
Role of colony-stimulating factor 1 in the neoplastic process of tenosynovial giant cell tumor
Abstract
Tenosynovial giant cell tumors (TGCTs) are rare, locally aggressive, mesenchymal neoplasms, most often arising from the synovium of joints, bursae, or tendon sheaths. Surgical resection is the first-line treatment, but recurrence is common, with resulting impairments in patients’ mobility and quality of life. Developing and optimizing the role of systemic pharmacologic therapies in TGCT management requires an understanding of the underlying disease mechanisms. The colony-stimulating factor 1 receptor (CSF1R) has emerged as having an important role in the neoplastic processes underlying TGCT. Lesions appear to contain CSF1-expressing neoplastic cells derived from the synovial lining surrounded by non-neoplastic macrophages that express the CSF1R, with lesion growth stimulated by both autocrine effects causing proliferation of the neoplastic cells themselves and by paracrine effects resulting in recruitment of CSF1 R-bearing macrophages. Other signaling pathways with evidence for involvement in TGCT pathogenesis include programmed death ligand-1, matrix metalloproteinases, and the Casitas B-cell lymphoma family of ubiquitin ligases. While growing understanding of the pathways leading to TGCT has resulted in the development of both regulatory approved and investigational therapies, more detail on underlying disease mechanisms still needs to be elucidated in order to improve the choice of individualized therapies and to enhance treatment outcomes.
1Classification and burden of tenosynovial giant cell tumors
Tenosynovial giant cell tumors (TGCTs) are rare, locally aggressive, mesenchymal neoplasms, classed as fibrohistiocytic tumors, and most often arise from the synovium of joints, bursae, or tendon sheaths [1, 2]. TGCT can be further classified into localized or diffuse disease. Localized TGCT is the most common type, occurring predominantly in the hand and presenting as a slow-growing, painless swelling close to the synovium of the tendon sheath or interphalangeal joint; it mainly occurs between the ages of 30 and 50 years, with a 1:0.5 predominance in women. The recurrence rate for localized TGCT is between 4% and 30% [1]. In contrast, diffuse TGCT is a destructive proliferation of cells (e.g., mononuclear) that may infiltrate surrounding soft tissues, affecting predominantly the knee, and presenting with pain, tenderness, swelling, and limitation of motion, commonly with hemorrhagic joint effusions; it occurs mainly in young adults (aged < 40 years) [1, 3]. Recurrences are common, occurring in up to two-thirds of cases following open synovectomy and up to 90% or more following arthroscopic synovectomy [4]. Repeated resection of diffuse TGCT can severely compromise joint function, with patients reporting joint instability and a reduced range of motion [1, 5]. While TGCT is a rare condition, with estimated incidence rates of up to 30 per million person years for localized disease and 8 per million person years for diffuse disease [6, 7], it imposes substantial personal burdens by impairing patients’ functioning and quality of life, as well as health care burdens from the costs of surgery, rehabilitation, and physical therapy [3, 7–10].
Surgery is the standard treatment for TGCT [4, 11, 12]. A systemic therapy, pexidartinib, is currently approved in the United States. Pexidartinib is a small-molecule tyrosine kinase inhibitor that targets colony-stimulating factor 1 receptor (CSF1R), KIT proto-oncogene receptor tyrosine kinase, and FMS-like tyrosine kinase 3 (FLT3) harboring an internal tandem duplication mutation. Pexidartinib is indicated in the United States and the Republic of Korea for the treatment of adult patients with symptomatic TGCT associated with severe morbidity or functional limitations and not amenable to improvement with surgery [13, 14]. In the United States, pexidartinib is only available through the Risk Evaluation and Mitigation Strategy (REMS) program to mitigate the risk of serious and potentially fatal hepatotoxicity. Optimizing the role of pharmacologic therapies in the management of TGCT to advance patient care requires an understanding of the underlying mechanisms of disease. The objective of this review is to examine the mechanism of action of the CSF1R and the pathways involved in the development of TGCT, clarifying the etiology of TGCT as a neoplastic process, distinguishing this from the resultant inflammatory aspect of the disease.
2TGCT mechanism of disease
Localized and diffuse forms of TGCT are both characterized by a proliferation of mononuclear cells, multinucleated giant cells, foamy macrophages, xanthoma cells, and inflammatory cells [1]. Synovial lining cells and fibroblasts are also active participants in the disease process, creating a mosaic of cell types. In the first published descriptions of TGCT, it was described as an inflammatory response of unknown etiology [15], but subsequent findings more strongly support a neoplastic origin [1].
TGCTs have been shown to express high levels of colony-stimulating factor 1 (CSF1) [16], also known as macrophage-CSF, a regulator of macrophage survival, proliferation, and differentiation that is known to have important roles in innate immunity, inflammatory conditions, and cancers [17]. One of the causes of this overexpression of CSF1 is a translocation of chromosomes 1 and 2 that links the CSF1 gene on chromosome 1p13 to the COL6A3 collagen promoter gene on chromosome 2q37 [18]. Only a small proportion of cells within each TGCT (2% –16%) show this translocation (see section 2.2 below) and subsequent overexpression of CSF1, with most cells not expressing CSF1 but instead expressing the CSF1R. Binding of CSF1 to the CSF1R stimulates activation of signaling pathways that modulate differentiation, proliferation, and chemotaxis toward the source of CSF1, leading to the recruitment of inflammatory cells to the joint cavity. Thus, TGCT lesions appear to contain CSF1-expressing neoplastic cells derived from synovial lining cells surrounded by non-neoplastic macrophages that express the CSF1R [18]. Growth of the lesion takes place by both autocrine effects causing proliferation of the neoplastic cells themselves and by paracrine effects resulting in recruitment of CSF1 R-bearing macrophages (Fig. 1) [18]. The CSF1/CSF1R axis provides a target for TGCT treatments (e.g., by targeting the extracellular or intracellular domains of the CSF1R; Fig. 2a and 2b).
Fig. 1
Role of the CSF1/CSF1R axis in the development of TGCT. Overexpression of CSF1R (e.g., through CSF1:COL6A3 gene translocation) promotes cell proliferation and accumulation in the synovium, while binding of the CSF1 ligand to the CSF1R leads to activation of signaling pathways that modulate differentiation, proliferation, and chemotaxis toward the source of CSF1, leading to the recruitment of inflammatory cells to the joint cavity.
CSF1, colony-stimulating factor 1; CSF1R, colony-stimulating factor 1 receptor; TGCT, tenosynovial giant cell tumor.
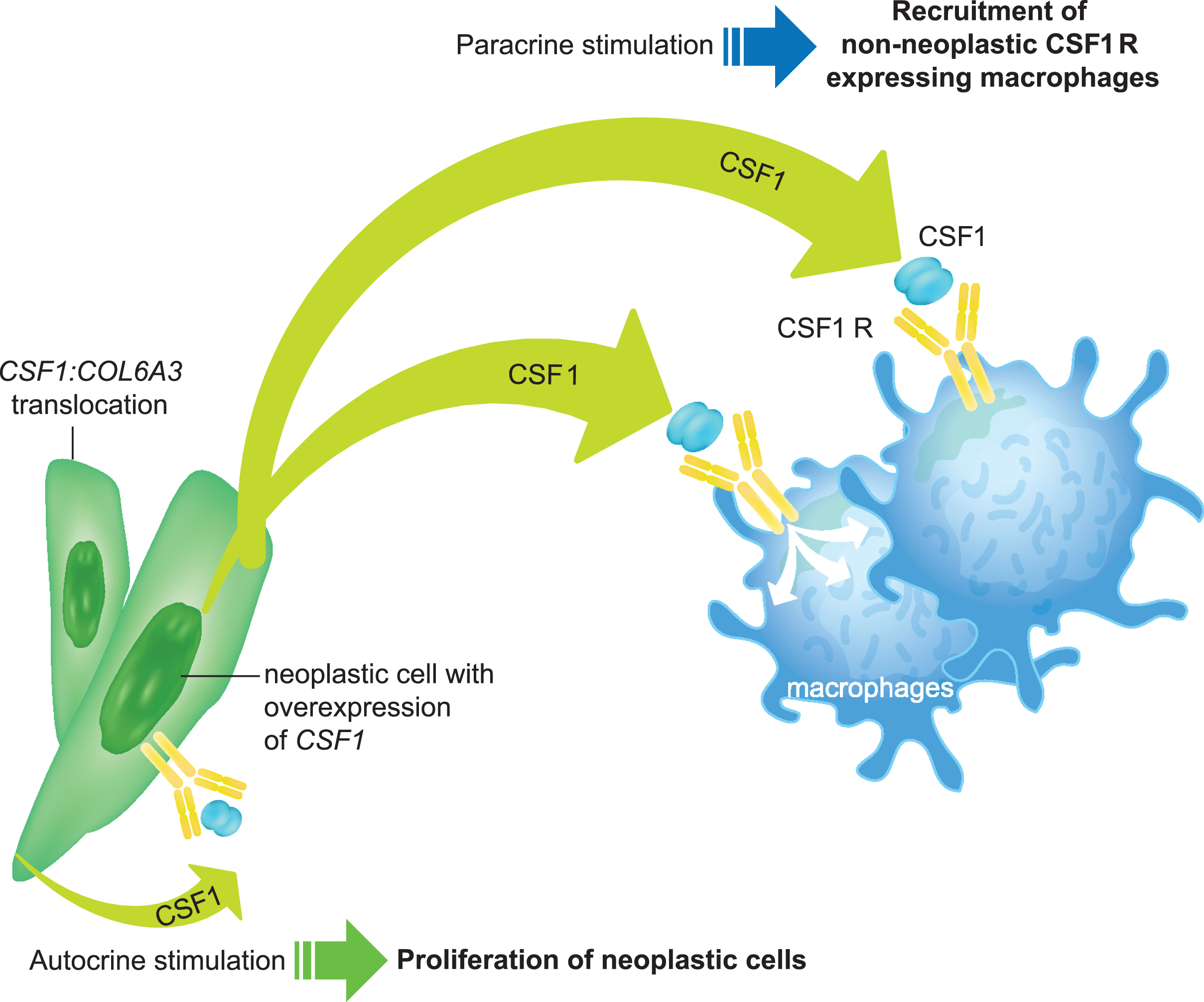
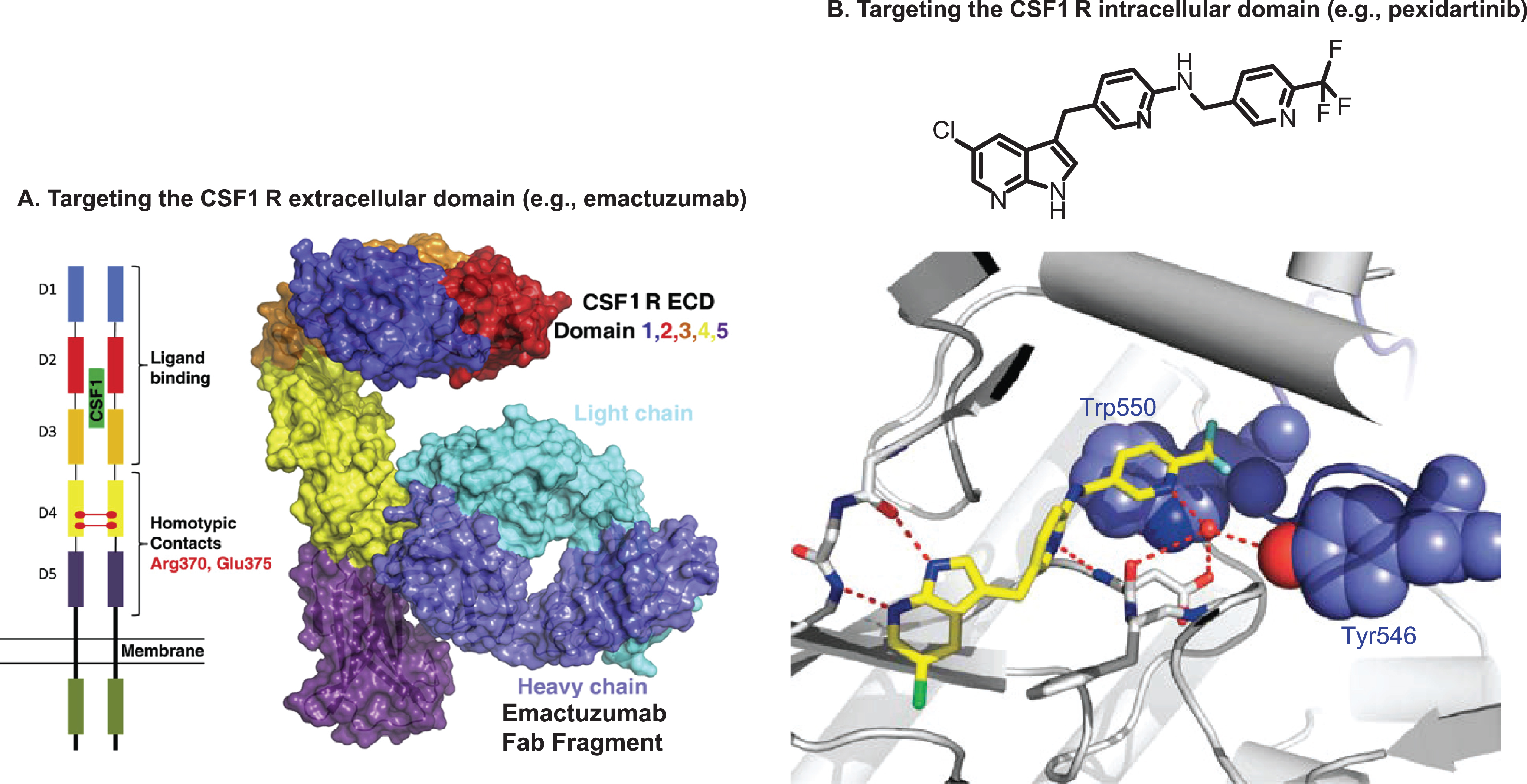
Fig. 2
Molecular targets for TGCT therapy. A. Schematic representation (left) of the CSF1R extracellular domain dimer. Key residues Arg370 and Glu375 most likely involved in dimerization in domain 4 are highlighted. The overall structure of the CSF1R extracellular domain monomer complex with fab fragment of emactuzumab is shown in surface representation (right). B. Chemical structure of pexidartinib (top) and conformation-specific inhibition of CSF1R (bottom). Pexidartinib binds the autoinhibited state of CSF1R and makes direct contact with the juxtamembrane region. The key hydrogen bond interactions are highlighted. The CSF1R kinase domain is shown in gray, the juxtamembrane region (including Tyr546 and Trp550) in purple, and PLX3397 in yellow (oxygen in red and chlorine in green).
CSF1R, colony-stimulating factor 1 receptor; ECD, extracellular domain; TGCT, tenosynovial giant cell tumor; Trp550, tryptophan residue 550; Tyr546, tyrosine residue 546.
Figure 2A is reprinted from Cancer Cell, vol 25, Carola H. Ries, Michael A. Cannarile, Sabine Hoves, et al., Targeting Tumor-Associated Macrophages with Anti-CSF-1 R Antibody Reveals a Strategy for Cancer Therapy, pages 846-859, Copyright 2014, with permission from Elsevier. Figure 2B is from The New England Journal of Medicine, William D. Tap, Zev A. Wainberg, Stephen P. Anthony, et al., Structure-Guided Blockade of CSF1R Kinase in Tenosynovial Giant-Cell Tumor, vol 373, pages 428-437. Copyright © 2015 Massachusetts Medical Society. Reprinted with permission from Massachusetts Medical Society.
2.1CSF1R signaling
The CSF1R belongs to the platelet-derived growth factor family and controls the development of cells such as monocytes and macrophages, playing a central role in neoplastic and inflammatory diseases. Like other members of this receptor family, the CSF1R comprises an extracellular domain, a transmembrane domain, and an intracellular domain made up of a juxtamembrane region and an intracellular kinase region [19]. The gene for CSF1R (CSF1R) is located on human chromosome 5 (5q32); its expression is usually low in hematopoietic stem cells but is substantially upregulated during differentiation of myeloid progenitor cells into macrophages [19].
Two different ligands, CSF1 and interleukin-34, act at the CSF1R [20]. Through differential binding to extracellular receptor domains, the two ligands show different patterns of biological activity and signal activation, resulting in complementary roles in regulating different cell populations [19, 21]. Downstream signaling pathways include the PI3K/Akt, phospholipase C (PLC), and FMS-interacting protein pathways mediating macrophage survival; the MEK, PI3K/Akt, JNK, and ERK1/2 pathways that contribute to macrophage proliferation; the PLC-γ2 pathway promoting macrophage differentiation; and the Wiskott-Aldrich syndrome protein (WASP) pathway triggering macrophage chemotaxis [19].
2.2CSF1 genetic abnormalities
Various genetic abnormalities have been found to play a role in the etiology of TGCT.
West et al. first demonstrated the involvement of rearrangements of the CSF1 gene in the pathogenesis of TGCT in a sample of nine tumors. In some cases, CSF1 was fused to the collagen promoter element COL6A3 (2q37), as previously discussed. Findings suggest a tumor-landscaping effect with aberrant CSF1 expression in the neoplastic cells, resulting in abnormal accumulation of non-neoplastic cells responding to CSF1, such as monocytes and macrophages, that form the bulk of the tumor [18]. Möller et al. subsequently confirmed that only TGCT cells with this COL6A3 translocation expressed CSF1 but found that the receptor-binding portion of the ligand was not retained in the fusion mRNA transcripts, suggesting that wild-type CSF1 expression is also required for receptor binding and activation [22]. Likewise, another study by Cupp et al. that included 60 cases of TGCT showed that all sampled cases expressed CSF1, with almost 40% lacking the CSF1 gene translocation but still having high CSF1 expression, suggesting again that alternative mechanisms other than gene translocation may be involved in the upregulation of CSF1 [23], or alternatively, that the sensitivity of the assay could not identify the translocation, given that the neoplastic clones form only a small proportion of the tumor. Panagopoulous et al. sought to further elucidate the mechanisms of altered CSF1 expression and identified a novel CSF1-S100A10 fusion gene with translocation t(1;1)(q21;p11) as well as another novel fusion of the coding region of CSF1 with a sequence located downstream of the CSF1 locus. Both fusions had the effect of replacing the 3’ untranslated region (UTR) of CSF1 with new sequences, resulting in expression of the protein-coding part of CSF1. The authors suggest that the CSF1-COL6A3 fusion gene might similarly result in the replacement of the 3’UTR of CSF1 with sequences from the COL6A3 gene rather than bringing CSF1 under the control of the collagen promotor, as had been previously assumed [24].
Most recently, Tsuda et al. further clarified the genomic abnormalities in TGCT [25]. Consistent with previous analyses, 13 of the 18 (72%) TGCT samples subjected to RNA sequencing had a CSF1 translocation, of which seven were intrachromosomal translocations and nine were interchromosomal translocations. Of the TGCT samples, 23% had CSF1-COL6A3 fusion genes. Several novel CSF1 fusion transcripts were also revealed, including CSF1-VCAM1, CSF1-FN1, and CSF1-CDH1 fusions. All CSF1 fusion transcripts were found to cause the deletion of CSF1 exon 9, which is a negative regulator of CSF1 expression. In addition, novel CBL mutations were found in 35% of the tumors, with or without the presence of CSF1 fusions, with CBL-mutated cases showing higher JAK2 expression than wild-type CBL cases [25]. CBL, which codes the Casitas B-cell lymphoma family of ubiquitin ligases, is involved in downregulation of tyrosine kinase receptor signaling and negatively regulates antitumor function of immune cells; CBL mutations are found in around 5% of myeloid neoplasms [26]. Overall, the findings of Tsuda’s study support the importance of CSF1 exon 9 deletion in the development of TGCT, and additionally suggest a role for CBL mutations, although further studies are required to elucidate how these mutations may impact the clinical presentation of the disease [25]. Finally, Ho et al. also investigated potential CSF1 rearrangements to determine the mechanism of CSF1 upregulation in TGCT samples from 39 patients [27]. The study found CSF1 rearrangements in 30 (83%) of the cases (13 translocations and 17 3’ deletions). In 92% of tumors, RNA and DNA sequencing found CSF1 3’ deletion or CSF1 translocation events resulting in deletion of the 3’UTR, which differs from the previously reported COL6A3-CSF1 fusion. The study also confirmed CBL mutations in six out of 21 cases, two of which had no identifiable CSF1 rearrangement. These findings support the hypothesis that CSF1 overexpression may be caused by the transcription of CSF1 lacking 3’ negative regulatory sequences. The presence of CBL mutations may also contribute to increased CSF1/CSF1R signaling [27]. While localized and diffuse types of TGCT have distinct clinical presentations, to date, no association has been found between CSF1 overexpression of rearrangement and clinical characteristics [28].
In a phase 1 multicenter study of patients with advanced solid tumors (ClinicalTrials.gov identifier NCT01004861), all tissue biopsy specimens from 25 patients with TGCT had detectable expression of CSF1, and 19 (76%) showed evidence of abnormal CSF1 transcripts. The sequencing data revealed two general patterns of CSF1 rearrangement, the most common being rearrangement near the exon 8/9 junction or within exon 9 (3’UTR) of CSF1 typically partnered with intergenic sequences, and the other being gene fusion of CSF1 exon 5/6 junction with exons of various other genes. Both patterns would result in loss of the 3’UTR regulatory sites, suggesting a loss of negative regulation of CSF1 gene expression, consistent with previous studies [29].
Emerging evidence suggests that various cell populations associated with TGCT express CSF1, such as increased expression in podoplanin-high, fibroblast-like synovial lining cells, the mechanisms for which are not yet defined [30].
Malignant giant cell tumor of tendon sheath is a very rare disease, defined by the coexistence of a benign giant cell tumor with overtly malignant areas or by the recurrence of a typical giant cell tumor as a sarcoma. [1]. Unbalanced rearrangement of the CSF1 locus has been found in up to 30% of cells in malignant TGCTs, demonstrated as retention of a single 3’CSF1 signal with loss of the 5’ signal on fluorescence in situ hybridization of cells from the sarcomatous focus of the tumors [31]. Huang et al. showed CSF1 expression in five out of six malignant TGCT tumors, with one tumor having CSF1 translocation, findings similar to those observed in benign TCGT lesions. In addition, the study showed that transformation from benign to malignant tumors appeared to involve alterations in cyclin A, p53, and chromosome arm 15q. In particular, deletions at 15q22-24 suggest the locations of tumor suppressor genes. Together, the results support the role of CSF1 dysregulation from the early stages of the disease process that may progress to malignancy once p53-interacting tumor suppressor genes are deleted, thereby compromising the tumor-suppressing function of p53 [32].
2.3Other TGCT molecular pathways
Some TGCTs exhibit unusual histologic features comprising areas with increased atypia, proliferative activity, irregular enlarged nuclei, and spindle cell morphology, accompanied by varying degrees of fibrosis and degenerative changes, in the absence of accepted features of malignant tumors. In a sample of three such tumors, novel gene fusions not involving CSF1, NIPBL-ERG, FN1-ROS1, and YAP1-MAML2 were identified, which contrasted with screening of three control TGCTs with conventional morphology that found translocations involving CSF1, with partner genes COL6A3, FN1, and KCNMA1. The atypical TGCTs were associated with late and multiple local recurrences compared with no recurrences of the conventional TGCTs. The findings suggest that the non-CSF1 fusions involving novel kinase-driven and transcription factor gene-driven pathways may be associated with a greater propensity for recurrences and local aggressiveness [33].
Programmed death ligand-1 (PD-L1) plays a role in regulating immune responses and tumor progression by suppressing the activation of T cells. Overexpression of PD-L1 has been associated with poor clinical outcomes in some cancers but better outcomes in others [34]. In a study involving 40 patients with TGCT, 21 (53%) showed PD-L1 expression in tumor mononuclear cells. PD-L1 expression was also found in multinucleated cells and foam cells in 25% and 18% of tumors, respectively. In addition, most cells with positive PD-L1 expression co-expressed the CD68 protein. Notably, positive expression of PD-L1 was associated with larger tumor sizes compared with patients with negative expression, suggesting that PD-L1 may play an important role in immune regulation by delivering inhibitory signals to maintain tumor growth [35].
Matrix metalloproteinases (MMPs) are a group of enzymes responsible for degradation of the extracellular matrix components, such as collagen and proteoglycans, that are involved in many disease processes, such as arthritis, cancer, periodontitis, and osteoporosis [36]. Studies involving tissues from diffuse TGCTs have shown that these tumors predominantly produce MMP-1 and MMP-9, with MMP-1 expressed in both mononuclear and multinuclear giant cells and MMP-9 expressed exclusively in multinucleated giant cells [37, 38]. This contrasted with synovial tissues from rheumatoid arthritis and osteoarthritis joints, which expressed MMP-1, MMP-2, and MMP-3 [37]. This expression of MMP-1 and MMP-9 may contribute to the invasive activity and bone and cartilage loss in diffuse TGCT [37]. However, TGCT and arthritis can share overlapping clinical and biologic features, including degenerative changes in the later stages of TGCT. Other factors, such as imbalances between MMP and tissue inhibitors of metalloproteinases, may be important contributors to joint destruction in both conditions [37].
3Future directions
Broadening our understanding of TGCT biology is important in order to effectively treat and manage patients with TGCT. Many questions currently remain unanswered. For example, it is unclear why certain morphologic features, such as the amount of hemosiderin in tumors, vary within and between patients and why certain tumors are painful and others are not, exclusive of the degree of associated degenerative disease or joint damage. In addition, the anatomic distribution of TGCT to the knee in more than half of cases remains unexplained, with interactions between disease location and the putative association with trauma and inflammation still to be elucidated. Other areas deserving of further research include the role of macrophage polarization, into type 1 subset with cytotoxic and antitumoral properties and type 2 subset with immunoregulatory and protumoral activities [39], in tipping the balance between proinflammatory and proneoplastic features, and the role of the host synovial lining cells and associated fibroblasts in governing the severity of disease and the likelihood of recurrence. Finally, the genetic and cell biology that underlies the difference between localized and diffuse disease begs for attention.
Growing evidence of the role of CSF1 in the pathogenesis of TGCT has resulted in the development of multiple investigational therapies; however, more detail on underlying disease mechanisms still needs to be elucidated in order to improve the choice of individualized therapies and treatment outcomes for patients with TGCT.
Acknowledgments
Medical writing and editorial assistance were provided by Michelle Hughes, BSc Hons, of Lumanity Scientific, Inc., and were financially supported by Daiichi Sankyo.
Author contributions
CONCEPTION: William D. Tap and John H. Healey
INTERPRETATION OF DATA: William D. Tap and John H. Healey
REVISION FOR IMPORTANT INTELLECTUAL CONTENT: William D. Tap and John H. Healey
SUPERVISION: William D. Tap and John H. Healey
Conflicts of interest
William D. Tap: reports a standard budget for site participation in a clinical trial from Plexxikon; personal fees for advisory board, consulting, and travel expenses from Eli Lilly and EMD Serono; personal fees for advisory board and consulting from Mundipharma, C4 Therapeutics, Daiichi Sankyo, Blueprint, GlaxoSmithKline, and Agios Pharmaceuticals; personal fees for advisory board from NanoCarrier and Deciphera; personal fees for consulting from Adcendo, Ayala Pharmaceuticals, Kowa, Servier, and AbMaxBio; a patent Companion Diagnostic for CDK4 inhibitors (14/854,329 pending to MSKCC/SKI); a patent Engima and CDH18 as Companion Diagnostics for CDK4 inhibitors (SKI2016-021-03 pending to MSKCC/SKI); participation on the Scientific Advisory Board for Certis Oncology Solutions and Innova Therapeutics; stock ownership in Certis Oncology Solutions; and stock ownership in and co-founder of Atropos Therapeutics.
John H. Healey: reports personal fees for consulting from Daiichi Sankyo, Stryker, and SynOx; serves as an editor for Clinical Orthopaedics and Related Research; and serves as a trustee for the Musculoskeletal Transplant Foundation and MIB Osteosarcoma Foundation.
References
[1] | WHO Classification of Tumours Editorial Board. Soft tissue and bone tumours. 5th ed. Lyon (France): International Agency for Research on Cancer; (2020) . |
[2] | Vilanova JC . WHO classification of soft tissue tumors. In: De Schepper AM, Vanhoenacker F, Parizel PM, Gielen J, editors. Imaging of soft tissue tumors. Germany: Springer; (2017) , p. 187–96. |
[3] | Bernthal NM , Spierenburg G , Healey JH , Palmerini E , Bauer S , Group TS , et al. The diffuse-type tenosynovial giant cell tumor (dt-tgct) patient journey: a prospective multicenter study. Orphanet J Rare Dis. (2021) ;16: (1):191. doi: 10.1186/s13023-021-01820-6 |
[4] | Healey JH , Bernthal NM , van de Sande M . Management of tenosynovial giant cell tumor: a neoplastic and inflammatory disease. J Am Acad Orthop Surg Glob Res Rev. (2020) ;4: (11):e20.00028. doi: 10.5435/JAAOSGlobal-D-20-00028 |
[5] | Gelhorn HL , Tong S , McQuarrie K , Vernon C , Hanlon J , Maclaine G , et al. Patient-reported symptoms of tenosynovial giant cell tumors. Clin Ther. (2016) ;38: (4):778–93. doi: 10.1016/j.clinthera.2016.03.008 |
[6] | Mastboom MJL , Verspoor FGM , Verschoor AJ , Uittenbogaard D , Nemeth B , Mastboom WJB , et al. Higher incidence rates than previously known in tenosynovial giant cell tumors. Acta Orthop. (2017) ;88: (6):688–94. doi: 10.1080/17453674.2017.1361126 |
[7] | Ehrenstein V , Andersen SL , Qazi I , Sankar N , Pedersen AB , Sikorski R , et al. Tenosynovial giant cell tumor: incidence, prevalence,patient characteristics, and recurrence. A registry-based cohortstudy in Denmark. J Rheumatol. (2017) ;44: (10):1476–83. doi: 10.3899/jrheum.160816 |
[8] | Lin F , Ionescu-Ittu R , Pivneva I , Wynant W , Shi S , Wu EQ , et al. The economic burden of tenosynovial giant cell tumors among employed workforce in the United States. J Occup Environ Med. (2021) ;63: (4):e197–e202. doi: 10.1097/JOM.0000000000002159 |
[9] | Burton TM , Ye X , Parker ED , Bancroft T , Healey J . Burden of illness associated with tenosynovial giant cell tumors. Clin Ther. (2018) ;40: (4):593–602 e1. doi: 10.1016/j.clinthera.2018.03.001 |
[10] | Mastboom MJ , Planje R , van de Sande M . The patient perspective on the impact of tenosynovial giant cell tumors on daily living: crowdsourcing study on physical function and quality of life. Interact J Med Res. (2018) ;7: (1):e4. doi: 10.2196/ijmr.9325 |
[11] | Staals EL , Ferrari S , Donati DM , Palmerini E . Diffuse-type tenosynovial giant cell tumour: current treatment concepts and future perspectives. Eur J Cancer. (2016) ;63: :34–40. doi: 10.1016/j.ejca.2016.04.022 |
[12] | NCCN Clinical Practice Guidelines in Oncology (NCCN guidelines®): Soft Tissue Sarcoma. Plymouth Meeting (PA): National Comprehensive Cancer Network; 2021 [updated 2020 Oct 30; cited 2022 Jan 27]. |
[13] | Turalio® (pexidartinib) capsules. Prescribing information. Basking Ridge (NJ): Daiichi Sankyo Inc; October 2021 [cited 2022 Feb 11]. Available from: https://daiichisankyo.us/prescribing-information-portlet/getPIContent?productName=Turalio&inline=true |
[14] | Turalio® capsules 200 milligrams (pexidartinib HCl). Prescribing information. Ministry of Food and Drug Safety of The Republic of Korea; 2021 [cited 2022 Feb 11]. Available from: https://nedrug.mfds.go.kr/pbp/CCBBB01/getItemDetail?itemSeq=202108140 |
[15] | Jaffe HL , Lichtenstein L , Sutro CJ . Pigmented villonodular synovitis, bursitis and tenosynovitis: a discussion of the synovial and bursal equivalents of the tenosynovial lesions commonly denoted as xanthoma, xanthogranuloma, giant cell tumor, or myeloma of the tendon sheath, with some consideration of the tendon sheath lesion itself. Arch Pathol. (1941) ;31: :731–65. |
[16] | Molena B , Sfriso P , Oliviero F , Pagnin E , Teramo A , Lunardi F , et al. Synovial colony-stimulating factor-1 mRNA expression in diffuse pigmented villonodular synovitis. Clin Exp Rheumatol. (2011) ;29: (3):547–50. |
[17] | Chitu V , Stanley ER . Colony-stimulating factor-1 in immunity and inflammation. Curr Opin Immunol. (2006) ;18: (1):39–48. doi: 10.1016/j.coi.2005.11.006 |
[18] | West RB , Rubin BP , Miller MA , Subramanian S , Kaygusuz G , Montgomery K , et al. A landscape effect in tenosynovial giant-cell tumor from activation of CSF1 expression by a translocation in a minority of tumor cells. Proc Natl Acad Sci U S A. (2006) ;103: (3):690–5. doi: 10.1073/pnas.0507321103 |
[19] | Stanley ER , Chitu V . CSF-1 receptor signaling in myeloid cells. Cold Spring Harb Perspect Biol. (2014) ;6: (6):a021857. doi: 10.1101/cshperspect.a021857 |
[20] | Lin H , Lee E , Hestir K , Leo C , Huang M , Bosch E , et al. Discovery of a cytokine and its receptor by functional screening of the extracellular proteome. Science. (2008) ;320: (5877):320807–11. doi: 10.1126/science.1154370 |
[21] | Chihara T , Suzu S , Hassan R , Chutiwitoonchai N , Hiyoshi M , Motoyoshi K , et al. Il-34 and M-CSF share the receptor Fms but are not identical in biological activity and signal activation. Cell Death Differ. (2010) ;17: (12):1917–27. doi: 10.1038/cdd.2010.60 |
[22] | Moller E , Mandahl N , Mertens F , Panagopoulos I . Molecular identification of COL6A3-CSF1 fusion transcripts in tenosynovial giant cell tumors. Genes Chromosomes Cancer. (2008) ;47: (1):21–5. doi: 10.1002/gcc.20501 |
[23] | Cupp JS , Miller MA , Montgomery KD , Nielsen TO , O’Connell JX , Huntsman D , et al. Translocation and expression of CSF1 in pigmented villonodular synovitis, tenosynovial giant cell tumor, rheumatoid arthritis and other reactive synovitides. Am J Surg Pathol. (2007) ;31: (6):970–6. doi: 10.1097/PAS.0b013e31802b86f8 |
[24] | Panagopoulos I , Brandal P , Gorunova L , Bjerkehagen B , Heim S . Novel CSF1-S100A10 fusion gene and CSF1 transcript identified by RNA sequencing in tenosynovial giant cell tumors. Int J Oncol. (2014) ;44: (5):1425–32. doi: 10.3892/ijo.2014.2326 |
[25] | Tsuda Y , Hirata M , Katayama K , Motoi T , Matsubara D , Oda Y , et al. Massively parallel sequencing of tenosynovial giant cell tumors reveals novel CSF1 fusion transcripts and novel somatic CBL mutations. Int J Cancer. (2019) ;145: (12):3276–84. doi: 10.1002/ijc.32421 |
[26] | Liyasova MS , Ma K , Lipkowitz S . Molecular pathways: Cbl proteins in tumorigenesis and antitumor immunity-opportunities for cancer treatment. Clin Cancer Res. (2015) ;21: (8):1789–94. doi: 10.1158/1078-0432.CCR-13-2490 |
[27] | Ho J , Peters T , Dickson BC , Swanson D , Fernandez A , Frova-Seguin A , et al. Detection of CSF1 rearrangements deleting the 3’ UTR in tenosynovial giant cell tumors. Genes Chromosomes Cancer. (2020) ;59: (2):96–105. doi: 10.1002/gcc.22807 |
[28] | Mastboom MJL , Hoek DM , Bovee J , van de Sande MAJ , Szuhai K . DoesCSF1 overexpression or rearrangement influence biological behaviourin tenosynovial giant cell tumours of the knee? Histopathology. (2019) ;74: (2):332–40. doi: 10.1111/his.13744 |
[29] | Tap WD , Singh AS , Anthony SP , Sterba M , Zhang C , Healey JH , et al. Results from phase 1 extension study assessing pexidartinib treatment in 6 cohorts with solid tumors including TGCT, and abnormal CSF1 transcripts in TGCT. Clin Cancer Res. (2022) ;28: (2):298–307. doi: 10.1158/1078-0432.CCR-21-2007 |
[30] | Chandler AC , Yakoub M , Fujiwara T , Donlin LT , Purdue PE , Healey JH . Neoplastic synovial lining cells that coexpress podoplanin and CD90 overproduce CSF-1, driving tenosynovial giant cell tumor. J Orthop Res. (2022) ;40: (8):1918–25. doi: 10.1002/jor.25216 |
[31] | Al-Ibraheemi A , Ahrens WA , Fritchie K , Dong J , Oliveira AM , Balzer B , et al. Malignant tenosynovial giant cell tumor: The true “synovial sarcoma?” A clinicopathologic, immunohistochemical, and molecular cytogenetic study of 10 cases, supporting origin from synoviocytes. Mod Pathol. (2019) ;32: :242–51. doi: 10.1038/s41379-018-0129-0 |
[32] | Huang HY , West RB , Tzeng CC , van de Rijn M , Wang JW , Chou SC , et al. Immunohistochemical and biogenetic features of diffuse-type tenosynovial giant cell tumors: the potential roles of cyclin A, P53, and deletion of 15q in sarcomatous transformation. Clin Cancer Res. (2008) ;14: (19):6023–32. doi: 10.1158/1078-0432.CCR-08-0252 |
[33] | Vougiouklakis T , Shen G , Feng X , Hoda ST , Jour G . Molecular profiling of atypical tenosynovial giant cell tumors reveals novel non-CSF1 fusions. Cancers (Basel). (2019) ;12: (1):100. doi: 10.3390/cancers12010100 |
[34] | Wang X , Teng F , Kong L , Yu J . PD-L1 expression in human cancers and its association with clinical outcomes. Onco Targets Ther. (2016) ;9: :5023–39. doi: 10.2147/OTT.S105862 |
[35] | Zheng B , Yu L , Hu J , Xu H , Wang J , Shi Y , et al. Expression of PD-L1 in mononuclear cells, multinucleated cells, and foam cells in tenosynovial giant cell tumors. Int J Clin Exp Pathol. (2019) ;12: (3):876–84. |
[36] | Woessner JF Jr. Matrix metalloproteinases and their inhibitors in connective tissue remodeling. FASEB J. (1991) ;5: (8):2145–54. |
[37] | Uchibori M , Nishida Y , Tabata I , Sugiura H , Nakashima H , Yamada Y , et al. Expression of matrix metalloproteinases and tissue inhibitors of metalloproteinases in pigmented villonodular synovitis suggests their potential role for joint destruction. J Rheumatol. (2004) ;31: (1):110–9. |
[38] | O’Keefe RJ , Rosier RN , Teot LA , Stewart JM , Hicks DG . Cytokine and matrix metalloproteinase expression in pigmented villonodular synovitis may mediate bone and cartilage destruction. Iowa Orthop J. (1998) ;18: :26–34. |
[39] | Mantovani A , Sica A , Sozzani S , Allavena P , Vecchi A , Locati M . The chemokine system in diverse forms of macrophage activation and polarization. Trends Immunol. (2004) ;25: (12):677–86. doi: 10.1016/j.it.2004.09.015 |


