
Novel Treatments for Polycystic Kidney Disease
Abstract
Polycystic kidney disease (PKD) and nephronophthisis are common manifestation of ciliopathies. PKD is the most common genetic renal condition; it affects 12.5 million people worldwide. PKD is a great example of decades of translational research leading to the discovery of novel treatments and significant number of clinical trials. This review will concentrate on the basic molecular and cellular pathophysiology that led to the development of therapeutic targets for PKD.
1Introduction
The most widely encountered renal ciliopathies include polycystic kidney disease (PKD), nephronophthisis (NPHP) and Bardet-Biedl, Joubert, and Meckel-Gruber syndromes. PKD refers to two specific genetic diseases, autosomal recessive polycystic kidney disease/congenital hepatic fibrosis (ARPKD/CHF, MIM *606702) and autosomal dominant polycystic kidney disease (ADPKD-OMIM *601313 and OMIM *173910). ARPKD/CHF and ADPKD have undergone decades of translational research resulting in the discovery of numerous therapeutic targets leading to a significant number of clinical trials. This review will provide a discussion of the basic molecular, and cellular pathophysiology that have led to the development of these therapies.
ARPKD/CHF is a dual organ disease characterized by cystic dilations of renal collecting ducts and hepatobiliary dysgenesis leading to varying degrees of congenital hepatic fibrosis [1, 2]. The incidence of ARPKD is estimated to be around one in 20,000 to one in 40,000 live births with a carrier rate of approximately 1/70 [3]. ARPKD is caused by mutations in PKHD1, which encodes fibrocystin or polyductin (FPC), a large protein with a single transmembrane spanning domain. Missense, frameshift, and truncating mutations have all been described [4], and nearly all patients are compound heterozygotes [5–7]. FPC colocalizes with polycystin-2 (PC2), the protein product of PKD2, at the basal bodies of primary cilia although the function of this complex is unknown.
ADPKD, the most common genetic renal disease occurs at a rate of 1/400 to 1/1000 live births [8] and affects 12.5 million individuals worldwide [9]. ADPKD classically presents with slow, progressive replacement of kidney tissue with fluid-filled cysts leading to end-stage renal disease (ESRD) most commonly by the sixth decade of life. ADPKD can manifest in utero, in infants, and in children and can be a significant cause of morbidity and mortality in this age group [10–12]. ADPKD is a heterogenic disease caused by mutations in two genes. PKD1 (OMIM *601313), encoding polycystin 1 (PC1) accounts for 80% of all ADPKD and PKD2 (OMIM *173910) responsible for 15% of ADPKD encoding PC2 [8]. The remaining 5% of ADPKD families do not map to either the PKD1 or PKD2 locus, suggesting the possibility of a third ADPKD gene; GANAB or DNAJB11 may account for some of these cases [13–15].
There is extreme allelic heterogeneity in PKD with over 1200 known pathogenic mutations in PKD1 (http://pkdb.mayo.edu). In contrast to PKD1, the PKD2 gene is much smaller and less complex and it has simpler features and structure [16]. However, a significant degree of allelic heterogeneity exists in PKD2 as well with over 200 pathogenic mutations identified to date (http://pkdb.mayo.edu). PC1 has a cytoplasmic C terminus that regulates multiple signaling cascades [17] and is also known to interact with PC2. PC2 is a calcium-permeable channel with six transmembrane spanning elements; both the amino and carboxy termini face the cytoplasmic compartment. Interaction with PC1 is required for full-length PC2 to leave the ER and reach the cilia [18, 19].
2Pathophysiology of cyst formation
The process of cyst formation and growth is multidimensional and dynamic with multiple independent contributing components. PKD proteins are localized to apical and basolateral plasma membranes, desmosomes, focal adhesions, basal bodies, and primary cilium. In mouse models of PKD, cyst initiation and expansion are regulated by Pkd1 gene dosage [20–22]. Tubular epithelial cells have a threshold expression level of Pkd1 that is necessary to maintain normal tubular size and non-proliferative state. When the level of functional PC1 falls below the cystogenic threshold, the proliferation of tubular epithelial cells and cyst formation follows. Recent studies have demonstrated that the rate of cyst growth is regulated and can be adjusted by altering levels of functional PC1 [22]. Overexpression of Pkd1 can rescue a mutant Pkhd1. Hence PC1 dosage modifies the severity of both ADPKD and ARPKD.
The role of the primary ciliary structure in PKD cyst formation has recently been interrogated using conditional and inducible mouse models that allow structural ciliary genes (Kif3a and Ift20) and PKD genes (Pkd1, Pkd2, and Pkhd1), to be inactivated or deleted in a spatiotemporal manner, either alone or in combination. These studies specifically examined the functional role of PC1 and PC2 proteins on the cilia and revealed a cilia-dependent proliferation (cyst-promoting) pathway that is inhibited by a normal PC1/PC2 complex on the cilia [23, 24]. Furthermore, it is shown that Tubby-related protein 3 (Tulp3) and Daz interacting protein-1 (DZIP1L) impact the efficiency of the PC1/PC2 insertion into the cilia. Murine knock-outs of either Tulp3 and Dzip1l leads to collecting duct cysts [25, 26].
The focal nature of cyst formation in ADPKD adds additional complexity to the pathophysiology. Despite all cells in the body having the same germ-line mutation, only 5 to 10% of the nephrons become cystic. This puzzling fact led to the development of the two-hit theory which hypothesizes that a second mutation (i.e., a somatic mutation) is required for the initiation of cyst formation. Hence ADPKD behaves as a recessive disease at the molecular level. Substantial evidence exists to support this theory [27–30] however, additional factors have been shown to influence disease progression and severity [31]. These include: 1) the developmental timing of PKD1 inactivation [32–35]; 2) reduction in functional PC1 levels [20, 22, 36, 37]; and 3) one cyst among neighboring cells and nephrons can create a toxic environment resulting in a “snowball effect” leading to cyst development in neighboring nephrons [38].
The complex interactions between the three PKD proteins make gene-targeted therapies a daunting task. As a result, most therapies at present are aimed at interrupting abnormal signaling pathways driving cyst growth and associated interstitial inflammation and fibrosis.
3Targeting the cystic phenotype
During cyst formation normal renal epithelial cells change from mature, differentiated, nonproliferative, absorptive cells to partially de-differentiated, secretory cystic cells characterized by specific polarity defects and increased rates of proliferation [8]. Cyst formation is associated with proliferation of cells lining the expanding cysts, fluid secretion into cysts that have lost their connection to the nephron and remodeling of the extracellular matrix surrounding the expanding tubules.
Multiple signaling molecules and pathways have been identified in the phenotypic change of normal tubular epithelial to the cystic phenotype [8]. The precise mechanisms by which defective PKD proteins alter these signaling pathways are not fully understood. Despite this knowledge gap, key pathogenic features common in ARPKD and ADPKD have been identified and form the basis of current therapeutic interventions and clinical trials. These features include:
• Altered intracellular cAMP levels coupled with decreased intracellular calcium leading to increased proliferation and fluid secretion.
• Abnormalities in expression, localization, and activity of the epidermal growth factor (EGFR) family of receptors and ligands (EGFR axis) leading to increased proliferation.
• Increased activity of cSrc, a critical mediator of cross-talk between the EGFR axis and G-protein-cAMP pathways which is also required for STAT3 activation by the PC1 tail.
• Alterations in cell-cell adhesion, and cell-matrix interactions.
• Chronic inflammation that leads to progressive fibrosis.
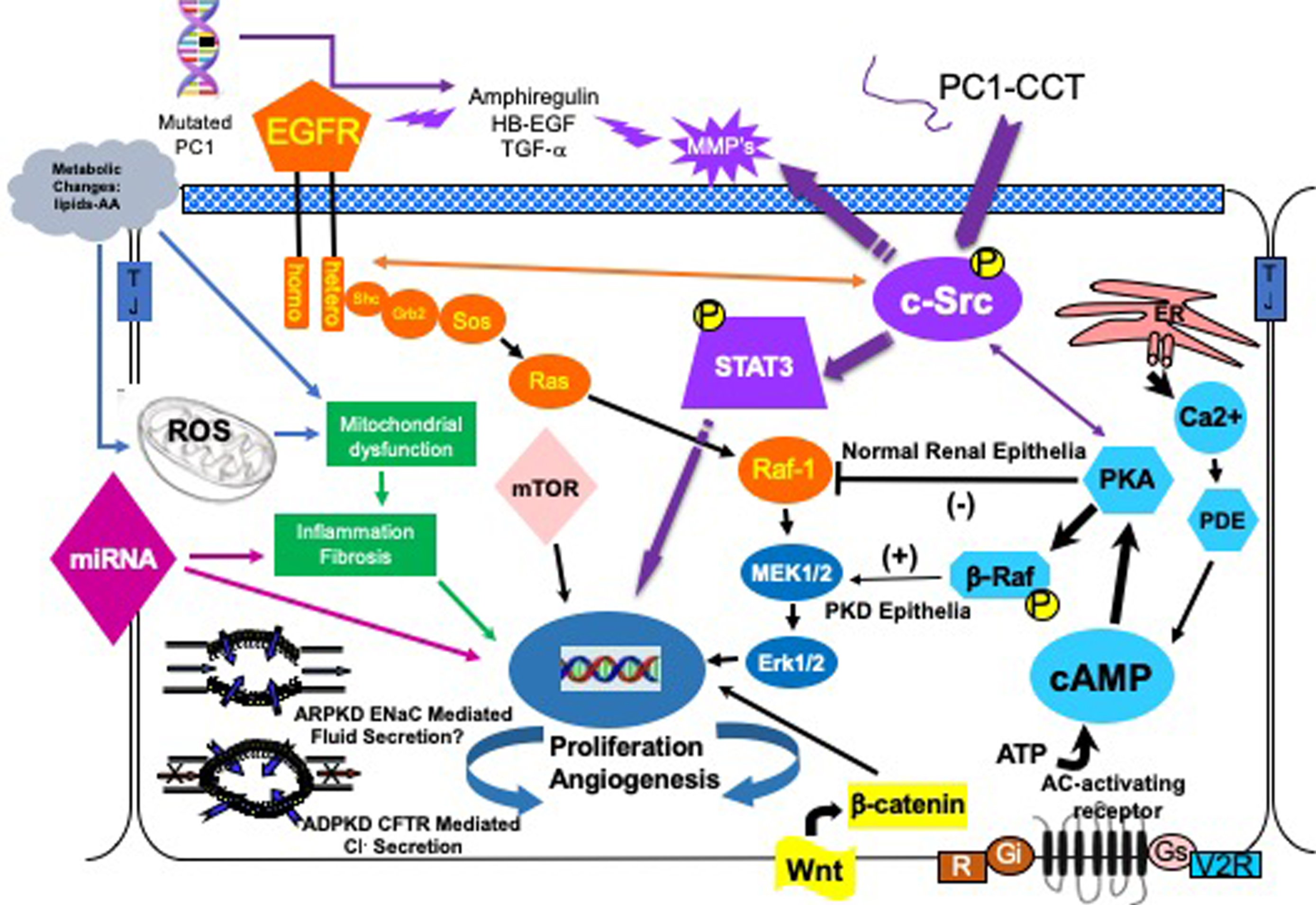
Clinical trials to date have been largely based on targeting these aberrant, complex cellular signaling elements. Therefore, a brief discussion of each along with the results of clinical trials targeting these elements to date will follow. Figure 1 depicts major signaling pathways that comprise the phenotype of the cystic cell. The main proliferative pathways are shown, but other entry points into these pathways are possible and likely. A list of current and completed clinical trials can be found at http://clinicaltrials.gov/
Fig.1
The cystic cellular phenotype. This cartoon is an abridged composite of the abnormal signal transduction pathways reported to be active in PKD. Two main conduits that lead to unchecked proliferation are 1) the EGFR axis (orange), and 2) a G-protein axis (aqua blue path) that leads to increased cAMP and a switch in the phenotypic response of renal epithelia to cAMP. The pathways suggest a possible pathogenic process as follows: in ADPKD (purple path) a mutated PC1 leads to increase amphiregulin, activating EGFR, resulting in increased cSrc phosphorylation; in both ARPKD and ADPKD, cSrc activation alters the cellular response of cAMP resulting in increased proliferation and secretion; in addition, the cytoplasmic tail, PC1-p30, is overexpressed. PC1-p30 interaction with Src causes Src-dependent activation of STAT3 by tyrosine phosphorylation; and in ARPKD, an apical EGFR becomes activated resulting in reciprocal phosphorylation of the nonreceptor tyrosine kinase cSrc leading to increased cAMP-mediated, and EGFR driven proliferation. Increased activity of EGFR and increased cAMP amplify the activation of Src-STAT3 pathway. There are multiple entry points that can augment these two main pathways including the WNT/B-Catenin pathway, metabolic changes leading to the generation of reactive oxygen species and mTOR activation and abnormalities of miRNAs.
3.1cAMP-mediated proliferation and secretion
The cAMP-dependent pathway, or the adenylate cyclase pathway, is a G-protein-coupled receptor signaling pathway. In normal renal epithelia, increased intracellular cAMP results in a signal to reduce epithelial proliferation, but in cystic renal epithelia this blocking signal is bypassed, and cAMP is needed to maintain a pro-proliferative phenotype. Both low intracellular calcium level, (theoretically due to a defective PC1/PC2 calcium channel) and cSrc dependent phosphorylation of β-Raf, allows the cell to bypass Raf-1 and increase ERK phosphorylation to stimulate cell proliferation [39, 40]. Increased cAMP in a cell with an active MAPK pathway also changes the cell from an absorptive to a secretory cell which contributes significantly to the progressive cyst enlargementin ADPKD [41].
Intracellular cAMP is pharmacologically reduced by vasopressin R2 receptor antagonists such as tolvaptan (Samsca or Jinarc®) or somatostatin (and its long-acting analogs such as lanreotide) that inhibit adenylate cyclase [42, 43]. Tolvaptan has undergone extensive human clinical trials. Tolvaptan has proven to slow the increase in total kidney volume and reduce the rate of decline in loss of renal function in ADPKD patients [44, 45]. However, in patients with later-stage ADPKD, treatment with lanreotide compared with standard care did not slow the decline in kidney function over 2.5 years of follow-up [46]. Jinarc® (Tolvaptan) has been approved in Europe, Japan, Canada and Australia for treatment of ADPKD in adults. Tolvaptan has recently been approved in the USA under the trade name JYNARQUE®. Due to FDA concerns regarding potentially serious adverse liver events, in the USA, JYNARQUE is only available through a restricted distribution program called the JYNARQUE Risk Evaluation and Mitigation Strategy designed to closely monitor patients for liver or other adverse events(www.fda.gov/safety; https://www.jynarque.com). Tolvaptan is not currently recommended for use in children due to concerns regarding the risk of fluid imbalance although clinical trials are underway in Europe [NCT02964273 Belgium and Italy].
3.2EGFR (ErbB) axis
EGFR axis consists of the epidermal growth factor/ErbB family of receptors and ligands. A large body of evidence indicates that one or more members of the ErbB receptor family play a role in renal cystic epithelial proliferation. This includes EGFR, as well as the related receptors, ErbB2 and ErbB4 [47–50]. In human ADPKD and ARPKD and every rodent model of PKD published to date, cystic kidneys display characteristic abnormalities in EGFR-axis. Inhibition or reduction of EGFR activity through genetic manipulation [51] or administration of tyrosine kinase inhibitors [52, 53], reduces cyst formation and enlargement in animal models.
Microarray profiling in human PKD1 cystic cells demonstrated the EGF/ErbB family receptor ErbB4, is a major driver of cyst growth in ADPKD. ErbB4 levels may also serve as a prognostic biomarker for disease progression [50]. Recent studies provide a direct link between PC1 and PC2 proteins and the EGFR axis; EGF reduces the threshold of PKD2 activation by releasing it from PIP(2)-mediated inhibition [54].
3.3cSrc
cSrc is a critical intermediate that integrates signals between two major proliferative pathways in a cystic epithelial cell, the EGFR axis and cyclic AMP pathway [55] (Fig. 1). Also, a proteolytic fragment of the PC1 cytoplasmic tail, PC1-p30, interacts with cSrc and activates STAT3 stimulating further increase in proliferation [56–58]. Src activation of STAT3 is amplified by increase of both EGFR axis activity and cAMP levels resulting in a self-perpetuating cycle that accelerates proliferation [58]. Further, a mutated PC1 increases amphiregulin expression, resulting in activation of the EGFR axis and reciprocal activation of Src which amplifies inputs from EGFR, and cAMP signaling pathways to activate STAT3 [58]. In a clinical trial on ADPKD patients, Bosutinib, a cSrc inhibitor, (ClinicalTrials.gov Identifier NCT01233869) was very effective in reducing total kidney volume but did not reduce the decline in renal function [59].
3.4mTOR
The mammalian target of rapamycin (mTOR) pathway integrates signals from growth factors (including EGFR), G-protein coupled receptors (which generate cAMP), cellular energy levels, and stress conditions to stimulate protein synthesis and cell growth through phosphorylation of S6K1 and eIF4E [60, 61]. In human ADPKD, ARPKD and a variety of animal models, cyst-lining epithelium reveals an increased activity of mTOR [61–64]. The mTOR inhibitors rapamycin and everolimus, therefore, have been tested in human clinical trials and were found to be ineffective in slowing total kidney volume or the progressive loss of renal function [65, 66].
In an effort to explain the disappointing results of the mTOR trials, it has been suggested that ineffective renal concentrations of mTOR inhibitors may be responsible for the unsatisfactory clinical results. Based on the finding that cystic cells express folate receptors, Shillingford et al. conjugated folate to the mTOR inhibitor rapamycin [67] and showed that in a Pkd1 animal model when rapamycin was conjugated with folate, target specificity was increased and the effectiveness of mTOR inhibition was dramatically improved [67]. This concept of linking a therapeutic agent with a protein that has a receptor on the cell of interest is not new but is one that deserves further investigation in the field of PKD therapy. In ARPKD, cystic lesions occur largely in collecting ducts. The ability to specifically target collecting duct cells allows for lower systemic exposure of the therapy while increasing the concentration at the site of disease.
3.5Multi-kinase inhibitor
Multi-kinase inhibitor therapy takes advantage of a compound that inhibits the activity of multiple proteins known to be aberrantly active in cystic disease process. One such compound Tesevatinib (TSV) previously known as KD-019, is a unique multi-kinase inhibitor that targets EGFR, c-Src and KDR (VEGFR2). TSV was effective in slowing the proliferation of renal epithelia and cyst growth, progression of fibrosis and the loss of renal function in the BPK and orthologous PCK rat models of ARPKD [68].
TSV is currently being tested on adults in two ADPKD Phase II-III clinical trials (ClinicalTrials.gov Identifier NCT01559363 and NCT03203642). TSV is also the first specific disease-targeted therapy to receive FDA approval for a clinical trial in pediatric ARPKD (ClinicalTrials.gov Identifier NCT03096080). All three clinical trials are still in progress.
3.620-HETE
Metabolic changes are known to occur during the development of ARPKD and ADPKD. Bioactive lipids such as 20-HETE, a P450-produced metabolite of arachidonic acid was shown to correlate with eGFR and TKV in ADPKD patients [69]. Inhibitors of 20-HETE have been effective in reducing cyst formation in animal models of PKD [70–72]. Therefore, inhibition of 20-HETE production may be a potential therapeutic agent for slowing PKD progression. In addition, urinary 20-HETE levels may serve as a non-invasive biomarker of disease severity [69].
4Future approaches
4.1miRNA
miRNAs are short non-coding RNA molecules (∼22 nucleotides) that regulate gene expression. There are emerging data that support the integral role of miRNAs in the pathogenesis of PKD [73–76]. Recent studies in human ADPKD have linked a single miRNA to the progression of disease [50], and more recently a unique interstitial miRNA signature was found to play a pivotal role in the pathogenesis of renal fibrosis in an orthologous animal model of ADPKD [77]. Inhibitors of miRNA are powerful candidates for therapeutic intervention against various pathological conditions. One advantage of such therapies is the small size that makes it easier to get to target disease tissue [78]. Pharmaceutical companies are actively pursuing the development of miRNA-based therapies for numerous human diseases [79].
4.2Targeting cilia
The primary cilium has an exceptionally high density of receptors on its membrane that are important for sensing and transducing extracellular stimuli. Moreover, the primary cilium exists as a separate cellular compartment from the cytosol, providing for unique spatial and temporal regulation of signaling molecules to initiate downstream events. Many cilia proteins are found at other sites and have non-ciliary functions. Therefore care must be exercised in attributing biological effects of a protein solely to the function of the cilia. The complexity of the spatial and temporal integration of proteins into the cilia must certainly be important in the proper function of the cilia and the efficiency in which signals are transmitted to the cell. Proteins and mechanisms that impact the efficiency or order of protein trafficking would theoretically be modifiable events and as such would present novel therapeutic targets.
Recent studies of cilia structure and function have identified proteins of interest, such as Tulp3 and Dzip1l, that may be targeted in PKD due to their ability to increase the efficiency of PC1/PC2 integration into the cilia. As the molecular biogenesis of the cilia becomes further characterized, opportunities to alter the efficiency or function of the cilia may become part of the therapeutic arsenal for PKD and other ciliopathies.
5Conclusion
The clinical trials to date have shown that targeting a single molecule of an aberrant pathway in PKD has been ineffective in preventing the loss of renal function. In the short term, the most promising therapies will likely target key signaling intermediates that appear to integrate multiple pathways. These might include Src, (Fig. 1) [55, 58] and/or a combination therapy approach where multiple compounds are used to target multiple pathways simultaneously or a single compound is used to target multiple pathways such as a multi-kinase inhibitors (MKI) like tesevatinib (TSV).
Rational therapies will require knowledge of both the extent of disease and the rate of disease progression. Therapeutic targets will be stage-specific and change as the disease progresses. Therapies will become increasingly focused on treating PKD in early childhood where they are likely to provide the greatest long-term benefit. Two clinical trials for children with PKD are underway: One for children with ADPKD [Tolvaptan [NCT02964273 Belgium and Italy] and one for the use of TSV in ARPKD (ClinicalTrials.gov Identifier NCT03096080). Therapies that target abnormal signaling pathways will need to be carefully tailored, so that pathway activity is reduced to normal basal levels rather than eliminated. Promising compounds will be modified to direct the molecule to the site of disease, making these therapies highly specific with low levels of toxicity [67]. Epigenetic and dietary factors that slow or accelerate the progression of PKD will likely be discovered, and adherence or avoidance of such factors may slow the rate of progression and eliminate the need for pharmacological intervention or renal replacement therapy for some.
The importance of gene dosage, modifier genes and somatic mutations in the clinical course of a disease in an individual provide a compelling rationale for personalized medicine. Personalized medicine will mean tailored approaches that modulate functional gene dosage and consider not only individual genotypes but also the response of the kidney to disease and the unanticipated response of the kidney to therapy. This unanticipated response to therapy may be active in tolvaptan treatment which reportedly results in increased shedding of HB-EGF [80]. The results of this shedding are unknown but would suggest that combining Tolvaptan with a tyrosine kinase inhibitor may improve clinical responses. Therapeutically improving the efficiency of PKD1 processing in the ER (neutral alpha-glucosidase designated AB, (GANAB)) or improving the efficiency of its insertion at the cilia (TULP3) may have significant impact on the disease expression.
Conflict of interest
The authors declare that they have no conflicts of interest concerning research, authorship and publication of this manuscript.
Acknowledgments
This work was supported, in part, by the Lillian Goldman Charitable Trust; the Amy P. Goldman Family Trust, the EllsworthFamily Fund; and the Children’s Research Institute at the Children’s Hospital of Wisconsin.
References
[1] | Gunay-Aygun M. . et al., Autosomal recessive polycystic kidney disease and congenital hepatic fibrosis: Summary statement of a first National Institutes of Health/Office of Rare Diseases conference, J Pediatr 149: (2) ((2006) ), 159–164. |
[2] | Sweeney W.E. Jr. , et al., Childhood Polycystic Kidney Disease, in Pediatric Nephrology, Avner E.D. , et al., Editors. (2016) , pp. 1–58. |
[3] | Zerres K. . et al., Mapping of the gene for autosomal recessive polycystic kidney disease (ARPKD) to chromosome 6p21-cen, Nat Genet 7: (3) ((1994) ), 429–432. |
[4] | Bergmann C. and Zerres K. Polycystic Kidney Disease:ADPKD and ARPKD, in Comprehensive Pediatric Nephrology, Geary D.F. and Schaefer F. , Editors, Mosby (Elsevier): Philadelphia, (2008) , pp. 155–178. |
[5] | Ward C.J. . et al., The gene mutated in autosomal recessive polycystic kidney disease encodes a large, receptor-like protein, Nat Genet 30: (3) ((2002) ), 259–269. |
[6] | Bergmann C. . et al., Spectrum of mutations in the gene for autosomal recessive polycystic kidney disease (ARPKD/PKHD1), J Am Soc Nephrol 14: (1) ((2003) ), 76–89. |
[7] | Rossetti S. and Harris P.C. , Genotype-phenotype correlations in autosomal dominant and autosomal recessive polycystic kidney disease, J Am Soc Nephrol 18: (5) ((2007) ), 1374–1380. |
[8] | Harris P.C. and Torres V.E. , Polycystic kidney disease, Annu Rev Med 60: ((2009) ), 321–337. |
[9] | Chapman A.B. . et al., Autosomal-dominant polycystic kidney disease (ADPKD): Executive summary from a Kidney Disease: Improving Global Outcomes (KDIGO) Controversies Conference, Kidney Int 88: (1) ((2015) ), 17–27. |
[10] | Chapman A.B. and Guay-Woodford L.M. , Renal volume in children with ADPKD: Size matters, Clin J Am Soc Nephrol 4: (4) ((2009) ), 698–699. |
[11] | Sweeney W.E. Jr and Avner E.D. , Pathophysiology of childhood polycystic kidney diseases: New insights into disease-specific therapy, Pediatr Res 75: (1-2) ((2014) ), 148–157. |
[12] | Cadnapaphornchai M.A. , Autosomal dominant polycystic kidney disease in children, Curr Opin Pediatr 27: (2) ((2015) ), 193–200. |
[13] | Porath B. . et al., Mutations in GANAB, encoding the glucosidase iIalpha subunit, cause autosomal-dominant polycystic kidney and liver disease, Am J Hum Genet 98: (6) ((2016) ), 1193–207. |
[14] | Cornec-Le Gall E. , et al., Monoallelic mutations to DNAJB11 cause atypical autosomal-dominant polycystic kidney disease, Am J Hum Genet 102: (5) ((2018) ), 832–844. |
[15] | Harris P.C. and Torres V.E. , Autosomal dominant polycystic kidney disease, in GeneClinics: Clinical Genetic Information Resource [database online]. Copyright, University of Washington, Seattle. Available from https://www.ncbi.nlm.nih.gov/books/NBK1246/. (2002) (updated Jul 19 2018), Adam MP, Ardinger HH, Pagon RA, et al. |
[16] | Mochizuki T. . et al., PKD2, a gene for polycystic kidney disease that encodes an integral membrane protein, Science 272: ((1996) ), 1339–1342. |
[17] | Hughes J. . et al., The polycystic kidney disease 1 (PKD1) gene encodes a novel protein with multiple cell recognition domains, Nat Genet 10: (2) ((1995) ), 151–160. |
[18] | Kim H. . et al., Ciliary membrane proteins traffic through the Golgi via a Rabep1/GGA1/Arl3-dependent mechanism, Nat Commun 5: ((2014) ), 5482. |
[19] | Gainullin V.G. . et al., Polycystin-1 maturation requires polycystin-2 in a dose-dependent manner, J Clin Invest 125: (2) ((2015) ), 607–620. |
[20] | Fedeles S.V. . et al., A genetic interaction network of five genes for human polycystic kidney and liver diseases defines polycystin-1 as the central determinant of cyst formation, Nat Genet 43: (7) ((2011) ), 639–647. |
[21] | Besse W. . et al., Isolated polycystic liver disease genes define effectors of polycystin-1 function, J Clin Invest 127: (5) ((2017) ), 1772–1785. |
[22] | Fedeles S.V. , Gallagher A.R. and Somlo S. , Polycystin-1: A master regulator of intersecting cystic pathways, Trends Mol Med 20: (5) ((2014) ), 251–260. |
[23] | Ma M. , Gallagher A.R. and Somlo S. , Ciliary mechanisms of cyst formation in polycystic kidney disease, Cold Spring Harb Perspect Biol 9: (11) ((2017) ). |
[24] | Ma M. . et al., Loss of cilia suppresses cyst growth in genetic models of autosomal dominant polycystic kidney disease, Nat Genet 45: (9) ((2013) ), 1004–1012. |
[25] | Legue E. and Liem K.F. Jr , Tulp3 is a ciliary trafficking gene that regulates polycystic kidney disease, Curr Biol 29: (5) ((2019) ), 803–812 e5. |
[26] | Lu H. . et al., Mutations in DZIP1L, which encodes a ciliary-transition-zone protein, cause autosomal recessive polycystic kidney disease, Nat Genet 49: (7) ((2017) ), 1025–1034. |
[27] | Qian F. . et al., The molecular basis of focal cyst formation in human autosomal dominant polycystic kidney disease type I, Cell 87: (6) ((1996) ), 979–987. |
[28] | Wu G. . et al., Somatic inactivation of Pkd2 results in polycystic kidney disease, Cell 93: ((1998) ), 177–188. |
[29] | Pei Y. , A “two-hit” model of cystogenesis in autosomal dominant polycystic kidney disease? Trends Mol Med 7: (4) ((2001) ), 151–156. |
[30] | Eccles M.R. and Stayner C.A. , Polycystic kidney disease - where gene dosage counts, F1000Prime Rep 6: ((2014) ), 24. |
[31] | Harris P.C. , What is the role of somatic mutation in autosomal dominant polycystic kidney disease? J Am Soc Nephrol 21: (7) ((2010) ), 1073–1076. |
[32] | Piontek K. . et al., A critical developmental switch defines the kinetics of kidney cyst formation after loss of Pkd1, Nat Med 13: (12) ((2007) ), 1490–1495. |
[33] | Shibazaki S. . et al., Cyst formation and activation of the extracellular regulated kinase pathway after kidney specific inactivation of Pkd1, Hum Mol Genet ((2008) ). |
[34] | Gallagher A.R. , Germino G.G. and Somlo S. , Molecular advances in autosomal dominant polycystic kidney disease, Adv Chronic Kidney Dis 17: (2) ((2010) ), 118–130. |
[35] | Rogers K.A. . et al., Differences in the timing and magnitude of Pkd1 gene deletion determine the severity of polycystic kidney disease in an orthologous mouse model of ADPKD, Physiol Rep 4: (12) ((2016) ). |
[36] | Lantinga-van Leeuwen I.S. , et al., Lowering of Pkd1 expression is sufficient to cause polycystic kidney disease, Hum Mol Genet 13: (24) ((2004) ), 3069–3077. |
[37] | Hopp K. . et al., Functional polycystin-1 dosage governs autosomal dominant polycystic kidney disease severity, J Clin Invest 122: (11) ((2012) ), 4257–4273. |
[38] | Leonhard W.N. . et al., Scattered deletion of PKD1 in kidneys causes a cystic snowball effect and recapitulates polycystic kidney disease, J Am Soc Nephrol ((2014) ). |
[39] | Hanaoka K. and Guggino W.B. , cAMP regulates cell proliferation and cyst formation in autosomal polycystic kidney disease cells, J Am Soc Nephrol 11: (7) ((2000) ), 1179–1187. |
[40] | Yamaguchi T. . et al., Calcium restriction allows cAMP activation of the B-Raf/ERK pathway, switching cells to a cAMP-dependent growth-stimulated phenotype, J Biol Chem 279: (39) ((2004) ), 40419–40430. |
[41] | Belibi F.A. . et al., Cyclic AMP promotes growth and secretion in human polycystic kidney epithelial cells, Kidney Int 66: (3) ((2004) ), 964–973. |
[42] | Gattone V.H. 2nd, et al., Inhibition of renal cystic disease development and progression by a vasopressin V2 receptor antagonist, Nat Med 9: (10) ((2003) ), 1323–1326. |
[43] | Wang X. . et al., Effectiveness of vasopressin V2 receptor antagonists OPC-0 and OPC-1 on polycystic kidney disease development in the PCK rat, J Am Soc Nephrol 16: (4) ((2005) ), 846–851. |
[44] | Torres V.E. . et al., Tolvaptan in patients with autosomal dominant polycystic kidney disease, N Engl J Med ((2012) ). |
[45] | Torres V.E. . et al., Effect of tolvaptan in autosomal dominant polycystic kidney disease by CKD stage: Results from the TEMPO 3:4 trial, Clin J Am Soc Nephrol 11: (5) ((2016) ), 803–811. |
[46] | Meijer E. . et al., Effect of lanreotide on kidney function in patients with autosomal dominant polycystic kidney disease: The DIPAK 1 randomized clinical trial, JAMA 320: (19) ((2018) ), 2010–2019. |
[47] | Du J. and Wilson P.D. , Abnormal polarization of EGF receptors and autocrine stimulation of cyst epithelial growth in human ADPKD, Am J Physiol 269: (2 Pt 1) ((1995) ), C487–495. |
[48] | Pugh J.L. , Sweeney W.E. Jr and Avner E.D. , Tyrosine kinase activity of the EGF receptor in murine metanephric organ culture, Kidney Int 47: ((1995) ), 774–781. |
[49] | Orellana S.A. . et al., Epidermal growth factor receptor expression is abnormal in murine polycystic kidney, Kidney Int 47: (2) ((1995) ), 490–499. |
[50] | Streets A.J. . et al., Parallel microarray profiling identifies ErbB4 as a determinant of cyst growth in ADPKD and a prognostic biomarker for disease progression, Am J Physiol Renal Physiol 312: (4) ((2017) ), F577–F588. |
[51] | Richards W.G. . et al., Epidermal growth factor receptor activity mediates renal cyst formation in polycystic kidney disease, J Clin Invest 101: (5) ((1998) ), 935–939. |
[52] | Sweeney W.E. . et al., Treatment of polycystic kidney disease with a novel tyrosine kinase inhibitor, Kidney Int 57: (1) ((2000) ), 33–40. |
[53] | Sweeney W.E. Jr , et al., Combination treatment of PKD utilizing dual inhibition of EGF-receptor activity and ligand bioavailability, Kidney Int 64: (4) ((2003) ), 1310–1319. |
[54] | Ma R. . et al., PKD2 functions as an epidermal growth factor-activated plasma membrane channel, Mol Cell Biol 25: (18) ((2005) ), 8285–8298. |
[55] | Sweeney W.E. Jr , et al., Src inhibition ameliorates polycystic kidney disease, J Am Soc Nephrol 19: (7) ((2008) ), 1331–1341. |
[56] | Talbot J.J. . et al., Polycystin-1 regulates STAT activity by a dual mechanism, Proc Natl Acad Sci U S A 108: (19) ((2011) ), 7985–7990. |
[57] | Silva C.M. , Role of STATs as downstream signal transducers in Src family kinase-mediated tumorigenesis, Oncogene 23: (48) ((2004) ), 8017–8023. |
[58] | Talbot J.J. . et al., The cleaved cytoplasmic tail of polycystin-1 regulates src-dependent STAT3 activation, J Am Soc Nephrol 25: ((2014) ), 1737–1748. |
[59] | Tesar V. . et al., Bosutinib versus placebo for autosomal dominant polycystic kidney disease, J Am Soc Nephrol 28: (11) ((2017) ), 3404–3413. |
[60] | Wullschleger S. , Loewith R. and Hall M.N. , TOR signaling in growth and metabolism, Cell 124: (3) ((2006) ), 471–484. |
[61] | Edelstein C.L. , Mammalian target of rapamycin and caspase inhibitors in polycystic kidney disease, Clin J Am Soc Nephrol 3: (4) ((2008) ), 1219–1226. |
[62] | Shillingford J.M. . et al., The mTOR pathway is regulated by polycystin-1, and its inhibition reverses renal cystogenesis in polycystic kidney disease, Proc Natl Acad Sci U S A 103: (14) ((2006) ), 5466–5471. |
[63] | Shillingford J.M. . et al., Rapamycin ameliorates PKD resulting from conditional inactivation of Pkd1, J Am Soc Nephrol 21: (3) ((2010) ), 489–497. |
[64] | Zafar I. . et al., Long-term rapamycin therapy in the Han:SPRD rat model of polycystic kidney disease (PKD), Nephrol Dial Transplant 24: (8) ((2009) ), 2349–2353. |
[65] | Walz G. . et al., Everolimus in patients with autosomal dominant polycystic kidney disease, N Engl J Med 363: (9) ((2010) ), 830–840. |
[66] | Serra A.L. . et al., Sirolimus and kidney growth in autosomal dominant polycystic kidney disease, N Engl J Med 363: (9) ((2010) ), 820–829. |
[67] | Shillingford J.M. . et al., Folate-conjugated rapamycin slows progression of polycystic kidney disease, J Am Soc Nephrol 23: (10) ((2012) ), 1674–1681. |
[68] | Sweeney W.E. , Frost P. and Avner E.D. , Tesevatinib ameliorates progression of polycystic kidney disease in rodent models of autosomal recessive polycystic kidney disease, World J Nephrol 6: (4) ((2017) ), 188–200. |
[69] | Klawitter J. . et al., Bioactive lipid mediators in polycystic kidney disease, J Lipid Res 55: ((2014) ), 1139–1149. |
[70] | Park F. . et al., Contribution of cytochrome P450 metabolites to the abnormal proliferation of renal epithelial cells in a murine model of PKD, J Am Soc Nephrol 18: ((2007) ), 129A. |
[71] | Park F. . et al., Chronic blockade of 20-HETE synthesis reduces polycystic kidney disease in an orthologous rat model of ARPKD, Am J Physiol Renal Physiol 296: (3) ((2009) ), F575–582. |
[72] | Park F. . et al., 20-HETE mediates proliferation of renal epithelial cells in polycystic kidney disease, J Am Soc Nephrol 19: (10) ((2008) ), 1929–1939. |
[73] | Chandrasekaran K. . et al., Role of microRNAs in kidney homeostasis and disease, Kidney Int 81: (7) ((2012) ), 617–627. |
[74] | Noureddine L. , Hajarnis S. and Patel V. , MicroRNAs and polycystic kidney disease, Drug Discov Today Dis Models 10: (3) ((2013) ), e137–e 1743. |
[75] | Yheskel M. and Patel V. , Therapeutic microRNAs in polycystic kidney disease, Curr Opin Nephrol Hypertens 26: (4) ((2017) ), 282–289. |
[76] | Hajarnis S. . et al., Suppression of microRNA activity in kidney collecting ducts induces partial loss of epithelial phenotype and renal fibrosis, J Am Soc Nephrol ((2017) ). |
[77] | Patil A. . et al., Unique interstitial miRNA signature drives fibrosis in a murine model of autosomal dominant polycystic kidney disease, World J Nephrol 7: (5) ((2018) ), 108–116. |
[78] | Shah M.Y. . et al., microRNA therapeutics in cancer - an emerging concept, EBioMedicine 12: ((2016) ), 34–42. |
[79] | Christopher A.F. . et al., MicroRNA therapeutics: Discovering novel targets and developing specific therapy, Perspect Clin Res 7: (2) ((2016) ), 68–74. |
[80] | Harskamp L.R. . et al., Urinary EGF receptor ligand excretion in patients with autosomal dominant polycystic kidney disease and response to tolvaptan, Clin J Am Soc Nephrol 10: (10) ((2015) ), 1749–1756. |


