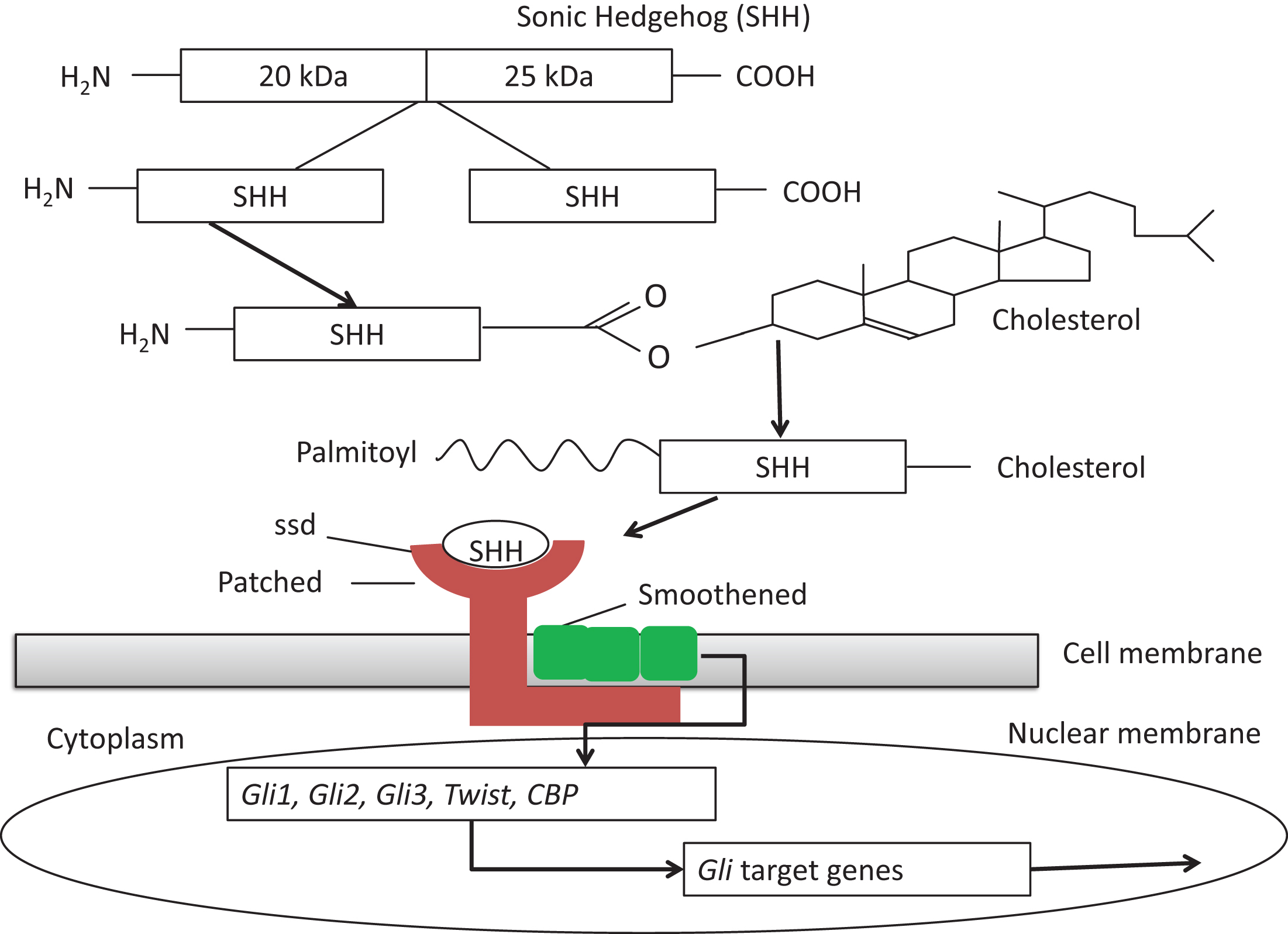
Fig.10
Cholesterol synthesis with indications of presumed site of action of its 5 teratogenic inhibitors and 8 known metabolic blocks. 1. Antley-Bixler syndrome (lanosterol-14α-demethylase). 2. Greenberg HEM dysplasia (sterol Δ-14-reductase). 3. CHILD syndrome of Happle (C-4 sterol-demethylase). 4. Conradi-Hünermann syndrome (sterol-Δ8-isomerase). 5. Lathosterolosis (sterol-Δ5-desaturase). 6. RSH/Smith-Lemli-Opitz syndrome (sterol-Δ7-reductase). 7. Desmosterolosis (sterol-Δ24-reductase). 8. Methyl sterol oxidase (SC4MOL) deficiency. Modified from Haas and Muenke (2010) [13] and Porter and Herman (2011) [42].
![Cholesterol synthesis with indications of presumed site of action of its 5 teratogenic inhibitors and 8 known metabolic blocks. 1. Antley-Bixler syndrome (lanosterol-14α-demethylase). 2. Greenberg HEM dysplasia (sterol Δ-14-reductase). 3. CHILD syndrome of Happle (C-4 sterol-demethylase). 4. Conradi-Hünermann syndrome (sterol-Δ8-isomerase). 5. Lathosterolosis (sterol-Δ5-desaturase). 6. RSH/Smith-Lemli-Opitz syndrome (sterol-Δ7-reductase). 7. Desmosterolosis (sterol-Δ24-reductase). 8. Methyl sterol oxidase (SC4MOL) deficiency. Modified from Haas and Muenke (2010) [13] and Porter and Herman (2011) [42].](https://content.iospress.com:443/media/trd/2016/1-2/trd-1-2-trd004/trd-1-trd004-g010.jpg)
Fig.11
Frontal facial view of a female patient diagnosed with Antley-Bixtler syndrome. showing turribrachycephaly with parietal bossing, midfacial hypoplasia, proptosis, and deformed auricles. (Courtesy from Malgorzata J.M. Nowaczyk, MD, McMaster University, Hamilton, Canada).

Fig.12
Radiographs of right and left upper limbs in the patient from Fig. 11 showing bilateral radiohumeral synostosis. (Courtesy from Malgorzata J.M. Nowaczyk, MD, McMaster University, Hamilton, Canada).
Fig.13
Skull radiographs in the patient from Fig. 11 showing synostosis of coronal and lambdoid sutures. (Courtesy from Malgorzata J.M. Nowaczyk, MD, McMaster University, Hamilton, Canada).
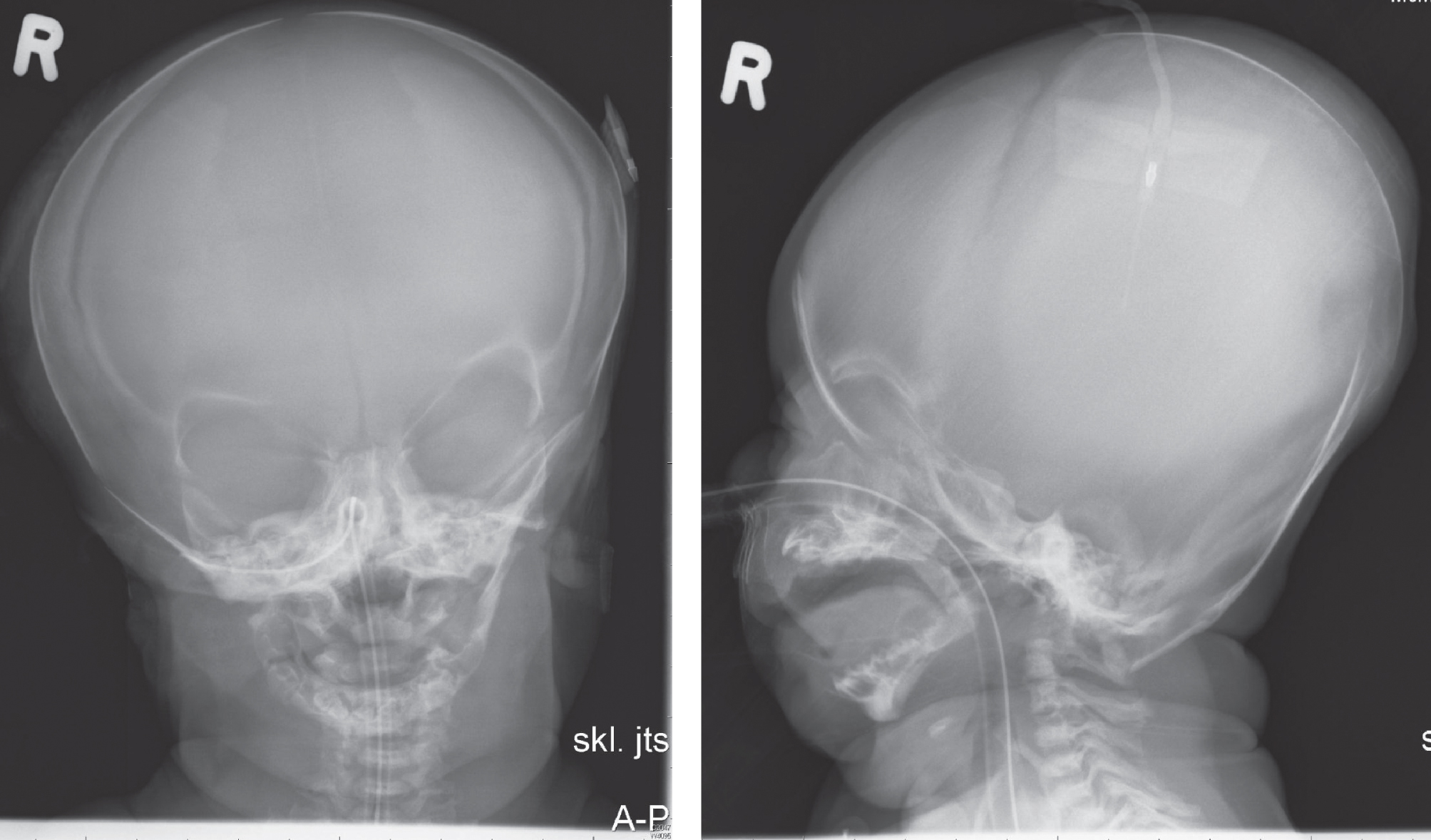
Fig.14
Radiographs of pelvis and lower limbs in the patient from Fig. 11 showing vertically slanting, narrow iliac bones and hypoplasia of pubic and ischial bones. (Courtesy from Malgorzata J.M. Nowaczyk, MD, McMaster University, Hamilton, Canada). Modified from Haas and Muenke [13] and Porter and Herman [42].
![Radiographs of pelvis and lower limbs in the patient from Fig. 11 showing vertically slanting, narrow iliac bones and hypoplasia of pubic and ischial bones. (Courtesy from Malgorzata J.M. Nowaczyk, MD, McMaster University, Hamilton, Canada). Modified from Haas and Muenke [13] and Porter and Herman [42].](https://content.iospress.com:443/media/trd/2016/1-2/trd-1-2-trd004/trd-1-trd004-g014.jpg)
Fig.15
a, b. 2-year-old girl with “mild” RSH/SLO syndrome diagnosed clinically (2/3 toe syndactyly!) at 17 months; confirmed biochemically by Dr. R.I. Kelley (7DHC: 86 mcg/nL, cholesterol: 102 mg/dL. Being followed by Dr. F.D. Porter at the NIH and Dr. R.D. Steiner at OHSU/Portland. The latter has determined 2 different mutations in the DHCR7 gene (R450L and N407K). She was born normally but developed reflux, failure to thrive, sleep disturbance, mild developmental and growth delay. Cholesterol treatment appeared to lead to a deterioration of behavior; Simvastatin treatment (Jira et al., 2000) did not appear to affect development. Now toilet-trained and in 3rd grade (special education).

Fig.16
Some mutations in the DHCR7 gene. TMD: transmembrane domains. From Yu and Patel, [140], with permission.
![Some mutations in the DHCR7 gene. TMD: transmembrane domains. From Yu and Patel, [140], with permission.](https://content.iospress.com:443/media/trd/2016/1-2/trd-1-2-trd004/trd-1-trd004-g016.jpg)


