
Disorders of phenylalanine and tyrosine metabolism
Abstract
This article provides a review of the inborn errors of phenylalanine and tyrosine metabolism including the diagnostic approach, dietary and pharmalogical management and emerging therapies. Hyperphenylalaninaemia results mainly from defects in either phenylalanine hydroxylase (PAH) (resulting in phenylketonuria (PKU)) or the production or recycling of tetrahydrobiopterin (BH4). Untreated PKU results in irreversible neurocognitive impairment. Five inherited disorders of tyrosine metabolism are known, which include tyrosinemia type I, type II, type III, hawkinsinuria and alkaptonuria. Newborn screening for these disorders has enabled their early detection and decreased the associated morbidity and mortality.
PHENYLKETONURIA
Phenylketonuria (PKU) is an autosomal recessive disorder resulting from a deficiency of phenylalanine hydroxylase (PAH), which converts phenylalanine (Phe), an essential amino acid, to tyrosine (Fig. 1) [1]. PAH is one of the three aromatic amino acid hydroxylases that utilizes tetrahydrobiopterin (BH4) as a cofactor. PKU occurs in all ethnic groups, although it is more common in individuals of Northern European and Native American ancestry. The incidence is lower in those of African American, Hispanic and Asian ancestry. The average incidence is about 1 in 10,000 in the United States and Europe, and it is even greater in other populations (∼1 : 2600 in Turkey and ∼1 : 4500 in Ireland) [2, 3].
Fig.1
Metabolic Pathway of Phenylalanine.
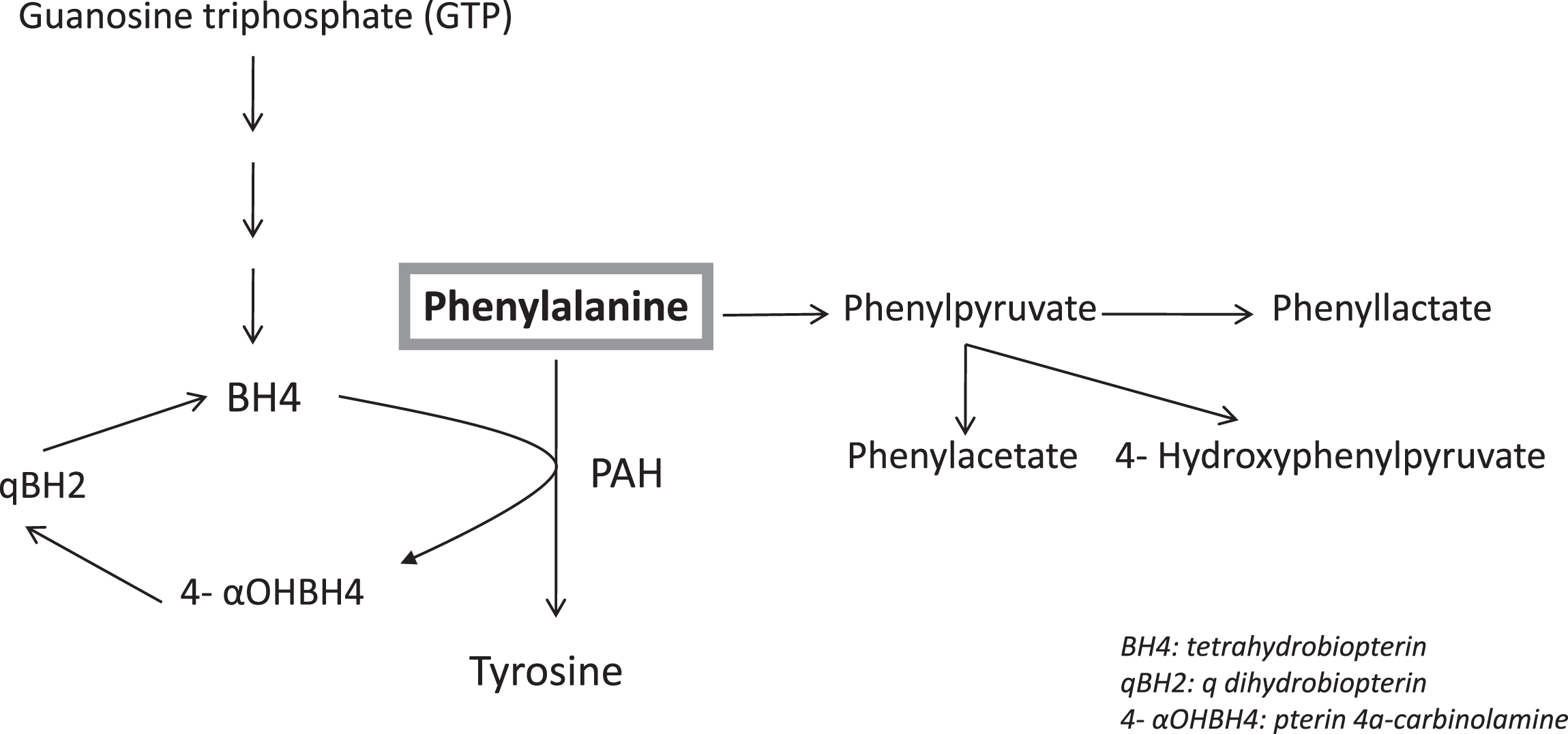
PKU is due to total or near total deficiency of PAH activity. Normally, PAH activity is detectable in the liver as early as the eighth week of fetal life, and it reaches adult levels by the second trimester [4–7]. The inability of the body to break down phenylalanine results in its accumulation as well as the accumulation of alternate pathway products including phenylacetate, phenyllactate, and phenylpyruvate.
PKU Classification
The clinical classification between its three subtypes is arbitrary and based on Phe levels on a normal diet and/or phenylalanine tolerance in diet (Table 1). Classical PKU: It is the most severe type, and phenylalanine levels are more than 1200 μmol/L (mean normal: 60 μmol/L) on a normal diet. These patients can tolerate less than 250–300 mg of dietary Phe per day. Moderate/Mild PKU: Phenylalanine levels are above 600 μmol/L but less than 1200 μmol/L on a normal diet. These patients can tolerate 350–600 mg of dietary Phe per day. Mild hyperphenylalaninemia: Phenylalanine levels ranges from 360–600 μmol/L on a normal diet.
Table 1
Inherited disorder of phenylalanine metabolism
| Enzyme defects | Tissue distribution | Chromosomal localization | Disorder | Characteristic | Treatment |
| PAH deficiency | Liver, kidney | 12q22–q24.1 | Classical PKU | Plasma phenylalanine > 1200μmol/L | Phenylalanine restricted diet |
| Moderate/Mild PKU | Plasma phenylalanine > 600 and < 1200μmol/L | Phenylalanine restricted diet | |||
| Mild hyperphenylalaninemia | Plasma phenylalanine < 600μmol/L | Regular diet* | |||
| GTP cyclohydrolase I (GTPCH) deficiency | Liver, brain, kidney, lymphocytes | 14q22.1–q22.2 | Tetrahydrobiopterin Deficiency | dopa-responsive dystonia | Levodopa/DCI |
| Autosomal dominant form | |||||
| Tetrahydrobiopterin Deficiency | Seizures, hypotonia, tremor, developmental delay, hyperphenylalaninemia, very low neopterin and biopterin | Low-phe diet, BH4, L-dopa/carbidopa,5-OH-tyrptophan, folinic acid | |||
| Autosomal recessive form | |||||
| 6-Pyruvoyl-tetrahydropterin synthase deficiency | Liver, brain, kidney, lymphocytes, erythrocytes, fibroblasts | 11q22.3–q23.3 | Tetrahydrobiopterin Deficiency | Mental retardation, microcephaly, seizures, irritability, abnormal movements, hyperthermia, hyperphenylalaninemia, high neopterin, trace biopterin | Low-phe diet, BH4, L-dopa/carbidopa,5-OH-tyrptophan, folinic acid |
| Sepiapterin reductase (SR) deficiency | All tissues | 2p14–p22 | Tetrahydrobiopterin Deficiency | dopa-responsive dystonia, sepiapterin is present in urine, CSF | L-dopa/carbidopa,5-OH-tyrptophan, folinic acid |
| Dihydropteridine reductase (DHPR) deficiency | All tissues | 4p15.3 | Tetrahydrobiopterin Deficiency | Microcephaly, hypotonia, mental retardation, seizures, hyperphenylalaninemia, normal/high neopterin, very high biopterin | Low-phe diet, BH4, L-dopa/carbidopa,5-OH-tyrptophan, folinic acid |
| Pterin-4-carbinolamine-dehydratase deficiency | Liver, kidney, intestine | 10q22 | Tetrahydrobiopterin Deficiency | Hyperphenylalaninemia, mild tremor, neonatal hypotonia, high neopterin, subnormal biopterin, Primapterin is present | Low-phe diet, BH4 |
History
Dr. A. Foelling, a Norwegian physician discovered PKU in 1934 [8]. He added ferric chloride to the urine of two children with intellectual disability and observed the deep green color change in their urine sample. He then used chemical analysis to determine the nature of the substance that caused that deep green color change and extracted phenylpyruvic acid from their urine. About 20 years later, a German professor, Horst Bickel, developed the first Phenylalanine- free formula that could decrease blood phenylalanine levels and prevent intellectual disability [9]. In 1962, Robert Guthrie developed a bacterial inhibition assay to measure phenylalanine level on a filter paper [10]. The test was fast, simple and inexpensive, and had acceptable specificity and sensitivity. Newborn screening for PKU was first started in Massachusetts in 1963. It was very successful, and many other states mandated the screening by the mid-1960s in the USA. Today, PKU is screened in all states of the USA and many other countries in all over the world allowing early diagnosis and better health outcome as well as resulting in less economic burden on the health system.
Diagnosis
Newborn screening
Phenylalanine levels are measured with tandem mass spectrometry (MS/MS) using dried blood spots in all newborns in US and many developed countries as early as 24 hours of life. Many laboratories also calculate and report the phenylalanine and tyrosine ratio to increase the specificity of the screening test. Newborns with Phe levels higher than 2 mg/dL (120 μmol/L) are reported as a positive screen. Elevated phenylalanine level can be caused not only by PKU but also defects of tetrahydrobiopterin, prematurity, liver disease and high protein intake. Elevated phenylalanine (Phe):tyrosine (Tyr) ratio over 2.5 mg/dL increases the likelihood of PKU [11]. A mean cutoff for Phe of 130 μmol/L (with a range of 65–234 μmol/L) and a Phe:Tyr ratio > 3 is abnormal [12]. A metabolic physician should see newborns with elevated phenylalanine levels as soon as possible. Blood amino acid quantification should be performed as a diagnostic follow-up of positive newborn screen. Elevated phenylalanine level and normal levels of other amino acids in plasma amino acid (PAA) analysis confirm the diagnosis of PKU. Further guidance on follow-up of an abnormal NBS test for Phe can be found in the ACMG ACT sheets for newborns along with confirmatory testing algorithms (www.acmg.net).
PAH gene sequencing is a clinically available test and also confirms the diagnosis of PKU.
Cofactor deficiency testing
It is essential to measure urine pterins and dihydropteridine reductase (DHPR) enzyme activity on a filter paper to rule out defect of tetrahydrobiopterin (BH4) synthesis and regeneration in all newborns with elevated Phe. If that will result in delay of diagnosis, then a 24-hour BH4 loading test can be performed in addition to the urine pterins and DHPR enzyme activity. The blood and urine samples should be collected before BH4 loading dose.
PAH enzyme activity
PAH enzymatic activity is detectable in hepatic and renal tissues only and is not appropriate for either screening or diagnostic testing. The diagnosis of PKU is established when the Phe concentration is above 120 μmol/L with altered Phe to tyrosine ratio and normal BH4 cofactor metabolism and/or finding biallelic pathogenic variants in PAH.
Genetics
The human PAH gene was mapped on chromosome 12 q22–q24.1 and contains 13 exons and 12 introns (Table 1). About 1,184 PAH variants (PAHvdb database; http://www.biopku.org; last accessed 06/22/2020) are known to be associated with PAH deficiency. The majority of the reported variants (60%) are missense, but deletions, duplications, and insertions are also observed. Missense variants usually result in abnormal folding of the PAH protein, increased protein turnover, decreased activity and/or impairment of catalytic functions. The genotype is clearly the best clinically available predictor of severity in PAH deficiency.
Genotyping patients with elevated Phe is not essential for the diagnosis of PKU but it may determine the degree of protein dysfunction, residual PAH activity and consequently the metabolic phenotype and therapy planning. Genotyping would help in determining the extent of dietary Phe restriction and the likelihood of response to BH4 supplementation. Alleles that are known to be responsive to treatment with BH4 are listed in the BIOPKU database. http://www.biopku.org/home/biopku.asp.
p.Arg408Trp (c.1222 C > T) in exon 12 is the most common pathogenic variant in Europe [13]. IVS12 + 1G>A (c.1315 + 1G>A) at the splice donor site of intron 12 is a common pathogenic variant in northern European ancestry. IVS10–11G>A is a splice site of intron 9, is the second most frequently reported pathogenic variant in PAHvdb and prevalent in southern Europe/Mediterranean populations and associated with the most severe phenotype [14].
While large numbers of variants have been reported as BH4 responsive, there is inconsistency in BH4 responsiveness in patients with the same variants [15].
Genetic counseling
PAH deficiency is inherited in an autosomal-recessive manner; in which the affected individual will inherit two copies of the pathogenic variants in the PAH gene, one from each parent. At each pregnancy, there is a 25% chance of having an affected child, a 50% of asymptomatic carrier, and a 25% chance of having unaffected and not a carrier child. Carrier testing for at-risk relatives and prenatal testing for pregnancies at increased risk are available if the PAH pathogenic variants have been identified in an affected family member.
Pathology
In morphologic descriptions of untreated PKU, both gray and white matter changes are noted [16, 17]. Minor architectural abnormalities of the cortex are described, such as lobar disproportion and gyral convolutional irregularities. The major changes are in the white matter, where vacuolation is seen in the central nervous system (CNS) of children and demyelination and astrogliosis in the CNS of adults. Vacuolation consistently involves the central white matter of the cerebrum and cerebellum, optic tracts and fornix. Demyelination follows the same pattern with a tendency for preservation of articulate fibers. Intense astrogliosis and sudanophylic and periodic acid-Schiff (PAS) - positive macrophages are also evident. One ultra-structural study describes membrane bound; parallel lamellar inclusions believed to be within oligodendrocytes [18]. Lipids and proteolipids associated with myelin are decreased [19]. A review of histopathology and neuroimaging studies shows that diffuse white matter pathology in untreated PKU patients likely reflects hypomyelination, while in early treated patients white matter abnormalities observed on magnetic resonance imaging (MRI) likely reflects intramyelinic edema [13].
The relationship between intellectual disability and morphologic and biochemical alterations is not clear. Some data points to impaired protein synthesis, perhaps because high blood levels of phenylalanine inhibit cerebral uptake of amino acids that share the same transporter with phenylalanine. Other data suggest a deficiency of brain dopamine, since this neurotransmitter forms from tyrosine that, in turn, derives from phenylalanine in the reaction (phenylalanine hydroxylase) that is defective in PKU [20, 21].
Evidence also indicates altered energy metabolism in the brain of the child with PKU [22–24]. Phenylalanine and its metabolites appear to impact several aspects of brain energetics including: (a) inhibition of glucose uptake; (b) diminished glycosylation of cytoskeletal proteins; (c) inhibition of hexokinase and pyruvate kinase; (d) reduced flux through the glycolysis and (e) inhibition of mitochondrial electron transport chain.
Clinical findings
PKU can cause severe intellectual disability that is preventable by the institution of a protein-restricted diet in early infancy [1, 25, 26]. Patients with untreated PKU appear normal in the first few months of life and then it is noticed that they fail to achieve developmental milestones in time and their head circumference does not grow [1]. Most untreated patients come to medical attention after 6–9 months of life because of developmental delay, musty body odor, microcephaly and seizures. Their complexion is usually much lighter than their parents and siblings secondary to the associated inhibition of tyrosinase and low tyrosine levels. Eczema occurs in about 30% of patients. In older untreated children, severe intellectual disability, epilepsy, aggressive behavior, self-mutilation, autistic features, schizophrenia-like symptoms, Parkinson-like features are common [1] (Table 2).
Table 2
Symptoms of untreated Classic PKU
| Symptoms |
| Odor (urine and body) |
| Lighter pigmentation in family constellation |
| Intellectual disability |
| Microcephaly |
| Irritability |
| Seizures |
| Hypertonia |
| Autism |
| Self mutilation |
| Rash, eczema |
Fortunately, early diagnosis and treatment through newborn screening has changed the clinical phenotype of PKU in many developed countries, allowing them to grow up with normal intelligence, however, lower than that in sib controls [27–31]. Implementation of a Phe-restricted diet shortly after birth significantly reduces blood Phe levels and prevents many of the neurologic manifestations of PKU [1, 25]. Lifelong adherence to a low phenylalanine diet is extremely difficult [32, 33]. If patients maintain good control as children and go off diet as teenagers or adults, significant difficulties with executive function concentration, attention deficit, emotional lability, anxiety and depression may occur [27, 29, 34]. Executive dysfunction may also occur in early treated children in spite of diet treatment [27, 34]. The severity of psychiatric symptoms correlates with elevation in blood Phe level, and reduction of Phe level generally results in symptom improvement. Given the increased risk for neurocognitive and psychological issues in patients with PAH deficiency, regular mental health monitoring is warranted [25].
It is important to closely monitor the bone health of individuals with PAH deficiency given the risk of osteopenia and low Z-scores [35].
Differential diagnosis
The differential diagnosis of hyperphenylalaninemia (HPA) includes high natural protein intake, prematurity, defects in BH4 metabolism and liver disease. Patients with disorders of BH4 metabolism including GTP cyclohydrolyase I (GTPCH) deficiency, 6-pyruvoyl-tetrahydropterin synthase (PTPS) deficiency, dihydropteridine reductase (DHPR) deficiency and pterin-4a-carbinolamine dehydratase (PCD) deficiency can present with any degree of HPA [36, 37].
Treatment
Dietary therapy
Treatment should be started immediately upon diagnosis to prevent neurological damage and it must be life long, with a goal of maintaining Phe in the range of 120–360 μmol/L. It was found that a 4-week delay in starting treatment caused a decline in IQ score by 4 points [38]. Based on the initial blood Phe levels, Phe may be excluded from the diet until levels approach the treatment range followed by initiation and titration of a Phe-restricted diet. The mainstay of treatment of PKU is a low–phenylalanine diet. The general consensus is to start diet treatment immediately in patients with Phe levels above 600 μmol/L [25]. Patients with blood Phe level <360 μmol/L should remain untreated, however, they will need to be monitored closely, especially in the first 2 years of life as there is a possibility of increase in the blood Phe level with age with protein intake [39, 40]. If treatment is not required before 2 years of age, then those patients need to be monitored on an annual or biennial basis.
Patients with Phe level between 360 and 600 μmol/L should be treated at least during the first 12 years of life to prevent cognitive impairment [41]. This is particularly important in women given the risks associated with maternal PKU and Phe level >360 μmol/L. However, there is no clear evidence of treating patients with Phe level between 360 and 600 μmol/L as previous studies showed mixed clinical outcomes of untreated patients ranging from normal cognition to subtle neurocognitive deficits [42–45].
Most physicians also advocate phenylalanine-restricted diet in patients with mild hyperphenylalaninemia whose levels are persistently above 360 μmol/L. The basic principle of the diet is to restrict the intake of phenylalanine while providing phenylalanine-free protein. This is achieved by replacing a certain quantity of phenylalanine containing foods (the amount replaced is adjusted according to each patient’s tolerance) with synthetic, phenylalanine – free formula and medical food to provide all other nutrients recommended for the patient’s age and necessary for normal growth and health maintenance [46]. A wide range of Phe-free formulas and low protein medical foods such as low protein-pastas and bread are available today to improve the compliance with the dietary Phe-restriction.
At the start of diet treatment, patients with phenylalanine levels above 800 μmol/L require phenylalanine free formula for 12–48 hours to bring their phenylalanine levels close to 360–420 μmol/L. To achieve smooth control, phenylalanine should be added once the phenylalanine level approaches the upper end of therapeutic range (about 400 μmol/L). Small changes in dietary phenylalanine allowance should be made based on blood phenylalanine levels that are monitored closely, ideally, every day in the first couple of days of treatment [46, 47]. It is important to have consistent testing methodology for better comparison over time. Long-term management, serial monitoring of Phe levels is essential to achieve good metabolic control. Although current best practices concerning the frequency of monitoring varies from one center to another, it is usually recommended once a week in the first 2 years, every other week by school age, once a month after 7 years of age [25].
In the early years of life, especially during rapid growth, infants with classical PKU may tolerate 300–400 mg/day phenylalanine; during linear growth, such children usually tolerate 200 mg/day Phe to keep their Phe levels within therapeutic range. There is no consensus regarding optimal levels of blood Phe across different countries and among treatment centers in a single country. In the USA, the most commonly advocated therapeutic range is 120 to 360 μmol/L (2 to 6 mg/dL) in the first 12 years and then 120–600 μmol/L (2–10 mg/dL) [25].
Dieticians should carefully follow patients and their 3-day-diet records should be reviewed regularly to prevent nutritional deficiencies. For further detailed dietary management of PAH deficiency, please refer to the nutrition guidelines by the Genetic Metabolic Dietitians International (www.gmdi.org) and Southeast Regional Genetics Network (www.southeastgenetics.org). Based on nutritional assessment and concern of suboptimal dietary intake, additional monitoring blood work should include PAA, transthyretin, albumin, complete blood count, ferritin, 25-OH vitamin D, vitamin B12, red blood cell essential fatty acids, trace minerals (zinc, copper, and selenium), vitamin A, comprehensive metabolic panel, and folic acid. Additionally, bone health should be considered given the suboptimal bone mineralization secondary to the low intake of calcium from the protein-restricted diet. However, the utility of routine DEXA scans to monitor bone density is controversial [35]. Additionally, adolescent with PKU are at risk of vitamin B12 deficiency if they are less adherent to their diet and amino acid supplementation and continue to choose low protein diet [48].
Although it is very well known that PKU patients should maintain a low protein diet for life, several factors can make it difficult to follow: many dislike the taste of the low-Phe medical food; it is costly and sometimes not covered by health insurance; many feel strong peer pressure to eat a normal diet [47].
Pharmacotherapy
Large neutral amino acids
Administration of large neutral amino acids (LNAA) has been introduced as another approach to diet therapy. LNAAs (tyrosine, tryptophan, arginine, leucine, isoleucine, valine, methionine, histidine, lysine, threonine and phenylalanine) use the same large neutral amino acid type 1(LAT-1) transporter to cross the intestinal cell membrane and blood brain barrier (BBB) [49, 50]. Binding of LNAA to the transporter is a competitive process. LAT- 1 transporter has different affinities and Km values for each LNAA, and phenylalanine has the lowest Km value indicating that it binds to the transporter more strongly than other LNAAs. In PKU, the phenylalanine level is much higher than that of other LNAAs, and phenylalanine readily crosses intestinal cell membrane and blood brain barrier and inhibits the transport of other LNAAs. The rationale for use of LNAA is that these molecules compete with phenylalanine for transporters across the BBB; therefore, large concentrations of other LNAAs in the blood reduce the uptake of phenylalanine into the brain. In addition to LNAAs blocking the influx of excess phenylalanine into the brain in PKU, LNAA may also promote protein synthesis [51].
The brain concentration of phenylalanine decreases with LNAA tablets despite increased natural protein intake [51]. Pietz at al reported that the LNAA blocked the phenylalanine transport to the brain by giving an oral phenylalanine challenge with or without LNAA and measuring the influx of phenylalanine into the brain [52]. Matalon et al. in 2006, showed about 40% reduction of blood Phe following substitution of a standard low Phe medical food with one supplemented with LNAA at a dose of 0.5 or 1.0 g/kg of body weight [53]. The use of the LNAA therapy has been shown to improve amino acids profiles as well as increase tyrosine and tryptophan levels in the blood, which are precursors for dopamine and serotonin [54].
LNAA are not offered to children or pregnant women due to a limited understanding of their effects on fetal growth and the developing central nervous system of the fetus and children but are considered appropriate for treating adults over 18 years of age, adolescents with poor dietary adherence and those with late diagnosed/treated PKUs. It is recommended that LNAA comprise 25–30% of a patient’s daily protein requirement and the remaining 70–75% comes from natural proteins [54].
Glycomacropeptide
Glycomacropeptide (GMP), a natural by-product of cheese that has very low Phe content, has been used in several medical foods as another approach of improving the nutritional management of PKU [55]. GMP is the only known dietary protein that is naturally free of phenylalanine in its pure form. GMP can be used as an alternative to amino acid formula as a source of low-phenylalanine protein for the PKU diet. However, there are some drawbacks to GMP. Isolation of GMP from cheese whey results in contamination with other whey proteins that contain phenylalanine and other amino acids not found in pure GMP. Commercially available GMP contains 2.5–5 mg phenylalanine/g of protein. GMP requires supplementation with amino acids to provide complete source of protein for PKU patients [56].
Sapropterin
In light of the challenges that many patients face in complying with the ongoing demands of a phenylalanine-restricted diet, a prescription medication that would permit better control of phenylalanine without the need for severe dietary restriction would significantly contribute to the management of PKU. Accordingly, researchers have sought alternate approaches to therapy. One strategy is the use of tetrahydrobiopterin (BH4) to enhance activity of pterin-dependent amino acid hydroxylases, one of which is phenylalanine hydroxylase, the others being tyrosine hydroxylase, and tryptophan hydroxylase. Each enzyme catalyzes the hydroxylation of the aromatic side chain of its respective amino acid substrate using molecular oxygen and a BH4 as substrates.
In 1975, Milstien and Kaufman suggested that BH4 might stimulate PAH in patients with residual activity [57]. Kure et al. confirmed this hypothesis in 4/5 patients with mild PKU in whom BH4 loading reduced blood phenylalanine by 40–70% while patients consumed a normal diet [58]. Since then, numerous studies have confirmed this finding [59–71]. Even patients with classic PKU occasionally respond to the point that it becomes possible to ease dietary restrictions or even replace the diet with a near-normal regimen. In December 2007, the FDA approved Sapropterin dihydrochloride (Kuvan), a synthetic form of the naturally occurring cofactor, tetrahydrobiopterin, at a recommended dose of 10 to 20 mg/kg/day. About 25–50% of the patients with PAH deficiency are sapropterin-responsive. In general, this treatment is considered successful when patients respond with at least a 30% decrease in phenylalanine level with the assumption that the diet remains stable throughout the testing period. The responsiveness is assessed based on frequent measurements of blood Phe level just before starting the sapropterin as a baseline, then obtaining follow up Phe level at 24 hours, 1 week, 2 weeks and sometimes 3 and 4 weeks. The recommended sapropterin testing dose is not less than 20 mg/Kg as it could underestimate the response rate. Sometimes, there is a delay in the effect of sapropterin on blood Phe up to 2–4 weeks. If there is no actual decrease in blood Phe level, but there is improvement in neuropsychiatric symptoms or increase in Phe tolerance, that should justify the sapropterin therapy as well. However, assessing the success of Kuvan with infants and small children on diet therapy requires more sensitive measurement. Such infants and children may be defined as responders to Kuvan if they can double their phenylalanine intake while keeping their Phe levels within the recommended range. For such children, Kuvan offers the opportunity to tolerate a less restricted diet allowing them to eat more natural protein while maintaining their blood Phe level in the desired range. Some patients could actually double and even triple their dietary Phe intake resulting in much better quality of life. Long-term therapy with sapropterin in responsive patients should be maintained lifelong. As mentioned previously, genotyping would help in predicting the response to BH4 supplementations. However, it is still difficult to predict the phenotype based on the genotype. So, the current guidelines recommend that every PAH-deficient patient should be offered a trial of sapropterin therapy to assess responsiveness except those with two pathogenic null variants in trans [25].
Enzyme substitution therapy
Polyethyleneglycol-conjugated phenylalanine ammonia lyase (PEG-PAL) or Pegvaliase is an FDA approved PAH enzyme substitution since May 2018 [25, 72]. It showed to be effective regardless of residual enzyme activity and has been only FDA- approved for use in adults who have uncontrolled blood Phe concentrations >600 μmol/L [73]. Phenylalanine ammonia lyase is a bacterial enzyme that degrades Phe to ammonia and trans-cinnamic acid; ammonia is metabolized by the liver while trans-cinnamic acid and its final product, benzoic acid, are conjugated with glycine and excreted in the urine [74, 75]. Pegvaliase therapy aims to provide maintenance of blood Phe concentrations while normalizing the diet in all adult patients with PKU who can adhere to the therapy and able to monitor for potential adverse events [73, 76].
Results of a phase III PRISM program support the efficacy of Pegvaliase for the treatment of adults with PKU regardless of residual enzyme activity, with a manageable safety profile in most participants [25, 77, 78]. It showed substantial reduction in blood Phe and improvement in inattention and mood symptoms, however serious adverse events have been reported such as anaphylaxis. The most common adverse events were arthralgia (70.5%), injection site reactions (62.1%), injection site erythema (47.9%), and headache (47.1%) [77, 78]. Therefore, premedication with antihistamine and/or antipyretics is recommended prior to administration of Pegvaliase as well as monitoring the patient during and at least 60 minutes following the administration of the treatment. Given the risk of anaphylaxis, it is important to educate the patients about the signs and symptoms as well as administration of epinephrine if needed [73].
It is recommended to obtain baseline blood Phe concentrations prior to the initiation of Pegvaliase [73]. The induction dosage for pegvaliase is 2.5 mg subcutaneously (SC) once weekly for 4 weeks under the supervision of a health care provider. The dosage then are titrated based on tolerability over at least 5 weeks to achieve a dosage of 20 mg SC once daily until the patient is titrated to an effective maintenance dose where therapeutic response is achieved. Therapeutic response is defined as 20% reduction in blood Phe concentration from baseline or blood Phe concentration concentration ≤600 μmol/L after 24 weeks. If this was not achieved, then consider increasing the Pegvaliase dose to a maximum dosage of 40 mg SC daily. If therapeutic response has not been achieved after 16 weeks on the maximum dosage, discontinuation of pegvaliase is recommended. However, if the patient develops very low blood Phe concentrations (<30 μmol/L) during titration and maintenance, the dosage of Pegvaliase may be reduced or the dietary protein and Phe intake may be adjusted [73]. Pegvaliase can be self-administered if patient shows adequate competency. Of note, the efficacy of pegvaliase without dietary therapy was not assessed in clinical trials and thus it is important to continue monitoring the Phe intake to avoid changing in the dosage of Pegvaliase [78]. Further, it is unclear whether the combination of sapropterin with Pegvaliase is more beneficial than either therapy alone as there are no clinical trials addressing their combined safety and efficacy [78].
Emerging therapies
Gene therapies
Both liver-directed and skeletal muscle directed gene therapy are being investigated in PKU murine models with good success [79–82]. There is an ongoing phase I/II clinical trial, which uses human hematopoietic stem cell-derived adeno-associated virus vectors (AAVHSCs) to deliver a functional copy of the PAH gene to the liver cells [83].
Cell-directed therapies
Liver repopulation with PAH-expressing cells has been investigated by Harding in 2006 to replace PAH-deficient hepatocytes of the affected individual with PAH-positive cells from some other source [82]. It could be a viable approach to the treatment of PKU if a selective growth advantage could be achieved for donor hepatocytes.
PKU treatment for life
The current recommendation is that the treatment should be lifelong for all patient with PAH deficiency maintaining Phe level between 120–360 μmol/L. Liberalization of the Phe-restricted diet and relaxation of Phe control is no longer acceptable practice given the known adverse neurocognitive and psychiatric outcomes that might develop later in life, which include deficits in executive functioning and psychiatric symptoms such as anxiety, depression, and phobias. This will ultimately lead to lower level of educational attainment and socioeconomic status. Additionally, it is very challenging to return to Phe-restricted diet after patients have tried high protein food and found it more palatable [25].
Surveillance
It is important to obtain regular monitoring of PAA concentrations in individuals with PKU. After Phe stabilization in newly diagnosed infants, it is essential to obtain PAA levels weekly in infants in the first year of life to foster optimal growth during the first year of life. Following the first year of life and until 12 years old, biweekly to monthly sampling may be adequate. In adolescents and adults who are stable and well controlled, blood level monitoring can be monthly. Further, regular assessment of growth and micronutrient needs as well as assessment of developmental progress and screening for mental illness are fundamental at every visit [3, 25].
Maternal PKU
Maternal hyperphenylalaninemia causes an embryopathy/fetopathy that compromises growth and causes congenital malformations, microcephaly, and intellectual disability in the fetus [84] (Table 3). It is a consequence of intrauterine phenylalanine excess derived from the positive transplacental gradient. All girls and women of childbearing age with hyperphenylalaninemia including those with classical PKU and milder forms of hyperphenylalaninemia, should be given reproductive counseling and should be on a treatment regimen with strict dietary phenylalanine restriction before conception and throughout the pregnancy; meticulous treatment may be compatible with a normal outcome for the fetus. Pregnant women without adequate blood Phe level will need intensive intervention, including hospitalization to start strict dietary control [25]. Additionally, frequent blood Phe testing and diet adjustments are required throughout pregnancy as maternal Phe requirements change significantly throughout gestation.
Table 3
Maternal PKU syndrome*
| Symptoms | Frequency |
| Mental retardation | 92% |
| Microcephaly | 73% |
| Low birth weight | 40% |
| Congenital heart disease | 12% |
*Plasma phenylalanine levels over 20mg/dl.
In a large collaboration study of 575 pregnant women with PKU, fetal outcome was significantly improved with early treatment and maintenance of phenylalanine concentrations of 120 μmol/L to 360 μmol/L (2–6 mg/dL) [85, 86]. Microcephaly, which occurred in almost 27% of offspring, was found only in offspring of women in whom treatment began after the first 8 weeks of pregnancy, i.e., late-treated pregnancies. Similarly, almost all cases of congenital heart disease and significant gastrointestinal abnormalities, including tracheoesophageal fistula, occurred in infants of mothers whose blood Phe concentrations were uncontrolled in the first trimester [26]. Hypoplasia of the left heart, infantile coarctation, and right heart outflow tract lesions were seen most frequently. The frequency of intrauterine growth restriction (IUGR) does not differ from that in the general population if maternal Phe levels are controlled by week 10 of gestation.
The best observed outcomes occur when strict control of maternal blood Phe concentration is instituted before pregnancy or by 8 weeks of gestation at the latest [87]. Elevated blood Phe levels in the first 8–10 weeks of gestation are associated with increased risk of congenital heart disease and poor fetal growth [25]. The currently recommended Phe concentrations during pregnancy are (120–360 μmol/L) [2–6 mg/dL] in the United States [87]. There is a linear relationship between maternal Phe levels >360 μmol/L throughout gestation and lower IQ of developing fetus. Maintaining maternal Phe levels <360 μmol/L prior to conception is recommended [25]. However, persistently low maternal Phe level (as low as 100 μmol/L) may be associated with increased risk of IUGR. Additionally, dietary over-restriction leading to inadequate protein and calorie intake, should be avoided as it may increase maternal Phe levels.
Some of the medications and dietary supplements used in PKU patients should not be used during pregnancy, this includes LNAA as they do not consistently affect maternal blood Phe. Sapropterin, which is a class C medication, can be used during pregnancy when the benefits of using it outweigh the risks. There is no evidence of sapropterin-associated teratogenicity; on the contrary, there are many reports of uneventful pregnancy outcomes with its use. Per the ACMG practice guidelines of PKU, women who are on sapropterin and become pregnant, should be offered the option of remaining on the medication and women who are not on it and may benefit from its use, should be offered the option of starting it.
Careful metabolic and nutritional monitoring is required in postpartum period as the Phe requirements decrease. Mothers should continue their Phe-restricted medical food to provide the required calories and protein to support breastfeeding (640 kcal/day and 25 g protein/day). Mothers with PAH deficiency can safely breastfeed [25].
Tetrahydrobiopterin (BH4) defects
Defects in the synthesis and recycle of the cofactor tetrahydrobiopterin (BH4) are responsible for a small fraction of cases with hyperphenylalaninemia, approximately 1–2% [1, 88]. Such cases are sometimes called “malignant” PKU because of the progressive deterioration in neurological function, which cannot be treated by a phenylalanine-restricted diet. These cases should be distinguished from PKU caused by PAH deficiency. BH4 defects cause decreased production of the neurotransmitters dopamine and serotonin. They also result in hyperphenylalaninemia with two exceptions: the autosomal dominant, GTP cyclohydrolase I deficiency (GTPCH), and sepiapterin reductase (SR) deficiency. BH4 metabolic enzymes include three synthetic enzymes: GTP cyclohydrolase I (GTPCH), 6-pyruvoyl-tetrahydropterin synthase (PTPS), and sepiapterin reductase (SR)-and two recycle enzymes: pterin-4cE-carbinolamine dehydratase (PCD) and dihydropteridine reductase (DHPR) (Fig. 2). Despite the low incidence of BH4 defects, all newborns with hyperphenylalaninemia detected through newborn screening should be screened for BH4 defects. With the exception of DHPR deficiency, which can be detected by determination of DHPR activity in dried blood spots (DBS), all other forms of BH4 deficiency (GTPCH, PTPS, and PCD deficiency) can be detected by specific pterin patterns in urine or DBS [36, 37, 89]. Prognosis and outcome strongly depend on the age when the diagnosis is made and treatment introduced, but also on the type of the enzyme defect and pathogenic variant [88, 90].
Fig.2
The Metabolic Pathway of Tetrahydrobiopterin (BH4).
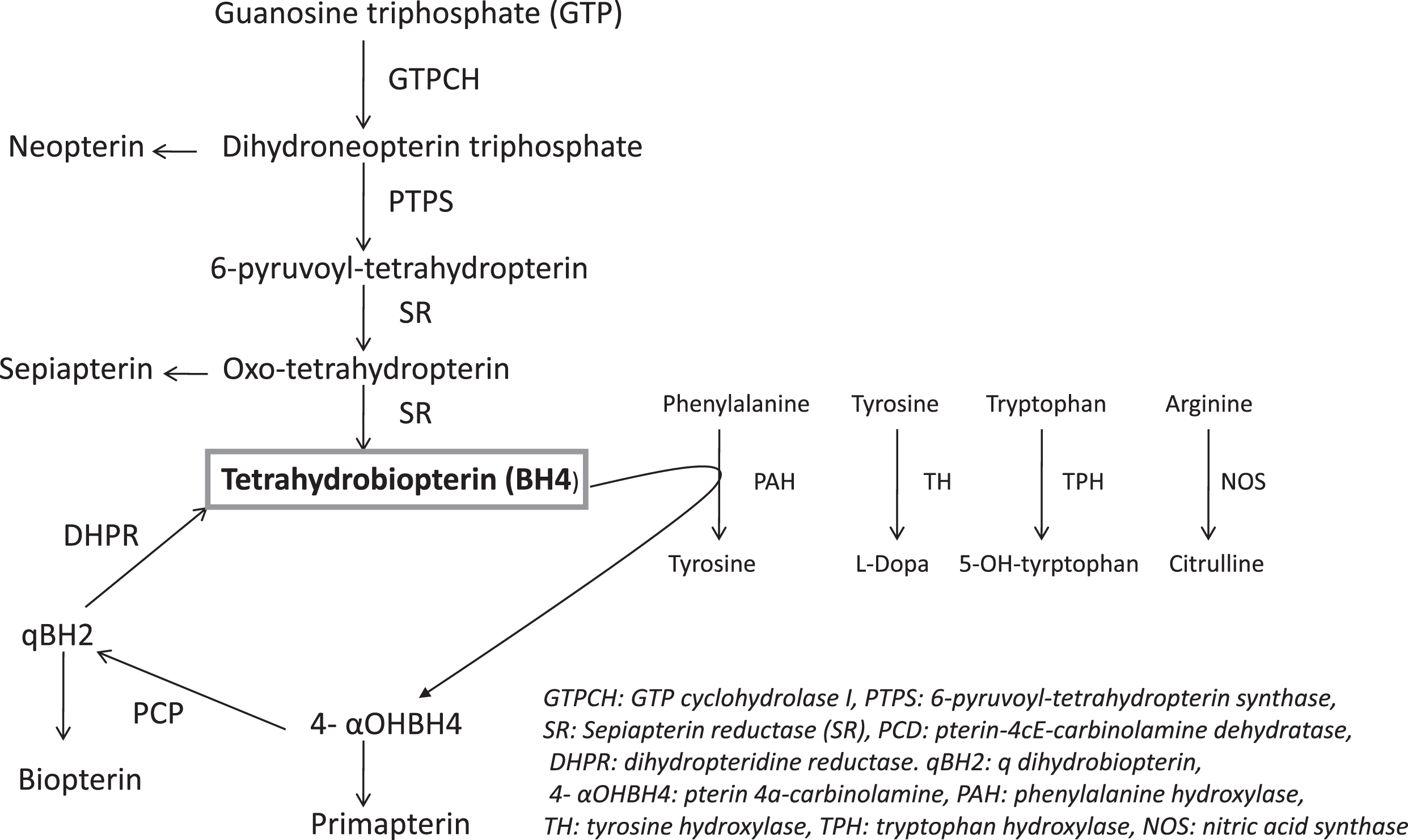
Early diagnosis of GTPCH, PTPS and DHPR deficiencies may prevent irreversible brain damage by pharmacological treatment. Those with PCD deficiency may be at risk of developing non-immune MODY-like diabetes or hypomagnesaemia and renal magnesium wasting [91, 92]. Evaluation for BH4 disorders for any neonate or infant with neurological problems of unknown origin is suggested even without increased Phe or negative NBS for increased Phe.
GTP cyclohydrolase (GTPCH) I deficiency
Deficiency of GTP cyclohydrolase I can occur in a recessive and dominant form (Table 1). The dominant form, with mutation in only one of the two alleles for GTP cyclohydrolase I, causes dopa-responsive dystonia [88]. Patients with the recessive form have mutations in both alleles for GTP cyclohydrolase I and are usually detected because of elevated phenylalanine on newborn screening and develop neurological dysfunction with axial hypotonia, hypertonia of the extremities, abnormal movements, tremors, seizures, and sometimes autonomic dysfunction [93, 94].
The GCH1 gene is composed of 6 exons on 14q22.1–22.2. More than one hundred different mutations have been identified in patients with different forms of GTP cyclohydrolase I deficiency [95]. Only 7 out of 110 mutant alleles are present in a homozygous or compound heterozygous state, causing the autosomal recessive form of GTP cyclohydrolase I deficiency [95]. All the others are present at the heterozygous state and cause dopa-responsive dystonia.
Autosomal dominant form of GTP cyclohydrolase 1-deficiency (dopa-responsive dystonia or Segawa syndrome) usually presents with gait disturbance caused by foot dystonia between ages one and 12 years and progresses to Parkinsonism [96]. Diagnosis is confirmed based on biochemical findings, response to oral administration of levodopa (L-dopa) and molecular testing. Concentrations of total biopterin and total neopterin in cerebrospinal fluid (CSF) are reduced in individuals with GTPCH1 deficiencies [96]. Patients are treated with levodopa/decarboxylase inhibitor (initial recommended dose ≤25 mg/day in children, 50 mg 1×–2×/day in adults). The dose should be gradually increased as needed.
AR form of GTP cyclohydrolase 1-deficiency presents with complex neurological dysfunction, including intellectual disability, global developmental delay, limb spasticity, truncal hypotonia, tremors, oculogyric episodes and seizures. While most patients have elevated plasma phenylalanine levels (300 and 1200 μmol/L) in the neonatal period, cases with normal phenylalanine levels also have been reported [93]. Diagnosis is based on elevated phenylalanine levels, decreased total biopterin and total neopterin levels in urine, and CSF. Molecular genetic testing can also help to confirm the diagnosis. The principles of treatment are low Phe diet, BH4 supplementation (5–10 mg/kg/day), and administration of L-dopa/carbidopa (5–10 mg/kg/day) and 5- hydroxy-tryptophan to improve neurotransmitter deficiency.
6-pyruvoyl-tetrahydropterin synthase (PTPS) deficiency
PTPS, an autosomal recessive disorder, is the most common BH4 defect causing BH4 deficiency [88]. PTPS causes not only hyperphenylalaninemia but also deficiency of dopamine and serotonin because of malfunctioning BH4-dependent tyrosine and tryptophan hydroxylases (Fig. 2). PTPS should be suspected in all newborns with elevated phenylalanine levels detected through newborn screening, especially if phenylalanine is moderately elevated [94]. The characteristic findings are intellectual disability, hypo or hypertonia, seizures, irritability, abnormal movements, recurrent hyperthermia, hypersalivation, failure to thrive, microcephaly and difficulty swallowing (Table 1) [97–99]. Symptoms are usually present at birth but become evident by 4–5 months of age. Brain MRI shows early brain atrophy [98]. Measuring pteridin levels in urine can make the diagnosis. 5-hydroxyindolacetic acid (5-HIAA) and homovanillic acid (HVA) levels are low in CSF. BH4 loading (20 mg/kg), which shows prompt decrease in phenylalanine level after BH4 supplementation, is commonly used as a diagnostic tool in Europe. Urine pteridin profile is characteristic for PTPS deficiency. There is decreased or undetectable biopterin and markedly elevated neopterin. RBC PTPS activity can also be measured and is very low in most typical patients.
The mainstay of treatment is to decrease phenylalanine level by protein restricted diet or BH4 supplementation (5–10 mg/kg/day), and to restore neurotransmitter deficiency by giving L-dopa/carbidopa (5–10 mg/kg/day) and 5- hydroxy-tryptophan (5–10 mg/kg/day). The doses of medications should be adjusted based on response to therapy, clinical improvement as well as adverse effects for each individual patient [98, 99]. There are no good biomarkers to measure the response to treatment except prolactin level [100]. Hyperprolactinemia is a good indicator of hypothalamic dopamine deficiency. Measuring neurotransmitter levels in CSF is a direct but not practical way to evaluate the response.
Sepiapterin reductase (SR) deficiency
SR deficiency, an autosomal recessively inherited disease, is the most recently discovered inherited defect in the synthesis of BH4. SR can be difficult to diagnose because patients have normal plasma phenylalanine levels and cannot be detected through newborn screening [94]. The SPR gene is located on chromosome 2p14–p22 (Table 1). The characteristic features are intellectual disability, dystonia with diurnal fluctuations, oculogyric crises, axial hypotonia, and spasticity [101, 102]. Diagnosis of SR deficiency requires CSF studies, which indicate the presence of sepiapterin as well as low concentrations of homovanillic acid, 5-hydroxyindol- acetic acid. It is best to supplement CSF studies with enzyme and genetic analyses. A marked decrease of sepiapterin reductase activity of the fibroblasts along with sepiapterin reductase gene (SPR gene) molecular analysis confirm the diagnosis [103].
The treatment is to administer L-dopa/carbidopa (1–10 mg/kg/day in 3–4 doses) and 5-hydroxytrptophan (1–8 mg/kg/day in 3–4 doses). The dose should be adjusted based on clinical response and adverse effects. Levodopa/carbidopa/5- hydroxy-tryptophan therapy may reduce cerebrospinal fluid folates (CH3-group trapping by levodopa to 3-metoxy- dopa) requiring folinic acid substitution (15 mg/day). Patients may also need BH4 supplementation [104].
Pterin-4cE-carbinolamine dehydratase (PCD) deficiency
PCD deficiency, an autosomal recessive disorder, has milder phenotype compared to other BH4 defects (Table 1) [90]. Patients have elevated phenylalanine levels that can be detected through newborn screening [94]. Neurodevelopment is good if phenylalanine levels are maintained within acceptable range. Some patients develop mild upper extremity tremors after stimulation. Hypotonia and irritability has been reported in the newborn period. In PCD deficiency neopterin is initially high, biopterin is in the subnormal range and primapterin is present in urine. Low phenylalanine diet and/or BH4 therapy (10 mg/kg/day) is recommended treatment for this disorder.
Dihydropteridine reductase (DHPR) deficiency
DHPR deficiency is an autosomal recessive disorder in the regeneration pathway of tetrahydrobiopterin (BH4). Individuals with DHPR deficiency present with hyperphenylalaninemia, microcephaly, hypotonia, intellectual disability and convulsions [90, 105] (Table 1). DHPR is encoded by the gene QPDR. Missense variants, deletions/insertions and splice site variants in the QPDR gene have been associated with DHPR deficiency [106–108]. Patients have elevated phenylalanine levels and can be detected through newborn screening [94]. DHPR enzyme activity can be measured on dry blood spot. Urine pteridin test shows very high biopterin levels.
Although there is no effective treatment for DHPR deficiency, low protein diet, L-dopa, and 5-hydroxytryptophan supplementations are the mainstay of treatment to keep phenylalanine levels within normal range and increase levels of neurotransmitters. Folinic acid supplementation is also utilized as DHPR is associated with the maintenance of appropriate folate levels and patients with DHPR deficiency have low folate (5-Methyltetrahydrofolate) levels in CSF studies [109]. BH4 supplementation is not routinely recommended because there is a concern that BH4 therapy may further increase BH2 production which will cause decreased BH4/BH2 ratio in patients with DHPR deficiency. In one case we showed that BH4 supplementation improved the clinical symptoms and normalized plasma phenylalanine level and no increase in BH2 level was found in CSF [110].
TYROSINEMIA
Tyrosine is derived either from dietary protein intake, tissue protein breakdown or hydroxylation of phenylalanine. Impaired degradation of the amino acid tyrosine is a feature of several acquired and genetic disorders [111]. Tyrosinemia is indicated by elevated blood levels of tyrosine above 200 μmol/L. Tyrosine is catalyzed by a series of five enzymatic reactions yielding acetoacetate (ketogenic) and fumarate (gluconeogenic). Only liver and renal proximal tubules express the complete pathway and contain all the enzymes required for tyrosine catabolism. The metabolic pathway of tyrosine is shown in Fig. 3. Tyrosine participates in the formation of thyroxin, dopamine, norepinephrine, and epinephrine (Fig. 2). Inborn errors of tyrosine degradation include (Table 4): (1) Tyrosine aminotransferase deficiency (Tyrosinemia type II), (2) 4-hydroxyphenylpyruvate dioxygenase deficiency (Tyrosinemia type III and Hawkinsinuria), (3) Homogentisic acid oxidase deficiency (Alkaptonuria), (4) Fumarylacetoacetase (FAH) deficiency (Tyrosinemia type I). Six patients with Maleylacetoacetate isomerase deficiency (the enzyme preceding FAH in tyrosine degradation) have been recently reported [112].
Fig.3
The Metabolic Pathway of Tyrosine.
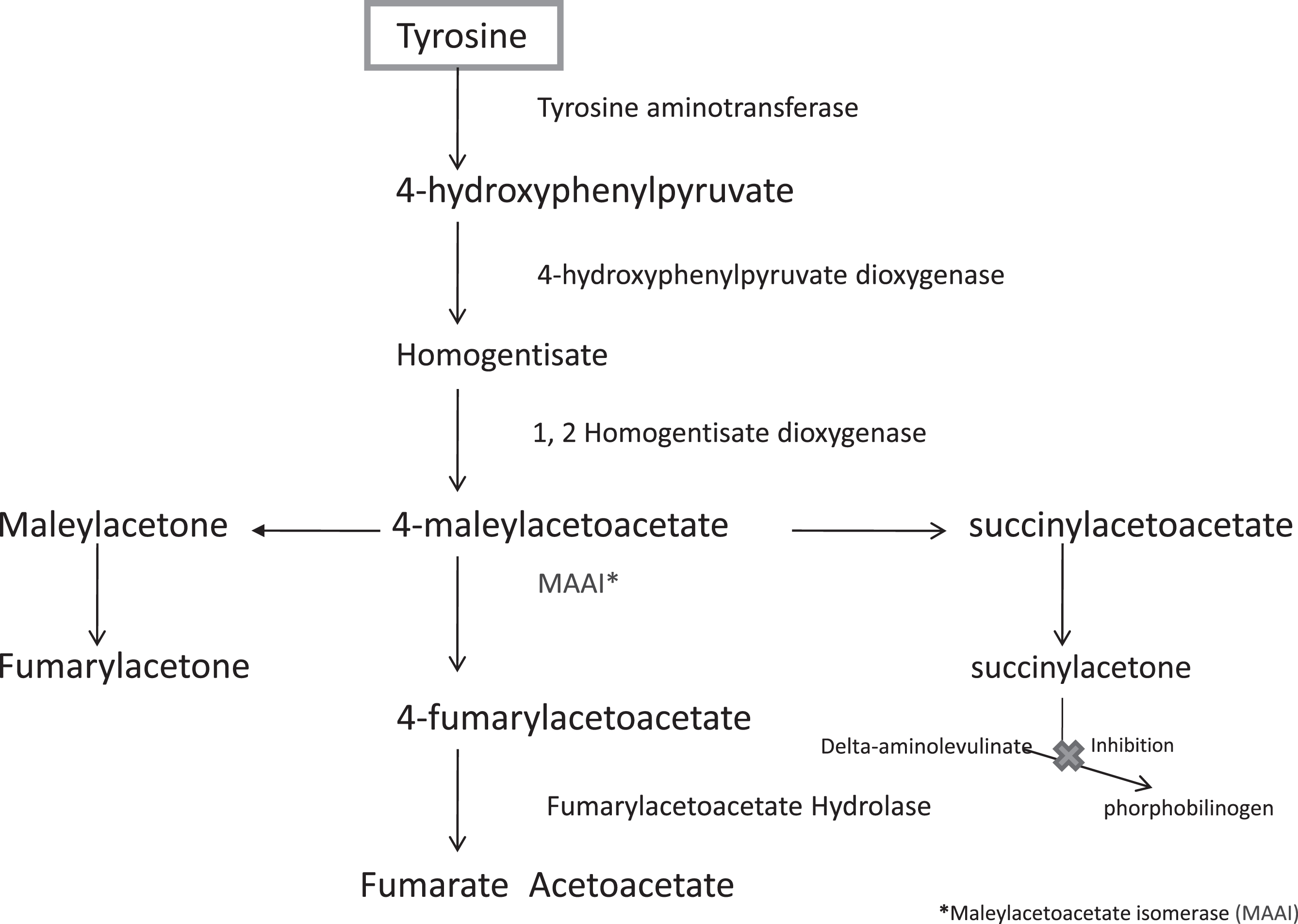
Table 4
Inherited disorder of tyrosine metabolism
| Enzyme defects | Tissue distribution | Chromosomal localization | Disorder | Characteristic | Laboratory findings | Treatment |
| Fumarylacetoacetase | Generalized | 15q23 | Tyrosinemia I | Acute liver failure, hepatomegaly, failure to thrive, vomiting, cirrhosis, renal tubular dysfunction | Succinylacetone in blood and urine, elevated alfa fetoprotein, elevated PT, PTT | Orfadin® (nitisinone), Tyrosine, Phenylalanine restricted diet |
| Tyrosine aminotransferase | Liver, kidney | 16q22.1–q22.3 | Tyrosinemia II | Bilateral pseudodendritic keratitis, painful palmoplantar hyperkeratotic lesions, photophobia, lacrimation, mild learning disability | Elevated plasma tyrosine (>500μmol/L), elevated urine 4-hydroxyphenyl-pyruvic acid, 4-hydroxyphenyllactate, and 4-hydroxyphenylacetate | Tyrosine, Phenylalanine restricted diet |
| 4-OH-phenylpyruvate dioxygenase | Liver, kidney | 12q14-qter | Tyrosinemia III | Mental retardation, ataxia | Elevated plasma tyrosine, elevated urine 4-hydroxyphenyl-pyruvic acid, 4-hydroxyphenyllactate, and 4-hydroxyphenylacetate | Low protein diet (?) |
| 4-OH-phenylpyruvate dioxygenase | Liver, kidney | Unknown | Hawkinsinuria | Failure to thrive, transient metabolic acidosis, mild mental retardation | Presence of Hawkinsin in urine organic acid analysis | Low protein diet, vitamin C (?) |
| Homogentisic acid oxidase | Liver, kidney | 3q2 | Alkaptonuria | Arthritis, ochronosis, aortic, mitral valve disease, aortic dilatation, myocardial infarction, dark urine | Presence of homogentisic acid | No specific therapy. Potential treatment: Orfadin® (nitisinone) |
Tyrosinemia type I
Tyrosinemia type I, also called hepatorenal tyrosinemia, is an autosomal recessive disorder of tyrosine metabolism, and is caused by deficiency of fumarylacetoacetate hydroxylase (FAH) enzyme (Fig. 3). The accumulation of succinylacetone and related metabolites are responsible for the disease that affects, in particular, liver and kidney function [113–115]. The estimated incidence of tyrosinemia type I is about 1 in 100,000. It is more common among specific ethnic enclaves such as the population in the Saguenay-Lac-St. Jean area of Quebec, where carrier rate and incidence are about 1 in 20 and 1 in 1,846, respectively [116].
Genetics
Fumarylacetoacetate hydroxylase (FAH) gene has been mapped to chromosome 15q23–q25. The gene span is about 35 kb and consists of 14 exons. In 1992, the first pathogenic variant causing tyrosinemia type I was reported [117]. It was a point mutation in which an isoleucine residue substituted for an asparagine at position 16. Many other pathogenic variants subsequently have been reported, and there is high degree of heterogeneity [118]. A variant in the intron 12-donor site (IVS12DSg+5a) has been found in patients of different ethnic origins (e.g., Iranians, French Canadians and Scandinavians), which indicates an ancient origin and a founder effect. The pathogenic variant was found in 100 % of the patients from Saguenay-Lac-St. Jean area, and 80% of the patients were homozygous for the variant [116].
The genotype data so far does not permit conclusions on genotype-phenotype correlations. Correlations may eventually emerge, but other factors clearly are important because both the acute form and chronic form have been found in patients with the same genotype.
Genetic counseling and prenatal testing
Genetic counseling should be provided to the parents of affected children with tyrosinemia type I as it is inherited in autosomal recessive pattern with 25% recurrence risk with each future pregnancy. This should include discussion of the available molecular testing for confirmation of the diagnosis, testing carriers as well as the possibility of prenatal testing and pre-implantation genetic testing once the FAH pathogenic variants have been identified.
Prenatal biochemical testing is also available by measuring succinylacetone in the amniotic fluid obtained by amniocentesis at 15–18 weeks of gestation. However, false negative results have been reported and thus molecular testing is always preferred for prenatal diagnosis [119].
Pathology and pathophysiology
Patients who are not detected through newborn screening and treated right away will show pathological changes and may suffer much more serious consequences.
Histologic examination usually reveals micronoduler cirrhosis, and quite often, marked bile duct proliferation within portal tracts and fibrotic septa with varying degrees of steatosis, and pseudoacinar or pseudoglandular formations around central canaliculus often containing prominent bile plugs [120–122]. A significant accumulation of iron pigment within Kupffer cells and hepatocytes and giant-cell transformation are also observed [123].
In older children with a more chronic course, the liver is characteristically coarsely nodular and frequently enlarged with mixed micronoduler cirrhosis [124]. Steatosis varies in extent between the various nodules, sometimes even within a single nodule. Fibrous septa vary in width and frequently contain a mild lymphoplasmatic infiltrate and little ductular proliferation. Intralobular cholestasis or inflammation is usually insignificant. A large grayish-white nodule of hepatoma (Fig. 4) can be easily distinguished from the surrounding nodules in the cirrhotic liver. Multiple microscopic foci of hepatocellular carcinoma and multiple areas of nuclear atypia and hyperchromatism qualifying as hepatocellular dysplasia may be present [124].
Fig.4
Gross appearance of liver after resection for transplantation showing macronodular and micronodular cirrhosis with large nodule of hepatocellular carcinoma (left) (Figure 7 in the first book).
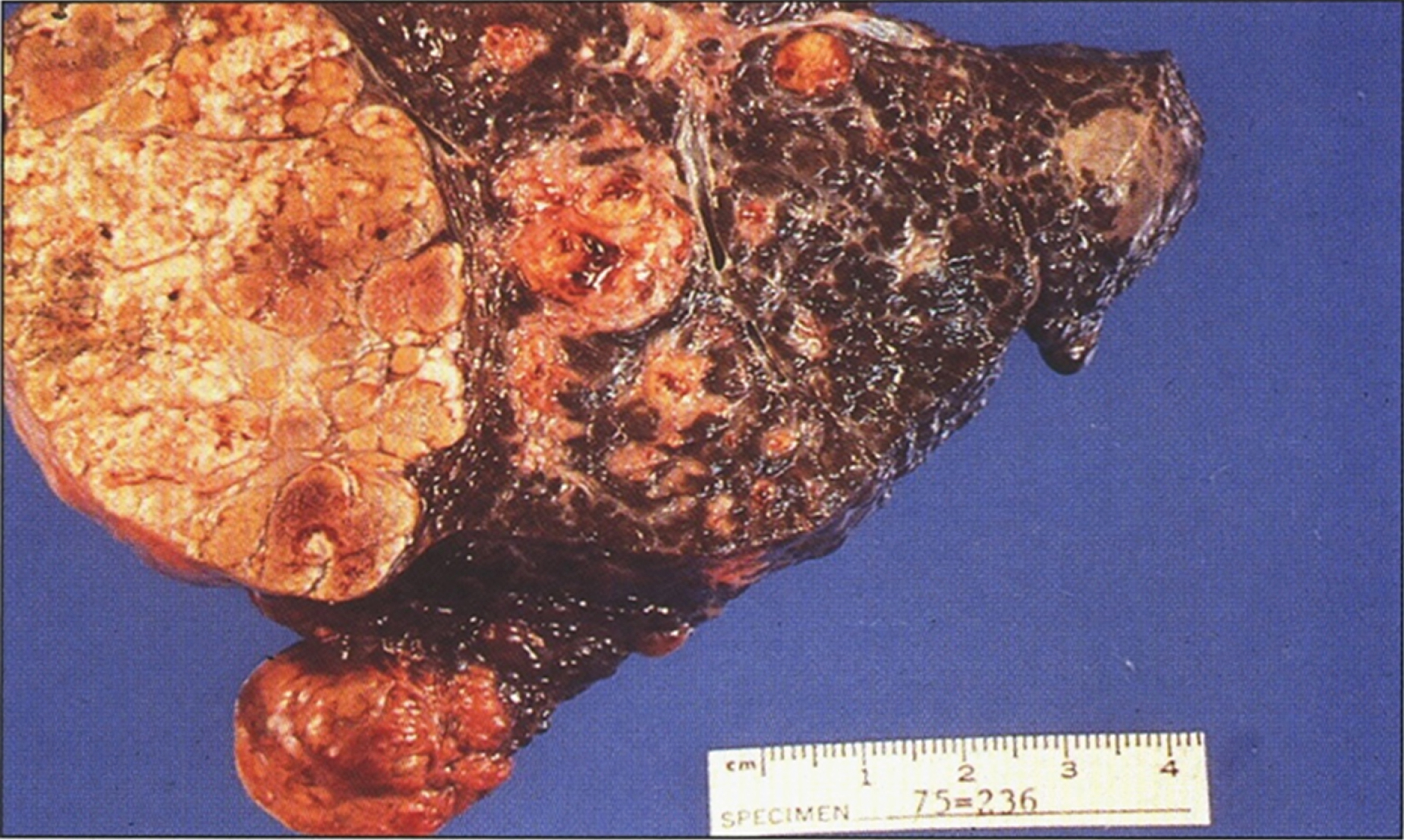
Liver cell dysplasia is a premalignant condition in patients who did not receive early diagnosis and treatment and such patients usually need liver transplantation. This is highly suggestive of an underlying powerful carcinogenic influence of some abnormal metabolites [125–128]. In dysplastic liver cells, irregular nuclear profiles with large nucleoli and reduction of cytoplasmic organelles are characteristic ultra-structural features [128].
Renal tubular dysfunction, characterized by Fanconi syndrome and hypophosphatemic rickets, can be a major manifestation of tyrosinemia type I. Typical morphologic changes are nephromegaly with microscopically irregular dilations of the proximal tubules and vacuolation of tubular epithelial cells [129]. Glycogen accumulation in collecting tubules has been reported [130], and nephrocalcinosis is fairly a constant feature. Aneuploidy in the renal tubular epithelial cells and pancreatic islet cells has been described as well as chromosomal breakage and rearrangements [131]. Ultrastructural examination reveals simplification of the epithelial cells, with loss of the brush border, and cytoplasmic vacuolation, especially in the peripheral area.
Hyperplasia and hypertrophy of the islets of Langerhans have been reported [120, 132, 133] and have rarely been associated with chronic hypoglycemia although most patients have normal blood glucose levels [129]. Hyalinization of the islets has also been reported in some patients [134].
There are several infrequent pathological findings in advanced cases. Myocardial hypertrophy is characterized by increased numbers of mitochondria in the myocytes [135]. Obstructive cardiomyopathy in 2 symptomatic cases has been described [136]. Axonal degeneration and secondary demyelination, similar to the changes seen in acute intermittent porphyria, were observed in 3 peripheral nerve specimens analyzed after paralytic crises [137].
In 1993, an unusual observation was made in tyrosinemia type 1 patients whose livers were removed at the time of liver transplantation. As is typical, multiple large and small nodules were found [138]; however, an immunohistological analysis with FAH antibody surprisingly, showed that the livers represented a mosaic of antibody positive and negative hepatic tissue. FAH enzyme activity was present in the cross-reacting material-positive nodules. Molecular analysis has revealed that they were generated by somatic reversion of the disease-causing inherited mutation followed by clonal selection of reverted hepatocytes [139]. This observation demonstrated the strong selective growth advantage of spontaneously reverted FAH positive hepatocytes in this disease.
Tyrosinemia type I provided the first example of spontaneous “gene therapy” in the liver. Interestingly, mutation reversion has been observed not only in compound heterozygotes, in which intragenic mitotic recombination could result in restoration of the enzyme activity, reversions also have been found in patients homozygous for the same mutant allele on both chromosomes. This phenomenon requires precise removal of the single base change mutation responsible for the disease. In most patients the amount of reverted tissue is too low to result in a cure of the disease, but it seems likely that some of the interfamilial variability in the clinical course of tyrosinemia type I may be due to somatic mosaicism. Patients with a significant mass of reverted hepatocytes may have a milder form of the disease.
Tyrosine is not toxic to the liver or kidney, but rather causes only dermatological, ophthalmological and possibly neurodevelopmental problems. What then causes liver and kidney damage in patients with Tyrosinemia type I? Fumarylacetoacetate (FAA), the compound that accumulates in FAH deficiency, is a potent alkylator, causing oxidative damage to the cells in which it is generated by reacting with glutathione and sulfhydryl groups of proteins [126, 140, 141]. Importantly, FAA acts on cell autonomously, directly damaging only the hepatocytes and renal proximal tubules in which it is produced and not adjacent cells. Because of its rapid reactivity, FAA itself is not found in body fluids of patients with tyrosinemia type I. Succinylacetoacetate and succinylacetone derived from the reduction of FAA are the principle metabolites of FAA [141]. These compounds are found systemically and testing for them diagnostically is routine. Succinylacetone (SA) is one of the main culprits in tyrosinemia type I. It interferes with the activity of two hepatic enzymes: Parahydroxyphenylpyruvic acid dioxygenase (p-HPPD), resulting in further increase in blood tyrosine, and porphobilinogen (PBG) synthase or enzyme δ-aminolevulinic acid (δ-ALA) dehydratase, with subsequent increase in δ-aminolevulinic acid (δ-ALA), which may induce acute neurologic (porphyria-like) crises in the patients.
The accumulation of FAA within the hepatocytes results in one of two outcomes; either apoptotic cell death or profound perturbation of gene expression. Recent studies have shown that acute accumulation of FAA triggers apoptosis in both hepatocytes and renal tubular cells [142]. Apoptosis is mediated by caspases 1 and 3 and is associated with cytochrome c release. Glutathione is protective, and its intracellular level can clearly modulate the threshold for FAA-induced apoptosis [142].
Of note, the elevation in blood tyrosine levels seen in other disorders of the tyrosine degradation pathway other than tyrosinemia type I are associated with dermatological, ophthalmological and possible neuro-mental problems, but with no manifestation of liver disease or renal tubular dysfunction. Tyrosinemia types II and III variably respond to phenylalanine and tyrosine-restricted therapies [143–145]. However, such dietary restriction in patients with tyrosinemia type I, even when started early in life, did not prevent the hepatic, renal or neurologic manifestations of the disease.
Clinical findings
Tyrosinemia type I presents either as an acute or chronic form. The acute form presents in the first months of life with acute liver failure, hepatomegaly, failure to thrive and vomiting [111, 113] (Table 5). The chronic form is characterized by chronic liver disease with a high incidence of hepatocellular carcinoma, renal tubular dysfunction with hypophosphatemic rickets, porphyria like episodes, and usually presents in early childhood (Table 5). Patients with the chronic form of the disease have mild hepatomegaly, failure to thrive due to renal tubular dysfunction and a history of easy bruising. All of these complications are proven to be preventable with the initiation of nitisinone treatment.
Table 5
Manifestations of Tyrosinemia Type I
| General Clinical | Failure to thrive |
| Bleeding/bruising | |
| Rickets | |
| Growth retardation in infancy | |
| Liver | Hepatomegaly |
| Cirrhosis | |
| Jaundice | |
| Ascites | |
| Hepatocellular carcinoma in childhood | |
| Kidney | Tubulopathy |
| Gastrointestinal Tract | Vomiting |
| Diarrhea | |
| Central Nervous System | Neurologic symptoms of acute intermittent porphyria (abdominal pain, hypertension, muscular weakness, painful paresthesias or progressive paralysis) in childhood and later |
Distinction between acute and chronic forms is not always clear-cut as some children have a stormy course in the first year of life typical of the acute form and subsequently develop a more indolent course compatible with the chronic form. However, children over 2 years of age who are considered to have the chronic form of tyrosinemia are nonetheless at risk for acute life-threatening liver and neurologic crisis [137].
Patients with the acute form usually present after one month of age with overt liver disease that may become evident with rapid increase liver size, ascites, anasarca and hemorrhagic diathesis secondary to loss of synthetic function for clotting factors resulting in marked prolongation of Prothrombin (PT) and partial thromboplastin times (PTT) [111, 146, 147] Unlike most forms of liver disease, the serum transaminase levels are modestly elevated, and bilirubin may be normal of slightly elevated.
Jaundice is usually a terminal event and is rarely observed in infants surviving an acute episode. Infants with liver decompensation emit a typical odor of boiled cabbage or rotten mushrooms. Untreated children rapidly develop cirrhosis with varying degrees of clinical severity. During the first 2 years of life, the child is particularly at risk for episodes of liver decompensation and neurologic crisis [148]. Thereafter, the dominant problems include renal involvement and risk of developing hepatocellular carcinoma. Hepatosplenomegaly occurs in approximately 70% of cases. Rickets may be apparent in those with moderate or severe renal disease [148].
Tyrosinemia type I should be suspected in any infant or child with evidence of hepatocellular disease, cirrhosis for which the cause is not evident [149] (Table 6). Rickets or characteristic renal or neurologic findings, especially if associated with abnormal hepatic function, also suggest this diagnosis.
Table 6
Metabolic Disorders That May Progress to Cirrhosis
| Hereditary tyrosinemia* |
| Galactosemia* |
| Wolman disease* |
| Peroxisomal biogenesis defects* |
| Glycogen storage disease type IV |
| Neonatal hemochromatosis* |
| Indian childhood cirrhosis |
| α1-antitrypsin deficiency |
| Wilson disease |
| Mitochondrial respiratory chain disorders |
| Familial cholestatic syndromes |
| Cystic fibrosis |
| Sialidosis |
| Gaucher disease |
| Niemann-Pick disease |
| Genetic hemochromatosis |
| *Very rapidly developing |
The incidence of hepatocellular carcinoma varies between 17% and 37% [150] in patients who were clinically diagnosed and not treated with Orfadin® (nitisinone/NTBC). Hepatocellular carcinoma can occur at a very young age and has been reported in a 2.5-year-old child [151]. Metabolic intermediates are natural alkylating agents with characteristics to make them primary candidates for carcinogenesis, and directly cause other cellular damage [126]. Neither CT nor Doppler ultrasound can discriminate between benign and malignant nodules, although the presence of low-attenuation nodules is highly suggestive of hepatocellular carcinoma.
Main findings and complications
Neurologic crisis
There are 2 phases of neurologic crises (1) an active period during which the child experiences painful paresthesias, autonomic signs such as hypertension and tachycardia, similar to acute intermittent porphyria and sometimes progressive paralysis and (2) a period of recuperation, seen in crises with weakness or paralysis. Painful crises are the most frequent neurologic sign, often localized to the legs, and frequently results in a position of extreme hyperextension of the trunk and neck that can be mistaken for opisthotonos or meningismus [111, 148]. Weakness necessitating respiratory support and hyponatremia with associated seizures can also be seen. The neurologic crises are thought to be secondary to axonal degeneration as a result of the demyelination [143].
Recuperation from paralytic crises is possible, although patients with repeated severe crises may have chronic weakness. Vomiting, ileus, and electrolyte imbalance occur frequently, usually prior to the pain crises. The mental development of children with tyrosinemia is normal, and during crises their level of consciousness is not diminished. The active phase of crisis usually lasts for 1 to 7 days.
Neurologic crisis is usually not associated with deterioration of the usual liver function tests. Urinary levels of δ-aminolevulinic acid (δ-ALA) tend to be higher during crisis secondary to inhibition of RBC δ-ALA hydratase, which is believed to explain such pain crises [152–154]. Treatment of the pain crises is mainly through dextrose containing fluids (dextrose 10% with normal saline) at 1.5 to 2 times maintenance rate for age and weight along with analgesics. In case of hypertension and hyponatremia, adequate rapid acting anti-hypertensives and normal saline fluids and sometimes 2% saline are required respectively. NTBC therapy has to be initiated immediately in order to inhibit the production of SA, which are responsible for inhibiting δ-ALA hydratase and resulting in the pain crises. Per the longitudinal study in Quebec in 2012, none of the patients with tyrosinemia type I developed an acute neurologic decompensation while compliant with NTBC therapy [155].
Renal disease
Renal tubular dysfunction and glomerular involvement occur secondary to ongoing presence of succinylacetone (SA) in the blood or urine, confirming the importance of NTBC treatment. Fanconi renal syndrome with generalized aminoaciduria, glycosuria, renal tubular acidosis and hypophosphatemic rickets can occur with normal serum calcium (Ca) [111, 113–118, 120–124, 126–130, 156]. Usually, patients show symptoms of renal involvement by age 6 months. Long-term complications include hypertension, glomerulosclerosis, nephrocalcinosis, and chronic renal failure. Majority of patients with tyrosinemia type 1 have some degree of abnormal renal architecture with dilated tubules, echogenicity, cysts and nephromegaly on ultrasound examination, and may have evidence of mild to moderate nephrocalcinosis. The glomerular filtration rate is generally decreased. Progressive renal disease occurs in untreated patients with tyrosinemia type I. SA is responsible for the renal tubular damage [157]. Soon after diagnosis of tyrosinemia type I, thorough evaluation of renal function should be initiated which include blood testing (BUN, Cr, electrolytes, bicarbonate, calcium, phosphate), urine analysis (glucose, amino acids, calcium, phosphate, Ca/Cr ratio, phosphate, protein, albumin), renal ultrasound, bone X-ray and DEXA scan for evidence of rachitic changes from hypophosphatemia. It is equally important to monitor the renal tubular function periodically, achieved via assessing urine amino acids, glucose, phosphate, protein, albumin or β2-microglobulin, and Ca/Cr ratios compared to plasma [158].
Ophthalmologic consequences
With the NTBC therapy and subsequent improvement in the survival rates in patients with tyrosinemia type I, ophthalmological complications have been raised. Elevated tyrosine levels have been associated with corneal crystals formation resulting in photophobia, lacrimation, pain and redness. The exact threshold of blood tyrosine level associated with corneal crystals is unclear, but the recommendation is to keep tyrosine level be low 600 μmol/L. Corneal crystals associated with photophobia are seen if blood tyrosine concentration is above 600 μmol/L (700–900), which can resolve with improvement in tyrosine level when patients are under strict control of tyrosine intake. The current recommendation is to obtain annual ophthalmologic slit-lamp evaluations patients with tyrosinemia I or when they are symptomatic or at increased risk [158].
Neurobehavioral issues
Neurodevelopmental consequences have come to attention after the improvement in the survival of patients treated with NTBC therapy. There are no studies measuring intelligence in individuals with tyrosinemia type I that were identified through NBS in the United States. However, there are other studies measuring intelligence in tyrosinemia type I patients treated with NTBC, which reported neuropsychological outcomes since the introduction of NTBC treatment. The IQ of patients ranged from 55–115. There is a concern that the hypertyrosinemia produced by the use of NTBC might be associated with long-term behavioral and neuropsychological effects. Accordingly, it is important to include neuropsychological assessment as part of routine follow-up of patients with tyrosinemia type I [158].
Cardiac complications
Hypertrophic cardiomyopathy has been reported in some untreated patients with tyrosinemia type I complicated with chronic liver disease or acute presentation in young infants. The exact mechanism is unclear, but there are different theories attributing it to the inhibition of myocardial porphobilinogen synthase resulting in elevated levels of δ-ALA, which can be cardiotoxic maybe secondary to mitochondrial dysfunction [159]. Further, advanced liver disease resulting in increased cardiac output may also play a role in the pathogenesis of cardiomyopathy. The cardiomyopathy is usually subclinical [160], however there are few patients presented initially with hypertrophic cardiomyopathy before they were diagnosed with tyrosinemia type I estimated as 30% [161]. Cardiomyopathy was significantly less common in those treated initially with NTBC [161]. The typical echocardiogram findings include concentric biventricular hypertrophy and/or interventricular septal hypertrophy. It resolved after treatment with NTBC for about 4 months [162] or liver transplant in severely affected patients.
Endocrine complications
Some may have episodes of hypoglycemia associated with islet-cell hypertrophy [133]. However, most patients are asymptomatic and pancreatic islets cell hyperplasia have been reported as pathologic finding in autopsies. Hypoglycemia could be attributed either to islet cell hyperplasia or to liver dysfunction [158].
Diagnosis
Demonstration of the presence of succinylacetone (SA) on dried filter paper of samples of plasma or urine is pathognomonic [163]. Urine organic acid analysis is an important diagnostic test to measure urine SA as well as tyrosine metabolites such as 4- hydroxyphenyllactate and pyruvate in clinical practice [111, 115, 148] SA and succinylacetoacetate are derived from reduction and subsequent decarboxylation of maleylacetoacetic acid and fumarlyacetoacetic acid. PAA analysis shows elevated levels of tyrosine and methionine. Urine amino acid analysis shows generalized aminoaciduria. Urinary δ-ALA can also be elevated.
The first laboratory indications of liver crisis are disproportionate prolongations of the coagulation time and a bleeding diathesis without other symptoms of liver disease [164]. PT and PTT can be alarmingly prolonged despite normal or near normal serum transaminase levels. Factor V levels, often used in other liver disorders as a marker of liver synthetic function, are usually preserved to within normal or close to normal range. In contrast, factors XI and XII and vitamin K dependent factors II, VII, IX, and X may be exceedingly low with normal factor VIII levels [111]. Highly elevated levels of α-fetoprotein (AFP) are seen, even before the elevation in tyrosine [165].
FAH can be assayed in lymphocytes and erythrocytes as well as on liver tissue. FAH activity is also measurable to some extent in kidney, lymphocytes, erythrocytes, fibroblasts, and chorionic tissue. Carrier detection by these techniques is imperfect, because some heterozygotes for the deficiency can have high levels of residual FAH activity and mosaicism of enzyme in liver.
High concentrations of AFP in umbilical cord blood of affected neonates suggest that liver disease may be present in late gestation.
Elevated levels of methionine and tyrosine in plasma and generalized aminoaciduria, large increase in tyrosine metabolites (4- hydroxyphenyllactic acid, 4- hydroxyphenylpyruvic acid) in urine may also develop in other metabolic disorders associated with hepatocellular disease such galactosemia, hereditary fructose intolerance, fructose 1,6 bisphosphatase deficiency, glycogenosis type IV, and alpha 1 – antitrypsin deficiency.
Newborn screening
The newborn screening (NBS) for tyrosinemia type I provided the most favorable outcomes for the patients. NBS allowed early diagnosis of tyrosinemia type I at an asymptomatic stage and subsequent initiation of treatment with NTBC in addition to dietary restriction of phenylalanine and tyrosine in the first few days of life, preventing hepatic and renal diseases and thus avoiding the need for liver transplant.
Tyrosine levels are measured with tandem mass spectrometry in all newborns born in the USA, but it has high false negative rate because many newborns with tyrosinemia type I do not have elevated tyrosine levels in the first days of life. Determination of blood SA is performed on the same filter paper in some newborn screening programs to increase the sensitivity of screening test [166]. All most all NBS programs in USA have used blood SA as a second-tier test when the initial blood tyrosine levels are above certain cutoff (150 μmol/L). The low threshold value chosen for blood tyrosine, combined with the specificity of SA as a marker can detect new cases with tyrosinemia type I while eliminating false positive results caused by transient tyrosinemia, liver disease, prematurity, concurrent use of total parental nutrition, mitochondrial depletion syndrome or other causes of tyrosinemia. The American College of Medical Genetics and Genomics (ACMG) recommends that blood SA should be the primary marker and thus be used to screen for tyrosinemia type I in USA.
Following the abnormal NBS for elevated tyrosine and/or SA, patients should be referred to metabolic centers for confirmatory testing that includes new sample for blood or urine SA level. Additional recommended testing includes PAA, liver function tests (PT, INR, PTT, AST, ALT), electrolytes, glucose and AFP. Once the diagnosis is confirmed, NTBC and dietary therapy should start immediately. Additionally, siblings of the affected patient should be tested immediately for the presence of SA in order to start treatment as soon as possible. Molecular testing of FAH should be performed, if possible, in all patients with confirmed positive NBS as well as in patients suspected to have tyrosinemia type I without clear positive SA results. Molecular testing however should not delay the treatment. Additionally, molecular testing is also warranted in cases where tyrosinemia type I is clinically suspected but without circulating SA or in cases where there is persistent SA without liver involvement or other clinical symptoms of tyrosinemia type I, which are secondary to maleylacetoacetic isomerase deficiency (pathogenic variants in GSTZ1) [112].
Treatment
\enlargethispage -12pt
Restriction of the intake of the precursor amino acids phenylalanine and tyrosine to the minimum necessary to allow for normal growth has been shown to improve renal tubular function and probably slows but does not prevent the progression of the liver disease [114, 165, 167–169].
Management of neurologic crisis includes analgesia for the severe pain. A high carbohydrate intake such as glucose inhibits δ-aminolevulinic acid synthase (the step prior to the δ-ALA hydratase) [111]. Hematin, which inhibits δ- aminolevulinic acid synthase, has been used with some success [154]. Symptomatic treatment for the hypertension, hyponatremia, hypokalemia, and hypophosphatemia should be provided [158].
Treatment with inhibition of 4-hydroxyphenylpyruvate dioxygenase
In 1992 Linstedt et al reported a new principle for treatment of tyrosinemia type I based on inhibition of 4-OH-phenylpyruvate dioxygenase with 2-(2-nitro-4trifluoro-methybenzoyl)-1,3-cyclohexanoidon (NTBC, Orfadin) [170]. This potent inhibitor of 4-OH-phenylpyruvate dioxygenase prevents the accumulation of maleylacetoacetate and fumarylacetoacetate compounds. The initial studies showed that plasma and urine succinylacetone, and urine 5-aminolevulinic acid levels decreased from initially high levels to levels within or slightly above the normal range, and red blood cell porphobilinogen synthase activity increased to normal values. Improved liver functions and dramatic decrease in initially high serum AFP concentrations were also reported [171, 172].
Orfadin® (nitisinone), also known as NTBC was designated an Orphan Drug in May 1995 by the Office for Orphan Product Development and approved by FDA in 2002. Initially there was a concern whether this drug would prevent long-term complications such as hepatocellular carcinoma. Now after 25 years of experience with Orfadin®, it is well known that if the treatment starts in the first few months of life, the outcome is good with no reported cases that developed hepatocellular carcinoma [155, 171–173]. Per FDA, the initial dose of Orfadin® (nitisinone/NTBC) is 1 mg/kg/day in two divided doses administered in the morning and evening for patients in their first year of life. In older children, one daily dose is as effective as twice a day and may be considered for better compliance [174]. If biochemical parameters are not normalized within one month of starting the treatment, NTBC dose should be increased to 1.5 mg/kg/day. The blood levels of NTBC can be adjusted to maintain plasma level 40–60 μmol/L, although there are no evidence-based guidelines about the target therapeutic goal. However, the lowest dose of NTBC that completely suppress the SA in plasma or urine with normalizing liver and kidney functions should be prescribed. NTBC doses as low as 0.55 mg/kg per day have been used with success [175]. NTBC has no effect on Cytochrome P450 enzymes and thus has no known interactions with other medications and therefore standard dosing and routing immunizations can be used with patients treated with NTBC.
Monitoring response to Orfadin® (Nitisinone/NTBC)
Regular liver monitoring by imaging (MRI) is recommended at least once a year. Urine SA and blood AFP levels should be monitored closely. Presence of SA is an indicator of inadequate treatment. Serum AFP levels are usually markedly elevated at the time of diagnosis, and gradually decrease during the course of NTBC treatment and should be within normal ranges. An increase in AFP level may be a sign of inadequate treatment but patients with increasing AFP or signs of nodules of the liver during treatment with NTBC should always be evaluated for hepatic malignancy. Therefore, patients with progressive liver disease, persistent or progressive elevation of AFP and/or abnormal liver imaging, should be referred immediately to the adequate specialists (oncologist/hepatologist/transplant center) for further management. Additionally, platelets and leukocytes should be monitored regularly because of the risk of thrombocytopenia and leukopenia.
NTBC increases tyrosine levels in the blood. Tyrosine and phenylalanine levels should be monitored closely. It is important to keep phenylalanine levels within normal range. Tyrosine levels should be less than 500 μmol/L in order to prevent toxic effects to the eyes (corneal ulcers, opacities, keratitis, conjunctivitis, and photophobia), skin (painful hyperkeratotic plaques on the soles and palms), and nervous system (variable degrees of mental retardation and developmental delay) [176, 177]. Slit-lamp examination of the eyes should be performed before initiation of NTBC treatment. Eye exam is recommended once a year. Patients who develop photophobia, eye pain or signs of inflammation such as redness, swelling, burning of the eyes should undergo slit-lamp examination [177]. Patients who have tyrosine crystals on their eyes must be on a more restricted diet to keep plasma tyrosine levels below 500 μmol/L. In most patients, eye symptoms are transient and improve with the diet therapy. NTBC dosage should not be decreased in order to lower the plasma tyrosine level.
Management of acute tyrosinemia type I
Upon the diagnosis of tyrosinemia type I, patient should be started immediately on NTBC and phenylalanine/tyrosine-restricted diet. Baseline labs to be obtained with the initiation of therapy include blood or urine SA, PAA, CBC, AFP, PT, PTT, AST/ALT, blood chemistries (BUN, Cr, bicarbonate, calcium, phosphate) and urine analysis. Also, liver and renal imaging should be performed as baseline that includes liver ultrasound or MRI with contrast and renal ultrasound. If patient presents acutely ill with liver failure and not through NBS, in that case, tyrosinemia type I should be in the differential diagnosis. This can progress to multi-organ failure requiring cardiorespiratory support and consultation of hepatologist. In addition to the supportive care, intravenous dextrose containing fluids (D10%), infused at rate 1.5–2.0 times maintenance rate in order to prevent tyrosine catabolism and the production of δ-ALA, which is associated with the neurologic crises. Treatment of metabolic acidosis secondary to renal tubular acidosis (RTA) should be considered as well as administration of fresh frozen plasma and vitamin K to correct the coagulopathy. NTBC must be initiated at 1 mg/Kg/day and can be increased to 2 mg/Kg in case there is no improvement. Clinical response is expected within 1 week of treatment and the urinary SA usually normalizes after 24 hours of treatment. Liver transplantation should be considered if there is no clinical improvement after 1 week of treatment.
Once the patient becomes clinically stable, a diet low in phenylalanine and tyrosine with the help of metabolic dietitian should be started by 36–48 hours even if via total parental nutrition, including small amount of complete protein (0.25–0.5 mg/Kg/day) to avoid ongoing catabolism and subsequent stimulation of tyrosine catabolic pathway [158].
Dietary management of NTBC-treated tyrosinemia type I
The goal of dietary therapy includes restriction of phenylalanine and tyrosine to maintain the levels within treatment range with restriction of the intact protein but still providing the amino acids needed for adequate growth and development.
Upon diagnosis, a formula (20–30 Kcal/oz) lacking phenylalanine and tyrosine and with no intact protein may be given for the first 48 hours to normalize the blood tyrosine level. Following the stabilization of the patient, a low intact dietary protein that is age-appropriate must be initiated. Many medical foods devoid of phenylalanine and tyrosine are now available (e.g., Tyrex 1, Tyros 1, Tyr coolers) in addition to modified low protein foods. With protein restriction, it is important to maintain blood tyrosine levels between 200–600 μmol/L and phenylalanine as well as other PAA within normal range even if it requires allowing more intact protein and/or phenylalanine supplementation. Additionally, patients on low-protein diet are prone to nutritional deficiencies and therefore it is important to provide age-appropriate daily requirement of vitamins and minerals to support growth [158]. It is equally important to monitor for bone health given the risk of osteopenia of individuals on restricted diet, especially with the presence of RTA.
Liver transplant
Before NTBC treatment became available, early liver transplant was the only option for most patients with tyrosinemia type I [151, 178–180]. For some patients who present with severe liver failure that is beyond the point where NTBC treatment is a cure, liver transplantation is still the only option [169]. There will also continue to be patients who at the time of diagnosis are already in urgent need of transplantation because of hepatocellular carcinoma, which has been described to occur as early as 1 year of age. It is recommended that patients with renal failure from tyrosinemia type I receive combined liver kidney transplants even in the face of normal hepatocellular function. It was noted that urine and plasma SA decrease but not completely suppressed following liver transplantation, presumably because of continued production in the kidneys [181]. Therefore, low dose NTBC treatment is recommended in patients who have persistent renal tubular dysfunction after liver transplant to prevent progression of tubular and glomerular dysfunction [182]. Additionally, periodic screening for renal disease post liver transplant is warranted in tyrosinemia type I given the pathophysiology of the disease [158].
Tyrosinemia type I and pregnancy
NTBC therapy is classified as pregnancy category C due to insufficient available experience and data. There were no reported adverse effects on fetal development in the three children of women with tyrosinemia type I treated with NTBC during pregnancy. Development and general health of all three infants during the first year were reported as normal [183–185]. However, developing fetus may be at risk because of alterations in tyrosine metabolism and thus follow-up should be performed for infants born to women treated with NTBC during pregnancy. On the other hand, breastfeeding is contraindicated while the mother is being treated with NTBC [158].
Surveillance
Suggested guidelines per Chinsky et al., 2017 and King et al., 2017 for the evaluation and follow-up of tyrosinemia type I patients that were diagnosed by newborn screening is presented in Table 7 [119, 158].
Table 7
Suggested guidelines per Chinsky et al., 2017 and King et al., 2017, for evaluation and follow-up of patients diagnosed with tyrosinemia type I by newborn screening
| Evaluation | Initiation of | First year of life | From 1 year to 5 years of age | After 5 years | ||||
| therapy | Monthly | Every | Every | Every | Yearly | of age | ||
| (baseline) | 3 months | 3 months | 6 months | |||||
| Tyrosinemia type I markers | Blood SAa | X | Xb | X | Every 6 months | |||
| Urine SA | X | X | X | Every 6 months | ||||
| Blood NTBC levela | X | X | Every 6 months | |||||
| PAA | X | Xb | X | Every 6 months | ||||
| CBC: HB, HCT, WBC, PLT | X | X | X | Yearly | ||||
| Liver evaluation | Serum AFP | X | X | X | Every 6 monthsd | |||
| PT/PTT | X | Xe | X | Yearly | ||||
| ALT/AST | X | Xe | X | Yearly | ||||
| Bilirubin | X | |||||||
| Imaging: CT or MRI (with | X | Xc | Yearlyc | |||||
| contrast) or USf . g | ||||||||
| Renal evaluation | BUN/Creatinine/bicarbonate/ | X | X | Yearly | ||||
| Calcium/Phosphate | X | |||||||
| Urinalysis | X | |||||||
| Imaging (Renal US) | X | |||||||
| Development evaluation/ | Before | |||||||
| neuropsychology assessment | school age | |||||||
AFP, α-fetoprotein; ALT, alanine transaminase; AST, aspartate transaminase; BUN, blood urea nitrogen; CBC, complete blood count; CT, computed tomography; MRI, magnetic resonance imaging; NTBC,2-[2-nitro-4-trifluoromethylbenzoyl]-1,3-cyclohexanedione; Prot, protein; PT, prothrombin time; PTT, partial thromboplastin time; US, ultrasound; WBC, white blood count. aPhysician has choice of blood NTBC or SA. bCan switch to filter paper monitoring if the same is used over time for the baseline. cAs needed based on compliance. dAnyincrease, immediate imaging. eUntil normal. fNeed to wait 1-week post initiation of NTBC treatment to avoid anesthetic-induced porphyric crisis. gUltrasound: validity isoperator-dependent. hWhile doing liver imaging, and then only as clinically indicated after this.
Long-term care and non-compliance
It is important to be aware of noncompliance to both the lifelong pharmacological and dietary therapy in some early treated patients with tyrosinemia type I. Further, the fact that brief periods of noncompliance to the medication or diet can be asymptomatic can encourage noncompliance in general. The occurrence of corneal crystals and paralytic neurological crises are commonly seen in periods of noncompliance. Additionally, elevated blood SA levels carry long-term risk of somatic mutation in the liver resulting in hepatocellular carcinoma [158].
Tyrosinemia type II
Tyrosinemia type II also known as Richner-Hanhart syndrome or oculocutaneous tyrosinemia is caused by a deficiency of the enzyme tyrosine amino-transferase (TAT). TAT is the rate-limiting enzyme of tyrosine catabolism (Fig. 3), and primarily expressed in the liver. There are two separate TAT, one in the cytoplasm and the other in the mitochondria. The cytosolic TAT is defective in tyrosinemia type II. Tyrosinemia type II is characterized by skin and eye lesions and, occasionally, learning disability [111, 113] (Table 4). Patients have elevated tyrosine blood levels and an increase in urinary tyrosine metabolites. Tyrosinemia type II was first described by Richner 1 in 1938 and later by Hanhart 2 in 1947 as an oculo-cutaneous syndrome.
Genetics
The disorder is inherited in an autosomal recessive manner. The TAT gene, assigned to chromosome 16q22.1–22.5, has 12 exons, encodes for an active protein containing 454 amino acids that is transported to the mitochondria and requires pyridoxal phosphate as a co-factor. The disorder is more common in Italy, where a common pathogenic variant, p.Arg57Ter, has been identified [186]. TAT gene sequencing is a clinically available test. No genotype-phenotype correlation has been observed so far.
Clinical findings
It is characterized by bilateral dendritic keratitis, painful palmoplantar hyperkeratotic lesions, and mild learning disability [187]. The corneal and skin lesions are thought to be an inflammatory response secondary to the deposition of tyrosine crystals [188, 189].
Plasma values of tyrosine are perhaps highest in this particular form of tyrosinemia and may exceed 1,000 μmol/L in untreated patients. Unlike tyrosinemia type I, there is no associated liver damage and elevation in methionine. Idiopathic keratitis is not typically seen in children, which necessitates checking plasma tyrosine level in any pediatric patient with keratitis.
Eye symptoms can develop as early as the first day of life, typically before skin lesions develop and as late as 38 years [190, 191]. Many reported patients had ocular manifestations without skin involvements and vice versa. The early ocular signs consist of excessive tearing, redness, pain, and photophobia [188, 192]. Slit lamp examination may reveal some degree of corneal ulceration, that poorly stains with fluorescein and occasionally birefringent crystals of tyrosine may be observed. The dendritiform corneal lesions are frequently misdiagnosed as herpetic keratitis until the cultures are negative or negative response to antiviral treatment. Late signs include corneal clouding with central or paracentral opacities, neovascularization, scarring and impaired vision. Glaucoma has been reported as well [193].
Skin lesions typically consist of hyperkeratotic plaques on the soles of the feet and palms of the hands, on the pressure-bearing areas. They can also occur near the tips of the digits and in the subungual regions. These lesions, which may begin as bullae and erosions that progress to crusted, hyperkeratotic plaques, are painful and often associated with hyperhidrosis, but not pruritic [187]. The plantar surface of the digits may show dramatic yellowish thickening associated with the hyperkeratosis [194]. Typical hyperkeratotic lesions are popular, well demarcated with irregular borders. When biopsied, these lesions show non-specific findings that include acanthosis, parakeratosis and hyperkeratosis, but unlike the cornea, no crystals were seen on skin biopsies of these patients. Age at onset of skin lesions can range from the first few weeks of life to the second decade.
Neurodevelopmental abnormalities are highly variable features of tyrosinemia type II [113, 188]. Developmental delay is a common but not ubiquitous finding for patients reported with tyrosinemia type II, it has been reported in about half of the patients, with different levels of severity [195]. Hyperactivity, convulsions, microcephaly and abnormal language development have been observed as well. Those patients with the highest values of tyrosine early in life may be more likely to develop neurologic problems.
Diagnosis
PAA and urine organic acid analyses are the two important initial tests in diagnosing a child with tyrosinemia type II. Tyrosine is the only amino acid that accumulates. Such children have plasma tyrosine levels above 500 μmol/L, while other amino acids including methionine and phenylalanine are normal, which differentiates it from transient tyrosinemia of the newborn where they will be elevated. The reported tyrosine levels ranged between 1100 and 3300 μmol/L (20 and 50 mg/dL). Symptoms usually do not occur when tyrosine is below 1,000 μmol/L. Urine organic acids show increased excretion of p-hydroxyphenylpyruvate, p-hydroxyphenyllactate, p-hydroxyphenylacetate, and small quantities of N-acetyltyrosine, and 4-tyramine. The p-OH-tyrosine metabolites are formed by the action of the mitochondrial tyrosine aminotransferase (aspartate aminotransferase/AST) at high concentrations of accumulated tyrosine that lead to the formation of p-hydroxyphenylpyruvic acid [113]. The absence of generalized aminoaciduria differentiates tyrosinemia type II from type I.
Newborn screening
Tyrosinemia type II can be detected by newborn screening even though it is not officially in the screening panel [196]. Tyrosinemia type I, II, III, transient tyrosinemia, prematurity or live disease are in the differential diagnosis of elevated tyrosine, which is measured mainly to detect newborns with tyrosinemia type I.
Treatment
A low-protein diet and the use of a special formula free of phenylalanine and tyrosine are currently the most effective treatment in lowering the plasma level of tyrosine to less than 600 μmol/L [187]. This allows resolution of both eye and skin symptoms within days to several weeks. Usually patients are free of symptoms when plasma tyrosine level is below 550–700 μmol/L. The recommended blood tyrosine level is <500 μmol/L. Other reports suggest the initiation of dietary treatment in infancy to maintain serum tyrosine concentrations between 300 and 800 μmol/L with occasional peaks <1,000 μmol/L is associated with normal psychomotor development [187, 197].
Tyrosinemia type III
Tyrosinemia type III is an autosomal recessive disorder caused by a deficiency in the activity of 4-hydroxyphenylpyruvic acid dioxygenase (HPD), and is characterized by elevated levels of blood tyrosine and massive excretion of its derivatives into urine, including 4-hydroxyphenylpyruvic acid, 4-hydroxyphenyllactic acid, and 4-hydroxyphenylacetic acid (Fig. 3, Table 4) [114]. It is one of the rarest defects of tyrosine metabolism. A few patients have been identified with the disorder with variable but rather mild phenotype including mild developmental delay, autism, seizures, and ataxia with the absence of liver damage [198, 199].
The human HPD gene is located at 12q24-qter and contains 14 exons and codes for a protein containing 392 amino acids [200, 201]. Its activity occurs in the cytoplasm and utilizes ascorbic acid as an apparent stabilizer and natural co-factor for enzymatic activity. The enzyme is expressed primarily in the liver and kidney and is inhibited by NTBC [170].
The first three patients were brought to medical attention because of neurologic symptoms of intellectual disability or ataxia, and the fourth was detected on routine screening [111]. These patients have neither liver involvement, nor reported skin or ocular changes. In two of the patients a deficiency of 4-HPPD activity was documented as the etiology of their hypertyrosinemia [198, 199]. It remains unclear whether tyrosinemia type III is a predisposing factor strongly associated with cognitive delays, or if the association with mental retardation is purely on the basis of ascertainment bias [202, 203]. Szymanska (2015) [204], reported an 11-years old girl who presented with recurrent proteinuria but normal mental development and was diagnosed with tyrosinemia type III through metabolic screening (tyrosine ranging 425 to 535 μmol, normal values: 29–86 μmol/L) and elevated urinary excretion of p-hydroxyphenyl derivatives confirmed genetically with the homozygous c.479ANG (p.Tyr160Cys) missense variant in the HPD gene [204]. Another case is 4 months old girl with recurrent seizures, was diagnosed with tyrosinemia type III; labs showed increased blood tyrosine level (737 μmol/L and 915 μmol/L), (normal range 88–204 μmol/L) with elevated urinary levels of 4-hydroxyphenyl lactic acid and 4-hydroxyphenyl pyruvic acid but normal liver and renal functions. She was treated with a diet low in tyrosine and phenylalanine plus ascorbic acid supplementation (50 mg/day in two doses) with normalization of blood tyrosine and improvement of her symptoms [205].
Diagnostically, tyrosinemia type III is associated with lower tyrosine levels than those seen in tyrosinemia type II. Tyrosine levels range from approximately 350 to 650 μmol/L, accompanied by increased excretion of 4-hydroxyphenyl-pyruvic acid, 4-hydroxyphenyllactate, and 4-hydroxyphenylacetate [202]. The diagnosis is confirmed by detection of mutations in the HPD gene. Treatment consists of a diet low in tyrosine and phenylalanine, although whether this diet can prevent or reverse the neurologic symptoms is uncertain [202, 203].
Hawkinsinuria
Hawkinsinuria is a variant of HPD deficiency and is an autosomal dominant inborn error of metabolism [111, 113]. The term “Hawkinsin” was applied to the unusual compound detected in the urine, with the name recognizing the original family in whom the condition was identified [206]. The condition is characterized by persistent metabolic acidosis, failure to thrive that usually start after weaning off breast milk. The clinical symptoms spontaneously resolved around the first year of life in all patients reported.
The defect is located at the HPD enzyme converting 4-hydroxy-phenylpyruvate to homogentisate, the second step in tyrosine degradation pathway (Fig. 3). The HPPD enzyme is highly expressed in the liver and kidney. The reaction involved decarboxylation then oxidation step, followed by rearrangement step to form homogentisate. In the presence of defective HPD that cannot catalyze the final rearrangement step, the highly reactive epoxide intermediate forms and reacts with cysteine or glutathione and hydride anions to give ninhydrin-positive (2-L-cystein-S-yl-1.4-dihydroxycyclohex-5-en-1-yl)-acetic acid, called hawkinsin [207], and 4-hydroxycyclohexylacetic acid [208, 209] respectively. Hawkinsin is excreted early in life; the ability to form and excrete the latter compound develops gradually and is observed not before the 11th month of age [210, 211].
Genetics
Tomoeda et al. [200] have identified a single variant (p.Ala33Thr) that is common in each of the families in which Hawkinsinuria has been identified. Hawkinsinuria is somewhat unique in that it is inherited as an autosomal dominant, while other variants of HPD cause the autosomal recessive tyrosinemia type III. This heterozygous missense variant (p.Ala33Thr) in the HPD gene may produce a partially ineffective enzyme that is capable of decarboxylation and oxidation, but not of rearrangement. Having even one copy of this variant allows translation for an abnormal enzyme and formation of a reactive epoxide, causing symptoms in heterozygotes [206]. The clinical manifestation of HPD gene mutations depends on whether the binding and activation of 4-hydoxyphenylpyruvate itself is degraded, resulting in tyrosinemia type III or the rest of the catalytic site is malformed resulting in hawkinsinuria [212]. In addition to the p.Ala33Thr variant reported, c.722A>G (p.Asn241Ser) is another variant reported in 3 patients with hawkinsinuria [212, 213].
Clinical findings
Children with this condition may demonstrate chronic acidosis and failure to thrive if fed standard infant formulas or cow’s milk as their primary nutritional source (Table 4) [206, 211, 214]. Such nutritional source contains higher amount of tyrosine and phenylalanine, suggesting that diets low in these amino acids may ameliorate symptoms. However, metabolic acidosis is transient in most cases, and symptoms usually improve within the first year of life. However, Gomez-Ospina et al. [213], reported fraternal twin with hawkinsinuria whose symptoms persisted well beyond the first year of life without spontaneous resolution. Infants nourished with breast milk can apparently escape symptoms during infancy. Patients with this disorder may also have sparse and fine hair, hypotonia, mild intellectual disability and/or convulsions, with the absence of liver damage [111]. Additional clinical and biochemical findings reported include hypoglycemia, hyperlipidemia, proteinuria, and hemolytic anemia, renal manifestations of RTA (hypophosphatemia) and non-nephrotic-range proteinuria[213].
Diagnosis
In addition to the typical tyrosine compounds identified by organic acid analysis (4-hydroxyphenyllactic, 4- hydroxyphenylpyruvic, 4-hydroxyphenylacetic, homogentisic acid), patients with hawkinsinuria excrete the compound (2-L-cystein-S-yl-1, 4-dihydroxycyclohex-5-en-1-yl) acetic acid identified as the “Hawkinsin” compound in their urine throughout their life, even after all clinical symptoms improve [206, 210, 211]. Hawkinsin compound was interpreted as an intermediate in the transformation of 4-hydroxyphenylpyruvic acid to homogentisic acid. Other urinary compounds include quinolacetic, 4- hydroxycyclohexylacetic and pyroglutamic (5-oxoproline) acids.
Pathophysiology
The pathogenesis of hawkinsinuria is not fully understood. Intracellular glutathione depletion and subsequent oxidative stress, disrupting the functions of multiple organs and tissues, are thought to play major role in the pathogenesis of hawkinsinuria. Quinolacetic acid is thought to underlie symptoms by reacting with glutathione-forming thiol conjugates and hawkinsin, thereby depleting intracellular pools of glutathione [213]. Additionally, patients with hawkinsinuria excrete large quantities of pyroglutamic acid, a metabolic abnormality also seen in glutathione synthetase deficiency, 5-oxoprolinase deficiency, and acetaminophen overdose. The elevated pyroglutamic acid suggests that treatment with N-acetyl-L-cysteine (NAC), a prodrug of the glutathione precursor L-cysteine may serve to replenish glutathione stores, which is usually used to correct glutathione depletion in cases of acetaminophen poisoning. Gomez-Ospina et al., 2016, demonstrated a therapeutic strategy using NAC supplementation in treating fraternal twins with hawkinsinuria [213].
Treatment
Therapy for affected children consists of administration of ascorbic acid and a low-protein diet [206]. This prevents clinical acidosis and restores normal rates of somatic growth. Protein restriction has not been necessary after early childhood. Treatment with NAC quickly improved endocrine, hematological, and growth abnormalities in 2 patients with hawkinsinuria [213] and resulted in rapid clinical and biochemical improvements despite a concomitant increase in protein intake. NAC was administered orally at a dose of 150 mg/Kg/day. Urinary excretion of pyroglutamic, 4-hydroxyphenylacetic, and 4-hydroxyphenylpyruvic acids decreased and eventually normalized over 12 months following treatment with NAC, while 4-hdyroxycyclohexylacetic acid levels remained variable. Further, hawkinsin excretion decreased slightly with NAC therapy. NAC was approved by FDA in 2004 for use of acute acetaminophen toxicity and thus was not available as treatment in previously reported patients with hawkinsinuria.
Alkaptonuria
Alkaptonuria is a rare metabolic disease caused by deficiency of homogentisic acid oxidase. Because of this defect, homogentisic acid, which is produced during the metabolism of tyrosine cannot be converted to maleylacetoacetate [215] (Fig. 3), accumulates in tissues, and is excreted in the urine. This enzyme primarily can be found in the liver and kidney. It requires oxygen, ferrous iron, and sulfhydryl groups to open the ring of homogentisic acid. The biochemical reactions in alkaptonuria are shown in Fig. 5.
Fig.5
Alkaptonuria-Metabolic Pathway.
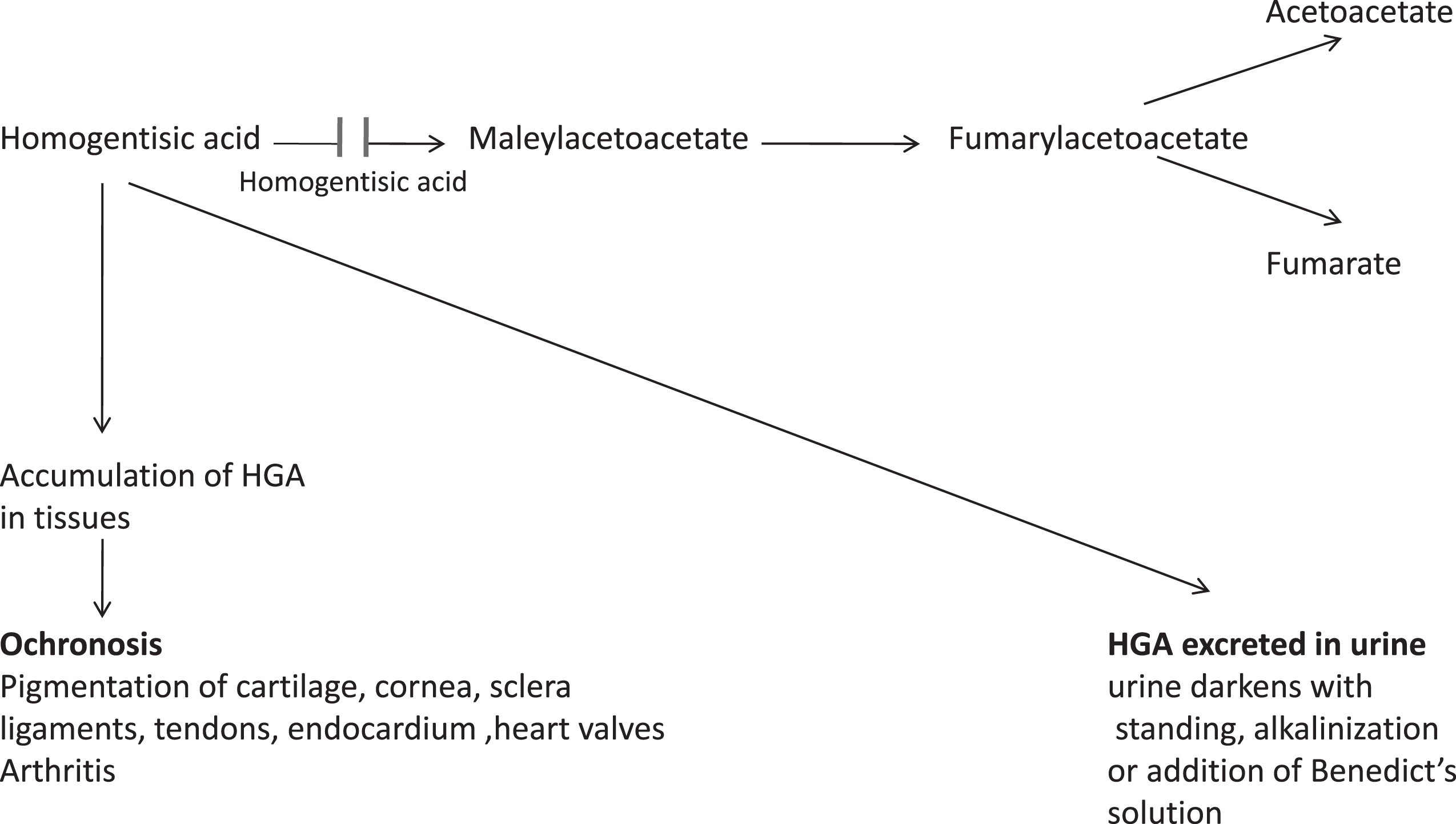
Accumulation of homogentisic acid and its metabolites causes ochronosis with darkening of cartilaginous tissues and bone, arthritis and joint destruction, and deterioration of cardiac valves [215, 216]. The estimated incidence of alkaptonuria is from 1 in 250,000 to 1 in 1,000,000 live births. The highest number of alkaptonuria was reported in Slovakia with incidence 1 in 19,000, and the Dominican Republic [217].
History
Despite its rarity, Alkaptonuria has long been described in the medical literature. An Egyptian mummy Harwa dated approximately 1500 BC showed the characteristic X-ray changes of alkaptonuria, extensive intervertebral disk calcification and narrowing of the hip and knee joints. Spectral analysis of pigment obtained from the hip region of the mummy resembled closely the pigment obtained by oxidation of homogentisic acid [218]. Several persons reported in the medical literature of the sixteenth and seventeenth centuries to continually pass dark urine are presumed to have had alkaptonuria [219]. The property of avid oxygen uptake in alkaline solution gave the substance the name “alcapton” [220] that was first used in 1859 to describe a patient’s urinary reducing compound. Two years later Boedker [221], spelled it “Alkapton”, and since then this condition has been known as “Alkaptonurie” in the German literature and as “Alcaptonurie” in the French literature. In 1891,”alcapton” was identified as 2, 5-dihydroxy-phenylacetic acid, or homogentisic acid [222].
Sir Archibald Garrod described alkaptonuria as the first disorder in humans to be found to conform to the principles of Mendelian autosomal recessive inheritance. He proposed that alkaptonuria results from a deficiency of an enzyme that normally splits the aromatic ring of homogentisic acid (HGA), a tyrosine-degradation product known to accumulate in patients with alkaptonuria [219].
Genetics
Mutation analysis in alkaptonuria patients has established defects of the homogentisate 1, 2-dioxygenase (HGD) gene as the cause of this metabolic disease [223]. It is inherited in autosomal recessive pattern. HGD gene is 54.3 kb in length, located on chromosome 3q21–23, contains 14 exons, coding for a 1715-bp transcript [224], most commonly missense variants. To date, more than 100 variants have been reported in the HGD gene [HGD mutation database] [217]. The majority of patients carry compound heterozygous variants; p.Met368Val is a common pathogenic variant in Europe except the Slovak population where mutational hot spots (e.g., c.342 + 1G>A) and a founder effect (e.g., the frequent pathogenic variant p.Gly161Arg) exist [215, 225, 226]. In the Dominican Republic, p.Cys120Trp is a founder variant [227]. No mutational hot spot or founder effect has been identified in the US [228]. There is no genotype-phenotype correlation observed between the HGD variant and disease severity. It is fully penetrant disease; elevated urinary HGA and ochronotic arthritis occur in all individuals with biallelic pathogenic variants in HGD gene.
Pathology
Cartilage in many areas, particularly the costal, laryngeal, and tracheal cartilage, is densely pigmented and is described as coal-black in some areas (Fig. 6). Pigmentation is also present in connective tissues, fibrocartilage, tendons, and ligaments and particularly in the intervertebral disk spaces (Fig. 7). To a lesser degree, it is also found in the endocardium, the intima of the larger vessels, and organs such as kidney (Fig. 8A), prostate (Fig. 8B), dura mater of the spinal cord (Fig. 9), lung, and epidermis. Microscopic examination shows the pigment to be deposited both intracellularly [229] and extracellularly, and it may be either granular or homogeneous. Electron microscopic analysis of alkaptonuric synovial membranes [230], hip joint tissue [231], and articular cartilage [232] shows in greater detail the fragments of cartilage stained with pigment derived from homogentisic acid (Fig. 10). Like melanin, the ochronotic pigment is bleached when treated for 24 hours with hydrogen peroxide, and it is soluble in alkali but only slightly soluble in hydrochloric acid. Thus, in many of its chemical characteristics, the ochronotic pigment resembles melanin arising from 3,4-dihydroxyphenylalanine (dopamine). No specific stain distinguishes the ochronotic pigment of alkaptonuria from melanin derived from other sources. The pigment deposited in ochronosis is presumably a polymer derived from homogentisic acid, but its exact chemical structure has not yet been determined.
Fig.6
Alkaptonuria-Black pigmental cartilage. (Figure 14 in the first book).
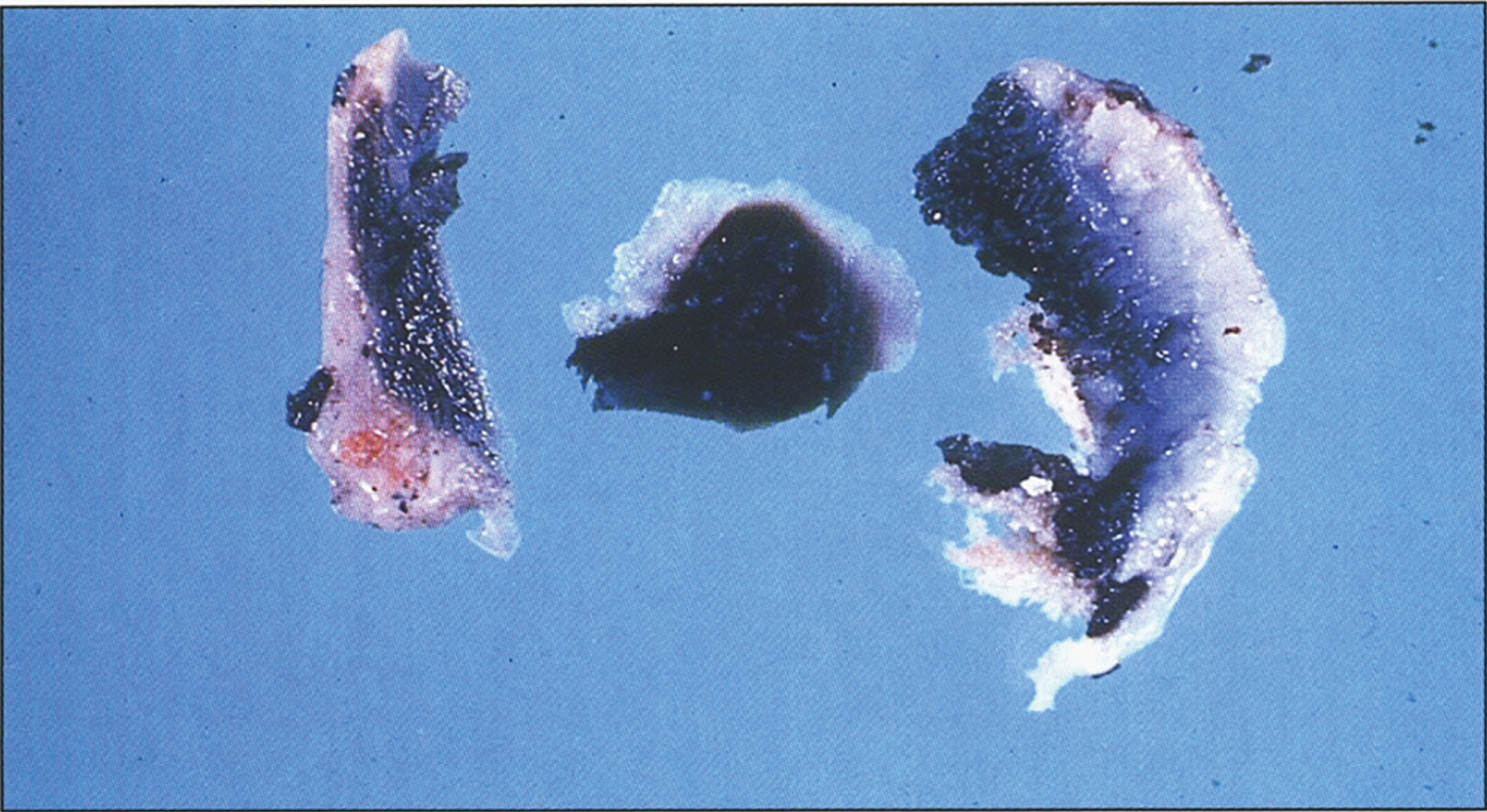
Fig.7
Alkaptonuria-Dense black pigmentation of the intervertebral disks of the vertebrae. (Figure 15 in the first book).

Fig.8
Alkaptonuria- (A) Microscopic section of kidney with pigment casts in the renal tubules. (B) Microscopic section of prostate with pigment casts in the prostatic glands. (Figure 17 in the first book).
Fig.9
Alkaptonuria- A segment of the spinal cord covered by densely pigmented dura mater. (Figure 18 in the first book).
Fig.10
Alkaptonuria- Electron micrography of cartilage with dense pigment deposit in the collagen. (Figure 20 in the first book).
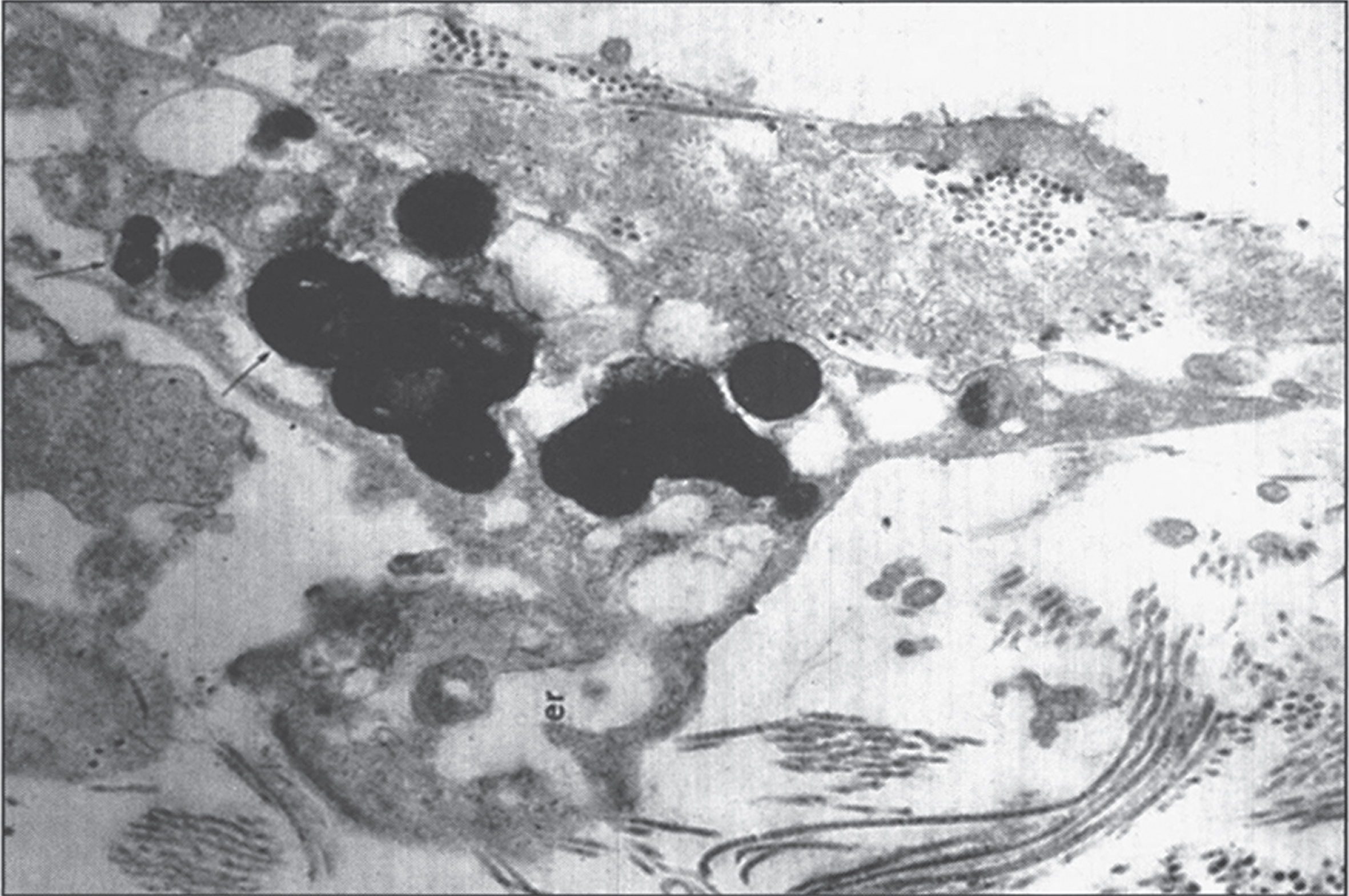
Skin and cartilage contain an enzyme, homogentisic acid polyphenol oxidase, which catalyzes the oxidation of homogentisic acid to an ochronotic-like pigment [233]. Benzoquinone acetic acid has been identified as an intermediary metabolite in the oxidation. The enzyme is a copper protein, but it is clearly distinguished from tyrosinase because tyrosine, DOPA, and other catechols are not substrates for the polyphenol oxidase. Binding and chemical reactions of benzoquinone with connective tissues are believed to produce important chemical changes that alter tissue constituents and lead to ochronosis and ochronotic arthritis [234].
Clinical findings
Ochronosis
Connective tissue ochronosis and arthritis of the spine and large joints (hips, knees, shoulders) characterize alkaptonuria. Small joint involvement is not significant. The pigmentary changes are usually seen after age 30 years. The first description of ochronosis was in 1866 when Virchow described a peculiar type of pigmentation of the connective tissue in a 67-year-old man [235]. The pigment was bluish black, and thus he named the condition ochronosis. Forty years later, Albrecht [236] demonstrated the connection between ochronosis and alkaptonuria. Osler diagnosed ochronosis clinically for the first time in 2 brothers with alkaptonuria [237]. Brown pigmentation of the sclera between the cornea and inner and outer canthi at insertion of recti muscles can be seen. This pigmentation does not affect vision. A more diffuse pigmentation may also involve the conjunctiva and cornea [238]. Slight pigmentation of the sclera or the ears is rarely noticeable before the patient is 20 or 30 years old. Typical pigmentary changes in the ear cartilages occur only in longstanding alkaptonuria and the ear cartilage feels irregular or thickened. The pigment appears in cerumen, perspiration; clothing near the axillary regions may be stained, and the skin may have a deep purple or black discoloration in the axillary and genital regions [216, 234] and on the skin of the hands corresponding to the underlying tendons, or in the web between the thumb and index finger.
Ochronotic arthritis is a manifestation of long-standing alkaptonuria [239]. It occurs at an earlier age and is more severe in males than in females [239], even though the sex incidence of alkaptonuria is roughly equal. Roentgenographically, it resembles osteoarthritis [240, 241]. The arthritis and other ochronotic manifestation are thought to be the result of from binding of highly reactive oxidation products of homogentisic acid to cartilage and other tissues. The earliest symptoms are usually some degree of limitation of motion of the hips, knee joints, or occasionally, the shoulders. There may be acute inflammation that resembles rheumatoid arthritis and later, marked limitation of motion and ankylosis in the lumbosacral region. Spine symptoms usually appear in the third decade, sparing sacroiliac joint. Lumbar and thoracic spine symptoms occur before cervical spine symptoms. The spinal involvement results in kyphosis, height loss, and decreased lumbar flexion, and the joint disease decreases the range of motion and causes effusions [242]. Limitation of spine flexion directly correlates with degree of disability and impaired function. About 50% of individuals with alkaptonuria require at least one joint replacement by age 55 years. Reduced renal function can accelerate the development of ochronosis and joint destruction as a result of accumulation of massive quantities of HGA. In addition to the joint involvement, tendons can also be involved resulting in thickened Achilles tendon, tendonitis, and rupture.
X-rays and Magnetic Resonance Imaging (MRI) reveal changes pathognomonic of alkaptonuria [243–245]. The vertebral bodies of the lumbar spine show degeneration of the intervertebral disks with a narrowing of the space and dense calcification of the remaining disk material with a variable degree by fusion of the vertebral bodies. In contrast to rheumatoid spondylitis, little osteophyte formation and minimal calcification of the intervertebral ligaments are present. The joints involved also differ from osteoarthritis in that the degenerative joint changes in ochronotic arthritis are most commonly in the shoulder and hip, whereas some joints such as the sacroiliac may be completely spared. Joint symptoms typically begin in the third or fourth decade of life and progress until chronic pain prompts a knee, hip, or shoulder replacement usually in mid 50s. Calcification of the ear cartilage may be observed on X-ray films, as well as calcified deposits most commonly in muscle and tendons around the large joints. The smaller joints usually show little or no abnormality.
MRI has proved more accurate than X-rays for individualizing the lesions and recognizing alterations, such as the thickness of the anterior longitudinal ligament. In the case of new diagnosis, an MRI is fundamental to recognize typical signs of the ochronotic arthropathy not well detected by X-rays [244].
Urinary changes
The urine is usually dark or turns dark upon standing (sometimes noticed several hours after voiding), or exposure to an alkaline agent.
Cardiac complications of alkaptonuria
There is a high incidence of heart disease [229, 246–248] secondary to the pigment deposition in the heart valves and blood vessels leading to valvular heart disease independent of standard cardiovascular risk factors. This include aortic or mitral valve calcification with stenosis or regurgitation and occasionally aortic dilatation. Aortic valve stenosis occurs at a high frequency in the sixth and seventh decades of life. Aortic stenosis may necessitate aortic valve replacement [249]. The autopsy findings in 11 patients with ochronosis indicated that 8 had chronic mitral and aortic valvulitis, 1 had an aortic aneurysm, and 1 had an aneurysm of the left ventricle. Generalized arteriosclerosis and calcification in the heart valves and of the annulus of the aortic and mitral valves have been noted [250, 251]. Phornphutkul et al showed that aortic dilatation or cardiac-valve involvement developed in 58 patients at around 50 years of age [216]. No patient had coronary-artery calcification before the age of 40 years, but 50 percent had computed tomographic (CT) evidence of coronary-artery calcification by the age of 59. There was no correlation between coronary-artery calcification and an elevated serum cholesterol level. Myocardial infarction is a common cause of death [252]. Deposition of the pigment in atherosclerotic plaques, particularly of the aorta (Fig. 11) and the coronary vessels augments and enhances the atherosclerotic process. CT and echocardiographic studies can reveal coronary-artery calcifications and cardiac-valve deterioration, respectively.
Fig.11
Alkaptonuria- A segment of the aorta showing pigmented atherosclerotic plaques. The presence of the pigment augments and accelerates the atherosclerotic process. (Figure 21 in the first book).
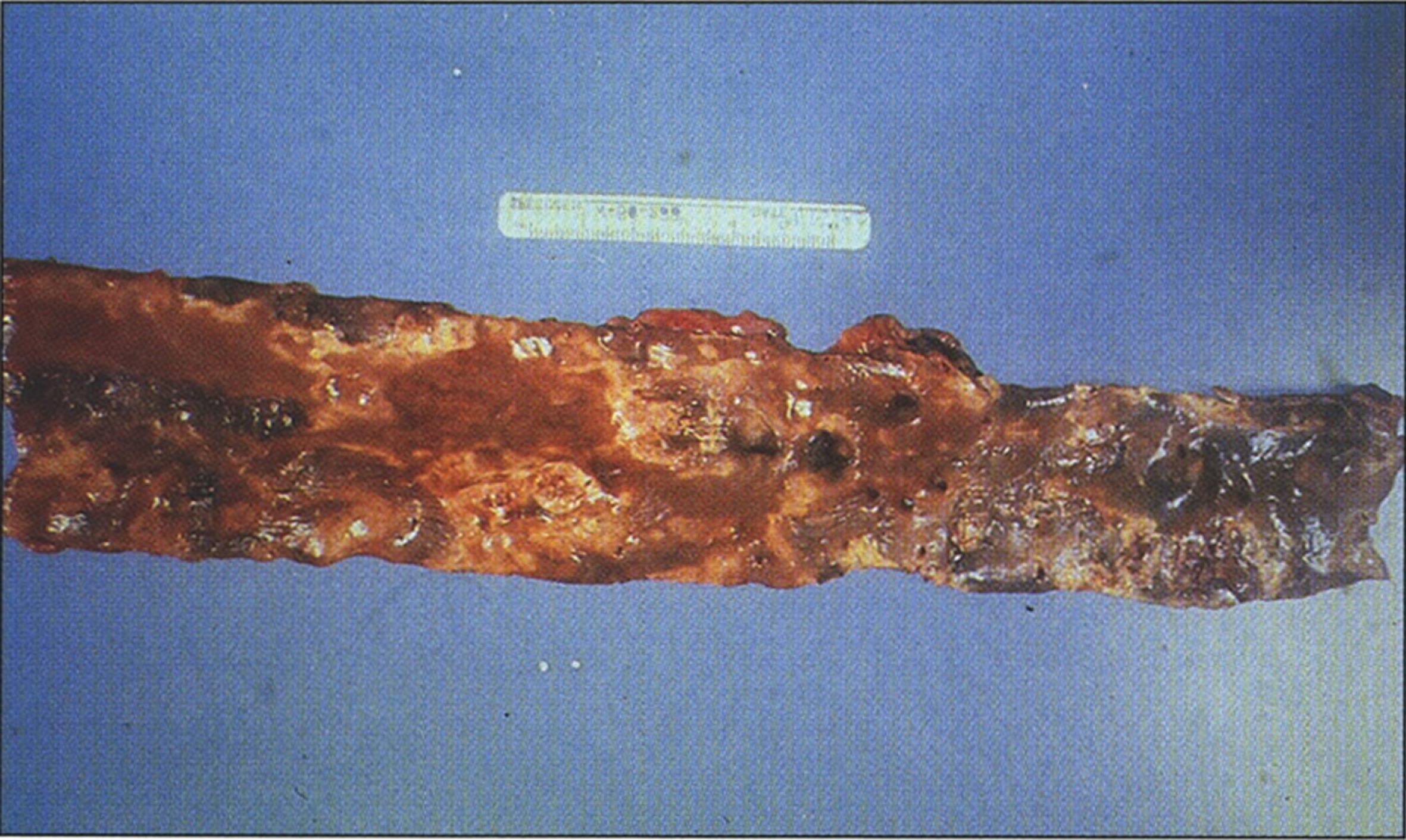
Other complications
These include ruptured intervertebral disks, prostate black stones, prostatitis, and renal stones 50% by age 64 [253–260]. Associations with polycythemia [261], nephrocalcinosis [262], and severe renal disease called “ochronotic nephrosis” have been reported.
Of note, alkaptonuria does not cause developmental delay or cognitive impairment and does not generally reduce the life span.
Diagnosis
Because of the presence of homogentisic acid, people with alkaptonuria have a history of dark urine or urine that turns dark on standing; however, a large number of patients have never noted any abnormality in the color of their urine during childhood [259, 263, 264], and diagnosis has been made only after they sought treatment for arthritis during their later years [263–267] with the unusual and distinctive X-ray changes in the spine. The disease may not be suspected until a surgical procedure reveals marked pigmentation of the cartilage.
If urine containing homogentisic acid is allowed to stand for some time, it gradually turns dark as the acid is oxidized to a melanin-like product. The polymerization is accelerated by alkali; this explains why washing diapers of infants with alkaptonuria with soaps tends to make the stains more intense instead of removing them. Vitamin C protects homogentisic acid against oxidation. With the addition of Benedict’s sugar reagent, homogentisic acid not only reduces the copper reagent to yield a yellow-orange precipitate (Fig. 12), but it also undergoes darkening because of the alkalinity of the reagent. The effect is an orange precipitate in a muddy brown solution [268, 269].
Fig.12
Alkaptonuria- The reaction of urine to oxidation, alkali, and Benedict’s solution. (Figure 13 in the first book).
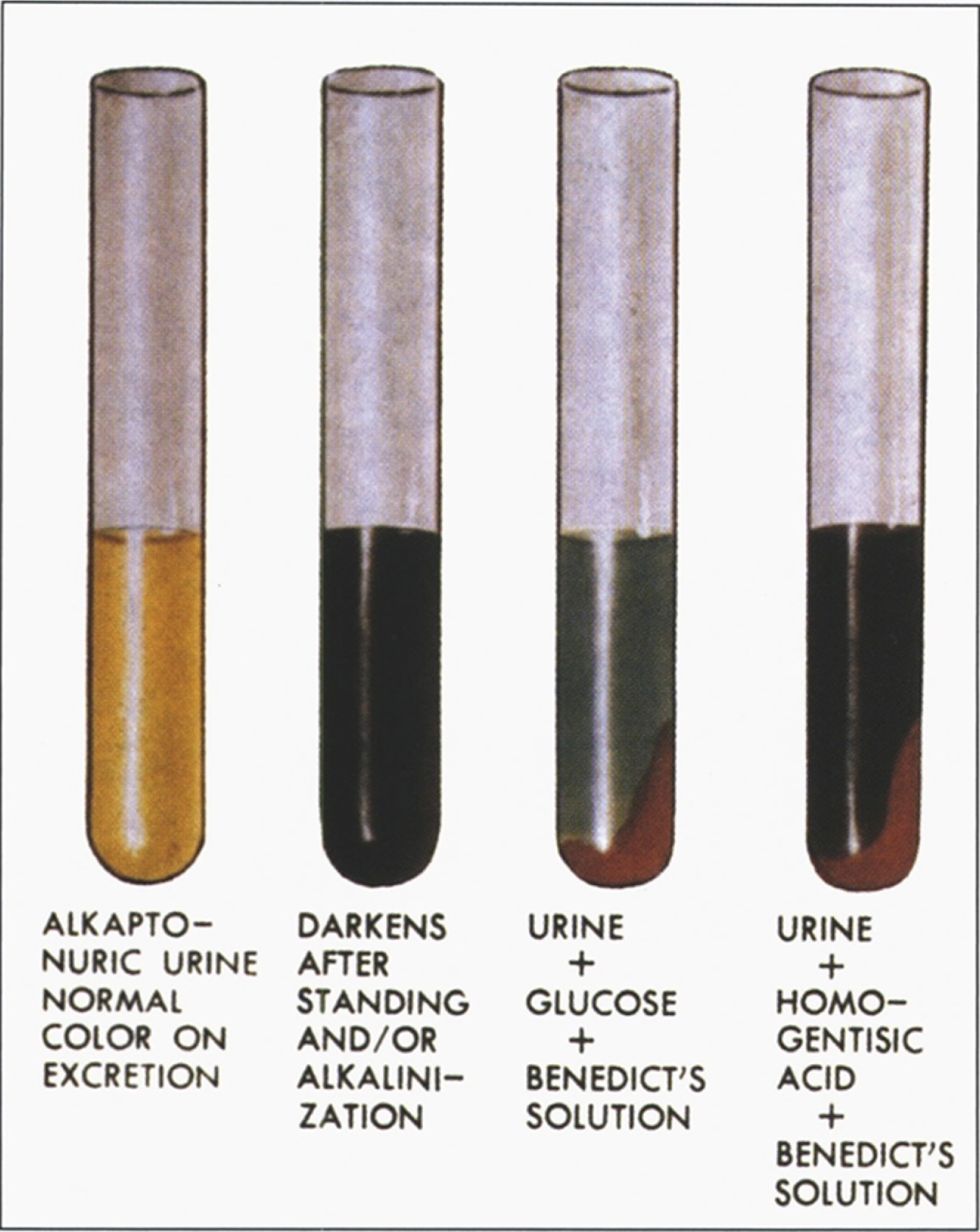
Biochemical diagnosis
Diagnosis of the disorder, which currently relies on documentation of an elevation in urinary HGA excretion by a factor of 100 to 600, is on the basis of gas chromatography–mass spectrometric analysis of organic acids. The amount of HGA excreted per day in individuals with alkaptonuria is usually between 1 and 8 grams. A normal 24-hour urine sample contains 20–30 mg of HGA [260]. Elevated HGA can be detected on a random urine sample.
Molecular diagnosis
Molecular diagnosis is not needed to establish the diagnosis but will help in determining carrier status and in prenatal testing.
Prenatal testing
Prenatal molecular genetic testing for at risk pregnancy is possible if the HGD pathogenic variants have been identified in an affected family member. No enough studies confirming the reliability of biochemical testing in affected fetus by measuring homogentisic acid in amniotic fluid.
Carrier testing and evaluation of relatives at risk
Molecular genetic carrier testing can be offered for at-risk relatives if the HGD pathogenic variant is known in the family. Biochemical testing is not reliable as a method of carrier detection. However, testing the urine for HGA in asymptomatic sibs of affected individual allows for early diagnosis of alkaptonuria and subsequent intervention.
Management
Evaluations following initial diagnosis of alkaptonuria
It is important to obtain thorough evaluation of newly diagnosed patients in order to establish the extent of disease. Such evaluation includes complete history and physical examination with particular attention to spine and large joints range of motion, physical medicine and rehabilitation evaluation in case of limited range of motion or joint pain, electrocardiogram and echocardiogram in individuals older than age 40 years, renal ultrasound examination or helical abdominal CT to evaluate for the presence of renal calculi and finally, consultation with a clinical geneticist and/or genetic counselor [260].
Treatment
There is no preventive or curative treatment available for alkaptonuria, however, several therapeutic approaches have been used. Surveillance for cardiac, renal, and prostate complications after the fourth decade of life and strict attention to pain control are advisable. Joint pain management is essential in patients with alkaptonuria. Both physical and occupational therapy are important to strengthen the muscles and maintain flexibility. Joint replacement surgeries of knee, hip and/or shoulder might be necessary for severe arthritis with the goal to relieve the pain rather than to increase range of movements. Prosthetic joint survival in patients with alkaptonuria is comparable to that found in individuals with osteoarthritis [270]. Renal and prostate stones might need surgical intervention. Aortic valve stenosis might require replacement.
In the natural history study, neither vitamin C supplementation nor protein restriction of phenylalanine and tyrosine showed a reduction in urinary HGA excretion [216]. Further, oral bisphosphonate therapy did not show any benefits in halting the progressive bone loss in four affected individuals [271].
Nitisinone, a triketone herbicide that inhibits 4-hydroxyphenylpyruvate dioxygenase, reduces HGA production, and theoretically could be an ideal therapy for alkaptonuria. Orfadin® (nitisinone), also known as NTBC, is approved by the Food and Drug Administration for the treatment of tyrosinemia type I (see above discussion of tyrosinemia type I) but not yet for alkaptonuria. The only known side effects of NTBC are elevated plasma tyrosine levels that could cause corneal irritation, photophobia and rarely corneal crystals. Patients on NTBC should be on a low protein diet to maintain tyrosine levels within acceptable range, which is less than 500 μmol/L. Oral NTBC (approximately 0.1 mg) reduced urinary HGA excretion by more than 80% in a murine model of alkaptonuria [272]. Phornphutkul et al. reported that two patients, who received small dose of nitisinone, showed dramatic decrease in their HGA extraction [216]. Twenty adult patients (age 38–68 years) with alkaptonuria received NTBC (2 mg/day) over a 36-month period in a prospective, randomized, placebo-controlled study. The study subjects showed 95% reduction of HGA in urine and plasma over the course of 3 years, but no clinical benefits were detected probably due to their advanced disease [273]. In an open label, single center study, low-dose NTBC (at a dosage of 1.05 mg bid) reduced urinary HGA by up to 94% in nine individuals with alkaptonuria. In the same study, seven individuals were treated for up to 15 weeks with NTBC while receiving normal protein intake; all had elevated plasma tyrosine concentrations but no ophthalmic, neurologic, or severe dermatologic complications were observed. Two individuals had transient transaminitis that resolved after stopping NTBC [274].
In a three-year therapeutic trial by conducted Introne et al 2011 [273], administration of 2 mg of NTBC daily resulted in reduction of urine and plasma HGA by 95%. Plasma tyrosine averaged 800 μmol/L without dietary restriction with minimal side effects. One affected individual developed corneal crystals that required discontinuation of NTBC, and one affected individual had elevated liver transaminases. There was no statistically significant improvement in hip range of motion and measurements of musculoskeletal function observed in the treatment group compared to the control group; however, there was a positive trend showing slowing of aortic stenosis [273]. Further clinical trials are currently underway to establish clinical benefit of NTBC in the treatment of alkaptonuria to assess the dose, efficacy, and the timing of initiation of the treatment.
Prevention of joint complications
Patients diagnosed with alkaptonuria should be counseled about maintaining joint range of motion through moderate non-weight-bearing exercise such as swimming and avoiding physical stress to the spine and large joints, contact and high impact sports. Additionally, patients should avoid any occupation that involves heavy manual labor. Further, instruction on joint strengthening and flexibility exercises as well as appropriate physical activity, can help preserve overall joint mobility and function [260].
Surveillance
Cardiac surveillance
For patients older than 40 years, surveillance for cardiac complications is advisable every one to two years and should include echocardiography to detect aortic dilation and aortic or mitral valve calcification and stenosis and CT scans to detect coronary artery calcification [260].
Urological surveillance
It is important to be aware of the urologic complications associated with alkaptonuria, which usually occur after the age of 40 years. However, routine surveillance for such complications is not recommended. Ochronotic prostate stones and kidney stones usually appear on radiography such as ultrasonography and helical abdominal CT [260].
References
[1] | Scriver C. and Kaufman S. . Hyperphenylalaninemia: phenylalanine hydroxylase deficiency, Metab. Mol. Bases Inherit. Dis. ((2001) ), 1667–1724. doi: 10.1036/ommbid.97. |
[2] | Aoki K. and Wada Y. . Outcome of the Patients Detected by Newborn Screening in Japan, Pediatr. Int. 30: ((1988) ), 429–424. doi: 10.1111/j.1442-200X.1988.tb02533.x. |
[3] | Regier G.C. . DS, Phenylalanine Hydroxylase Deficiency - GeneReviews® –NCBI Bookshelf, GeneReviews, 2017. https://www.ncbi.nlm.nih.gov/books/NBK/1504/ (accessed June 24 2020). |
[4] | Räihä N.C.R. . Phenylalanine hydoxylase in human liver during development, Pediatr. Res. 7: ((1973) ), 1–4. doi: 10.1203/00006450-197301000-00001. |
[5] | Jakubovič A. . Phenylalanine-hydroxylating system in the human fetus at different developmental ages, BBA - Gen. Subj. 237: ((1971) ), 469–475. doi: 10.1016/0304-4165(71)90265-0. |
[6] | Ryan W.L. and Orr W. . Phenylalanine conversion to tyrosine by the human fetal liver, Arch. Biochem. Biophys. 113: ((1966) ), 684–686. doi: 10.1016/0003-9861(66)90248-7. |
[7] | Friedman P.A. and Kaufman S. . A study of the development of phenylalanine hydroxylase in fetuses of several mammalian species, Arch. Biochem. Biophys. 146: ((1971) ), 321–326. doi: 10.1016/S0003-9861(71)80070-X. |
[8] | Følling I. . The discovery of phenylketonuria, Acta Pædiatrica. 83: ((1994) ), 4–10. doi: 10.1111/j.1651-2227.1994.tb13440.x. |
[9] | Bickel H. . The effects of a phenylalanine-free and phenylalanine-poor diet in phenylpyruvic oligophrenis, Exp. Med. Surg. 12: ((1954) ), 114–117. doi: 10.1159/000382329. |
[10] | Guthrie S.A. . R, A simple phenylalanine method for detecting phenylketonuria in large populations of newborn infants - pubmed, Pediatric. ((1963) ) 338–343. https://pubmed.ncbi.nlm.nih.gov/14063511/(accessed June 25 2020). |
[11] | Schulze A. , Kohlmueller D. and Mayatepek E. . Sensitivity of electrospray-tandem mass spectrometry using the phenylalanine/tyrosine-ratio for differential diagnosis of hyperphenylalaninemia in neonates, Clin. Chim. Acta. 283: ((1999) ), 15–20. doi: 10.1016/S0009-8981(99)00016-9. |
[12] | Norman R. , Haas M. and Wilcken B. . International perspectives on the cost-effectiveness of tandem mass spectrometry for rare metabolic conditions, Health Policy (New. York). 89: ((2009) ), 252–260. doi: 10.1016/j.healthpol.2008.08.003. |
[13] | Anderson P.J. and Leuzzi V. . White matter pathology in phenylketonuria, Mol. Genet. Metab. 99: ((2010) ), doi: 10.1016/j.ymgme.2009.10.005. |
[14] | Guzzetta V. . Phenylketonuria in Italy: Distinct distribution pattern of three mutations of the phenylalanine hydroxylase gene, J. Inherit. Metab. Dis. 20: ((1997) ), 619–624. doi: 10.1023/A:1005315106604. |
[15] | Trefz F.K. , Scheible D. , Götz H. , Frauendienst-Egger G. . Significance of genotype in tetrahydrobiopterin-responsive phenylketonuria, in J Inherit Metab Dis. (2009) : pp. 22–23, doi: 10.1007/s10545-008-0940-8. |
[16] | Malamud N. . Neuropathology of phenylketonuria, J. Neuropathol. Exp. Neurol. 25: ((1966) ), 254–268. doi: 10.1097/00005072-196604000-00006. |
[17] | Alvord E.C. , Stevenson L.D. , Vogel F.S. , Engle R.L. . Neuropathological Findings in Phenyl-Pyruvic Oligophrenia (Phenyl-Ketonuria), J. Neuropathol. Exp. Neurol. 9: ((1950) ), 298–310. doi: 10.1097/00005072-195007000-00004. |
[18] | Oteruelo F.T. . “PKU Bodies”: Characteristic inclusions in the brain in phenylketonuria, Acta Neuropathol. 36: ((1976) ), 298–305. doi: 10.1007/BF00699635. |
[19] | Agrawal D.A. , HC, Biochemistry of the Developing Brain: Myelination and amino acid imbalance in the develoing brain, 1973 https://books.google.com/books?id=YpfzDQAAQBAJ&pg=PA99&lpg=PA99&dq=20.% 09 Agrawal+HC,+Davison+AN:+Myelination+and+amino+acid+imbalance+in+the+developing+brain.+In+Himwich+WA,+editor,+Biochemistry+of+the+Developing+Brain,+vol+1,+New+York,++Dekker,+p+14 (accessed June 25, 2020). |
[20] | Landvogt C. , Mengel E. , Bartenstein P. , Buchholz H.G. , Schreckenberger M. , Siessmeier T. , Scheurich A. , Feldmann R. , Weglage J. , Cumming P. , Zepp F. and Ullrich K. . Reduced cerebral fluoro-L-dopamine uptake in adult patients suffering from phenylketonuria, J. Cereb. Blood Flow Metab. 28: ((2008) ), 824–831. doi: 10.1038/sj.jcbfm.9600571. |
[21] | de Groot M.J. , Hoeksma M M. , Blau N , Reijngoud D.J and van Spronsen F.J. , Pathogenesis of cognitive dysfunction in phenylketonuria: Review of hypotheses, Mol. Genet. Metab. 99: ((2010) ), doi: 10.1016/j.ymgme.2009.10.016. |
[22] | Rech V.C. , Feksa L.R. , Dutra-Filho C.S. , De Souza Wyse A.T. , Wajner M. and Wannmacher C.M.D. , Inhibition of the mitochondrial respiratory chain by phenylalanine in rat cerebral cortex, Neurochem. Res. 27: ((2002) ), 353–357. doi: 10.1023/A:1015529511664. |
[23] | Wasserstein M.P. , Snyderman S.E. , Sansaricq C. , Buchsbaum M.S. . Cerebral glucose metabolism in adults with early treated classic phenylketonuria, Mol. Genet. Metab. 87: ((2006) ), 272–277. doi: 10.1016/j.ymgme.2005.06.010. |
[24] | Pietz J. , Rupp A. , Ebinger F. , Rating D. , Mayatepek E. , Boesch C. and Kreis R. . Cerebral energy metabolism in phenylketonuria: Findings by quantitative 31P MR spectroscopy, Pediatr. Res. 53: ((2003) ), 354–662. doi: 10.1203/01.PDR.0000055867.83310.9E. |
[25] | Vockley J. , Andersson H.C. , Antshel K.M. , Braverman N.E. , Burton B.K. , Frazier D.M. , Mitchell J. , Smith W.E. , Thompson B.H. , Berry S.A. . Phenylalanine hydroxylase deficiency: Diagnosis and management guideline, Genet. Med. 16: ((2014) ), 188–200. doi: 10.1038/gim.2013.157. |
[26] | Cockburn F. , Barwell B.E. , Brenton D.P. , Chapple J. , Clark B. , Curzon G. , Davidson D.C. , Heeley A.F. , Laing S.C. , Lister-Cheese I.A.F. , McDonald I. , Malcolm S. , Pollitt R.J. , Quinn D. , Rylance G. , Smith I. , Stewart A. , Surtees R. , Thompson A.J. . Recommendations on the dietary management of phenylketonuria, Arch. Dis. Child. 68: ((1993) ), 426–427. doi: 10.1136/adc.68.3.426. |
[27] | White DA N.M.M.T.H.K.S.R . Deficits in Memory Strategy Use Related to Prefrontal Dysfunction During Early Development: Evidence From Children With Phenylketonuria - PubMed, Neuropsychology. 2: ((2001) ), 221–229. https://pubmed.ncbi.nlm.nih.gov/11324865/ (accessed June 25, 2020). |
[28] | Anastasoaie V. , Kurzius L. , Forbes P. and Waisbren S. . Stability of blood phenylalanine levels and IQ in children with phenylketonuria, Mol. Genet. Metab. 95: ((2008) ), 17–20. doi: 10.1016/j.ymgme.2008.06.014. |
[29] | Koch R. , Azen C. , Friedman E.G. , Williamson M.L. . Paired comparisons between early treated PKU children and their matched sibling controls on intelligence and school achievement test results at eight years of age, J. Inherit. Metab. Dis. 7: ((1984) ), 86–90. doi: 10.1007/BF01805813. |
[30] | Liu S.R , Zuo Q.H . Newborn Screening for Phenylketonuria in Eleven Districts - PubMed, Chin Med J. 2: ((1986) ), 113–118. https://pubmed.ncbi.nlm.nih.gov/1573093157/ (accessed June 25, 2020). |
[31] | Rickel H , Bachmann C and Beckers R . Neonatal mass screening for metabolic disorders, Eur J Pediatr. ((1981) ), 133. https://books.google.com/books?id=YpfzDQAAQBAJ&pg=PA100&lpg=PA100&dq=29.% 09Rickel+H,+Bachman n+C,+Beckers+R:+Neonatal+mass+screening+for+metabolic+disorders.+Eur+J+Pediatr+137:133,+&source= bl&ots=HEOb8siCmC&sig=ACfU3U1nlU7AqXQrPr4joGHL_SLYWwErPQ&hl=en&sa=X&ved=2ahUKEwirz_O UoZzqAhV8g3IEHSXMDF4Q6AEwBHoECAMQAQ#v=onepage&q=29.% 09Rickel+H% 2C+Bachmann+C% 2C+Beckers+R% 3A+Neonatal+mass+screening+for+metabolic+disorders.+Eur+J+Pediatr+137% 3A133% 2C+&f=false (accessed June 25, 2020). |
[32] | Fisch R.O , Comments on diet and compliance in phenylketonuria, in: Eur. J.ediatr. Suppl., Springer Verlag, (2000) . doi: 10.1007/pl00014378 |
[33] | Walter J.H. , White F.J. , Hall S.K. , MacDonald A. , Rylance G. , Boneh A. , Francis D.E. , Shortland G.J. , Schmidt M. and Vail A. . How practical are recommendations for dietary control in phenylketonuria? Lancet. 360: ((2002) ), 55–57. doi: 10.1016/S0140-6736(02)09334-0. |
[34] | Waisbren S.E. , Noel K. , Fahrbach K. , Cella C. , Frame D. , Dorenbaum A. and Levy H. . Phenylalanine blood levels and clinical outcomes in phenylketonuria: A systematic literature review and meta-analysis, ((2007) ). doi: 10.1016/j.ymgme.2007.05.006. |
[35] | Coakley K.E. , Douglas T.D. , Goodman M. , Ramakrishnan U. , Dobrowolski S.F. , Singh R.H. . Modeling correlates of low bone mineral density in patients with phenylalanine hydroxylase deficiency, J. Inherit. Metab. Dis. 39: ((2016) ), 363–372. doi: 10.1007/s10545-015-9910-0. |
[36] | Opladen T. , Hoffmann G.F. and Blau N. . An international survey of patients with tetrahydrobiopterin deficiencies presenting with hyperphenylalaninaemia, J. Inherit. Metab. Dis. 35: ((2012) ), 963–973. doi: 10.1007/s10545-012-9506-x. |
[37] | Dhondt J.L. . Strategy for the screening of tetrahydrobiopterin deficiency among hyperphenylalaninaemic patients: 15-Years experience, J. Inherit. Metab. Dis. 14: ((1991) ), 117–127. doi: 10.1007/BF01800581. |
[38] | Smith I. , Beasley M.G. , Ades A.E. . Intelligence and quality of dietary treatment in phenylketonuria, Arch. Dis. Child. 65: ((1990) ), 472–478. doi: 10.1136/adc.65.5.472. |
[39] | Schulze A. , Mayatepek E. , Hoffmann G.F. . Evaluation of 6-year application of the enzymatic colorimetric phenylalanine assay in the setting of neonatal screening for phenylketonuria, Clin. Chim. Acta. 317: ((2002) ), 27–37. doi: 10.1016/S0009-8981(01)00736-7. |
[40] | Berlin C.M. , Levy H.L. , Hanley W.B. . Delayed increase in blood phenylalanine concentration in phenylketonuric children initially classified as mild hyperphenylalaninemia, Screening. 4: ((1995) ), 38–39. doi: 10.1016/0925-6164(95)00105-U. |
[41] | Van Wegberg A.M.J. , MacDonald A. , Ahring K. , Bélanger-Quintana A. , Blau N. , Bosch A.M. , Burlina A. , Campistol J. , Feillet F. , Giżewska M. , Huijbregts S.C. , Kearney S. , Leuzzi V. , Maillot F. , Muntau A.C. , Van Rijn M. , Trefz F. , Walter J.H . Van Spronsen F.J. , The complete European guidelines on phenylketonuria: Diagnosis and treatment, Orphanet J. Rare Dis. 12: ((2017) ), doi: 10.1186/s13023-017-0685-2. |
[42] | Weglage J. , Pietsch M. , Feldmann R. , Koch H.G. , Zschocke J. , Hoffmann G. , Muntau-Heger A. , Denecke J. , Guldberg P. , GüTtler F. , MLler H. , Wendel U. , Ullrich K. and Harms E. . Normal clinical outcome in untreated subjects with mild hyperphenylalaninemia, Pediatr. Res. 49: ((2001) ), 532–536. doi: 10.1203/00006450-200104000-00015. |
[43] | Diamond A. , Prevor M.B. , Callender G. , Druin D.P. . Prefrontal Cortex Cognitive Deficits in Children Treated Early and Continuously for PKU, Monogr. Soc. Res. Child Dev. ((1997) )–doi: 10.2307/1166208. |
[44] | Gassió R. , Artuch R. , Vilaseca M.A. , Fusté E. , Boix C. , Sans A. and Campistol J. . Cognitive functions in classic phenylketonuria and mild hyperphenyl-alaninaemia: experience in a paediatric population, Dev. Med. Child Neurol. 47: ((2007) ), 443–448. doi: 10.1111/j.1469-8749.2005.tb01169.x. |
[45] | Costello P.M. , Beasley M.G. , Tillotson S.L. and Smith I. . Intelligence in mild atypical phenylketonuria, Eur. J. Pediatr. 153: ((1994) ), 260–263. doi: 10.1007/BF01954515. |
[46] | Ross product division, Nutrition support protocols, 4th editio, (2001) . https://books.google.com/books?id=YpfzDQAAQBAJ&pg=PA100&lpg=PA100&dq=38.% 09Nutrition+Support+protocols.+Phenylketonuria.+Ross+product+ division.+4th+edition.++1-32,+2001.&source=bl&ots=HEOb8sjArH&sig=ACfU3U1nnPSMhlhwSVI0_mv7wi9Vt7_oIw&hl=en&sa=X&ved=2ahUKE (accessed June 25, 2020). |
[47] | MacLeod E.L. , Ney D.M. . Nutritional management of phenylketonuria, Ann. Nestle. 68: ((2010) ), 58–69. doi: 10.1159/000312813. |
[48] | Robinson M. , White F.J. , Cleary M.A. , Wraith E. , Lam W.K. , Walter J.H. . Increased risk of vitamin B12 deficiency in patients with phenylketonuria on an unrestricted or relaxed diet, J. Pediatr. 136: ((2000) ), 545–547. doi: 10.1016/S0022-3476(00)90022-2. |
[49] | Shershen H. , Debler E.A. and Lajtha A. . Amino acids in health and disease: New perspectives. Alterations of cerebral amino acid transport processes, Wiley, ((1987) )–doi: 10.1016/0014-5793(88)81393-0. |
[50] | Smith Q.R. . Amino acids health and disease: Kinetic analysis of neutral amino acid transport across the blood-brain barrier. New Perspectives, New York, 1987. https://books.google.com/books?id=YpfzDQAAQBAJ&pg=PA100& lpg=PA100&dq=40.% 09Smith+QR:+Kinetic+analysis+of+neutral+amino+acid+transport+across+the+blood-brain +barrier.+In+Kaufman+S,+editor:+Amino+Acids+in+Health+and+Disease:+New+Perspectives,+New+York,+198 7,+Alan+R-.2pt+-.2pt+p-.2pt+65.-.2pt&source-.2pt=bl-.2pt&ots-.2pt=-.2ptHEOb8skusE-.2pt&sig-.2pt=-.2ptACfU3U1qUEaUKA-.2pt–.2ptPxjD4cFH5CN2-.2pt–.2pt5xQMMg-.2pt&hl=en&sa=X&ved=2ahUKEwjEr-jupZzqAhUglnIEHfu2A1gQ6AEwAHoECAoQAQ#v=onepage&q=40.% 09Smith QR% 3A Kinetic analysis of neutral amino acid transport across the blood-brain barrier. In Kaufman S% 2C editor% 3A Amino Acids in Health and Disease% 3A New Perspectives%2C New York% 2C 1987% 2C Alan R Liss% 2C p 65.&f=false (accessed June 25, 2020). |
[51] | Koch R. , Moseley K.D. , Yano S. , Nelson M. , Moats R.A. . Large neutral amino acid therapy and phenylketonuria: A promising approach to treatment, Mol. Genet. Metab. 79: ((2003) ), 110–113. doi: 10.1016/S1096-7192(03)00078-7. |
[52] | Pietz J. , Kreis R. , Rupp A. , Mayatepek E. , Rating D. , Boesch C. , Bremer H.J. . Large neutral amino acids block phenylalanine transport into brain tissue in patients with phenylketonuria, J. Clin. Invest. 103: ((1999) ), 1169–1178. doi: 10.1172/JCI5017. |
[53] | Matalon R. , Michals-Matalon K. , Bhatia G. , Grechanina E. , Novikov P. , McDonald J.D. , Grady J. , Tyring S.K. and Guttler F. . Large neutral amino acids in the treatment of phenylketonuria (PKU), J. Inherit. Metab. Dis. 29: ((2006) ), 732–738. doi: 10.1007/s10545-006-0395-8. |
[54] | Van Spronsen F.J. , De Groot M.J. , Hoeksma M. , Reijngoud D.J. and Van Rijn M. , Large neutral amino acids in the treatment of PKU: From theory to practice, J. Inherit. Metab. Dis. 33: ((2010) ), 671–676. doi: 10.1007/s10545-010-9216-1. |
[55] | Lim K. , van Calcar S.C. , Nelson K.L. . , Gleason S.T. and Ney D.M. , Acceptable low-phenylalanine foods and beverages can be made with glycomacropeptide from cheese whey for individuals with PKU, Mol. Genet. Metab. 92: ((2007) ), 176–178. doi: 10.1016/j.ymgme.2007.06.004. |
[56] | Van Calcar S.C. , MacLeod E.L. , Gleason S.T. , Etzel M.R. , Clayton M.K. , Wolff J.A. and Ney D.M. , Improved nutritional management of phenylketonuria by using a diet ontaining glycomacropeptide compared with amino acids, Am. J. Clin. Nutr. 89: ((2009) ), 1068–1077. doi: 10.3945/ajcn.2008.27280. |
[57] | Milstien S. and Kaufman S. . Studies on the Phenylalanine Hydroxylase System in Liver Slices - PubMed, J Biol Chem 12: ((1975) ), 4777–81. https://pubmed.ncbi.nlm.nih.gov/124732/ (accessed June 25, 2020). |
[58] | Kure S. , Hou D.C. , Ohura T. , Iwamoto H. , Suzuki S. , Sugiyama N. , Sakamoto O. , Fujii K. , Matsubara Y. and Narisawa K. . Tetrahydrobiopterin-responsive phenylalanine hydroxylase deficiency, J. Pediatr. 135: ((1999) ), 375–378. doi: 10.1016/S0022-3476(99)70138-1. |
[59] | Weglage J. , Grenzebach M. , Teeffelen-Heithoff A.V. , Marquardt T. , Feldmann R. , Denecke J. , Gödde D. , Koch H.G. . Tetrahydrobiopterin responsiveness in a large series of phenylketonuria patients, J. Inherit. Metab. Dis. 25: ((2002) ), 321–322. doi: 10.1023/A:1016514727870. |
[60] | Trefz F.K. , Scheible D. , Frauendienst-Egger G. , Korall H. and Blau N. . Long-term treatment of patients with mild and classical phenylketonuria by tetrahydrobiopterin, Mol. Genet. Metab. 86: ((2005) ), 75–80. doi: 10.1016/j.ymgme.2005.06.026. |
[61] | Blau N. , Trefz F.K. . Tetrahydrobiopterin-responsive phenylalanine hydroxylase deficiency: Possible regulation of gene expression in a patient with the homozygous L48S mutation, Mol. Genet. Metab. 75: ((2002) ), 186–187. doi: 10.1006/mgme.2001.3294. |
[62] | Bernegger C. and Blau N. . High frequency of tetrahydrobiopterin-responsiveness among hyperphenylalaninemias: A study of 1919 patients observed from 1988 to 2002, Mol. Genet. Metab. 77: ((2002) ), 304–313. doi: 10.1016/S1096-7192(02)00171-3. |
[63] | Bélanger-Quintana A. , García M.J. , Castro M. , Desviat L.R. , Pérez B. , Mejía B. , Ugarte M. , Martínez-Pardo M. . Spanish BH4-responsive phenylalanine hydroxylase-deficient patients: Evolution of seven patients on long-term treatment with tetrahydrobiopterin, Mol. Genet. Metab. 86: ((2005) ), 61–66. doi: 10.1016/j.ymgme.2005.07.024. |
[64] | Steinfeld R. , Kohlschütter A. , Ullrich K. and Lukacs Z. . Efficiency of long-term tetrahydrobiopterin monotherapy in phenylketonuria, J. Inherit. Metab. Dis. 27: ((2004) ), 449–453. doi: 10.1023/B:BOLI.0000037351.10132.99. |
[65] | Shintaku H. , Kure S. , Ohura T. , Okano Y. , Ohwada M. , Sugiyama N. , Sakura N. , Yoshida I. , Yoshino M. , Matsubara Y. , Suzuki K. , Aoki K. and Kitagawa T. . Long-Term Treatment and Diagnosis of Tetrahydrobiopterin-Responsive Hyperphenylalaninemia with A Mutant Phenylalanine Hydroxylase Gene, Pediatr. Res, 55: ((2004) ), 425–430. doi: 10.1203/01.PDR.0000111283.91564.7E. |
[66] | Muntau A.C. , Röschinger W. , Habich M. , Demmelmair H. , Hoffmann B. , Sommerhoff C.P. , Roscher A.A. . Tetrahydrobiopterin as an alternative treatment for mild phenylketonukia, N. Engl. J. Med. 347: ((2002) ), 2122–2132. doi: 10.1056/NEJMoa021654. |
[67] | Matalon R. , Michals-Matalon K. , Koch R. , Grady J. , Tyring S. , Stevens R.C. . Response of patients with phenylketonuria in the US to tetrahydrobiopterin, Mol. Genet. Metab. 86: ((2005) ), 17–21. doi: 10.1016/j.ymgme.2005.06.024. |
[68] | Lindner M. , Steinfeld R. , Burgard P. , Schulze A. , Mayatepek E. and Zschocke J. . Tetrahydrobiopterin sensitivity in German patients with mild phenylalanine hydroxylase deficiency, Hum. Mutat. 21: ((2003) ), 400–400. doi: 10.1002/humu.9117. |
[69] | Lässker U. , Zschocke J. , Blau N. and Santer R. . Tetrahydrobiopterin responsiveness in phenylketonuria. Two new cases and a review of molecular genetic findings, J. Inherit. Metab. Dis. 25: ((2002) ), 65–70. doi: 10.1023/A:1015194002487. |
[70] | Koch R. , Guttler F. and Blau N. . Mental illness in mild PKU responds to biopterin, Mol. Genet. Metab. 75: ((2002) ), 284–286. doi: 10.1006/mgme.2001.3289. |
[71] | Danks D.M. , Cotton R.G.H. and Schlesinger P. . Tetrahydrobiopterin treatment of variant form of phenylketonuria, Lancet. 306: ((1975) ), 1043. doi: 10.1016/S0140-6736(75)90335-9. |
[72] | FDA approves a new treatment for PKU, a rare and serious genetic disease/, Case Med. Res. ((2018) ). doi: 10.31525/fda2-ucm608835.htm. |
[73] | Home | Palynziq HCP, (n.d.). https://www.palynziq.com/hcp/ (accessed June 28, 2020). |
[74] | Bell S.M. , Wendt D.J. , Zhang Y. , Taylor T.W. , Long S. , Tsuruda L. , Zhao B. , Laipis P. , Fitzpatrick P.A. . Formulation and PEGylation optimization of the therapeutic PEGylated phenylalanine ammonia lyase for the treatment of phenylketonuria, PLoS One. 12: ((2017) ). doi: 10.1371/journal.pone.0173269. |
[75] | Sarkissian C.N. , Shao Z. , Blain F. , Peevers R. , Su H. , Heft R. , Chang T.M.S. , Scriver C.R. . A different approach to treatment of phenylketonuria: Phenylalanine degradation with recombinant phenylalanine ammonia lyase, Proc. Natl. Acad. Sci. U. S. A. 96: ((1999) ), 2339–2344. doi: 10.1073/pnas.96.5.2339. |
[76] | Of H. , Of H. , Information P. and Information P. . Highlights of prescribing information, Metab. Clin. Exp. ((2008) ). www.fda.gov/medwatch. |
[77] | Thomas J. , Levy H. , Amato S. , Vockley J. , Zori R. , Dimmock D. , Harding C.O. , Bilder D.A. , Weng H.H. , Olbertz J. , Merilainen M. , Jiang J. , Larimore K. , Gupta S. , Gu Z. and Northrup H. . Pegvaliase for the treatment of phenylketonuria: Results of a long-term phase 3 clinical trial program (PRISM), Mol. Genet. Metab. 124: ((2018) ), 27–38. doi: 10.1016/j.ymgme.2018.03.006. |
[78] | Hydery T. , Coppenrath V.A. . A Comprehensive Review of Pegvaliase, an Enzyme Substitution Therapy for the Treatment of Phenylketonuria, Drug Target Insights. 13: ((2019) ). doi: 10.1177/1177392819857089. |
[79] | Grisch-Chan H.M. , Schlegel A. , Scherer T. , Allegri G. , Heidelberger R. , Tsikrika P. , Schmeer M. , Schleef M. , Harding C.O. , Häberle J. , Thöny B. . Low-Dose Gene Therapy for Murine PKU Using Episomal Naked DNA Vectors Expressing PAH from Its Endogenous Liver Promoter, Mol. Ther. –Nucleic Acids. 7: ((2017) ), 339–349. doi: 10.1016/j.omtn.2017.04.013. |
[80] | Rebuffat A. , Harding C.O. , Ding Z. , Thöny B. . Comparison of adeno-associated virus pseudotype 1, 2, and 8 vectors administered by intramuscular injection in the treatment of murine phenylketonuria, Hum. Gene Ther, 21: ((2010) ), 463–477. doi: 10.1089/hum.2009.127. |
[81] | Ding Z. , Harding C.O. , Rebuffat A. , Elzaouk L. , Wolff J.A. , Thöny B. . Correction of murine PKU following AAV-mediated intramuscular expression of a complete phenylalanine hydroxylating system, Mol. Ther. 16: ((2008) ), 673–681. doi: 10.1038/mt.2008.17. |
[82] | Harding C.O. , Gillingham M.B. , Hamman K. , Clark H. , Goebel-Daghighi E. , Bird A. , Koeberl D.D. . Complete correction of hyperphenylalaninemia following liver-directed, recombinant AAV2/8 vector-mediated gene therapy in murine phenylketonuria, Gene Ther. 13: ((2006) ), 457–462. doi: 10.1038/sj.gt.3302678. |
[83] | Homology Medicines Presents Preclinical Data from PKU Gene Therapy Program Demonstrating Restored Metabolic Pathway and Increased Neurotransmitter Production: Homology Medicines, (n.d.). https://www.homologymedicines.com/news-story/homology-medicines-presents-preclinical-data-from-pku-gene-therapy-program (accessed June 28, 2020). |
[84] | Levy H.L. and Ghavami M. . Maternal phenylketonuria: A metabolic teratogen, Teratology. 53: ((1996) ), 176–184. doi: 10.1002/(SICI)1096-9926(199603)53:3<176::AID-TERA5>3.0.CO;2-2. |
[85] | Koch R. , Levy H.L. , Matalon R. , Rouse B. , Hanley W. and Azen C. . The North American Collaborative Study of Maternal Phenylketonuria: Status Report –https://, Am. J. Dis. Child. 147: ((1993) ), 1224–1230. doi: 10.1001/archpedi.1993.02160350098015. |
[86] | Preventing Birth Defects from Maternal Phenylketonuria, Am J Public Heal. ((1989) ), 228. https://www.apha.org/Policies-and-Advocacy/Public-Health-Policy-Statements/Policy-Database/2014/07/29/12/24/Preventing-Birth-Defects-from-Maternal-Phenylketonuria (accessed June 25, 2020). |
[87] | Cunniff C. , Frias J.L. , Kaye C. , Moeschler J.B. , Panny S.R. , Trotter T.L. , De La Cruz F. , Williams J. , Hanson J.W. , Moore C.A. , Lloyd-Puryear M. , Hoyme H.E. , Wappner R.S. and Hall L. , Maternal phenylketonuria, Pediatrics. 122: ((2001) ), 445. doi: 10.1542/peds.107.2.427. |
[88] | Blau N. , Thoeny B. , Cotton R.G.H. and Hyland K. . Disorders of Tetrahydrobiopterin and Related Biogenic Amines | The Online Metabolic and Molecular Bases of Inherited Disease | OMMBID | McGraw-Hill Medical, ((2001) ), 1725–77. https://ommbid.mhmedical.com/content.aspx?bookid=2709§ionid=225082340 (accessed June 25, 2020). |
[89] | Niederwieser A. , Ponzone A. , Curtius H.C. . Differential diagnosis of tetrahydrobiopterin deficiency, J. Inherit. Metab. Dis. 8: ((1985) ), 34–38. doi: 10.1007/BF01800657. |
[90] | Blau N. , Thony B. , Spada M. and Ponzone A. . Tetrahydrobiopterin and Inherited Hyperphenylalaninemias -, PubMe. 38: ((2020) ) 19–35. https://pubmed.ncbi.nlm.nih.gov/8819618/ (accessed June 25 , 2020). |
[91] | Ferrè S. , De Baaij J.H.F. , Ferreira P. , Germann R. R. , De Klerk J.B.C. , Lavrijsen M. , Van Zeeland F. , Venselaar H. , Kluijtmans L.A.J. , Hoenderop J.G.J. and Bindels R.J.M. , 574-586 Mutations in PCBD1 cause hypomagnesemia and renal magnesium wasting, J. Am. Soc. Nephrol. 25: ((2014) ), 574–586. doi: 10.1681/ASN.2013040337. |
[92] | Simaite D. , Kofent J. , Gong M. , Rüschendorf F. , Jia S. , Arn P. , Bentler K. , Ellaway C. , Kühnen P. , Hoffmann G.F. , Blau N. , Spagnoli F.M. , Hübner N. and Raile K. . Recessive mutations in PCBD1 cause a new type of early-onset diabetes, Diabetes. 63: ((2014) ), 3557–3564. doi: 10.2337/db13-1784. |
[93] | Opladen T. , Hoffmann G. , Hörster F. , Hinz A.B. , Neidhardt K. , Klein C. and Wolf N. . Clinical and biochemical characterization of patients with early infantile onset of autosomal recessive GTP cyclohydrolase I deficiency without hyperphenylalaninemia, Mov. Disord. 26: ((2011) ), 157–161. doi: 10.1002/mds.23329. |
[94] | Opladen T. , Hoffmann G.F. and Blau N. . An international survey of patients with tetrahydrobiopterin deficiencies presenting with hyperphenylalaninaemia, J. Inherit. Metab. Dis. 35: ((2012) ), 963–973. doi: 10.1007/s10545-012-9506-x. |
[95] | Thöny B. and Blau N. . Mutations in the BH4-metabolizing genes GTP cyclohydrolase I, 6-pyruvoyl-tetrahydropterin synthase, sepiapterin reductase, carbinolamine-4a-dehydratase, and dihydropteridine reductase, Hum. Mutat. 27: ((2006) ), 870–878. doi: 10.1002/humu.20366. |
[96] | Segawa M. , Nomura Y. and Nishiyama N. . Autosomal dominant guanosine triphosphate cyclohydrolase I deficiency (Segawa disease), Ann. Neurol. 54: ((2003) ), S32–S45. doi: 10.1002/ana.10630. |
[97] | Jäggi L. , Zurflüh M.R. , Schuler A. , Ponzone A. , Porta F. , Fiori L. , Giovannini M. , Santer R. , Hoffmann G.F. , Ibel H. , Wendel U. , Ballhausen D. , Baumgartner M.R. and Blau N. . Outcome and long-term follow-up of 36 patients with tetrahydrobiopterin deficiency, Mol. Genet. Metab. 93: ((2008) ), 295–305. doi: 10.1016/j.ymgme.2007.10.004. |
[98] | Wang L. , Yu W.M. , He C. , Chang M. , Shen M. , Zhou Z. , Zhang Z. , Shen S. , Liu T.T. , Hsiao K.J. . Long-term outcome and neuroradiological findings of 31 patients with 6-pyruvoyltetrahydropterin synthase deficiency, J. Inherit. Metab. Dis. 29: ((2006) ), 127–134. doi: 10.1007/s10545-006-0080-y. |
[99] | Leuzzi V. , Carducci C. , Carducci C. , Pozzessere S. , Burlina A. , Cerone R. , Concolino D. , Donati M.A. , Fiori L. , Meli C. , Ponzone A. , Porta F. , Strisciuglio P. , Antonozzi I. and Blau N. . Phenotypic variability, neurological outcome and genetics background of 6-pyruvoyl-tetrahydropterin synthase deficiency, Clin. Genet. 77: ((2010) ), 249–257. doi: 10.1111/j.1399-0004.2009.01306.x. |
[100] | Birnbacher R. , Scheibenreiter S. , Blau N. , Bieglmayer C. , Frisch H. and Waldhauser F. . Hyperprolactinemia, a tool in treatment control of tetrahydrobiopterin deficiency: Endocrine studies in an affected girl, Pediatr. Res, 43: ((1998) ), 472–477. doi: 10.1203/00006450-199804000-00006. |
[101] | Friedman J. , Roze E. , Abdenur J.E. , Chang R. , Gasperini S. , Saletti V. , Wali G.M. , Eiroa H. , Neville B. , Felice A. , Parascandalo R. , Zafeiriou D.I. , Arrabal-Fernandez L. , Dill P. , Eichler F.S. , Echenne B. , Gutierrez-Solana L.G. , Hoffmann G.F. , Hyland K. , Kusmierska K. , Tijssen M.A.J. , Lutz T. , Mazzuca M. , Penzien J. , Poll-The B.T. , Sykut-Cegielska J. , Szymanska K. , Thöny B. and Blau N. . Sepiapterin reductase deficiency: A Treatable Mimic of Cerebral Palsy, Ann. Neurol. 71: ((2012) ), 520–530. doi: 10.1002/ana.22685. |
[102] | Abeling N.G. , Duran M. , Bakker H.D. , Stroomer L. , Thöny B. , Blau N. , Booij J. , Poll-The B.T. . Sepiapterin reductase deficiency an autosomal recessive DOPA-responsive dystonia, Mol. Genet. Metab. 89: ((2006) ), 116–120. doi: 10.1016/j.ymgme.2006.03.010. |
[103] | Arrabal L. , Teresa L. , Sánchez-Alcudia R. , Castro M. , Medrano C. , Gutiérrez-Solana L. , Roldán S. , Ormazábal A. , Pérez-Cerdá C. , Merinero B. , Pérez B. , Artuch R. , Ugarte M. , Desviat L.R. . Genotype-phenotype correlations in sepiapterin reductase deficiency. A splicing defect accounts for a new phenotypic variant, Neurogenetics. 12: ((2011) ), 183–191. doi: 10.1007/s10048-011-0279-4. |
[104] | Smith I. , Hyland K. , Kendall B. and Leeming R. . Clinical role of pteridine therapy in tetrahydrobiopterin deficiency, J. Inherit. Metab. Dis. 8: ((1985) ), 39–45. doi: 10.1007/BF01800658. |
[105] | Ponzone A. , Spada M. , Ferraris S. , Dianzani I. and De L. . Sanctis, Dihydropteridine Reductase Deficiency in Man: From Biology to Treatment, Med. Res. Rev. 24: ((2004) ), 127–150. doi: 10.1002/med.10055. |
[106] | Howells D.W. , Forrest S.M. , Dahl H.H. , Cotton R.G. . Insertion of an Extra Codon for Threonine Is a Cause of Dihydropteridine Reductase Deficiency - PubMed, Am J Hum Genet. 47: ((2020) ), 279–85. https://pubmed.ncbi.nlm.nih.gov/2116088/ (accessed June 25, 2020). |
[107] | I D. . de S. L. , PM S. , TJ G. , C A. , A B. , M S. , N B. , D M. , HP Z. , N Y. , P A. , WL A. and RG C. , Dihydropteridine Reductase Deficiency: Physical Structure of the QDPR Gene, Identification of Two New Mutations and Genotype-Phenotype Correlations, Hum. Mutat. 12: ((1998) ). doi: 10.1002/(SICI)1098-1004(1998)12:4<267::AID-HUMU8>3.0.CO;2-C. |
[108] | Dahl H.H. , Hutchison W. , McAdam W. , Wake S. , Morgan F.J. , Cotton R.G. . Human Dihydropteridine Reductase: Characterisation of a cDNA Clone and Its Use in Analysis of Patients With Dihydropteridine Reductase Deficiency - PubMed, Nucleic Acids Res. 15: ((1987) ), 1921–32. https://pubmed.ncbi.nlm.nih.gov/3031582/ (accessed June 25, 2020). |
[109] | Irons M. , Levy H.L. , O’Flynn M.E. , Stack V.C. , Langlais P.J. , Butler I.J. , Milstien S. and Kaufman S. , Folinic acid therapy in treatment of dihydropteridine reductase deficiency, J. Pediatr. 110: ((1987) ), 61–67. doi: 10.1016/S0022-3476(87)80289-5. |
[110] | Coughlin C.R. , Hyland K. , Randall R. , Ficicioglu C. ,;Dihydropteridine reductase deficiency and treatment with tetrahydrobiopter: A case reort, in: JIMD Rep., Springer, (2013) : pp. 53–56. doi: 10.1007/8904_2012_202A |
[111] | Mitchell G.A. , Lambert M. , Tanguay R.M. . Hypertyrosinemia | The Online Metabolic and Molecular Bases of Inherited Disease | OMMBID | McGraw-HillMedical, ((2001) ) https://ommbid.mhmedical.com/content.aspx?bookid=2709§ionid=225082825 (accessed June 25, 2020). |
[112] | Yang H. , Al-Hertani W. , Cyr D. , Laframboise R. , Parizeault G. , Wang S.P. , Rossignol F. , Berthier M.T. , Giguère Y. , Waters P.J. , Mitchell G.A. , Alvarez F. , Brunel-Guitton C. , Buhas D. , Dubois J. , Gosselin M. , Halac U. , Maranda B. , Mitchell J. , Turcotte J.F. . Hypersuccinylacetonaemia and normal liver function in maleylacetoacetate isomerase deficiency, J. Med. Genet. 54: ((2017) ), 241–247. doi: 10.1136/jmedgenet-2016-104289. |
[113] | Scott C.R. . The genetic tyrosinemias, Am. J. Med. Genet. - Semin. Med. Genet. 142 C: ((2006) ), 121–126. doi: 10.1002/ajmg.c.30092. |
[114] | Grompe M. . The pathophysiology and treatment of hereditary tyrosinemia type 1, Semin. Liver Dis. 21: ((2001) ), 563–571. doi: 10.1055/s-2001-19035. |
[115] | Berger R. . Biochemical aspects of type 1 hereditary tyrosinemia: Inherited Disease of Amino Acid Metabolism, 1985. https://books.google.com/books?id=YpfzDQAAQBAJ&pg=PA102&lpg=PA102&dq=berger+r:+biochemical+aspe cts+of+type+1+hereditary+tyrosinemia.+in+bickel+h,+wachtel+u,+editors:+inherited+disease+of+amino+acid+metabolism,+stuttgart,+1985+thiémeverlag,+p+192.&source=bl&ots=HEOb9nkBnH&sig=ACfU3U3eTmcpuGjskJL AUV3FM_TgUNq06Q&hl=en&sa=X&ved=2ahUKEwjr2fnH4p3qAhXPl3IEHTmvDVQQ6AEwAHoECAUQAQ#v=onepage&q=berger r% 3A biochemical aspects of type 1 hereditary tyrosinemia. in bickel h% 2C wachtel u% 2C editors% 3A inherited disease of amino acid metabolism% 2C stuttgart% 2C 1985% 2C thiémeverlag% 2C p 192.&f=false (accessed June 25, 2020). |
[116] | Demers S.I. , Phaneuf D. , Tanguay R.M. . Hereditary Tyrosinemia Type I: Strong Association With Haplotype 6 in French Canadians Permits Simple Carrier Detection and Prenatal Diagnosis - PubMed, Am J Hum Genet. 55: ((1994) ), 327–33. https://pubmed.ncbi.nlm.nih.gov/7913582/ (accessed June 25, 2020). |
[117] | Phaneuf D. , Lambert M. , Laframboise R. , Mitchell G. , Lettre F. , Tanguay R.M. . Type 1 hereditary tyrosinemia. Evidence for molecular heterogeneity and identification of a causal mutation in a French Canadian patient, J. Clin. Invest. 90: ((1992) ), 1185–1192. doi: 10.1172/JCI115979. |
[118] | M S.-L. and RM T. . Mutations in the Fumarylacetoacetate Hydrolase Gene Causing Hereditary Tyrosinemia Type I: Overview, Hum. Mutat. 9: ((1997) ). doi: 10.1002/(SICI)1098-1004(1997)9:4<291::AID-HUMU1>3.0.CO;2-9. |
[119] | King LS M. MSc, CGC , Trahms C MS, RD and Ronald Scott C . Tyrosinemia Type I - GeneReviews® –NCBI Bookshelf, GeneReviews® [Internet].(2017). https://www.ncbi.nlm.nih.gov/books/NBK1515/#tyrosinemia.Genetic_Counseling (accessed June 23, 2020). |
[120] | Halvorsen S. , Pande H. , Løken A.C. , Gjessing L.R. and A Tyrosinosis. . study of 6 cases, Arch. Dis. Child. 41: ((1966) ), 238–249. doi: 10.1136/adc.41.217.238. |
[121] | Carson N.A.J. , Biggart J.D. , Bittles A.H. and Donovan D. . Hereditary tyrosinaemia. Clinical, enzymatic, and pathological study of an infant with the acute form of the disease, Arch. Dis. Child. 51: ((1976) ), 106–113. doi: 10.1136/adc.51.2.106. |
[122] | Prive L. . Pathological findings in patients with tyrosinemia, Can. Med. Assoc. J. 97: ((1967) ), 1054. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC1923568/ (accessed June 25, 2020). |
[123] | Russo P. , O’Regan S. . Visceral Pathology of Hereditary Tyrosinemia Type I, Am J Hum Genet. 47: ((1990) ), 317–24. https://pubmed.ncbi.nlm.nih.gov/2378357/ (accessed June 25, 2020). |
[124] | Dehner L.P. , Snover D.C. , Sharp H.L. , Ascher N. , Nakhleh R. , Day D.L. . Hereditary tyrosinemia type I (chronic form): Pathologic findings in the liver, Hum. Pathol. 20: ((1989) ), 149–158. doi: 10.1016/0046-8177(89)90179-2. |
[125] | Lindblad B. , Lindstedt S. and Steen G. . On the enzymic defects in hereditary tyrosinemia, Proc. Natl. Acad. Sci. U. S. A. 74: ((1977) ), 4641–4645. doi: 10.1073/pnas.74.10.4641. |
[126] | Laberge C. , Lescault A. , Tanguay R.M. , Hereditary Tyrosinemias (Type I): A New Vista on Tyrosine Toxicity and Cancer, in: Essent. Nutr. Carcinog., Sringer US, (1986) : pp. 209–221. https://.doi: 10.1007/978-1-4613-1835-4_17 |
[127] | Manowski Z. , Sliver M.M. , Roberts E.A. , Superina R.A. , Phillips M.J. . Liver Cell Dysplasia and Early Liver Transplantation in Hereditary Tyrosinemia, Mod Pathol. 3: ((1990) ), 694–701. https://pubmed.ncbi.nlm.nih.gov/2175899/ (accessed June 25, 2020). |
[128] | Watanabe S. , Okita K. , Harada T. , Kodama T. , Numa Y. , Takemoto T. and Takahashi T. . Morphologic studies of the liver cell dysplasia, Cancer. 51: ((1983) ), 2197–2205. doi: 10.1002/1097-0142(19830615)51:12<2197::AID-CNCR2820511208>3.0.CO;2-5. |
[129] | Jevtic M.M. , Thorp F.K. and Hruban Z. . Hereditary tyrosinemia with hyperplasia and hypertrophy of juxtaglomerular apparatus, Am. J. Clin. Pathol. 61: ((1974) ), 423–437. doi: 10.1093/ajcp/61.3.423. |
[130] | Bendon R.W. and Hug G. . Glycogen accumulation in the pars recta of the proximal tubule in fanconi syndrome, Fetal Pediatr. Pathol. 6: ((1986) ), 411–429. doi: 10.3109/15513818609041556. |
[131] | Gilbert-Barness E. , Barness L.A. , Meisner L.F. . Chromosomal instability in hereditary tyrosinemia type i, Fetal Pediatr. Pathol. 10: ((1990) ), 243–252. doi: 10.3109/15513819009067111. |
[132] | Larochelle J. , Privé L. , Bélanager M. , Bélanager L. , Tremlay M. , Claveau J.C. , Aubin G. and Paradis D. . [Hereditary Tyrosinemia. I. Clinical and Biological Study of 62 Cases] - PubMed, Pediatrie. 28: ((1973) ), 5–18. https://pubmed.ncbi.nlm.nih.gov/4715467/ (accessed June 25, 2020). |
[133] | Perry T.L. . Tyrosinemia Associated With Hypermethioninemia and Islet Cell Hyperplasia - PubMed, Can Med Assoc J. 97: ((1967) ), 1067–75. https://pubmed.ncbi.nlm.nih.gov/4862180/ (accessed June 25, 2020). |
[134] | Hardwick D.F. , Dimmick J.E. . Metabolic cirrhoses of infancy and early childhood, Perspect. Pediatr. Pathol. 3: ((1976) ), 103–144. https://pubmed.ncbi.nlm.nih.gov/823524/ (accessed June 25, 2020). |
[135] | Lindblad B. , Fällström S.P. , Höyer S. , Nordborg C. , Solymar L. and Velander H. . Cardiomyopathy in fumarylacetoacetase deficiency (hereditary tyrosinaemia): a new feature of the disease, J. Inherit. Metab. Dis. 10: ((1987) ), 319–322. doi: 10.1007/BF01811439. |
[136] | Edwards M.A. , Green A. , Colli A. and Rylance G. . Tyrosinaemia type I and hypertrophic obstructive cardiomyopathy, Lancet. 329: ((1987) ), 1437–1438. doi: 10.1016/S0140-6736(87)90631-3. |
[137] | Mitchell G. , Larochelle J. , Lambert M. , de Weerd A.W. , Gianella-Borradori A. , Michaud J. , Grenier A. , Ogier H. , Gauthier M. , Lacroix J. , Vanasse M. , Larbrisseau A. , Paradis K. , Weber A. , Lefevre Y. , Melançon S. and Dallaire L. , Neurologic Crises in Hereditary Tyrosinemia, N. Engl. J. Med. 322: ((1990) ), 432–437. doi: 10.1056/NEJM199002153220704. |
[138] | Kvittingen E.A. , Rootwelt H. , Brandtzaeg P. , Bergan A. and Berger R. . Hereditary tyrosinemia type I: Self-induced correction of the fumarylacetoacetase defect, J. Clin. Invest. 91: ((1993) ), 1816–1821. doi: 10.1172/JCI116393. |
[139] | Kvittingen E.A. , Rootwelt H. , Berger R. and Brandtzaeg P. . Self-induced correction of the genetic defect in tyrosinemia type I, J. Clin. Invest. 94: ((1994) ), 1657–1661. doi: 10.1172/JCI117509. |
[140] | Kvittingen E. . Hereditary Tyrosinemia Type I –an Overview, Scand J Clin Lab Invest Suppl. 184: ((1986) ) ,27. https://pubmed.ncbi.nlm.nih.gov/3296130/ (accessed June 25, 2020). |
[141] | Manabe S. , Sassa S. and Kappas A. . Hereditary tyrosinemia: Formation o f succinylacetone-amino acid adducts, J. Exp. Med. 162: ((1985) ), 1060–1074. doi: 10.1084/jem.162.3.1060. |
[142] | Tanguay R.M. , Jorquera R. , Poudrier J. , St-Louis M. . Tyrosine and Its Catabolites: From Disease to Cancer, Acta Biochim Pol. 43: ((1996) ) , 209–16. https://pubmed.ncbi.nlm.nih.gov/8790725/ (accessed June 25, 2020). |
[143] | Endo F. , Katoh H. and Matsuda I. . Putative genetic deficiency of 4-hydroxyphenylpyruvic acid dioxygenase in mice: A murine model for hereditary tyrosinaemia type III, J. Inherit. Metab. Dis. 13: ((1990) ), 780–782. doi: 10.1007/BF01799587. |
[144] | Bienfang D.C. , Kuwabara T. , Pueschel S.M. . The Richner-Hanhart Syndrome: Report of a Case with Associated Tyrosinemia, Arch. Ophthalmol. 94: ((1976) ), 1133–1137. doi: 10.1001/archopht.1976.03910040045009. |
[145] | Rehák A. , Selim M.M. and Yadav G. . Richner-Hanhart syndrome (tyrosinaemia-II), (report of four cases without ocular involvement), Br. J. Dermatol. 104: ((1981) ), 469–475. doi: 10.1111/j.1365-2133.1981.tb15320.x. |
[146] | Gray R.G.F. , Patrick A.D. , Preston F.E. , Whitfield M.F. . Acute hereditary tyrosinaemia type I: Clinical, biochemical and haematological studies in twins, J. Inherit. Metab. Dis. 4: ((1981) ), 37–40. doi: 10.1007/BF02263580. |
[147] | Evans D.I.K. , Sardharwalla I.B. . Coagulation defect of congenital tyrosinaemia, Arch. Dis. Child. 59: ((1984) ), 1088–1090. doi: 10.1136/adc.59.11.1088. |
[148] | Russo P.A. , Mitchell G.A. , Tanguay R.M. . Tyrosinemia: A review, Pediatr. Dev. Pathol. 4: ((2001) ), 212–221. doi: 10.1007/s100240010146. |
[149] | Castilloux J. , Laberge A.M. , Martin S.R. , Lallier M. and Marchand V. . “Silent” tyrosinemia presenting as hepatocellular carcinoma in a 10-year-old girl, J. Pediatr. Gastroenterol. Nutr. 44: ((2007) ), 375–377. doi: 10.1097/MPG.0b013e31802f640c. |
[150] | van Spronsen F.J. , Thomasse Y. , Smit . , Leonard J.V. , Clayton P.T. . , Fidler V. V. , Berger R. and Heymans H.S. , Hereditary Tyrosinemia Type I: A new clinical classification with difference in prognosis on dietary treatment, Hepatology. 20: ((1994) ), –1187–91. https://pubmed.ncbi.nlm.nih.gov/7927251/ (accessed June 25, 2020). |
[151] | Mieles L.A. , Esquivel C.O. , Van Thiel D.H. ., Koneru B. , Makowka L. , Tzakis A.G. and Starzl T.E. , Liver transplantation for tyrosinemia - A review of 10 cases from the university of Pittsburgh, Dig. Dis. Sci. 35: ((1990) ), 153–157. doi: 10.1007/BF01537237. |
[152] | Strife C.F. , Zuroweste E.L. , Emmett E.A. , Finelli V.N. , Petering H.G. , Berry H.K. . Tyrosinemia with acute intermittent porphyria: Aminolevulinic acid dehydratase deficiency related to elevated urinary aminolevulinic acid levels, J. Pediatr. 90: ((1977) ), 400–404. doi: 10.1016/S0022-3476(77)80701-4. |
[153] | Cole D.E.C. , Tithecott G.A. , Crocker J.F.S. , Moss M.A. . Alphaxalone/alphadolone and porphyria, Lancet. 323: ((1984) ), 690. doi: 10.1016/S0140-6736(84)92212-8. |
[154] | Rank J.M. , Pascual-Leone A. , Payne W. , Glock M. , Freese D. , Sharp H. and Bloomer J.R. . Hematin therapy for the neurologic crisis of tyrosinemia, J. Pediatr. 118: ((1991) ), 136–139. doi: 10.1016/S0022-3476(05)81867-0. |
[155] | Larochelle J. , Alvarez F. , Bussières J.F. , Chevalier I. , Dallaire L. , Dubois J. , Faucher F. , Fenyves D. , Goodyer P. , Grenier A. , Holme E. , Laframboise R. , Lambert M. , Lindstedt S. , Maranda B. , Melançon S. , Merouani A. , Mitchell J. , Parizeault G. , Pelletier L. , Phan V. , Rinaldo P. , Scott C.R. , Scriver C. and Mitchell G.A. , Effect of nitisinone (NTBC), treatment on the clinical course of hepatorenal tyrosinemia in Québec, Mol. Genet. Metab. 107: ((2012) ), 49–54. doi: 10.1016/j.ymgme.2012.05.022. |
[156] | J P. , M S.-L. , F L. , K G. , C P. , J L. and RM T. . Frequency of the IVS12+5G–>A Splice Mutation of the Fumarylacetoacetate Hydrolase Gene in Carriers of Hereditary Tyrosinaemia in the French Canadian Population of Saguenay-Lac-St-Jean, Prenat. Diagn. 16: ((1996) ). doi: 10.1002/(SICI)1097-0223(199601)16:1<59::AID-PD810>3.0.CO;2-D. |
[157] | Endo F. and Sun M.S. . Tyrosinaemia type I and apoptosis of hepatocytes and renal tubular cells, J. Inherit. Metab. Dis. 25: ((2002) ), 227–234. doi: 10.1023/A:1015646400182. |
[158] | Chinsky J.M. , Singh R. , Ficicioglu C. , van Karnebeek C.D.M. , Grompe M. , Mitchell G. , Waisbren S.E. , Gucsavas-Calikoglu M. , Wasserstein M.P. , Coakley K. and Scott C.R. , Diagnosis and treatment of tyrosinemia type I: a US and Canadian consensus group review and recommendations,https://, Genet. Med. 19: ((2017) ). doi: 10.1038/gim.2017.101. |
[159] | Hermes-Lima M. , Valle V.G.R. , Vercesi A.E. , Bechara E.J.H. . Damage to rat liver mitochondria promoted by δ-aminolevulinic acid-generated reactive oxygen species: connections with acute intermittent porphyria and lead-poisoning, BBA - Bioenerg. 1056: ((1991) ), 57–63. doi: 10.1016/S0005-2728(05)80072-6. |
[160] | Lindblad B. , Fällström S.P. , Höyer S. , Nordborg C. , Solymar L. and Velander H. . Cardiomyopathy in fumarylacetoacetase deficiency (hereditary tyrosinaemia): a new feature of the disease, J. Inherit. Metab. Dis. 10: ((1987) ), 319–322. doi: 10.1007/BF01811439. |
[161] | Arora N. , Stumper O. , Wright J. , Kelly D.A. , McKiernan P.J. . Cardiomyopathy in tyrosinaemia type I is common but usually benign, J. Inherit. Metab. Dis. 29: ((2006) ), 54–57. doi: 10.1007/s10545-006-0203-5. |
[162] | Mohamed S. , Kambal M.A. , Al Jurayyan N.A. , Al-Nemri A. , Babiker A. , Hasanato R. and Al-Jarallah A.S. , Tyrosinemia type 1: A rare and forgotten cause of reversible hypertrophic cardiomyopathy in infancy,https://, BMC Res. Notes. 6: ((2013) ). doi: 10.1186/1756-0500-6-362. |
[163] | Weigel J.F. , Janzen N. , Pfäffle R.W. , Thiery J. , Kiess W. and Ceglarek U. . Tandem mass spectrometric determination of succinylacetone in dried blood spots enables presymptomatic detection in a case of hepatorenal tyrosinaemia, J. Inherit. Metab. Dis. 30: ((2007) ), 610. doi: 10.1007/s10545-007-0608-9. |
[164] | Croffie J.M. . Tyrosinemia type 1 should be suspected in infants with severe coagulopathy even in the absence of other signs of liver failure, Pediatrics. 103: ((1999) ), 675–678. doi: 10.1542/peds.103.3.675. |
[165] | Holme E. and Lindstedt S. . Diagnosis and management of tyrosinemia type I, Curr. Opin. Pediatr. 7: ((1995) ), 726–732. doi: 10.1097/00008480-199512000-00017. |
[166] | Magera M.J. , Gunawardena N.D. , Hahn S.H. , Tortorelli S. , Mitchell G.A. , Goodman S.I. , Rinaldo P. and Matern D. . Quantitative determination of succinylacetone in dried blood spots for newborn screening of tyrosinemia type I, Mol. Genet. Metab. 88: ((2006) ), 16–21. doi: 10.1016/j.ymgme.2005.12.005. |
[167] | Sass-Kortsak A. , Ficici S. , Paunier L. , Kooh S.W. , Fraser D. and Jackson S.H. . Observations on treatment in patients with tyrosyluria, Can Med Assoc. 97: ((1967) ), 1089–95. https://pubmed.ncbi.nlm.nih.gov/6050911/ (accessed June 25, 2020). |
[168] | Suzuki Y. , Konda M. , Imai I. , Imamure H. , Shimao S. and Okaka T. . Effect of Dietary Treatment on the Renal Tubular Function in a Patient With Hereditary Tyrosinemia, Int J Pediatr Nephrol. 8: ((1987) ), 171–6. https://pubmed.ncbi.nlm.nih.gov/3429140/ (accessed June 25, 2020). |
[169] | Mohan N. , McKiernan P. , Preece M.A. , Green A. , Buckels J. , Mayer A.D. , Kelly D.A. . Indications and outcome of liver transplantation in tyrosinaemia type 1,Suppl. 158 https://, Eur. J. Pediatr. suppl. 158: ((1999) ). doi: 10.1007/pl00014321. |
[170] | Lindstedt S. , Holme E. , Lock E.A. , Hjalmarson O. and Strandvik B. . Treatment of hereditary tyrosinaemia type I by inhibition of 4-hydroxyphenylpyruvate dioxygenase, Lancet. 340: ((1992) ), 813–817. doi: 10.1016/0140-6736(92)92685-9. |
[171] | Santra S. and Baumann U. . Experience of nitisinone for the pharmacological treatment of hereditary tyrosinaemia type 1, Expert Opin. Pharmacother. 9: ((2008) ), 1229–1236. doi: 10.1517/14656566.9.7.1229. |
[172] | Holme E. , Lindstedt S. . Tyrosinaemia type I and NTBC (2-(2-nitro-4-trifluoromethylbenzoyl)-1,3- cyclohexanedione) in: J. Inherit. Metab. Dis., J Inherit Metab Di. (1998) : pp. 507–517, doi: 10.1023/A:1005410820201. |
[173] | Masurel-Paulet A. , Poggi-Bach J. , Rolland M.O. , Bernard O. , Guffon N. , Dobbelaere D. , Sarles J. , Baulny O.O.H. and Touati G. . NTBC treatment in tyrosinaemia type I: Long-term outcome in French patients, J. Inherit. Metab. Dis. 31: ((2008) ), 81–87. doi: 10.1007/s10545-008-0793-1. |
[174] | Schlune A. , Thimm E. , Herebian D. and Spiekerkoetter U. . Single dose NTBC-treatment of hereditary tyrosinemia type I, J. Inherit. Metab. Dis. 35: ((2012) ), 831–836. doi: 10.1007/s10545-012-9450-9. |
[175] | El-Karaksy H. , Rashed M. , El-Sayed R. , El-Raziky M. , El-Koofy N. , El-Hawary M. and Al-Dirbashi O. . Clinical practice: NTBC therapy for tyrosinemia type 1: How much is enough? Eur. J. Pediatr. 169: ((2010) ), 689–693. doi: 10.1007/s00431-009-1090-1. |
[176] | Thimm E. , Richter-Werkle R. , Kamp G. , Molke B. , Herebian D. , Klee D. , Mayatepek E. and Spiekerkoetter U. . Neurocognitive outcome in patients with hypertyrosinemia type i after long-term treatment with NTBC, J. Inherit. Metab. Dis. 35: ((2012) ), 263–268. doi: 10.1007/s10545-011-9394-5. |
[177] | Gissen P. , Preece M.A. , Willshaw H.A. and McKiernan P.J. . Ophthalmic follow-up of patients with tyrosinaemia type I on NTBC, J. Inherit. Metab. Dis. 26: ((2003) ), 13–16. doi: 10.1023/A:1024011110116. |
[178] | Tuchman M. , Freese D.K. , Sharp H.L. , Ramnaraine M.L.R. , Ascher N. , Bioomer J.R. . Contribution of extrahepatic tissues to biochemical abnormalities in hereditary tyrosinemia type I: Study of three patients after liver transplantation, J. Pediatr. 110: ((1987) ), 399–403. doi: 10.1016/S0022-3476(87)80501-2. |
[179] | Freese D.K. , Tuchman M. , Schwarzenberg S.J. , Sharp H.L. , Rank J.M. , Bloomer J.R. , Ascher N.L. , Payne W.D. . Early Liver Transplantation Is Indicated for Tyrosinemia Type I, J. Pediatr. Gastroenterol. Nutr. 13: ((1991) ), 10–15. doi: 10.1097/00005176-199107000-00002. |
[180] | Halvorsen S. , Kvittingen E.-A. and Flatmark A. . Outcome of therapy of hereditary tyrosinemia, Pediatr. Int. 30: ((1988) ), 425–428. doi: 10.1111/j.1442-200X.1988.tb02532.x. |
[181] | Maiorana A. , Malamisura M. , Emma F. , Boenzi S. , Di Ciommo V.M. and Dionisi-Vici C. , Early effect of NTBC on renal tubular dysfunction in hereditary tyrosinemia type 1, Mol. Genet. Metab. 113: ((2014) ), 188–193. doi: 10.1016/j.ymgme.2014.07.021. |
[182] | Pierik L.J.W.M. , van Spronsen F.J. , Bijleveld C.M.A. , van Dael C.M.L. , Renal function in tyrosinaemia type I after liver transplantation: A long-term follow-up, J. Inherit. Metab. Dis. 28: ((2005) ), 871–876. doi: 10.1007/s10545-005-0059-0. |
[183] | Kassel R. , Sprietsma L. and Rudnick D.A. . Pregnancy in an NTBC-treated patient with hereditary tyrosinemia type I, J. Pediatr. Gastroenterol. Nutr. 60: ((2015) ), e5–e7. doi: 10.1097/MPG.0b013e3182a27463. |
[184] | Garcia Segarra N. , Roche S. , Imbard A. , Benoist J.F. , Grenèche M.O. , Davit-Spraul A. and Ogier de Baulny H. , Maternal and fetal tyrosinemia type I, J. Inherit. Metab. Dis. 33: ((2010) ), S507–S510. doi: 10.1007/s10545-012-9569-8. |
[185] | Vanclooster A. , Devlieger R. , Meersseman W. , Spraul A. , Vande Kerckhove K. , Vermeersch P. , Meulemans A. , Allegaert K. and Cassiman D. , Pregnancy during nitisinone treatment for tyrosinaemia type I: First human experience, in: JIMD Rep, Springer, (2012) : pp. 27–33. doi: 10.1007/8904_2011_88. |
[186] | Hühn R. , Stoermer H. , Klingele B. , Bausch E. , Fois A. , Farnetani M. , Di Rocco M. , Boué J. J. , Kirk J.M. , Coleman R. and Scherer G. . Novel and recurrent tyrosine aminotransferase gene mutations in tyrosinemia type II, Hum. Genet. 102: ((1998) ), 305–313. doi: 10.1007/s004390050696. |
[187] | Rabinowitz L.G. , Williams L.R. , Anderson C.E. , Mazur A. and Kaplan P. . Painful keratoderma and photophobia: Hallmarks of tyrosinemia type II, J. Pediatr. 126: ((1995) ), 266–269. doi: 10.1016/S0022-3476(95)70558-9. |
[188] | Macsai M.S. , Schwartz T.L. , Hinkle D. , Hummel M.B. , Mulhern M.G. and Rootman D. . Tyrosinemia type II: Nine cases of ocular signs and symptoms, Am. J. Ophthalmol. 132: ((2001) ), 522–527. doi: 10.1016/S0002-9394(01)01160-6. |
[189] | Charlton K.H. , Binder P.S. , Wozniak L. and Digby D.J. . Pseudodendritic keratitis and systemic tyrosinemia, Ophthalmology. 88: ((1981) ), 355–360. doi: 10.1016/S0161-6420(81)35036-2. |
[190] | Gounod N. , Ogier H. , Dufier J.L. , Larrègue M. , Saudubray J.M. and de Prost Y. . [Type II oculo-cutaneous tyrosinosis], Ann. Dermatol. Venereol. 111: ((2020) ), 697–8. http://www.ncbi.nlm.nih.gov/pubmed/6152137. (accessed June 24, 2020). |
[191] | Chitayat D. , Balbul A. , Hani V. , Mamer O.A. , Clow C. , Scriver C.R. . Hereditary tyrosinaemia type II in a consanguineous Ashkenazi Jewish family: Intrafamilial variation in phenotype; absence of parental phenotype effects on the fetus, J. Inherit. Metab. Dis. 15: ((1992) ), 198–203. doi: 10.1007/BF01799631. |
[192] | Al-Hemidan A.I. and, Al-Hazzaa S.A.F. . Richner-hanhart syndrome (tyrosinemia type II), case report and literature review, Ophthalmic Genet. 16: ((1995) ), 21–23. doi: 10.3109/13816819509057850. |
[193] | Pelet B. , Antener I. , Faggioni R. , Spahr A. and Gautier E. . Tyrosinemia without liver or renal damage with plantar and palmar keratosis and keratitis (hypertyrosinemia type II), Helv. Paediatr. Acta. 34: ((1979) ), 177–83. http://www.ncbi.nlm.nih.gov/pubmed/156708. (accessed June 24, 2020). |
[194] | Rehak A. , Selim M.M. and Yadav G. . Richner-Hanhart syndrome (tyrosinaemia-II), (report of four cases without ocular involvement, Br. J. Dermatol. 104: ((1981) ), 469–475. doi: 10.1111/j.1365-2133.1981.tb15320.x. |
[195] | B.B.. W. , Nyhan A.-A.A. . Atlas of Inherited Metabolic Diseases 3E, Third Edit, CRC Press, Boca Raton, 2012. https://online.vitalsource.com/#/books/1/cfi/6/54!/4/18/4/2/48/4/2/2@0:0 (accessed June 24, 2020). |
[196] | Meissner T. , Betz R.C. , Pasternack S.M. , Eigelshoven S. , Ruzicka T. , Kruse R. , Laitenberger G. and Mayatepek E. . Richner-Hanhart syndrome detected by expanded newborn screening, Pediatr. Dermatol. 25: ((2008) ), 378–380. doi: 10.1111/j.1525-1470.2008.00687.x. |
[197] | Machino H. , Miki Y. , Kawatsu T. , Kida K. and Matsuda H. . Successful dietary control of tyrosinemia II, J. Am. Acad. Dermatol. 9: ((1983) ), 533–539. doi: 10.1016/S0190-9622(83)70165-9. |
[198] | Endo F. , Kitano A. , Uehara I. , Nagata N. , Matsuda I. , Shinka T. , Kuhara T. and Matsumoto I. . Four-hydroxyphenylpyruvic acid oxidase deficiency with normal fumarylacetoacetase: A new variant form of hereditary hypertyrosinemia, Pediatr. Res. 17: ((1983) ), 92–96. doi: 10.1203/00006450-198302000-00002. |
[199] | Giardini O. , Cantani A. , Kennaway N.G. , D’Eufemia P. . Chronic tyrosinemia associated with 4-hydroxyphenylpyruvate dioxygenase deficiency with acute intermittent ataxia and without visceral and bone involvement, Pediatr. Res. 17: ((1983) ), 25–29. doi: 10.1203/00006450-198301000-00005. |
[200] | Tomoeda K. , Awata H. , Matsuura T. , Matsuda I. , Ploechl E. , Milovac T. , Boneh A. , Scott C.R. , Danks D.M. and Endo F. . Mutations in the 4-hydroxyphenylpyruvic acid dioxygenase gene are responsible for tyrosinemia type III and hawkinsinuria, Mol. Genet. Metab. 71: ((2000) ), 506–510. doi: 10.1006/mgme.2000.3085. |
[201] | Rüetschi U. , Cerone R. , Pérez-Cerda C. , Schiaffino M.C. , Standing S. , Ugarte M. and Holme E. . Mutations in the 4-hydroxyphenylpyruvate dioxygenase gene (HPD), in patients with tyrosinemia type III, Hum. Genet. 106: ((2000) ), 654–662. doi: 10.1007/s004390000307. |
[202] | Cerone R. , Holme E. , Schiaffino M. , Caruso U. , Maritano L. and Romano C. . Tyrosinemia type III: diagnosis and ten-year follow-up, Acta Paediatr. 86: ((1997) ), 1013–1015. doi: 10.1111/j.1651-2227.1997.tb15192.x. |
[203] | Ellaway C.J. , Holme E. , Standing S. , Preece M.A. , Green A. , Ploechl E. , Ugarte M. , Trefz F.K. , Leonard J.V. . Outcome of tyrosinaemia type III, J. Inherit. Metab. Dis. 24: ((2001) ), 824–832. doi: 10.1023/A:1013936107064. |
[204] | Szymanska E. , Sredzinska M. , Ciara E. , Piekutowska-Abramczuk D. , Ploski R. , Rokicki D. , Tylki-Szymanska A. . Tyrosinemia type III in an asymptomatic girl, Mol. Genet. Metab. Reports. 5: ((2015) ), 48–50. doi: 10.1016/j.ymgmr.2015.10.004. |
[205] | Najafi R. , Mostofizadeh N. and Hashemipour M. . A Case of Tyrosinemia Type III with Status Epilepticus and Mental Retardation, Adv. Biomed. Res. 7: ((2018) ), 7. doi: 10.4103/2277-9175.223740. |
[206] | Wilcken B. , Hammond J.W. , Howard N. , Bohane T. , Hocart C. and Halpern B. . Hawkinsinuria: A Dominantly Inherited Defect of Tyrosine Metabolism with Severe Effects in Infancy, N. Engl. J. Med. 305: ((1981) ), 865–869. doi: 10.1056/NEJM198110083051505. |
[207] | Niederwieser A. , Matasovic A. , Tippett P. , Danks D.M. . A new sulfur amino acid, named hawkinsin, identified in a baby with transient tyrosinemia and her mother, Clin. Chim. Acta. 76: ((1977) ), 345–356. doi: 10.1016/0009-8981(77)90161-9. |
[208] | Niederwieser A. , Wadman S.K. , Danks D.M. . Excretion of cis- and trans-4-hydroxycyclohexylacetic acid in addition to hawkinsin in a family with a postulated defect of 4-hydroxyphenylpyruvate dioxygenase, Clin. Chim. Acta. 90: ((1978) ), 195–200. doi: 10.1016/0009-8981(78)90522-3. |
[209] | Niederwieser A. , Matasović A. , Neuheiser F. and Wetzel E. . New tyrosine metabolites in humans: Hawkinsin and cis- and trans-4-hydroxycyclohexylacetic acids. Unusual adsorption of deuterated and non-deuterated hawkinsin during gas chromatography, J. Chromatogr. B Biomed. Sci. Appl. 146: ((1978) ), 207–212. doi: 10.1016/S0378-4347(00)81886-1. |
[210] | Lehnert W. , Stögmann W. , Engelke U. , Wevers R.A. , Van G.B. . Den Berg, Long-term follow up of a new case of hawkinsinuria, Eur. J. Pediatr. 158: ((1999) ), 578–582. doi: 10.1007/s004310051151. |
[211] | Borden M. , Holm J. , Leslie J. , Sweetman L. , Nyhan W.L. , Fleisher L. , Nadler H. , Lewis D. , Scott C.R. . Hawkinsinuria in two families, Am. J. Med. Genet. 44: ((1992) ), 52–56. doi: 10.1002/ajmg.1320440113. |
[212] | Item C.B. , Mihalek I. , Lichtarge O. , Jalan A. , Vodopiutz J. , Muhl A. , Bodamer O.A. . Manifestation of hawkinsinuria in a patient compound heterozygous for hawkinsinuria and tyrosinemia III, Mol. Genet. Metab. 91: ((2007) ), 379–383. doi: 10.1016/j.ymgme.2007.04.008. |
[213] | Gomez-Ospina N. , Scott A.I. , Oh G.J. , Potter D. , Goel V.V. , Destino L. , Baugh N. , Enns G.M. , Niemi A.K. , Cowan T.M. . Expanding the phenotype of hawkinsinuria: new insights from response to N-acetyl-L-cysteine, J. Inherit. Metab. Dis. 39: ((2016) ), 821–829. doi: 10.1007/s10545-016-9963-8. |
[214] | Danks D.M. , Tippett P. and Rogers J. . A new form of prolonged transient tyrosinemia presenting with severe metabolic acidosis,diatrica.} 209–214. https://, Acta Pæ. 64: ((1975) ), 209–206. doi: 10.1111/j.1651-2227.1975.tb03823.x. |
[215] | LA DU BN, Alkaptonuria | The Online Metabolic and Molecular Bases of Inherited Disease | OMMBID | McGraw-Hill Medical, (n.d.). https://ommbid.mhmedical.com/content.aspx?bookid=2709§ionid=225085636 (accessed June 26, 2020). |
[216] | Phornphutkul C. , Introne W.J. , Perry M.B. , Bernardini I. , Murphey M.D. , Fitzpatrick D.L. , Anderson P.D. , Huizing M. , Anikster Y. , Gerber L.H. , Gahl W.A. . Natural history of alkaptonuria, N. Engl. J. Med. 347: ((2002) ), 2111–2121. doi: 10.1056/NEJMoa021736. |
[217] | Nemethova M. , Radvanszky J. , Kadasi L. , Ascher D.B. , Pires D.E.V. , Blundell T.L. , Porfirio B. , Mannoni A. , Santucci A. , Milucci L. , Sestini S. , Biolcati G. , Sorge F. , Aurizi C. , Aquaron R. , Alsbou M. , Lourenço C.M. , Ramadevi K. , Ranganath L.R. , Gallagher J.A. , Van Kan C. , Hall A.K. , Olsson B. , Sireau N. , Ayoob H. , Timmis O.G. , Le Quan Sang K.H. , Genovese F. , Imrich R. , Rovensky J. , Srinivasaraghavan R. , Bharadwaj S.K. , Spiegel R. and Zatkova A. . Twelve novel HGD gene variants identified in 99 alkaptonuria patients: Focus on “black bone disease” in Italy, Eur, J. Hum. Genet. 24: ((2016) ), 66–72. doi: 10.1038/ejhg.2015.60. |
[218] | Stenn F.F. , Milgram J.W. , Lee S.L. , Weigand R.J. and Veis A. . Biochemical identification of homogentisic acid pigment in an ochronotic Egyptian mummy, Scienc (80-.). 197: ((1977) ), 566–568. doi: 10.1126/science.327549. |
[219] | Garrod A.E. . The Croonian Lectureson inborn errors of metabolism. Lecture II. Alkaptonuria, Lancet. ((1908) ), 73. https://link.springer.com/article//10.1007/s10545-008-0984–9 (accessed June 25, 2020). |
[220] | Boedeker C. . Ueber das Alcapton; ein neuer Beitragzure Frage: Welche Stoffe des Harns können Kupfer reduction bewirken? Rat Med. 7: ((1859) ), 130. https://books.google.com/books?id=YpfzDQAAQBAJ&pg=PA105&lpg=PA105&dq=% 3DBoedeker+C:+Ueber+das+Alcapton;+ein+neuer+Beitrag+Are+Frage:&source=bl&ots=HEOb9qlypB&sig=ACfU3U38Uom2QU8xFyr3Ipy0pOKZuYWVYg&hl=en&sa=X&ved=2ahUKEwiC0rr91J7qAhX2lXIEHRwJDVEQ6AEwAXoECAwQAQ#v=onepage&q=% 3DBoedeker C% 3A Ueber das Alcapton% 3B ein neuer Beitrag Are Frage% 3A&f=false (accessed June 26, 2020). |
[221] | Boedeker C. . Das Alkapton; ein Beitrag zur Frage: Welche Stoffe des Harnesönnen aus einer alkalischen Kupferoxydlösung Kupferoxydulreduciren? Ann Chem Pharmacol.117: ((2020) ), 98. https://books.google.com/books?id=YpfzDQAAQBAJ&pg=PA105&lpg=PA105&dq=Boedeker+C:+Das+Alkapton;+ein+Beitrag+zur+ Frage:+Welche+Stoffe+des+Harnesönnen+aus+einer+alkalischen+Kupferoxydlösung+Kupferoxydulreduciren?+Ann+Chem+Pharmacol+117:98,+&source=bl&ots=HEOb9qlztC&sig=ACfU3U2t6zNhQ-X4F2ehcLbO_16tihTtog&hl=en&sa=X&ved=2ahUKEwiBgfzA1Z7qAhUhmHIEHfizDlIQ6AEwAHoECAoQAQ#v=onepage&q=Boedeker C% 3A Das Alkapton% 3B ein Beitrag zur Frage% 3A Welche Stoffe des Harnesönnen aus einer alkalischen Kupferoxydlösung Kupferoxydulreduciren% 3F Ann Chem Pharmacol 117% 3A98% 2C &f=false (accessed June 26, 2020). |
[222] | Wolkow M. and Baumann E. . Ueber das Wesen der Alkaptonurie, Z Physiol Chem. 15: ((1891) ), 225. https://books.google.com/books?id=OPr_VCXvF1kC&pg=PA229&lpg=PA229&dq=178.% 09Wolkow+M,+Baumann+E:+Ueber+das+Wesen+der+Alkaptonurie.+Z+Physiol+Chem+15:228,+&source=bl&ots=HwsprDt2Lv&sig=ACfU3U3aUkDRuYyE7UEAtzWk0vvzqu3RDA&hl=en&sa=X&ved=2ahUKEwiekcz81Z7qAhWcgnIEHRisBbEQ6AEwAXoECAgQAQ#v=onepage&q=178.% 09Wolkow M% 2C Baumann E% 3A Ueber das Wesen der Alkaptonurie. Z Physiol Chem 15% 3A228% 2C &f=false (accessed June 26, 2020). |
[223] | Müller C.R. , Fregin A. , Srsen S. , Srsnova K. , Halliger-Keller B. , Felbor U. , Seemanova E. and Kress W. . Allelic heterogeneity of alkaptonuria in Central Europe, Eur. J. Hum. Genet. 7: ((1999) ), 645–651. doi: 10.1038/sj.ejhg.5200343. |
[224] | Granadino B. , De Bernabé D.B.V. , Fernández-Cañón J.M. . , M.A Peñalva. and S.R De Córdoba. . The human homogentisate 1,2-dioxygenase (HGO) gene, Genomics. 43: ((1997) ), 115–122. doi: 10.1006/geno.1997.4805. |
[225] | Srsen S. , Müller C.R. , Fregin A. and Srsnova K. . Alkaptonuria in Slovakia: Thirty-two years of research on phenotype and genotype, Mol. Genet. Metab. 75: ((2002) ), 353–359. doi: 10.1016/S1096-7192(02)00002-1. |
[226] | Zatkova A. , Chmelikova A. , Polakova H. , Ferakova E. and Kadasi L. . Rapid detection methods for five HGO gene mutations causing alkaptonuria, Clin. Genet. 63: ((2003) ), 145–149. doi: 10.1034/j.1399-0004.2003.00027.x. |
[227] | Goicoechea De Jorge E. , Lorda I. I. , Gallardo M.E. , Pérez B. , Peréz De Ferrán C. , Mendoza H. and Rodríguez De Córdoba S. . Alkaptonuria in the Dominican Republic: identification of the founder AKU mutation and further evidence of mutation hot spots in the HGO gene, J. Med. Genet. 39: ((2002) ). doi: 10.1136/jmg.39.7.e40. |
[228] | Phornphutkul C. , Introne W.J. , Perry M.B. , Bernardini I. , Murphey M.D. , Fitzpatrick D.L. , Anderson P.D. , Huizing M. , Anikster Y. , Gerber L.H. and Gahl W.A. . Natural history of alkaptonuria, N. Engl. J. Med. 347: ((2002) ), 2111–2121. doi: 10.1056/NEJMoa021736. |
[229] | Gaines J.J. Jr , Pai G.M. . Cardiovascular Ochronosis –PubMed, Arch Pathol Lab Med. 111: ((1987) ), 991–4. https://pubmed.ncbi.nlm.nih.gov/3632274/ (accessed June 25, 2020). |
[230] | Kutty M.K. , Iqbal Q.M. , Teh E.C. and Lumpur K. . Ochronotic Arthropathy: An Electron Microscopical Study With a View on Pathogenesis, Arch Pathol. 98: ((1974) ), 55–7. https://pubmed.ncbi.nlm.nih.gov/4829777/. (accessed June 25, 2020). |
[231] | Gaucher A. , Faure G. , Netter P. , Floguet J. and Duheille J. . Synovial membrane from ochronotic arthropathy of the hip joint. Scanning electron microscopic study, Z Rheumatol. 39: ((1980) ), 231–5. https://pubmed.ncbi.nlm.nih.gov/7456770/ (accessed June 25, 2020). |
[232] | Mohr W. , Wessinghage D. and Lenschow E. . Die Ultrastruktur von hyaline Knorpel und Glenk kapselgewebe bei der aldaptonischen Ochronose, Z Rheumatol. 39: ((1980) ), 39. https://books.google.com/books?id=YpfzDQAAQBAJ&pg=PA106&lpg=PA106&dq=183.% 09Mohr+W,+Wessinghage+D,+Lenschow+E:+Die+Ultrastruktur+von+hyaline+Knorpel+und+Glenk+kapselgewebe+bei+der+aldaptonischen+Ochronose.+Z+Rheumatol+39:55,+&source=bl&ots=HEOb9qmzlA&sig=ACfU3U3Q9yfdZf-HkADhY5oAx2_fNcfXzA&hl=en&sa=X&ved=2ahUKEwjms-b22J7qAhXlmXIEHU4MAd0Q6AEwAHoECAoQAQ#v=onepage&q=183.% 09Mohr W% 2C Wessinghage D% 2C Lenschow E% 3A Die Ultrastruktur von hyaline Knorpel und Glenk kapselgewebe bei der aldaptonischen Ochronose. Z Rheumatol 39% 3A55% 2C &f=false (accessed June 26, 2020). |
[233] | Zannoni V.G. , Lomtevas N. and Goldfinger S. . Oxidation of homogentisic acid to ochronotic pigment in connective tissue, BBA - Gen. Subj. 177: ((1969) ), 94–105. doi: 10.1016/0304-4165(69)90068-3. |
[234] | La Du B.N. , Zannoni V.G. and Wolman M. Pigments in Pathology. Ochronosis, Academic Press, New York, (1969) . https://books.google.com/books/about/Pigments_in_pathology.html?id=mGZrAAAAMAAJ. (accessed June 25, 2020). |
[235] | Virchow R. . Ein Fall von allgemeiner Ochronose der Knorpel und knorpelähnlichen Theile,r Pathol. Anat. Und Physiol. Und Für Klin. Med, Arch. Für. 37: ((1866) ), 212–219. doi: 10.1007/BF01935634. |
[236] | Albrecht H. . Ueber Ochronose, Heilkdunde 23 (1902) 366. https://books.google.com/books?id=AqenBwAAQBAJ& pg=PA38&lpg=PA38&dq=Uber+Ochronosis.+Heilkunde+23:366,+&source=bl&ots=WQQjP9dYxm&sig=ACfU3U3NEI_0AO2rW5PJFQ-dIeAWoV0SUA&hl=en&sa=X&ved=2ahUKEwieqfSGlZ7qAhUGlXIEHSlzAdAQ6AEwAHoECAsQAQ#v=onepage&q=Uber Ochronosis. Heilkunde 23% 3A366% 2C &f=false (accessed June 25, 2020). |
[237] | Osler W. . Ochronosis: the pigmentation of cartilages, sclerotics, and skin in alkaptonuria, Lance. 163: ((1904) ), 10–11. doi: 10.1016/S0140-6736(00)97815-2. |
[238] | Smith J.W. . Ochronosis of the sclera and cornea complicating alkaptonuria: Review of the literature and report of four cases, J. Am. Med. Assoc. 120: ((1942) ), 1282–1288. doi: 10.1001/jama.1942.02830510020005. |
[239] | Van Offel J.F. . De Clerck L.S. . , Francx L.M. . and W.J Stevens. . The clinical manifestations of ochronosis: A review, Acta Clin. Belg. 50: ((1995) ), 358–362. doi: 10.1080/17843286.1995.11718475. |
[240] | Hench P.S. and Bauer W. . Rheumatism and arthritis; a review of American and English literature, Ann. Intern. Med. 28: ((1948) ), 309–451. doi: 10.7326/0003-4819-28-2-309. |
[241] | Hamdulay S.S. , Finegold J. , Boyer L. , Khanna M. , Akmal M. , Walker A. and Kinderlerer A. . Clinical images: Magnetic resonance imaging appearance of alkaptonuria, Arthritis Rheum. 64: ((2012) ), 129. doi: 10.1002/art.33357. |
[242] | Al-Mahfoudh R. , Clark S. and Buxton N. . Alkaptonuria presenting with ochronotic spondyloarthropathy, Br. J. Neurosurg. 22: ((2008) ), 805–807. doi: 10.1080/02688690802226368. |
[243] | Pomeranz M.M. , Friedman L.J. , Tunick I.S. . Roentgen Findings in Alkaptonuric Ochronosis, Radiology. 37: ((1941) ), 295–303. doi: 10.1148/37.3.295. |
[244] | Perrone A. , Impara L. , Bruni A. , Primicerio P. and Marini M.R. . Radiographic and MRI Findings in Ochronosis, Radiol Med. 110: ((2020) ), 349–58. https://pubmed.ncbi.nlm.nih.gov/16292242/ (accessed June 25, 2020). |
[245] | Justesen P. and Andersen P.E. . Radiologic manifestations in alcaptonuria, Skeletal Radiol. 11: ((1984) ), 204–208. doi: 10.1007/BF00349495. |
[246] | Kenny D. , Ptacin M.J. , Bamrah V.S. and Almagro U. . Cardiovascular ochronosis: A case report and review ofthe medical literature, Cardiol. 77: ((1990) ), 477–483. doi: 10.1159/000174641. |
[247] | Fisher A.A. and Davis M.W. . Alkaptonuric ochronosis with aortic valve and joint replacements and femoral fracture: a case report and literature review, Clin. Med. Res. 2: ((2004) ), 209–215. doi: 10.3121/cmr.2.4.209. |
[248] | Concistrè G. , Fiorani B. , Ranocchi F. , Casali G. , Loforte A. and Musumeci F. . Black aorta in a patient with alkaptonuria (ochronosis), J. Cardiovasc. Med. 12: ((2011) ), 444–445. doi: 10.2459/JCM.0b013e328344e5d2. |
[249] | Hannoush H. , Introne W.J. , Chen M.Y. , Lee S.J. , O’Brien K. , Suwannarat P. , Kayser M.A. , Gahl W.A. and Sachdev V. . Aortic stenosis and vascular calcifications in alkaptonuria, Mol. Genet. Metab. 105: ((2012) ), 198–202. doi: 10.1016/j.ymgme.2011.10.017. |
[250] | Lichtenstein L. and Kaplan L. . Hereditary ochronosis; pathologic changes observed in two necropsied cases, Am J Pathol. 30: ((1954) ), 99–125. https://pubmed.ncbi.nlm.nih.gov/13124462/ (accessed June 2, 2020). |
[251] | Vlay S.C. , Hartman A.R. , Culliford A.T. . Alkaptonuria and aortic stenosis, Ann. Intern. Med. 104: ((1986) ), 446. doi: 10.7326/0003-4819-104-3-446_1. |
[252] | Ptacin M. , Sebastian J. , Bamrah V.S. . Ochronotic cardiovascular disease, Clin. Cardiol. 8: ((1985) ), 441–445. doi: 10.1002/clc.4960080807. |
[253] | Sener R.N. . Prostatic and renal stones and unilateral obstruction of the urinary tract caused by ochronosis [9],, Am. J. Roentgenol. 158: ((1992) ), 214–215. doi: 10.2214/ajr.158.1.1727353. |
[254] | Young H.H. . Calculi of the prostate associated with ochronosis and alkaptonuria, J. Urol. 51: ((1944) ), 48–58. doi: 10.1016/S0022-5347(17)70325-4. |
[255] | Oppenheimer B.S. , Kline B.S. . Ochronosis: With a study of an additional case, Arch. Intern. Med. 29: ((1922) ), 732–747. doi: 10.1001/archinte.1922.00110060024002. |
[256] | Akeda K. , Kasai Y. , Kawakita E. , Matsumura Y. , Kono T. , Murata T. and Uchida A. . Thoracic myelopathy with alkaptonuria, Spine (Phila. Pa. 1976). 33: ((2008) ). doi: 10.1097/BRS.0b013e31816046ea. |
[257] | Eisenberg H. . Alkaptonuria, ochronosis, arthritis and ruptured intervertebral disk: complicated by homologous serum reaction, Arch. Intern. Med. 86: ((1950) ), 79–86. doi: 10.1001/archinte.1950.00230130101006. |
[258] | Smith H.P. , Smith H.P. . Ochronosis: report of two cases, Ann. Intern. Med. 42: ((1955) ), 171. doi: 10.7326/0003-4819-42-1-171. |
[259] | Cooper P.A. . Alkaptonuria With Ochronosis, Proc R Soc Med. 44: ((2020) ), 917. https://pubmed.ncbi.nlm.nih.gov/14900184/ (accessed June 25, 2020). |
[260] | Introne W.J. , Gahl W.A. . Alkaptonuria, University of Washington, Seattle, 2017. http://www.ncbi.nlm.nih.gov/pubmed/20301627 (accessed June 24, 2020). |
[261] | Rosenbaum H. and Reveno W.S. . Polycythemia and Alkaptonuria; case report, Harper Hosp Bull. 10: ((2020) ), 36–8. https://pubmed.ncbi.nlm.nih.gov/14906911/ (accessed June 25, 2020). |
[262] | Goldberg B.H. , Penso J.S. , Stern L.M. and Bergstein J.M. . Alkaptonuria with nephrocalcinosis, J. Pediatr. 88: ((1976) ), 518–519. doi: 10.1016/S0022-3476(76)80285-5. |
[263] | Minno A.M. , Rogers J.A. . Ochronosis: report of a case, Ann. Intern. Med. 46: ((1957) ), 179–183. doi: 10.7326/0003-4819-46-1-179. |
[264] | Yules J.H. . Ochronotic arthritis: report of a case, Ochronotic Arthritis; report of a case, Bull N Engl Med. 16: ((2020) ), 168–73. https://pubmed.ncbi.nlm.nih.gov/13219449/ (accessed June 25, 2020). |
[265] | Hammond G. and Powers H.W. . Alkaptonuric arthritis; report of a case,Lahey Clin Found Bull. 11: ((1958) ), 18–22. https://pubmed.ncbi.nlm.nih.gov/13612138/ (accessed June 25, 2020). |
[266] | Crissey R.E. , Day A.J. . Ochronosis: A case report, J Bone Jt. Surg Am. 32: ((1950) ), 688. https://journals.lww.com/jbjsjournal/Citation/1950/32030/OCHRONOSIS__A_Case_Report.26.aspx (accessed June 25, 2020). |
[267] | Martin W.J. , Underdahl L.O. , Mathieson D.R. . Alkaptonuria: Report of 3 Cases, Proc Staff Meet Mayo Clin. 27: ((2020) ), 193 –200. https://pubmed.ncbi.nlm.nih.gov/14920558/ (accessed June 25, 2020). |
[268] | Lustberg T.J. , Schulman J.D. , Seegmiller J.E. . The preparation and identification of various adducts of oxidized homogentisic acid and the development of a new sensitive colorimetric assay for homogentisic acid, Clin. Chim. Acta. 35: ((1971) ), 325–333. doi: 10.1016/0009-8981(71)90202-6. |
[269] | Bory C. , Boulieu R. , Chantin C. and Mathieu M. . Diagnosis of alcaptonuria: Rapid analysis of homogentisic acid by HPLC, Clin. Chim. Acta. 189: ((1990) ), 7–11. doi: 10.1016/0009-8981(90)90228-K. |
[270] | Spencer J.M.F. , Gibbons C.L.M.H. , Sharp R.J. , Carr A.J. , Athanasou N.A. . Arthroplasty for ochronotic arthritis: No failure of 11 replacements in 3 patients followed 6–12 years, Acta Orthop. Scand. 75: ((2004) ), 355–358. doi: 10.1080/00016470410001321. |
[271] | Aliberti G. , Pulignano I. , Pisani D. , March M.R. , Del Porto F. and Proietta M. . Bisphosphonate treatment in ochronotic osteoporotic patients, Clin. Rheumatol. 26: ((2007) ), 729–735. doi: 10.1007/s10067-006-0390-3. |
[272] | Suzuki Y. , Oda K. , Yoshikawa Y. , Maeda T. and Suzuki T. . A novel therapeutic trial of homogentisic aciduria in a murine model of alkaptonuria, J. Hum. Genet. 44: ((1999) ), 79–84. doi: 10.1007/s100380050114. |
[273] | Introne W.J. , Perry M.B. , Troendle J. , Tsilou E. , Kayser M.A. , Suwannarat P. , O’Brien K.E. , Bryant J. , Sachdev V. , Reynolds J.C. , Moylan E. , Bernardini I. , Gahl W.A. . A 3-year randomized therapeutic trial of nitisinone in alkaptonuria, Mol. Genet. Metab. 103: ((2011) ), 307–314. doi: 10.1016/j.ymgme.2011.04.016. |
[274] | Suwannarat P. , O’Brien K. , Perry M.B. , Sebring N. , Bernardini I. , Kaiser-Kupfer M.I. , Rubin B.I. , Tsilou E. , Gerber L.H. and Gahl W.A. . Use of nitisinone in patients with alkaptonuria, Metabolis. 54: ((2005) ), 719–728. doi: 10.1016/j.metabol.2004.12.017. |


