
Using human urine-derived renal epithelial cells to model kidney disease in inherited ciliopathies
Abstract
The extreme heterogeneity of renal ciliopathies warrants the use of personalised, patient-specific disease models. Kidney tubular epithelia are exposed to continuous passage of filtrate and viable renal tubular cells are excreted daily in the urine, representing a non-invasive source of patient primary material. These cells can be isolated, cultured and employed for a range of applications, from disease modelling to ex vivo drug testing.
1Introduction
Genetic disorders of the kidney include cystic kidney diseases, tubulopathies, podocytopathies, inherited forms of immune glomerulonephritis and inherited metabolic disorders. A unifying pathogenic concept for inherited cystic kidney diseases arose from the discovery that almost all the known causative genes for cystic kidney disease encode proteins that localise at the primary cilium and related structures [1]. Dysfunctional primary cilia may represent the primary pathogenic defect in kidney tissue development and/or homeostasis, ultimately leading to cystogenesis.
Several cell and animal models have been used to study inherited renal disease and inherited ciliopathies in particular. These include the single-celled green alga C. reinhardtii, the ciliated protozoan T. thermophile, invertebrate and vertebrate models such as C. elegans and D. rerio (zebrafish) respectively and mammalian models (mice, rats and mammalian cell cultures) [2].
Zebrafish and rodents have been employed as pre-clinical models in several studies aimed to investigate novel therapeutic strategies for inherited ciliopathies. However, with the exception of tolvaptan in the treatment of autosomal dominant polycystic kidney disease, none of the promising therapeutic approaches identified in these preclinical studies have so far led to satisfactory results in clinical trials [3]. The extreme genetic and clinical heterogeneity that characterizes renal ciliopathy patients may contribute to the poor translatability of therapeutic findings from pre-clinical models and highlights the need for personalised, patient-specific models.
Kidney tubular epithelia are exposed to continuous passage of filtrate and daily shed several thousands of renal tubular cells that can be found in the urine. Therefore, human urine represents a limitless and non-invasive source of patient-specific viable cells that can be isolated and cultured to enable detailed phenotyping and molecular studies to be undertaken [4, 5].
2Isolation and culture of urine-derived renal epithelial cells
The mammalian urinary system is composed of two kidneys, two ureters, the bladder and the urethra. Approximately 2,000 to 7,000 cells exfoliate daily from the epithelia of different parts of the urinary tract and can be found in the urinary sediment [6]. These include transitional cells from the bladder, urethra and renal pelvis, squamous epithelia cells from the anterior urethra and renal tubular epithelial cells. Since the first report in 1972, scientists have been implementing protocols to isolate cells from urine and culture exfoliated cells of the urinary tract [4–11].
Following collection of 50 ml of fresh urine, a centrifugation step for 10 minutes at 400 g is required to form a urinary sediment pellet that contains cellular material. This pellet can be washed with antibiotics to decrease the risk of contamination and then re-suspended in DMEM supplemented with 10% foetal bovine serum and plated on a standard, uncoated tissue culture plate with a surface area≤4 cm2. To increase chances of harvesting viable cells, urine sample should be processed within 4 h from collection [4, 5].
If the isolation step is successful, the formation of small cell colonies will be observed between day 3 to day 14 after urine sample collection (Fig. 1A). Once these cells reach ∼80% confluence in the original well of the tissue culture plate, they can be detached and sub-cultured for a few passages (typically between 4 and 10) before they visibly transform, acquiring an elongated and flattened shape, which suggests that cells are undergoing senescence.
Fig.1
Methodology and application of URECs. (A) Schematic of human urine processing to isolate URECs. Within 4 hours from collection, urine is transferred into 50 ml tubes and is centrifuged. Urine cell pellet is re-suspended in medium and plated on a well of an uncoated tissue culture 12-well plate. Formation of small cell colonies will be observed within 1–2 weeks. When cells reach at least 80% confluence, they can be detached and sub-cultured. Scale bar, 100μm. (B) Scanning electron microscopy (SEM) of wild type and JBTS type 5 patient URECs. SEM reveals that the primary cilium in a JBTS patient carrying the homozygous mutation G1890* in CEP290 is elongated compared to wild type cilium. Treatment with an antisense oligonucleotide (ASO), designed to skip the mutation-carrying exon 41, restores normal cilia length in JBTS 5 URECs. Scale bar, 2μm.
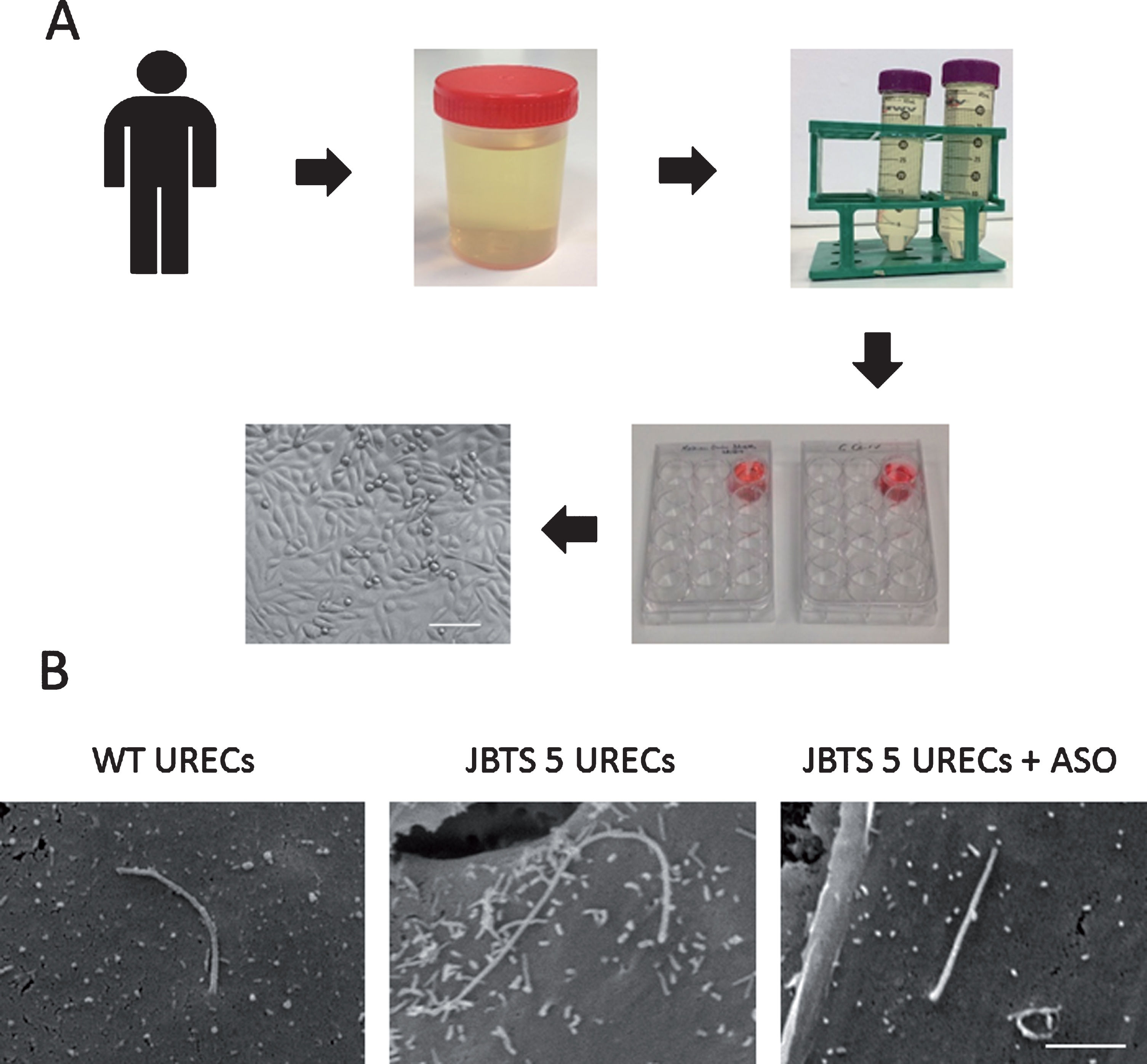
Urine-derived cells display a certain degree of morphological heterogeneity, with the majority of cells corresponding to two main morphologies that have been previously described as type 1 and type 2 shapes [10]. Type 1 cells are the most common, with a bipolar shape, they appear less elongated than fibroblast cells and are characterised by a relatively close cell-to-cell contact. Type 2 cells are less common, have a cobblestone-like shape, appear slightly smaller than type 1 cells and form very compact colonies.
Several attempts of characterization of these cells by immunofluorescence, western blot or quantitative RT-PCR indicated that urine-derived cells are a heterogeneous population, with predominant epithelial origin. The identity of urine-derived cells was inferred on the basis of the expression of markers for epithelial cells of the urinary tract. Cultures of urine-derived cells were positive for markers known to be expressed in collecting duct cells (AQP2 and L1-CAM), distal tubule cells (KRT7 and ENaC) and proximal tubule cells (CD13, LRP2 and sodium-glucose transporters), with little staining for the fibroblastic-like markers fibronectin and vimentin [5, 6, 12], indicating that urine-derived cells are predominantly of renal origin. Therefore, from now on, we shall refer to them as urine-derived renal epithelial cells (URECs). Conflicting evidence (perhaps due to differences in medium composition) exists regarding the expression of the bladder cell marker UPK3 [4, 6]. Cultures enriched for one of the two morphologies (type 1 or type 2) display similar expression of the analysed markers, so differences in cell shape don’t seem to correspond to a difference in cell identity, at least at the resolution allowed by the techniques employed for the characterization [12].
Success of the isolation step, defined as the appearance of cell colonies after 14 days of culture, is variable and, in our hands, the success rate is around 80%. Occasionally, even when cell colonies appear, they cannot be sub-cultured, due to low duplication rates. We attain cells that are amenable to sub-culture from around 70% of samples processed. High variability exists also in terms of growth rate, cell culture yield and ratios between the two main cell morphologies among cultures derived from different individuals and even among different collections from the same individual.
3Proposed applications of URECs
Despite the intrinsic variability, URECs represent an invaluable, non-invasive source of primary patient material. Using urine as a source of patient-specific cells represents an alternative to blood samples or skin biopsies that doesn’t cause any physical or psychological discomfort and can be attained without medical assistance, making it particularly suitable for paediatric patients.
URECs were shown to be amenable to manipulations such as gene silencing by siRNA transfection [13, 14] and can be cultured in a 3-dimensional setting in Matrigel. In Matrigel, URECs form spheroidal structures made of ciliated cells, with cilia that protrude towards the lumen of the spheroid, thus mimicking the renal tubular 3-dimensional structure [5, 15].
The versatility of URECs (Table 1) makes them an ideal patient-specific model, especially for renal disease and a powerful tool in the validation of the pathogenicity of novel candidate variants and for personalised, ex vivo drug testing. Indeed, human URECs were recently shown to be amenable to be directly reprogrammed to generate muscle-like cells [16] or reprogrammed to induced pluripotent stem cells that can be in turn differentiated to virtually any tissue [4, 12, 17–19]. Thus, the relevance of URECs culture is not restricted to the investigation of renal physiology and pathology but extends to the study of a wide range of organs and tissues.
Table 1
Cellular and molecular biology techniques that may be applied to URECs
| Nucleic acid extraction and analysis | (Rahmoune et al. 2005; Zhou et al. 2011; Srivastava et al. 2017; Ramsbottom et al. 2018; Molinari et al. 2018) |
| Immunocytochemistry | (Rahmoune et al. 2005; Zhou et al. 2011; Slaats et al. 2014; Ajzenberg et al. 2015; Slaats et al. 2015; Srivastava et al. 2017; Ramsbottom et al. 2018; Oud et al. 2018) |
| Scanning electron microscopy (SEM) | (Srivastava et al. 2017; Ramsbottom et al. 2018) |
| Protein expression analysis (Western blot and proteomics) | (Slaats et al. 2015; Bartram et al. 2016; Srivastava et al. 2017; Ramsbottom et al. 2018; Slaats et al. 2018) |
| Cell reprogramming | (Zhou et al. 2011, 2012; Kim et al. 2016; Deng et al. 2018; Gaignerie et al. 2018) |
| 3-dimensional culture in Matrigel | (Hynes et al. 2014; Ajzenberg et al. 2015) |
| Gene silencing (siRNA transfection) | (Slaats et al. 2015; Srivastava et al. 2017) |
| Drug testing | (Hynes et al. 2014; Srivastava et al. 2017) |
| Fluorescence-activated cell sorting | (Srivastava et al. 2017) |
4URECs to model renal disease
Kidney biopsy is an extremely invasive procedure and alternative approaches should be favoured, when available, for diagnostic and prognostic purposes. URECs were shown to retain features of kidney disease and can therefore represent a potential alternative to renal biopsies.
Studies of URECs from a cohort of 7 patients with Fabry disease, a hereditary lysosomal storage disorder caused by mutations in the alpha-galactosidase A gene (GLA), showed that these cells were characterised by typical features of the disease, such as decreased alpha-galactosidase A enzyme activity and globotriaosylceramide (Gb3) accumulation. Therefore, URECs may be used as a diagnostic tool and to monitor treatment efficacy in Fabry disease patients, which are often diagnosed late due unspecific symptoms that complicate the clinical evaluation. Moreover, URECs can be used to identify pathways involved in the disease; proteomic analysis of patients’ URECs highlighted for the first time an overall upregulation of lysosomal proteins in Fabry disease.
Proteomic analysis of URECs was also employed to study a novel de-novo p.G195D variant in the ACTN4 gene, in an autosomal dominant familial focal segmental glomerulosclerosis (FSGS) patient. Proteomic analysis revealed pathways potentially altered in the patient cells, including actin cytoskeleton regulation and a destabilization of ACTN4 interactome [22].
URECs have been also extensively employed in the study of renal ciliopathies, a heterogeneous class of disorders characterised by high clinical and phenotypical variability, with no rigorous genotype-phenotype correlation. Individuals affected by the same mutation can display divergent phenotypes, suggesting the involvement of genetic modifiers [23]. Considering the potential importance of genetic background in these disorders, it is essential to study them in a model that is patient-specific.
URECs are typically ciliated cells and will express a single primary cilium of ∼4μm in length [13, 24], following serum starvation for 48 h. These cells are therefore an ideal cell model for the study of renal ciliopathies and the role of cilia in their pathogenesis.
The use of URECs has proven to be particularly efficacious for the study of Joubert syndrome type 5 (JBTS5), a cerebello-oculo-renal ciliopathy caused by mutations in CEP290. Using URECs, it was shown that two different JBTS5 patients displayed abnormally elongated cilia, identifying the elongation of primary cilia as a pathophysiological feature of CEP290-associated JBTS [13, 24]. This was confirmed by studying renal biopsies from patients with CEP290 mutations and a JBTS mouse carrying a Cep290 genetrap mutation, importantly validating the use of URECs as a cellular model for renal ciliopathies [24]. This ciliary phenotype in JBTS5 URECs was in contrast with the phenotype of URECs derived from a patient affected by Leber congenital amaurosis (LCA) secondary to a CEP290 mutation. LCA URECs did not display cilia length abnormality, suggesting that this is a specific feature of JBTS5 with renal phenotypes. Interestingly, the finding of an abnormal URECs ciliary phenotype correlated with the reduction in levels of CEP290 expression [13]. Indeed, URECs can be employed to get information on the expression levels of the gene of interest in the renal tissue. Western blot from URECs lysates revealed that JBTS5 patients, but not LCA patients, had reduced levels of full-length CEP290 protein [13, 14, 24]. The correlation between ciliary abnormalities and levels of CEP290 was recapitulated in wild type URECs by CEP290 silencing; following siRNA mediated knockdown of CEP290, wild type cells displayed abnormally long and disorganised cilia, reminiscent of the phenotype observed in JBTS5 patients’ URECs [13].
The use of URECs to model renal ciliopathies has also contributed to shed light on the pathogenic mechanisms of the renal ciliopathy condition known as nephronophthisis, an autosomal recessive cause of chronic interstitial nephropathy, cystic kidney disease and childhood kidney failure. Recent evidences suggested an association between nephronophthisis (NPHP) and DNA damage response (DDR) signalling [25–28]. In particular, it was shown that RPE and IMCD3 cells depleted for NPHP genes CEP164 and CEP290 accumulate the DNA damage marker phospho-H2AX (γH2AX). Importantly, these results were confirmed by analysing the levels of γH2AX in URECs derived from CEP164 and CEP290 patients, validating replication stress, defects in cell cycle progression and/or DNA damage as a contributing pathomechanism in NPHP kidney degeneration [14, 29].
Moreover, CEP290 URECs displayed an increased activity of cyclin A- and cyclin B-associated kinases, supporting the possible role of cyclin dependent kinases (CDKs) as mediators of replication stress, thus providing possible therapeutic targets for NPHP [14].
5URECs as an ex vivo model to test novel treatments for inherited renal ciliopathies
Treatment with selective inhibitors of cyclin-dependent kinases led to reduced renal cystogenesis in murine models of cystic kidney disease [30–32] and rescued the DNA damage phenotypes in Cep290LacZ/LacZ murine kidney cells. However, the poor translational applicability of therapeutic findings from animal models is a major drawback of such preclinical studies [33, 34]. In addition, the heterogeneity of renal ciliopathies, where the primary genetic lesion can only partially explain the phenotypical features [1, 35], warrants the employment of human, personalised models for drug testing.
Recently, we demonstrated the suitability of URECs as a human, patient-specific model for ex vivo drug testing. We showed that CDK inhibition rescues the elongated ciliary phenotype in URECs derived from a JBTS5 patient. Purmorphamine, a Hedgehog (HH) agonist, similarly leads to a significant reduction in cilia length in JBTS5 URECs. Moreover, purmorphamine treatment restores the ability to form 3-dimensional spheroidal structures in Matrigel in JBTS5 URECs. Interestingly, the observation that purmorphamine reduces CDK5 protein levels in URECs suggests a possible convergence between HH and CDK pathways to promote the ciliary phenotype, providing new mechanist insights for the pathogenesis of renal ciliopathies [13].
Moreover, URECs have proven to be a valuable tool for personalised medicine in renal ciliopathies. In a recent paper, Ramsbottom et al. showed that antisense oligonucleotide-induced splicing of the mutated exon (41, G1890*) restores CEP290 protein expression and rescues increased cilia length phenotype in URECs from a JBTS5 patient, providing a rationale for a genetic therapy approach in the treatment of JBTS [24].
6URECs to validate novel variants in human disease
At least 50% of patients with symptoms attributable to an inherited kidney disease lack a genetic diagnosis. Next-generation sequencing has considerably increased the discovery rate of novel variants. However, functional studies may be required to prove the clinical significance of such variants.
Recently, urine was reported to be a non-invasive source of primary cells to study the pathogenicity of variants detected by genetic testing. Oud et al. employed URECs derived from a patient with mild skeletal abnormalities and vision and hearing impairment to investigate the pathogenicity of two novel variants in IFT140, identified by whole exome sequencing. The IFT140 gene encodes a subunit of the intraflagellar transport complex A (IFTA) that mediates retrograde transport in the cilium. Analysis of ciliary phenotype in patient URECs didn’t show a defect in ciliogenesis or ciliary length, but revealed an accumulation of the IFT-B protein IFT88 at the ciliary tip, indeed suggesting impaired retrograde ciliary transport. These results, together with rescue experiments in Ift140 knock out mouse cells transfected with wild type or mutant IFT140, confirmed a Mainzer–Saldino syndrome diagnosis [36].
Furthermore, URECs have proven to be extremely useful for the validation of variants that affect kidney-specific splicing. Notably, splicing variants can be synonymous gene coding variants that require validation at the RNA level to be taken forward as disease-causing mutations.
We recently utilised URECs to study RNA splicing of NPHP3, a genetic cause of nephronophthisis. A heterozygous synonymous SNV (c.2154 C > T, p.Phe781=) was identified 18 bp from an exon-intron boundary in NPHP3, in combination with a second heterozygous frameshift mutation in two siblings. Biallelic mutations in NPHP3 would be consistent with the patients’ clinical features, suggestive of nephronophthisis. The synonymous variant was predicted within ClinVar as “likely benign”, however in silico tool Human Splicing Finder implicated this variant in aberrant splicing, resulting in mis-splicing of exon 15 of NPHP3. Interestingly, within whole blood RNA, a transcript that included exon 15 was not expressed in healthy controls. URECs RNA was therefore used and allowed the demonstration of normal splicing of exon 15 in healthy controls and alternative splicing with loss of exon 15 of NPHP3 in the presence of the heterozygous synonymous variant [20]. Similar effects of synonymous variants affecting pre-mRNA splicing have been shown in PKD1 [37]. This finding of tissue specific transcripts of genes (such as NPHP3, which is associated with prominent renal phenotypes) highlights the importance of using a relevant source of RNA for assessing putative splicing variants. Whole blood RNA studies may be misleading if the transcript of interest is not expressed or it is expressed as an alternate transcript. Recently, the Genotype-Tissue Expression (GTEx) project has reported variations in levels of expression across 44 human tissues, with the important finding that local genetic variants affect gene expression in the majority of genes [38]. Data of gene expression using normal kidney tissue is under-represented due to the demands for kidney organ donation and transplantation from healthy donors. URECS provide a non-invasive way to explore renal tubular cell tissue expression, and using RNA-seq approaches could help refine wild type and disease specific renal tubular cell tissue expression patterns.
7Concluding remarks
Ciliopathy animal models have been extensively employed to study disease mechanisms, to test novel treatments and to validate novel variants in candidate genes. However, interspecies differences and the poor predictive power of the treatment efficacy when translated to humans limits the translational value of such animal models. Moreover, genetic background is proposed to be a strong contributor to the phenotypic outcome in ciliopathies, warranting the use of patient-specific human models of the disease.
Fibroblasts derived from skin biopsies retain the information related to the patient-specific genetic background but dissection of molecular pathomechanisms is limited by gene expression differences between these cells and the affected tissue. Primary patient material from the affected tissue is often not available, especially in the case of renal disease, where kidney biopsy is an extremely invasive procedure. The use of URECs can overcome these limitations and provide a human tissue-specific and patient-specific model for renal disease. Indeed, URECs have proven to be a valuable platform to dissect molecular mechanisms in renal disease. In particular, they can be interrogated to identify tissue-specific isoforms and assess expression levels for a certain protein of interest in the kidney, holding a great diagnostic and prognostic potential that remains to be fully explored. However, information is lacking regarding the progressive transformation that these cells likely undergo during sub-culture. Full characterisation of heterogeneity of URECs population and gene expression pattern variations during cell culture is warranted to avoid misinterpretation of cell culture artefacts. Furthermore, to date, URECs can be derived only from fresh urine samples, limiting their employment to locally-based patients. In order to expand the cohort of patients from which it is possible to derive URECs, it is important to devise a method to prolong the viability of cells in urine, to allow the shipment of samples from various locations to the processing hub.
Several studies have shown the value of URECs as a platform for disease modelling and drug testing. Further investigation may open up the possibility to routinely use URECs as a non-invasive tool for diagnostic and prognostic applications such as the analysis of biomarkers for kidney disease. The extreme non-invasiveness of URECs derivation and the possibility to reprogram them directly to other cell lineages or to iPSCs also makes them a powerful source of primary patient material whose applicability extends beyond the study of kidney disease.
Acknowledgments
We thank all the patient and family members who contributed to this study. This work is funded by The Medical Research Council (Award MR/M012212/1), Kids Kidney Research, Kidney Research UK and Northern Counties Kidney Research Fund.
References
[1] | D.A. Braun and F. Hildebrandt , Ciliopathies, Cold Spring Harb Perspect Biol. 9: (3) ((2017) ), a028191. |
[2] | J.M. Brown and G.B. Witman , Cilia and diseases, Bioscience 64: (12) ((2014) ), 1126–37. |
[3] | E. Molinari and J.A. Sayer , Emerging treatments and personalised medicine for ciliopathies associated with cystic kidney disease, Expert Opin Orphan Drugs. 5: (10) ((2017) ), 785–98. |
[4] | T. Zhou , C. Benda , S. Dunzinger , Y. Huang , J.C. Ho and J. Yang , et al. Generation of human induced pluripotent stem cells from urine samples, Nat Protoc. 7: (12) ((2012) ), 2080–9. |
[5] | H. Ajzenberg , G.G. Slaats , M.F. Stokman , H.H. Arts , I. Logister , H.Y. Kroes , et al. Non-invasive sources of cells with primary cilia from pediatric and adult patients, Cilia. 4: ((2015) ), 8. |
[6] | H. Rahmoune , P.W. Thompson , J.M. Ward , C.D. Smith , G. Hong and J. Brown , Glucose transporters in human renal proximal tubular cells isolated from the urine of patients with non-insulin-dependent diabetes, Diabetes 54: (12) ((2005) ), 3427–34. |
[7] | G.R. Sutherland and A.D. Bain , Culture of cells from the urine of newborn children, Nature. 239: (5369) ((1972) ), 231. |
[8] | D. Linder , Culture of cells from the urine and bladder washings of adults, Somatic Cell Genet. 2: (3) ((1976) ), 281–3. |
[9] | F. Herz , Culture of urinary cells, Birth Defects Orig Artic Ser. 16: (2) ((1980) ), 85–93. |
[10] | J.S. Felix , T.T. Sun and J.W. Littlefield , Human epithelial cells cultured from urine: Growth properties and keratin staining, In Vitro 16: (10) ((1980) ), 866–74. |
[11] | A. Dörrenhaus , J.I. MÜller , K. Golka , P. Jedrusik , H. Schulze and W. Föllmann , Cultures of exfoliated epithelial cells from different locations of the human urinary tract and the renal tubular system, Arch Toxicol. 74: (10) ((2000) ), 618–26. |
[12] | T. Zhou , C. Benda , S. Duzinger , Y. Huang , X. Li and Y. Li , et al. Generation of induced pluripotent stem cells from urine, J Am Soc Nephrol JASN 22: (7) ((2011) ), 1221–8. |
[13] | S. Srivastava , S.A. Ramsbottom , E. Molinari , S. Alkanderi , A. Filby and K. White , et al. A human patient-derived cellular model of Joubert syndrome reveals ciliary defects which can be rescued with targeted therapies, Hum Mol Genet. 26: (23) ((2017) ), 4657–67. |
[14] | G.G. Slaats , J.C. Saldivar , J. Bacal , M.K. Zeman , A.C. Kile and A.M. Hynes , et al. DNA replication stress underlies renal phenotypes in CEP290-associated Joubert syndrome. J Clin Invest. 125: (9) ((2015) ), 3657–66. |
[15] | A.M. Hynes , R.H. Giles , S. Srivastava , L. Eley , J. Whitehead and M. Danilenko , et al. Murine Joubert syndrome reveals Hedgehog signaling defects as a potential therapeutic target for nephronophthisis, Proc Natl Acad Sci. 111: (27) ((2014) ), 9893–8. |
[16] | E.Y. Kim , P. Page , L.M. Dellefave-Castillo , E.M. McNally , E.J. Wyatt , Direct reprogramming of urine-derived cells with inducible MyoD for modeling human muscle disease, Skelet Muscle. 6: (1) ((2016) ), 32. |
[17] | K. Takahashi , K. Tanabe , M. Ohnuki , M. Narita , T. Ichisaka and K. Tomoda , et al. Induction of pluripotent stem cells from adult human fibroblasts by defined factors, Cell. 131: (5) ((2007) ), 861–72. |
[18] | W-L. Deng , M-L. Gao , X-L. Lei , J-N. Lv , H. Zhao and K-W. He , et al. Gene correction reverses ciliopathy and photoreceptor loss in iPSC-derived retinal organoids from retinitis pigmentosa patients, Stem Cell Rep. 10: (4) ((2018) ), 1267–81. |
[19] | A. Gaignerie , N. Lefort , M. Rousselle , V. Forest-Choquet , L. Flippe , and V. Francois-Campion , et al. Urine-derived cells provide a readily accessible cell type for feeder-free mRNA reprogramming, Sci Rep. 8: (1) ((2018) ), 14363. |
[20] | E. Molinari , E. Decker , H. Mabillard , J. Tellez , S. Srivastava and S. Raman , et al. Human urine-derived renal epithelial cells provide insights into kidney-specific alternate splicing variants, Eur J Hum Genet EJHG. 12: ; (2018) . |
[21] | G.G. Slaats , F. Braun , M. Hoehne , L.E. Frech , L. Blomberg and T. Benzing , et al. Urine-derived cells: a promising diagnostic tool in Fabry disease patients, Sci Rep. 8: (1) ((2018) ), 11042. |
[22] | M.P. Bartram , S. Habbig , C. Pahmeyer , M. Höhne , L.T. Weber and H. Thiele , et al. Three-layered proteomic characterization of a novel ACTN4 mutation unravels its pathogenic potential in FSGS, Hum Mol Genet. 25: (6) ((2016) ), 1152–64. |
[23] | S. Ramsbottom , C. Miles and J. Sayer , Murine Cep290 phenotypes are modified by genetic backgrounds and provide an impetus for investigating disease modifier alleles, F1000Research. 4: ((2015) ), 590. |
[24] | S.A. Ramsbottom , E. Molinari , S. Srivastava , F. Silberman , C. Henry and S. Alkanderi , et al. Targeted exon skipping of a CEP290 mutation rescues Joubert syndrome phenotypes in vitro and in a murine model, Proc Natl Acad Sci U S A. 115: (49) ((2018) ), 12489–94. |
[25] | S. Sivasubramaniam , X. Sun , Y-R. Pan , S. Wang and EY-HP. Lee , Cep164 is a mediator protein required for the maintenance of genomic stability through modulation of MDC1, RPA, and CHK1, Genes Dev. 22: (5) ((2008) ), 587–600. |
[26] | Y-R. Pan and EY-HP. Lee , UV-dependent interaction between Cep164 and XPA mediates localization of Cep164 at sites of DNA damage and UV sensitivity, Cell Cycle Georget Tex. 8: (4) ((2009) ), 655–64. |
[27] | M. Chaki , R. Airik , A.K. Ghosh , R.H. Giles , R. Chen and G.G. Slaats , et al. Exome Capture Reveals ZNF423 and CEP164 Mutations, Linking Renal Ciliopathies to DNA Damage Response Signaling, Cell. 150: (3) ((2012) ), 533–48. |
[28] | H.J.C. Choi , J-R. Lin , J-B. Vannier , G.G. Slaats , A.C. Kile and R.D. Paulsen , et al. NEK8 Links the ATR-Regulated Replication Stress Response and S Phase CDK Activity to Renal Ciliopathies, Mol Cell. 51: (4) ((2013) ), 423–39. |
[29] | G.G. Slaats , A.K. Ghosh , L.L. Falke , S.L. Corre , I.A. Shaltiel and Hoek G van de , et al. Nephronophthisis-Associated CEP164 Regulates Cell Cycle Progression, Apoptosis and Epithelial-to-Mesenchymal Transition, PLOS Genet. 10: (10) ((2014) ), e1004594. |
[30] | N.O. Bukanov , L.A. Smith , K.W. Klinger , S.R. Ledbetter and O. Ibraghimov-Beskrovnaya , Long-lasting arrest of murine polycystic kidney disease with CDK inhibitor roscovitine, Nature. 444: (7121) ((2006) ), 949–52. |
[31] | H. Husson , S. Moreno , L.A. Smith , M.M. Smith , R.J. Russo and R. Pitstick , et al. Reduction of ciliary length through pharmacologic or genetic inhibition of CDK5 attenuates polycystic kidney disease in a model of nephronophthisis, Hum Mol Genet. 25: (11) ((2016) ), 2245–55. |
[32] | N.O. Bukanov , S.E. Moreno , T.A. Natoli , K.A. Rogers , L.A. Smith and S.R. Ledbetter , et al. CDK inhibitors R-roscovitine and S-CR8 effectively block renal and hepatic cystogenesis in an orthologous model of ADPKD, Cell Cycle Georget Tex. 11: (21) ((2012) ), 4040–6. |
[33] | I.W. Mak , N. Evaniew and M. Ghert , Lost in translation: animal models and clinical trials in cancer treatment, Am J Transl Res. 6: (2) ((2014) ), 114–8. |
[34] | T. Ehret , F. Torelli , C. Klotz , A.B. Pedersen and F. Seeber , Translational rodent models for research on parasitic protozoa-a review of confounders and possibilities, Front Cell Infect Microbiol 7: ((2017) ), 238. |
[35] | V.E. Torres , P.C. Harris and Y. Pirson , Autosomal dominant polycystic kidney disease, Lancet Lond Engl. 369: (9569) ((2007) ), 1287–301. |
[36] | M.M. Oud , B.L. Latour , Z. Bakey , S.J. Letteboer , D. Lugtenberg and K.M. Wu , et al. Cellular ciliary phenotyping indicates pathogenicity of novel variants in IFT140 and confirms a Mainzer-Saldino syndrome diagnosis, Cilia. 7: (1) ((2018) ), 1. |
[37] | F. Claverie-Martin , F.J. Gonzalez-Paredes and E. Ramos-Trujillo , Splicing defects caused by exonic mutations in PKD1 as a new mechanism of pathogenesis in autosomal dominant polycystic kidney disease, RNA Biol. 12: (4) ((2015) ), 369–74. |
[38] | GTEx Consortium, Laboratory, Data Analysis & Coordinating Center (LDACC)-Analysis Working Group, Statistical Methods groups-Analysis Working Group, Enhancing GTEx (eGTEx) groups, NIH Common Fund, NIH/NCI, et al. Genetic effects on gene expression across human tissues, Nature. 550: (7675) ((2017) ), 204–13. |


