
Retinal disease in ciliopathies: Recent advances with a focus on stem cell-based therapies
Abstract
Ciliopathies display extensive genetic and clinical heterogeneity, varying in severity, age of onset, disease progression and organ systems affected. Retinal involvement, as demonstrated by photoreceptor dysfunction or death, is a highly penetrant phenotype among a vast majority of ciliopathies. Photoreceptor cells possess a specialized and modified sensory cilium with membrane discs where efficient photon capture and ensuing signaling cascade initiate the visual process. Disruptions of cilia biogenesis and protein transport lead to impairment of photoreceptor function and eventually degeneration. Despite advances in elucidation of ciliogenesis and photoreceptor cilia defects, we have limited understanding of pathogenic mechanisms underlying retinal phenotype(s) observed in human ciliopathies. Patient-derived induced pluripotent stem cell (iPSC)-based approaches offer a unique opportunity to complement studies with model organisms and examine cilia disease relevant to humans. Three-dimensional retinal organoids from iPSC lines feature laminated cytoarchitecture, apical-basal polarity and emergence of a ciliary structure, thereby permitting pathogenic modeling of human photoreceptors in vitro. Here, we review the biology of photoreceptor cilia and associated defects and discuss recent progress in evolving treatment modalities, especially using patient-derived iPSCs, for retinal ciliopathies.
1Introduction
Ciliopathies encompass a broad group of genetically and phenotypically diverse diseases, caused by mutations in almost 200 genes that contribute to cilia biogenesis and transport [1–4]. The cilium is an intricately-structured organelle that protrudes from the extracellular surface of most mammalian cells. Since the first documentations by Otto Frederik Muller in 1768 and long thought to be vestigial, the cilia are now recognized to have a prominent role in cell physiology [5–7]. The cilia perform divergent tasks in mechanical processes, such as cerebrospinal fluid flow as motile cilia, or in signal transduction in response to environmental cues as in case of non-motile primary (sensory) cilia. Significant progress has been made to further characterize cilia structure, particularly through the application of advanced techniques and the use of animal models, to better define cilia function in the normal and diseased states [8, 9]. Nonetheless, unravelling clinical manifestations of ciliopathies [10, 11] have been complicated, particularly because of disparate syndromic and non-syndromic phenotypes, affecting different tissues and cell types, by distinct mutations even in a single gene.
Retinal degeneration is a frequently-observed clinical entity in ciliopathies, mainly caused by dysfunction of proteins involved in biogenesis, function, or maintenance of photoreceptor primary cilium [12–15]. As an easily accessible tissue and immune privileged site, the retina is an ideal tissue to investigate molecular and cellular pathways implicated in ciliopathies and evaluate novel treatment modalities [16–18]. Application of model organisms to investigate pathophysiology of retinal ciliopathies has greatly advanced our understanding and provided valuable proof-of-concept evidence to evaluate therapeutics [5, 19–21]. However, model organisms do not faithfully recapitulate many characteristics of human pathophysiology, such as clinical progression, and a key challenge for developing treatments has been to robustly replicate the retinal photoreceptor-associated manifestations of ciliopathies. Human pluripotent stem cells (PSCs) provide a valuable resource for generating specific somatic cell types. Advent of induced pluripotent stem cell (iPSC) technology [22] and differentiation systems have made it possible to examine the relationship of specific mutations with disease phenotypes. Three-dimensional (3-D) retinal organoids derived from PSCs [23, 24] can effectively mimic in vivo development and are a powerful complementary platform to model retinal degeneration caused by ciliary gene defects, interrogate pathogenic mechanisms in vitro using patient-derived cells, and develop relevant treatment strategies.
Retinal ciliopathies are an active area of research since almost 25% of all retinal degeneration are caused by genes associated with photoreceptor cilia structure and/or function [14, 25, 26]. In this review, we provide a brief overview of photoreceptor cilia biology and retinal ciliopathies but have attempted to cite as many relevant references and reviews as possible. We then illustrate recent advances in stem cell-based technologies pertaining to retinal organoids and discuss their applications in modeling human retinal ciliopathies for translational therapeutics.
2Photoreceptor cilia
2.1Structure
Primary cilia are extracellular microtubule-based protrusions present in almost every quiescent mammalian cell and serve as cellular antenna to execute crucial roles in response to exogenous cues including light, mechanical stimulation and morphogens [7, 15, 27]. The cilium exhibits a complex structure with extensive multiprotein assemblies, originating from the basal body, which, similar to other cells, serves as the primary microtubule organizing center in photoreceptors. The basal body, analogous to the centrioles, possesses a specialized structural organization comprising of 9-fold symmetry of microtubule arrays. The microtubule organization of the cilium changes from triplets at the basal body to a doublet structure in the transition zone and the axoneme, becoming singlet near the tip. The transition zone contains additional Y-linkers that connect the doublet microtubules to the ciliary membrane. The distal and subdistal appendages are required for docking the basal body to the cellular membrane and to facilitate the transport of cargo proteins [4]. The ciliary axoneme is constructed and maintained by intraflagellar transport (IFT) [5, 28]. IFT proteins form an intricate network and interact with numerous proteins to regulate ciliary length, composition and cargo trafficking [4, 29, 30]. The IFT process involves IFT complex B, including kinesin motor proteins, to transfer proteins from the ciliary base to the tip (anterograde transport) and IFT complex A for moving cargos from the tip down to the base via dynein motor proteins by retrograde transport [31]. The trafficking of ciliary cargo proteins, including building blocks of the axoneme (e.g., α and β tubulin) and signaling molecules associated with multiple pathways (such as Sonic Hedgehog, Wnt and Notch), are crucial for cellular functions [32].
2.2Ciliogenesis
In post-mitotic cells such as photoreceptors, apical anchoring of the mother centriole with distal appendages assembles the basal body [33, 34]. During early ciliogenesis, dynamic remodeling occurs at the distal appendages of the basal body by docking and fusion of membrane vesicles to produce a larger ciliary vesicle, which in turn leads to uncapping by removal of the negative regulators of ciliogenesis such as CP110 [34]. Interestingly, CP110 also promotes proper docking of the ciliary vesicles as well as the assembly of subdistal appendages [35]. Subsequent accumulation and complex assemblies of the transition zone facilitated by IFT proteins lead to elongation of the axonemal microtubules, and eventually the emergence of ciliary axoneme through the cell membrane [36–40]. Photoreceptor cilia biogenesis and function may require over 1000 proteins [41]; defects in several of these have been associated with blindness in Leber congenital amaurosis (LCA), Bardet Biedl Syndrome (BBS), nephronophthisis (NPHP) and other cilioapthies.
2.3The transition zone
The specialized region between the basal body and the ciliary axoneme is called the transition zone, which serves as a gate to control the entry to and the exit from the ciliary shaft [30, 42] (Fig. 1). In photoreceptors, the formation of outer segment (OS) membrane discs (see below, section 2.4) is initiated just distal to the transition zone (also called connecting cilium). Given its unique placement, the transition zone proteins exhibit complex interaction dynamics with other cilia associated proteins (see, for example, the mutant phenotype modifications reflected by CEP290 and BBS alleles [43, 44].
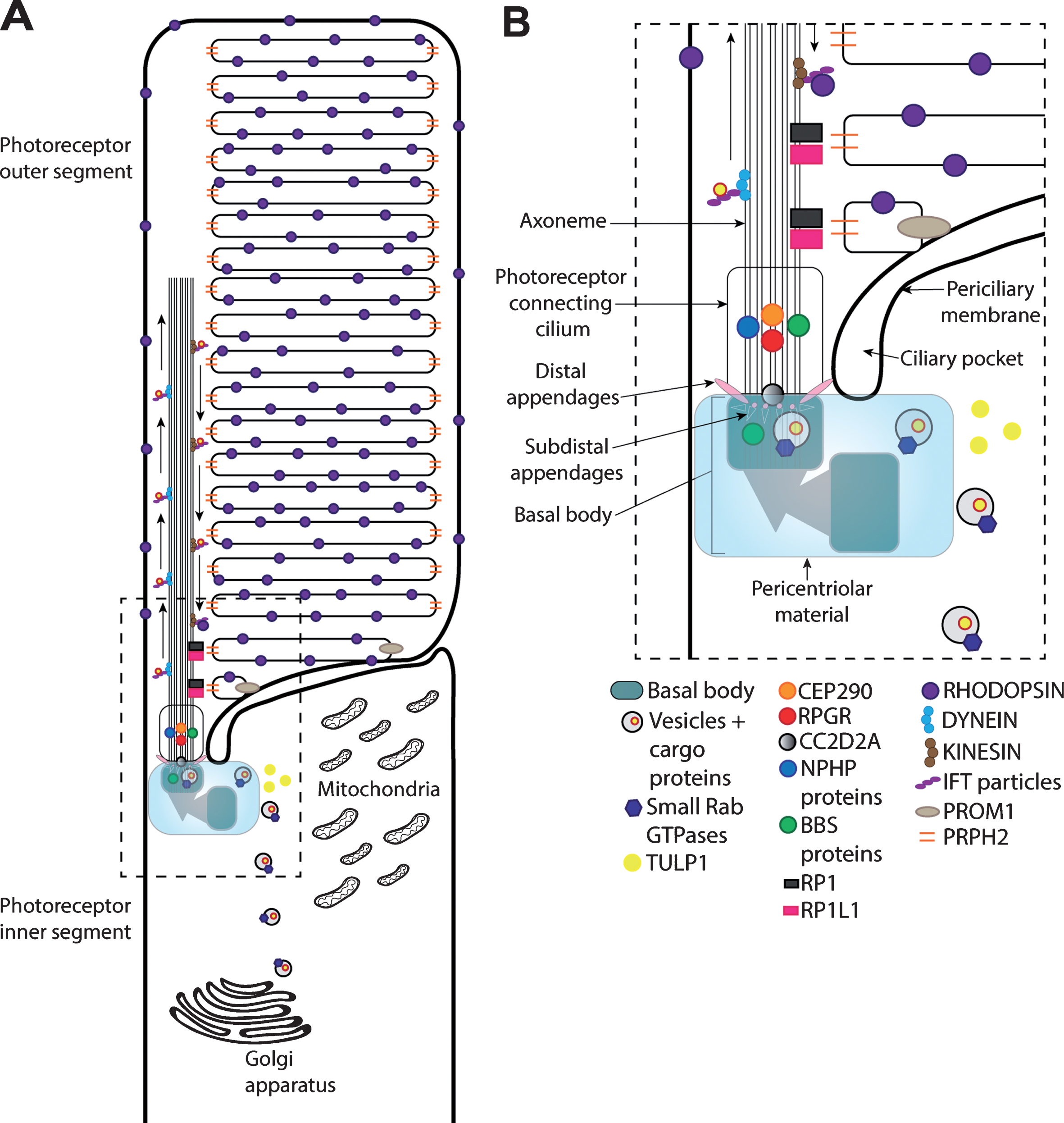
Fig.1
Schematic of the rod photoreceptor primary cilium. (A) The rod primary cilium is highly adapted to efficiently capture photons of light that initiate the phototransduction cascade through rhodopsin GPCR activation (in the photoreceptor outer segment) and facilitate the transportation of proteins involved cilia maintenance, structure and function from the inner segment to the outer segment (photoreceptor connecting cilium). The photoreceptor inner segment is crucial for photoreceptor proteins and lipid synthesis. (B) Within the inner segment, vesicles containing cargo proteins are transported from the Golgi apparatus to the basal body via small Rab GTPases together with proteins involved in trafficking cargo to the cilia (e.g., TULP1). The basal body is formed of the mother centriole and daughter centriole, the mother centriole docks onto the apical photoreceptor cell membrane to supports ciliogenesis and axoneme development. The basal body also contains additional structural elements, including distal appendages, with subdistal appendages and associated proteins (e.g., CC2D2A) to support vesicular transportation.The BBSome complex, including a number of BBS proteins, and multiprotein complexes in the pericentriolar material play critical functions in ciliogenesis and ciliary trafficking. Cargo transport to the outer segment is further regulated by proteins within the photoreceptor connecting cilium, such as CEP290 and NPHP proteins. Other proteins, such as prominin-1 (PROM1), peripherin (PRPH2), RP1 and RP1L1 regulate more structural components of the cilia, such as outer segment disk stacking and organization. Proteins are transported along the axoneme within the photoreceptor outer segment through anterograde and retrograde transportation, involving kinesin and dynein motor proteins, respectively. IFT particles help to support the protein transportation along the axoneme.
2.4The outer segment
The primary cilium of the retinal photoreceptor is modified to maximize photon capture and facilitate phototransduction [45] to become a unique and highly specialized structure, called OS, which are densely packed with hundreds of membranous discs that are highly enriched with proteins essential for phototransduction (Fig. 1A). The two types of photoreceptors, rods and cones, exhibit distinct OS features, reflecting their respective contributions to dim-light and day-light vision. The rod OS discs are separated from the ciliary membrane and are typically more elongated in contrast to cone cells, which possess a shorter, wider OS tapered towards the tip and features discs continuous with the plasma membrane [46–48]. Due to their similarities in initiating the visual process, rods and cones express numerous common proteins for maintaining their OS integrity and function; however, several rod- and cone-specific versions of proteins are tailored towards the requirement of each photoreceptor type. Another unique feature of this modified cilium is the high rate of OS disc renewal; 8–10% of the membrane discs at the distal end are shed and engulfed by retinal pigment epithelium daily, with new discs formed at the cilia base to compensate for the loss and maintain OS homeostasis [49]. The OS renewal process involves ciliary transport, which is critical for photoreceptor survival.
2.5Phototransduction
The photoreceptor cilia, like other primary cilia, perform sensory function and capture visual information via the visual pigment (rhodopsin in rods and opsin of distinct wavelength excitation in different cone subtypes), which constitutes over 90% of the protein in membrane discs [50] (Fig. 1). The light (photon) leads to photoisomerization of the chromophore 11-cis retinal to all-trans retinal and triggers a conformational change within the opsin protein, which in turn activates the G-protein transducin leading to the dissociation of subunit Gα from βγ [51]. cGMP phosphodiesterase is then activated and catalyzes the hydrolysis of cGMP to GMP, resulting in the closure of cyclic-nucleotide-gated (CNG) channels on the plasma membrane. Thus, unlike other neurons, the photoreceptor is hyperpolarized in response to light and signals are transmitted to interneurons by modulation of neurotransmitter release. Termination of phototransduction is initiated in part by reduced influx, and consequently lower intracellular Ca2 + levels due to the closure of CNG channels. At low Ca2 +, guanylyl cyclase activating proteins activate guanylyl cyclase to promote cGMP synthesis and restore its levels to reopen CNG channels. The termination process also requires inactivation of rhodopsin by phosphorylation and binding of arrestin. The movement of transducing and arrestin along the cilium in response to visual pigment activation and subsequent regeneration process is critical for phototransduction and consequently vision.
3Retinal disease in ciliopathies
Depending on the gene function, the mutation type, the location of mutation within a gene, and/or other genetic modifiers, ciliopathies comprise a broad spectrum of clinical entities that affect ciliogenesis and ciliary function [4, 10]. Functional defects when confined primarily to the photoreceptor cilia result in non-syndromic ciliopathies [14, 25, 26] (RetNet, http://sph.uth.edu/retnet), such as LCA and retinitis pigmentosa (RP), with occasional manifestation of ciliary dysfunction in other cell types [52–54]. LCA is a severe, early onset retinal disease with mutations identified in at least 20 different genes; of these, CEP290 is the most common LCA gene, accounting for 20–25% of disease [55, 56]. RP is the most common form of inherited retinal dystrophy caused by mutations in almost 50 distinct genes; mutations in RPGR are a major cause of X-linked forms of RP [57–60] and account for a significant proportion of simplex male RP patients [61]. Photoreceptor cilia disease also manifests as part of a broad range of syndromic ciliopathies with involvement of multiple cell types and tissues; these include BBS, Senior Loken syndrome, Joubert syndrome (JBTS), and Alstrom syndrome (Table 1) [13, 14, 16, 26, 62]. Most ciliopathies affect cilia function by disrupting key steps in photoreceptor cilia biogenesis or transport.
Table 1
Ciliopathies featuring retinal degeneration, including associated gene mutations and clinical phenotypes (Based on Retnet http://sph.uth.edu/retnet, and National Library of Medicine Genetics Home Reference: https://ghr.nlm.nih.gov/resources#databases)
| Selected ciliopathies | Associated gene mutation | Major phenotypes |
| Leber Congenital Amaurosis (LCA) | CEP290, GUCY2D, RPE65, AIPL1, SPATA7, RPGRIP1, CRX, LCA5, NMNAT1, RD3, LRAT, IMPDH1, RDH12, TULP1, MYO7A, NPHP5, ALMS1, CNGA3, CABP4, GDF6, KCNJ13, CRB1, NPHP4, NPHP1, NPHP5, NPHP3, CC2D2A, DTHD1, AHI1, GDF6, NPHP2, INPP5E, TMEM216, CCT2, POC1B, OTX2, RDH12, IFT140, CLUAP1, AIPL1 | Retinal degeneration (rod and cone degeneration) |
| Retinitis Pigmentosa (RP) | RP1, RP2, RP9, RPE65, RPGR, ABCA4, BEST1, CA4, ZNF513, WDR19, USH2A, TULP1, CERKL, CLRN1, CNGA1, CRX, TTC8, TOPROS, SPATA7, CNGB1, CRB1, CRX, EYS, FAM161A, SNRNP200, SEMA4A, SAG, ROM1, RLBP1, RHO, RGR, FSCN2, GUCA1B, IDH3B, IMPDH1, IMPG2, KLHL7, LRAT, RDH12, RBP3, PRPH2, PRPF31, MERTK, MT-TS2, NR2E3, NRL, PCARE, PRPF8, PRPF3, PROM1, PRCD, PDE6 G, PDE6B, PDE6A | Retinal degeneration (primarily rod degeneration) |
| Senoir Loken Syndrome (SLS) | NPHP1, 2 (INVS), 3, 4, 5 (IQCB1), 6 (CEP290), 7 (GLIS2), 8 (NEK8), 13 (WDR19) | Retinal degeneration, nephronophthisis |
| Bardet Biedl Syndrome (BBS) | BBS1, 2, 3 (ARL6), 4, 5, 6 (MKKS), 7, 8 (TTC8), 9, 10, 11 (TRIM32), 12, 13 (MKS1), 14 (CEP290), 15 (WDPCP), 16 (SDCCAG8), 17 (LZTFL1), 18 (BBIP1), 19 (ITF27), 20 (ITF172), 21 (C8orf37), NPHP1, CEP19 | Retinal degeneration, obesity, polycystic kidney disease, polydactyly, cognitive impairment, hypogonadism, anosmia |
| Joubert Syndrome (JBTS) | MKS1, 2, 3, 4, 5, 6, NPHP1, TMEM237, CC2D2A, AHI1, CSPP1, INPP5E, TMEM216, CEP290, POC1B, ZNF423, RPGRIP1 L, TMEM67, ARL13B, TCTN1, TCTN2, TCTN3, TMEM231, C5orf42, CEP41, KIF7 | Retinal degeneration, polycystic kidney disease, polydactyly, cognitive impairment, brain development abnormalities, muscle hypotonia, ataxia |
| Meckel Syndrome (MSK) | TMEM216, TMEM67, RPGRIPL, MKS1, CEP290, CC2D2A, B9D1, B9D2, WDPCP, CSPP1 | Retinal degeneration, encephalocele, polydactyly, polycystic kidney disease, cognitive impairment |
| Jeune Syndrome | IFT80, WDR19, IFT140, IFT172 | Retinal degeneration, polydactyly, abnormal bone growth |
| Alstrom Syndrome (AS) | ALMS1 | Retinal degeneration, hearing loss, dilated cardiomyopathy, obesity, acanthosis nigricans |
Defects in early stages of cilia formation can also lead to severe clinical manifestations. For example, mutations in CC2D2A which encodes a subdistal appendage protein associated with vesicular transport [63, 64] (Fig. 1B) can cause divergent phenotypes including JBTS and Meckel syndrome (MKS) [65–67], with the latter being the most severe form of ciliopathy. Genetic mutations that influence transition zone structure or function are a prominent cause of ciliopathies with divergent clinical spectrum from the least severe form in LCA to early lethality in MKS [4]. Photoreceptor dysfunction and death are commonly observed in ciliopathies caused by mutations in transition zone proteins [14, 68]. In addition, unique to photoreceptors, disruptions in the formation and/or daily renewal of OS membrane discs, requiring transport of large amounts of nascent macromolecules to the cilia, can also result in photoreceptor dysfunction or death. For example, the tubby family proteins serve as adapters for ciliary trafficking [69, 70]; specifically, Tulp1 (tubby-like protein 1) is implicated in rhodopsin transport to OS and is essential for rod function [71] (Fig. 1B). Mutations in TULP1 lead to RP [72] and LCA [73].
Given the genetic diversity and functional complexities associated with retinal ciliopathies, we will focus on two transition zone proteins – CEP290 and RPGR. Genetic defects in numerous cilia-associated CEP290 and RPGR interacting proteins also cause retinal degeneration [13, 14]. However, mutations in CEP290 and RPGR constitute a major cause of LCA and RP phenotypes, respectively (https://sph.uth.edu/retnet/), and these proteins have been investigated extensively in thephotoreceptors.
3.1CEP290 and ciliopathies including retinal disease
The 290-kDa centrosome-cilia protein CEP290 tethers microtubules in the transition zone to the ciliary membrane, where it interacts with multiple ciliary proteins, including NPHP and BBS proteins, and is proposed to be a gatekeeper of ciliary protein transport [74] (Fig. 1B). CEP290 protein includes multiple distinct domains that can interact with numerous ciliary proteins [13]. In photoreceptors, CEP290 is precisely localized in Y-linkers of the transition zone [75]. CEP290 mutations have been associated with most ciliopathy phenotypes [76, 77]. In case of LCA, mutations in CEP290 account for 20–25% of all patients [78, 79]. Most CEP290-LCA patients also exhibit additional sensory deficits [53]. How different CEP290 mutations contribute to a broad spectrum of human ciliopathies is still poorly understood though mouse models have begun to provide some insights [75, 80].
The rd16 mouse carries an in-frame deletion in the myosin tail of Cep290 and phenocopies the human CEP290-LCA phenotype [81], demonstrating early-onset retinal degeneration, failure to develop outer segments and mis-localization of rhodopsin and RPGR [81, 82]. However, CEP290-LCA patients typically retain cone cell bodies in the macular region even at an advanced age; therefore, rd16/Nrl-/- mice represent a better model for the disease by recapitulating extended survival of dysfunctional cone photoreceptors [83]. The ciliary defects can partially be replicated in vitro, as demonstrated by studies using CEP290-LCA patient fibroblasts and iPSCs, which exhibit lower CEP290 expression, shorter ciliary length and/or less ciliated cells upon serum starvation [84–87].
3.2RPGR and non-syndromic retinal ciliopathies
The RPGR protein is localized to photoreceptor transition zone [88] and interacts with CEP290 and multiple centrosome-cilia proteins [89] (Fig. 1B). The retina of Rpgr-knockout mouse shows normal development, but eventually the mislocalization of opsins within the photoreceptor cells results in progressive degeneration at later stages [90]. The rd9 mouse model of RPGR-disease harbors a frameshift mutation and demonstrates a similar rate of photoreceptor degeneration [91]. Two RP dog models exhibit frameshift deletions in RPGR, with the XLPRA1 model resembling Rpgr-knockout mouse and the XLPRA2 model showing somewhat rapid photoreceptor degeneration [92, 93]. Despite extensive studies, we still have poor understanding of the precise ciliary role of the RPGR protein and pathogenesis of RPGR disease [60].
4Stem cell– based modeling of retinal ciliopathies
Model organisms have provided invaluable insights in unravelling pathogenesis of retinal ciliopathies (Fig. 2). Studies in Chlamydomonas reinhardtii, Tetrahymena thermophila and Caenorhabditis elegans have permitted fundamental understanding of cilia structure and diversity [94, 95], ciliary proteome [96, 97], cilia biology and compartments [74, 98, 99], and association of cilia defects to human disease [100–102]. Xenopus [103], Zebrafish [104] and mice [20, 105] (https://www.jax.org/) have also been extensively employed for investigating cilia function and ciliopathies, due to their easy and inexpensive management in a laboratory environment and availability of genetic mutants. The findings across multiple models have untangled complex aspects of ciliogenesis and ciliopathies, including the role of CEP290 and other transition zone proteins in regulating cargo proteins into the cilium [74, 81, 83, 99, 106]. However, many human disease-associated phenotypes are not faithfully replicated in model organisms; e.g., in case of Usher syndrome, the Myo7a knockout mouse model fails to present retinal degeneration [107, 108]. Notably, a number of common ciliopathy-causing mutations are specific to humans, whereby human cells adopt unique mechanisms to largely preserve the functions of mutated proteins, such as nonsense-mediated alternative splicing in CEP290-LCA [109, 110]. Hence, underlying mechanisms of human pathology remain largely unclear for retinal ciliopathies and development of useful therapies is still in infancy.
Fig.2
Modeling human retinal ciliopathies using model organisms (such as mice), human-derived somatic cells, and human iPSC-derived retinal organoids.
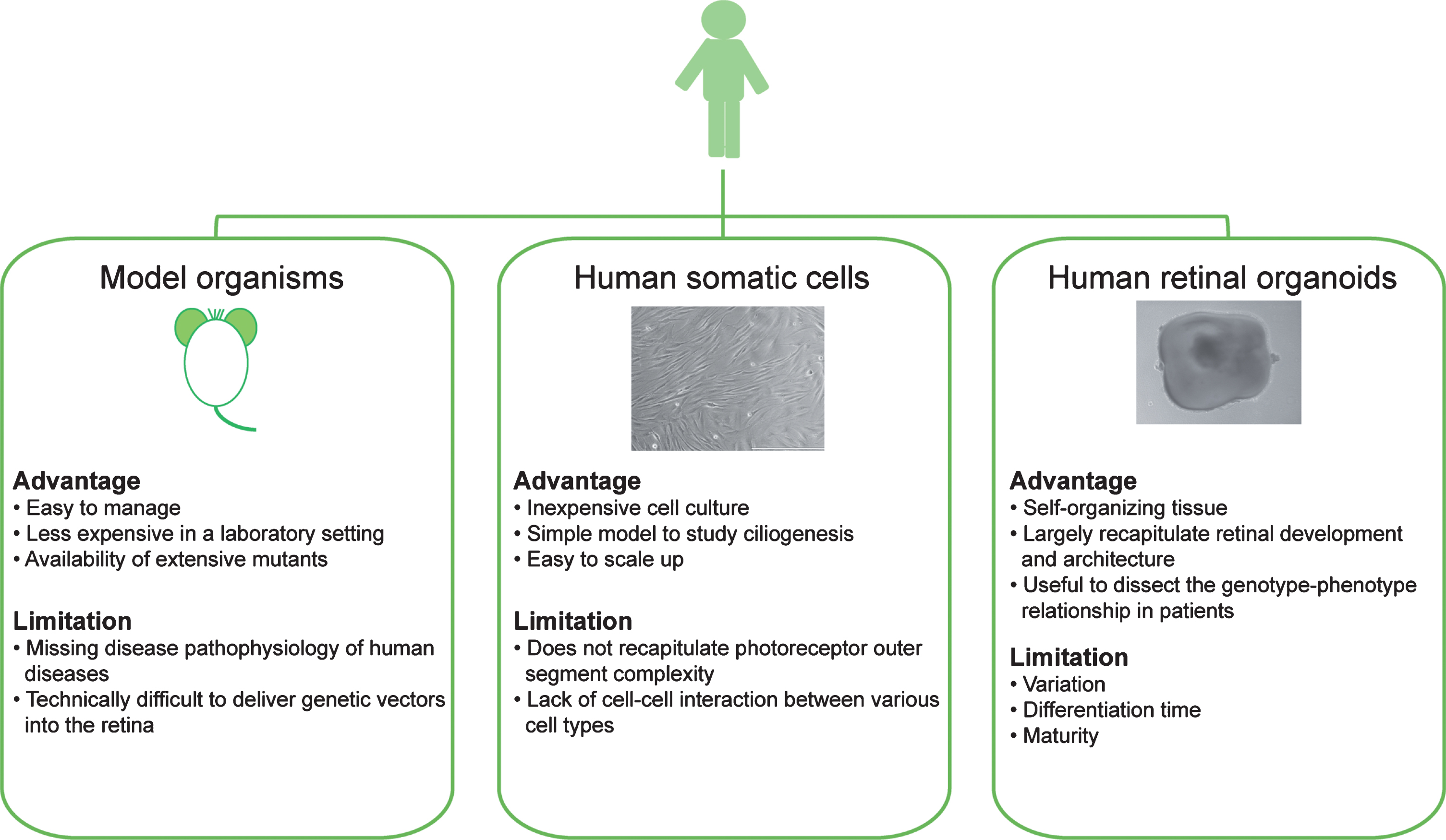
One complementary approach to model organisms is to use patient-derived cell lines, which can allow evaluation of cilia phenotypes in relevant human genetic background [85–87]. Fortunately, terminally-differentiated somatic cells from patients can now be reprogrammed to iPSCs with the capacity of self-renewal and differentiation into three germ layers by delivery of Yamanaka factors [111]. Patient iPSC lines have been successfully used to produce photoreceptors in 2-D culture and in 3-D retinal organoids [23, 112–116], thereby presenting a more relevant platform for understanding genotype-phenotype relationship in retinal ciliopathies and for developing therapies for human disease (Fig. 2).
4.1Photoreceptor differentiation and disease modeling using stem cells
Early efforts to generate photoreceptors and retinal cells from embryonic stem cells relied on the use of co-culturing systems with mouse retina [117] or using telencephalic serum-free floating culture of embryoid body-like aggregates systems [118] in order to induce forebrain cell fate before promoting retinal differentiation. These systems modulate key growth signaling pathways involved in driving early neural differentiation, such as Wnt (Dickkopf-1), Nodal (LeftyA), early retinal differentiation (TGFβ (activin A), Noggin, IGF-1) and photoreceptor generation (retinoic acid, Shh, Notch (DAPT), FGF), in addition to adjusting medium compositions, serum content and basal lamina components [112, 119–121]. These 2-D adherent methods have been used to generate photoreceptor cells from patient iPSCs in order to characterize the RP disease phenotype caused by photoreceptor cilia/OS genes such as RP1, PRPH2 and RHO [122]. In addition to providing initial insights into disease mechanisms, these adherent cultures are also relatively easy to scale up, suggesting their use in high throughput screening of small molecules to develop new therapies. Although patient-derived photoreceptors can reveal disease-associated phenotypes, 2-D iPSC-derived retinal cultures do not replicate cell-cell and cell-matrix interactions that are critical for organogenesis [123], and even more importantly, photoreceptors in 2-D culture lack cell polarity, making them less desirable for studying ciliogenesis in vitro.
With the advances in 3-D culture system, aggregates of stem cells can self-organize into retinal organoids, forming optic vesicles or optic cups, with major cell types self-patterning into a polarized, laminated cytoarchitecture highly reminiscent of the human retina in vivo [23, 24, 124–126]. Further improvements in retinal organoid cultures are reported to facilitate the emergence of a rudimentary outer segment-like structure [126, 127], possible light-sensitive properties [24] and response to cGMP and GABA stimulation [128], suggesting at least partial maturation and functioning of photoreceptor cells. The human stem cell-derived retinal organoids are now being used to model LCA and RP [86, 87, 129, 130]. Here, we will highlight the progress in using 3-D retinal organoids for modeling CEP290-LCA and RPGR-RP disease.
4.2Modeling LCA and RP disease in retinal organoids
A deep intronic mutation identified in most CEP290-LCA patients was one of the first retinal degenerations to be modeled using 3-D retinal organoids; in this case, patient iPSC-derived photoreceptors demonstrated reduced cilia length and mis-localization of ciliary proteins [86]. Notably, patient iPSC-derived photoreceptors also demonstrated a high level of aberrant splicing compared to patient fibroblast and iPSC-derived RPE cells, suggesting tissue-specific splicing can have a direct effect on the phenotype presentation [86]. The tissue-specific disease-associated phenotypes were also observed in another study using CEP290-LCA cells, where ciliary defects were detected in patient iPSC-derived organoid photoreceptors but not in patient fibroblasts, further illustrating the value of retinal organoids in disease modeling [87]. Similarly, cilia defects in photoreceptors, such as abnormal cell morphology with shorter outer segments and opsin mis-localization have also been demonstrated in RPGR patient-derived retinal organoids [129]. This study also included the use of RNA-seq to show reduced levels of opsin gene expression in RP patient organoids compared to the control organoids [129]. A transcriptomic approach was also applied to PRPF31-RP patient iPSC-derived photoreceptors, which revealed defects in splicing, cilia biogenesis and phototransduction [130].
4.3Limitations of current organoid cultures
Despite exciting advances, several limitations exist in the current 3-D organoid culture systems. First, although the neural retina in organoids structurally recapitulates in vivo human development, it is unclear whether similar cellular machineries are involved in vivo and in vitro. With the availability of human fetal transcriptome data [131], a comparison of developing organoid transcriptomes to the in vivo developing retina would be helpful and yield valuable insights. Second, retinal development in human organoids requires as many as 200 days to achieve photoreceptor maturation which includes the formation of inner segment, CC and rudimentary OS structures, making it expensive, labor intensive and difficult to use for routine screening of potential therapies. Initial bioreactor works demonstrate that efficient exchange of nutrients and waste might facilitate a more rapid differentiation of retinal organoids [132, 133]. However, due to the positioning of photoreceptor cilia to the exterior in human organoids, the developed cilia are vulnerable to shear force or mechanical damage in the bioreactor. Development of new types of bioreactors or incorporation of extracellular matrix to protect the cilia may facilitate human organoid culture in bioreactors [134]. Third, in vitro differentiation system for human organoids exhibits high variability because of different protocols, reagent batches, iPSC clones, and clonal variations, contributing to non-disease-associated phenotypes and false interpretation of data. Observed abnormalities in organoids must be concordant with natural history of disease pathogenesis in humans and caused by the targeted mutations and not by organoid culture variability. Though time- and resource-intensive, the use of different clones from the same patient or different patients with same mutations to perform multiple experiments is recommended to confirm disease-causing phenotypes in organoids. Fourth, as elaborated previously, the photoreceptor primary cilium features a more intricate sensory structure with OS membrane discs that are not present in cilia of other cell types. Transmission electron microscopy studies do not reveal long cilia and OS disc formation in the retinal organoids [24, 126, 127, 129]. This could be due to the lack of additional exogenous cues, such as those provided by the retinal pigment epithelium, which is needed for OS disc morphogenesis in vivo [135]. This limitation greatly hinders the application of organoid cultures to model retinal ciliopathies. Nonetheless, retinal organoids derived from patients with mutations in transition zone proteins display disease-relevant phenotypes and thus provide valuable experimental platform to investigate pathophysiology of ciliopathies and evaluation of treatments. As the field continues to advance, we believe these systems will impart greater insight into the pathology of retinal ciliopathies.
5Development of stem cell-based therapies for retinal ciliopathies
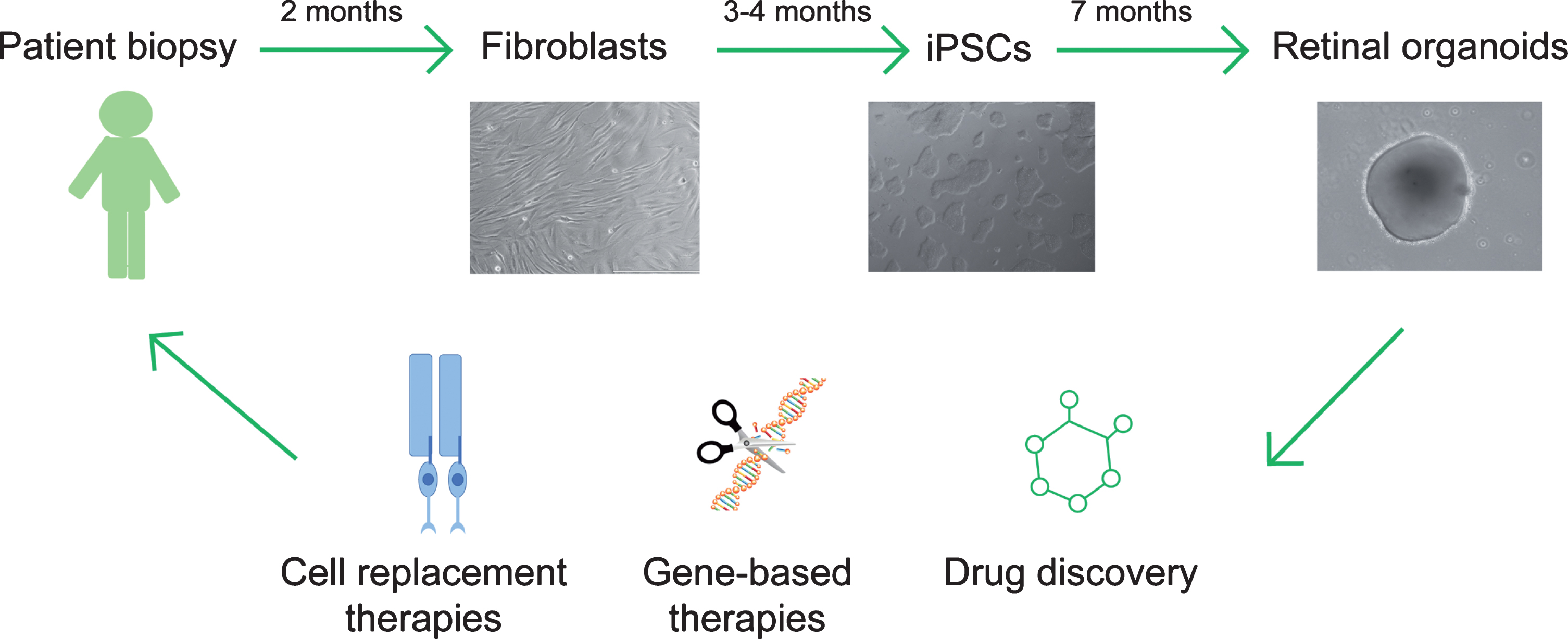
Given that the retina is readily accessible, the subretinal space and the intravitreal cavity offer convenient conduits for delivery of drugs, genes or cells for evaluation of therapies. We will now discuss the use of stem cell-derived retinal organoid cultures as a renewable cell source for photoreceptor replacement or as a platform to assess small molecule and gene-based therapies of retinal ciliopathies (Fig. 3).
Fig.3
Translational therapeutics using patient iPSC-derived retinal organoids. Patient fibroblasts are obtained from skin biopsy samples and subsequently reprogramed to generate induced pluripotent stem cells (iPSCs). Retinal organoids can be differentiated from iPSCs and applied as a cell source to replace dysfunctional or dead photoreceptors in retinal ciliopathy patients, or as an in vitro platform to evaluate gene-based treatments or drug candidates.
5.1Cell replacement
Replacement of dying or dysfunctional rods and cones is a highly relevant therapeutic option as photoreceptors are presynaptic and functional connections may be easier to form with the host interneurons [136]. Notably, interneurons and ganglion cells are preserved during retinal degeneration even though retinal remodeling can be extensive [137, 138]. The ideal outcomes of a successful photoreceptor cell replacement therapy would include the survival and maturation of transplanted cells and formation of functional connectivity with the host inner retina. One of the original methods used to achieve photoreceptor replacement involved subretinal transplantation of developing and immature transgenic mouse (Nrl-GFP) rods [139] into mouse recipient retina, whereby the transplanted cells appear to integrate into the host retina, allowing for partial restoration of light-sensitivity [140]. Later, cell transplantations were conducted using mouse or human stem cell-derived photoreceptors [141–146]. However, recent evidence suggests that the majority of transplanted photoreceptors engage in cytoplasmic material (RNA and/or protein) transfer with host photoreceptor cells, and only a small number of donor cells may integrate into the recipient retina depending on the host environment [147–151]. For late stage retinal degeneration where the host photoreceptor layer has completely degenerated, transplantation of dissociated donor cells in the host retina may still be feasible [152] using improved strategies including enrichment of human photoreceptor cells prior to transplantation by targeting surface antigens and cell sorting [153–155] and making use of better animal models [156].
Another strategy for replacing lost photoreceptors is to transplant retinal sheets into the degenerating retina [157]. Proof-of-principle studies have demonstrated survival of the graft following the transplantation of stem cell-derived retinal sheets into mouse or monkey models and possible interaction with the host inner retina [158, 159]. Improvement of light response was also reported in a late stage retinal degeneration model following transplantation of a iPSC-derived retinal sheet [160]. Transplantation of retinal sheets maintains correct cell polarity, interaction with neighboring cells and state of differentiation in the donor cells, thereby providing an advantage over dissociated photoreceptors. However, a major problem in transplanting retinal sheets is establishment of connectivity between the donor graft and host cells, which is required to restore visual response.
Cell replacement therapy requires further assessment of donor/host interaction, especially with regards to cytoplasmic material transfer, and assessment of the appropriate time window for therapeutic intervention since the outcome of grafted cell integration and functional recovery are likely related to the type of disease, stage of disease progression, glial scar formation and/or activation of immune responses in the recipient retina [161–164].
5.2Drug discovery
Progress towards drug discovery for the treatment of retinal ciliopathies has been hampered by the lack of appropriate models for high throughput screening (HTS). Proof-of-concept studies have been performed to target disease-relevant molecular pathways using human iPSC-derived somatic cells with promising results [165]. Stem cell-based 2-D photoreceptors and 3-D retinal organoids provide a valuable clinically relevant resource for HTS to develop effective drugs for retinal ciliopathies [166, 167].
Previous efforts in drug development predominantly focused on target-based drug discovery (TDD), which is based on defined molecular targets from prior knowledge of disease pathogenesis [168]. However, TDD may not be suitable for retinal ciliopathies, which harbor complex pathological phenotypes with largely unknown disease mechanisms. An alternative approach is to employ phenotypic drug discovery (PDD), which does not require prior knowledge of underlying mechanisms and screens for compounds to reverse disease-associated phenotypes [169]. This approach has already been developed and tested with kidney organoids, in which multiple organoids are cultured in a fully automated, HTS-compatible platform for individual compounds [170]. Thus, retinal organoids can be employed for a similar PDD screening to identify novel therapeutics for retinal ciliopathies by assessing photoreceptor dysfunction, death or survival as final outcomes of drug application.
Several issues should be addressed to fully utilize retinal organoids for HTS. Heterogeneity of the retinal organoids can lead to false positives or negative hits in a PDD screen. Application of multiple organoids for a single compound might be able to compensate for the variability among organoids and reduce false positives or negative readouts. However, human retinal organoids are not suitable for an automated HTS platform due to their size, tissue rigidity and long time needed for differentiation. Though mini-retinal organoids from mouse PSCs [171] have been attempted, the process is labor-intensive and time-consuming. One possible approach is to dissociate the organoids into single cells, which would ensure cell homogeneity for testing of compounds before isolating the desired cell type [166]. This however eliminates the apical-basal polarity of photoreceptors after dissociation, which is needed to examine the pathology of retinal ciliopathies. The design of secondary assays on 3-D retinal organoids to test positive candidate hits would be crucial to facilitate the identification of effective drug candidates. Another downside of human retinal organoids is the long differentiation time, which extends the treatment window and makes it difficult for HTS due to issues related to expense and media change. An alternate strategy is to use organoids from mouse models of CEP290-LCA or RPGR-RP, which would require a shorter differentiation period [172], for HTS and then validate promising drug candidates using the patient-derived retinal organoids. RNA-seq analysis has been used for assessing drug efficacy for retinal degeneration [173] and could help in identifying molecular phenotypes before the appearance of photoreceptor cilia defects in patient organoids.
5.3Gene-based therapies
CRISPR/Cas system can be exploited to correct the mutant gene in patient iPSCs as a proof-of-concept approach for therapeutics; for example, correction of a RPGR mutation in patient-derived iPSCs has been reported [129]. Photoreceptors in retinal organoids from corrected patient iPSCs appear to show improved ciliogenesis and survival, and transcriptome analysis revealed somewhat restored gene expression pattern. Although gene profiles from corrected photoreceptors still deviated from the control ones raising off-target concerns, this study demonstrates the feasibility of genome-editing-based treatment paradigms. However, as is the case for CEP290-LCA, many ciliopathies are caused by single nucleotide mutations and the base editing efficiency by CRISPR system is far below therapeutic needs (<10% in monolayered cell culture) in such scenarios [174].
Additional approaches are being considered as treatment modelities for retinal ciliopathies, such as the delivery of CEP290 into patient-derived cells using lenti- or adeno-associated virus (AAV). However, one must be cautious as overexpression of CEP290 may lead to cytotoxicity [85]. AAV is a non-integrating virus with low immunogenicity, largely reducing the probability of insertional mutagenesis [175], and has been a vector of choice for the retina [176, 177]. However, the packaging limit of AAV is less than 5 kb and thus AAV can not be used for delivery of large genes such as CEP290. Some studies propose to use AAV dual vectors which recombine in vivo but the efficiency and feasibility remain uncertain [178]. As different domains of CEP290 have relatively diverse and independent functions [84], a recent study innovatively delivered a small region of CEP290 into rd16 mouse to complement the in-frame deletion in the myosin tail and largely restore structure and function of photoreceptors [179]. However, LCA-associated mutations are distributed throughout CEP290 and cause disease pathogenesis probably through different mechanisms. Evaluation of discrete CEP290 domains to rescue photoreceptor ciliogenesis in different patient-derived retinal organoids, though labor intensive, could provide useful information for designing AAV-based gene therapies.
Over 80% of CEP290-LCA patients carry at least one copy of IVS26 + 1665A>G mutation, which leads to aberrant alternative splicing and subsequent inclusion of a cryptic exon with premature stop codon [110]. Thus, a possible therapeutic approach can be to target disease-causing splicing in patient cells/organoids using antisense oligonucleotides (AONs) [86, 180]. AON therapeutic strategy is shown to be effective in CEP290-LCA fibroblasts and more disease-relevant patient iPSC-derived photoreceptors, where aberrant CEP290 splicing is corrected and ciliary defects are rescued [86, 181, 182].
6Conclusions
Biogenesis of photoreceptor modified cilium, including the OS membrane discs, relies on complex interactions of hundreds of proteins and dozens of multiprotein complexes. The phenotypes of retinal ciliopathies are determined by specific disease-causing mutations and by modifying factors including variations in interacting genes. Model organisms have contributed greatly to our fundamental understanding of ciliogenesis and retinal biology. More recently, patient-derived iPSCs have been used to complement animal models by in vitro generation of photoreceptors and retinal organoids to investigate disease-relevant cilia defects and design of therapies. Rapid advances of human iPSC-based approaches provide a real opportunity to interrogate genotype-phenotype relationships, high throughput screening of small molecules, and evaluation of personalized mutation-based treatments. Stem cell-based in vitro modeling and therapeutics of retinal ciliopathies are still in its infancy. We, however, are confident that better understanding of human retina development and advances in bioengineering platforms for retinal organoids would overcome current barriers for stem cell-based design of therapies for retinal ciliopathies.
Acknowledgments
We are grateful to Lina Zelinger for comments on the manuscript and Hiroko Shimad-Ishii for the human fibroblast image. The research in our laboratory is supported by the Intramural Research Program of the National Eye Institute (EY000450 and EY000474).
References
[1] | Benmerah A. , et al., The more we know, the more we have to discover: An exciting future for understanding cilia and ciliopathies, Cilia 4: ((2015) ), 5. |
[2] | Schueler M. , et al., Large-scale targeted sequencing comparison highlights extreme genetic heterogeneity in nephronophthisis-related ciliopathies, J Med Genet 53: (3) ((2016) ), 208–214. |
[3] | Shaheen R. , et al., Characterizing the morbid genome of ciliopathies, Genome Biol 17: (1) ((2016) ), 242. |
[4] | Reiter J.F. and Leroux M.R. , Genes and molecular pathways underpinning ciliopathies, Nat Rev Mol Cell Biol 18: (9) ((2017) ), 533–547. |
[5] | Rosenbaum J.L. and Witman G.B. , Intraflagellar transport, Nat Rev Mol Cell Biol 3: (11) ((2002) ), 813–825. |
[6] | Nigg E.A. and Raff J.W. , Centrioles, centrosomes, and cilia in health and disease, Cell 139: (4) ((2009) ), 663–678. |
[7] | Bangs F. and Anderson K.V. , Primary Cilia and Mammalian Hedgehog Signaling, Cold Spring Harb Perspect Biol 9: (5) ((2017) ). |
[8] | Ibanez-Tallon I. , Heintz N. and Omran H. , To beat or not to beat: Roles of cilia in development and disease, Hum Mol Genet 12: (Spec No 1) ((2003) ), R27–35. |
[9] | Quinlan R.J. , Tobin J.L. and Beales P.L. , Modeling ciliopathies: Primary cilia in development and disease, Curr Topics Dev Biol 84: ((2008) ), 249–310. |
[10] | Hildebrandt F. , Benzing T. and Katsanis N. , Ciliopathies, N Engl J Med 364: (16) ((2011) ), 1533–1543. |
[11] | Zhang W. , et al., Expanding the genetic architecture and phenotypic spectrum in the skeletal ciliopathies, Hum Mutat 39: (1) ((2018) ), 152–166. |
[12] | Besharse J.C. , et al., Photoreceptor intersegmental transport and retinal degeneration: A conserved pathway common to motile and sensory cilia, Adv Exp Med Biol 533: ((2003) ), 157–164. |
[13] | Rachel R.A. , Li T. and Swaroop A. , Photoreceptor sensory cilia and ciliopathies: Focus on CEP290, RPGR and their interacting proteins, Cilia 1: (1) ((2012) ), 22. |
[14] | Bujakowska K.M. , Liu Q. and Pierce E.A. , Photoreceptor Cilia and Retinal Ciliopathies, Cold Spring Harb Perspect Biol 9: (10) ((2017) ). |
[15] | May-Simera H. , Nagel-Wolfrum K. and Wolfrum U. , Cilia - The sensory antennae in the eye, Prog Retin Eye Res 60: ((2017) ), 144–180. |
[16] | Mockel A. , et al., Retinal dystrophy in Bardet-Biedl syndrome and related syndromic ciliopathies, Prog Retin Eye Res 30: (4) ((2011) ), 258–274. |
[17] | Sorusch N. , et al., Usher syndrome protein network functions in the retina and their relation to other retinal ciliopathies, Adv Exp Med Biol 801: ((2014) ), 527–33. |
[18] | Uytingco C.R. , Green W.W. and Martens J.R. , Olfactory loss and dysfunction in ciliopathies: Molecular mechanisms and potential therapies, Curr Med Chem (2018) . |
[19] | Vincensini L. , Blisnick T. and Bastin P. , 1001 model organisms to study cilia and flagella, Biol Cell 103: (3) ((2011) ), 109–130. |
[20] | Veleri S. , et al., Biology and therapy of inherited retinal degenerative disease: Insights from mouse models, Dis Model Mech 8: (2) ((2015) ), 109–129. |
[21] | Scholl H.P. , et al., Emerging therapies for inherited retinal degeneration, Sci Transl Med 8: (368) ((2016) ), 368rv6. |
[22] | Takahashi K. , et al., Induction of pluripotent stem cells from adult human fibroblasts by defined factors, Cell 131: (5) ((2007) ), 861–872. |
[23] | Nakano T. , et al., Self-formation of optic cups and storable stratified neural retina from human ESCs, Cell Stem Cell 10: (6) ((2012) ), 771–785. |
[24] | Zhong X. , et al., Generation of three-dimensional retinal tissue with functional photoreceptors from human iPSCs, Nat Commun 5: ((2014) ), 4047. |
[25] | Wright A.F. , et al., Photoreceptor degeneration: Genetic and mechanistic dissection of a complex trait, Nat Rev Genet 11: (4) ((2010) ), 273–284. |
[26] | Khanna H. and Baehr W. , Retina ciliopathies: From genes to mechanisms and treatment, Vis Res 75: ((2012) ), 1. |
[27] | Oh E.C. and Katsanis N. , Cilia in vertebrate development and disease, Development 139: (3) ((2012) ), 443–448. |
[28] | Mourao A. , Christensen S.T. and Lorentzen E. , The intraflagellar transport machinery in ciliary signaling, Curr Opin Struct Biol 41: ((2016) ), 98–108. |
[29] | Taschner M. and Lorentzen E. , The Intraflagellar Transport Machinery, Cold Spring Harb Perspect Biol 8: (10) (2016) . |
[30] | Goncalves J. and Pelletier L. , The Ciliary Transition Zone: Finding the Pieces and Assembling the Gate, Mol Cells 40: (4) ((2017) ), 243–253. |
[31] | Hirokawa N. , Kinesin and dynein superfamily proteins and the mechanism of organelle transport, Science 279: (5350) ((1998) ), 519–526. |
[32] | Wheway G. , Nazlamova L. and Hancock J.T. , Signaling through the Primary Cilium, Front Cell Dev Biol 6: ((2018) ), 8. |
[33] | Izawa I. , et al., Current topics of functional links between primary cilia and cell cycle, Cilia 4: ((2015) ), 12. |
[34] | Sanchez I. and Dynlacht B.D. , Cilium assembly and disassembly, Nat Cell Biol 18: (7) ((2016) ), 711–717. |
[35] | Yadav S.P. , et al., Centrosomal protein CP110 controls maturation of the mother centriole during cilia biogenesis, Development 143: (9) ((2016) ), 1491–1501. |
[36] | Ishikawa H. and Marshall W.F. , Ciliogenesis: Building the cell’s antenna, Nat Rev Mol Cell Biol 12: (4) ((2011) ), 222–234. |
[37] | Bornens M. , The centrosome in cells and organisms, Science 335: (6067) ((2012) ), 422–4226. |
[38] | Tsang W.Y. and Dynlacht B.D. , CP110 and its network of partners coordinately regulate cilia assembly, Cilia 2: (1) ((2013) ), 9. |
[39] | Tanos B.E. , et al., Centriole distal appendages promote membrane docking, leading to cilia initiation, Genes Dev 27: (2) ((2013) ), 163–168. |
[40] | Wang L. and Dynlacht B.D. , The regulation of cilium assembly and disassembly in development and disease, Development 145: (18) (2018) . |
[41] | Liu Q. , et al., The proteome of the mouse photoreceptor sensory cilium complex, Mol Cell Proteomics 6: (8) ((2007) ), 1299–1317. |
[42] | Reiter J.F. , Blacque O.E. and Leroux M.R. , The base of the cilium: Roles for transition fibres and the transition zone in ciliary formation, maintenance and compartmentalization, EMBO Rep 13: (7) ((2012) ), 608–618. |
[43] | Rachel R.A. , et al., Combining Cep290 and Mkks ciliopathy alleles in mice rescues sensory defects and restores ciliogenesis, J Clin Invest 122: (4) ((2012) ), 1233–1245. |
[44] | Zhang Y. , et al., BBS mutations modify phenotypic expression of CEP290-related ciliopathies, Hum Mol Genet 23: (1) ((2014) ), 40–51. |
[45] | Lamb T.D. , Collin S.P. and Pugh E.N. Jr. , Evolution of the vertebrate eye: Opsins, photoreceptors, retina and eye cup, Nat Rev Neurosci 8: (12) ((2007) ), 960–976. |
[46] | Anderson D.H. , Fisher S.K. and Steinberg R.H. , Mammalian cones: Disc shedding, phagocytosis, and renewal, Invest Ophthalmol Vis Sci 17: (2) ((1978) ), 117–133. |
[47] | Carter-Dawson L.D. and LaVail M.M. , Rods and cones in the mouse retina. I. Structural analysis using light and electron microscopy, J Comp Neurol 188: (2) ((1979) ), 245–262. |
[48] | Gilliam J.C. , et al., Three-dimensional architecture of the rod sensory cilium and its disruption in retinal neurodegeneration, Cell 151: (5) ((2012) ), 1029–1041. |
[49] | Young R.W. , The renewal of photoreceptor cell outer segments, J Cell Biol 33: (1) ((1967) ), 61–72. |
[50] | Lamb T.D. , Evolution of phototransduction, vertebrate photoreceptors and retina, Prog Retin Eye Res 36: ((2013) ), 52–119. |
[51] | Arshavsky V.Y. , Lamb T.D. and Pugh E.N. Jr. , G proteins and phototransduction, Annu Rev Physiol 64: ((2002) ), 153–187. |
[52] | Iannaccone A. , et al., Clinical and immunohistochemical evidence for an X linked retinitis pigmentosa syndrome with recurrent infections and hearing loss in association with an RPGR mutation, J Med Genet 40: (11) ((2003) ), e118. |
[53] | McEwen D.P. , et al., Hypomorphic CEP290/NPHP6 mutations result in anosmia caused by the selective loss of G proteins in cilia of olfactory sensory neurons, Proc Natl Acad Sci U S A 104: (40) ((2007) ), 15917–15922. |
[54] | Papon J.F. , et al., Abnormal respiratory cilia in non-syndromic Leber congenital amaurosis with CEP290 mutations, J Med Genet 47: (12) ((2010) ), 829–34. |
[55] | Coussa R.G. , Lopez Solache I. and Koenekoop R.K. , Leber congenital amaurosis, from darkness to light: An ode to Irene Maumenee, Ophthalmic Genet 38: (1) ((2017) ), 7–15. |
[56] | Thompson J.A. , et al., Leber Congenital Amaurosis: Identification of Disease-Causing Variants and Personalised Therapies, Adv Exp Med Biol 1074: ((2018) ), 265–271. |
[57] | Breuer D.K. , et al., A comprehensive mutation analysis of RP2 and RPGR in a North American cohort of families with X-linked retinitis pigmentosa, Am J Hum Genet 70: (6) ((2002) ), 1545–1554. |
[58] | Sharon D. , et al., RP2 and RPGR mutations and clinical correlations in patients with X-linked retinitis pigmentosa, Am J Hum Genet 73: (5) ((2003) ), 1131–1146. |
[59] | Shu X. , et al., RPGR mutation analysis and disease: An update, Hum Mutat 28: (4) ((2007) ), 322–328. |
[60] | Megaw R.D. , Soares D.C. and Wright A.F. , RPGR: Its role in photoreceptor physiology, human disease, and future therapies, Exp Eye Res 138: ((2015) ), 32–41. |
[61] | Branham K. , et al., Mutations in RPGR and RP2 account for 15% of males with simplex retinal degenerative disease, Invest Ophthalmol Vis Sci 53: (13) ((2012) ), 8232–8237. |
[62] | Estrada-Cuzcano A. , et al., BBS1 mutations in a wide spectrum of phenotypes ranging from nonsyndromic retinitis pigmentosa to Bardet-Biedl syndrome, Arch Ophthalmol 130: (11) ((2012) ), 1425–1432. |
[63] | Veleri S. , et al., Ciliopathy-associated gene Cc2d2a promotes assembly of subdistal appendages on the mother centriole during cilia biogenesis, Nat Commun 5: ((2014) ), 4207. |
[64] | Bachmann-Gagescu R. , et al., The Ciliopathy Protein CC2D2A Associates with NINL and Functions in RAB8-MICAL3-Regulated Vesicle Trafficking, PLoS Genet 11: (10) ((2015) ), e1005575. |
[65] | Gorden N.T. , et al., CC2D2A is mutated in Joubert syndrome and interacts with the ciliopathy-associated basal body protein CEP290, Am J Hum Genet 83: (5) ((2008) ), 559–571. |
[66] | Noor A. , et al., CC2D2A, encoding a coiled-coil and C2 domain protein, causes autosomal-recessive mental retardation with retinitis pigmentosa, Am J Hum Genet 82: (4) ((2008) ), 1011–1018. |
[67] | Tallila J. , et al., Identification of CC2D2A as a Meckel syndrome gene adds an important piece to the ciliopathy puzzle, Am J Hum Genet 82: (6) ((2008) ), 1361–1367. |
[68] | Estrada-Cuzcano A. , et al., Non-syndromic retinal ciliopathies: Translating gene discovery into therapy, Hum Mol Genet 21: (R1) ((2012) ), R111–24. |
[69] | Mukhopadhyay S. and Jackson P.K. , The tubby family proteins, Genome Biology 12: (6) ((2011) ), 225. |
[70] | Badgandi H.B. , et al., Tubby family proteins are adapters for ciliary trafficking of integral membrane proteins, J Cell Biol 216: (3) ((2017) ), 743–760. |
[71] | Hagstrom S.A. , et al., A role for the Tubby-like protein 1 in rhodopsin transport, Invest Ophthalmol Vis Sci 42: (9) ((2001) ), 1955–1962. |
[72] | Banerjee P. , et al., TULP1 mutation in two extended Dominican kindreds with autosomal recessive retinitis pigmentosa, Nat Genet 18: (2) ((1998) ), 177–179. |
[73] | Hanein S. , et al., Leber congenital amaurosis: Comprehensive survey of the genetic heterogeneity, refinement of the clinical definition, and genotype-phenotype correlations as a strategy for molecular diagnosis, Hum Mutat 23: (4) ((2004) ), 306–317. |
[74] | Craige B. , et al., CEP290 tethers flagellar transition zone microtubules to the membrane and regulates flagellar protein content, J Cell Biol 190: (5) ((2010) ), 927–940. |
[75] | Rachel R.A. , et al., CEP290 alleles in mice disrupt tissue-specific cilia biogenesis and recapitulate features of syndromic ciliopathies, Hum Mol Genet 24: (13) ((2015) ), 3775–3791. |
[76] | Coppieters F. , et al., CEP290, a gene with many faces: Mutation overview and presentation of CEP290base, Hum Mut 31: (10) ((2010) ), 1097–1108. |
[77] | Drivas T.G. and Bennett J. , CEP290 and the primary cilium, Adv Exp Med Biol 801: ((2014) ), 519–525. |
[78] | Perrault I. , et al., Spectrum of NPHP6/CEP290 mutations in Leber congenital amaurosis and delineation of the associated phenotype, Hum Mutat 28: (4) ((2007) ), 416. |
[79] | den Hollander A.I. , et al., Leber congenital amaurosis: Genes, proteins and disease mechanisms, Prog Retin Eye Res 27: (4) ((2008) ), 391–419. |
[80] | Hynes A.M. , et al., Murine Joubert syndrome reveals Hedgehog signaling defects as a potential therapeutic target for nephronophthisis, Proc Natl Acad Sci U S A 111: (27) ((2014) ), 9893–9898. |
[81] | Chang B. , et al., In-frame deletion in a novel centrosomal/ciliary protein CEP290/NPHP6 perturbs its interaction with RPGR and results in early-onset retinal degeneration in the rd16 mouse, Hum Mol Genet 15: (11) ((2006) ), 1847–1857. |
[82] | Cideciyan A.V. , et al., Centrosomal-ciliary gene CEP290/NPHP6 mutations result in blindness with unexpected sparing of photoreceptors and visual brain: Imlications for therapy of Leber congenital amaurosis, Hum Mutat 28: (11) ((2007) ), 1074–1083. |
[83] | Cideciyan A.V. , et al., Cone photoreceptors are the main targets for gene therapy of NPHP5 (IQCB1) or NPHP6 (CEP290) blindness: Generation of an all-cone Nphp6 hypomorph mouse that mimics the human retinal ciliopathy, Hum Mol Genet 20: (7) ((2011) ), 1411–1423. |
[84] | Drivas T.G. , Holzbaur E.L. and Bennett J. , Disruption of CEP290 microtubule/membrane-binding domains causes retinal degeneration, J Clin Invest 123: (10) ((2013) ), 4525–4539. |
[85] | Burnight E.R. , et al., CEP290 gene transfer rescues Leber congenital amaurosis cellular phenotype, Gene Ther 21: (7) ((2014) ), 662–672. |
[86] | Parfitt D.A. , et al., Identification and Correction of Mechanisms Underlying Inherited Blindness in Human iPSC-Derived Optic Cups, Cell Stem Cell 18: (6) ((2016) ), 769–781. |
[87] | Shimada H. , et al., Modeling Using Ciliopathy-Patient-Derived Cells Reveals Distinct Cilia Dysfunctions Caused by CEP290 Mutations, Cell Rep 20: (2) ((2017) ), 384–396. |
[88] | Hong D.H. , et al., RPGR isoforms in photoreceptor connecting cilia and the transitional zone of motile cilia, Invest Ophthalmol Vis Sci 44: (6) ((2003) ), 2413–2421. |
[89] | Khanna H. , et al., RPGR-ORF15, which is mutated in retinitis pigmentosa, associates with SMC1, SMC3, and microtubule transport proteins, J Biol Chem 280: (39) ((2005) ), 33580–33587. |
[90] | Hong D.H. , et al., A retinitis pigmentosa GTPase regulator (RPGR)-deficient mouse model for X-linked retinitis pigmentosa (RP3), Proc Natl Acad Sci USA 97: (7) ((2000) ), 3649–3654. |
[91] | Thompson D.A. , et al., Rd9 is a naturally occurring mouse model of a common form of retinitis pigmentosa caused by mutations in RPGR-ORF15, PLoS One 7: (5) ((2012) ), e35865. |
[92] | Zhang Q. , et al., Different RPGR exon ORF15 mutations in Canids provide insights into photoreceptor cell degeneration, Hum Mol Genet 11: (9) ((2002) ), 993–1003. |
[93] | Beltran W.A. , et al., A frameshift mutation in RPGR exon ORF15 causes photoreceptor degeneration and inner retina remodeling in a model of X-linked retinitis pigmentosa, Invest Ophthalmol Vis Sci 47: (4) ((2006) ), 1669–16681. |
[94] | Witman G.B. , et al., Chlamydomonas flagella. I. Isolation and electrophoretic analysis of microtubules, matrix, membranes, and mastigonemes, J Cell Biol 54: (3) ((1972) ), 507–539. |
[95] | Perkins L.A. , et al., Mutant sensory cilia in the nematode Caenorhabditis elegans, Dev Biol 117: (2) ((1986) ), 456–487. |
[96] | Pazour G.J. , et al., Proteomic analysis of a eukaryotic cilium, J Cell Biol 170: (1) ((2005) ), 103–13. |
[97] | Keller L.C. , et al., Proteomic analysis of isolated chlamydomonas centrioles reveals orthologs of ciliary-disease genes, Curr Biol 15: (12) ((2005) ), 1090–1098. |
[98] | Brown J.M. , et al., Kinesin-II is preferentially targeted to assembling cilia and is required for ciliogenesis and normal cytokinesis in Tetrahymena, Mol Biol Cell 10: (10) ((1999) ), 3081–3096. |
[99] | Williams C.L. , et al., MKS and NPHP modules cooperate to establish basal body/transition zone membrane associations and ciliary gate function during ciliogenesis, J Cell Biol 192: (6) ((2011) ), 1023–1041. |
[100] | Pazour G.J. , et al., Chlamydomonas IFT88 and its mouse homologue, polycystic kidney disease gene tg737, are required for assembly of cilia and flagella, J Cell Biol 151: (3) ((2000) ), 709–718. |
[101] | Brown J.M. and Witman G.B. , Cilia and Diseases, Bioscience 64: (12) ((2014) ), 1126–1137. |
[102] | Fliegauf M. , Benzing T. and Omran H. , When cilia go bad: Cilia defects and ciliopathies, Nat Rev Mol Cell Biol 8: (11) ((2007) ), 880–893. |
[103] | Blum M. , Ott T. , Xenopus: An Undervalued Model Organism to Study and Model Human Genetic Disease, Cells Tissues Organs 7: ((2018) ), 1133.1–11. |
[104] | Molinari E. , et al., Using zebrafish to study the function of nephronophthisis and related ciliopathy genes, F1000Res ((2018) ), 1133. |
[105] | Chang B. , Mouse models for studies of retinal degeneration and diseases, Methods Mol Biol 935: ((2013) ), 27–39. |
[106] | Garcia-Gonzalo F.R. , et al., A transition zone complex regulates mammalian ciliogenesis and ciliary membrane composition, Nat Genet 43: (8) ((2011) ), 776–784. |
[107] | Gibson F. , et al., A type VII myosin encoded by the mouse deafness gene shaker-1, Nature 374: (6517) ((1995) ), 62–64. |
[108] | Weil D. , et al., Defective myosin VIIA gene responsible for Usher syndrome type 1B, Nature 374: (6517) ((1995) ), 60–61. |
[109] | Garanto A. , et al., Unexpected CEP290 mRNA splicing in a humanized knock-in mouse model for Leber congenital amaurosis, PLoS One 8: (11) ((2013) ), e79369. |
[110] | Drivas T.G. , et al., Basal exon skipping and genetic pleiotropy: A predictive model of disease pathogenesis, Sci Transl Med 7: (291) ((2015) ), 291ra97. |
[111] | Takahashi K. and Yamanaka S. , Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors, Cell 126: (4) ((2006) ), 663–676. |
[112] | Osakada F. , et al., Toward the generation of rod and cone photoreceptors from mouse, monkey and human embryonic stem cells, Nat Biotechnol 26: (2) ((2008) ), 215–224. |
[113] | Hirami Y. , et al., Generation of retinal cells from mouse and human induced pluripotent stem cells, Neuroscience letters 458: (3) ((2009) ), 126–131. |
[114] | Lamba D.A. , Karl M.O. and Reh T.A. , Strategies for retinal repair: Cell replacement and regeneration, Prog Brain Res 175: ((2009) ), 23–31. |
[115] | Gamm D.M. and Meyer J.S. , Directed differentiation of human induced pluripotent stem cells: A retina perspective, Regen Med 5: (3) ((2010) ), 315–317. |
[116] | Phillips M.J. , et al., Blood-derived human iPS cells generate optic vesicle-like structures with the capacity to form retinal laminae and develop synapses, Invest Ophthalmol Vis Sci 53: (4) ((2012) ), 2007–2019. |
[117] | Zhao X. , Liu J. and Ahmad I. , Differentiation of embryonic stem cells into retinal neurons, Biochem Biophys Res Commun 297: (2) ((2002) ), 177–1784. |
[118] | Watanabe K. , et al., Directed differentiation of telencephalic precursors from embryonic stem cells, Nat Neurosci 8: (3) ((2005) ), 288–296. |
[119] | Ikeda H. , et al., Generation of Rx+/Pax6+ neural retinal precursors from embryonic stem cells, Proc Natl Acad Sci U S A 102: (32) ((2005) ), 11331–11336. |
[120] | Lamba D.A. , et al., Efficient generation of retinal progenitor cells from human embryonic stem cells, Proc Natl Acad Sci U S A 103: (34) ((2006) ), 12769–12774. |
[121] | Mellough C.B. , et al., Efficient stage-specific differentiation of human pluripotent stem cells toward retinal photoreceptor cells, Stem Cells 30: (4) ((2012) ), 673–686. |
[122] | Jin Z.B. , et al., Modeling retinal degeneration using patient-specific induced pluripotent stem cells, PLoS One 6: (2) ((2011) ), e17084. |
[123] | Yin X. , et al., Engineering Stem Cell Organoids, Cell Stem Cell 18: (1) ((2016) ), 25–38. |
[124] | Meyer J.S. , et al., Optic vesicle-like structures derived from human pluripotent stem cells facilitate a customized approach to retinal disease treatment, Stem Cells 29: (8) ((2011) ), 1206–1218. |
[125] | Kuwahara A. , et al., Generation of a ciliary margin-like stem cell niche from self-organizing human retinal tissue, Nat Commun 6: ((2015) ), 6286. |
[126] | Wahlin K.J. , et al., Photoreceptor Outer Segment-like Structures in Long-Term 3D Retinas from Human Pluripotent Stem Cells, Sci Rep 7: (1) ((2017) ), 766. |
[127] | Gonzalez-Cordero A. , et al., Recapitulation of Human Retinal Development from Human Pluripotent Stem Cells Generates Transplantable Populations of Cone Photoreceptors, Stem Cell Reports 9: (3) ((2017) ), 820–837. |
[128] | Hallam D. , et al., Human-Induced Pluripotent Stem Cells Generate Light Responsive Retinal Organoids with Variable and Nutrient-Dependent Efficiency, Stem Cells 36: (10) ((2018) ), 1535–1551. |
[129] | Deng W.L. , et al., Gene Correction Reverses Ciliopathy and Photoreceptor Loss in iPSC-Derived Retinal Organoids from Retinitis Pigmentosa Patients, Stem Cell Reports 10: (4) ((2018) ), 1267–1281. |
[130] | Buskin A. , et al., Disrupted alternative splicing for genes implicated in splicing and ciliogenesis causes PRPF31 retinitis pigmentosa, Nat Commun 9: (1) ((2018) ), 4234. |
[131] | Hoshino A. , et al., Molecular Anatomy of the Developing Human Retina, Dev Cell 43: ((2017) ), 763–779. |
[132] | DiStefano T. , et al., Accelerated and Improved Differentiation of Retinal Organoids from Pluripotent Stem Cells in Rotating-Wall Vessel Bioreactors, Stem Cell Reports 10: (1) ((2018) ), 300–313. |
[133] | Ovando-Roche P. , et al., Use of bioreactors for culturing human retinal organoids improves photoreceptor yields, Stem Cell Res Ther 9: (1) ((2018) ), 156. |
[134] | Phelan M.A. , Lelkes P.I. and Swaroop A. , Mini and customized low-cost bioreactors for optimized high-throughput generation of tissue organoids, Stem Cell Investig 5: ((2018) ), 33. |
[135] | Nasonkin I.O. , et al., Conditional knockdown of DNA methyltransferase 1 reveals a key role of retinal pigment epithelium integrity in photoreceptor outer segment morphogenesis, Development 140: (6) ((2013) ), 1330–1341. |
[136] | Zhao B.S. , Roundtree I.A. and He C. , Post-transcriptional gene regulation by mRNA modifications, Nat Rev Mol Cell Biol 18: (1) ((2017) ), 31–42. |
[137] | Marc R.E. , et al., Neural remodeling in retinal degeneration, Prog Retin Eye Res 22: (5) ((2003) ), 607–655. |
[138] | Mazzoni F. , Novelli E. and Strettoi E. , Retinal ganglion cells survive and maintain normal dendritic morphology in a mouse model of inherited photoreceptor degeneration, J Neurosci 28: (52) ((2008) ), 14282–14292. |
[139] | Akimoto M. , et al., Targeting of GFP to newborn rods by Nrl promoter and temporal expression profiling of flow-sorted photoreceptors, Proc Natl Acad Sci U S A 103: (10) ((2006) ), 3890–3895. |
[140] | MacLaren R.E. , et al., Retinal repair by transplantation of photoreceptor precursors, Nature 444: (7116) ((2006) ), 203–207. |
[141] | Lamba D.A. , Gust J. and Reh T.A. , Transplantation of human embryonic stem cell-derived photoreceptors restores some visual function in Crx-deficient mice, Cell Stem Cell 4: (1) ((2009) ), 73–79. |
[142] | Lamba D.A. , et al., Generation, purification and transplantation of photoreceptors derived from human induced pluripotent stem cells, PLoS One 5: (1) ((2010) ), e8763. |
[143] | Tucker B.A. , et al., Transplantation of adult mouse iPS cell-derived photoreceptor precursors restores retinal structure and function in degenerative mice, PLoS One 6: (4) ((2011) ), e18992. |
[144] | Homma K. , et al., Developing rods transplanted into the degenerating retina of crx-knockout mice exhibit neural activity similar to native photoreceptors, Stem Cells 31: (6) ((2013) ), 1149–1159. |
[145] | Gonzalez-Cordero A. , et al., Photoreceptor precursors derived from three-dimensional embryonic stem cell cultures integrate and mature within adult degenerate retina, Nat Biotechnol 31: (8) ((2013) ), 741–747. |
[146] | Barnea-Cramer A.O. , et al., Function of human pluripotent stem cell-derived photoreceptor progenitors in blind mice, Sci Rep 6: ((2016) ), 29784. |
[147] | Pearson R.A. , et al., Donor and host photoreceptors engage in material transfer following transplantation of post-mitotic photoreceptor precursors, Nat Commun 7: ((2016) ), 13029. |
[148] | Santos-Ferreira T. , et al., Retinal transplantation of photoreceptors results in donor-host cytoplasmic exchange, Nat Commun 7: ((2016) ), 13028. |
[149] | Singh M.S. , et al., Transplanted photoreceptor precursors transfer proteins to host photoreceptors by a mechanism of cytoplasmic fusion, Nat Commun 7: ((2016) ), 13537. |
[150] | Ortin-Martinez A. , et al., A Reinterpretation of Cell Transplantation: GFP Transfer From Donor to Host Photoreceptors, Stem Cells 35: (4) ((2017) ), 932–939. |
[151] | Waldron P.V. , et al., Transplanted Donor- or Stem Cell-Derived Cone Photoreceptors Can Both Integrate and Undergo Material Transfer in an Environment-Dependent Manner, Stem Cell Reports 10: (2) ((2018) ), 406–421. |
[152] | Singh R. , et al., iPS cell modeling of Best disease: Insights into the pathophysiology of an inherited macular degeneration, Hum Mol Genet 22: (3) ((2013) ), 593–607. |
[153] | Welby E. , et al., Isolation and Comparative Transcriptome Analysis of Human Fetal and iPSC-Derived Cone Photoreceptor Cells, Stem Cell Reports 9: (6) ((2017) ), 1898–1915. |
[154] | Lakowski J. , et al., Isolation of Human Photoreceptor Precursors via a Cell Surface Marker Panel from Stem Cell-Derived Retinal Organoids and Fetal Retinae, Stem Cells 36: (5) ((2018) ), 709–722. |
[155] | Gagliardi G. , et al., Characterization and Transplantation of CD73-Positive Photoreceptors Isolated from Human iPSC-Derived Retinal Organoids, Stem Cell Reports 11: (3) ((2018) ), 665–680. |
[156] | Wang W. , et al., Swine cone and rod precursors arise sequentially and display sequential and transient integration and differentiation potential following transplantation, Invest Ophthalmol Vis Sci 55: (1) ((2014) ), 301–309. |
[157] | Seiler M.J. and Aramant R.B. , Cell replacement and visual restoration by retinal sheet transplants, Prog Retin Eye Res 31: (6) ((2012) ), 661–687. |
[158] | Assawachananont J. , et al., Transplantation of embryonic and induced pluripotent stem cell-derived 3D retinal sheets into retinal degenerative mice, Stem Cell Reports 2: (5) ((2014) ), 662–674. |
[159] | Shirai H. , et al., Transplantation of human embryonic stem cell-derived retinal tissue in two primate models of retinal degeneration, Proc Natl Acad Sci U S A 113: (1) ((2016) ), E81–90. |
[160] | Mandai M. , et al., iPSC-Derived Retina Transplants Improve Vision in rd1 End-Stage Retinal-Degeneration Mice, Stem Cell Reports 8: (4) ((2017) ), 1112–1113. |
[161] | Suzuki T. , et al., Chondroitinase ABC treatment enhances synaptogenesis between transplant and host neurons in model of retinal degeneration, Cell Transplant 16: (5) ((2007) ), 493–503. |
[162] | Langmann T. , Microglia activation in retinal degeneration, J Leukoc Biol 81: (6) ((2007) ), 1345–1351. |
[163] | West E.L. , et al., Pharmacological disruption of the outer limiting membrane leads to increased retinal integration of transplanted photoreceptor precursors, Exp Eye Res 86: (4) ((2008) ), 601–611. |
[164] | Barber A.C. , et al., Repair of the degenerate retina by photoreceptor transplantation, Proc Natl Acad Sci U S A 110: (1) ((2013) ), 354–359. |
[165] | Avior Y. , Sagi I. and Benvenisty N. , Pluripotent stem cells in disease modelling and drug discovery, Nat Rev Mol Cell Biol 17: (3) ((2016) ), 170–182. |
[166] | Kaewkhaw R. , et al., Treatment Paradigms for Retinal and Macular Diseases Using 3-D Retina Cultures Derived From Human Reporter Pluripotent Stem Cell Lines, Invest Ophthalmol Vis Sci 57: (5) ((2016) ), ORSFl1–ORSFl11. |
[167] | Llonch S. , Carido M. and Ader M. , Organoid technology for retinal repair, Dev Biol 433: (2) ((2018) ), 132–143. |
[168] | Moffat J.G. , et al., Opportunities and challenges in phenotypic drug discovery: An industry perspective, Nat Rev Drug Discov 16: (8) ((2017) ), 531–543. |
[169] | Soldner F. and Jaenisch R. , Stem Cells, Genome Editing, and the Path to Translational Medicine, Cell 175: (3) ((2018) ), 615–632. |
[170] | Czerniecki S.M. , et al., High-Throughput Screening Enhances Kidney Organoid Differentiation from Human Pluripotent Stem Cells and Enables Automated Multidimensional Phenotyping, Cell Stem Cell 22: (6) ((2018) ), 929–940e4. |
[171] | Volkner M. , et al., Retinal Organoids from Pluripotent Stem Cells Efficiently Recapitulate Retinogenesis, Stem Cell Reports 6: (4) ((2016) ), 525–538. |
[172] | Chen H.Y. , Kaya K. D. , Dong L. and Swaroop A. , Three-dimensional retinal organoids from mouse pluripotent stem cells mimicdevelopment with enhanced stratification and rod photoreceptor differentiation, Mol Vis 22: ((2016) ), 1077–1094. |
[173] | Chen Y. , et al., Synergistically acting agonists and antagonists of G protein-coupled receptors prevent photoreceptor cell degeneration, Sci Signal 9: (438) ((2016) ), ra74. |
[174] | Gapinske M. , et al., CRISPR-SKIP: Programmable gene splicing with single base editors, Genome Biol 19: (1) ((2018) ), 107. |
[175] | Chandler R.J. , Sands M.S. and Venditti C.P. , Recombinant Adeno-Associated Viral Integration and Genotoxicity: Insights from Animal Models, Hum Gene Ther 28: (4) ((2017) ), 314–322. |
[176] | Day T.P. , et al., Advances in AAV vector development for gene therapy in the retina, Adv Exp Med Biol 801: ((2014) ), 687–693. |
[177] | Naso M.F. , et al., Adeno-Associated Virus (AAV) as a Vector for Gene Therapy, BioDrugs 31: (4) ((2017) ), 317–334. |
[178] | McClements M.E. and MacLaren R.E. , Adeno-associated Virus (AAV) Dual Vector Strategies for Gene Therapy Encoding Large Transgenes, Yale J Biol Med 90: (4) ((2017) ), 611–623. |
[179] | Mookherjee S. , et al., A CEP290 C-Terminal Domain Complements the Mutant CEP290 of Rd16 Mice In Trans and Rescues Retinal Degeneration, Cell Rep 25: (3) ((2018) ), 611–623e6. |
[180] | Rinaldi C. and Wood M.J.A. , Antisense oligonucleotides: The next frontier for treatment of neurological disorders, Nat Rev Neurol 14: (1) ((2018) ), 9–21. |
[181] | Collin R.W. , et al., Antisense Oligonucleotide (AON)-based Therapy for Leber Congenital Amaurosis Caused by a Frequent Mutation in CEP290, Mol Ther Nucleic Acids 1: ((2012) ), e14. |
[182] | Gerard X. , et al., AON-mediated Exon Skipping Restores Ciliation in Fibroblasts Harboring the Common Leber Congenital Amaurosis CEP290 Mutation, Mol Ther Nucleic Acids 1: ((2012) ), e29. |


