
Induction of alternative NF-κB within TAg-induced basal mammary tumors in activation-resistant inhibitor of κ-B kinase (IKKα) mutant mice
Abstract
BACKGROUND:
The alternative NF-κB pathway is activated by the NF-κB-inducing kinase (NIK) mediated phosphorylation of the inhibitor of κ-B kinase α (IKKα). IKKα then phosphorylates p100/NFKB2 to result in its processing to the active p52 subunit. Evidence suggests that basal breast cancers originate within a subpopulation of luminal progenitor cells which is expanded by signaling to IKKα.
OBJECTIVE:
To determine the role of IKKα in the development of basal tumors.
METHODS:
Kinase dead IkkαAA/AA mice were crossed with the C3(1)-TAg mouse model of basal mammary cancer. Tumor growth and tumor numbers in WT and IkkαAA/AA mice were assessed and immunopathology, p52 expression and stem/progenitor 3D colony forming assays were performed. Nik-/- mammary glands were isolated and mammary colonies were characterized.
RESULTS:
While tumor growth was slower than in WT mice, IkkαAA/AA tumor numbers and pathology were indistinguishable from WT tumors. Both WT and IkkαAA/AA tumors expressed p52 except those IkkαAA/AA tumors where NIK, IKKαAA/AA and ErbB2 were undetectable. Colonies formed by WT and IkkαAA/AA mammary cells were nearly all luminal/acinar however, colony numbers and sizes derived from IkkαAA/AA cells were reduced. In contrast to IkkαAA/AA mice, virgin Nik-/- mammary glands were poorly developed and colonies were primarily derived from undifferentiated bipotent progenitor cells.
CONCLUSIONS:
C3(1)-TAg induced mammary tumors express p100/p52 even without functional IKKα. Therefore the development of basal-like mammary cancer does not strictly rely on IKKα activation. Signal-induced stabilization of NIK may be sufficient to mediate processing of p100NFKB2 which can then support basal-like mammary tumor formation. Lastly, in contrast to the pregnancy specific role of IKKα in lobuloalveogenesis, NIK is obligatory for normal mammary gland development.
1Introduction
The mammary epithelium consists of both stem and progenitor cells that have been defined immunophenotypically and functionally and include a stem-like basal population (MaSc), a luminal progenitor (LP) population and a mature cell population [1–3]. Progenitors with proliferative capacity are thought to be the cells of origin for the different subtypes of breast cancers where luminal breast cancers constitute approximately 80–85% of all tumors and basal-type breast cancers that include the triple negative subtype make up the majority of remaining tumors although rare metaplastic tumors are also found [4, 5]. Both basal and luminal breast cancers are derived from the LP cells consistent with a spectrum of fate potential within this population [6–9].
SV40 large TAg promotes the development of cancer through inactivation of both p53 and Rb tumor suppressor proteins [10, 11]. C3(1)-promoter directs gene expression in the terminal ductal lobular unit (TLDU) [12] which is thought to be the site of origin of breast cancers in humans [13] and contains stem and progenitor cells [14]. The C3(1)/SV40 T-antigen transgenic mouse model (C3(1)-TAg mice) develops mammary hyperplasia by 3 months of age which progress to adenocarcinomas by 5–6 months of age [15]. Tumors derived from this model are highly similar to human basal-like breast cancer expressing both basal and luminal markers [16, 17]. The majority of C3(1)-TAg tumors express genes characteristic of human basal-like breast cancer while about 10% are similar to metaplastic lesions [18–20].
Numerous publications in the past 20 years have demonstrated the important role of NF-κB in the normal mammary gland [21–23] and our lab and others in mammary tumorigenesis [24–27]. NF-κB consists of two pathways termed the canonical pathway consisting of p50/p65(RelA) heterodimers and the alternative pathway mediated by p52/RelB heterodimers. Both pathways can be regulated through receptor mediated signaling. The alternative pathway is induced by receptors including CD40, BAFF and RANK which, upon ligand binding, recruit the cIAP/TRAF3 complex preventing the ubiquitination and proteasome mediated degradation of the NF-κB inducing kinase (NIK/MAP3K14). NIK phosphorylation of p100 at Ser866 and Ser870, and complexes and phosphorylates IKKα homodimers resulting in IKKα-mediated phosphorylation of p100 at several other serines. This promotes recognition by the SCF/βTRCP ubiquitin ligase complex, resulting in partial degradation of the C-terminal ankyrin repeat domain of p100 by the 26S proteasome [28–31]. Since ankyrin repeat domains mask the nuclear localization signal, processed p52 binds to RelB and undergoes nuclear translocation. Both NIK and IKKα have critical roles in p100 processing although neither kinase appears sufficient for optimal phosphorylation and proteolytic processing to p52.
The alternative pathway is critical to expansion of the mammary gland during pregnancy when hormonal stimulation results in production of receptor activator of NF-κB kinase ligand (RANKL) which primarily stimulates the activity of IKKα through RANK. Indeed, while C57BL/6 IkkαAA/AA mice, which are homozygous for an activation-resistant mutant of IKKα through replacement of the serines 176/180 in the kinase activation loop with alanines, develop normal mammary glands, they fail to undergo lubuloalveolar development and demonstrate a lactational defect [21]. The number of secretory alveoli was dramatically reduced in comparison to WT glands of same developmental stage. In addition, the MaSC pool increases markedly when exposed to maximal progesterone levels at the luteal/dioestrus phase of the mouse [32]. Evidence shows that progesterone-initiated signaling in luminal cells activates production of Wnt4 and RANKL signals, in turn, relayed to basal cells resulting in expansion of this population. In previous work we have shown that alternative NF-κB activity predominates in patient basal tumors [25]. Based on this, we surmised that basal tumorigenesis in IkkαAA/AA mice might be diminished. To determine whether IKKα activation is requisite for tumorigenesis and progression of basal-type tumors we have crossed IkkαAA/AA mice with mice carrying the C3(1)-SV40T-Ag transgene. Our results revealed that IkkαAA/AA T-Ag expressing mice develop similar numbers of tumors but with a small but significantly increased latency. Tumor pathology was indistinguishable from control IkkαWT/WT mice. While p52 was strongly reduced in virgin IkkαAA/AA mammary glands, robust expression of p52 was present in all tumors that was correlated in part with ErbB2 and Nik detection in most tumors. Together, these results suggest that the luminal progenitor population that gives rise to basal tumors is independent of IKKα activity, and that the activating phosphorylation of IKKα is not necessary to promote sufficient alternative NF-κB to support basal tumor progression in this model.
2Results
2.1Characterization of IkkαAA/AA alternative NF-κB pathway activation and mammary progenitor cell colony forming ability
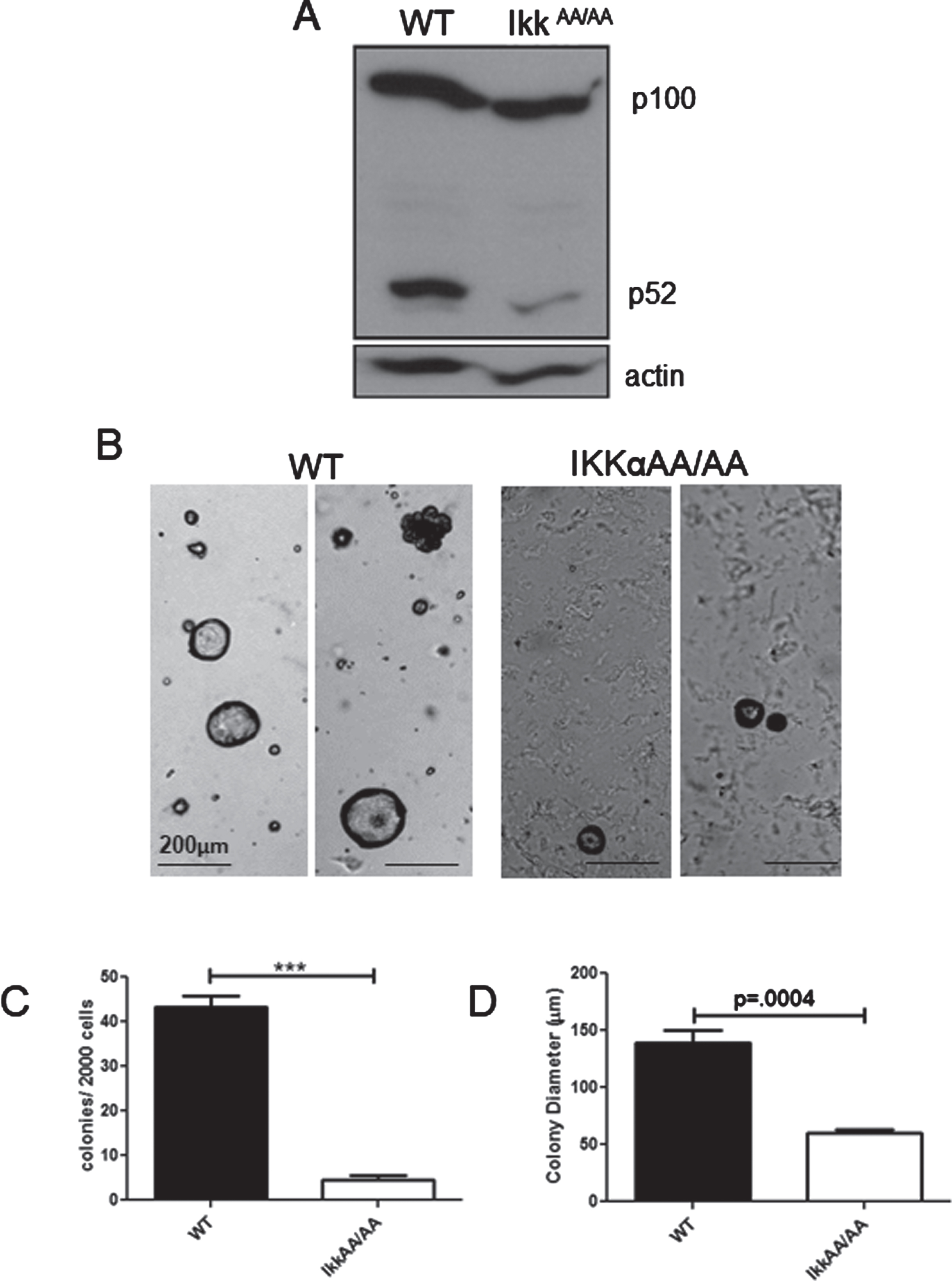
To assess the status of p100/p52 we performed an immunoblot analysis to detect steady state levels of p100/p52 in WT and IkkαAA/AA virgin mammary glands. Figure 1A shows that both mice produce similar levels of p100, but a strong diminution of p52 was evident in the IkkαAA/AA glands. The mammary epithelium consists of a spectrum of stem/basal cells and luminal progenitor cells that can be enriched by immunophenotype using FACS. It was previously reported that the relative abundance of each population is not altered in IkkαAA/AA mouse mammary glands [33]. And, although virgin glands are indistinguishable from WT mice, the number of secretory alveoli was dramatically reduced in comparison to WT glands during pregnancy rendering dams with a lactational defect [21]. The colony forming cell (CFC) assay provides an in vitro readout for progenitor cells that can form discrete colonies in matrigel [34]. Acinar structures are characteristic of structures formed by luminal progenitor cells in Matrigel [35]. The relative sizes of these colonies reflects the proliferative potential within these progenitor populations [36]. To test the role of kinase activity deficient IKKαAA/AA on colony formation we plated lin cells (CD31-; Ter119-; CD45-;(L-)) from IkkαAA/AA and WT mammary glands in Matrigel supplemented with B27 which contains progesterone, and analyzed colonies after 12 days. Examples of colonies are shown in Fig. 1B. As expected, the large majority of colonies (>90%) in both WT and IkkαAA/AA cell cultures were acinar luminal colonies. WT cells produced more than 2-fold more colonies compared to IkkαAA/AA mice (Fig. 1C). Colony sizes formed by IkkαAA/AA cells were also significantly (∼50%) smaller than WT colonies indicating a proliferative deficiency despite factor/hormone supplementation of the media (Fig. 1D). Overall, these data are commensurate with the inability of IkkαAA/AA mice to expand the lobuloalveolar network during pregnancy and demonstrate defective proliferative capacity of IkkαAA/AA luminal progenitor cells.
Fig. 1
Attenuated colony formation by IkkαAA/AA mammary cells: A, Whole mammary glands from virgin WT and IkkαAA/AA mice were isolated and protein extracts immunoblotted to detect p100/p52. Actin was used as a loading control. B, WT and IkkαAA/AA mammary glands were isolated from 3 each 7-8 week old mice and linneg cells were subjected to Matrigel colony formation assays. Examples of luminal and basal (arrow) colonies are shown form both mice. Bars are 200μm. C, Colonies per 2000 seeded cells were counted and D, colony diameters measured. Statistical analyses: unpaired t-tests ***p < 0.0001.
2.2IkkαAA/AA C3(1)-TAg tumors grow slower than WT tumors but are pathologically indistinguishable from WT tumors
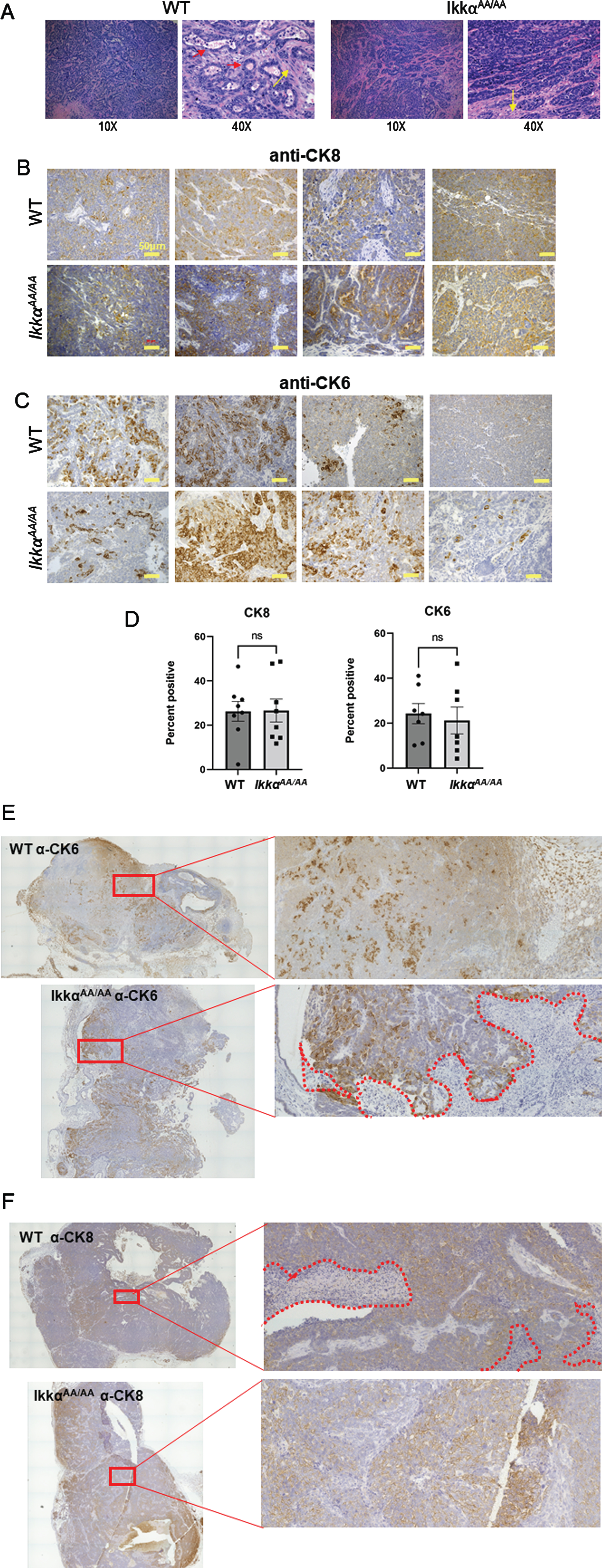
We previously demonstrated that human basaloid/TNBC differentially expresses components of the alternative NF-κB pathway [25]. To test the ability of mice deficient in IKKα kinase activity to produce basal tumors we crossed IkkαAA/AA mice with the C3(1)-TAg model of basal breast cancer. WT/WT and AA/AA genotypes positive for the TAg transgene were confirmed by PCR (Fig. 2A). The Kaplan-Meier analysis in Fig. 2B shows that there was a significant increase in latency of tumor formation in the IkkαAA/AA mice compared to WT controls however, tumor numbers per mouse and tumor sizes at endpoint were not significantly different (Fig. 2C, D). The C3(1)-driven TAg generates basaloid tumors that typically heterogeneously express the basal cytokeratin CK6 and luminal cytokeratin CK8 [37]. H&E sections in Fig. 3A are representative images of basaloid adenocarcinomas with regions of squamous tumor in WT and IkkαAA/AA mammary glands. IHC for CK8 (Fig. 3B) and CK6 (Fig. 3C) expression in tumors from WT (n = 10) and IkkαAA/AA (n = 12) showed heterogeneous expression of both cytokeratins. Immunostained slides were scanned and analyzed using ImageJ for expression of CK8 (n = 8 WT and n = 9 IkkαAA/AA) and CK6 (n = 7 each WT and IkkαAA/AA) and revealed no difference between expression levels of either cytokeratin between tumor genotypes (Fig. 3D). Representative whole tumor IHC scans are shown in Figs. 3E and 3F which also illustrate the varying levels of necrotic tissue in tumors from both genotypes.
Fig. 2
IkkαAA/AA tumors grow slower than C3(1)-TAg mice but tumor numbers and sizes are similar to WT. A, PCR genotyping to identify IkkαAA/AA mice lacking the IKKα-kinase activation domain harbouring the C3(1)-TAg transgene. Mice used in this study were WT and IkkαAA/AA; C3(1)TAg+ females. B, Kaplan Meier analysis of survival of mice to endpoint determined by largest tumor size of ∼2mm3, p = 0.0134 (log rank test). C, The number of tumors per mouse was calculated from mice of each genotype (n = 9 per genotype). No significant difference was obtained (unpaired t-test). D, Volumes of all tumors from each mouse were measured using calipers (n = 11 mice per genotype).
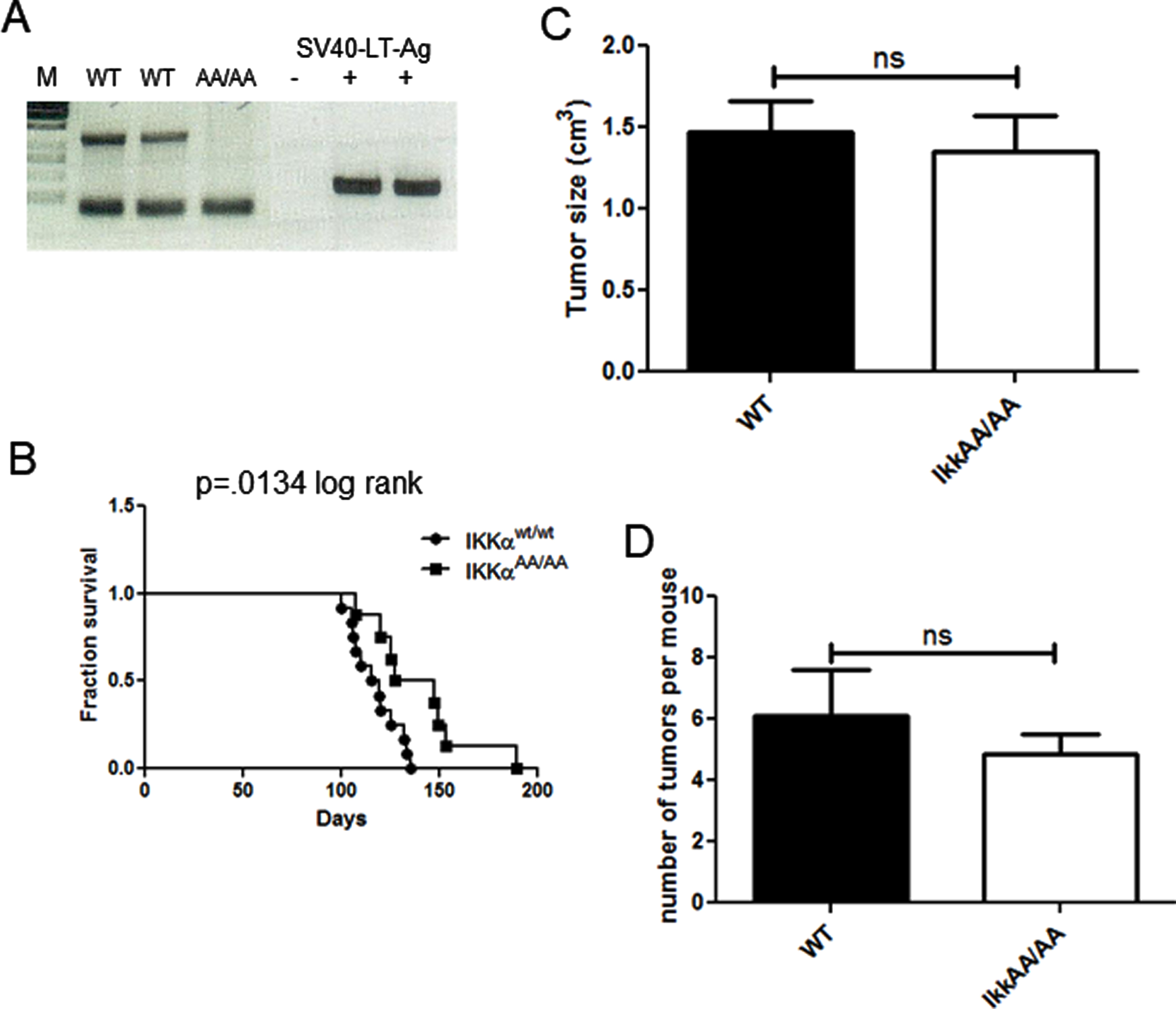
Fig. 3
Histopathology of C3(1)-TAg tumors from WT and IkkαAA/AA mice. A, Examples of H&E sections from different IkkαAA/AA and WT mice showing typical solid adenocarcinomas with varying regions of squamous (metaplastic) differentiation (yellow arrows). Some tumors had tumor surrounding residual lobular and ductal structures (red arrows). Image magnification is shown below. B, Tumor sections from the indicated mouse genotypes were immunostained with anti-CK8 (luminal cytokeratin) and C, anti-CK6 (basal cytokeratin) as described in Methods. Bars are 50μm. D, Full tumor immunohistochemistry sections were scanned on an EVOS FL Auto 2 microscope and analyzed using FIJI for expression of CK8 (n = 8 WT and n = 9 IkkαAA/AA tumors) and CK6 (n = 7 WT and IkkαAA/AA tumors). Representative C(3)1-TAg whole tumor scans from WT and IkkαAA/AA mice are shown immunostained with CK6, E and CK8, F (20X objective). Boxed regions are expanded on the right. Necrotic areas are outlined by dotted lines.
2.3Expression of C3(1)-TAg occurs in subsets of cells in the LP and MaSC-enriched populations
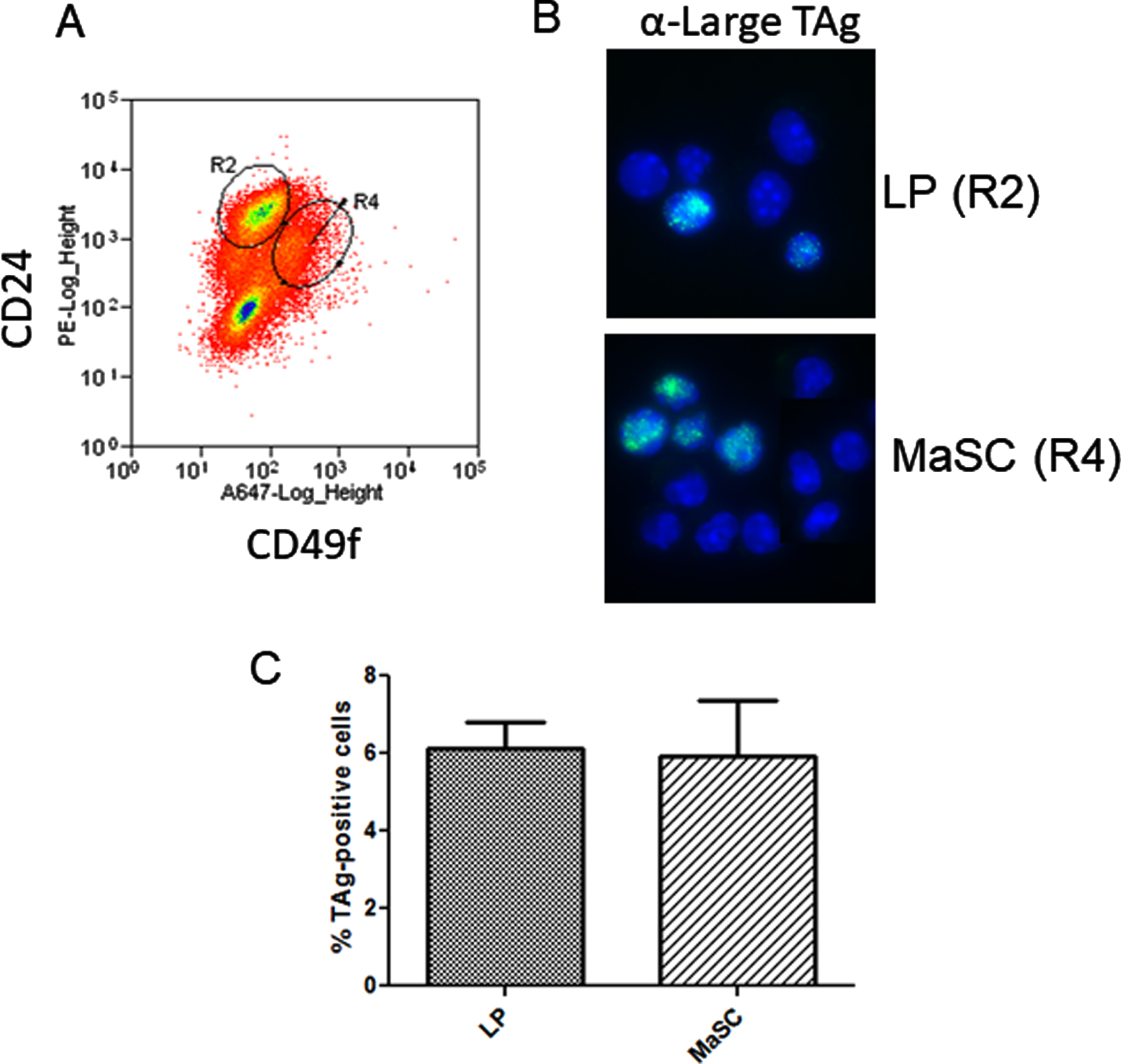
The development of basal tumors in the C3(1)-TAg model is reported to be augmented by estrogen stimulation in the early phase of tumor formation primarily by increasing the number of target cells capable of activation of the C3(1) promoter [38]. In vivo, estrogen is permissive for progesterone-induced expansion of the basal/MaSC population through the activity of RANKL [32]. Basal tumors have been shown to originate in a subpopulation of luminal mammary progenitor cells. Since IkkαAA/AA mammary cell colony formation was reduced relative to WT and tumor growth rate was decreased, we wondered if C3(1)-promoter might drive expression of TAg within populations that are independent of IKKα activity for proliferation. Figure 4A shows the FACS enrichment plot of IkkαAA/AA mouse luminal and basal MaSC cells. Immunofluorescence of FACS-sorted cells from C3(1)-TAg mammary glands showed that TAg was present in both luminal (CD24+/CD49fl ∘) and basal/MaSc (CD24+/CD49fhi)-enriched progenitor populations at relatively equal levels (Fig. 4 B,C). Therefore at least a subset of the spectrum of cells within both the basal/MaSC and luminal progenitor populations could contribute to C3(1)-TAg tumors. Combined with tumor numbers and basal and luminal keratin expression that phenocopies WT mice, lack of IKKα activity does not appear to effect the cell populations that are competent to activate the C3(1) promoter.
Fig. 4
C3(1)-SV40-TAg is expressed in basal and luminal mammary progenitors: A, Linneg mammary gland cells from 8 week old virgin C3(1)TAg transgenic mice were isolated and subjected to FACS analysis to enrich mammary luminal progenitors and basal/MaSc populations using linp ∘s exclusion and CD24/CD49f immunophenotype. B, FACS-sorted cells were cytospun and IF performed to detect TAg. C, The percentage of TAg-expressing cells was calculated from counting total GFP-positive cells/ total DAPI nuclei. >200 cells per slide were counted. No significant difference between populations was obtained (unpaired t-test).
2.4p100 processing to p52 in IkkαAA/AA mammary tumors correlates with IKKα protein expression
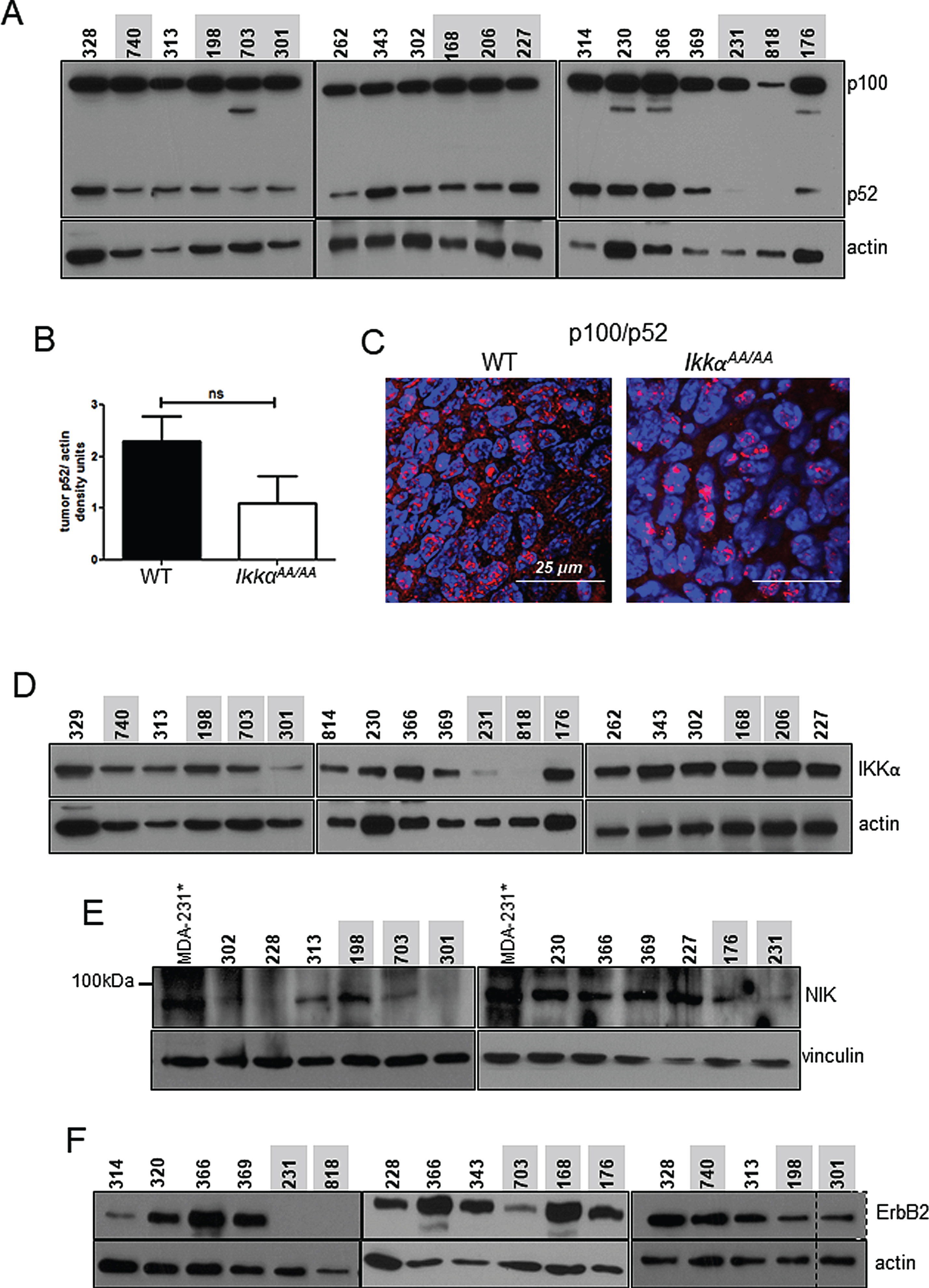
As described, IKKα, considered integral to phosphorylation of p100 to signal processing to p52 C3(1)-TAg-driven mammary basal tumor formation, in IkkαAA/AA mice resulted in a mild but significant latency phenotype. Remarkably, immunoblots to determine the status of p100 processing in WT vs IkkαAA/AA tumors showed that tumors from both animals produced a range of levels of p52, and only a few IkkαAA/AA tumors were deficient in p52 (Fig. 5A). When p52 protein was normalized to actin levels, p52 protein levels were lower in IkkαAA/AA tumors but not significantly lower than that in WT tumors (Fig. 5B). To further characterize p100/p52 activation in tumors we performed immunofluorescence on tumor sections and confirmed the presence of nuclear p52 in both IkkαAA/AA and WT tumors (Fig. 5C). IKKα protein has been shown to associate with the NIK:p100 complex to facilitate p100 phosphorylation [28]. We therefore immunoblotted for IKKα to detect either WT or the mutant IKKαAA/AA kinase in tumors (Fig. 5D) to determine if there was a correlation between IKKα expression and p52. Interestingly, 2 tumors (301 and 818) that did not express p52 also did not express IKKα. Immunoblot for NIK (Fig. 5E) was also performed where MDA-MB-231 cells treated with SM164 to stabilize NIK through inhibition of cIAP-mediated ubiquitination were used as a positive control. Again tumors that do not express p52 had low or undetectable levels of NIK. Thus, at least some C3(1)-TAg tumors do require activation of alternative NF-κB. Conversely, IkkαAA/AA tumors 227 and 168 contained abundant p52 which was associated with robust expression of both IKKα and NIK. Thus the absence of the mutant IKKα protein expression correlated with a lack of p100 processing.
ErbB2 expressing breast cancer cell lines contain p52 at varying levels, yet, knockdown of IKKα has no effect on the presence of p52 [39]. This suggests that ErbB2 signaling may provide an alternate IKKα-independent means of signaling p100/p52 processing. Immunoblot for ErbB2 in the TAg tumors in Fig. 5F shows that ErbB2 was readily detected in tumors from both genotypes with the exception of tumors 231 and 818 corresponding to the absence of p52. Thus, in some IkkαAA/AA tumors expressing high levels of ErbB2, such as 168 and 176, it is possible that ErbB2 signaling may have contributed to the activation of p100 processing.
Fig. 5
WT and IkkαAA/AA C3(1)-TAg tumors contain p100 and processed/nuclear p52: A, Immunoblot to detect p100/p52 in WT and IkkαAA/AA tumors. Actin was used as a loading control.Mouse identification numbers are shown above each sample and were used to track tumor genotype and protein samples (WT tumors are indicated in black and IkkαAA/AA tumors are indicated in red). B, p52 densitometry compiled from each tumor sample normalized to actin. Bars are mean +/- SEM. ns- not significant by upaired t-test. C, Representative immunofluorescence detecting p100/p52 on sections of tumors from WT and IkkαAA/AA mice. Red, p100/p52; blue, DAPI. D, Immunoblots detecting IKKα expression and E, NIK in the indicated WT and IkkαAA/AA mice. MDA-MB-231* cells treated with SM164 were used as a positive control for NIK protein stabilization and detection. F, Immunoblots detecting ErbB2. Vinculin and actin respectively were used as loading controls. Actin served as a lysate loading control.
2.5NIK is critical for normal mammary development
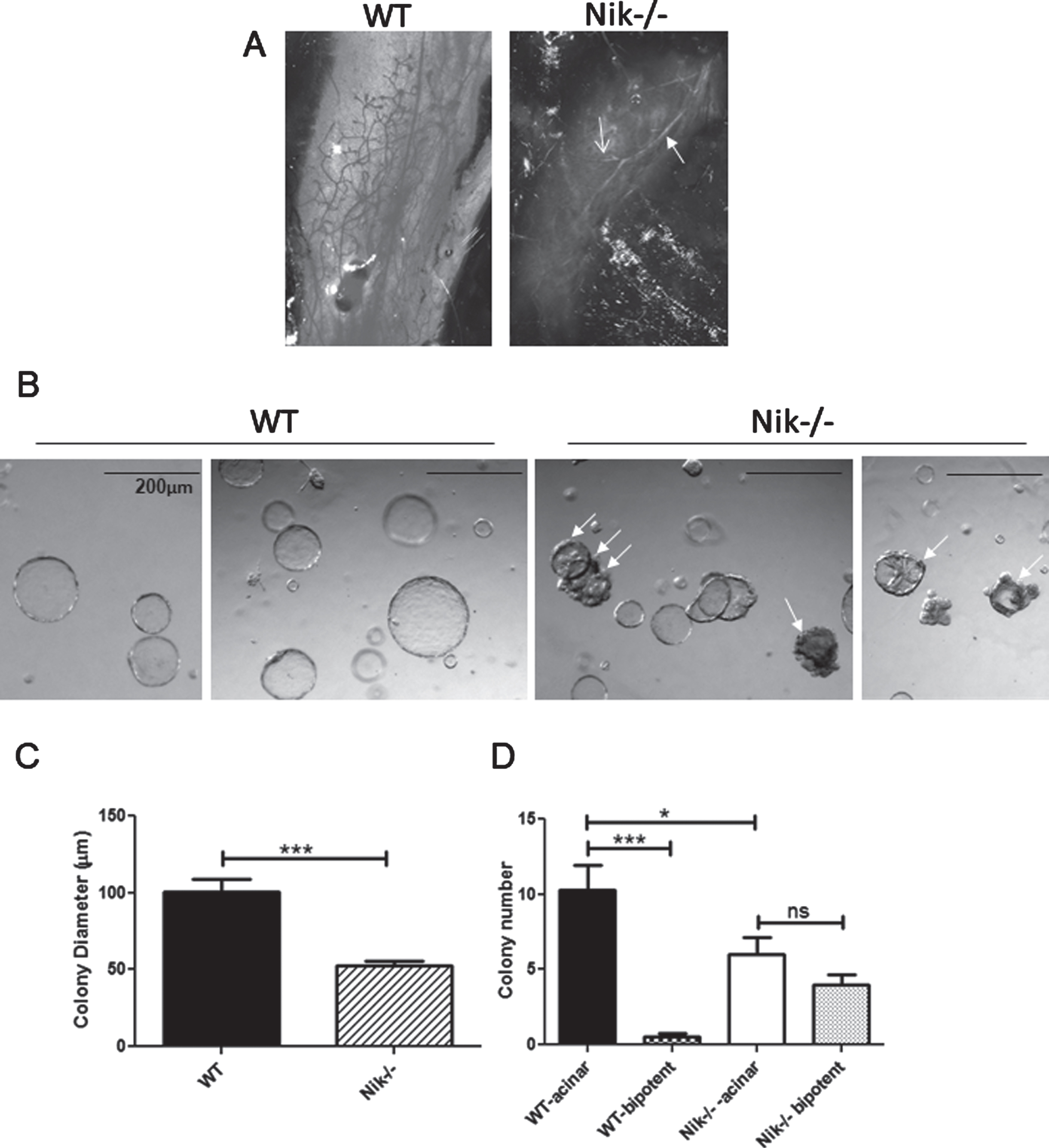
The presence of NIK protein in many of the tumors that contained both IKKα and processed p100 suggests that at least in tumors NIK can activate the alternative NF-κB pathway, even in the presence of a non-inducible IKKα. As noted above, IkkαAA/AA mice do not display a deficit in normal mammary development or alterations in stem/progenitor cell profiles, but cannot respond to pregnancy hormone induction of RANKL signaling and fail to form sufficient secretory alveoli [21, 33]. In contrast, assessment of whole mammary glands from WT and Nik-/- mice showed a marked lack of mammary development beyond the primary duct at post-natal 6 weeks in the absence of NIK (Fig. 6A). Examples of colony formation by WT and Nik-/- MECs are shown in Fig. 6B. Measurement of Nik-/- colonies revealed a significantly smaller diameter relative to WT colonies although not as large a reduction as IkkαAA/AA MEC colonies (27% vs 48% smaller than WT colonies) (Fig. 6C). The majority of colonies formed from single-cell suspensions of MECs are derived from luminal-specific progenitor cells which form tight smooth-edged acinar colonies. Colonies derived from basal cells are filled with a smooth surface while a subset derived from bipotent progenitors form colonies with a central core of luminal cells that are surrounded by an uneven periphery of basal cells [40]. The most striking difference between WT and Nik-/- colony formation was the presence of almost 50% of bipotent like colonies compared to ∼2% in WT cultures quantified in Fig. 6D. Thus the mouse mammary phenotype associated with loss of NIK function involves a block to differentiation, ostensibly at the level of bipotent progenitor cells, whereas the activation-resistant mutant of IKKα has no effect on pubertal mammary epithelial cell differentiation or branching morphogenesis.
Fig. 6
Nik-/- mice have high levels of undifferented bipotent progenitors and poorly differentiated mammary glands: A, Linneg mammary cells from WT and Nik-/- mice were used for Matrigel colony assays. Acinar colonies are luminal and bipotent colonies are partially filled and have uneven cellular borders (arrows). B, Graph showing all colony diameters (mean +/- SEM) from WT and Nik-/- colony assays derived from 3 separate experiments. C, Graph depicting luminal and bipotent colonies from mice as indicated. ***p < 0.0001; *p < 0.05; ns-not significant (One-way ANOVA). D, Images of whole mammary glands from 6 week old virgin WT and Nik-/- mice. Solid arrow indicates the primary duct, open arrow indicates a side-branch.
3Discussion
In this study we have examined the role of IKKα in the generation of basaloid TAg-induced tumors and the activation of the alternative NF-κB pathway. Our results indicate that the main phenotype associated with the IkkαAA/AA mutant is an increased latency to tumor formation, however tumor frequency, end-point size and pathology were not affected. Importantly, although p100/p52 processing was on average lower than in WT C3(1)-TAg tumors, p52 was still detected in nearly all IkkαAA/AA tumors examined.
The signal-induced activation of the alternative NF-κB pathway provides tight control over the function of p52/RelB activity. Receptor-mediated activation occurs through ligand-activated CD40, BAFF, RANK and TLRs [41]. These pathways all involve the stabilization of NIK through receptor-mediated TRAF3-cIAP recruitment thus preventing NIK ubiquitination and degradation (reviewed in ref [41]. While S866 and S870 are phosphorylated in a NIK and IKKα-dependent manner [30], and are essential for p100 ubiquitination and proteasomal processing [30, 42], the kinase responsible for this phosphorylation is not known. In the normal mammary gland RANK signaling plays a key role in lobuloalveogenesis during pregnancy in response to estrogen/progesterone stimulation. However, given that C(3)1-TAg IkkαAA/AA tumors grew more slowly, the RANK-IKKα-dependent signaling cascade may participate in progression of tumors from early neoplastic lesions to carcinomas. NIK is in a constitutively active conformation [43, 44] and a NIK mutant lacking the TRAF3-binding motif is stabilized and strongly stimulates the non-canonical NF-κB pathway [45, 46]. As tumors progress, signaling through RANK and possibly other TNF family receptors would continue to promote accumulation of NIK. Moreover epigenetic alteration of the gene encoding NIK has been reported to be involved in enhanced NIK expression in basal-like breast cancer [47]. The presence of p52 in IkkαAA/AA tumors suggest that NIK and possibly another kinase may form a complex with this mutant form of IKKα to promote p100 phosphorylation. NIK may also induce additional signaling factor(s) that cooperate with IKKα in the induction of effective p100 processing [41]. To this point, unlike IkkαAA/AA mice, the IKKα KO mouse has a severe morphogenic/epidermal dysregulation and perinatal lethality phenotype [48] accompanied by impaired B-cell maturation and lymphoid organogenesis [31]. Notably, IKKα is less effective in inducing p100 processing relative to NIK [42] and NIK is required to promote binding of IKKα to p100.
Aside from classical receptor signaling to the alternative pathway, other pathways may also contribute to p52 generation in TAg-induced tumors. Basal tumors derive from a population of ER–luminal progenitor cells [7, 8] that are maintained by SOX9 and NOTCH1-mediated signaling [49–51]. In the C3(1)-TAg tumor model, the inactivation of RB and p53 leads to upregulation of SOX9, which then drives luminal-to-basal reprogramming of transformed cells in vivo [52]. Moreover, Notch signaling promotes tumor cell self-renewal and aggressive tumor behavior in TNBC where it is hyperactivated (reviewed in refs [53, 54]). Sox9 increases expression of NF-κB proteins from both the canonical and alternative pathway in basal breast cancers [52], and, although the mechanism is not clear, Notch 1 inhibition reduces p52 in lymphoma cells [55]. Interestingly, NIK knockdown but not IKKα knockdown in T lymphoma cells results in a reduction in p52 levels [56], further suggesting that IKKα activity may be dispensable for phosphorylation of p100 under circumstances when NIK protein is stabilized.
Overall these studies suggest that, in vivo, NIK has an obligate role in activation of the alternative pathway during pubertal mammary development, such that absence results in profound changes in mammary cell differentiation and gland formation. This function of NIK may explain why p100/p52 processing was not eliminated in IkkαAA/AA tumors in which NIK protein was detected. Based on the fact that NIK has been shown to directly induce p100 processing and, other than IKKα, is the only enzyme robustly demonstrated to perform this function, we propose that, in vivo, NIK kinase activity, may be sufficient to induce p100 processing in basaloid mammary cancer cells.
4Methods
4.1Mice and genotyping
IkkαAA/AA FVB/n mice were the generous gift of Dr. Michael Karin. C3(1)-TAg mice were purchased from Jackson labs (FVB-Tg(C3-1-TAg) cJeg/JegJ #013591). IkkαAA/AA and C3(1)TAg mice were crossed to generate IkkαAA/AA;C3(1)TAg progeny. Nik-/+ mice were purchased from Jackson Labs (B6N.129-Map3k14tm1Rds/J Strain #025557) and were bred to homozygosity. Animal husbandry was conducted in accordance with Policy 31 of the University of Ottawa and the Canadian Council on Animal Care guidelines. Mice were genotyped using PCR. Primers used for genotyping IkkαAA/AA mice were: p392 (mutant)(5′-ggatccgatatctgcatgaaaac), pWT (5′-caatgttcccacaaaagatgtacagagact), pAA (5′-caatgttcccacaaacgctgtacagagcgc), and p776 (mutant)(5′-ggtaaatggctactaaaccgaacttctcc). Primers to detect the TAg transgene were:TAg1:5’-gacctgtggctgagtttgcta and TAg2:5’-gctttatttgtaaccattataag. Primers for genotyping Nik-/- mice were 5’-agtccaattccatgttgctgctgt and 5’-tctgagataggcatatccctggct.
4.2Immunoblots and antibodies
Mouse tumors were isolated at necropsy and portions subjected to flash freezing in liquid N2 or fixed in formalin for paraffin-embedding. Frozen tumors were pulverized under liquid N2, sonicated in RIPA buffer and centrifuged to remove insoluble material. Supernatants were aliquoted and stored at –80°C. Immunoblotting was performed as previously described [27]. Quantification of immune-reactive bands on immunoblots was performed using ImageJ software (www.rsb.info.nih.gov/ij/). The area under curve of the specific signal was corrected for the AUC of actin. Immunoblot antibodies: anti ErbB2 (abcam #ab2428-1); Actin (Sigma #A-2066); vinculin (abcam #ab129002); IKKα (Cell Signaling #2682).
4.3Mouse stem and progenitor cell isolation and assays
Isolation of mammary epithelial subpopulations was performed as described previously [57]. Purified mammary epithelial cells were stained with biotin-conjugated anti-CD31 (catalogue no. 553371; BD) and biotinylated CD45+ and Ter119+ (StemSep murine chimaera cocktail, catalogue no. 13058C; Stem Cell Technologies) to label endothelial and hematopoietic cells (Lineage neg (linneg). Linneg cells were excluded by FACS with streptavidin-conjugated allophycocyanin (catalogue no. 554067; BD). Staining with anti-CD49f (catalogue no. 551129; BD) and CD24 (catalogue no. 553261; BD) was used to identify the mammary-stem/progenitor cell-enriched populations [34, 58]. Cell sorting was performed on a Beckman MoFlo Astrios EQ FACS sorter. To detect TAg expression FACS-sorted cells were cytospun onto slides, fixed and stained with anti-TAg (SV40 Large T Antigen (Cell Signaling (D1E9E) #15729).
Colony forming assays were performed as previously described [27]. Briefly, single-cell suspensions of EasySep-derived (STEMCELL Technologies) linneg mammary epithelial cells were seeded in 20μl Matrigel (BD Biosciences) in Epicult-B medium (STEMCELL Technologies) supplemented with B27 (Gibco). Colony formation was scored after 12 days. The diameters of at least 30 of the largest acini were measured using Northern Eclipse software to calculate the mean and SEM.
4.4Immunohistochemistry and analysis
For IHC immuno-peroxidase staining, paraffin-embedded sections were dehydrated and antigenic epitopes were exposed by using a 10 mM citrate buffer and microwaving. Sections were incubated with antibodies against cytokeratin 6, cytokeratin 8, and p100/p52 followed by detection using peroxidase-conjugated secondary antibodies and DAB substrate (Dako). Antibodies were cytokeratin 6 (Abcam #ab24646), cytokeratin 8 (Thermo Scientific #pa5-92607), NF-κB p52 (Millipore #06-413).
The slides were imaged using an EVOS inverted microscope (ThermoFisher FL Auto 2) with 20X Brightfield imaging. Tile stitching and colour correction were performed using the corresponding software (EVOS FL Auto 2 Software Revision 2.0.2094.0). FIJI was used to perform percent analysis of positive staining for each IHC marker on total tissue sections. Colour deconvolution was selected under “image” menu. “Hematoxylin + DAB” was selected for the corresponding colour separation. Using the “Threshold” tool, the total areas stained separately with hematoxylin and DAB were calculated. Percent of positive tumor epithelium was then acquired by dividing the DAB area by Hematoxylin. A standard unpaired T-test was performed for each marker using Graphpad Prism software 9.
Acknowledgments
This work was supported by a grant to MACP from the Canadian Breast Cancer Foundation.
Author contributions
CONCEPTION: MACP
DATA CURATION: FO, AS and DC
ANALYSIS OF DATA: FO, AS, DC, TJ and MACP
PREPARATION OF THE MANUSCRIPT: MACP
SUPERVISION: MACP
Conflict of interest
FO has no conflict of interest to report.
AS has no conflict of interest to report.
DC has no conflict of interest to report.
TJ has no conflict of interest to report.
MACP has no conflict of interest to report
References
[1] | Lloyd-Lewis B , Harris OB , Watson CJ , Davis FM , Mammary Stem Cells: Premise, Properties, and Perspectives, Trends Cell Biol (2017) ;27: (8):556–67. doi: 10.1016/j.tcb.2017.04.001. |
[2] | Visvader JE , Keeping abreast of the mammary epithelial hierarchy and breast tumorigenesis, Genes Dev (2009) ;23: (22):2563–77. doi: 10.1101/gad.1849509. |
[3] | Fu NY , Nolan E , Lindeman GJ , Visvader JE , Stem Cells and the Differentiation Hierarchy in Mammary Gland Development, Physiol Rev (2020) ;100: (2):489–523. doi: 10.1152/physrev.00040.2018. |
[4] | Asselin-Labat ML , Vaillant F , Shackleton M , Bouras T , Lindeman GJ , Visvader JE , Delineating the epithelial hierarchy in the mouse mammary gland, Cold Spring Harb Symp Quant Biol (2008) ;73: :469–78. doi: 10.1101/sqb.2008.73.020. |
[5] | Sreekumar A , Roarty K , Rosen JM , The mammary stem cell hierarchy: a looking glass into heterogeneous breast cancer landscapes, Endocr Relat Cancer (2015) ;22: (6):T161–176. doi: 10.1530/ERC-15-0263. |
[6] | Lim E , Wu D , Pal B , Bouras T , Asselin-Labat ML , Vaillant F , et al., Transcriptome analyses of mouse and human mammary cell subpopulations reveal multiple conserved genes and pathways, Breast Cancer Res. (2010) ;12: (2):R21. doi: 10.1186/bcr2560. |
[7] | Lim E , Vaillant F , Wu D , Forrest NC , Pal B , Hart AH , et al., Aberrant luminal progenitors as the candidate target population for basal tumor development in BRCA1 mutation carriers, Nat Med (2009) ;15: (8):907–13. doi: 10.1038/nm.2000. |
[8] | Molyneux G , Geyer FC , Magnay FA , McCarthy A , Kendrick H , Natrajan R , et al., BRCA1 Basal-like Breast Cancers Originate from Luminal Epithelial Progenitors and Not from Basal Stem Cells, Cell Stem Cell (2010) ;7: (3):403–17. doi: 10.1016/j.stem.2010.07.010. |
[9] | Proia TA , Keller PJ , Gupta PB , Klebba I , Jones AD , Sedic M , et al., Genetic Predisposition Directs Breast Cancer Phenotype by Dictating Progenitor Cell Fate, Cell Stem Cell (2011) ;8: (2):149–63. doi: 10.1016/j.stem.2010.12.007. |
[10] | Mietz JA , Unger T , Huibregtse JM , Howley PM , The transcriptional transactivation function of wild-type p53 is inhibited by SV40 large T-antigen and by HPV-16 E6 oncoprotein, EMBO J (1992) ;11: (13):5013–20. |
[11] | Dyson N , Buchkovich K , Whyte P , Harlow E , The cellular 107K protein that binds to adenovirus E1A also associates with the large T antigens of SV40 and JC virus, Cell (1989) ;58: (2):249–55. doi: 10.1016/0092-8674(89)90839-8. |
[12] | Green JE , Shibata MA , Yoshidome K , Liu ML , Jorcyk C , Anver MR , et al., The C-7/SV40 T-antigen transgenic mouse model of mammary cancer: ductal epithelial cell targeting with multistage progression to carcinoma. Oncogene (2000) ;3: (1):1020–7. doi: 10.1038/sj.onc.1203280. |
[13] | Wellings SR . A hypothesis of the origin of human breast cancer from the terminal ductal lobular unit, Pathol Res Pract (1980) ;166: (4):515–35. doi: 10.1016/S0344-0338(80)80248-2. |
[14] | Tharmapalan P , Mahendralingam M , Berman HK , Khokha R , Mammary stem cells and progenitors: targeting the roots of breast cancer for prevention, EMBO J. (2019) ;38: (14):e100852. doi: 10.15252/embj.2018100852. |
[15] | Maroulakou IG , Anver M , Garrett L , Green JE . Prostate and mammary adenocarcinoma in transgenic mice carrying a rat simian virus 40 large tumor antigen fusion gene C3(1) Proc Natl Acad Sci USA. (1994) ;91: (23). :6–40. doi: 10.1073/pnas.91.23.11236. |
[16] | Dontu G , Ince TA . Of Mice and Women: A Comparative Tissue Biology Perspective of Breast Stem Cells and Differentiation, J Mammary Gland Biol Neoplasia (2015) ;20: (1-2):51–62. doi: 10.1007/s10911-015-9341-4. |
[17] | Gusterson B , Eaves CJ , Basal-like Breast Cancers: From Pathology to Biology and Back Again, Stem Cell Rep (2018) ;10: (6):1676–86. doi: 10.1016/j.stemcr.2018.04.023. |
[18] | Herschkowitz JI , Simin K , Weigman VJ , Mikaelian I , Usary J , Hu Z , et al., Identification of conserved gene expression features between murine mammary carcinoma models and human breast tumors, Genome Biol (2007) ;8: (5):R76. doi: 10.1186/gb-2007-8-5-r76. |
[19] | Pfefferle AD , Herschkowitz JI , Usary J , Harrell J , Spike BT , Adams JR , et al., Transcriptomic classification of genetically engineered mouse models of breast cancer identifies human subtype counterparts, Genome Biol (2013) ;14: (11):. doi: 10.1186/gb-2013-14-11-r125. |
[20] | Usary J , Darr DB , Pfefferle AD , Perou CM , Overview of Genetically Engineered Mouse Models of Distinct Breast Cancer Subtypes, Current Protocols in Pharmacology (2016) ;72: (1). doi: 10.1002/0471141755.ph1438s72. |
[21] | Cao Y , Bonizzi G , Seagroves TN , Greten FR , Johnson R , Schmidt EV , et al., IKKalpha provides an essential link between RANK signaling and cyclin D1 expression during mammary gland development, Cell (2001) ;107: (6):763–75. doi: 10.1016/s0092-8674(01)00599-2. |
[22] | Brantley DM , Chen CL , Muraoka RS , Bushdid PB , Bradberry JL , Kittrell F , et al., Nuclear factor-kappaB (NF-kappaB) regulates proliferation and branching in mouse mammary epithelium, Mol Biol Cell (2001) ;12: (5):1445–55. doi: 10.1091/mbc.12.5.1445. |
[23] | Baxter FO , Came PJ , Abell K , Kedjouar B , Huth M , Rajewsky K , et al., IKKbeta/2 induces TWEAK and apoptosis in mammary epithelial cells, Development (2006) ;133: (17):3485–94. doi: 10.1242/dev.02502. |
[24] | Cao Y , Luo JL , Karin M , IkappaB kinase alpha kinase activity is required for self-renewal of ErbB2/Her2-transformed mammary tumor-initiating cells, Proc Natl Acad Sci U S A (2007) ;104: (40):15852–7. doi: 10.1073/pnas.0706728104. |
[25] | Pratt M aC , Tibbo E , Robertson SJ , Jansson D , Hurst K , Perez-Iratxeta C , et al., The canonical NF-kappaB pathway is required for formation of luminal mammary neoplasias and is activated in the mammary progenitor population, Oncogene (2009) ;28: (30):2710–22. doi: 10.1038/onc.2009.131. |
[26] | Song K , Farzaneh M , Signaling pathways governing breast cancer stem cells behavior, Stem Cell Res Ther (2021) ;12: (1):245. doi: 10.1186/s13287-021-02321-w. |
[27] | Sau A , Lau R , Cabrita MA , Nolan E , Crooks PA , Visvader JE , et al., Persistent Activation of NF-κB in BRCA1-Deficient Mammary Progenitors Drives Aberrant Proliferation and Accumulation of DNA Damage, Cell Stem Cell (2016) ;19: (1):52–65. doi: 10.1016/j.stem.2016.05.003. |
[28] | Xiao G , Fong A , Sun SC , Induction of p100 processing by NF-kappaB-inducing kinase involves docking IkappaB kinase alpha (IKKalpha) to p100 and IKKalpha-mediated phosphorylation, J Biol Chem (2004) ;279: (29):30099–105. doi: 10.1074/jbc.M401428200. |
[29] | Shih VFS , Tsui R , Caldwell A , Hoffmann A , A single NFκB system for both canonical and non-canonical signaling, Cell Res (2011) ;21: (1):86–102. doi: 10.1038/cr.2010.161. |
[30] | Liang C , Zhang M , Sun SC , beta-TrCP binding and processing of NF-kappaB2/p100 involve its phosphorylation at serines 866 and 870, Cell Signal (2006) ;18: (8):1309–17. doi: 10.1016/j.cellsig.2005.10.011. |
[31] | Senftleben U , Cao Y , Xiao G , Greten FR , Krähn G , Bonizzi G , et al., Activation by IKKα of a Second, Evolutionary Conserved, NF-κB Signaling Pathway, Science (2001) ;293: (5534):1495–9. doi: 10.1126/science.1062677. |
[32] | Joshi PA , Jackson HW , Beristain AG , Di Grappa MA , Mote PA , Clarke CL , et al., Progesterone induces adult mammary stem cell expansion, Nature (2010) ;465: (7299):803–7. doi: 10.1038/nature09091. |
[33] | Zhang W , Tan W , Wu X , Poustovoitov M , Strasner A , Li W , et al., A NIK-IKKα module expands ErbB2-induced tumor-initiating cells by stimulating nuclear export of p27/Kip1, Cancer Cell (2013) ;23: (5):647–59. doi: 10.1016/j.ccr.2013.03.012. |
[34] | Stingl J , Eirew P , Ricketson I , Shackleton M , Vaillant F , Choi D , et al., Purification and unique properties of mammary epithelial stem cells, Nature (2006) ;439: (7079):993–7. doi: 10.1038/nature04496. |
[35] | Petersen OW , Ronnov-Jessen L , Howlett AR , Bissell MJ , Interaction with basement membrane serves to rapidly distinguish growth and differentiation pattern of normal and malignant human breast epithelial cells, Proc Natl Acad Sci USA (1992) ;89: (19):9064–8. doi: 10.1073/pnas.89.19.9064. |
[36] | Williams CM , Engler AJ , Slone RD , Galante LL , Schwarzbauer JE , Fibronectin expression modulates mammary epithelial cell proliferation during acinar differentiation, Cancer Res (2008) ;68: (9):3185–92. doi: 10.1158/0008-5472.CAN-07-2673. |
[37] | Hoenerhoff MJ , Shibata MA , Bode A , Green JE , Pathologic progression of mammary carcinomas in a C247-/SV40 T/t-antigen transgenic rat model of human triple-negative and Her2-positive breast cancer5920(2). Transgenic Res (2011) ;3: (1):247–50. doi: 10.1158/0008-5472.CAN-07-2673. |
[38] | Yoshidome K , Shibata MA , Couldrey C , Korach KS , Green JE , Estrogen promotes mammary tumor development in C3(1)/SV40 large T-antigen transgenic mice: paradoxical loss of estrogen receptoralpha expression during tumor progression. Cancer Res. (2000) ;60: (24):6901–10. doi:. |
[39] | Merkhofer EC , Cogswell P , Baldwin AS , Her2 activates NF-kappaB and induces invasion through the canonical pathway involving IKKalpha, Oncogene (2010) ;29: (8):1238–48. doi: 10.1038/onc.2009.410. |
[40] | Stingl J , Eaves CJ , Zandieh I , Emerman JT , Characterization of bipotent mammary epithelial progenitor cells in normal adult human breast tissue, Breast Cancer Res Treat (2001) ;67: (2):93–109. doi: 10.1023/a:1010615124301. |
[41] | Sun SC , Non-canonical NF-κB signaling pathway, Cell Res (2011) ;21: (1):71–85. doi: 10.1038/nri.2017.52. |
[42] | Xiao G , Harhaj EW , Sun SC , NF-kappaB-inducing kinase regulates the processing of NF-kappaB2 p100, Mol Cell (2001) ;7: (2):401–9. doi: 10.1016/s1097-2765(01)00187-3. |
[43] | de Leon-Boenig G , Bowman KK , Feng JA , Crawford T , Everett C , Franke Y , et al., The crystal structure of the catalytic domain of the NF-κB inducing kinase reveals a narrow but flexible active site, Structure (2012) ;20: (10):1704–14. doi: 10.1016/j.str.2012.07.013. |
[44] | Liu J , Sudom A , Min X , Cao Z , Gao X , Ayres M , et al., Structure of the nuclear factor κB-inducing kinase (NIK) kinase domain reveals a constitutively active conformation, J Biol Chem (2012) ;287: (33):27326–34. doi: 10.1074/jbc.M112.366658. |
[45] | Liao G , Zhang M , Harhaj EW , Sun SC , Regulation of the NF-kappaB-inducing kinase by tumor necrosis factor receptor-associated factor 3-induced degradation, J Biol Chem (2004) ;279: (25):26243–50. doi: 10.1074/jbc.M403286200. |
[46] | Sasaki Y , Calado DP , Derudder E , Zhang B , Shimizu Y , Mackay F , et al., NIK overexpression amplifies, whereas ablation of its TRAF3-binding domain replaces BAFF:BAFF-R-mediated survival signals in B cells, Proc Natl Acad Sci U S A (2008) ;105: (31):10883–8. doi: 10.1073/pnas.0805186105. |
[47] | Yamamoto M , Ito T , Shimizu T , Ishida T , Semba K , Watanabe S , et al., Epigenetic alteration of the NF-κB-inducing kinase (NIK) gene is involved in enhanced NIK expression in basal-like breast cancer, Cancer Sci (2010) ;101: (11):2391–7. doi: 10.1111/j.1349-7006.2010.01685.x. |
[48] | Takeda K , Takeuchi O , Tsujimura T , Itami S , Adachi O , Kawai T , et al., Limb and skin abnormalities in mice lacking IKKalpha, Science (1999) ;284: (5412):313–6. doi: 10.1126/science.284.5412.313. |
[49] | Van Keymeulen A , Fioramonti M , Centonze A , Bouvencourt G , Achouri Y , Blanpain C , Lineage-Restricted Mammary Stem Cells Sustain the Development, Homeostasis, and Regeneration of the Estrogen Receptor Positive Lineage, Cell Rep (2017) ;20: (7):1525–32. doi: 10.1016/j.celre2017.07.066. |
[50] | Rodilla V , Dasti A , Huyghe M , Lafkas D , Laurent C , Reyal F , et al., Luminal Progenitors Restrict Their Lineage Potential during Mammary Gland Development, Eaves CJ, editor. PLoS Biol (2015) ;13: (2):e1002069. doi: 10.1371/journal.pbio.1002069. |
[51] | Wang C , Christin JR , Oktay MH , Guo W , Lineage-Biased Stem Cells Maintain Estrogen-Receptor-Positive and -Negative Mouse Mammary Luminal Lineages, Cell Rep (2017) ;18: (12):2825–35. doi: 10.1016/j.celre2017.02.071. |
[52] | Christin JR , Wang C , Chung CY , Liu Y , Dravis C , Tang W , et al., Stem Cell Determinant SOX9 Promotes Lineage Plasticity and Progression in Basal-like Breast Cancer, Cell Rep (2020) ;31: (10):107742. doi: 10.1016/j.celre2020.107742. |
[53] | Chen W , Wei W , Yu L , Ye Z , Huang F , Zhang L , et al., Mammary Development and Breast Cancer: a Notch Perspective, J Mammary Gland Biol Neoplasia (2021) ;26: (3):309–20. doi: 10.1007/s10911-021-09496-1. |
[54] | Stoeck A , Lejnine S , Truong A , Pan L , Wang H , Zang C , et al., Discovery of biomarkers predictive of GSI response in triple-negative breast cancer and adenoid cystic carcinoma, Cancer Discov (2014) ;4: (10):1154–67. doi: 10.1158/2159-8290.CD-13-0830. |
[55] | Schwarzer R , Dörken B , Jundt F , Notch is an essential upstream regulator of NF-κB and is relevant for survival of Hodgkin and Reed-Sternberg cells, Leukemia (2012) ;26: (4):806–13. doi: 10.1038/leu.2011.265. |
[56] | Odqvist L , Sánchez-Beato M , Montes-Moreno S , Martín-Sánchez E , Pajares R , Sánchez-Verde L , et al., NIKcontrols classical and alternative NF-κB activation and isnecessary for the survival of human T-cell lymphoma cells, ClinCancer Res (2013) ;19: (9):2319–30. doi: 10.1158/1078-0432.CCR-12-3151. |
[57] | Schramek D , Leibbrandt A , Sigl V , Kenner L , Pospisilik JA , Lee HJ , et al., Osteoclast differentiation factor RANKL controls development of progestin-driven mammary cancer, Nature (2010) ;468: (7320):98–102. doi: 10.1038/nature09387. |
[58] | Shackleton M , Vaillant F , Simpson KJ , Stingl J , Smyth GK , Asselin-Labat ML , et al., Generation of a functional mammary gland from a single stem cell, Nature (2006) ;439: (7072):84–8. doi: 10.1038/nature04372. |


