
Current Status of Developmental Encephalopathies: Rett Syndrome, MECP2 Duplication Disorder, CDKL5 Deficiency Disorder and FOXG1 Disorder
Abstract
This review describes the similarities and differences between the developmental encephalopathies including Rett syndrome (RTT), MECP2 Duplication Disorder (MDD), CDKL5 Deficiency Disorder (CDD), and FOXG1 Disorder (FD). RTT and MDD represent opposite ends of deficiency and duplication of MECP2 whereas CDD and FD were initially described as variants of RTT before their specific genetic variants were identified and they were recognized as distinct disorders. The subsequent discussions outline the clinical features, phenotype-genotype relationships, and potential for therapeutic intervention including gene replacement. Natural history studies involving each disorder are providing the essential underpinnings for developing outcome measures and biomarkers to enhance the advancement of such treatment efforts.
Rett syndrome was first recognized by Andreas Rett [1], predominantly in females, more than 50 years ago and first termed Rett syndrome (RTT) in the landmark 1983 paper of Hagberg and colleagues in the Annals of Neurology [2], launching this disorder worldwide. Clinical and genetic studies progressed rapidly culminating in identification of pathogenic variants in the gene, Methyl-CpG-binding-protein 2 (MECP2) in 1999 [3]. However, variant forms of RTT had already been described by Hagberg (atypical RTT) [4], Hanefeld (early infantile seizure variant) [5], and Rolando (congenital RTT) in 1985 [6]. Shortly after the identification of variants in MECP2, a disorder was recognized predominantly in males associated with duplications of MECP2, proving to be as difficult clinically as RTT [7]. In 2003, the US Natural History Study (NHS) of RTT began enrolling individuals with both classic and atypical RTT (NCT00299312), including some individuals with MECP2 Duplication disorder and some newly recognized individuals with mutations in CDKL5 and FOXG1, heretofore regarded as those with atypical RTT as described by Hanefeld and by Rolando. The inclusion criteria for entry into the US NHS were very simple and straightforward: the individual, female or male, should meet the clinical criteria for RTT [8] and should have proof of genetic testing. Copies of such testing, whether positive or negative, were essential for enrollment. If results were unavailable, provision was made through the Greenwood Genetic Center, Greenwood, South Carolina for obtaining the specific test results. Individuals could enroll in the study if they met clinical criteria for RTT, even if they did not have a genetic variant in MECP2, or if they had a genetic variant in MECP2, even if they did not meet the clinical criteria for RTT.
In 2014, the NHS study then began enrolling individuals representing all four disorders (NCT02738281): RTT, MECP2 Duplication Disorder (MDD), CDKL5 Deficiency Disorder (CDD), and FOXG1 Disorder (FD). Significant natural history data has developed across the world in each disorder, leading to their collective categorization as Developmental Encephalopathies [9]. In addition, other disorders that could fit within this rubric, such as the severe form of Epileptic-Aphasia Disorder (GRIN2A) or Pitt-Hopkins (TCF4), are beginning to emerge.
Thus, it is timely to consider the current understanding of this group of disorders, their similarities and their differentiating features. This will include the current diagnostic strategies, the array of clinical, neurophysiologic and neuroimaging features, and their developmental trajectories across the age range. With the identification of specific variants (or different duplications in the MECP2 Duplication Disorder) in each disorder, consideration of phenotype-genotype correlations has become important in developing an understanding for the range of developmental difficulties in each. In addition, the identification of the specific genes involved has allowed important basic and translational studies to progress with an eye to treatment strategies in each. The US NHS has provided the opportunity to develop comparative analyses of these four disorders, which provide clear points of differentiation to allow for accurate diagnosis and subsequent care. Finally, effective disease-modifying agents are already undergoing initial trials and potentially disease-reversing strategies are under careful consideration. These strategies will be considered in each presentation. Originally planned to address each of these disorders individually with a concluding presentation summarizing comparative features, the program was modified to combine the description of CDD with final discussion of comparison features in these four disorders.
1Statement of goals
The Developmental Encephalopathies have emerged as a relevant diagnostic group over the past decade or more. Initially described as variants of RTT, the diagnostic picture clarified rapidly in the past twenty years with identification of MECP2 variants in RTT, duplications of the same gene in MDD, and the identification of CDKL5 and FOXG1 variants in the respective disorders, CDD and FD. Current understanding now recognizes the importance of denoting these disorders as such, rather than variants of RTT. Substantial progress has occurred in the identification and diagnosis of individuals with these disorders and of developing natural history data in preparation for on-going or future clinical trials. In this symposium, each disorder will be described in detail, their core features compared, and the prospects for meaningful trials discussed.
2Statement of objectives
Understanding the similarities and unique features of each disorder is critical to advance both clinical and basic research. The subsequent presentations will review the diagnostic strategies for establishing the specific diagnosis and propose proper treatment guidelines. Fundamental to successful treatment of each of these disorders is the development of effective remediation. As these disorders have different timelines of discovery and subsequent study, comparison of the disease-modifying or gene replacement clinical trial protocols will have different timelines from clinical trials currently in place to current concepts being planned.
Competing interests
Dr. Percy has no competing interests.
Author contributions
Dr. Percy is solely responsible for this review.
References
[1] | Rett A. , [On a unusual brain atrophy syndrome in hyperammonemia in childhood], Wien Med Wochenschr 116: (37) ((1966) ), 723–6. |
[2] | Hagberg B. , Aicardi J. , Dias K. and Ramos O. , A progressive syndrome of autism, dementia, ataxia, and loss of purposeful hand use in girls: Rett’s syndrome: report of 35 cases, Ann Neurol 14: (4) ((1983) ), 471–9. |
[3] | Amir R.E. , Van den Veyver I.B. , Wan M. , Tran C.Q. , et al., Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2, Nat Genet 23: (2) ((1999) ), 185–8. |
[4] | Hagberg B.A. and Skjeldal O.H. , Rett variants: a suggested model for inclusion criteria, Pediatr Neurol 11: (1) ((1994) ), 5–11. |
[5] | Hanefeld F. , The clinical pattern of the Rett syndrome, Brain Dev 7: (3) ((1985) ), 320–5. |
[6] | Rolando S. , Rett syndrome: report of eight cases, Brain Dev 7: (3) ((1985) ), 290–6. |
[7] | Friez M.J. , Jones J.R. , Clarkson K. , Lubs H. , et al., Recurrent infections, hypotonia, and mental retardation caused by duplication of MECP2 and adjacent region in Xq28, Pediatrics 118: (6) ((2006) ), e1687–95. |
[8] | Neul J.L. , et al., Rett syndrome: revised diagnostic criteria and nomenclature, Ann Neurol 68: (6) ((2010) ), 944–50. |
[9] | Cutri-French C. , et al. Comparison of core features in four developmental encephalopathies in the Rett Natural History Study, Ann Neurol 88: ((2020) ), 396–406. |
First recognized more than 60 years ago by Andreas Rett in Austria and Bengt Hagberg in Sweden, Rett syndrome (RTT) burst on the international scene following the publication of Hagberg and colleagues in 1983 [1, 2]. Thereafter, significant clinical research commenced resulting in the identification of pathogenic variants in MECP2 (methyl-CpG-binding protein 2) as the cause of this rare neurodevelopmental disorder [3]. This led to increased efforts to understand the natural history of RTT and the fundamental issues in brain development underlying this disorder. RTT, primarily affecting females, has an incidence of ∼1 : 10,000 female births, >95% having mutations in the MECP2 gene [9, 10]. Clinical criteria were elaborated first in 1985 [11], revised in 2002 [12], and revised again in 2010 to account for advances in understanding of RTT [8]. The current criteria (Table 1) represent a simplification of prior efforts as well as an effort to create more easily understandable terminology. For classic RTT, the supportive features originally included in the initial criteria are no longer required. For atypical RTT, individuals must meet 2 of 4 main criteria and 5 of 11 supportive criteria. Those with atypical RTT may have either more or less significant difficulties than those with the classic form.
Table 1
Rett syndrome: diagnostic criteria
| Classic Rett Syndrome | Atypical Rett Syndrome |
| Main criteria | Main criteria |
| Regression of development | Regression of development |
| Partial or complete loss of acquired purposeful hand skills | Must meet 2 of 4 main criteria stated above |
| Partial or complete loss of acquired spoken language | Must meet 5 of 11 supportive criteria |
| Gait abnormalities: Impaired or absent ability | |
| Stereotypic hand movements | |
| Supportive criteria: not required | |
| Supportive Criteria | |
| Breathing disturbances when awake | Scoliosis/kyphosis |
| Bruxism when awake | Small, cold hands and feet |
| Impaired sleep pattern | Inappropriate laughing/screaming spells |
| Abnormal muscle tone | Peripheral vasomotor disturbances |
| Diminished response to pain | Growth retardation |
| Intense eye communication –eye pointing | |
In 2003, a multi-center longitudinal natural history study (NHS) was begun in the US with funding by the NIH/ORDR/NICHD, focusing on RTT and individuals with MECP2 Duplication Syndrome (MDS). The inclusion criteria for the US NHS were outlined in the opening section of this symposium. In 2014, this study was extended to its current 14 sites across the US with emphasis expanded to add individuals with CDKL5 Deficiency Disorder (CDD) and FOXG1 Disorder (FD), previously considered to have more severe forms of atypical RTT. Together, these studies have enrolled more than 1700 individuals with one of these disorders including individuals with MECP2 variants representing females who do not meet criteria for classic or atypical RTT or males who range from very severe infantile encephalopathy to those with signs of mild to moderate developmental delay.
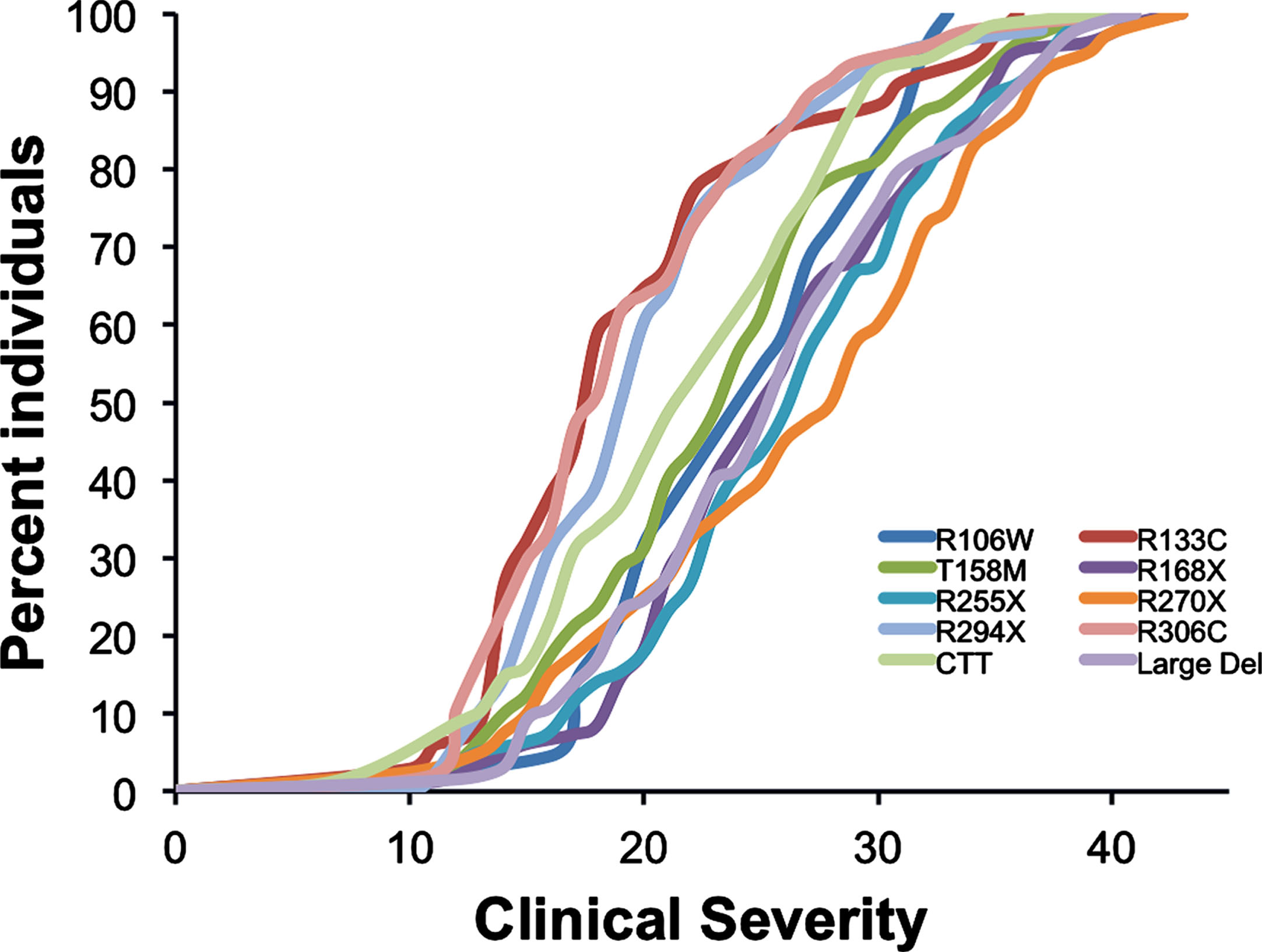
In classic RTT, significant phenotype-genotype differences are observed across specific mutations [13, 14]. Although these group-wise differences are significant (Fig. 1), in specific individuals, differences may be less apparent. This is probably due to a number of factors including variations in X-chromosome inactivation (XCI), clonal distribution of mutant genes across brain regions, background variation in other genes, and environmental factors. Aside from differences in XCI, evidence is lacking to support these suppositions.
Fig. 1
Genotype-phenotype relationship.
1The case for effective treatment
Mouse and rat models of RTT share many features with the human disorder despite some differences including later onset in the animal models [15–17]. In 2007, Jacky Guy and colleagues presented convincing evidence in a mouse model of RTT that inserting a normal MECP2 gene regulated by a specific controller yielded marked improvement in both male and female mutants and markedly prolonged survival [18]. This provided the necessary encouragement to investigate the potential for disease-modifying agents including gene replacement in RTT animal models.
2Pharmaceutical treatment in RTT
The first substantive effort utilized the amino-terminal tripeptide of the insulin growth factor-1 (IGF-1) in a mouse model, showing partial reversal of specific features including prolongation of survival [19]. This led to human trial of the full length, recombinant IGF-1 (Mecasermin) in children with RTT at Boston Children’s Hospital [20, 21]. While a phase I trial demonstrated safety and some evidence of efficacy, the subsequent phase II study demonstrated no clear evidence of improvement, perhaps, related to limited ability of this agent to transit the blood-brain barrier (BBB). Subsequently, Neuren Pharmaceuticals launched randomized placebo-controlled study of a modified amino-terminal tripeptide (Trofinetide), which crosses the BBB. Trofinetide was utilized in two phase II studies, one in adolescents and adults, the other in children 5-15 years [22, 23]. The trial in the older age group demonstrated good safety profiles and suggestive evidence for effectiveness in a four-week exposure. The subsequent (and somewhat longer trial) in children again demonstrated safety and tolerability and at the highest dose of 200 mg/kg showed improvements in the parent-reported Rett Syndrome Behavioral Questionnaire (RSBQ) and in two physician scales, the Clinical Global Impression –Improvement (CGI-I) and Rett Syndrome Domain Specific Concerns. Currently, Acadia Pharmaceuticals has completed a phase III, 12-week placebo-controlled trial (Lavender) followed by a 40-week open label extension (Lilac) utilizing two of the outcome measures noted above, the RSBQ and the CGI-I. Neuren Pharmaceuticals is planning a similar phase III trial in Europe for next year.
Following the demonstration that chronic administration of the N-Methyl-D-Aspartate receptor antagonist, ketamine, improved the phenotype of RTT animal mice [24], a double blind, placebo-controlled crossover trial with escalating doses is on-going in the US (NCT03633058). This is an investigator-initiated phase II trial with safety as the primary endpoint. Secondary endpoints include measurements of breathing and the same two outcome measures described above: RSBQ and CGI-I. This trial is being conducted in children ages 6-12. The first cohort (0.75 mg/kg) is completed, and the next escalating dose is ongoing.
Two additional trials are ongoing in the US. The GW Pharmaceuticals trial of Epidiolex (cannabidiol) oral solution in children with RTT is a phase III double blind, placebo-controlled study of efficacy with the same two outcome measures. Anavex Life Sciences is engaged in a phase II placebo controlled, double-blind study on older adolescents and adults studying the muscarinic and Sigma-1 receptor agonist, Anavex2-73. This agent was shown to be effective in a mouse model of RTT. The primary outcome is safety with the same two measures as secondary outcomes.
Additional trials have failed to establish efficacy. These include Sarizotan (Newron Pharmaceuticals) and glatiramer acetate.
3Gene therapy in RTT
Two more recent RTT mouse model studies in female animals demonstrated that both systemic and cisternal delivery of an optimized MeCP2 transgene rescued both motor, behavioral, and cellular deficits leading to the development an AAV vector for gene therapy [25, 26]. Two companies, Taysha Gene Therapies and Neurogene, are preparing to launch early phase safety trials in optimized MECP2 platforms.
4Current challenges for clinical trials in RTT
Biomarkers: Biomarkers could broaden the links to disease severity, identify treatment responders, and be early predictors of treatment responses. Current efforts include neurophysiological and metabolic biomarkers. Neurophysiological biomarkers have been examined including a project in the US NHS to evaluate visual and auditory evoked potentials [27]. Visual evoked responses in RTT mutant mice and individuals with RTT show a marked reduction in amplitudes compared to normal controls [28]. In the NHS, both parameters were markedly reduced in individuals with RTT versus typically developing children. In female RTT heterozygous mice, an age dependent effect was demonstrated as well as a strong correlation with phenotypic severity [29]. These studies need to be extended in individuals with RTT including the evaluation of power and coherence [30]. Similarly, longitudinal characterization of these features in heterozygous RTT mice should be coupled with the possible responses in mice who have received MeCP2 reconstitution.
Metabolic biomarkers have also been examined in the US NHS [31]. Significant differences were noted between normal siblings in a number of important metabolic pathways including links to changes in gut microflora, aging, and oxidative stress. While much work remains to validate these findings, additional correlations with overall severity and specific clinical issues such as epilepsy, abnormal breathing patterns, movement disorders, and others will be evaluated. As with the neurophysiologic biomarkers, examining the same metabolic parameters in heterozygous RTT mice needs to be evaluated versus disease progression and examined after gene therapy.
Outcome Measures: Clinician and parent-reported outcome measures are areas of importance for clinical trials. The need for reliable and valid measures is of major concern for investigators and trial developers. The currently utilized measures are the Clinical Global Impression-Improvement (CGI-I) and the Rett Syndrome Behavioral Quotient (RSBQ). Additional markers under consideration are a Revised Motor Behavioral Assessment, video assessments of motor function, breathing assessment, and adaptation of the Mullen Scales of Early Learning. Both the CGI-I and the RSBQ are currently being utilized in several human trials. The CGI scales were developed initially in RTT for the first Trofinetide trials [32]. This is a clinician’s assessment of global function which is divided into two forms, Clinical Global Impression –Severity (CGI-S) and Clinical Global Impression –Improvement (CGI-I), both as 7-point Likert scales. These are both accepted by the US (FDA) and European (EMA) regulatory agencies. Specific anchors were developed for the CGI-S based on clinical features of RTT. The CGI-I does not provide details on specific domains of improvement, rather indicating graded levels of both positive and negative effects. In order to utilize these scales in a multi-site clinical trial, it is critical that all investigators are trained on these disease-specific anchors and measures of improvement and provide evidence of training currently using written vignettes. Even better rater training and reliability could be assessed utilizing video-based vignettes.
The RSBQ is a 45-item caregiver completed rating scale that was not established initially as an outcome measure. Although the RSBQ focuses on aberrant behavior, it does contain measures of other disabling features in RTT. It has undergone psychometric evaluation for factor analysis and test-retest reliability, is relatively simple to complete, and is accepted by the FDA and EMA. However, it does not cover all domains of interest equally, has a limited three-point response range (not at all, sometimes, always), and contains items that require training to explain the intent. Further, a recent publication expressed concerns about its reliability [33].
The development of a new clinician reported outcome has emerged from a psychometric analysis of the Motor Behavioral Assessment (MBA) score originally developed by Fitzgerald, Percy, and Jankovic [34, 35]. This was initially designed as a 37 item, 5-point Likert scale with a range of 0-148, higher scores representing increased severity. The scale was divided into three conceptual domains, Behavioral-Social, Orofacial-Respiratory, and Motor-Physical signs. This has been collected consistently over 17 years in the US NHS. Some items were known to have marked floor or ceiling effects and they did not factor well into the three conceptual domains. It was never assessed psychometrically until last year when the cross-sectional responses from 1075 individuals were submitted to formal psychometric analysis in collaboration with Research Triangle Institute (RTI), a research institute in Durham, North Carolina [36]. Two-thirds of the sample were used as a development sample, the remaining one-third was used as a confirmatory sample. Factor analysis reduced the scale to 21 items in five factors: Motor dysfunction, Functional Skills, Social Skills, Aberrant Behavior, and Rett-specific Behavior. The total revised MBA (R-MBA) score included 21 items plus 3 additional clinically important items: stereotypical hand movements/hand mouthing, truncal rocking, and seizures. Internal consistency was reasonable for the subscales, yielding Cronbach’s α=0.55-0.8 for the subscales and 0.78 for the total. Comparing these factors with a separate parent report of clinical domains for validity demonstrated excellent correlation. The R-MBA also showed excellent correlation with the Clinical Severity Scale (CSS), significant correlation by genotype group, and significant correlation with the CGI-S. Interestingly, the R-MBA increased overall with age, similar to the full-scale MBA. Further assessment of the R-MBA will include longitudinal assessment of NHS data, establishment of internal reliability using video-based assessment, intra- and inter-rater reliability, external validity, and formal evaluation of sensitivity and responsivity to establish the minimally clinically important difference.
Other potential outcome measures under consideration are breathing assessments using band plethysmography, adapting the Mullen Scales of Early Learning for a standard assessment of development in RTT, and development of two video-based evaluation tools to assess hand and gait function.
5Concluding remarks
RTT is a rare genetic neurodevelopmental disorder with a variety of clinical features found in other disorders. Mouse models of RTT reproduce many of the features seen in humans and provide evidence for modification and even reversibility. Clinical trials are underway or proposed to provide disease-modifying therapies. Problems still remain in terms of improving clinical outcome measures for which the US NHS data can be critical. Biomarker development and implementation is needed both for clinical assessment and for use in clinical trials. Animal models can provide useful avenues for identifying and characterizing these.
Competing interests
Dr. Neul has no competing interests.
Author contributions
Dr. Neul is solely responsible for this review.
Conflicts of interest
For the respective Trofinetide trials, Dr. Benke, Dr. Marsh, Dr. Neul, and Dr. Percy did not have any conflict of interest. These authors were blinded as to participant involvement, whether as placebo or pharmacologic agent and did not participate in the data analysis.
References
[1] | Rett A. , [On a unusual brain atrophy syndrome in hyperammonemia inchildhood], Wien Med Wochenschr 116: (37) ((1966) ), 723–6. |
[2] | Hagberg B. , et al., A progressive syndrome of autism, dementia,ataxia, and loss of purposeful hand use in girls: Rett’s syndrome:report of 35 cases, Ann Neurol 14: (4) ((1983) ), 471–9. |
[3] | Amir R.E. , et al., Rett syndrome is caused by mutations in X-linkedMECP2, encoding methyl-CpG-binding protein 2, Nat Genet 23: (2) ((1999) ), 185–8. |
[4] | Laurvick C.L. , et al., Rett syndrome in Australia: a review of theepidemiology, J Pediatr 148: (3) ((2006) a), 347–52. |
[5] | Fehr S. , et al., Trends in the diagnosis of Rett syndrome inAustralia, Pediatr Res 70: (3) ((2011) ), 313–9. |
[6] | Hagberg B. , et al., Rett syndrome: criteria for inclusion andexclusion, Brain Dev 7: (3) ((1985) ), 372–3. |
[7] | Hagberg B. , et al., An update on clinically applicable diagnostic criteria in Rett syndrome. Comments to Rett Syndrome Clinical Criteria Consensus Panel Satellite to European Paediatric NeurologySociety Meeting, Baden Baden, Germany, 11 September 2001, Eur J Paediatr Neurol 6: (5) ((2002) ), 293–7. |
[8] | Neul J.L. , et al., Rett syndrome: revised diagnostic criteria andnomenclature, Ann Neurol 68: (6) ((2010) ), 944–50. |
[9] | Neul J.L. , et al., Specific mutations in methyl-CpG-binding protein2 confer different severity in Rett syndrome, Neurology 70: (16) ((2008) ), 1313–21. |
[10] | Cuddapah V.A. , et al., Methyl-CpG-binding protein 2 (MECP2) mutationtype is associated with disease severity in Rett syndrome, JMed Genet 51: (3) ((2014) ), 152–8. |
[11] | Veeraragavan S. , et al., Loss of MeCP2 in the rat models regression,impaired sociability and transcriptional deficits of Rett syndrome, Hum Mol Genet 25: (15) ((2016) ), 3284–3302. |
[12] | Pitcher M.R. , et al., Rett syndrome like phenotypes in the R255XMecp2 mutant mouse are rescued by MECP2 transgene, Hum MolGenet 24: (9) ((2015) ), 2662–72. |
[13] | Merritt J.K. , et al., Pharmacological read-through of R294X Mecp2 ina novel mouse model of Rett syndrome, Hum Mol Genet 29: (15) ((2020) ), 2461–2470. |
[14] | Guy J. , et al., Reversal of neurological defects in a mouse model ofRett syndrome, Science 315: ((2007) ), 1143–7. |
[15] | Tropea D. , et al., Partial reversal of Rett Syndrome-like symptomsin MeCP2 mutant mice, Proc Natl Acad Sci U S A 106: (6) ((2009) ), 2029–34. |
[16] | Khwaja O.S. , et al., Safety, pharmacokinetics, and preliminaryassessment of efficacy of mecasermin (recombinant human IGF-1) forthe treatment of Rett syndrome, Proc Natl Acad Sci U S A 111: (12) ((2014) ), 4596–601. |
[17] | O’Leary H.M. , et al., Placebo-controlled crossover assessment ofmecasermin for the treatment of Rett syndrome, Ann Clin TranslNeurol 5: (3) ((2018) ), 323–332. |
[18] | Glaze D.G. , et al., A double-blind, randomized, placebo-controlledclinical study of trofinetide in the treatment of rett syndrome, Pediatr Neurol 76: ((2017) ), 37–46. |
[19] | Glaze D.G. , et al., Double-blind, randomized, placebo-controlledstudy of trofinetide in pediatric Rett syndrome, Neurology 92: (16) ((2019) ), e1912–e1925. |
[20] | Patrizi A. , et al., Chronic administration of then-methyl-d-aspartate receptor antagonist ketamine improves rettsyndrome phenotype, Biol Psychiatry 79: (9) ((2016) ), 755–764. |
[21] | Garg S.K. , et al., Systemic delivery of MeCP2 rescues behavioral andcellular deficits in female mouse models of Rett syndrome, JNeurosci 33: (34) ((2013) ), 13612–20. |
[22] | Gadalla K.K.E. , et al., Development of a novel AAV gene therapycassette with improved safety features and efficacy in a mouse modelof rett syndrome, Mol Ther Methods Clin Dev 5: ((2017) ), 180–190. |
[23] | Saby J.N. , et al., Multisite study of evoked potentials in rett syndrome, Ann Neurol (2021). |
[24] | LeBlanc J.J. , et al., Visual evoked potentials detect corticalprocessing deficits in Rett syndrome, Ann Neurol 78: (5) ((2015) ), 775–86. |
[25] | Dong H.W. , et al., Detection of neurophysiological features infemale R255X MeCP2 mutation mice, Neurobiol Dis 145: ((2020) ), 105083. |
[26] | Roche K.J. , et al., Electroencephalographic spectral power as amarker of cortical function and disease severity in girls with Rettsyndrome, J Neurodev Disord 11: (1) ((2019) ), 15. |
[27] | Neul J.L. , et al., Metabolic signatures differentiate rett syndromefrom unaffected siblings, Front Integr Neurosci 14: ((2020) ), 7. |
[28] | Neul J.L. , et al., Improving treatment trial outcomes for rettsyndrome: the development of rett-specific anchors for the clinicalglobal impression scale, J Child Neurol 30: (13) ((2015) ), 1743–8. |
[29] | Hou W. , et al., Assessment of a clinical trial metric for rettsyndrome: critical analysis of the rett syndrome behaviouralquestionnaire, Pediatr Neurol 107: ((2020) ), 48–56. |
[30] | FitzGerald P.M. , et al., Extrapyramidal involvement in Rett’ssyndrome, Neurology 40: (2) ((1990) ), 293–5. |
[31] | FitzGerald P.M. , Jankovic J. and Percy A.K. , Rett syndrome andassociated movement disorders, Mov Disord 5: (3) ((1990) ), 195–202. |
[32] | Raspa M. , et al., A psychometric evaluation of the motor-behavioralassessment scale for use as an outcome measure in rett syndromeclinical trials, Am J Intellect Dev Disabil 125: (6) ((2020) ), 493–509. |
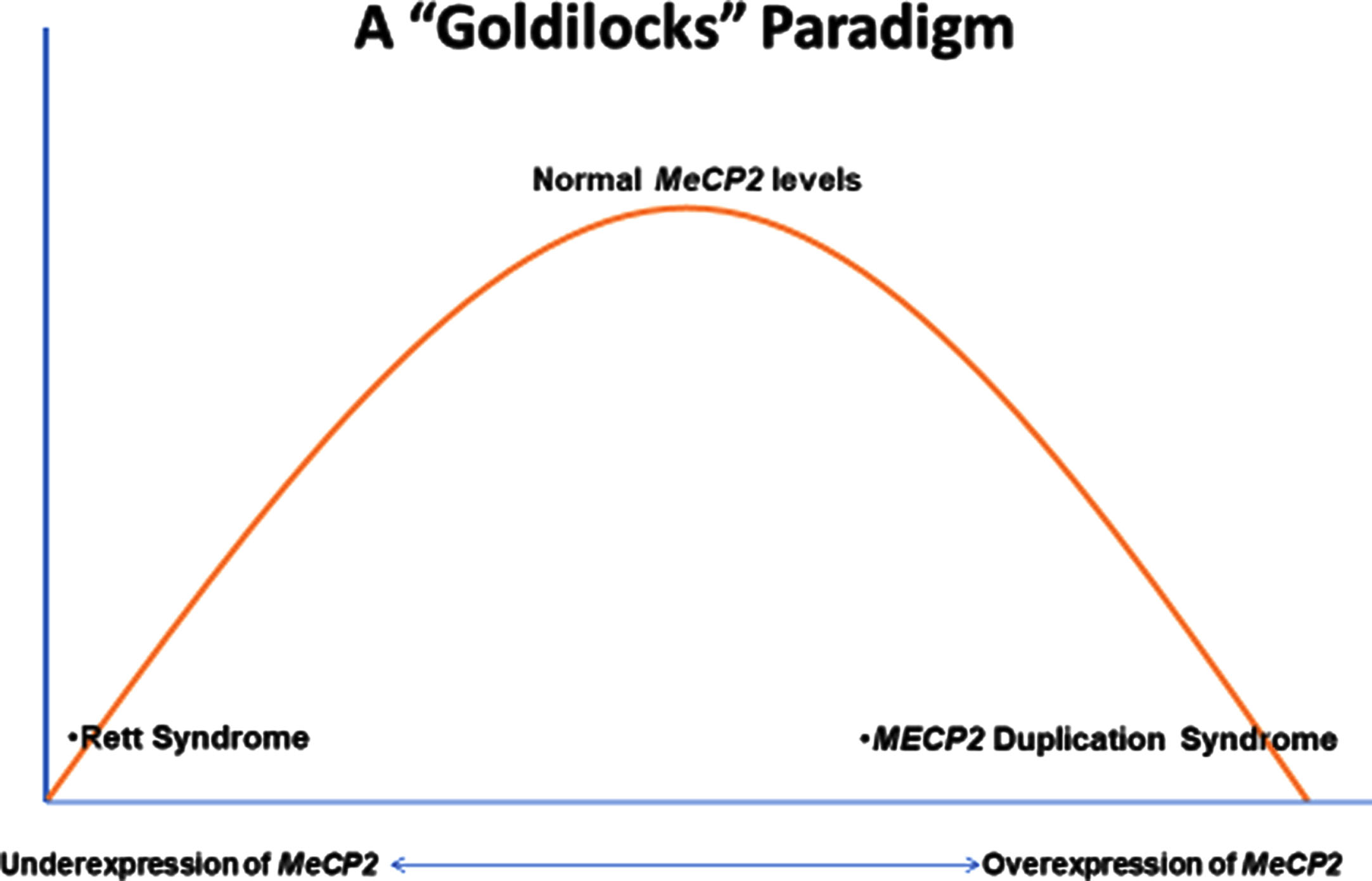
MECP2 Duplication Syndrome (MDS) represents the antithesis of Rett Syndrome (RTT). To understand this idea, one must examine the MECP2 (methyl-CpG-binding protein 2) gene. MECP2 was well-studied in cancer genetics as a methylation binding regulator prior to its identification as the principal cause of RTT in 1999 [3]. Located at Xq28, MeCP2 functions in the cell nucleus as an epigenetic gene regulator by binding to methylated DNA nucleotides. It is recognized that MeCP2 regulates genes critical for normal brain development including the maturation and maintenance of synapses. Following the identification of MECP2 as causative in RTT, an animal model was engineered to over-express MECP2 producing an equally devastating condition [37, 38]. Subsequently, duplications of MECP2, principally in males, were associated with significant neurodevelopmental delay, hypotonia, and recurrent infections [7, 39-44]. The duplication was mainly transmitted by the mother who had marked skewing of X chromosome inactivation and was regarded as cognitively normal. Evidence was presented that these carrier females do have increased evidence of anxiety and depression [45]. Thus, the Goldilocks Paradigm (Fig. 1) emerged. The under-expression of MECP2 yields RTT, the “normal” expression is just right, and the over-expression produces MDS.
Fig. 1
The goldilocks paradigm.
To reiterate, MDS is generally identified in males accounting for 1-2% of X-linked intellectual disability [46]. Females have been identified, but much less frequently, often as de novo mutations from the paternal allele or autosomal duplications. The typical clinical features of MDS include infantile hypotonia, developmental delays, epilepsy, choreiform movements, progressive spasticity, and recurrent infections. Thus, MDS is quite different from RTT and probably under-diagnosed due to relative absence of distinctive features such as deceleration in rate of head growth, regression in early childhood, or stereotypic hand movements.
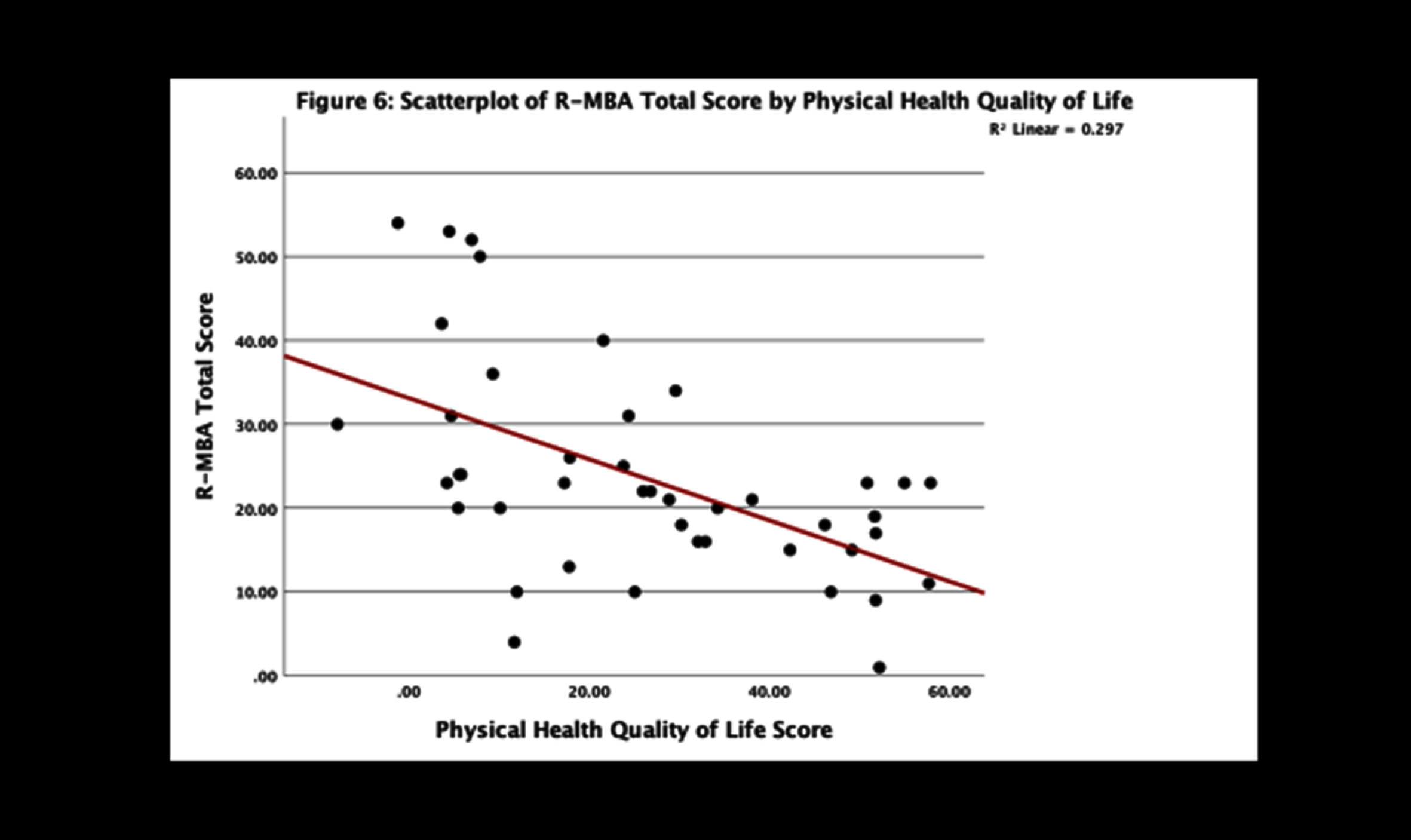
Important questions regarding regression and epilepsy have been addressed in the US Natural History Study (NHS). Among 69 participants (62 males and 7 females; age range 1-28 yr.) with MDS, the most common features (Table 1) included 70% with developmental delay in the first six months, 88% with hypotonia, 81% with constipation, 77% with drooling, and 73% with vasomotor disturbances. Other features such as bruxism and sleep issues were less prominent. Regression, impacting motor skills predominantly and seen at a much older age than RTT, was typically associated with the onset of significant, often refractory, seizures. Among those with seizures, 73% had developmental regression. The Revised Motor Behavioral Assessment (R-MBA) revealed an increased severity with age (Fig. 2). When assessing the parental concerns, the most prominent were lack of effective communication, abnormal walking or balance, constipation, seizures, and ineffective chewing and swallowing. Recurrent infections were sixth most prominent (<6%). Repetitive hand movements, virtually 100% in RTT, were recorded in less than 5%. When evaluating these concerns in those with seizures, the top three concerns were seizures (60%), abnormal walking or balance issues (17%), and ineffective communication (11%). Parent rating of overall functioning by age revealed improved function prior to 10 years, but unchanged or worsening function after age 12 years. Comparing the R-MBA with the Physical Health Quality of Life (from the Child Health Questionnaire) revealed an inverse relationship (Fig. 3), that is, higher scores on the R-MBA, which indicate greater severity, were associated with lower scores on the physical quality of life (QOL), indicating poorer QOL. Conversely, lower scores on the R-MBA were associated with higher scores on the physical QOL.
Table 1
Common features of MECP2 duplication syndrome
 |
Fig. 2
Change in revised motor behavioral assessment with age.
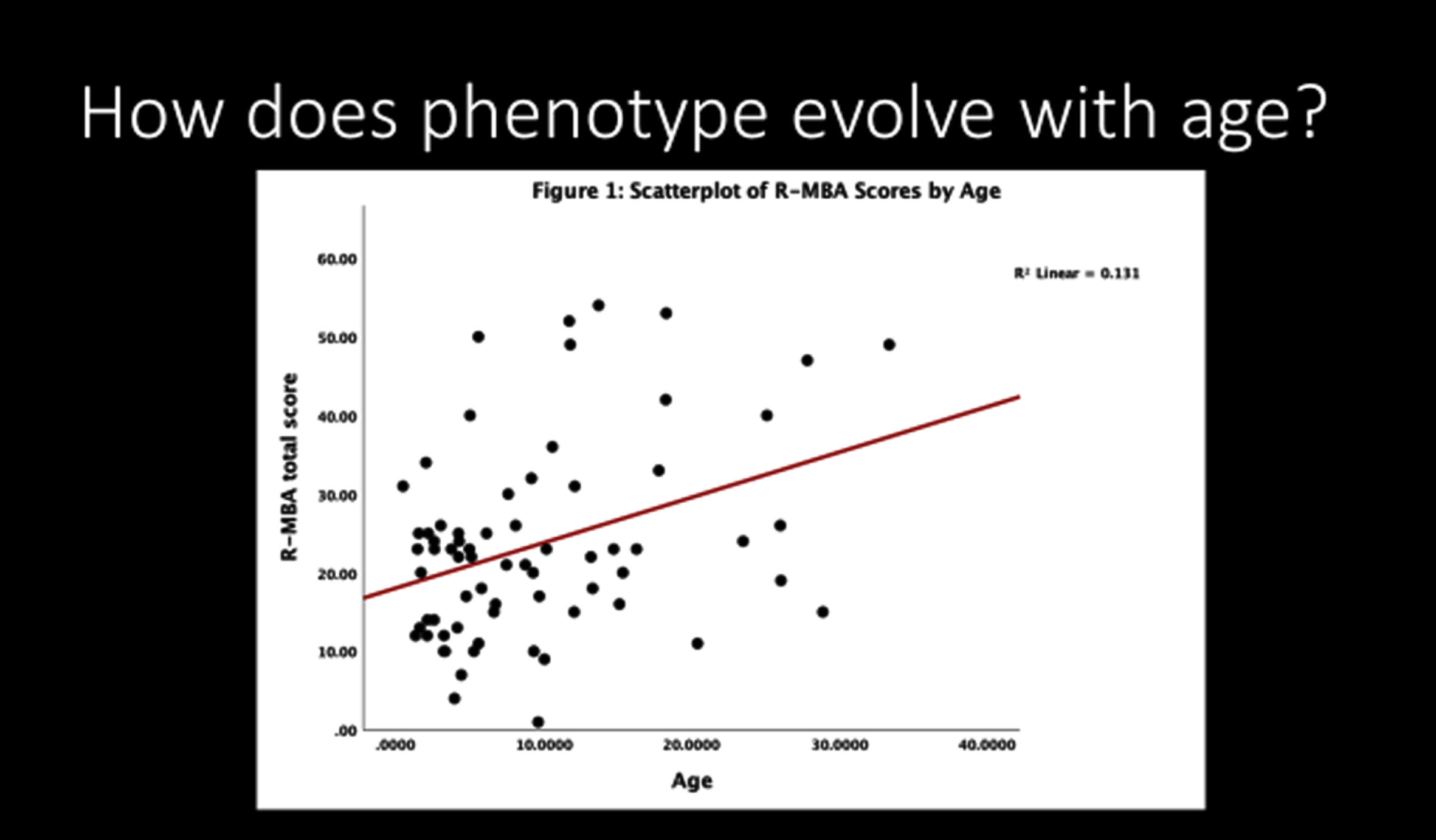
Fig. 3
Comparison of revised motor behavioral assessment with physical health quality of life.
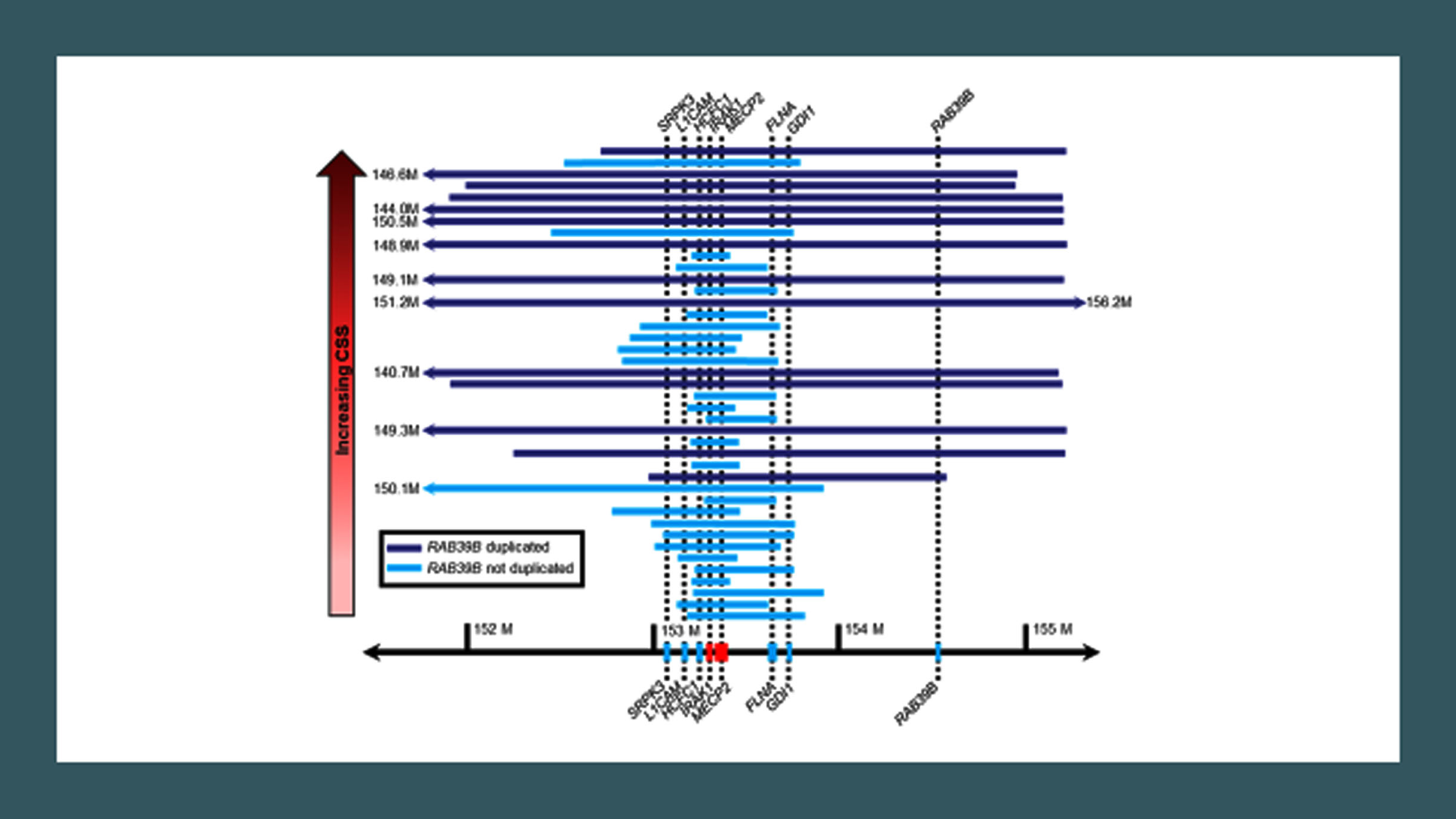
A second question regards what factors lead to the correct diagnosis? This is coupled with a rather wide variability in phenotypic characteristics and what could account for this variability. Possible factors include duplication size, presence or absence of epilepsy, and immunologic or inflammatory markers. Comparing duplication size and composition with the Clinical Severity Score (higher scores representing greater severity) showed a close correlation overall with duplication size and composition [47], particularly those including HCFC1, IRAK1, FLNA, GDI1, and RAB398, in addition to MECP2 (Fig. 4).
Fig. 4
Duplication size in MECP2 duplication syndrome.
1Advancing toward clinical trials
Important progress in reaching human clinical trials has been suggested in a mouse model of MECP2 duplication using antisense oligonucleotides (ASO). These studies demonstrated improvements in hypoactivity, anxiety-like behaviors, motor abnormalities, and social behavior.
This raises the related issue of targeting specific symptoms versus correcting the duplication. An important issue regarding correcting the duplication is how to modulate the gene therapy so as to reduce MeCP2 expression correctly without overshooting and creating an RTT phenotype. Further, the reproducibility of preclinical models and improvements in QOL is critical to ensuring the development of drug development for humans.
This translation of preclinical results to human clinical trials is a universal problem when moving from animal models to humans. The animal models often fail to predict efficacy of potential therapies in humans. Thus, the third question regards the development of possible biomarkers for assessing disease progression. The absence of high-quality outcome measures including the ability to measure improvements across a broad functional range can lead to failure. In addition, absence of validated biomarkers and the inability to examine the target organ, that is, the brain make direct observation difficult. In order to obviate these problems, validated clinical measures and biomarkers need to be developed, and clinical trial readiness needs to be standardized across the clinical trial sites.
The key endpoints required are the establishment of typical developmental trajectories, development of measures sufficiently sensitive to detect improvement, and ability to detect minimally clinically significant differences.
2Concluding remarks
Research in MDS has evolved steadily over the past nearly twenty years. Nonetheless, significant questions remain. How do the duplication size and the contiguous genes involved contribute to the clinical features and developmental trajectory? Can effective outcome measures and biomarkers (neurophysiologic or immunologic for example, be developed? Can we improve clinical assessments by capturing data from wearable devices?
Overall, these are the keys to development and assessment of effective therapies for conducting meaningful clinical trials.
Competing interests
Dr. Peters has no competing interests.
Author contributions
Dr. Peters is solely responsible for this review.
References
[1] | Amir R.E. , et al., Rett syndrome is caused by mutations in X-linkedMECP2, encoding methyl-CpG-binding protein 2, Nat Genet 23: (2) ((1999) ), 185–8. |
[2] | Collins A.L. , et al., Mild overexpression of MeCP2 causes aprogressive neurological disorder in mice, Hum Mol Genet 13: (21) ((2004) ), 2679–89. |
[3] | del Gaudio D. , et al., Increased MECP2 gene copy number as theresult of genomic duplication in neurodevelopmentally delayed males, Genet Med 8: (12) ((2006) ), 784–92. |
[4] | Van Esch H. , et al., Duplication of the MECP2 region is a frequentcause of severe mental retardation and progressive neurologicalsymptoms in males, Am J Hum Genet 77: (3) ((2005) ), 442–53. |
[5] | Meins M. , et al., Submicroscopic duplication in Xq28 causesincreased expression of the MECP2 gene in a boy with severe mentalretardation and features of Rett syndrome, J Med Genet 42: (2) ((2005) ), e12. |
[6] | Friez M.J. , et al., Recurrent infections, hypotonia, and mentalretardation caused by duplication of MECP2 and adjacent region inXq28, Pediatrics ((2006) ) 118: (6), e1687–95. |
[7] | Samaco R.C. , et al., Crh and Oprm1 mediate anxiety-related behaviorand social approach in a mouse model of MECP2 duplication syndrome, Nat Genet 44: (2) ((2012) ), 206–11. |
[8] | Pai G.S. , et al., A new X linked recessive syndrome of mentalretardation and mild dysmorphism maps to Xq28, J Med Genet 34: (7) ((1997) ), 529–34. |
[9] | Lubs H. , et al., XLMR syndrome characterized by multiple respiratoryinfections, hypertelorism, severe CNS deterioration and early deathlocalizes to distal Xq28, Am J Med Genet 85: (3) ((1999) ), 243–8. |
[10] | Sanlaville D. , et al., Functional disomy of the Xq28 chromosomeregion, Eur J Hum Genet 13: (5) ((2005) ), 579–85. |
[11] | Ramocki M.B. , et al., Autism and other neuropsychiatric symptoms areprevalent in individuals with MeCP2 duplication syndrome, AnnNeurol 66: (6) ((2009) ), 771–82. |
[12] | Lugtenberg D. , et al., Structural variation in Xq28: MECP2duplications in 1% of patients with unexplained XLMR and in 2% ofmale patients with severe encephalopathy, Eur J Hum Genet 17: (4) ((2009) ), 444–53. |
[13] | Peters S.U. , et al., Characterizing the phenotypic effect of Xq28duplication size in MECP2 duplication syndrome, Clin Genet 95: (5) ((2019) ), 575–581. |
FOXG1 disorder (FD) was initially described as the congenital variant of Rett syndrome (RTT). The overlap between the two disorders was based on clinical features which included severe neurodevelopmental delay, postnatal microcephaly, absent language, epilepsy, a hyperkinetic-dyskinetic movement disorder, and features of autism. In addition, specific neuroimaging features emerged consisting of hypogenesis of the corpus callosum, simplification of the gyral pattern with mild frontal pachygyria, and hypomyelination with particularly reduced white matter volume in the frontal lobes. The clear distinction between the two disorders was the early developmental delay and the neuroimaging abnormalities in FD. The original description of the congenital variant of RTT by Rolando et al. [6] was followed up in 2008 by Ariani and colleagues in 2008 [48] and Kortüm et al. [49] in 2011 linking these clinical features with pathogenic variants in the autosomal dominant FOXG1 gene at chromosome 14q12. In addition to the neuroimaging findings, FD differs from RTT by the striking presence of dyskinesias, a lack of regression, and lack of irregular breathing patterns, especially during wakefulness.
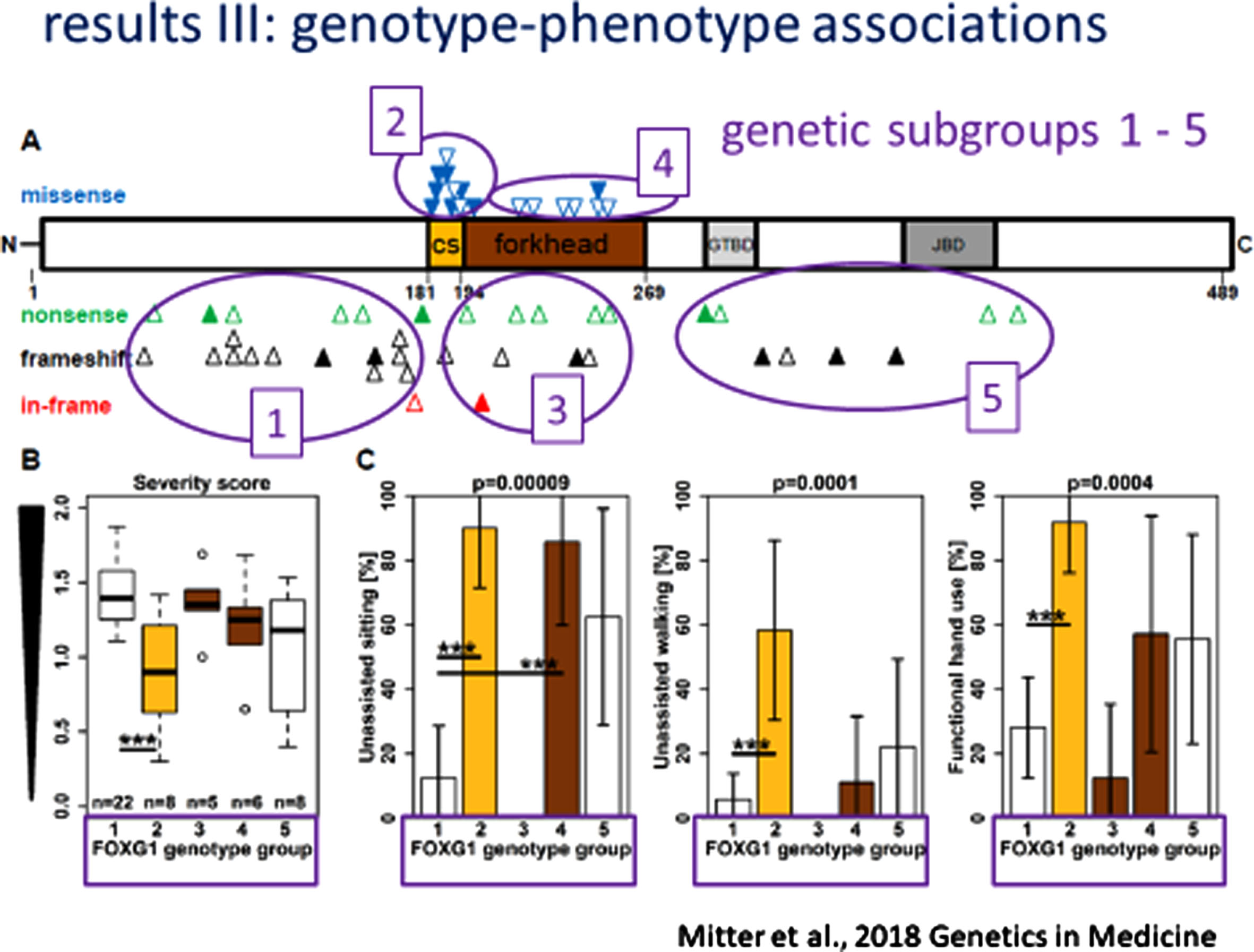
Subsequent efforts to delineate FD involved detailed analysis of the clinical and neuro-imaging phenotype and the development of clear genotype-phenotype correlation [50]. This arose from an analysis of 30 new individuals with pathogenic/likely pathogenic variants in FOXG1 and absent microaberrations at 14q12 together with 53 previously reported indivduals with FD aided by the application of a FOXG1 severity score. From these two groups, 76 individuals were assigned to one of 5 genetic subgroups according to the location and type of their variant: 1) N-terminal domain frameshift or nonsense variants (n = 37); 2) forkhead domain conserved site 1 missense variants (n = 12); 3) forkhead domain excluding conserve site 1, frameshift and nonsense variants (n = 9); 4) forkhead domain except conserved site 1, missense variants (n = 9), and C-terminal domain frameshift and nonsense variants (n = 9). The phenotypic features included microcephaly in 24% at birth and 84% at last follow-up, sitting without support in 45% (5-108 months of life), walking without support in 15% (24-132 months of life), functional hand use in 40%, loss of motor skills in 18%, verbal expression in 21% with a maximum of 100 words, and epilepsy in 68% with onset ranging from 3-168 months (mean = 25 months of life). Among the entire cohort of 83 individuals, 54 different heterozygous variants were noted: 20 frameshift variants (37%); 17 missense variants (31%); 15 nonsense variants (28%); and 2 in-frame variations (4%). The framshift and nonsense variants were distributed across the FOXG1 protein domains (Fig. 1). The resulting genotype-phenotype correlations were quite evident. (Fig. 2) More severe clinical phenotypes were associated with truncating variants in the N-terminal domain and forkhead domain except conserved site 1 (genetic subgroups 1 and 3). Milder phenotypes were seen in individuals with missense variants in the forkhead conserved site 1 (genetic subgroup 2).
Fig. 1
FOXG1 variants across the protein domains.
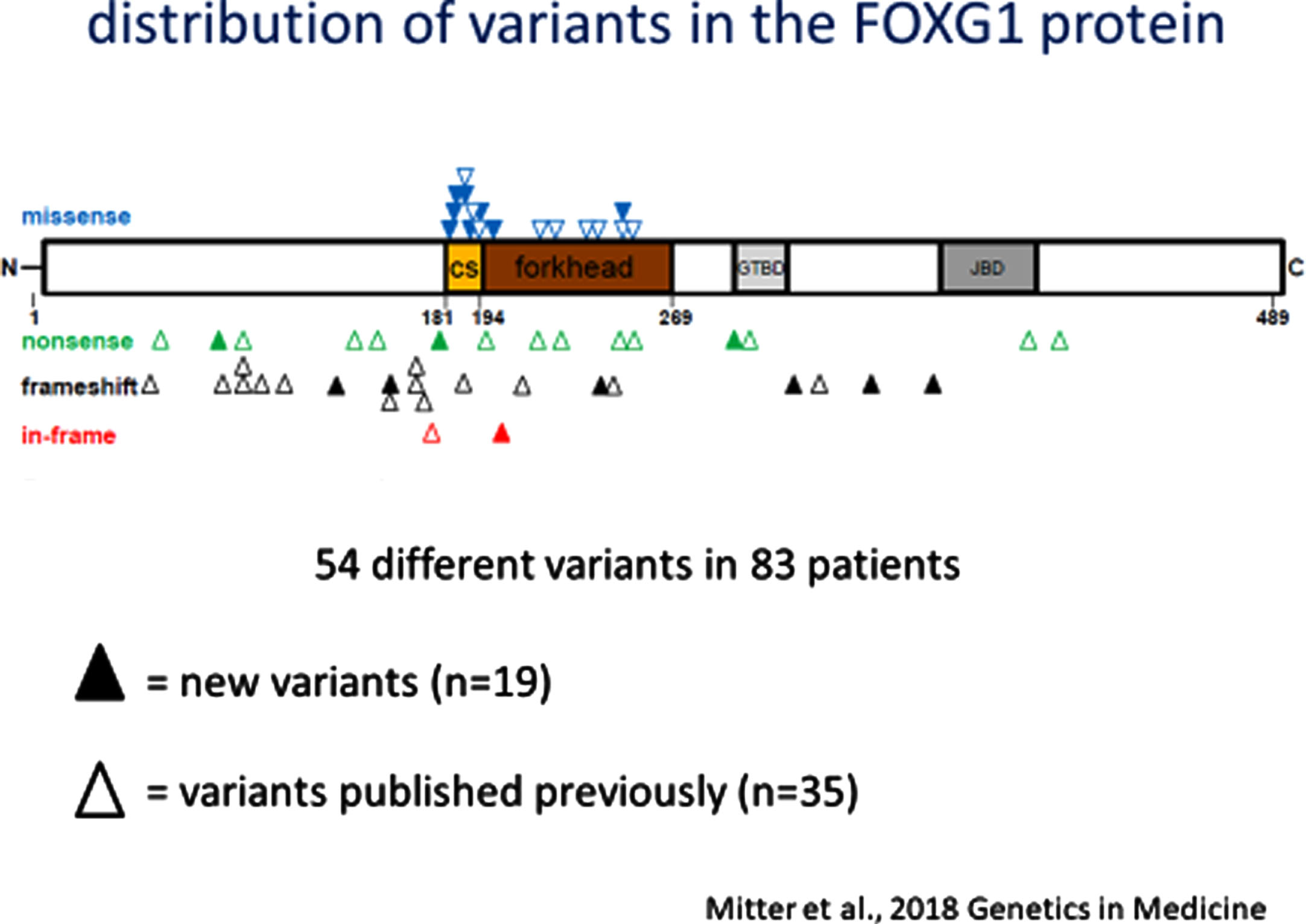
Fig. 2
FOXG1 Genotype-Phenotype Correlations.
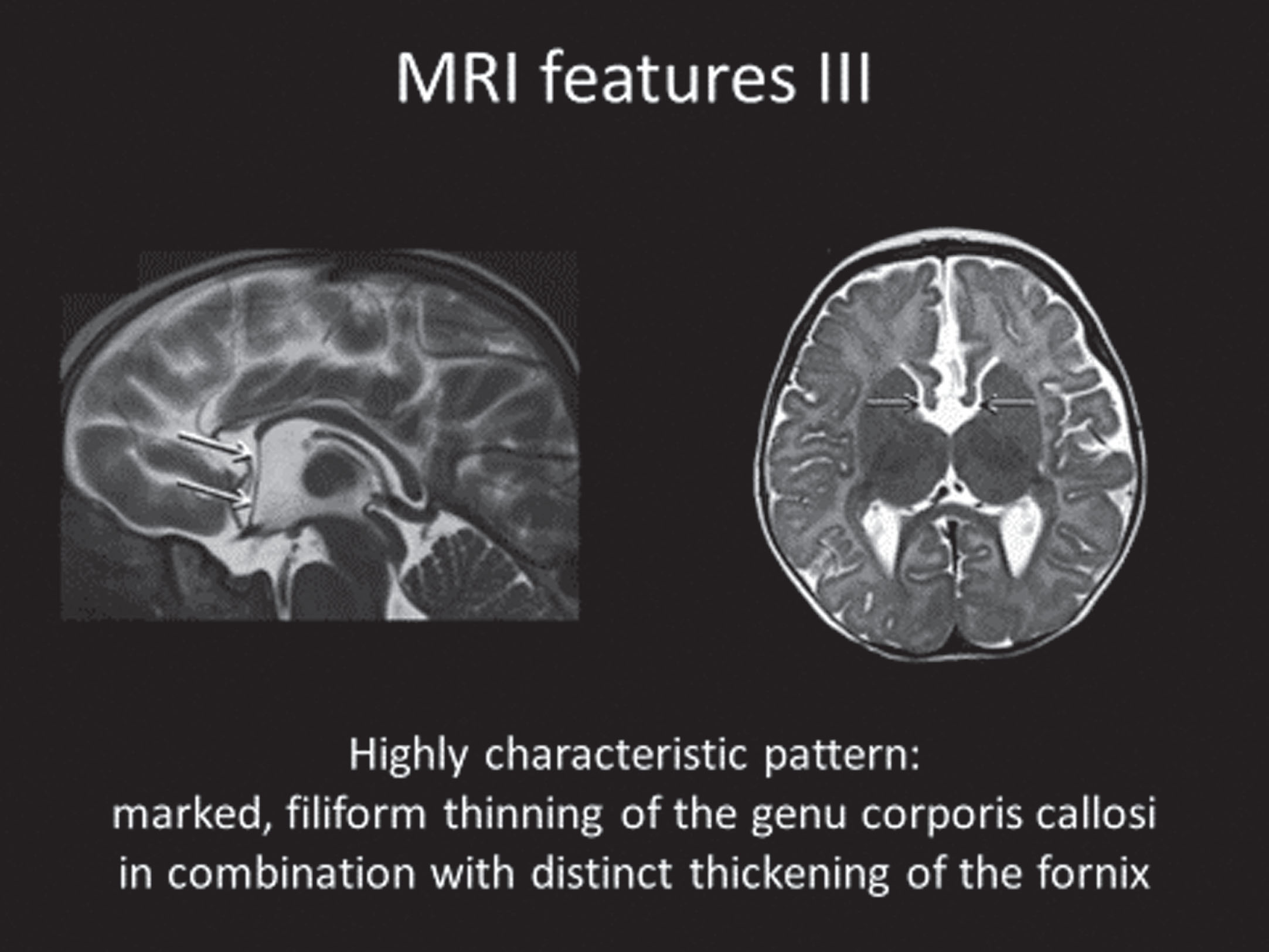
Analysis of the neuroimaging findings were also quite informative [51, 52]. Cranial MRIs were available from 34 individuals with heterogeneous variants. Systematic evaluation of these neuroimages were made according to assignment to one of the five genetic subgroups. The MRI features of FD was described in six specific domains (Fig. 3A-3D: 1) corpus callosum anomalies (82%); 2) thickening of the fornices (74%); 3) simplified gyral pattern (56%); 4) enlargement of inner CSF spaces (44%); 5) hypoplasia of basal ganglia (38%); and 6) hypoplasia of frontal lobes (29%).
Fig. 3A
MRI Features of FOXG1 Disorder: Fornix Cerebri.
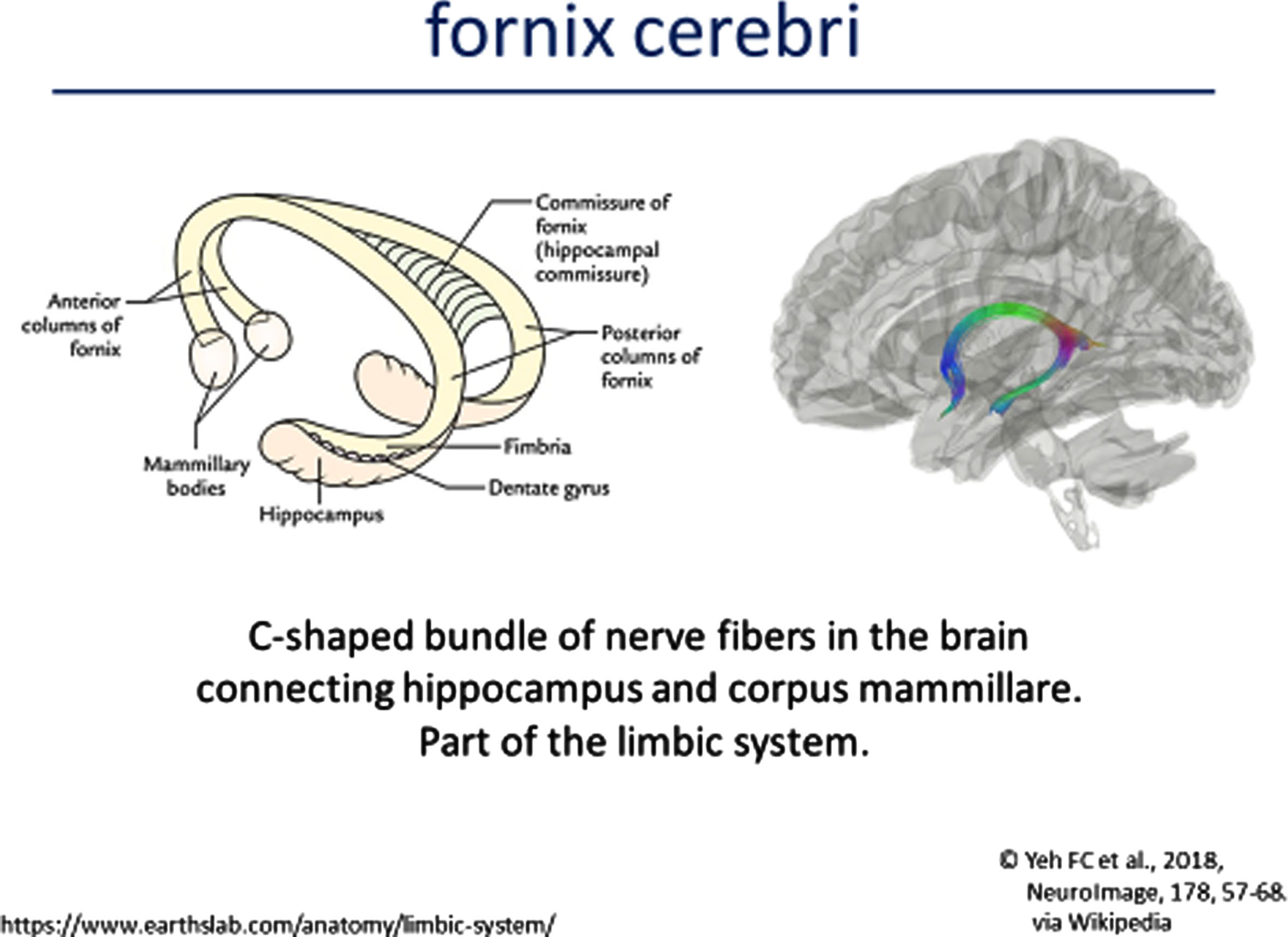
Fig. 3B
MRI Features of FOXG1 Disorder: Gyral Patterns.
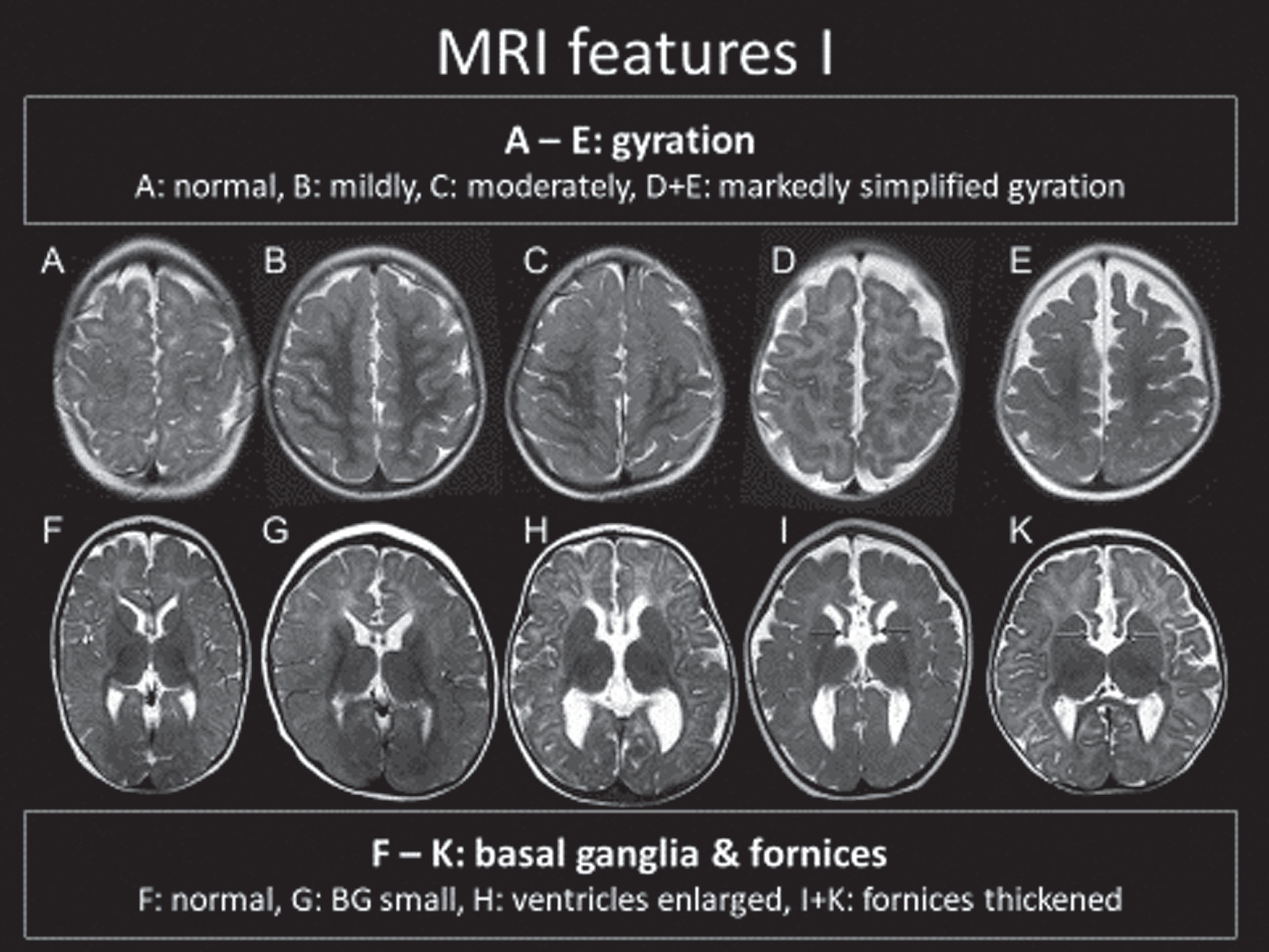
Fig. 3C
MRI Features of FOXG1 Disorder: Corpus Callosum Alterations.
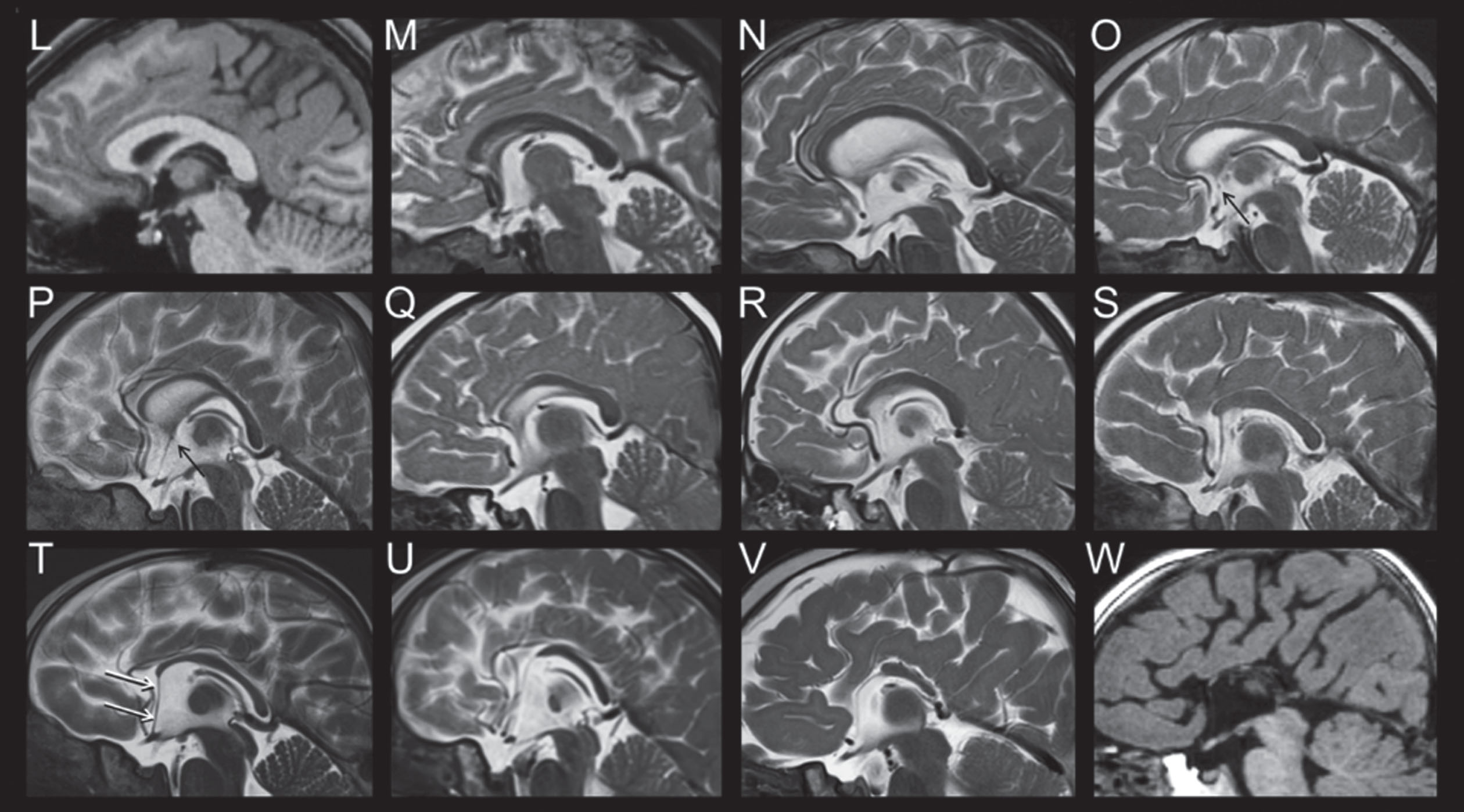
Fig. 3D
MRI Features of FOXG1 Disorder: Thickening of the Fornix.
Utilizing a FOXG1 severity score based on quantitation of the cerebral anomalies, statistical analysis of their genotype-MRI phenotypes was performed. The simplified gyral pattern was more frequent in those with early truncating variants. Higher FD MRI severity scores were associated with greater Clinical Severity scores.
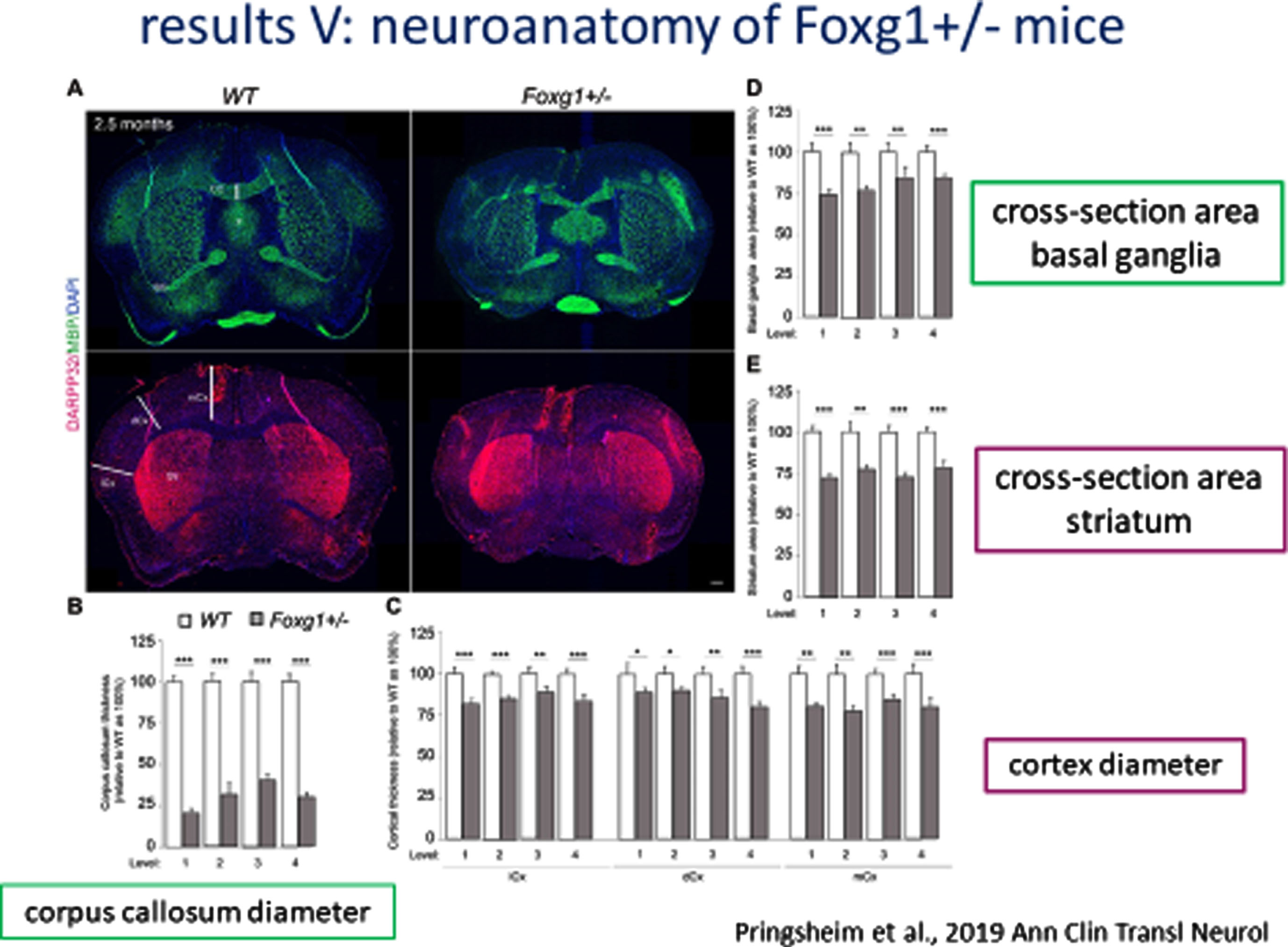
Similar neuranatomic features were found in a mouse model of Foxg1 including a nearly 25% reduction in cross-sectional area of the telencephalon and a progressive increase (anterior to posterior) in the cross-sectional area of the fornix (Fig. 4A) and reductions in the corpus callosum diameter, cross-sectional area of the basal ganglia and striatum, and diameter of the cortex (Fig. 4B).
Fig. 4A
Neuroanatomy of FOXG1 Mouse Model: Cross-sectional area of Fornix.

Fig. 4B
Neuroanatomy of FOXG1 Mouse Model: Cross-sectional area of Basal Ganglia and Striatum and Diameter of Cortex.
1Concluding remarks
These results demonstrate more clearly that FD should be considered in infants and children with global neurocognitive delays, microcephaly, and hyper-/dyskinesias. Further, better understanding of the clinical consequences of different FOXG1 variants is demonstrated along with the ability to provide more informed genetic counseling information to parents of a child with FD. The neuroimaging features reveal a combination of corpus callosal anomalies, simplified gyral patterns, and thickening of the fornix which seem to be pathognomonic. All three features were noted in 16/34 (47%) and two of the three features in an additional 9 (26%). Thus, 73% of the group demonstrated at least two. Further, these features provide a strong biologic biomarker for clinical trials in reversing the phenotype in FD.
Competing interests
Dr. Brockmann has no competing interests.
Author contributions
Dr. Brockmann is solely responsible for this review.
References
[1] | Rolando S. , Rett syndrome: report of eight cases, Brain Dev 7: (3) ((1985) ), 290–6. |
[2] | Ariani F. , et al., FOXG1 is responsible for the congenital variantof Rett syndrome, Am J Hum Genet 83: (1) ((2008) ), 89–93. |
[3] | Kortum F. , et al., The core FOXG1 syndrome phenotype consists of postnatal microcephaly, severe mental retardation, absent language, dyskinesia, and corpus callosum hypogenesis, J Med Genet 48: (6) ((2011) ), 396–406. |
[4] | Mitter D. , et al., FOXG1 syndrome: genotype-phenotype association in83 patients with FOXG1 variants, Genet Med 20: (1) ((2018) ), 98–108. |
[5] | Pringsheim M. , et al., Structural brain anomalies in patients withFOXG1 syndrome and in Foxg1+/- mice, Ann Clin Transl Neurol 6: (4) ((2019) ), 655–668. |
[6] | Yeh F.C. , et al., Population-averaged atlas of the macroscale humanstructural connectome and its network topology, Neuroimage 178: ((2018) ), 57–68. |
Comparison of the core clinical feature of Rett syndrome (RTT), MECP2 Duplication Syndrome (MDS), FOXG1 Disorder (FD, and CDKL5 Deficiency Disorder (CDD) is critical to improve our understanding of the shared and distinct characteristics of these disorders. In this way, outcome measure development for assessing disease-modifying therapies can occur that both address unique but also shared features of these distinct clinical entities.
The three previous discussions dealt with RTT, MDS, and FD. CDD is described here in order to lay the groundwork prior to comparing and contrasting the characteristics of each.
CDD was first described as an infantile epilepsy variant of RTT with neurodevelopmental abnormalities [1]. While individuals with CDD have some features in common with RTT, the disorder was sufficiently distinct to warrant further molecular studies that resulted in identification of pathogenic variants in the CDKL5 gene at Xp22.13 [2]. This finding allowed CDD to be regarded as a unique entity with an incidence of 1 : 41,000 births [3]. Unlike RTT, CDD occurs in both males and females with equal severity. Since 2014, 70 individuals with CDD have been followed in the US Natural History Study (NHS). The molecular genetic analyses of CDKL5 (Fig. 1) indicate that the majority of mutations occur in the catalytic domain as missense mutations [4]. CDKL5 is localized in the nucleus and the dendritic spines of excitatory glutamatergic synapses [5]. It is regarded as a putative nuclear kinase for MeCP2 and DNMT1. As a synaptic kinase, additional substrates are considered to be EB2, NGL-1, and amphiphysin. It stabilizes synapse formation and interacts with PSD-95, collybisin, lsyn1, NGL-1, and actin. Studies in animal models reveal altered behavior, excitatory synapses, and visual function.
Fig. 1
Molecular Genetics of CDKL5 Deficiency Disorder.
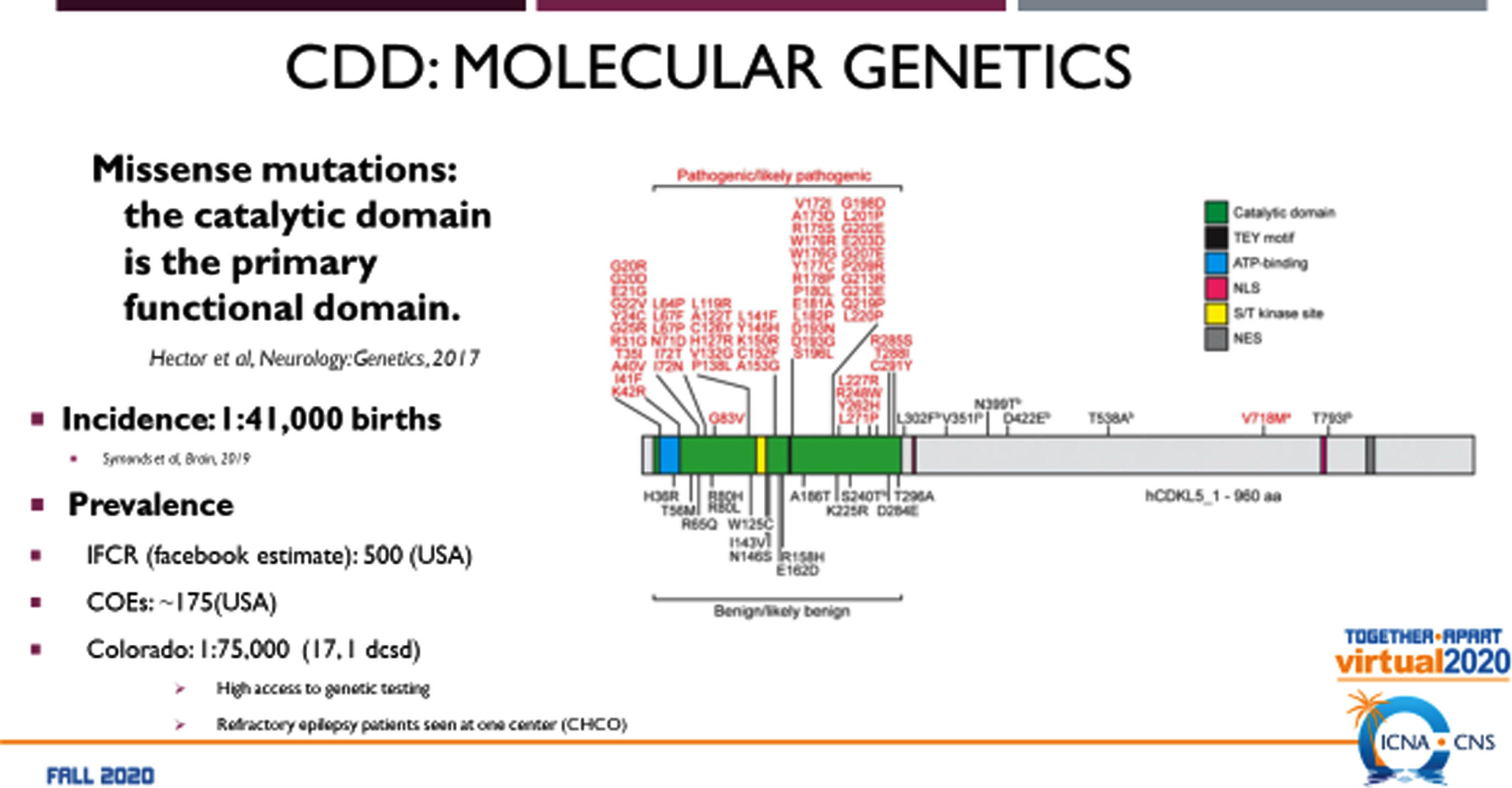
Clinically, CDD is principally recognized as an infantile epileptic encephalopathy with medically refractory tonic seizures occurring as early as 2 weeks of life and infantile spasms without the EEG findings of hypsarrhythmia [6, 7]. Spasms and tonic-spasm sequences become prominent after 12 months and are persistent. A honeymoon period has been noted between ages 1-2 years that may last 12 months. In addition, prominent cortical visual impairment is evident with roving or dysconjugate eye movements and abnormal fixation [8, 9]. Global developmental delay is notable for markedly delayed fine motor development [10, 11]. Unlike RTT, regression is uncommon. Additional features include hypotonia, movement disorders (dyskinesia), sleep disturbances, autonomic abnormalities including circulatory and breathing irregularities, gastrointestinal issues including swallowing dysfunction, gastroesophageal reflux, and constipation, chronic aspiration, and scoliosis. The difficulty swallowing and recurrent aspiration often require placement of a gastrostomy tube. Overall, the spectrum of involvement can be very broad, particularly with walking, functional hand use, and talking [7]. Phenotype-genotype analyses have not revealed different severity by variant group [6] (but see [12]). Epilepsy remains a chronic problem in most and with aging, parkinsonian features emerge, particularly in those who are ambulatory [13–15].
A clinical severity assessment (CDD-SA) has been developed [16, 17]. This assessment requires both parental input about activity in the 30 days prior to the assessment as well as a physician deployed evaluation. The CDD-SA is divided into 7 categories including epilepsy, motor, cognition/behavior/vision/speech, autonomic, overall caregiver impression, therapies, and scoring. This assessment has not yet been evaluated psychometrically to establish its reliability and validity or to conduct a factor analysis as an outcome measure.
1Comparison of developmental encephalopathies
Developmental encephalopathy is defined as a severe, genetically based disorder with postnatal onset of neurologic features including developmental delay, epilepsy, autonomic dysfunction, motor and communication impairments, mood disorders, and systemic symptoms involving the GI tract, scoliosis, and other orthopedic issues [18]. This is contrasted with an epileptic encephalopathy [19] in which neurodevelopmental dysfunction results from ongoing epileptic activity and may or not be reversible even if the epileptic abnormality is stopped [20]. A developmental, epileptic encephalopathy involves a disorder arising early in development that may be made worse by on-going epileptic activity and may not be modified by effective treatment of the epilepsy.
Fig. 2
The RTT-Related Developmental Encephalopathies: RTT. MDS. CDD, and FD.
Fig. 3
Comparison of the RTT-related Developmental Encephalopathies: Participants.
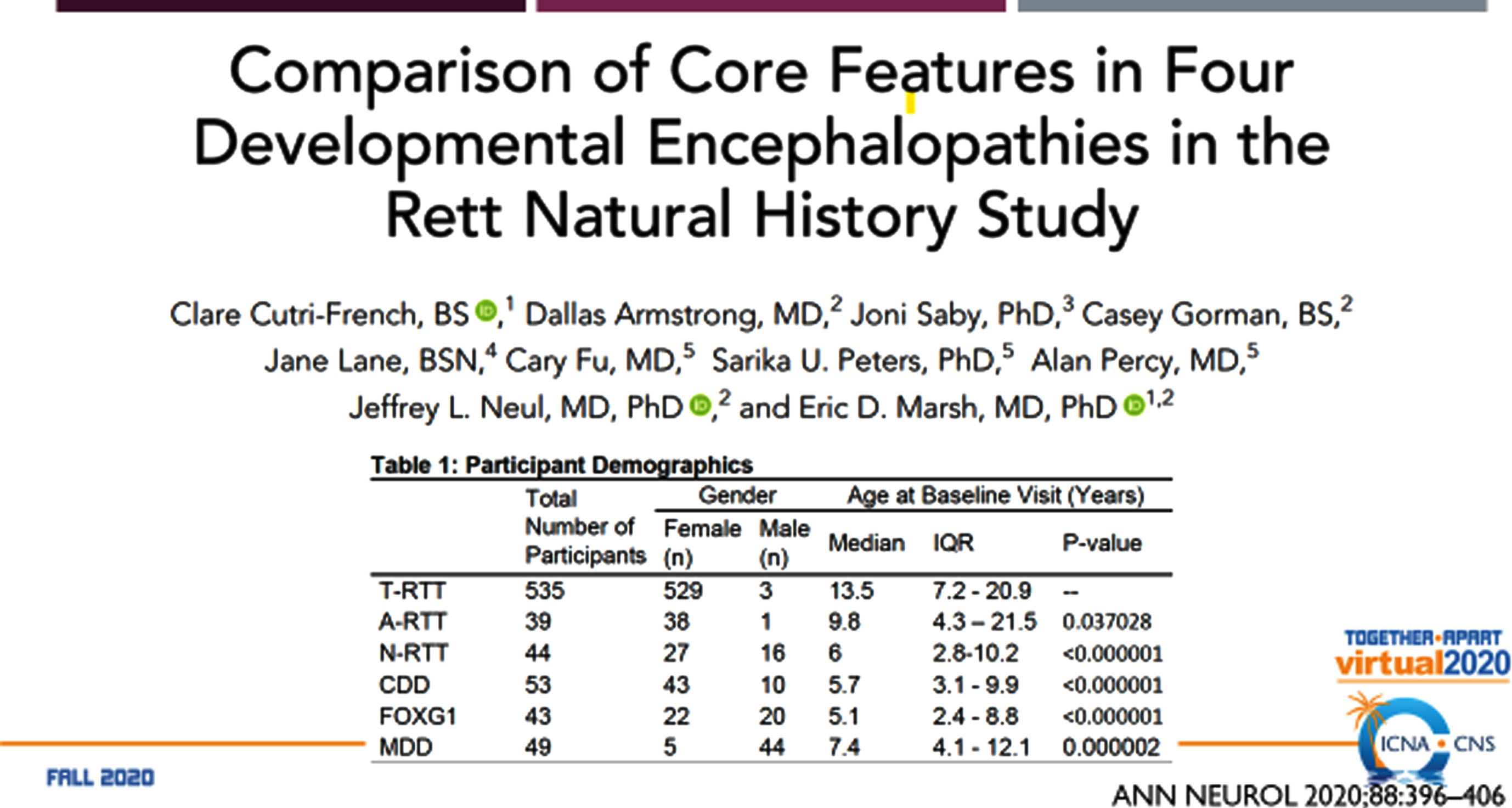
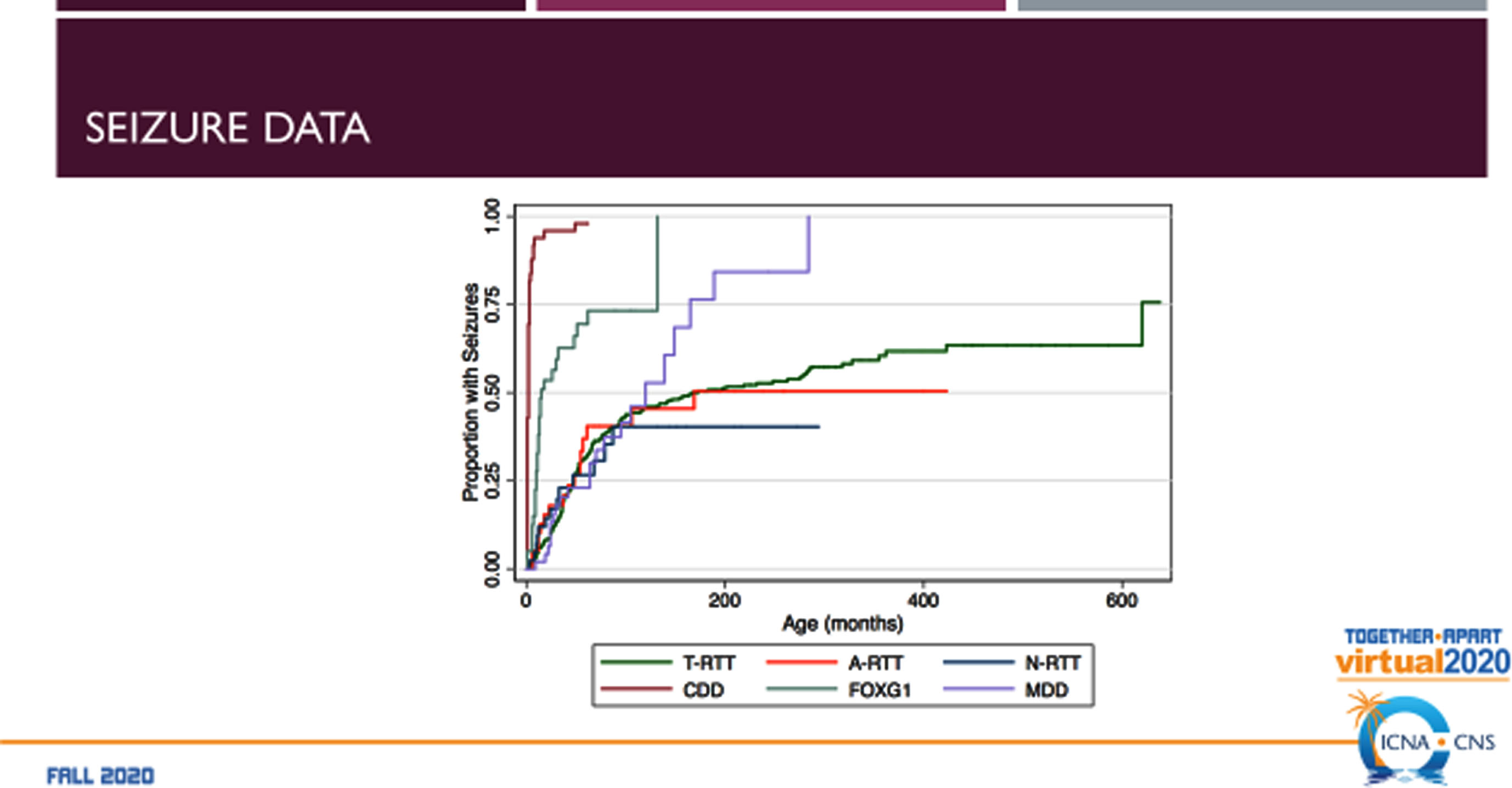
The RTT-related developmental encephalopathies (Fig. 2) are RTT, MDS, CDD (originally early onset seizure variant of RTT, and FD (originally congenital RTT). Comparison of the four developmental encephalopathies was facilitated by the data collection from the US NHS involving 763 individuals (Fig. 3). By collecting the same data elements across these disorders, a direct and meaningful comparison could be performed [21]. The population of the NHS included 535 typical or classic RTT, 39 atypical RTT, 44 with MECP2 mutations lacking features of RTT, 53 CDD, 43 FD, and 39 MDS. Overall severity assessed by the Clinical Severity Scale (CSS) (Fig. 4A) revealed a rank order (greater to lesser severity) of CDD, FD, RTT, atypical RTT and MDS, and N-RTT. Comparing the occurrence of regression in these disorders (Fig. 4B) revealed a quite different pattern with typical RTT (100%), atypical RTT (94.7%), FD (35.7%), CDD (34.0%), Non-RTT (30.2), and MDS (22.5%). Seizures represent yet another variable (Fig. 4C) occurring most prominently in CDD (98.1%) and FD (69.8%) with smaller numbers in typical RTT (47.5%), MDS (44.9%), and atypical RTT (41.0%), and fewer in Non-RTT (29.5%). Early onset seizures were almost exclusively in CDD (88.5%), seen in only 10% of FD, and not at all in typical and atypical RTT and MDS. Average age at onset was also quite distinct with CDD and FD less than one year, two years for Non-RTT, nearly forty months for atypical RTT and nearly 4 years for typical RTT, but greater than 5 years in MDS. Overall occurrence is depicted in Fig. 4C.
Fig. 4A
Comparison of the RTT-related Developmental Encephalopathies: Overall Severity.

Fig. 4B
Comparison of the RTT-related Developmental Encephalopathies: Occurrence of Regression.
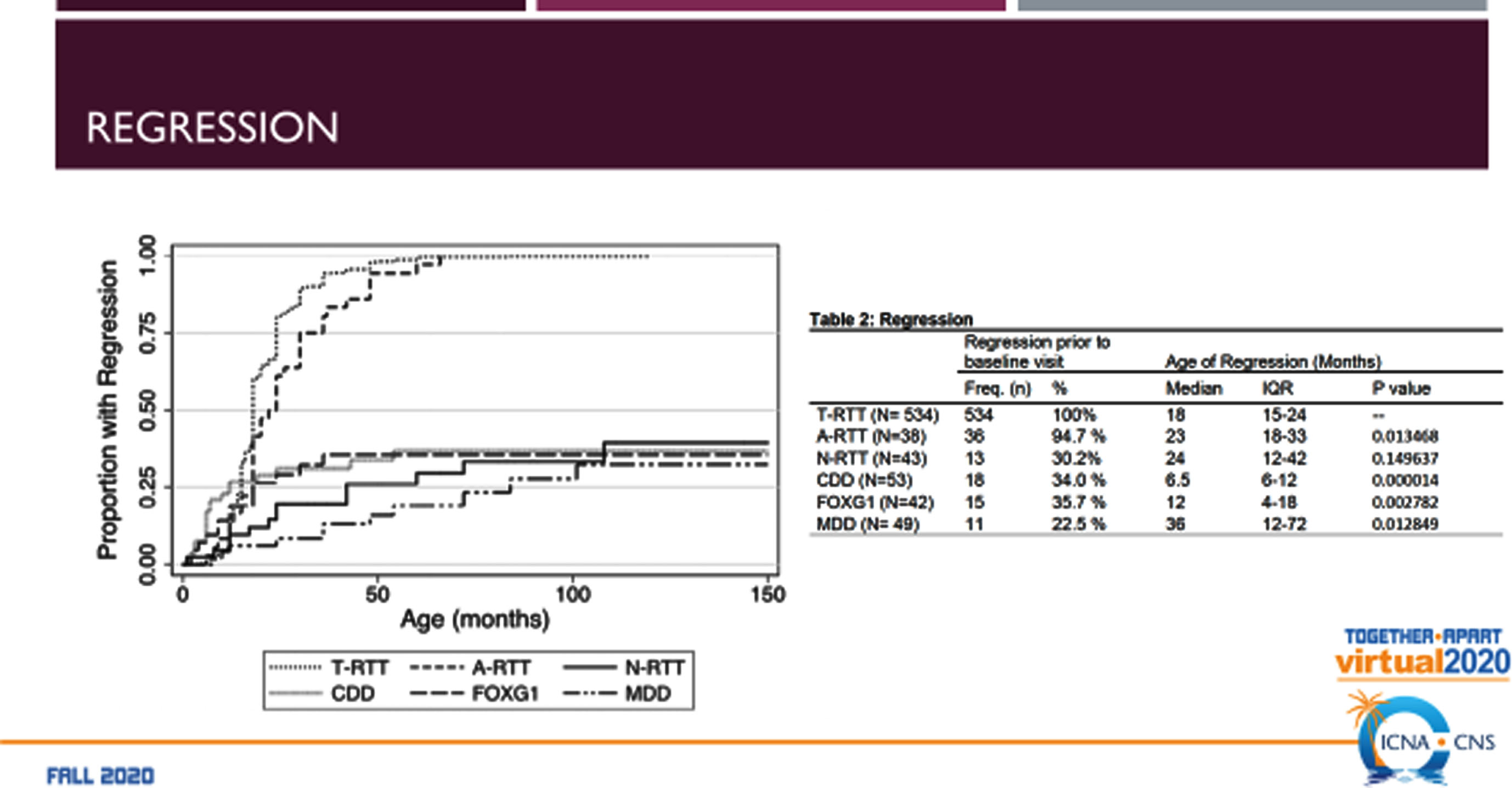
Fig. 4C
Comparison of the RTT-related Developmental Encephalopathies: Seizure Occurrence.
The mean value of head circumference Z-scores was also distinctive being markedly reduced in FD, typical RTT, and CDD and essentially normal in MDS. Comparing the head circumference Z-scores with the CSS across all disorders revealed a significant inverse relationship.
2Clinical trials
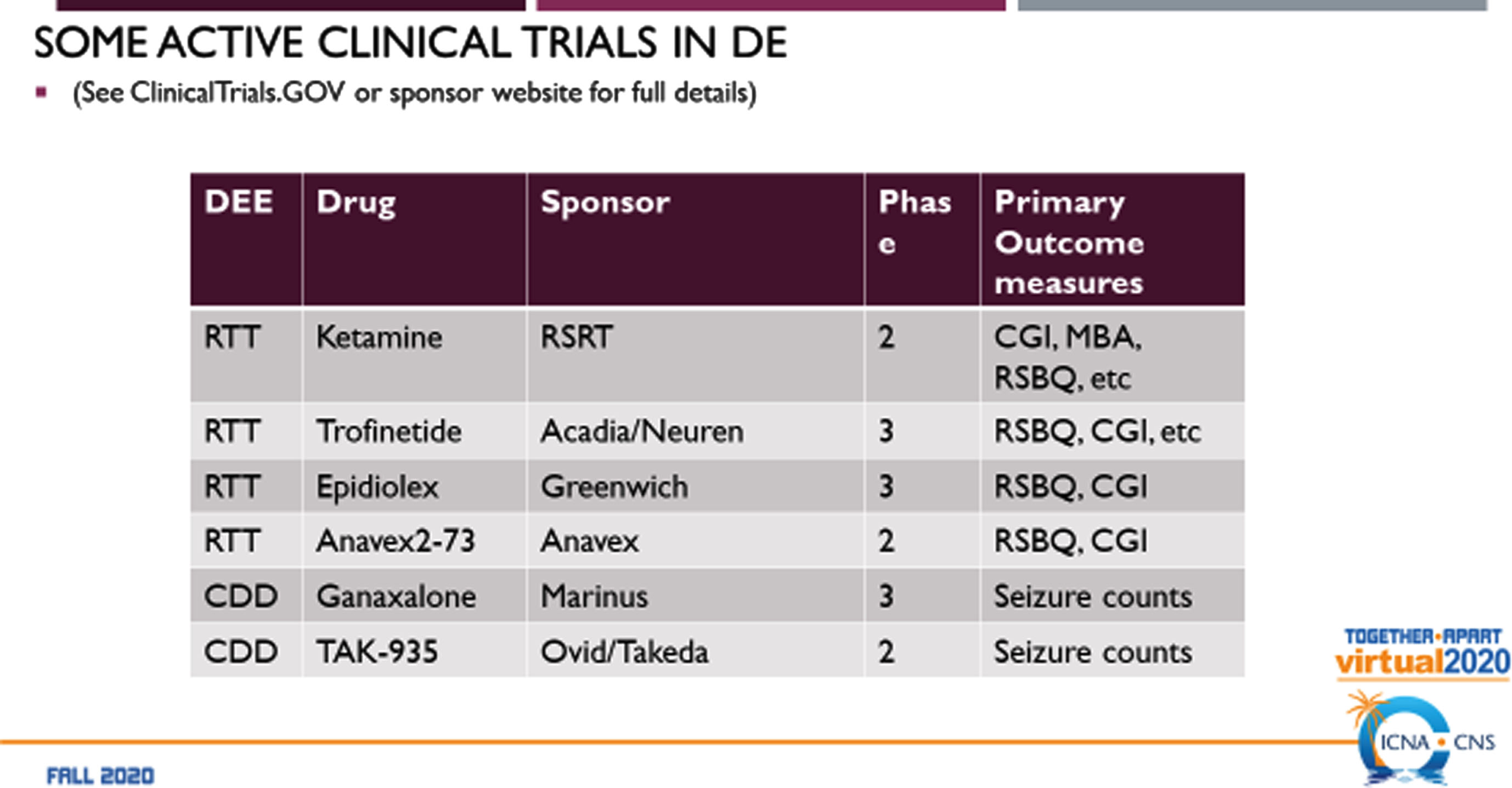
Before clinical trials can be considered, it is critical to align the Natural History data for each disorder and generate outcome measures and biomarkers that can be effective within each disorder. Comparison of these measures across disorders is important as well to understand where convergence exists and to separate out those that are divergent. Examples of some on-going clinical trials in RTT and CDD demonstrate the distinct differences in outcome measures (Fig. 5). For RTT, the measures of Global Impression-Improvement and the Rett Syndrome Behavioral Quotient are accepted by the FDA in each trial. For CDD, similar measures are not used, instead utilizing seizure counts as a measure of efficacy as done recently for the first FDA approved drug for CDD, ganaxolone [22]. Clearly, much work remains to establish effective outcome measures and biomarkers [23] for the developmental encephalopathies in order to demonstrate the effectiveness of potential disease-modifying therapies [15].
Fig. 5
Current clinical trials in Neurodevelopmental Encephalopathies.
Competing interests
Tim A. Benke: Consultancy for AveXis, Ovid, GW Pharmaceuticals, International Rett Syndrome Foundation, Takeda, Neurogene, Ultragenyx, Zogenix/UCB, GrinTherapeutics, Alcyone and Marinus; Clinical Trials with Acadia, Ovid, GW Pharmaceuticals, Marinus and RSRT; all remuneration has been made to his department.
Eric Marsh: Consultancy for Stoke therapeutics, Cipla pharmaceuticals. Clinical trials with Acadia, GW pharma, Marinus, RSRT, Biopharm, Stoke therapeutics, Zogenix Pharmaceuticals.
Author contributions
Dr. Marsh and Dr. Benke are solely responsible for this review.
References
[1] | Hanefeld F. , The clinical pattern of the Rett syndrome, Brain Dev. 7: (3) ((1985) ), 320–5. |
[2] | Weaving L.S. , Christodoulou J. , Williamson S.L. , Friend K.L. , McKenzie O.L. , Archer H. , et al. Mutations of CDKL5 cause a severeneurodevelopmental disorder with infantile spasms and mentalretardation, AmJ HumGenet. 75: (6) ((2004) ), 1079–93. |
[3] | Symonds J.D. , Zuberi S.M. , Stewart K. , McLellan A. , O’Regan M. , MacLeod S. , et al. Incidence and phenotypes of childhood-onset geneticepilepsies: a prospective population-based national cohort, Brain. 142: (8) ((2019) ), 2303–18. |
[4] | Hector R.D. , Kalscheuer V.M. , Hennig F. , Leonard H. , Downs J. , Clarke A. , et al. CDKL5 variants: Improving our understanding of a rareneurologic disorder, Neurol Genet. 3: (6) ((2017) ), e200. |
[5] | Zhu Y.C. and Xiong Z.Q. , Molecular and synaptic bases of CDKL5 disorder, Dev Neurobiol. (2018). |
[6] | Demarest S.T. , Olson H.E. , Moss A. , Pestana-Knight E. , Zhang X. , Parikh S. , et al. CDKL5 deficiency disorder: Relationship between genotype, epilepsy, cortical visual impairment, and development, Epilepsia. (2019). |
[7] | Olson H.E. , Demarest S.T. , Pestana-Knight E.M. , Swanson L.C. , Iqbal S. , Lal D. , et al. Cyclin-Dependent Kinase-Like 5 DeficiencyDisorder: Clinical Review, Pediatr Neurol. 97: ((2019) ), 18–25. |
[8] | Brock D. , Fidell A. , Thomas J. , Juarez-Colunga E. , Benke T.A. and Demarest S. , Cerebral Visual Impairment in CDKL5 Deficiency DisorderCorrelates With Developmental Achievement, J Child Neurol. 36: (11) ((2021) ), 974–80. |
[9] | Olson H.E. , Costantini J.G. , Swanson L.C. , Kaufmann W.E. , Benke T.A. , Fulton A.B. , et al. Cerebral visual impairment in CDKL5 deficiency disorder: vision as an outcome measure. Dev Med Child Neurol. (2021). |
[10] | Fehr S. , Leonard H. , Ho G. , Williams S. , de Klerk N. , Forbes D. , et al. There is variability in the attainment of developmentalmilestones in the CDKL5 disorder, J Neurodev Disord. 7: (1) ((2015) ), 2. |
[11] | Fehr S. , Downs J. , Ho G. , de Klerk N. , Forbes D. , Christodoulou J. , et al. Functional abilities in children and adults with the CDKL5disorder, Am J Med Genet A. 170: (11) ((2016) ), 2860–9. |
[12] | Wong K. , Junaid M. , Demarest S. , Saldaris J. , Benke T.A. , Marsh E.D. , et al. Factors influencing the attainment of major motor milestones in CDKL5 deficiency disorder, Eur J Hum Genet. (2022). |
[13] | Hong W. , Haviland I. , Pestana-Knight E. , Weisenberg J.L. , Demarest S. , Marsh E.D. , et al. CDKL5 Deficiency Disorder-Related Epilepsy: A Review of Current and Emerging Treatment, CNS Drugs. 36: (6) ((2022) ), 591–604. |
[14] | Olson H.E. , Daniels C.I. , Haviland I. , Swanson L.C. , Greene C.A. , Denny A.M.M. , et al. Current neurologic treatment and emergingtherapies in CDKL5 deficiency disorder, J Neurodev Disord. 13: (1) ((2021) ), 40. |
[15] | Leonard H. , Downs J. , Benke T.A. , Swanson L. , Olson H. and Demarest S. , CDKL5 deficiency disorder: clinical features, diagnosis,and management, Lancet Neurol. 21: (6) ((2022) ), 563–76. |
[16] | Demarest S. , Pestana-Knight E.M. , Olson H.E. , Downs J. , Marsh E.D. , Kaufmann W.E. , et al. Severity Assessment in CDKL5 DeficiencyDisorder, Pediatr Neurol. 97: ((2019) ), 38–42. |
[17] | Saldaris J. , Weisenberg J. , Pestana-Knight E. , Marsh E.D. , Suter B. , Rajaraman R. , et al. Content validation of clinician-reported itemsfor a severity measure for CDKL5 deficiency disorder, J ChildNeurol. 36: (11) ((2021) ), 998–1006. |
[18] | Paciorkowski A.R. , Seltzer L.E. and Neul J.L. , Developmental Encephalopathies. In: Swaiman KF, Ashwal S, Ferriero DM, Schor NF, Finkel RS, Gropman AL, et al., editors. Swaiman’s Pediatric Neurology. 6 ed. Philadelphia: Mosby (2018), 242–8. |
[19] | Scheffer I.E. , Berkovic S. , Capovilla G. , Connolly M.B. , French J. , Guilhoto L. , et al. ILAE classification of the epilepsies: Positionpaper of the ILAE Commission for Classification and Terminology, Epilepsia. 58: (4) ((2017) ), 512–21. |
[20] | Kalser J. and Cross J.H. , The epileptic encephalopathy jungle –from Dr West to the concepts of aetiology-related and developmental encephalopathies, Curr Opin Neurol. 31: (2) ((2018) ), 216–22. |
[21] | Cutri-French C. , Armstrong D. , Saby J. , Gorman C. , Lane J. , Fuet C. , et al., Comparison of core features in four developmental encephalopathies in the rett natural history study, Ann Neurol. 88: (2) ((2020) ), 396–406. |
[22] | Knight E.M.P. , Amin S. , Bahi-Buisson N. , Benke T.A. , Cross J.H. , Demarest S.T. , et al. Safety and efficacy of ganaxolone in patients with CDKL5 deficiency disorder: results from the double-blind phaseof a randomised, placebo-controlled, phase 3 trial, Lancet Neurol. 21: (5) ((2022) ), 417–27. |
[23] | Saby J.N. , Mulcahey P.J. , Zavez A.E. , Peters S.U. , Standridge S.M. , Swanson L.C. , et al. Electrophysiological biomarkers of brainfunction in CDKL5 deficiency disorder, Brain Commun. 4: (4) ((2022) ), fcac197. |


