
Clinical significance of TP53 alterations in advanced NSCLC patients treated with EGFR, ALK and ROS1 tyrosine kinase inhibitors: An update
Abstract
The development of targeted therapies for non-small cell lung cancer (NSCLC), such as the epidermal growth factor receptor (EGFR), anaplastic lymphoma receptor tyrosine kinase (ALK), and ROS proto-oncogene 1 (ROS1), has improved patients’ prognosis and significantly extended progression-free survival. However, it remains unclear why some patients do not benefit from the treatment as much or have a rapid disease progression. It is considered that, apart from the oncogenic driver gene, molecular alterations in a number of caretaker and gatekeeper genes significantly impact the efficacy of targeted therapies.
The tumor protein 53 (TP53) gene is one of the most frequently mutated genes in NSCLC. To date, numerous studies have investigated the influence of various TP53 alterations on patient prognosis and responsiveness to therapies targeting EGFR, ALK, or ROS1. This review focuses on the latest data concerning the role of TP53 alterations as prognostic and/or predictive biomarkers for EGFR, ALK, and ROS1 tyrosine kinase inhibitors (TKIs) in advanced NSCLC patients. Since the presence of TP53 mutations in NSCLC has been linked to its decreased responsiveness to EGFR, ALK, and ROS1 targeted therapy in most of the referenced studies, the review also discusses the impact of TP53 mutations on treatment resistance.
It seems plausible that assessing the TP53 mutation status could aid in patient stratification for optimal clinical decision-making. However, drawing meaningful conclusions about the clinical value of the TP53 co-mutations in EGFR-, ALK- or ROS1-positive NSCLC is hampered mainly by an insufficient knowledge regarding the functional consequences of the TP53 alterations. The integration of next-generation sequencing into the routine molecular diagnostics of cancer patients will facilitate the detection and identification of targetable genetic alterations along with co-occurring TP53 variants. This advancement holds the potential to accelerate understanding of the biological and clinical role of p53 in targeted therapies for NSCLC.
1Introduction
Lung cancer remains one of the main causes of cancer-related deaths worldwide [1]. The World Health Organization (WHO) has employed pathological criteria to categorize lung cancer into two main types: non-small cell lung cancer (NSCLC) and small cell lung cancer (SCLC). NSCLC accounts for over 85% of all lung cancer cases and is histopathologically subdivided into four types: 1) adenocarcinoma (LUAD), which comprises 40% of all primary lung cancer diagnoses; 2) squamous cell carcinoma (LUSC), accounting for approximately 30% of all lung cancer cases; 3) large cell carcinoma (LCC), accounting for about 10% of all lung cancer cases; and 4) other rare morphological types (about 5%) [2].
Clinical outcomes directly depend on the cancer stage at the time of diagnosis. According to Workman et al. [3], for patients with stage I lung cancer, the estimated five-year survival is 68.4%, while for stage IV lung cancer, 5.8%. The majority of patients are diagnosed with advanced-stage disease at stage IIIB-IV and have a correspondingly poor life expectancy. However, the introduction of molecular targeted therapies has improved the prognosis for patients with advanced NSCLC and greatly extended progression-free survival time (PFS) compared to chemotherapy. Currently, targeted treatment therapies for advanced NSCLC (stage IIIB-IV) approved by the US Food and Drug Administration (FDA) and/or the European Medicines Agency (EMA) are offered to patients with non-squamous subtypes carrying molecular alterations of the epidermal growth factor receptor (EGFR), the anaplastic lymphoma receptor tyrosine kinase (ALK), the ROS Proto-Oncogene 1, Receptor Tyrosine Kinase (ROS1), the Ret Proto-Oncogene (RET), the MET Proto-Oncogene, Receptor Tyrosine Kinase (MET), the Neurotrophic Receptor Tyrosine Kinases 1–3 (NTRK1-3), the Erb-B2 Receptor Tyrosine Kinase 2 (ERBB2, HER2), the KRAS Proto-Oncogene, GTPase (KRAS) or the B-Raf Proto-Oncogene, Serine/Threonine Kinase (BRAF) [4, 5]. For patients treated with EGFR, ALK and ROS1 tyrosine kinase inhibitors (TKIs), the efficacy of treatment is improving with consecutive TKI generations, however the average objective response rate (ORR) is about 70%, and progression-free survival (PFS) usually does not exceed 18 months [6–12]. It remains unclear why some patients do not respond to TKI-based treatment or have rapid disease progression. The molecular mechanisms responsible for drug resistance are not sufficiently clarified, making it difficult to predict treatment outcome. Since next-generation sequencing (NGS) technologies have been widely used, the importance of assessing the clinical value of co-occurring gene alterations has increased. The tumor protein 53 gene (TP53) is frequently mutated in NSCLC (in approximately 40% of LUAD and 51% of LUSC) [13], resulting in the loss of its tumor suppressor function (impaired protein expression) or the gain of oncogenic activity (aberrant protein expression). Importantly, TP53-mutated NSCLC in patients with a history of smoking present an increased risk of distant metastases [14]. Recent studies emphasize the influence of TP53 gene alterations on the prognosis and the overall responsiveness to targeted therapies in NSCLC [15] (Table 1).
Table 1
EGFR, ALK and ROS1 tyrosine kinase inhibitors used in the studies evaluating the TP53 status in NSCLC
| Drug Class | Target genes | Gene aberrations approved for the NSCLC targeted treatment | Tyrosine kinase inhibitor | Approval Status | Source with TP53 mutation status analysis |
| EGFR-TKIs | |||||
| first-generation | EGFR inhibitor | EGFR activating mutations | Erlotinib | FDA, EMA | [25, 29–31, 34] |
| Gefitinib | FDA, EMA | [25, 29–31, 34, 36] | |||
| Icotinib | CFDA | [29, 34] | |||
| second-generation | pan-HER inhibitor: EGFR, HER2, HER3 and HER4 | EGFR mutations | Afatinib | FDA, EMA, CFDA | [25, 30, 34] |
| Dacomitinib | FDA, EMA | [25, 34] | |||
| third-generation | EGFR inhibitor | EGFR mutations | Osimertinib | FDA, EMA | [25, 30, 34] |
| Olmutinib | KFDA (Conditional) | [30] | |||
| ALK-TKIs | |||||
| first-generation | multi targeted MET, ALK, and ROS1 inhibitor | ALK or ROS1 rearrangement | Crizotinib | FDA, EMA | [44–49] |
| second-generation | highly potent ALK inhibitor | ALK rearrangement | Ceritinib | FDA, EMA | [45] |
| Alectinib | FDA, EMA | [45, 47, 50] | |||
| highly potent, selective ALK and ROS1 inhibitor | ALK rearrangement | Brigatinib | FDA, EMA | [45] | |
| third-generation | highly potent, selective ALK and ROS1 inhibitor | ALK rearrangement | Lorlatinib | FDA, EMA | [51, 62] |
| Ensartinib | FDA | [74] | |||
| ROS1-TKIs | |||||
| first-generation | multi targeted MET, ALK, and ROS1 inhibitor | ALK or ROS1 rearrangement | Crizotinib | FDA, EMA | [56, 59, 61, 63] |
| second-generation | highly potent ALK inhibitor | ALK rearrangement (does not have approval for ROS1) | Ceritinib | FDA, EMA | [45] |
| third-generation | highly potent, selective ALK and ROS1 inhibitor | ALK rearrangement and mutations, (does not have approval for ROS1) | Lorlatinib | FDA, EMA | [51, 56, 63] |
| potent, pan-TRK inhibitor: NTRK, ROS1 and ALK | ROS1 gene fusion, NTRK gene fusion | Entrectinib | FDA, EMA | [9, 55] |
Abbreviations: EMA, the European Medicines Agency; CFDA, the China Food and Drug Administration; KFDA, the Korea Food and Drug Administration.
This review focuses on the latest studies evaluating the prognostic and/or the predictive significance of TP53 gene alterations in NSCLC patients treated with EGFR-, ALK-, and ROS1-TKIs, providing a critical overview of important clinical findings, drawbacks, and controversies on this topic.
2TP53 gene: Functions, structure and mutations in NSCLC
The TP53 gene encodes the p53 protein, which acts as a transcription factor and a key regulator of diverse cellular processes (Fig. 1), such as cell-cycle, autophagy, apoptosis, proliferation, differentiation, metabolism, and DNA repair [16]. The TP53 gene is located on the short arm of the chromosome 17 (17p13.1) and consists of 11 exons that code for the 393 amino acid p53 protein localized in the nucleus and cytoplasm. The p53 comprises functional domains: the N-terminal transactivation domain (TAD, residues 1–67), the proline-rich domain (PRD, residues 68–98), the DNA-binding domain (DBD, residues 99–303), the tetramerization domain (TET, residues 323–363), and the C-terminal regulatory domain (CTD, residues 364–393) [17–19]. Briefly, the TAD is subdivided into two regions: TAD1 and TAD2, which allow p53 to bind to different cofactors, and are required for p53-mediated suppression of tumorigenesis in response to stress. The PRD is essential for p53 stability, efficient transcription function, and activation of apoptosis and cell cycle arrest. The DBD is a protease-resistant region containing a zinc ion, which allows p53 to act as a transcription factor that is crucial for the direct regulation of genes involved in DNA repair, apoptosis, and cell-cycle regulation. The TET is responsible for the p53 protein’s oligomerization as a tetramer, conferring the appropriate conformation that binds to DNA, which is important for its full transcriptional function. The CTD is a regulator domain subjected to post-translational modifications that regulate the ability of p53 to bind to specific DNA sequences, and contains the nuclear export and nuclear localization signals.
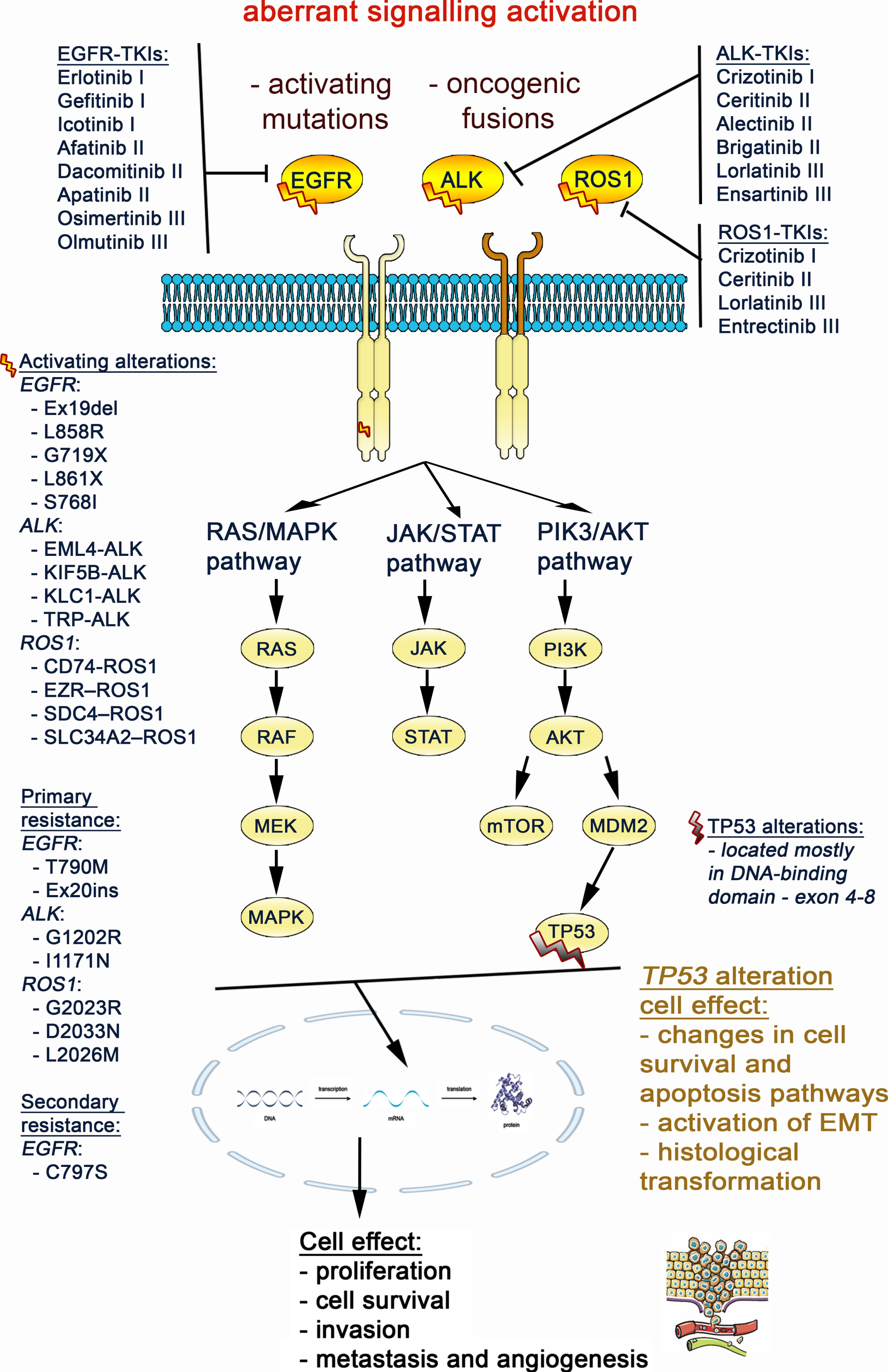
Fig. 1
The detailed scheme of the main cancer signaling pathways associated with EGFR, ALK, ROS1 and TP53 genes, based on KEGG pathway maps database [75]. (https://www.genome.jp/kegg/pathway.html).
![The detailed scheme of the main cancer signaling pathways associated with EGFR, ALK, ROS1 and TP53 genes, based on KEGG pathway maps database [75]. (https://www.genome.jp/kegg/pathway.html).](https://content.iospress.com:443/media/tub/2024/46-s1/tub-46-s1-tub230034/tub-46-tub230034-g001.jpg)
Through nearly 50 years of extensive research, the TP53 gene has been recognized to act as either a gatekeeper or caretaker tumor suppressor [20]. Gatekeeper genes control cell growth by inhibiting proliferation and leading to apoptosis and/or promotion of terminal differentiation. Caretaker tumor suppressor genes maintain the genome stability by reducing the mutation rates of several genes involved in DNA repair, including gatekeepers and oncogenes. In fact, by inducing apoptosis, TP53 may be considered a progression gatekeeper whereas by preventing genomic instability, TP53 acts as a caretaker, and appears to be ‘a guardian of the genome’. The p53 protein has a short half-life and it is constitutively expressed at low levels. However, it may accumulate in the cell as a result of various types of stress, such as DNA damage, hypoxia, loss of normal growth and survival signals, and inflammatory processes, which may occur in different physiological or pathological conditions, including tumorigenesis [21, 22]. Accordingly, more than 50% of NSCLC tumors show p53 protein overexpression as evaluated by immunohistochemistry [23].
The TP53 alterations, frameshifts – deletions and insertions, nonsense, silent, and missense mutations, occur throughout the entire gene sequence and may result in a variety of oncogenic effects on the p53 protein (Fig. 2). Missense variants, the most common TP53 alterations, occur mainly in the DBD domain (exons 5– 8, accounting for about 70% of all TP53 alterations), often leading to p53 accumulation in the cell, which may result in the loss of regulatory functions and promote uncontrolled cellular proliferation. Approximately 10% of the TP53 alterations are nonsense variants, yielding truncated proteins that are usually degraded shortly after translation [19, 24]. Additionally, TP53 mutations can be classified as disruptive and non-disruptive variants, as previously described [25], based on the their impact on the protein structure. Disruptive mutations result in a complete or almost complete loss of function of the p53 protein, typically caused by stop codons, missense mutations occurring outside of the L2 or L3 p53 loops, and in-frame deletions in the L2 or L3 loops. Non-disruptive mutations encompass missense mutations and in-frame deletions that occur outside the L2 or L3 loops, as well as missense mutations inside the L2 or L3 loops that lead to a substitution of one amino acid residue with another of the same polarity/charge.
Fig. 2
The scheme of the p53 protein sequence, functional domains, and exons of the TP53 gene, with the locations of amino acids changes introduced by pathogenic missense (green pin), in-frame (brown pin), splice (orange pin), and nonsense single nucleotide variants, SNVs (black pin), commonly detected in lung adenocarcinoma (6294 samples from 5844 patients in 13 studies, cBioPortal v. 4.0.2 database [76, 77].
![The scheme of the p53 protein sequence, functional domains, and exons of the TP53 gene, with the locations of amino acids changes introduced by pathogenic missense (green pin), in-frame (brown pin), splice (orange pin), and nonsense single nucleotide variants, SNVs (black pin), commonly detected in lung adenocarcinoma (6294 samples from 5844 patients in 13 studies, cBioPortal v. 4.0.2 database [76, 77].](https://content.iospress.com:443/media/tub/2024/46-s1/tub-46-s1-tub230034/tub-46-tub230034-g002.jpg)
The TP53 gene abnormalities result in p53 protein loss-of-function or gain-of-function, leading to promotion of cancer cell proliferation, invasion, metastasis and drug resistance [18]. To date, many studies have indicated varied impact of TP53 co-mutations on clinical outcomes in EGFR-, ALK-, and ROS1-positive NSCLC patients. Nonetheless, the precise molecular mechanisms underlying the influence of altered p53 on responses of NSCLC patients to EGFR-, ALK- and ROS1-targeted therapies remain poorly understood. TP53 mutations appear to play a significant role in the primary, adaptive, and acquired resistance of cancer cells to TKIs through increased genomic instability, stimulation of tumor angiogenesis (via the Hypoxia inducible factor 1 subunit alpha (HIF1)/Vascular endothelial growth factor A (VEGFA) pathway), or down-regulation of apoptosis and cell-cycle arrest (as discussed in dedicated sections).
3Clinical importance of TP53 mutations in NSCLC patients treated with EGFR-, ALK-, and ROS1-targeted therapies
3.1EGFR-TKIs
The EGFR tyrosine kinase (RTK) family includes four distinct receptors encoded by genes: EGFR (ErbB-1, HER1), ErbB-2 (Neu, HER2), ErbB-3 (HER3), and ErbB-4 (HER4). All receptors have a similar structure and contain intracellular, extracellular, and transmembrane regions. Epidermal growth factor (EGF) ligand binding leads to dimerization of EGFR, followed by its activation through sequential autophosphorylation of tyrosine residues. Activated EGFR initiates intracellular signaling pathways, including Phosphatidylinositol 3-kinase (PI3K) / AKT Serine/Threonine Kinase (AKT), and mitogen-activated protein kinase (MAPK), which are involved in cell proliferation, differentiation, division, survival, and cancer development [26], Figs. 1 and 3.
As EGFR is an important oncogenic driver that is commonly altered in NSCLC (up to 16% non-squamous tumors), it has become a crucial therapeutic target for treating this malignancy. To date, several generations of oral small-molecule inhibitors targeting the tyrosine kinase structural domain of the EGFR protein have been developed, becoming the fundamental therapeutic approach in NSCLC treatment. The somatic activating EGFR mutations located in the gene sequence coding for the tyrosine kinase structural domain of the protein (mainly exon 19 deletions and exon 21 substitutions) present the predictive value in the targeted treatment based on the first-, second-, and third-generation of EGFR-TKIs in NSCLC [27]. For non-squamous-NSCLC patients treated with EGFR-TKIs, the average objective response rate (ORR) is 70%, and the progression-free survival (PFS) varies from 12 to 20 months [28]. Molecular mechanisms underlying primary, adaptive, and acquired resistance to TKIs await better understanding.
Recently, the association between TP53 mutations and patient responses to single-agent treatment with EGFR-TKIs have been studied, demonstrating the negative impact of those alterations on survival outcomes in advanced NSCLC, as shown in Table 2. For example, Huo et al. [29] showed significantly shorter median PFS (6.5 vs. 14 months, P = 0.025) and median overall survival (OS, 28 vs. 52 months, P = 0.023) in advanced NSCLC patients with the TP53 co-mutations treated with the first-generation TKIs (gefitinib, erlotinib, and icotinib) with respect to the patients with the wild-type TP53. In this study, TP53 alterations, particularly in exon 7, had the greatest impact on patients’ prognosis when compared with wild-type TP53 (mPFS: 5.0 vs. 14.0 months, P = 0.002, HR 3.98, P = 0.002; mOS: 14 vs. 52.5 months, P = 0.008; HR 5.29, P = 0.009). However, no statistically significant difference was observed in the objective response rate (ORR) between the TP53-mutated and the TP53-wild-type patients (25% vs. 28%, P = 0.374). Similar results were reported by Kim et al. [30], who showed that TP53 alterations were associated with worse PFS in the EGFR-mutated NSCLC patients treated with both first-/second-generation EGFR-TKIs (cohort 1: HR: 2.02, P = 0.038) and third-generation EGFR-TKIs (cohort 2: HR: 2.23, P = 0.017). Additionally, Canale et al. [25] demonstrated that only TP53 mutations located in exon 8 were significantly associated with a shorter median PFS and poorer OS, when compared to non-exon 8-mutated or wild-type TP53 (PFS: 5.8 vs. 14.4 and vs. 12.4 months; OS: 18.53 vs. 34.8 and vs. 27.3 months, respectively) in NSCLC patients treated with both first- or second-generation TKIs (especially in the subgroup with EGFR exon 19 deletion). No statistically significant associations between TP53 mutations and ORR were found. The same phenomenon was later observed by Li et al. [31] for exon 4 or 7 of TP53, TP53 mutations except in exons 4 and 7, or wild-type TP53 (ORR, 59.1%, 53.3%, and 57.4%, respectively, P = 0.788). Nonetheless, TP53 variants located in exon 7 and exon 4 were independent negative prognostic factors for PFS (HR 0.55, P = 0.001) and OS (HR 0.594, P = 0.004) [31]. The impact of TP53 mutations was also investigated in patients with EGFR-mutated NSCLC carrying the T790M resistance co-mutation, and treated in 2nd/further lines with osimertinib [32]. The TP53 mutations were confirmed as a negative predictive factor for PFS (9 vs. 14 months, P < 0.008) and OS (16 vs. 24 months, P < 0.025).
Table 2
Associations between TP53 mutations and responses to EGFR-, ALK- and ROS1-TKIs in NSCLC
| Tyrosine kinase inhibitor | TP53 mutations | PFS (months) | OS (months) | ORR | Source |
| EGFR-TKIs | |||||
| gefitinib, erlotinib, icotinib | TP53 any mutations vs wild type | 6.5 vs. 14.0, P = 0.025 | 28.0 vs. 52.0, P = 0.023 | nd 25% vs. 28%, P = 0.374 | Huo et al. [29] |
| gefitinib, erlotinib, icotinib | TP53 exon 7 mutations vs wild type | 5.0 vs. 14.0, P = 0.002, | 14.0 vs. 52.5, P = 0.008 | na | Huo et al. [29] |
| gefitinib, erlotinib, afatinib, osimertinib, olmutinib | TP53 any mutations vs wild type | 8.9 vs 12.8, P = 0.029 | 17.8 vs 26.6, P = 0.007 | na | Kim et al. [30] |
| erlotinib, gefitinib, afatinib, osimertinib | TP53 exon 8 mutations vs other TP53 mutations vs wild type | 5.8 vs. 14.4 vs. 12.4, P < 0.05 | nd 18.53 vs. 34.8 vs. 27.3, P > 0.05 | nd, | Canale et al. [25] |
| gefitinib, erlotinib | TP53 exon 4 or 7 mutations vs other TP53 mutations vs wild type | 9.4 vs. 11.0, vs. 14.5, P = 0.009 | 5.8 vs. 20.0 vs. 26.1, P = 0.004 | nd 59.1% vs. 53.3% vs. 57.4%, P = 0.788 | Li et al. [31] |
| osimertinib | TP53 any mutations vs wild type | 9.0 vs. 14.0, P < 0.008 | 16.0 vs. 24.0, P < 0.025 | nd 38% vs 49 % | Roeper et al [32] |
| ALK-TKIs | |||||
| crizotinib proceeded by chemotherapy | TP53 exon 4–8 mutations vs wild type | 5.0 vs. 14.0, P < 0.001 | 17.0 vs. not reached, P = 0.002 | na | Kron et al. [45] |
| crizotinib followed by a ceritinib | TP53 exon 4–8 mutations vs wild type | 5.4 vs. 9.9, P = 0.039 | 7.0 vs. 50.0, P = 0.001 | na | Kron et al. [45] |
| crizotinib | TP53 any mutations vs wild type | PFS longer in TP53 wild-type vs TP53 mutation, HR = 1.96, P = 0.045 | nd | 40% vs 81.6%, P = 0.003 | Song et al. [44] |
| crizotinib | TP53 non-disruptive vs wild type | PFS shorter in non-disruptive TP53 mutations vs TP53 wild-type HR = 3.27, P = 0.003 | Nd | na | Song et al. [44] |
| crizotinib | TP53 any mutations vs wild type | 3.3 vs. 10.4, P = 0.038 | 21.4 vs. 34.2, P = 0.021 | 37.50% vs. 76.92% | Wang et al. [46] |
| crizotinib | TP53 any mutations vs wild type | nd P > 0.05 | 22.5 vs. 30.0, P = 0.046 | na | Li et al. (2021b) [48] |
| alectinib | TP53 any mutations vs wild type | 3.8 vs. 9.6 | na | 35% vs 65% | Wolf et al. [50] |
| lorlatinib | TP53 any mutations vs wild type | 3.7 vs. 10.8, P = 0.003 | nd 10.9 vs 24.7, P = 0.24 | na | Frost et al. [51]. |
| ROS1-TKIs | |||||
| crizotinib | concomitant mutations including TP53 any mutations vs wild type | 8.5 vs. 15.5, P = 0.021 | na | na | Zeng et al. [61] |
| crizotinib | TP53 any mutations vs wild type | PFS not reached in both groups, P = 0.0417 | na | na | Lin et al. [59] |
| lorlatinib | nd | nd | 27.3% vs. 50% | Landi et al. [62] | |
| 8.5 vs. 26.3 | 14.6 vs. 30.7 |
nd – no statistically significant difference; na – not analysed.
Despite the unfavorable effect of TP53 variants on single-agent treatment outcomes, promising efficacy emerged when combining EGFR-TKIs with angiogenesis inhibitors in NSCLC with concurrent TP53 mutations. For instance, Zhao et al. [33] showed that the median PFS for the EGFR-mutant NSCLC with concomitant TP53 mutation in exon 8 was favorable in the cohort receiving gefitinib plus apatinib (a vascular endothelial growth factor receptor (VEGFR2) inhibitor) compared to the gefitinib plus placebo group (12.5 vs. 1.1 months, respectively; HR 0.56). Similarly, Sun et al. [34] demonstrated that combination therapy based on the EGFR-TKIs (gefitinib, icotinib, erlotinib, afatinib, dacomitinib, osimertinib, or almonertinib) and anti-angiogenic drugs or chemotherapy, had higher efficacy than EGFR-TKIs alone in NSCLC patients with concomitant EGFR and TP53 mutations. The median PFS was significantly longer in the combination group than in the EGFR-TKIs-treated group with TP53 exon 4/7 mutations (18 vs. 7 months). Also, the RELAY trial (ramucirumab plus erlotinib in the untreated metastatic EGFR-mutant NSCLC) demonstrated that patients with concomitant TP53 mutations benefit more from combination therapy than placebo plus erlotinib (median PFS: 15.2 vs. 10.6 months, respectively; HR 0.54) [35]. In a recent study, Hou et al. [36] reported that EGFR and TP53 co-mutated NSCLC patients with brain metastases treated with gefitinib plus chemotherapy, achieved a better intracranial objective response rate (85% vs. 63%; P = 0.002), and had a longer median PFS (15.6 months vs. 9.1 months; HR 0.36), as compared to those treated with gefitinib alone.
The available data highlight the importance of determining the TP53 co-occurrence status in predicting the responsiveness to EGFR-TKIs. Since the TP53 mutation status affects the clinical efficacy of EGFR-TKIs, a more comprehensive assessment of these alterations could improve therapeutic outcome.
3.2ALK-TKIs
The ALK gene encodes a tyrosine kinase that belongs to the insulin receptor superfamily. The ALK protein comprises an extracellular domain of 1030 amino acides (aa), a transmembrane domain of 28 aa, and an intracellular kinase domain of 561 aa. Activation of ALK is based on the binding of its ligands to the extracellular domain, which promotes dimerization and activates downstream cell signaling pathways, including the PI3K, Janus kinase (JAK)/ Signal transducer and activator of transcription (STAT), and small GTPase (RAS)/ Mitogen-activated protein kinase (MEK)/Extracellular signal-regulated kinases (ERK) pathways, and promotes cell growth [37], Fig. 3.
Numerous studies have identified oncogenic ALK rearrangements present in 3–7% of NSCLC patients [38, 39]. The majority of these rearrangements are chromosomal translocations, resulting in the formation of chimeric fusion oncoproteins coded by ALK and another partner gene, which become overexpressed in cancers. The most common (∼90%) ALK fusion partner in NSCLC is the EMAP-like 4 gene (EML4). The EML4-ALK fusion consists of the basal domain encoded by the EML4 gene and the kinase region encoded by the ALK gene [39]. To date, numerous distinct ALK fusion partners, including EML4, Kinesin family member 5B (KIF5B), Kinesin Light Chain 1 (KLC1), and Translocated promoter region, nuclear basket protein (TPR), have been identified [40]. Among ALK-rearranged NSCLC, both the fusion partner and the EML4-ALK fusion variants have been considered candidate modifiers of transforming potential and response biomarkers to ALK-TKIs [41]. However, clinical studies in the ALK-positive NSCLC population have demonstrated differences in ORR (49–54.5%) and PFS (9.4–48.5 months), based on different ALK fusion subtypes and treatment protocols [7, 8, 10–12, 42, 43]. Therefore, understanding the underlying molecular interplay between ALK fusions and ALK-TKIs is necessary to guide clinical treatment.
Fig. 3
Aberrant activation of the EGFR, ALK and ROS1 gene triggers signaling through the MAPK, JAK/STAT and PI3K/AKT/mTOR pathways, that results in promoting lung cancer cell migration and proliferation. The TP53 mutation impact on cell signaling was also marked at crossing point with the EGFR, ALK, ROS1 pathways.
To date, TP53 alterations have been considered the most frequent genetic co-alterations (20.3–38.1%) in NSCLC with ALK fusions [44–48]. These alterations have been identified as negative biomarkers of ALK-TKI therapy outcome, as shown in Table 2. For example, Kron et al. [45] revealed that concomitant TP53 mutations (exons 4–8) were adverse prognostic factors for patients with ALK-rearranged NSCLCs treated with: 1) a first-generation TKI, multitargeted MET, ALK, and ROS1 inhibitor - crizotinib followed by chemotherapy (median PFS: 5.0 vs. 14.0 months, p < 0.001; median OS: 17.0 vs. not reached, P = 0.002); 2) and crizotinib followed by a second-generation ALK inhibitor (ceritinib) (median PFS: 5.4 vs. 9.9 months, P = 0.039; median OS: 7.0 vs. 50.0 months, P = 0.001). Similarly, Song et al. [44] showed that TP53 mutations (mostly in exons 6 and 8) negatively affected the response to crizotinib (ORR: 40%, P = 0.003) and correlated with shorter PFS (HR: 1.96, P = 0.045) compared to wild-type TP53 in ALK-rearranged NSCLC. Additionally, non-disruptive TP53 mutations had a negative impact on prognosis (PFS: HR: 3.27, P = 0.003). However, no significant difference in OS according to the TP53 mutation status was observed. Another study confirmed that mutated TP53 reduced responsiveness to crizotinib (ORR: 37.50% vs. 76.92%) and worsened prognosis (PFS: 3.3 vs. 10.4 months, P = 0.038; OS: 21.4 vs. 34.2 months, P = 0.021), as compared to the wild-type group [46]. In contrast to others, Li et al. (2021b) [48] found that patients with concomitant TP53 mutations had a shorter median OS (22.5 vs. 30 months, P = 0.046), while no significant difference in PFS was reported. Interestingly, Shi et al. [49] presented a case report of a patient with a new ALK- LIM zinc finger domain containing 1 (LIMS1) fusion variant and a TP53 N310Tfs*35 co-mutation, presenting a PFS of only two months, possibly due to p53-mediated primary resistance to crizotinib. Additional point mutations were identified (KIT proto-oncogene, receptor tyrosine kinase (KIT) E561K, AXL receptor tyrosine kinase (AXL) E66K, NTRK3 L835M, Interleukin 6 cytokine family signal transducer (IL6ST) D171N) at the time of progression, with retention of the TP53 mutation and ALK-LIMS1 fusion.
The presence of a mutant TP53 was also a negative prognostic factor in terms of PFS for second- and third-generation ALK-TKIs (for alectinib HR of 1.88 [50]; for lorlatinib PFS of 3.7 vs. 10.8 months, HR 3.3, P = 0.003) [51]. Furthermore, Canale et al. [47] identified a link between non-disruptive TP53 mutations and worse treatment efficacy, regardless of the ALK-TKIs generation and line. However, no statistically significant association between TP53 mutations and ORR was demonstrated.
Interestingly, it has been suggested that individual genomic breakpoints of EML4 and ALK may have a different impact on the response to ALK-TKIs [40, 52]. Nevertheless, a negative impact of TP53 mutations on PFS and OS, regardless of the EML4-ALK variant type (1 or 3a/b), was shown [45, 44].
In recent studies, TP53 mutations have emerged as an important intrinsic factor that impacts treatment outcomes in NSCLC patients with ALK fusions. However, the diversity of results emphasizes the need for further studies to create comprehensive evidence to find the most appropriate treatment strategy.
3.3ROS1-TKIs
The ROS proto-oncogene 1 belongs to the subfamily of tyrosine kinase insulin receptor genes and generates two dominant splice variants of ROS1 encoded by either 43 or 44 exons. It has a large extracellular N-terminal domain (coded by exons 1–34) composed of nine fibronectin type III repeats and three β-propeller domains, a single transmembrane domain, an intracellular tyrosine kinase domain, and C-terminal domains [53]. ROS1 activation leads to autophosphorylation of specific tyrosine residues and activation of downstream cell signaling pathways, including the small GTPase (RAS)–serine/threonine protein kinase (RAF)–MEK–ERK, PI3K–AKT–Mechanistic target of rapamycin kinase (mTOR), and JAK–STAT3, which broadly regulate cell survival, growth, and proliferation [53], Fig. 3. Somatic ROS1 rearrangements, mostly fusions with numerous gene partners, are strong oncogenic drivers, identified in approximately 0.9–2.6% of NSCLC tumors. The ROS1 fusion protein, though lacking the extracellular structural domain, becomes continuously activated and capable of stimulating distinct downstream signaling pathways [54]. ROS1-TKIs are characterized by high clinical efficacy [8, 51]. For example, Dziadziuszko et al. [55] reported median progression-free survival (PFS) values of 15.7 months (range: 11.0 to 21.1) and a response rate of 67.1% (range: 59.3 to 74.3) for entrectinib, while Drilon et al. [9] reported a median PFS of 29.1 months (range: 21.8–35.9) and a response rate of 68% (range: 60.2–74.8). Yet, similarly to other TKIs, the secondary resistance mechanisms are not well understood, besides ROS1 kinase domain mutations [56] or MAPK-mediated bypass signaling [57, 58].
While ∼36% of ROS1-positive NSCLCs present with oncogenic co-mutations, several studies have indicated that concurrent TP53 gene mutations adversely impact the efficacy of ROS1-TKI [59, 60], as shown in Table 2. In the two studies evaluating crizotinib efficacy, ROS1-positive, NSCLC patients with co-mutated TP53 showed worse outcome than the wild-type TP53 group, with median PFS of 8.5 vs. 15.5 months; P = 0.021 [61], and PFS not reached in both groups, P = 0.0417 [59]. Likewise, later-line lorlatinib treatment was less effective in ROS1-positive NSCLC patients with co-mutated TP53 compared to the wild-type TP53 group [51], with a median PFS of 8.5 months vs. 26.3 months and a response rate of 27.3% vs. 50% [62]. Begum et al. [56] and Balázs et al. (2022) [63] reported two NSCLC patient cases with the CD74 molecule (CD74)-ROS1 fusion and concomitant TP53 mutations (H193Y, exon 5) and (R306*, exon 7) who rapidly progressed on first-line crizotinib, and were subsequently treated with lorlatinib. In the NGS analysis of the tumor or liquid biopsy specimens collected at the three time points (baseline, 1st progression on crizotinib, 2nd progression on lorlatinib), TP53 mutations were detected in all three analyses along with the CD74-ROS1 translocation, indicating the importance of TP53 alterations in maintaining the aggressive phenotype.
Despite limited data, the potential predictive significance of TP53 mutations for ROS1-TKI treatment is noteworthy. TP53 alterations might help identify ROS1-positive patients at a higher risk of resistance to ROS1-TKIs. Further analyses to explore alternative treatment strategies for this subpopulation of patients are needed.
4Impact of TP53 mutations on treatment resistance
Secondary resistance to EGFR, ALK, and ROS1 tyrosine kinase inhibitors is expected to develop in any NSCLC patient on targeted treatment, yet the mechanisms underlying this phenomenon have not been fully elucidated.
The available clinical data highlight the association of TP53 exon 4– 8 mutations with an unfavorable outcome, identifying the DNA-binding domain as the mutation hotspot for TP53. This is in line with in vitro studies linking TP53 DBD alterations to primary and acquired resistance to TKIs. Accordingly, the p53 silencing in the HCC827 (EGFR-Del19, p53-v218del) cell line resulted in a reduction of EGFR activity and expression, and induced primary resistance to gefitinib through the enhancement of AXL [64], while the TP53 R273H alteration was instrumental in induction of the epithelial-to-mesenchymal transition (EMT). However, the regulation of EMT in acquired resistance to EGFR-TKIs is still not fully understood. Furthermore, the H1975 (EGFR-L858R + T790M, p53-R273H) p53 knockout cell line was more prone to acquiring the epithelial phenotype with defined cell-cell contacts when treated with osimertinib.
Additionally, mutations in DBD of TP53 have also been identified as predictors of a favorable response to combination therapy involving EGFR-TKIs and anti-angiogenic VEGFR2 inhibitors (such as anlotinib or bevacizumab) or chemotherapies (pemetrexed and carboplatin or cisplatin) [34]. Previously, it has been demonstrated that the administration of VEGF/VEGFR inhibitors was significantly associated with improved outcomes in cancer patients with TP53 mutations (P = 0.005) [65]. Apparently, the expression of TP53 hotspot mutant proteins (R175H, R248W, and R273H) might stimulate tumor angiogenesis through the up-regulation of reactive oxygen species formation and subsequent activation of the HIF1/VEGFA pathway [66]. In contrast to lung squamous cell carcinomas, in lung adenocarcinoma, TP53 mutations were associated with an increased VEGFA expression, the major ligand of the VEGF/VEGFR pathway and a key regulator of angiogenesis [67]. To date, the role of EGFR and TP53 in regulating chemosensitivity has not been fully elucidated. It has been suggested that in chemosensitive NSCLC, the cisplatin-caused p53 activation occurs through increased phosphorylation of p53 at the sites of Ser 15 and Ser 20, leading to the stabilization of the p53 protein, a subsequent blockade of p53-Mdm2 interaction and suppression of p53 degradation. This further leads to EGFR signaling suppression, intracellular reactive oxygen species generation, which results in cell cycle progression inhibition and apoptosis induction [68].
Notably, the negative prognostic role of non-disruptive TP53 alterations, leading to a partial loss of p53 functions while retaining its functional protein properties, has been reported. These variants are often associated with p53 gain-of-function (GOF), which affects the function of p53-related proteins by modulating transcriptional output. For example, downregulation of apoptotic Fas cell surface death receptor (FAS) and cell-arrest Cyclin dependent kinase inhibitor 1A (CDKN1A) genes, as well as upregulation of immortalizing Telomerase reverse transcriptase (TERT), mitogenic (Early growth response 1 (EGR1), MYC Proto-Oncogene, BHLH Transcription Factor (MYC)), stress-protective (Heat Shock 70 KDa Protein, HSPA1A), angiogenic (Angiopoietin 1, ANGPT1), or drug resistance (ATP Binding Cassette Subfamily B Member 1 (ABCB1), AXL Receptor Tyrosine Kinase (AXL)) genes, have been reported [69].
Interestingly, the effect of TP53 mutations is also reflected in their tumorigenic ability. It has been shown that most of the TP53 exon 4–8 alterations play a crucial role in NSCLC transformation into small-cell lung cancer (SCLC) [70–72]. A TP53 mutation can induce genomic instability in cancer cells, facilitating cell plasticity and phenotype reprogramming in approximately 3– 5% of adenocarcinomas with preserved primary EGFR mutation and ALK or ROS1 fusions. As SCLC is resistant to targeted therapies [73], this transformation results in TKIs failure, despite the initial presence of primary activating alterations in NSCLC. Additionally, transformation from adenocarcinoma to squamous cell carcinoma has been observed in NSCLC patients with pre-existing alterations of RB Transcriptional Corepressor 1 (RB1) and TP53 who progressed on EGFR-TKIs [73].
5Conclusion
Our review demonstrate that the presence of TP53 mutations in NSCLC has been linked to decreased responsiveness to targeted therapy with oral EGFR, ALK, and ROS1 tyrosine kinase inhibitors in most of the referenced studies. However, in some studies, concomitant TP53 mutations were associated with a favorable effect on targeted treatment outcomes, particularly when EGFR-TKIs were used in combination with angiogenesis inhibitors. Yet, the exact mechanisms, as well as the clinical significance of this phenomenon, have not been fully understood.
Currently, the integration of next-generation sequencing technologies into the routine molecular diagnostics of cancer patients facilitates the detection and identification of targetable genetic alterations, along with co-occurring TP53 variants, in well characterized prospective cohorts. This advancement may translate into rapid progress in understanding the biological and clinical role of p53 in targeted therapies for NSCLC. It seems plausible that assessing the TP53 mutation status could potentially aid in patient stratification for optimal clinical decision-making.
Importantly, drawing meaningful conclusions about the actual clinical value of TP53 co-mutations in EGFR- or ALK- or ROS1-positive NSCLC is hampered by insufficient knowledge of the functional consequences of the TP53 alterations, as well as a lack of detailed information regarding the exact TP53 variant location according to the Human Genome Variation Society (HGVS) recommendations in some papers. Moreover, studies based on the targeted sequencing of appointed TP53 gene regions (hot-spot) may still miss some important variants leading to significant bias and incorrect conclusions.
ACKNOWLEDGMENTS
Statutory activity project no. 3.9/2022.
Author contributions
CONCEPTION: JM-S, AS and JC-W.
DATA CURATION: JM-S.
ANALYSIS OF DATA: JM-S.
PREPARATION OF THE MANUSCRIPT: JM-S, AS and JC-W.
REVISION FOR IMPORTANT INTELLECTUAL CONTENT: JM-S, AS and JC-W.
SUPERVISION: JM-S.
Conflict of interest
J. Moes-Sosnowska, A. Szpechcinski, and J. Chorostowska-Wynimko have no conflicts of interest to report.
References
[1] | Siegel RL , Miller KD , Fuchs HE , Jemal A . CA: A Cancer Journal for Clinicians. (2022) ;72: :7–33. doi: 10.3322/caac.21708. |
[2] | Travis WD , Brambilla E , Burke AP , Marx A , Nicholson AG . Introduction to the 2015 world health organization classification of tumors of the lung, pleura, thymus, and heart. J Thorac Oncol. (2015) ;10: (9):1240–2. doi: 10.1097/JTO.0000000000000663. |
[3] | Workman S , Jabbour SK , Deek MP . A narrative review of genetic biomarkers in non-small cell lung cancer: An update and future perspectives. AME Med J. (2023) ;8: :6–6. doi: 10.21037/amj-2022-01. |
[4] | Melosky B , Wheatley-Price P , Juergens RA , Sacher A , Leighl NB , Tsao M-S , et al. The rapidly evolving landscape of novel targeted therapies in advanced non-small cell lung cancer. Lung Cancer. (2021) ;160: :136–51. doi: 10.1016/j.lungcan.2021.06.002. |
[5] | Liu SV . Novel targets in advanced non-small cell lung cancer. Clin Adv Hematol Oncol. (2022) ;20: (7):426–9. |
[6] | Marin-Acevedo JA , Pellini B , Kimbrough EO , Hicks JK , Chiappori A . Treatment strategies for non-small cell lung cancer with common EGFR mutations: A review of the history of EGFR TKIs approval and emerging data. Cancers. (2023) ;15: (3):629. doi: 10.3390/cancers15030629. |
[7] | Gibson AJW , Box A , Dean ML , Elegbede AA , Hao D , Sangha R , et al. Retrospective real-world outcomes for patients with ALK-rearranged lung cancer receiving ALK receptor tyrosine kinase inhibitors. JTO Clin Res Rep. (2021) ;2: (4):100157. doi: 10.1016/j.jtocrr.2021.100157. |
[8] | Bernabé-Caro R , Garrido P , García-Campelo R , Palmero R , Artal Á , Bayona C , et al. Alectinib after failure to crizotinib in patients with ALK-positive non-small cell lung cancer: Results from the Spanish early access program. Oncotarget. (2022) ;13: (1):812–27. doi: 10.18632/oncotarget.28244. |
[9] | Drilon A , Chiu C-H , Fan Y , Cho BC , Lu S , Ahn M-J , et al. Long-term efficacy and safety of entrectinib in ROS1 fusion-positive NSCLC. JTO Clin Res Rep. (2022) ;3: (6):100332. doi: 10.1016/j.jtocrr.2022.100332. |
[10] | Ou S-HI , Nishio M , Yoshida T , Kumagai T , Ahn M-J , Mok T , et al. MA13.03 integrated efficacy and safety of brigatinib following alectinib treatment in the ALTA-2 and J-ALTA studies. J Thorac Oncol. (2022) ;17: (9):S89. doi: 10.1016/j.jtho.2022.07.149. |
[11] | Shaw AT , Solomon BJ , Besse B , Bauer TM , Lin C-C , Soo RA , et al. ALK resistance mutations and efficacy of lorlatinib in advanced anaplastic lymphoma kinase-positive non-small-cell lung cancer. J Clin Oncol. (2019) ;37: (16):1370–9. doi: 10.1200/JCO.18.02236. |
[12] | Felip E , Shaw AT , Bearz A , Camidge DR , Solomon BJ , Bauman JR , et al. Intracranial and extracranial efficacy of lorlatinib in patients with ALK-positive non-small-cell lung cancer previously treated with second-generation ALK TKIs. Ann Oncol. (2021) ;32: (5):620–30. doi: 10.1016/j.annonc.2021.02.012. |
[13] | Mitsudomi T , Hamajima N , Ogawa M , Takahashi T . Prognostic significance of p53 alterations in patients with non-small cell lung cancer: A meta-analysis. Clin Cancer Res. (2000) ;6: (10):4055–63. |
[14] | Van Egeren D , Kohli K , Warner JL , Bedard PL , Riely G , Lepisto E , et al. Genomic analysis of early-stage lung cancer reveals a role for TP53 mutations in distant metastasis. Sci Rep. (2022) ;12: (1):19055. doi: 10.1038/s41598-022-21448-1. |
[15] | Jiao X-D , Qin B-D , You P , Cai J , Zang Y-S . The prognostic value of TP53 and its correlation with EGFR mutation in advanced non-small cell lung cancer, an analysis based on cBioPortal data base. Lung Cancer. (2018) ;123: :70–5. doi: 10.1016/j.lungcan.2018.07.003. |
[16] | Voskarides K , Giannopoulou N . The role of TP53 in adaptation and evolution. Cells. (2023) ;12: (3). doi: 10.3390/cells12030512. |
[17] | Chillemi G , Kehrloesser S , Bernassola F , Desideri A , Dötsch V , Levine AJ , et al. Structural evolution and dynamics of the p53 proteins. Cold Spring Harb Perspect Med. (2017) ;7: (4):a028308. doi: 10.1101/cshperspect.a028308. |
[18] | Capuozzo M , Santorsola M , Bocchetti M , Perri F , Cascella M , Granata V , et al. P From fundamental biology to clinical applications in cancer. Biology. (2022) ;11: (9):1325. doi: 10.3390/biology11091325. |
[19] | Kulesza M , Dansonka-Mieszkowska A , Pieńkowska-Grela B . Repair or perish – the role of p53 protein in a cell’s life. Journal of Oncology. (2019) ;69: (5–6):168–78. doi: 10.5603/njo.2019.0031. |
[20] | Fanale D , Maragliano R , Bazan V , Russo A . Caretakers and Gatekeepers. eLS. Wiley; (2017) , pp. 1–10. doi: 10.1002/9780470015902.a0006048.pub2. |
[21] | Flöter J , Kaymak I , Schulze A . Regulation of metabolic activity by p53. Metabolites. (2017) ;7: (2):21. doi: 10.3390/metabo7020021. |
[22] | Yue X , Zhao Y , Xu Y , Zheng M , Feng Z , Hu W . Mutant p53 in cancer: Accumulation, gain-of-function, and therapy. J Mol Biol. (2017) ;429: (11):1595–606. doi: 10.1016/j.jmb.2017.03.030. |
[23] | Mogi A , Kuwano H . TP53 mutations in nonsmall cell lung cancer. J Biomed Biotechnol. (2011) ;2011: :583929. doi: 10.1155/2011/583929. |
[24] | Donehower LA , Soussi T , Korkut A , Liu Y , Schultz A , Cardenas M , et al. Integrated analysis of TP53 gene and pathway alterations in the cancer genome atlas. Cell Rep. (2019) ;28: (11):3010. doi: 10.1016/j.celre2019.08.061. |
[25] | Canale M , Petracci E , Delmonte A , Bronte G , Chiadini E , Ludovini V , et al. Concomitant TP53 mutation confers worse prognosis in EGFR-mutated non-small cell lung cancer patients treated with TKIs. J Clin Med. (2020) ;9: (4):1047. doi: 10.3390/jcm9041047. |
[26] | Wee P , Wang Z . Epidermal growth factor receptor cell proliferation signaling pathways. Cancers. (2017) ;9: (5):52. doi: 10.3390/cancers9050052. |
[27] | Robichaux JP , Le X , Vijayan RSK , Hicks JK , Heeke S , Elamin YY , et al. Structure-based classification predicts drug response in EGFR-mutant NSCLC. Nature. (2021) ;597: (7878):732–7. doi: 10.1038/s41586-021-03898-1. |
[28] | Peng W , Yao C , Pan Q , Zhang Z , Ye J , Shen B , et al. Novel considerations on EGFR-based therapy as a contributor to cancer cell death in NSCLC. Front Oncol. (1120) ;13: :278. doi: 10.3389/fonc.2023.1120278. |
[29] | Hou H , Qin K , Liang Y , Zhang C , Liu D , Jiang H , et al. Concurrent TP53 mutations predict poor outcomes of EGFR-TKI treatments in Chinese patients with advanced NSCLC. Cancer Manag Res. (2019) ;11: :5665–75. doi: 10.2147/CMAR.S201513. |
[30] | Kim Y , Lee B , Shim JH , Lee S-H , Park W-Y , Choi Y-L , et al. Concurrent genetic alterations predict the progression to target therapy in EGFR-mutated advanced NSCLC. J Thorac Oncol. (2019) ;14: (2):193–202. doi: 10.1016/j.jtho.2018.10.150. |
[31] | Li X-M , Li W-F , Lin J-T , Yan H-H , Tu H-Y , Chen H-J , et al. Predictive and prognostic potential of TP53 in patients with advanced non-small-cell lung cancer treated with EGFR-TKI: Analysis of a phase III randomized clinical trial (CTONG 0901). Clin Lung Cancer. (2021) ;22: (2):100–109.e3. doi: 10.1016/j.cllc.2020.11.001. |
[32] | Roeper J , Christopoulos P , Falk M , Heukamp LC , Tiemann M , Stenzinger A , et al. TP53 co-mutations as an independent prognostic factor in 2nd and further line therapy-EGFR mutated non-small cell lung cancer IV patients treated with osimertinib. Transl Lung Cancer Res. (2022) ;11: (1):4–13. doi: 10.21037/tlcr-21-754. |
[33] | Zhao H , Yao W , Min X , Gu K , Yu G , Zhang Z , et al. Apatinib plus gefitinib as first-line treatment in advanced EGFR-mutant NSCLC: The phase III ACTIVE study (CTONG1706). J Thorac Oncol. (2021) ;16: (9):1533–46. doi: 10.1016/j.jtho.2021.05.006. |
[34] | Sun H , Ren P , Chen Y , Lan L , Yan Z , Yang Y , et al. Optimal therapy for concomitant EGFR and TP53 mutated non-small cell lung cancer: A real-world study. BMC Cancer. (2023) ;23: (1):198. doi: 10.1186/s12885-023-10637-4. |
[35] | Nishio M , Paz-Ares L , Reck M , Nakagawa K , Garon EB , Popat S , et al. RELAY, ramucirumab plus erlotinib (RAM+ERL) in untreated metastatic EGFR-mutant NSCLC (EGFR+NSCLC): Association between TP53 status and clinical outcome. Clin Lung Cancer. (2023) ). doi: 10.1016/j.cllc.2023.02.010. |
[36] | Hou X , Li M , Wu G , Feng W , Su J , Jiang H , et al. Gefitinib plus chemotherapy vs gefitinib alone in untreated EGFR-mutant non-small cell lung cancer in patients with brain metastases: The GAP BRAIN open-label, randomized, multicenter, phase 3 study. JAMA Netw Open. (2023) ;6: (2):e2255050. doi: 10.1001/jamanetworkopen.2022.55050. |
[37] | Della Corte CM , Viscardi G , Di Liello R , Fasano M , Martinelli E , Troiani T , et al. Role and targeting of anaplastic lymphoma kinase in cancer. Mol Cancer. (2018) ;17: (1). doi: 10.1186/s12943-018-0776-2. |
[38] | Golding B , Luu A , Jones R , Viloria-Petit AM . The function and therapeutic targeting of anaplastic lymphoma kinase (ALK) in non-small cell lung cancer (NSCLC). Mol Cancer. (2018) ;17: (1). doi: 10.1186/s12943-018-0810-4. |
[39] | Lei Y , Lei Y , Shi X , Wang J . EML4-ALK fusion gene in non-small cell lung cancer. Oncol Lett. (2022) ;24: (2);277. doi: 10.3892/ol.2022.13397. |
[40] | Wang Z , Han Y , Tao H , Xu M , Liu Z , Zhu J , et al. Molecular characterization of genomic breakpoints of ALK rearrangements in non-small cell lung cancer. Mol Oncol. (2023) ;17: (5):765–78. doi: 10.1002/1878-0261.13348. |
[41] | Xiang Y , Zhang S , Fang X , Jiang Y , Fang T , Liu J , et al. Therapeutic advances of rare ALK fusions in non-small cell lung cancer. Curr Oncol. (2022) ;29: (10):7816–31. doi: 10.3390/curroncol29100618. |
[42] | Li M , Tang Q , Chen S , Wang Y . A novel HIP1-ALK fusion variant in lung adenocarcinoma showing resistance to Crizotinib. Lung Cancer. (2021) ;151: :98–100. doi: 10.1016/j.lungcan.2020.11.014. |
[43] | Camidge DR , Dziadziuszko R , Peters S , Mok T , Noe J , Nowicka M , et al. Updated efficacy and safety data and impact of the EML4-ALK fusion variant on the efficacy of alectinib in untreated ALK-positive advanced non-small cell lung cancer in the global phase III ALEX study. J Thorac Oncol. (2019) ;14: (7):1233–43. doi: 10.1016/j.jtho.2019.03.007. |
[44] | Song P , Zhang F , Li Y , Yang G , Li W , Ying J , et al. Concomitant TP53 mutations with response to crizotinib treatment in patients with ALK-rearranged non-small-cell lung cancer. Cancer Med. (2019) ;8: (4):1551–7. doi: 10.1002/cam4.2043. |
[45] | Kron A , Alidousty C , Scheffler M , Merkelbach-Bruse S , Seidel D , Riedel R , et al. Impact of TP53 mutation status on systemic treatment outcome in ALK-rearranged non-small-cell lung cancer. Ann Oncol. (2018) ;29: (10):2068–75. doi: 10.1093/annonc/mdy333. |
[46] | Wang W-X , Xu C-W , Chen Y-P , Liu W , Zhong L-H , Chen F-F , et al. TP53 mutations predict for poor survival in ALK rearrangement lung adenocarcinoma patients treated with crizotinib. J Thorac Dis. (2018) ;10: (5):2991–8. doi: 10.21037/jtd.2018.04.98. |
[47] | Canale M , Petracci E , Cravero P , Mariotti M , Minuti G , Metro G , et al. Prognosis of ALK-rearranged non-small-cell lung cancer patients carrying TP53 mutations. Transl Oncol. (2023) ;23: (101471):101471. doi: 10.1016/j.tranon.2022.101471. |
[48] | Li J , Zhang B , Zhang Y , Xu F , Zhang Z , Shao L , et al. Concomitant mutation status of ALK-rearranged non-small cell lung cancers and its prognostic impact on patients treated with crizotinib. Transl Lung Cancer Res. (2021) ;10: (3):1525–35. doi: 10.21037/tlcr-21-160. |
[49] | Shi J , Jia Z , Zhou Z , Zhao L , Meng Q , Liu Y . Ineffectiveness of crizotinib in a non-small-cell lung cancer with novel ALK- LIMS1 fusion: A case report. Onco Targets Ther. (2023) ;16: :109–14. doi: 10.2147/OTT.S388962. |
[50] | Wolf J , Helland Å , Oh I-J , Migliorino MR , Dziadziuszko R , Wrona A , et al. Final efficacy and safety data, and exploratory molecular profiling from the phase III ALUR study of alectinib versus chemotherapy in crizotinib-pretreated ALK-positive non-small-cell lung cancer. ESMO Open. (2022) ;7: (1):100333. doi: 10.1016/j.esmoo2021.100333. |
[51] | Frost N , Christopoulos P , Kauffmann-Guerrero D , Stratmann J , Riedel R , Schaefer M , et al. Lorlatinib in pretreated ALK- or ROS1-positive lung cancer and impact of TP53 co-mutations: Results from the German early access program. Ther Adv Med Oncol. (2021) ;13: :1758835920980558. doi: 10.1177/1758835920980558. |
[52] | Song Z , Lian S , Mak S , Chow MZ-Y , Xu C , Wang W , et al. Deep RNA sequencing revealed fusion junctional heterogeneity may predict crizotinib treatment efficacy in ALK-rearranged NSCLC. J Thorac Oncol. (2022) ;17: (2):264–76. doi: 10.1016/j.jtho.2021.09.016. |
[53] | Drilon A , Jenkins C , Iyer S , Schoenfeld A , Keddy C , Davare MA . ROS1-dependent cancers - biology, diagnostics and therapeutics. Nat Rev Clin Oncol. (2021) ;18: (1):35–55. doi: 10.1038/s41571-020-0408-9. |
[54] | Gendarme S , Bylicki O , Chouaid C , Guisier F . ROS-1 fusions in non-small-cell lung cancer: Evidence to date. Curr Oncol. (2022) ;29: (2):641–58. doi: 10.3390/curroncol29020057. |
[55] | Dziadziuszko R , Krebs MG , De Braud F , Siena S , Drilon A , Doebele RC , et al. Updated integrated analysis of the efficacy and safety of entrectinib in locally advanced or metastatic ROS1 fusion-positive non-small-cell lung cancer. J Clin Oncol. (2021) ;39: (11):1253–63. doi: 10.1200/JCO.20.03025. |
[56] | Begum P , Cui W , Popat S . Crizotinib-resistant ROS1 GA mutation associated with sensitivity to lorlatinib in ROS1-rearranged NSCLC: Case report. JTO Clin Res Rep. (2022) ;3: (9):100376. doi: 10.1016/j.jtocrr.2022.100376. |
[57] | Guaitoli G , Bertolini F , Bettelli S , Manfredini S , Maur M , Trudu L , et al. Deepening the knowledge of ROS1 rearrangements in non-small cell lung cancer: Diagnosis, treatment, resistance and concomitant alterations. Int J Mol Sci. (2021) ;22: (23):12867. doi: 10.3390/ijms222312867. |
[58] | Priest K , Le A , Gebregzabheir A , Nijmeh H , Reis GB , Mandell M , et al. Evolution of acquired resistance in a ROS1+KRAS G12C+NSCLC through the MAPK pathway. NPJ Precis Oncol. (2023) ;7: (1):9. doi: 10.1038/s41698-023-00349-0. |
[59] | Lin G , Xu H , Zhao J , Kong J , Ai X , Yu F , et al. P2.14-09 concurrent TP53 mutation adversely impact the efficacy of crizotinib in ROS1-rearranged lung cancer patients. J Thorac Oncol. (2019) ;14: (10):S832. doi: 10.1016/j.jtho.2019.08.1794. |
[60] | Wiesweg M , Eberhardt WEE , Reis H , Ting S , Savvidou N , Skiba C , et al. High prevalence of concomitant oncogene mutations in prospectively identified patients with ROS1-positive metastatic lung cancer. J Thorac Oncol. (2017) ;12: (1):54–64. doi: 10.1016/j.jtho.2016.08.137. |
[61] | Zeng L , Li Y , Xiao L , Xiong Y , Liu L , Jiang W , et al. Crizotinib presented with promising efficacy but for concomitant mutation in next-generation sequencing-identified ROS1-rearranged non-small-cell lung cancer. Onco Targets Ther. (2018) ;11: :6937–45. doi: 10.2147/OTT.S176273. |
[62] | Landi L , Tiseo M , Heukamp LC , Menon R , de Marinis F , Minuti G , et al. P2.14-02 TP53 mutations affect sensitivity to lorlatinib in ROS1 positive NSCLC: Final results of the PFROST trial. J Thorac Oncol. (2022) ;17: (9):S157. doi: 10.1016/j.jtho.2022.07.258. |
[63] | Jóri B , Falk M , Hövel I , Weist P , Tiemann M , Heukamp LC , et al. Acquired GR resistance mutation in ROS1 to lorlatinib therapy detected with liquid biopsy. Curr Oncol. (2022) ;29: (9):6628–34. doi: 10.3390/curroncol29090520. |
[64] | Jung S , Kim DH , Choi YJ , Kim SY , Park H , Lee H , et al. Contribution of p53 in sensitivity to EGFR tyrosine kinase inhibitors in non-small cell lung cancer. Sci Rep. (2021) ;11: (1):19667. doi: 10.1038/s41598-021-99267-z. |
[65] | Wheler JJ , Janku F , Naing A , Li Y , Stephen B , Zinner R , et al. TP53 alterations correlate with response to VEGF/VEGFR inhibitors: Implications for targeted therapeutics. Mol Cancer Ther. (2016) ;15: (10):2475–85. doi: 10.1158/1535-7163.MCT-16-0196. |
[66] | Khromova NV , Kopnin PB , Stepanova EV , Agapova LS , Kopnin BP . p53 hot-spot mutants increase tumor vascularization via ROS-mediated activation of the HIF1/VEGF-A pathway. Cancer Lett. (2009) ;276: (2):143–51. doi: 10.1016/j.canlet.2008.10.049. |
[67] | Li AM , Boichard A , Kurzrock R . Mutated TP53 is a marker of increased VEGF expression: Analysis of 7,525 pan-cancer tissues. Cancer Biol Ther. (2020) ;21: (1):95–100. doi: 10.1080/15384047.2019.1665956. |
[68] | Zhang Y , Han CY , Duan FG , Fan X-X , Yao X-J , Parks RJ , et al. p53 sensitizes chemoresistant non-small cell lung cancer via elevation of reactive oxygen species and suppression of EGFR/PI3K/AKT signaling. Cancer Cell Int. (2019) ;19: (1):188. doi: 10.1186/s12935-019-0910-2. |
[69] | Molina-Vila MA , Bertran-Alamillo J , Gascó A , Mayo-de-las-Casas C , Sánchez-Ronco M , Pujantell-Pastor L , et al. Nondisruptive p53 mutations are associated with shorter survival in patients with advanced non-small cell lung cancer. Clin Cancer Res. (2014) ;20: (17):4647–59. doi: 10.1158/1078-0432.CCR-13-2391. |
[70] | Lee J-K , Lee J , Kim S , Kim S , Youk J , Park S , et al. Clonal history and genetic predictors of transformation into small-cell carcinomas from lung adenocarcinomas. J Clin Oncol. (2017) ;35: (26):3065–74. doi: 10.1200/JCO.2016.71.9096. |
[71] | Marcoux N , Gettinger SN , O’Kane G , Arbour KC , Neal JW , Husain H , et al. EGFR-mutant adenocarcinomas that transform to small-cell lung cancer and other neuroendocrine carcinomas: Clinical outcomes. J Clin Oncol. (2019) ;37: (4):278–85. doi: 10.1200/JCO.18.01585. |
[72] | Lin JJ , Langenbucher A , Gupta P , Yoda S , Fetter IJ , Rooney M , et al. Small cell transformation of ROS1 fusion-positive lung cancer resistant to ROS1 inhibition. NPJ Precis Oncol. (2020) ;4: (1):21. doi: 10.1038/s41698-020-0127-9. |
[73] | Schoenfeld AJ , Chan JM , Kubota D , Sato H , Rizvi H , Daneshbod Y , et al. Tumor analyses reveal squamous transformation and off-target alterations as early resistance mechanisms to first-line osimertinib in EGFR-mutant lung cancer. Clin Cancer Res. (2020) ;26: (11):2654–63. doi: 10.1158/1078-0432.CCR-19-3563. |
[74] | Yang Y , Huang J , Wang T , Zhou J , Zheng J , Feng J , et al. Decoding the evolutionary response to ensartinib in patients with ALK-positive NSCLC by dynamic circulating tumor DNA sequencing. J Thorac Oncol. (2021) ;16: (5):827–39. doi: 10.1016/j.jtho.2021.01.1615. |
[75] | Kanehisa M , Furumichi M , Sato Y , Kawashima M , Ishiguro-Watanabe M . KEGG for taxonomy-based analysis of pathways and genomes. Nucleic Acids Res. (2023) ;51: :D587–92. doi.org/10.1093/nar/gkac963. |
[76] | Cerami E , Gao J , Dogrusoz U , Gross BE , Sumer SO , Aksoy BA , et al. The cBio cancer genomics portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov. (2012) ;2: (5):401–4. doi: 10.1158/2159-8290.CD-12-0095. |
[77] | Gao J , Aksoy BA , Dogrusoz U , Dresdner G , Gross B , Sumer SO , et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal. (2013) ;6: (269):l1. doi: 10.1126/scisignal.2004088. |


