
Clinical characteristics of individual organ system disease in non-motile ciliopathies
Abstract
Non-motile ciliopathies (disorders of the primary cilia) include autosomal dominant and recessive polycystic kidney diseases, nephronophthisis, as well as multisystem disorders Joubert, Bardet-Biedl, Alström, Meckel-Gruber, oral-facial-digital syndromes, and Jeune chondrodysplasia and other skeletal ciliopathies. Chronic progressive disease of the kidneys, liver, and retina are common features in non-motile ciliopathies. Some ciliopathies also manifest neurological, skeletal, olfactory and auditory defects. Obesity and type 2 diabetes mellitus are characteristic features of Bardet-Biedl and Alström syndromes. Overlapping clinical features and molecular heterogeneity of these ciliopathies render their diagnoses challenging. In this review, we describe the clinical characteristics of individual organ disease for each ciliopathy and provide natural history data on kidney, liver, retinal disease progression and central nervous system function.
1Introduction
Ciliopathies are an expanding group of inherited disorders caused by defects in proteins required for normal structure and function of the cilia [1–4]. Cilia are essential components of almost all cells in the human body. Therefore, ciliary dysfunction can manifest in almost any tissue and result in a wide range of phenotypic consequences varying from single organ diseases to multisystem disorders. The cumulative prevalence of ciliopathies is approximately 1 in every 2000 individuals [1]. These disorders are associated with significant morbidity and mortality. Hence, accurate and timely diagnosis is essential for management and genetic counseling [3].
This review focuses on disorders of the primary (non-motile) cilia. (For a review of diseases caused by defects of the motile cilia, please refer to the article entitled “Primary Ciliary Dyskinesia (PCD): A genetic disorder of motile cilia” by Margaret W. Leigh et al. in this issue). The most commonly affected organ systems in non-motile ciliopathies include the kidneys, liver, brain, and retina [2, 3]. Depending on the nature of the defective protein’s function, the number of organs affected may range from only one to more than five. For example, in autosomal recessive polycystic kidney disease (ARPKD), the disease process is limited to the kidneys and liver because the genetic defect impairs the function of only one ciliary protein in the context of otherwise normal ciliary structure and function [5]. In contrast, multisystem ciliopathies such as Joubert (JS), Meckel-Gruber (MKS), and Bardet-Biedl syndromes (BBS) are caused by defects in proteins with critical roles in ciliary biogenesis or in maintenance of general ciliary structure and function [3].
Four characteristics of multisystem ciliopathies make their diagnosis challenging; 1) Clinical heterogeneity 2) Molecular heterogeneity, 3) Overlap of clinical features 4) Overlap of causative genes. Historically, these multisystem ciliopathies were described based on their most typical set of clinical characteristics, and the etiology of their pleiotropic and overlapping nature was unknown. Now it is known that these disorders are caused by defects in subunit components of multiprotein complexes including large complexes at the ciliary transition zone (Joubert/Meckel-Gruber and nephronophthisis complexes) and the intraflagellar transport machinery (IFT A and B and Bardet-Biedl complexes) [3]. These findings explain some of the heterogeneity and overlap of phenotypes and genotypes in multisystem ciliopathies. Gene-based or gene function-based reclassification of these conditions such as “MKS3-related ciliopathy” [6] or “ciliary gate disorders” [7] may help overcome some of these diagnostic difficulties.
In this review, we describe the clinical characteristics of individual organ disease for each ciliopathy and, when available, provide natural history data on kidney, liver, retinal disease progression and central nervous system function.
2Autosomal recessive polycystic kidney disease (ARPKD)
ARPKD is the most common hepatorenal fibrocystic disease in childhood, with an incidence of approximately 1 in 20,000 live births [5, 8–11]. ARPKD is caused by bi-allelic pathological variants in PKHD1, which encodes fibrocystin, a membrane protein localized to non-motile cilia in addition to other parts of the cell [11]. All ARPKD patients have congenital hepatic fibrosis (CHF) which can also present in other ciliopathies such as JS and MKS (Fig. 1). Diagnostic criteria for ARPKD require absence of any other congenital anomalies such as polydactyly or cerebellar vermis hypoplasia [5, 11]. The severities of kidney and liver disease in ARPKD are independent from one another. More explicitly, patients may present with various combinations of organ system disease such as severe kidney-mild liver, severe liver-mild kidney, severe liver-severe kidney and mild liver-mild kidney [8]. Both kidney and liver disease in ARPKD are progressive in nature [8, 10, 11].
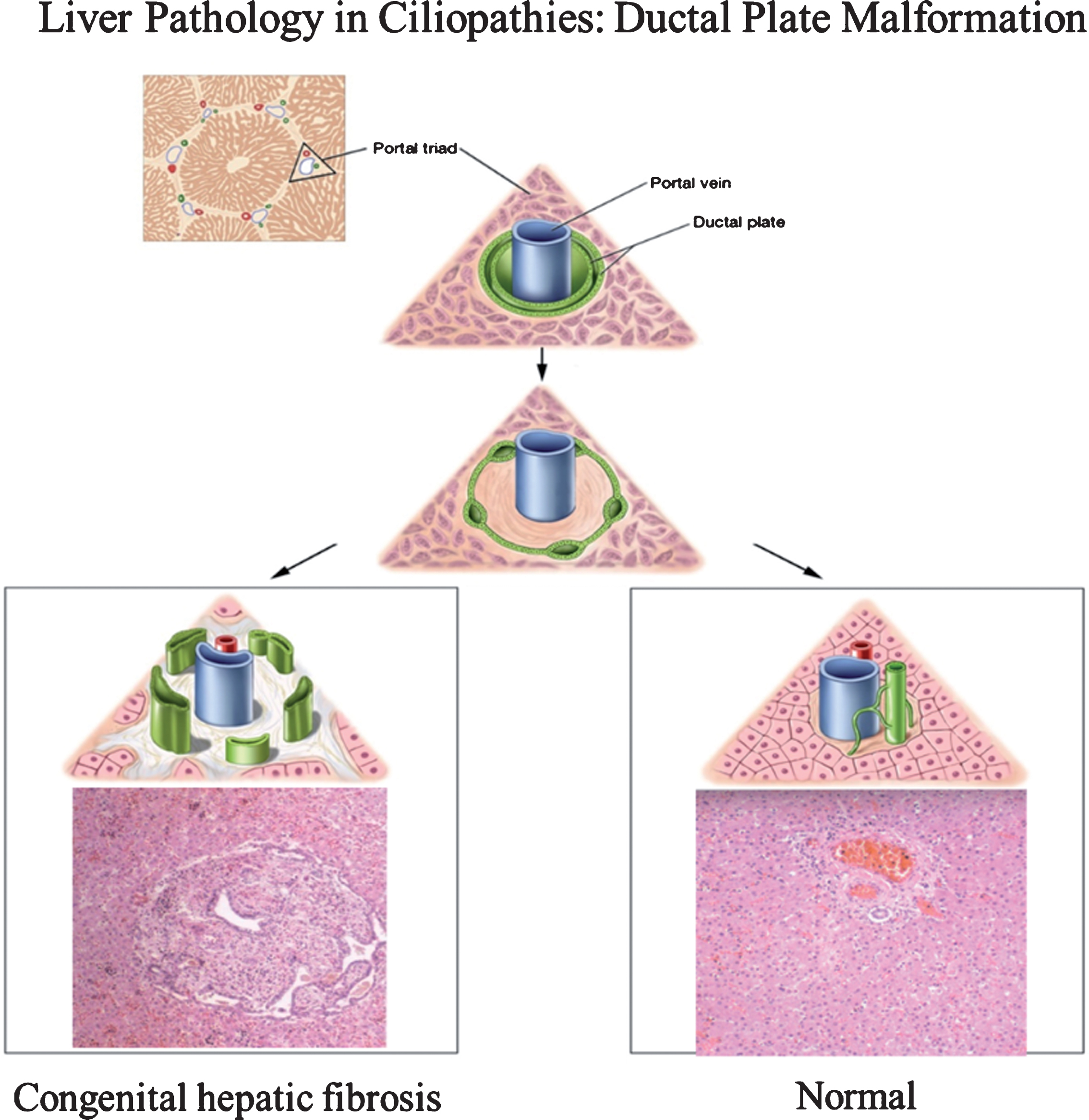
Fig.1
Drawings of ductal plate malformation in comparison to normal bile duct development and corresponding liver histopathology showing congenital hepatic fibrosis (CHF) and normal portal triad. The ductal plate initially forms as a sleeve-like structure around the portal vein branches. Normal remodeling of the ductal plate involves resorption of parts of this structure and migration of the remodeled ducts centrally closer to the portal vein (right panel). The liver biopsy at 10×magnification shows a normal portal tract with sections of portal vein, bile duct, and hepatic artery. Defective remodeling, termed the ductal plate malformation, is characterized by retention of excessive numbers of bile duct remnants in their original peripheral interrupted ring-like position (left panel). The biopsy with CHF shows persistence of bile duct remnants (magnification = 40×). (Liver biopsies are from Potter’s pathology of the fetus, infant and child, 2nd edition, Ed: Gilbert-Barness E. Mosby Elsevier.)
2.1Kidney disease in ARPKD
Kidney pathology in ARPKD is characterized by non-obstructive fusiform dilations of the renal collecting ducts [5, 11]. The severity of kidney disease in ARPKD is variable (Fig. 2A-D). On the most severe end of the spectrum, the cystic pathology affects almost all parts of the renal medulla and cortex (Fig. 2D and E). These patients typically present prenatally or at birth with markedly enlarged kidneys and oligohydramnios [5, 8, 12] (Fig. 3A). On ultrasonography, the kidneys appear diffusely hyperechogenic with loss of the typical corticomedullary distinction, but they preserve their reniform contour [13]. Common features in newborns with ARPKD include anuria/oliguria, hyponatremia and complications of oligohydramnios such as pulmonary hypoplasia [11]. Feeding difficulties are seen frequently because the massive volume of the kidneys leaves little space for the stomach [11, 12]. Most of these newborns develop severe hypertension which requires multiagent therapy[8, 11].
Fig.2
Artist’s rendering, ultrasound, and MRI findings showing the spectrum of kidney abnormalities in ARPKD. Percentages refer to the frequency of each pattern within our study population of 62 clinically and molecularly diagnosed ARPKD patients followed at the NIH Clinical Center [8]. (We had originally published this figure in the Clinical Journal of the American Society of Nephrology [8]; it is republished here with permission). A) Normal-sized kidneys with hyperechogenicity and ductal dilations involving parts of the medulla (white dots on artist’s rendering). B) Mildly enlarged kidneys with hyperechogenicity and ductal dilations involving most of the medulla but sparing the cortex. C) Enlarged kidneys with diffuse hyperechogenicity and ductal dilations sparing only parts of the cortex. Some macrocysts (black) are present. D) Massively enlarged kidneys with complete involvement of medulla and cortex and numerous macrocysts. MRI: magnetic resonance imaging; USG: ultrasonography; HR-USG: high-resolution ultrasonography.
![Artist’s rendering, ultrasound, and MRI findings showing the spectrum of kidney abnormalities in ARPKD. Percentages refer to the frequency of each pattern within our study population of 62 clinically and molecularly diagnosed ARPKD patients followed at the NIH Clinical Center [8]. (We had originally published this figure in the Clinical Journal of the American Society of Nephrology [8]; it is republished here with permission). A) Normal-sized kidneys with hyperechogenicity and ductal dilations involving parts of the medulla (white dots on artist’s rendering). B) Mildly enlarged kidneys with hyperechogenicity and ductal dilations involving most of the medulla but sparing the cortex. C) Enlarged kidneys with diffuse hyperechogenicity and ductal dilations sparing only parts of the cortex. Some macrocysts (black) are present. D) Massively enlarged kidneys with complete involvement of medulla and cortex and numerous macrocysts. MRI: magnetic resonance imaging; USG: ultrasonography; HR-USG: high-resolution ultrasonography.](https://content.iospress.com:443/media/trd/2019/4-1-2/trd-4-1-2-trd190033/trd-4-trd190033-g002.jpg)
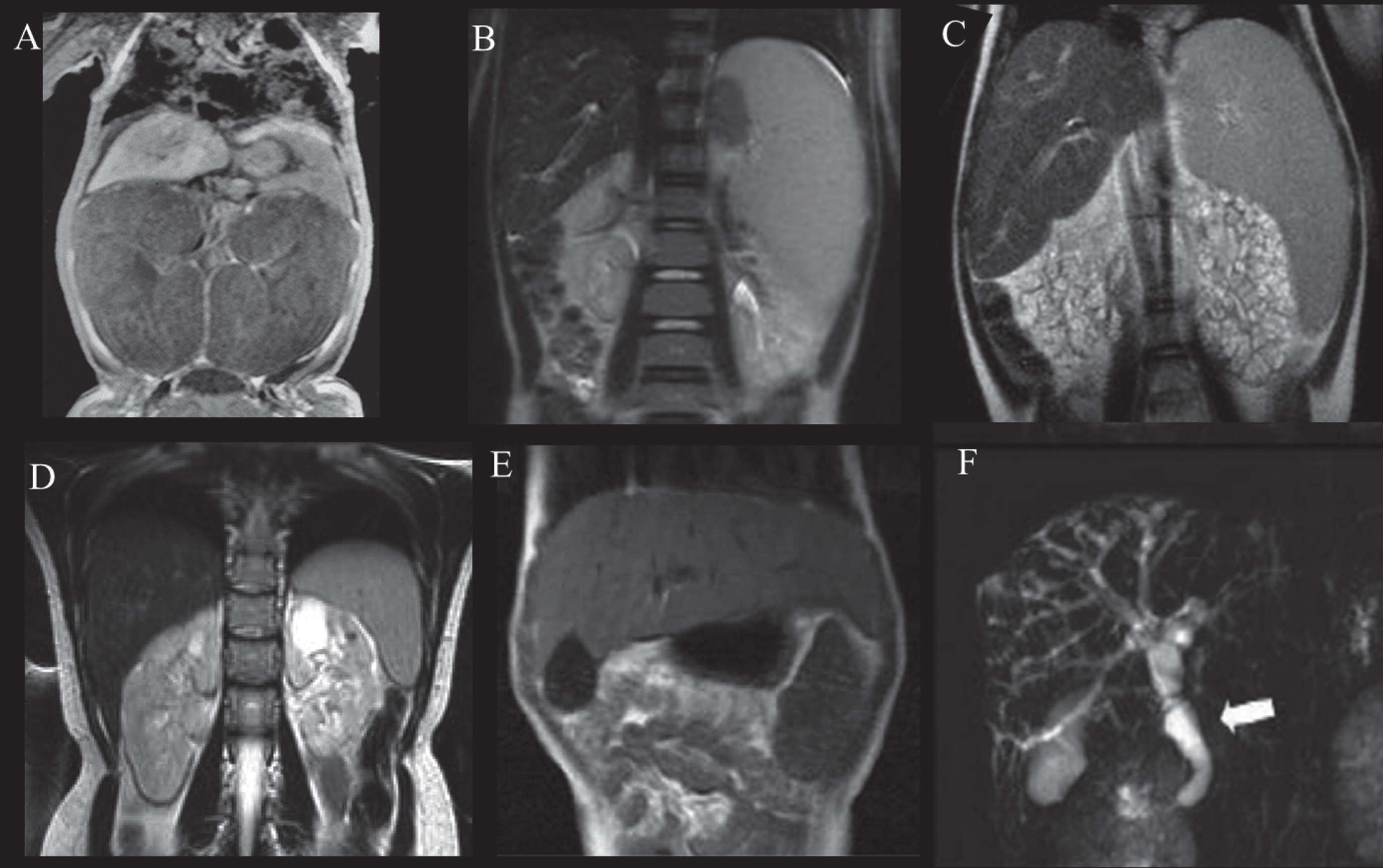
Fig.3
Abdominal MRI of patients with ARPKD displaying variable severities of liver and kidney disease. A) Newborn with ARPKD with massively enlarged kidneys. B) Seven-year-old boy with markedly enlarged spleen and mild kidney disease with imaging findings limited to parts of the medulla, and normal kidney function. C) Six-year-old girl with markedly enlarged spleen and enlarged cystic end stage kidneys with glomerular filtration rate at 22 mL/min/1.73 m2. D) 24-year-old female with mildly enlarged spleen in association with moderate renal disease affecting the entire medulla but only parts of the cortex and decreased glomerular filtration rate at 55 mL/min/1.73 m2. E) Enlarged left lobe of the liver in congenital hepatic fibrosis extending to the left sub-diaphragmatic area. F) ARPKD patient with Caroli’s syndrome. Fusiform and small cystic dilatations of peripheral and central intrahepatic bile ducts as well as fusiform dilatation of the extrahepatic common bile duct (arrow) and large gallbladder.
On the other hand, ARPKD patients at the milder end of the kidney disease spectrum present in childhood or adulthood most commonly with liver-related complications [10, 14] (Fig. 2A and B, Fig. 3B). In these patients, diagnosis of asymptomatic mild kidney involvement usually occurs with screening ultrasonography ordered after the diagnosis of CHF [14]. In most patients with mild disease, the kidneys are normally sized or only mildly enlarged with imaging abnormalities mostly limited to the renal medulla (Fig. 2A and B) [8]. In those with the mildest kidney disease affecting only parts of the medulla, visualization of the abnormalities is only possible with high resolution ultrasonography probes (7–9 MHz); images obtained using standard probes (4 MHz) appear normal (Fig. 2A) [8]. In the majority of the patients at the mildest end of the kidney disease spectrum, glomerular function is normal or only slightly decreased with polyuria and polydipsia due to decreased urinary concentration capacity [8].
Overall, approximately 60–70% of ARPKD patients with perinatal presentation require kidney transplantation by approximately 15 years of age [12, 15]. Correlation of ultrasonography findings with functional data demonstrates that the progression rate of kidney disease is faster in patients with enlarged kidneys with corticomedullary disease in comparison to patients with normally sized or mildly enlarged kidneys without cortical involvement [8]. Perinatally symptomatic patients with markedly enlarged kidneys and corticomedullary disease require renal transplantation significantly earlier (25% require transplantation by age 11) when compared to those with medullary-only disease and only minimal or no cortex involvement (25% require transplantation at age 32) [8].
2.2Liver disease in ARPKD
CHF, the liver pathology in ARPKD, does not primarily affect the hepatocytes; rather, it is a developmental defect of the bile ducts and the portal venous system [9, 16] (Fig. 1). All ARPKD patients have some degree of CHF at least at the microscopic level. Histopathology of CHF is characterized by three features: 1) remnants of embryonic forms of bile ducts due to defective remodeling (ductal plate malformation), often described as “bile duct proliferation” in pathology reports, 2) abnormal branching of the intrahepatic portal vein tree, and 3) portal tract fibrosis which progressively gets worse as patients get older [9, 17] (Fig. 1). A liver with CHF is enlarged and abnormally shaped with a disproportionately large left lobe often palpable under the xiphoid process (Fig. 3E). In newborns and young children with ARPKD, ultrasonography of the liver is typically normal because it takes many years before the microscopic fibrosis in the liver becomes detectable on imaging [10, 13]. In older children and adults, ultrasonography shows increased liver echogenicity and coarsened parenchymal echotexture. In addition to CHF, 70% of ARPKD patients have cystic dilatations of the biliary system (Caroli’s syndrome) with a predisposition to cholangitis (Fig. 3F) [10]. Enlarged common bile duct and enlarged gall bladder are other common biliary abnormalities in ARPKD (Fig. 3F). CHF is characterized by fibrosis, but its natural history is different from cirrhosis because in CHF hepatocellular function is largely spared [9, 18]. The main clinical manifestation of CHF is non-cirrhotic portal hypertension [18, 19]. Portal hypertension in ARPKD is common (65%) and of early onset resulting in splenomegaly in 60% of the patients before 5 years of age [10] (Fig. 3B-C). Presence or severity of portal hypertension does not correlate with kidney function [10]. ARPKD patients develop typical complications of portal hypertension including bleeding from gastroesophageal varices and decreased cell counts due to hypersplenism [10, 18]. In fact, in ARPKD, platelet count is the best predictor of the severity of portal hypertension [10].
Newborns with ARPKD with bi-allelic null mutations with no functional fibrocystin do not survive; at least one missense mutation (compound heterozygous missense and null, or bi-allelic missense) is required for survival [12]. Outside of the correlation of bi-allelic null mutations with neonatal mortality, neither the type (null or missense) nor the location of the PKHD1 mutations explains the variability of disease severity among the neonatal survivors and later onset patients [8]. Variability in disease severity is seen even among siblings within the same family carrying the same PKHD1 mutations, suggesting contribution from modifying genes [8, 20].
3Autosomal dominant polycystic kidney disease (ADPKD)
ADPKD is the most common type of polycystic kidney disease with a prevalence of approximately 1 in 1,000 at birth [21]. It affects approximately 300,000 individuals in the United States. ADPKD is caused by heterozygous pathogenic variants in PKD1 (78%), PKD2 (15%), GANAB (< 1%), or DNAJB11 (<1%). The genetic cause remains unknown in approximately 5% of patients [21, 22]. Approximately 10% of ADPKD is caused by de novo mutations. Polycystic liver disease is the most common extrarenal manifestation [23]. Intracranial vascular aneurysms and cardiovascular manifestations are the most significant extrarenal features [23]. ADPKD patients may also have cysts in other organs including the pancreas, seminal vesicles, and arachnoid membranes [24–26]. Diagnosis is made based on age-specific cyst number criteria [27, 28] and molecular genetic testing [21, 22].
3.1Kidney disease in ADPKD
Manifestations of ADPKD include hypertension, renal pain and a progressive decrease in kidney function resulting in end stage renal disease (ESRD) in 50% of the patients by the age of 60 [21, 29]. Renal pain and an uncomfortable sensation of fullness (due to renal enlargement/anatomical distortion, cyst hemorrhage or infection, and nephrolithiasis) are frequent complaints [30]. Gross hematuria may result from cyst hemorrhage, or nephrolithiasis. Decline in renal glomerular function starts later in the disease process, on average approximately 12 years before reaching ESRD [21]. However, once renal function starts to decline, the glomerular function rate tends to decrease relatively rapidly, at an average rate of 4–6 ml/min/year [31]. PKD1-related ADPKD is associated with more severe disease (mean age at ESRD, 58.1 years) in comparison to PKD2 (79.7 years) [32, 33]. Among patients with the PKD1 genotype, truncating mutations are associated with earlier onset of ESRD (median age at ESRD, 52–55 years) in comparison to non-truncating mutations (median age at ESRD, 66–71 years) [22, 33]. In addition, males with ADPKD have more severe renal disease than females [21]. Differing from ARPKD, cysts in ADPKD can originate from any part of the nephron, and they are true closed cysts that continue increasing in number and in size throughout the lifespan of the patient (Fig. 4). Kidney volume is a good predictor of future decline in kidney function [34]. Notably, when the height-adjusted total kidney volume reaches ≥600 mL/m, there is a strong likelihood that the patient will develop renal insufficiency within 8 years [35].
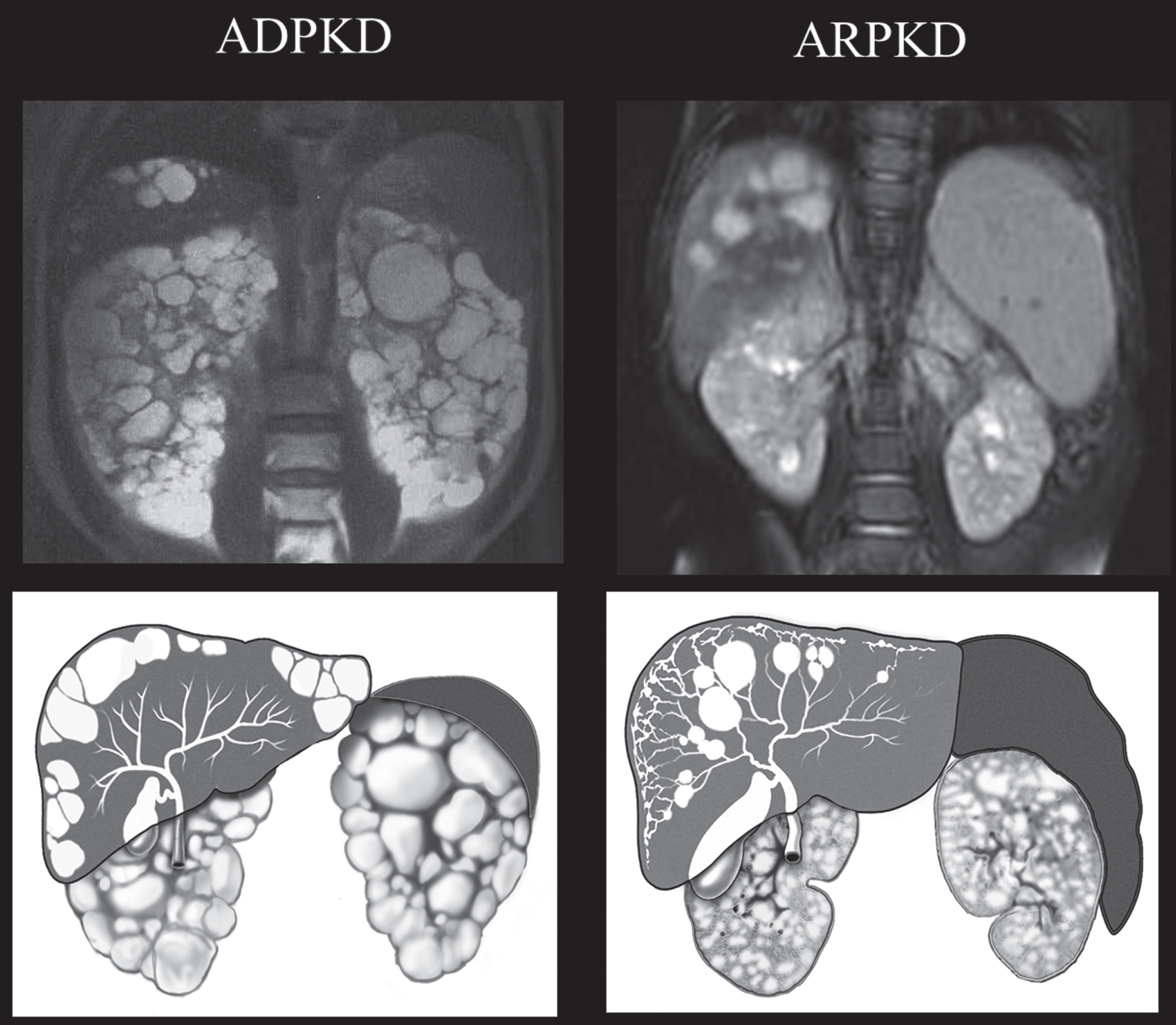
Fig.4
Drawings and MRI images of kidneys, liver, and spleen in ADPKD and ARPKD. In ADPKD portal hypertension is not typical. Therefore, the spleen is not enlarged. Liver cysts in ADPKD are isolated cysts that are not contiguous with the biliary tree. ADPKD kidneys often have distorted contours due to macrocysts. In contrast, in ARPKD, kidneys preserve their reniform contour. CHF in ARPKD is often complicated by portal hypertension evidenced by the enlarged spleen on the MRI. Seventy percent of ARPKD patients also have dilatations of the intra- and extra-hepatic biliary system. (ADPKD MRI is from Heptinstall’s pathology of the kidney, Ed: Jennette J., Olson J., Schwartz M., Silva F.)
Approximately 2–5% of patients with ADPKD present in early childhood or prenatally (<1%) similar to ARPKD with enlarged diffusely echogenic kidneys with or without distinct cysts [36, 37]. The terms “very early onset ADPKD” (diagnosed prenatally or before 18 months) and “early onset ADPKD” (diagnosed between 18 months to 15 years) are suggested for early onset patients. These classifications exclude children diagnosed early based on imaging performed because of family history [22]. Patients with bi-allelic ultra-low penetrance mutations in PKD1 or PKD2 present with very early onset ADPKD [22]. These patients develop ESRD earlier than those with adult onset disease [36].
3.2Liver disease in ADPKD
Unlike the liver cysts in ARPKD/Caroli’s syndrome, liver cysts in ADPKD are closed cysts that are not in continuity with the biliary tree [16] (Fig. 4). In general, they remain asymptomatic; when symptoms do occur, they are typically secondary to the mass effect of the cysts (early satiety, dyspnea, abdominal distention, abdominal pain or back pain) or complications such as a cyst infection, hemorrhage, or rupture [38]. The frequency of liver cysts increases with age and they are uncommon in children. Polycystic liver disease in ADPKD is earlier onset and more severe in females, especially after multiple pregnancies; however, cysts tend to get smaller after menopause [39]. CHF complicated with portal hypertension occurs in very rare ADPKD families [40].
3.3Cardiac and central nervous system involvement in ADPKD
Approximately 10% of patients with ADPKD are diagnosed with cerebrovascular aneurysms [41]. The occurrence is higher (∼22%) in those with a positive family history of intracranial hemorrhage. In ADPKD patients with intracranial aneurysm, the mean age of aneurysm rupture is significantly younger (39 years), than in the general population (51 years) [21, 42]. As for cardiac manifestations in ADPKD, mitral valve prolapse seen in 25% of patients, is the most common finding [43]. ADPKD patients may also exhibit slowly progressive aortic regurgitation in association with aortic root dilation which typically does not require valve replacement [21]. Similar to ARPKD, there is notable variation in the severity of renal disease as well as the other disease manifestations in ADPKD, even in patients within the same family [21].
4Joubert syndrome (JS)
JS is a rare (∼1 in 80,000) multisystem ciliopathy characterized by a distinct set of mid-hind brain anomalies [44]. Similar to other syndromic ciliopathies, JS is genetically heterogeneous with 35 causative genes [44]. The inheritance pattern is autosomal recessive, except in those with JS due to mutations in OFD1, which is X-linked. (For a review of molecular genetics of JS, please refer to the article entitled “The Molecular Genetics of Joubert Syndrome and Related Ciliopathies: The Challenges of Genetic and Phenotypic Heterogeneity” by Melissa Parisi in this issue). Neuroimaging plays an essential role in identifying features of JS required for diagnosis: cerebellar vermis hypoplasia in association with the “molar tooth sign” [45] (Fig. 5A). The “molar tooth sign” is the result of thickened, elongated, and horizontally oriented superior cerebellar peduncles with an abnormally deep interpeduncular fossa. These features are best visualized at the ponto-mesencephalic junction on axial MRI. JS displays marked clinical heterogeneity. By definition, all JS patients have the typical brain abnormalities. Approximately 25% of patients with JS do not develop extra-neurological organ system disease; various combinations of retinal, liver and kidney disease and polydactyly occur in subsets of the remaining patients [46]. This clinical heterogeneity called for the use of the umbrella term “Joubert syndrome and related disorders”, which includes Senior-Løken syndrome (nephronophthisis and retinal degeneration) and COACH (colobomas, oligophrenia, ataxia, cerebellar vermis hypoplasia, and hepatic fibrosis). More recently, for simplicity, the term Joubert syndrome is used to refer to all patients with the “molar tooth sign” including COACH and Senior- Løken syndrome.
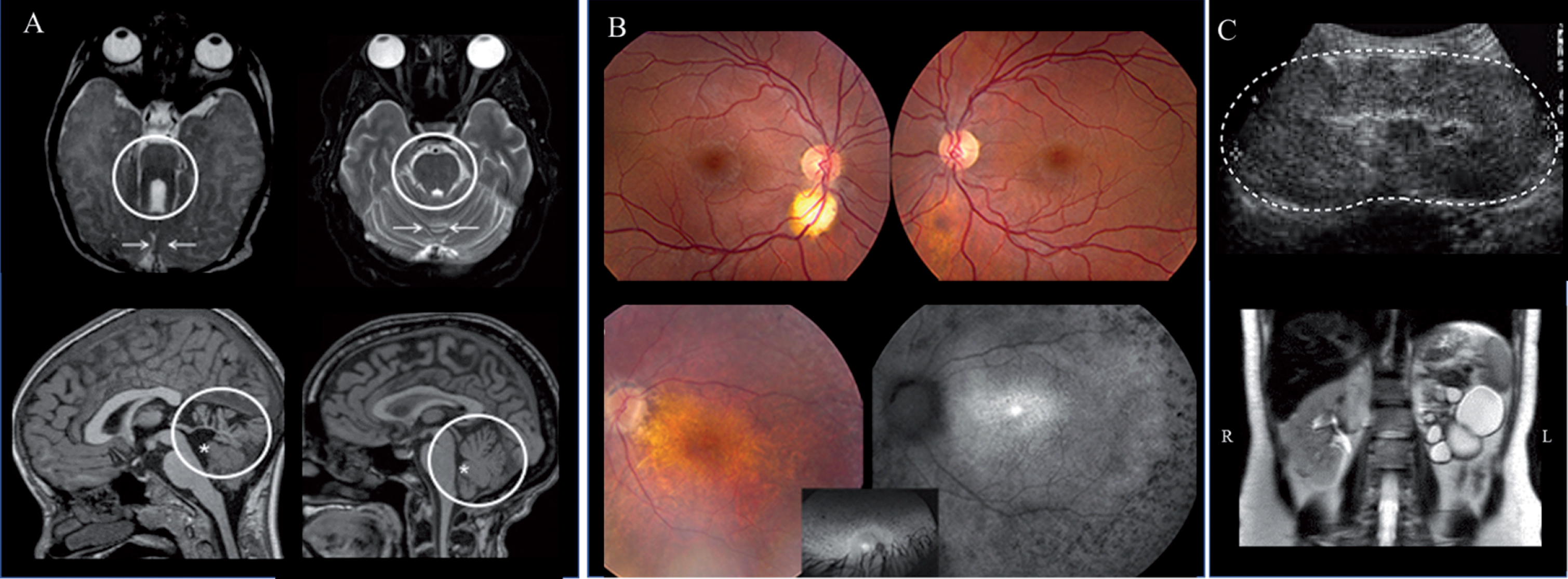
Fig.5
Brain (A), retina (B) and kidney (C) findings in JS. A) Axial brain MRI in Joubert syndrome (JS) (left upper panel) showing the molar tooth sign (circle) and hypoplastic cerebellar vermis (arrows) compared to normal (right upper panel). Hypoplasia and dysplasia of the cerebellar vermis (circle) and enlarged fourth ventricle (asterisk) with rostral displacement of the fastigium in JS (left lower panel) compared to normal (right lower panel). B) A small (left) and forme fruste (right) coloboma in a patient with Joubert syndrome caused by mutations in TMEM67 (upper panel). Advanced retinal degeneration in a 17-year-old patient with JS with AHI1 genotype (lower panel). C) Kidney ultrasonography image of a 4-year-old with TMEM67-related Joubert syndrome showing enlarged and hyperechogenic left kidney (outlined by the dots) with loss of corticomedullary differentiation (upper image). Abdominal MRI of patient with AHI1-related Joubert syndrome demonstrating a left multicystic dysplastic kidney and normal right kidney (lower image).
Almost all patients with JS display hypotonia at birth. Abnormal eye movements and abnormalities in the regulation of respiration manifesting as tachypnea and/or apnea are often noted within the first days of life [44]. Other features recognized within the first years include developmental delays, oculomotor apraxia, speech ataxia, and truncal ataxia [47]. Oculomotor apraxia describes difficulty in moving eyes quickly and smoothly in a desired horizontal direction away from straight-ahead gaze, often compensated by head titubation [47].
4.1Brain and cognitive function in JS
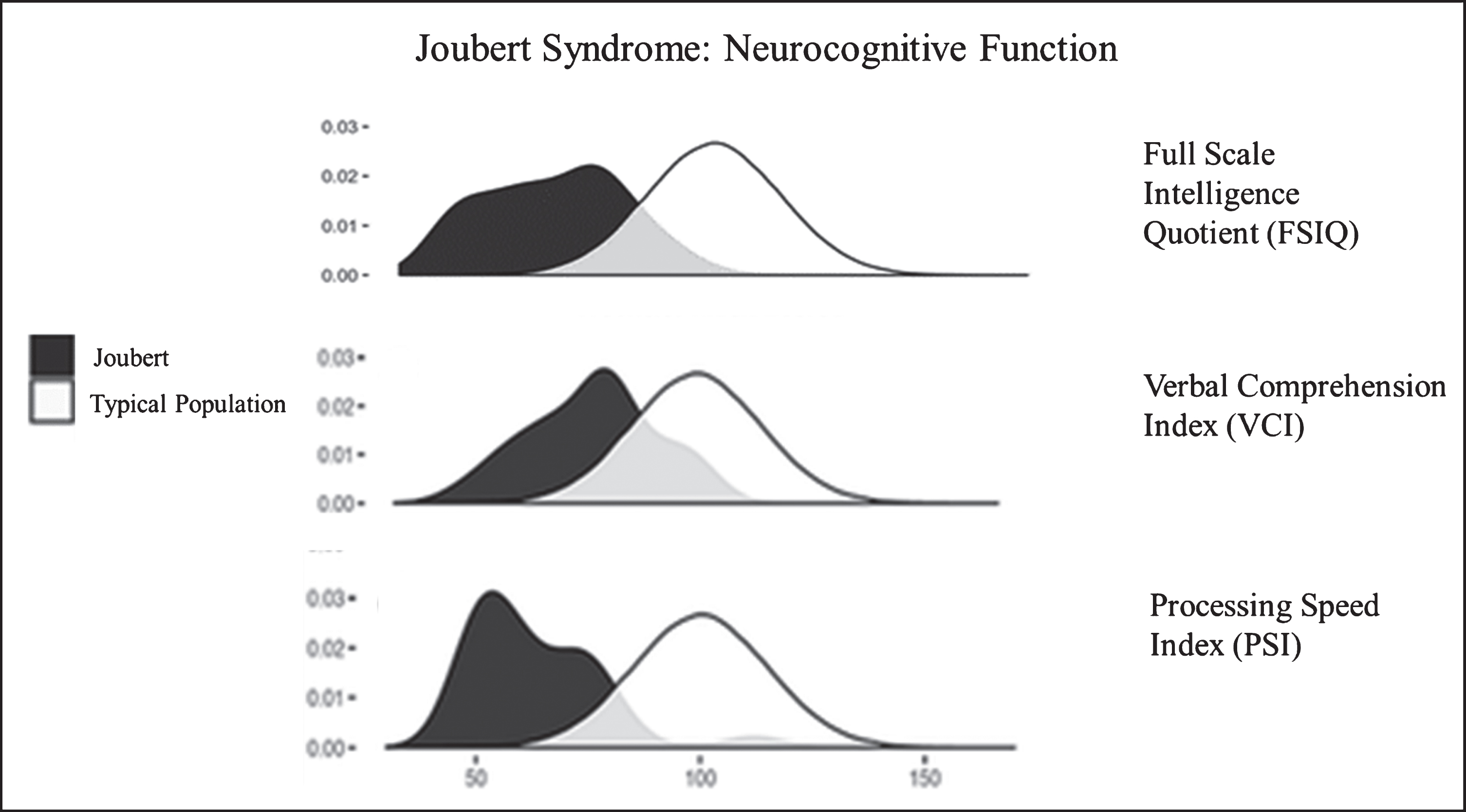
Neurocognitive function in JS varies from extremely low to normal [48, 49]. Approximately two thirds of individuals with JS function in the intellectually disabled range [49]. Relative strengths in JS are verbal comprehension and reasoning abilities while speed of information processing, visual scanning, and visual discrimination are weaker [49] (Fig. 6). Most individuals with JS require special education and benefit from speech, occupational, and physical therapies and augmentative and assistive communication devices [48, 49]. In addition to the molar tooth sign and cerebellar vermis hypoplasia required for diagnosis, some JS patients display other brain anomalies including enlarged cerebellar hemispheres, enlarged posterior fossa (resembling Dandy-Walker malformation), brainstem abnormalities, mild ventriculomegaly (generally does not necessitate shunting), and malrotation of the hippocampi [45]. A systematic re-analysis of the brain MRIs of more than 120 JS patients in the context of neurocognitive function identified cerebellar vermis hypoplasia as the best predictor of neurodevelopmental outcome in JS; the more severe the vermis hypoplasia the more severe the neurocognitive deficits [45]. Seizures occur in a minority (less than 10%) of individuals with JS [49]. Notably, diffuse background slowing in EEG, which is seen more commonly (28%, including those without seizures), is associated with worse cognitive function [49]. In addition, high rates of behavioral/psychiatric concerns are observed in individuals with JS, but these rates are relatively low when compared to other developmentally delayed populations [49].
Fig.6
Neurocognitive function in JS in comparison to the unaffected population. Density plots comparing the bell curves for Full Scale Intelligence Quotient (FSIQ) (upper panel), Verbal Comprehension Index (VCI) (middle panel) and Processing Speed Index (PSI) (lower panel) in JS (dark shaded) and in the unaffected population. Processing speed is a relative weakness, while verbal comprehension and receptive language are relative strengths in JS.
4.2Ophthalmologic disease in JS
Ophthalmologic involvement is very common in JS and varies from relatively mild to severe and is often predictable based on genotype [44, 47]. Oculomotor apraxia (78%), strabismus (72%), nystagmus (67%) and ptosis (31%) are common features observed across most JS genotypes [47]. In addition, subsets of JS patients are affected by chorioretinal colobomas (28%), retinal degeneration (24 %), and less commonly, optic nerve atrophy (9%) [47] (Fig. 5B). Among individuals with JS, coloboma and retinal degeneration appear to be almost mutually exclusive; i.e., JS patients who have a coloboma are less likely to develop retinal degeneration, and colobomas are not observed in JS patients with retinal degeneration [47]. Severe, early-onset retinal degeneration occurs in specific JS genotypes, most notably, in those due to CEP290 and AHI1 mutations(44, 47, 50). On the other hand, severe retinal degeneration is less likely to occur in patients with INPP5E, MKS1, and NPHP1-related JS [44, 47]. Moreover, in JS patients with TMEM67, C5orf52, and KIAA0586 genotypes, no evidence for retinal degeneration has been identified thus far [47]. Among 68 JS patients with quantifiable vision, visual acuity ranged from a Snellen score of 20/20 to 20/632 [47]. The main causes of severe reduction in visual acuity in JS are retinal degeneration and central visual impairment. However, optic atrophy, refractive errors, and amblyopia are also less frequent causes. Colobomas are typically located inferior to the optic nerve and do not impair vision in most patients [47]. Infants and toddlers with JS display delayed visual maturation. Children with JS reach complete visual ability at ages 4–6 in comparison to the normal age of 2-3 [47]. The amblyogenic time frame in children with JS may be notably longer than that of the general population.
4.3Kidney disease in JS
Kidney disease affects 1 in every 3 to 4 patients with JS [46, 50, 51]. Specific types of kidney disease observed in JS include 1) nephronophthisis, 2) ARPKD-like disease associated with severe early onset hypertension and 3) unilateral multicystic dysplastic kidneys (Fig. 5C). Kidney disease is more prevalent in JS due to mutations in CEP290, TMEM67, and AHI1 [51]. On the other hand, kidney disease has not been observed to date in C5orf42 or KIAA0586-related JS [51]. Among JS patients with kidney disease, the genotypes have not been predictive of the severity or progression of renal disease [51]. The rate of decline in kidney function is similar across the genotypes with need for renal transplantation occurring in childhood or early adulthood (n = 13, range 6 to 24 years, mean 11.3±4.8 years) [51]. Prenatal ultrasonography is a poor predictor of kidney disease in JS, appearing normal in 72% of patients diagnosed with renal disease postnatally [51]. In addition, in some patients, fetal ultrasonography findings may not be distinguishable from ARPKD [6]. Therefore, it is important for fetuses found to have hyperechogenic kidneys to have thorough imaging to evaluate for other anatomical anomalies, particularly findings in the brain. Polydactyly occurs in only 13% of patients with JS; hence, in a fetus with ARPKD-like kidneys, absence of extra digits cannot exclude a multisystem ciliopathy such as JS.
4.4Liver disease in JS
Unlike ARPKD, only a proportion of patients with JS display liver disease [46]. Based on the criteria used, the frequency of liver involvement in JS varies between 15 to 45% [52]. Up to 45% of patients display at least one of the following findings; 1) elevated liver enzymes, 2) increased liver echogenicity on ultrasonography, and 3) splenomegaly. However, significant liver disease complicated by portal hypertension occurs in approximately 15% of JS patients overall [52]. Similar to CHF in ARPKD, liver ultrasonography shows hyperechogenicity (except for infants and young children) and liver histopathology is consistent with CHF with ductal plate malformation and portal fibrosis [52]. Most JS patients with significant liver disease have mutations in TMEM67 [52, 53]. The majority of patients with JS due to TMEM67 also have retinal colobomas, and approximately 50% have renal disease [52]. Elevations in liver enzymes including alkaline phosphatase, alanine aminotransferase, aspartate aminotransferase, and gamma-glutamyl transferase (GGT) are common findings in the liver disease of JS with GGT showing the highest relative elevation [52]. Similar to ARPKD, synthetic liver function is preserved. However, in contrast to ARPKD, macroscopic liver cysts or bile duct dilatation (Caroli’s syndrome) are not observed in JS [52].
5Meckel-Gruber syndrome (MKS)
MKS is a severe ciliopathy with an incidence of approximately 1 in 135,000 births worldwide. Higher incidences are observed in Gujarati Indians, Tatars, Hutterites, and Finnish (1 in 9,000 births) and in Saudi Arabia and among Kuwaiti Bedouin tribes (1 in 3,530 births) [54, 55]. MKS is classically characterized by the triad of occipital encephalocele, enlarged cystic kidneys, and postaxial polydactyly (70–80%) [55]. Congenital hepatic fibrosis [56] is another consistent finding [56]. Additional abnormalities, described in 25–40% of cases, include cleft lip and/or palate, congenital heart defects, bowing and shortening of long bones, and abnormal male genitalia [54, 55]. Less frequent (<20%) features include situs defects, cystic dysplasia of the thyroid or lungs, and retinal colobomas [54, 55]. Due to the severity of its features, MKS is typically intrauterine lethal. As expected from its allelic overlap with JS, rare cases of MKS are described to have survived the first few years of life.
5.1Brain abnormalities in MKS
Severity of central nervous system anomalies in MKS varies widely, ranging from total craniorachischisis to partial defects of the corpus callosum [57] and typically include components from 3 categories; 1) prosencephalic dysgenesis (arhinenecephaly-holoprosencephaly, small optic nerves, microphthalmia,) 2) occipital exencephalocele (extrusion through a large posterior fontanelle) 3) rhombic roof dysgenesis with variable severity of posterior fossa anomalies (agenesis/dysgenesis of cerebellar vermis, absent brainstem tectum, Dandy-Walker-like anomalies) [58]. Polymicrogyria and pachygyria are very common.
5.2Kidney disease in MKS
The kidneys are grossly enlarged, resulting in a protuberance of the abdomen [59]. Oligohydramnios is common and often results in Potter’s sequence with the typical secondary facial deformities and multiple joint contractures. Kidney histopathology shows diffuse microcystic changes associated with dysplastic features [59]. However, in contrast to “multicystic dysplastic kidneys” characterized by poorly differentiated and disorganized nephron segments, renal tissue in MKS displays some normal appearing nephrons [59]. Prenatal ultrasonography shows enlarged echogenic kidneys without any demarcation between the medulla and the cortex. The diagnosis of MKS is often made when occipital encephalocele and/or polydactyly are detected in addition to enlarged echogenic kidneys on transabdominal prenatal ultrasonography (at gestational ages of 10–14 weeks). Follow up imaging may include transvaginal ultrasonography and fetal MRI. DNA testing is useful only if it is positive because the currently known genes account for 50–60% of MKS cases.
5.3Liver disease in MKS
CHF is a constant feature of MKS; all patients display ductal plate malformation associated with cystic dilatation of primitive biliary structures, portal fibrosis and an abnormal pattern of portal veins [56].
5.4Retinal disease in MKS
The few publications that describe the retinal findings in MKS report retinal dysplasia. In addition, the MKS mouse model displays markedly abnormal retina [60]. Therefore, retinal dysplasia is probably a typical but under-evaluated pathology in MKS.
6Bardet-Biedl Syndrome (BBS)
Bardet-Biedl syndrome (BBS) is an autosomal recessive ciliopathy characterized by retinal dystrophy, obesity, renal malformations and/or renal parenchymal disease, cognitive impairment, postaxial polydactyly, male hypogonadotropic hypogonadism, and female genitourinary malformations [61, 62]. To date, more than 21 genes are known to cause BBS. BBS affects 1 in 100,000 to 1 in 160,000 North Americans and Europeans. It is more prevalent in Newfoundland (1 in 17,000 newborns) and among the Bedouin population in Kuwait (1 in 13,500 newborns). The diagnosis of BBS is made based on clinical features; either four primary features (retinal dystrophy, obesity, postaxial polydactyly, renal malformations and/or renal parenchymal disease, hypogonadism, and cognitive impairment) or three primary and two secondary features are required. Secondary features include cardiovascular anomalies, anosmia, developmental delay, speech abnormalities, liver disease, subtle craniofacial dysmorphism, Hirschsprung disease, brachydactyly/syndactyly, ataxia/poor coordination/imbalance, oral/dental abnormalities, mild hypertonia, type 2 diabetes mellitus (T2DM), behavioral abnormalities, and eye abnormalities such as strabismus, cataracts, and astigmatism [62]. BBS exhibits variable expressivity and both inter-and intrafamilial phenotypic variation.
6.1Cognitive function in BBS
Cognitive impairment in BBS is mild in the majority of patients [63–65]. Formal neurodevelopmental evaluation of 24 molecularly diagnosed BBS patients showed that the mean intellectual ability of participants fell 1.5 standard deviations below normal expectations [64]. However, only the minority (25–30%) of participants fulfilled the criteria for “intellectual disability”. Verbal fluency was within the average range for 55–60% of participants. However, the majority of individuals with BBS were severely impaired in perceptual reasoning (53%), attentional capacity (69%), and functional independence (74%) [64].
6.2Urogenital anomalies and kidney disease in BBS
Renal disease is a hallmark of BBS [66]. Structural abnormalities of kidneys and urinary tract as well as renal parenchymal disease occur in 53% to 82% of patients [66, 67]. Complex urogenital anomalies in females with BBS include hypoplastic or duplex uterus, septate vagina, hydrometrocolpos, persistent urogenital sinus and vesico-vaginal fistula [61]. On ultrasonography, abnormalities of kidneys or the urinary tract are detected in approximately 50% of BBS patients and include increased echogenicity and/or loss of corticomedullary differentiation (28% of those with abnormal ultrasonography) and cysts ranging from unilateral single cysts to multiple bilateral cystic disease (30%). Developmental abnormalities include horseshoe, ectopic, duplex, and absent kidneys (20%), hydronephrosis (12%) and scarred, atrophic kidneys (10%) [67]. Similar to other ciliopathies, urinary concentration defects are common in BBS. Consistently, histopathology shows nephronophthisis-like tubulointerstitial disease. Dysplastic changes and glomerulosclerosis are reported in some cases [67, 68]. Prenatal ultrasonography is not a good predictor of disease in BBS; it was normal in 36 % of those who had renal anomalies detected postnatally [67]. Advanced chronic kidney disease (stage 2–5) is present in 31% of children and 42% of adults with BBS [67]. The onset of primary renal disease in BBS occurs in infancy. In the majority of pediatric BBS patients with advanced (stage 4-5) chronic kidney disease, the initial diagnosis of renal disease is made within the first year of life and almost all are diagnosed by 5 years of age [67]. BBS patients either develop advanced chronic kidney disease in childhood or maintain normal or almost normal renal function into adulthood [67]. Other manifestations of BBS such as hypertension and T2DM, may also contribute to renal disease. In addition, urological complications including neurogenic bladder, and bladder outflow obstructions, occur in approximately 5–10% of adults with BBS [62, 67]. Patients with mutations in BBS2, BBS10, and BBS12 are more likely to develop severe renal disease in comparison to those with the BBS1 genotype [67]. In addition, bi-allelic truncating mutations and truncating/missense compound heterozygous mutations are more frequent in patients with severe renal disease when compared to those with bi-allelic missense mutations [67]. The most common mutation in BBS1 (M390R) is associated with lower risk of ESRD in comparison to other BBS genotypes. In addition, other urogenital abnormalities are common in BBS including absent urethral orifice, absent vaginal orifice, persistent urogenital sinus, hypoplastic uterus and ovaries, vaginal atresia and hydrometrocolpos in females and hypogonadism in males [61]. Structural renal/urinary tract anomalies do not show association with any specific BBS genotype [67].
6.3Retinal disease in BBS
Retinal cone-rod dystrophy is the most consistent feature in BBS, affecting more than 95% of patients [69, 70]. Most children with BBS first come to medical attention due to retinal dysfunction. Within the first decade of life, patients present with gradual onset of night blindness and decreased peripheral vision, followed by photophobia and loss of central and color vision [71]. Electroretinography is more likely to show significant findings after 5 years of life, although some patients may show early findings within the first two years of life [62]. Although rare patients with milder disease exist, patients with BBS often become legally blind between the second and third decades of life [62]. Other eye abnormalities, including cataracts and strabismus, also occur.
6.4Obesity and endocrine-metabolic abnormalities and liver disease in BBS
Early childhood onset central obesity is another hallmark of BBS [72]. Birth weight is normal. Abnormal weight gain starts within the first year of life. Obesity and its comorbidities including hypertriglyceridemia, hypercholesterolemia, T2DM and metabolic syndrome (defined as having three or more of the following: abdominal obesity, hypertriglyceridemia, low level of high-density lipoprotein (HDL)-cholesterol, hypertension, impaired glucose tolerance) commonly affect children and adults with BBS [72–74]. The exact cause(s) of obesity in BBS is not well understood; multiple factors including hyperphagia; leptin resistance [73]; and lower levels of physical activity [74] may be contributing. Abnormalities of the leptin– melanocortin signaling axis caused by mis-localization of key G-protein-coupled receptors of the ciliary membrane of the hypothalamic neurons are documented in animal models of BBS [75]. Metabolic syndrome, insulin resistance and subsequent T2DM are significantly more common in BBS in comparison to similarly obese individuals without BBS [72].
Primary liver disease in BBS is characterized by the histopathological features in the spectrum of CHF including bile duct abnormalities and periportal fibrosis [76, 77]. However, portal hypertension is uncommon in BBS [78]. In addition, many patients with BBS display fatty liver disease as a part of the typical endocrine-metabolic manifestations of the syndrome [78].
6.5Other findings in BBS
Cardiac abnormalities (including structural heart defects and cardiomyopathy) occur in 7–15% of individuals with BBS [61, 79].
Diverse laterality defects including situs inversus totalis, situs ambiguus, and heterotaxy affect a small percentage of patients with BBS; they occur in 1.6% of individuals with BBS, representing a 170-fold increase in frequency in comparison to individuals without BBS (1.1 per 10,000 livebirths) [80].
7Alström syndrome
Alström syndrome (AS) is a monogenic form of obesity and metabolic syndrome associated with retinal degeneration, hearing loss, cardiomyopathy and progressive liver and kidney disease. AS patients have normal cognitive function [81, 82]. AS is likely underdiagnosed; prevalence estimates range from 1 in 100,000 to 1 in 1,000,000 [81]. It is caused by loss-of-function mutations in ALMS1 which encodes the protein ALMS1 [83]. The subcellular location of ALMS1 includes the cytoplasm, cytoskeleton, microtubule organizing center, centrosomes and ciliary basal bodies, and it is thought to play roles in the formation and maintenance of cilia, cell cycle regulation, and endosomal trafficking [81]. Fibrosis seen in many organs of patients with AS, including heart, lungs, kidneys and liver [82], suggests a common causal relationship, possibly directly related to the absence of the ALMS1 protein or to a secondary chronic inflammatory response to a cellular insult caused by loss of ALMS1function.
7.1Retinal disease in AS
Retinal disease in AS is an early-onset, severe, progressive retinal cone-rod dystrophy that begins in infancy and leads to blindness in childhood [82, 84, 85]. Marked photophobia and horizontal nystagmus develop within the first weeks to months of life. These infants are often misdiagnosed as having “congenital achromatopsia” or “Leber congenital amaurosis”. Given the absence of most other features of AS at this age, the correct diagnosis of AS in this age group can only be made based on molecular genetic testing. Typically, retinal dystrophy progresses relatively rapidly in early childhood, resulting in blindness in the second decade of life, with no light perception by 20 years of age [86].
7.2Cardiomyopathy in AS
Two different forms of myocardial disease occur in AS [82, 87]. A subset of AS patients present in early infancy with severe infantile cardiomyopathy characterized by mitogenic histopathological features including cardiomyocyte hyperplasia and proliferation [88]. This mitogenic cardiomyopathy is caused by abnormal persistence of cardiomyocyte replication because ALMS1 is required for regulation of postnatal cardiomyocyte cell cycle arrest [89]. Many of these infants succumb to congestive heart failure before the diagnosis of AS is made. Hence, sequencing of ALMS1 is warranted in “idiopathic” infantile cardiomyopathy because there may be a larger number of unrecognized patients with AS among these infants who do not recover from cardiomyopathy. Interestingly, in those infants who survive, myocardial function recovers to low-normal within a few years. This acute mitogenic cardiomyopathy does not re-occur. The second form of cardiomyopathy in AS is chronic restrictive in nature and characterized by slowly progressive fibrosis of the myocardium [90, 91]. Almost all AS patients develop some degree of this slow restrictive cardiomyopathy [87, 92].
7.3Obesity, Endocrine-Metabolic Abnormalities and Liver Disease in AS
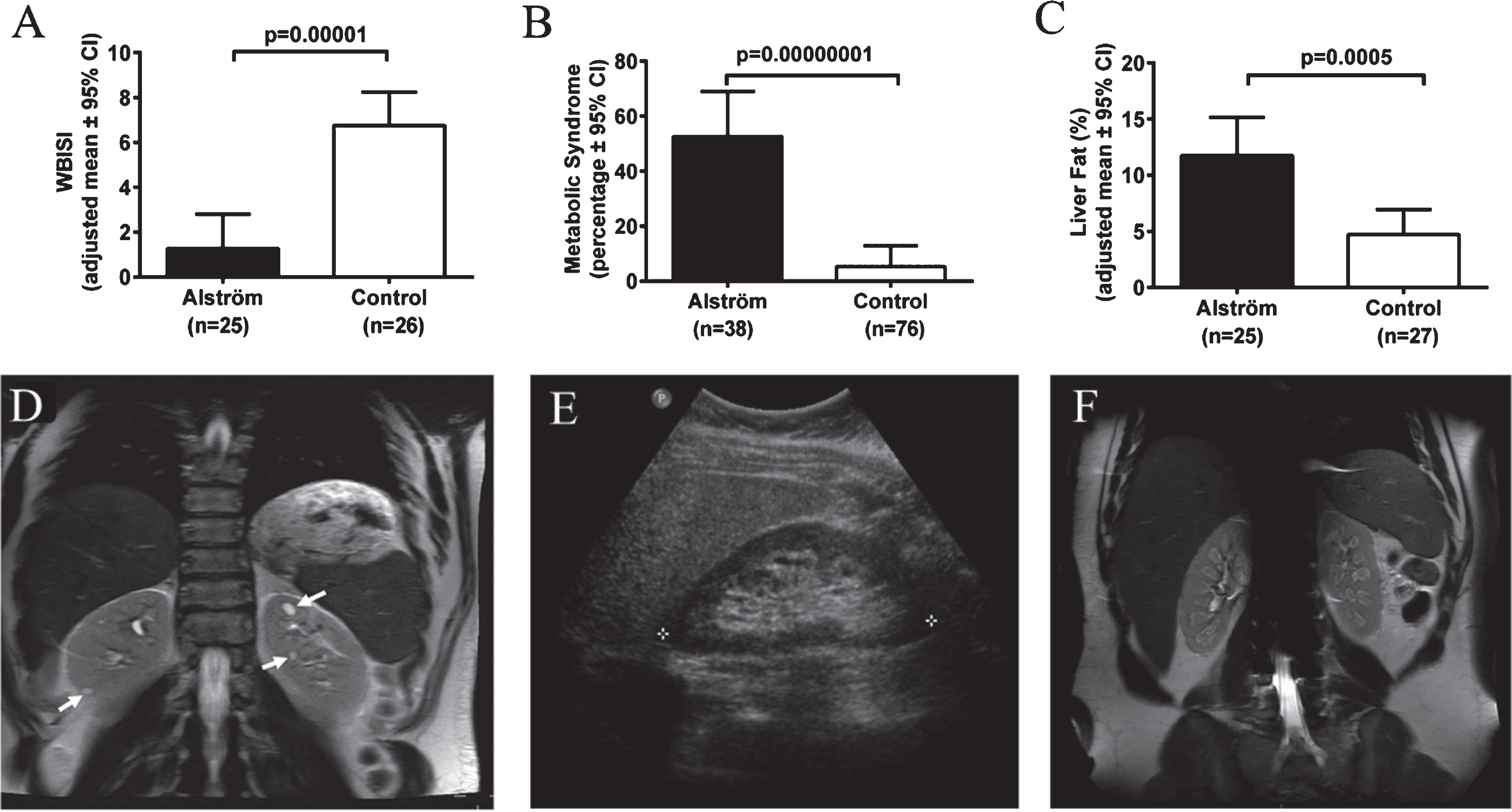
Severe insulin resistance, T2DM and metabolic syndrome are the hallmarks of AS [93, 94]. In most children with AS, obesity and insulin resistance begin to develop during the first year [85, 93, 95]. More than two thirds of children and 85% of adults with AS are obese [93]. In comparison to similarly obese controls, patients with AS have significantly greater insulin resistance indices [93] (Fig. 7A), consistent with impaired insulin-induced trafficking of the insulin receptor GLUT4 to the plasma membrane in cells lacking ALMS1 [96]. Insulin resistance, present in approximately 60% of children and 100% of adults with AS, progresses to T2DM in approximately 20% of children and 70% of adults [93]. Marked hypertriglyceridemia is typical and it is complicated by pancreatitis in some cases. In comparison to equally obese controls, patients with AS are 10 times more likely to develop metabolic syndrome (Fig. 7B). Hepatic steatosis is almost universal [93, 94]. Portal hypertension occurs in some adults. While they have similar percentage of total body fat to equally obese controls, AS patients have significantly higher liver fat, suggesting a direct role of loss of ALMS1 function in the pathogenesis of liver disease in AS [93, 94] (Fig. 7C). Other endocrine abnormalities frequently observed in AS include adult hypogonadism, hypothyroidism, and female hyperandrogenism [93].
Fig.7
Endocrine-metabolic abnormalities and kidney findings in Alstrom syndrome (AS). A) Patients with AS have markedly lower insulin sensitivity in comparison to body mass index-matched controls. WBISI: whole-body insulin sensitivity index. B) Patients with AS are at increased risk for metabolic syndrome in comparison to body mass index-matched controls. Metabolic syndrome is defined as three or more of the following: abdominal obesity, hypertriglyceridemia, low HDL-cholesterol, elevated BP, or impaired glucose tolerance. C) Patients with AS have higher liver fat compared to equally obese controls. D) Abdominal MRI of 34-year-old male patient with AS showing multiple discrete cysts (arrows). E) Ultrasonography image of right kidney of 35-year-old female with AS showing hyperechogenicity of renal medulla. F) Abdominal MRI of 18-year-old male patient with AS showing hyperintensities in the medulla of both kidneys.
7.4Kidney disease in AS
Chronic progressive nephropathy is one of the primary manifestations of AS [97–99]. Renal failure was found to be the cause of death in 23% of AS patients between the ages of 22 and 48 years [82]. Nephropathy in AS is early onset; decline in glomerular function may start as early as 10 years [98]. In adults with AS, renal function declines rapidly, at an average rate of 17 mL/min/1.73 m2 per decade in males, and 11 mL/min/1.73 m2 per decade in females [98]. Approximately two thirds of adults with AS (median age 22 years) meet the criteria for stage 3 or worse chronic kidney disease based on eGFR <60 mL/min/1.73 m2 or evidence of albuminuria [98]. Renal histopathology shows interstitial fibrosis and tubular atrophy associated with varying degrees of glomerular sclerosis but no features of diabetic or reflux nephropathy [82, 98, 100, 101]. On imaging, solitary renal cysts are observed in less than 10% of AS patients (Fig. 7D). The most frequent renal imaging finding is parenchymal hyperechogenicity which is limited to the medulla in most patients (Fig. 7E and 7F) [98, 99]. This hyperechogenicity in AS, is not true “nephrocalcinosis” because parathyroid hormone, serum calcium, phosphorus, and vitamin D levels and urinary calcium excretion are normal in AS [99]. Interestingly, hyperuricemia is documented in approximately 80% of patients with AS, a finding which may be causally related to the medullary hyperechonegicity [98]. In addition to the lack of histopathological features of diabetic or reflux nephropathy, kidney dysfunction in AS does not show a significant association with T2DM, dyslipidemia, hypertension, cardiomyopathy or portal hypertension, suggesting that kidney disease is a primary manifestation of the syndrome due to lack of ALMS1 protein [98, 99]. However, it is possible that diabetes and hypertension have an additive effect on renal disease progression in AS. In addition, lower urinary tract symptoms such as urgency, intermittency, straining to start urination, and incomplete bladder emptying are seen in approximately 70% of AS patients [98]. These urological findings do not correlate with renal function [82, 98].
7.5Hearing loss in AS
Hearing loss in AS is preceded by retinal dystrophy in all cases [95, 102]. The majority of newborns with AS pass the newborn hearing screening [102]. The rate of progression of hearing loss in AS is estimated at approximately 10–15 dB per decade [102]. Absent otoacoustic emissions, intact speech discrimination, and disproportionately normal auditory brainstem responses map the auditory defect in AS to the outer hair cells of the cochlea. Therefore, AS patients are good candidates for traditional aural amplification or, in cases of severe to profound hearing loss, cochlear implantation [102].
7.6Respiratory disease in AS
Recurrent respiratory infections and fibrosis on lung biopsies are reported in AS [82]. Otitis media is very common among children with AS (92%), with 50% requiring pressure equalization tube placement [103]. A history of bronchitis/pneumonia and sinusitis is reported in 61% and 50% of individuals, respectively. These reports of frequent infections raise the possibility of dysfunction of the respiratory motile cilia in AS. However, characteristic manifestations of primary ciliary dyskinesia (laterality defects, unexplained neonatal respiratory distress, year-round nasal congestion, and wet cough) are far less prevalent in AS. Furthermore, decreased nasal nitric production which is a characteristic of primary ciliary dyskinesia, is not observed in AS [103]. Hence, the oto-sino-respiratory complications in AS are prominent enough to warrant increased clinical attention, but patients with AS are unlikely to have significantly impaired respiratory cilia function as seen in primary ciliary dyskinesias [103].
8Oral-facial-digital syndrome type 1 (OFD-1)
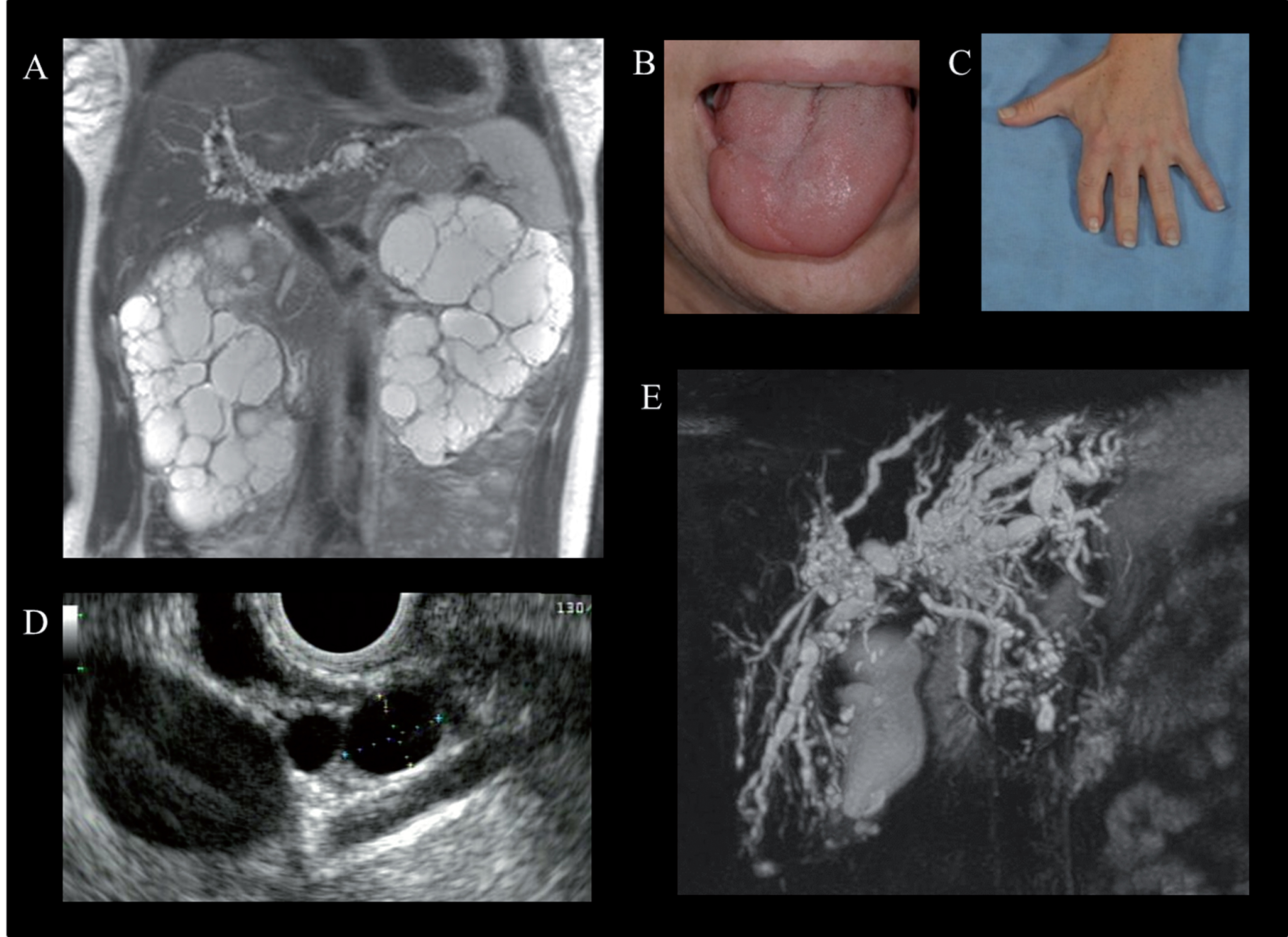
Oral-facial-digital syndrome type 1 (OFD-1) is the most common of the OFD syndromes, a heterogeneous group of disorders characterized by abnormalities of the oral cavity, face and digits [104]. OFD-1 displays an X-linked dominant pattern of inheritance with embryonic male lethality [104]. OFD1, the gene mutated in OFD-1, encodes a protein that localizes to the centrosome and basal body of primary cilia [105]. External findings include hypertelorism, facial asymmetry, hypoplasia of the alae nasi, median cleft or pseudo-cleft of the upper lip, oral clefts and abnormal frenulae, tongue anomalies (cysts, clefts, hamartomas), anomalous dentition, and brachydactyly and/or syndactyly of the fingers and toes (Fig. 8B and C) [104]. Some degree of intellectual disability is observed in approximately 50% of patients. In addition, structural central nervous system anomalies such as agenesis of the corpus callosum, cerebellar agenesis, or a Dandy-Walker-like malformation occur in some patients.
Fig.8
Physical examination and imaging findings in Oral-facial-digital syndrome type 1 (OFD-1). A) Abdominal MRI of a 37-year-old female with OFD-1 showing enlarged polycystic kidneys and beaded dilatations of the intrahepatic bile ducts. Cleft tongue (B) and brachydactyly and repaired syndactyly (C). Endoscopic ultrasound showing multiple pancreatic cysts (D) (courtesy of Dr. Phuong Nguyen, Hoag Memorial Hospital, Newport Beach, CA) and magnetic resonance cholangiopancreatography demonstrating cystic dilatations of the intrahepatic bile ducts (E) in a 38-year-old female with OFD-1.
8.1Kidney disease in OFD-1
Polycystic kidney disease is a hallmark visceral manifestation in OFD-1 [106–108] (Fig. 8A). Therefore, patients with OFD-1 should be routinely evaluated for kidney disease. However, in approximately 25% of affected females, the diagnosis of OFD-1 is made late after that of polycystic kidney disease [108]. In fact, some patients with OFD-1 are misdiagnosed as having ADPKD because craniofacial and digital features may be quite subtle. Two features that are potentially helpful to distinguish X-linked OFD-1-related PKD from ADPKD are the absence of affected males in multiple generations and imaging findings of preserved renal contour in OFD-1 in contrast to the distorted renal contour in ADPKD. Renal cysts in OFD-1 are derived from the glomeruli [107]. Typically, renal imaging in OFD-1 shows mild to moderately enlarged kidneys with findings of numerous cysts (from a few mm to 4-5 cm in diameter) involving the cortex and medulla [107]. The rate of kidney function decline in OFD-1 is faster than ADPKD, with the mean age at ESRD in patients with OFD-1 being approximately 36 years (range:11 to 64 years) [107, 108]. In addition, the typical complications of kidney cysts in ADPKD, including lumbar pain, lithiasis, and hematuria are not common in OFD-I [107].
8.2Liver and pancreas involvement in OFD-1
In addition to PKD, OFD-1 is also associated with fibrocystic disease of the liver and pancreas, which are other under-evaluated and under-diagnosed manifestations of this disorder [108] (Fig. 8D and E). Multiple hepatic cysts and dilated intrahepatic bile ducts occur in 45% and 25% of patients with ODF-1, respectively [108] (Fig. 8E). Multiple pancreatic cysts were identified in approximately 30% (Fig. 8D).
9Skeletal ciliopathies
Skeletal ciliopathies are a group of disorders caused by defects in more than 20 genes which encode components of the ciliary transport machinery, most commonly the retrograde IFT complexes (IFT-A complex and dynein 2 complex motor) which bring cargo from the tip of the cilium back to the cytoplasm [109]. Specific disorders in this group include Jeune chondrodysplasia, cranioectodermal dysplasia [110] and various types of short rib-polydactyly syndromes. Characteristic features include abnormal bone growth that results in a narrow chest due to short ribs, short stature associated with disproportionately short limbs and digit patterning defects that lead to polydactyly [109, 110]. Variable non-skeletal features include nephronophthisis, cystic kidney disease, sclerosing glomerulopathy, CHF, retinal degeneration, cerebellar abnormalities, cardiac anomalies and cleft lip/palate and other oral defects [112]. Other rare features may include fibrocystic changes in the pancreas, ambiguous genitalia, anal atresia, polyhydramnios, malrotation, and hydrops fetalis [112]. Many of these skeletal ciliopathies have severe forms associated with perinatal lethality due to pulmonary insufficiency [112].
10Treatment of non-motile ciliopathies
Currently no specific therapy exists for ciliopathies; symptoms are treated by standard therapies as in the general population. However, remarkable progress has been made in understanding the molecular pathophysiology underlying the retinopathy, renal and hepatic fibrocystic disease and the endocrine-metabolic manifestations of ciliopathies. While malformations in the brain and skeletal system are determined prenatally, the disease process in the retina, liver, kidneys and the endocrine-metabolic system progresses as the patients get older, providing a window of opportunity for treatments that may ameliorate or reverse the course of disease. Targeted drugs and gene-based therapies such as gene replacement, exon skipping, readthrough and gene editing are being developed for specific aspects of ciliopathies including retinopathy [111, 112], polycystic kidney disease [113, 114], and obesity [115]. In this issue, “Retinal disease in ciliopathies: recent advances with a focus on stem cell-based therapies review” by Chen, H et al., “Novel Treatments for Polycystic Kidney Disease” by Patil, A et al., and “Using human urine-derived renal epithelial cells to model kidney disease in inherited ciliopathies” by Sayer, J and Molinari, E., focus on organ-specific targeted therapeutic approaches to ciliopathies.
References
[1] | Kagan K.O. , Dufke A. and Gembruch U. , Renal cystic disease and associated ciliopathies, Curr Opin Obstet Gynecol 29: (2) ((2017) ), 85–94. |
[2] | Braun D.A. and Ciliopathies H.F. , Cold spring harb perspect biol, 9: (3) ((2017) ). |
[3] | Mitchison H.M. and Valente E.M. , Motile and non-motile cilia in human pathology: From function to phenotypes, J Pathol 241: (2) ((2017) ), 294–309. |
[4] | Shapiro A.J. , Davis S.D. , Polineni D. , Manion M. , Rosenfeld M. , Dell S.D. , et al., Diagnosis of primary ciliary dyskinesia. An official american thoracic society clinical practice guideline. Am J Respir Crit Care Med 197: (12) ((2018) ), e24–e39. |
[5] | Zerres K. , Volpel M.C. and Weiss H. , Cystic kidneys, Genetics, pathologic anatomy, clinical picture, and prenatal diagnosis, Human genetics 68: (2) ((1984) ), 104–135. |
[6] | Gunay-Aygun M. , Parisi M.A. , Doherty D. , Tuchman M. , Tsilou E. , Kleiner D.E. , et al. MKS3-related ciliopathy with features of autosomal recessive polycystic kidney disease, nephronophthisis, and Joubert Syndrome, J Pediatr 155: (3) ((2009) ), 386–392el. |
[7] | Li C. , Jensen V.L. , Park K. , Kennedy J. , Garcia-Gonzalo F.R. , Romani M. , et al. MKS5 and CEP290 dependent assembly pathway of the ciliary transition zone, PLoS Biol 14: (3) ((2016) ), e1002416. |
[8] | Gunay-Aygun M. , Font-Montgomery E. , Lukose L. , Tuchman M. , Graf J. , Bryant J.C. , et al., Correlation of kidney function, volume and imaging findings, and PKHD1 mutations in 73 patients with autosomal recessive polycystic kidney disease, Clin J Am Soc Nephrol 5: (6) ((2010) ), 972–984. |
[9] | Gunay-Aygun M. , Gahl W.A. and Heller T. Congenital Hepatic Fibrosis Overview. In: Adam M.P. , Ardinger H.H. , Pagon R.A. , Wallace S.E. , Bean L.J.H. , Stephens K. , et al., editors. GeneReviews((R)). Seattle (WA) (1993) . |
[10] | Gunay-Aygun M. , Font-Montgomery E. , Lukose L. , Tuchman M. , Piwnica-Worms K. , Choyke P. ,et al. Characteristics of congenital hepatic fibrosis in a large cohort of patients with autosomal recessive polycystic kidney disease, Gastroenterology 144: (1) ((2013) ), 112–21 e2. |
[11] | Sweeney W.E. and Avner E.D. , Polycystic Kidney Disease, Autosomal Recessive.In: Adam M.P. , Ardinger H.H. , Pagon R.A. , Wallace S.E. , Bean L.J.H. , Stephens K. , et al., editors. GeneReviews((R)). Seattle WA (1993) . |
[12] | Bergmann C. , Senderek J. , Windelen E. , Kupper F. , Middeldorf I. , Schneider F. , et al., Clinical consequences of PKHD1 mutations in 164 patients with autosomal-recessive polycystic kidney disease (ARPKD), Kidney Int 67: (3) ((2005) ), 829–848. |
[13] | Turkbey B. , Ocak I. , Daryanani K. , Font-Montgomery E. , Lukose L. , Bryant J. , et al., Autosomal recessive polycystic kidney disease and congenital hepatic fibrosis (ARPKD/CHF), Pediatr Radiol 39: (2) ((2009) ), 100–111. |
[14] | Adeva M. , El-Youssef M. , Rossetti S. , Kamath P.S. , Kubly V. , Consugar M.B. , et al., Clinical and molecular characterization defines a broadened spectrum of autosomal recessive polycystic kidney disease (ARPKD), Medicine 85: (1) ((2006) ), 1–21. |
[15] | Roy S. , Dillon M.J. , Trompeter R.S. and Barratt T.M. , Autosomal recessive polycystic kidney disease: Long-term outcome of neonatal survivors, Pediatr Nephrol 11: (3) ((1997) ), 302–306. |
[16] | Gunay-Aygun M. , Liver and kidney disease in ciliopathies, Am J Med Genet C Semin Med Genet 151C: (4) ((2009) ), 296–306. |
[17] | Desmet V.J. , Ludwig symposium on biliary disorders–part I, Pathogenesis of ductal plate abnormalities, Mayo Clin Proc 73: (1) ((1998) ), 80–89. |
[18] | Srinath A. and Shneider B.L. , Congenital hepatic fibrosis and autosomal recessive polycystic kidney disease, J Pediatr Gastroenterol Nutr 54: (5) ((2012) ), 580–587. |
[19] | Capisonda R. , Phan V. , Traubuci J. , Daneman A. , Balfe J.W. and Guay-Woodford L.M. , Autosomal recessive polycystic kidney disease: Outcomes from a single-center experience, Pediatr Nephrol 18: (2) ((2003) ), 119–126. |
[20] | Gunay-Aygun M. , Tuchman M. , Font-Montgomery E. , Lukose L. , Edwards H. , Garcia A. , et al., PKHD1 sequence variations in 78 children and adults with autosomal recessive polycystic kidney disease and congenital hepatic fibrosis, Mol Genet Metab 99: (2) ((2010) ), 160–173. |
[21] | Harris P.C. , Torres V.E. , Polycystic Kidney Disease, Autosomal Dominant. In: Adam M.p. , Ardinger H.H. , Pagon R.A. , Wallace S.E. , Bean L.J.H. , Stephens K. , et al., editors. GeneReviews((R)). Seattle WA, (1993) . |
[22] | Cornec-Le Gall E. , Torres V.E. and Harris P.C. , Genetic complexity of autosomal dominant polycystic kidney and liver diseases, J Am Soc Nephrol 29: (1) ((2018) ), 13–23. |
[23] | Pirson Y. , Extrarenal manifestations of autosomal dominant polycystic kidney disease, Adv Chronic Kidney Dis 17: (2) ((2010) ), 173–180. |
[24] | Torra R. , Sarquella J. , Calabia J. , Marti J. , Ars E. , Fernandez-Llama P. , et al., Prevalence of cysts in seminal tract and abnormal semen parameters in patients with autosomal dominant polycystic kidney disease, Clin J Am Soc Nephrol 3: (3) ((2008) ), 790–793. |
[25] | Danaci M. , Akpolat T. , Bastemir M. , Sarikaya S. , Akan H. , Selcuk M.B. , et al., The prevalence of seminal vesicle cysts in autosomal dominant polycystic kidney disease, Nephrol Dial Transplant 13: (11) ((1998) ), 2825–2828. |
[26] | Basar O. , Ibis M. , Ucar E. , Ertugrul I. , Yolcu O.F. , Koklu S. , et al., Recurrent pancreatitis in a patient with autosomal-dominant polycystic kidney disease, Pancreatology 6: (1-2) ((2006) ), 160–162. |
[27] | Pei Y. , Obaji J. , Dupuis A. , Paterson A.D. , Magistroni R. , Dicks E. , et al., Unified criteria for ultrasonographic diagnosis of ADPKD, J Am Soc Nephrol 20: (1) ((2009) ), 205–212. |
[28] | Pei Y. , Hwang Y.H. , Conklin J. , Sundsbak J.L. , Heyer C.M. , Chan W. , et al., Imaging-based diagnosis of autosomal dominant polycystic kidney disease, J Am Soc Nephrol 26: (3) ((2015) ), 746–753. |
[29] | Gabow P.A. , Johnson A.M. , Kaehny W.D. , Kimberling W.J. , Lezotte D.C. , Duley I.T. , et al., Factors affecting the progression of renal disease in autosomal-dominant polycystic kidney disease, Kidney Int 41: (5) ((1992) ), 1311–1319. |
[30] | Bajwa Z.H. , Sial K.A. , Malik A.B. and Steinman T.I. , Pain patterns in patients with polycystic kidney disease, Kidney Int 66: (4) ((2004) ), 1561–1569. |
[31] | Klahr S. , Breyer J.A. , Beck G.J. , Dennis V.W. , Hartman J.A. , Roth D. , et al., Dietary protein restriction, blood pressure control, and the progression of polycystic kidney disease, modification of diet in renal disease study group, J Am Soc Nephrol 5: (12) ((1995) ), 2037–2047. |
[32] | Hateboer N. , v Dijk M.A. , Bogdanova N. , Coto E. , Saggar-Malik A.K. , San Millan J.L. , et al., Comparison of phenotypes of polycystic kidney disease types 1 and 2, European PKD1-PKD2 Study Group Lancet 353: (9147) ((1999) ), 103–107. |
[33] | Cornec-Le Gall E. , Audrezet M.P. , Chen J.M. , Hourmant M. , Morin M.P. , Perrichot R. , et al., Type of PKD1 mutation influences renal outcome in ADPKD, J Am Soc Nephrol 24: (6) ((2013) ), 1006–1013. |
[34] | Grantham J.J. , Torres V.E. , Chapman A.B. , Guay-Woodford L.M. , Bae K.T. , King B.F. Jr , et al., Volume progression in polycystic kidney disease, N Engl J Med 354: (20) ((2006) ), 2122–2130. |
[35] | Chapman A.B. , Bost J.E. , Torres V.E. , Guay-Woodford L. , Bae K.T. , Landsittel D. , et al., Kidney volume and functional outcomes in autosomal dominant polycystic kidney disease, Clin J Am Soc Nephrol 7: (3) ((2012) ), 479–486. |
[36] | Zerres K. , Rudnik-Schoneborn S. and Deget F. , Childhood onset autosomal dominant polycystic kidney disease in sibs: Clinical picture and recurrence risk, German working group on paediatric nephrology (arbeitsgemeinschaft fur padiatrische nephrologie, Journal of Medical Genetics 30: (7) ((1993) ), 583–588. |
[37] | Audrezet M.P. , Corbiere C. , Lebbah S. , Moriniere V. , Broux F. , Louillet F. , et al., Comprehensive PKD1 and PKD2 mutation analysis in prenatal autosomal dominant polycystic kidney disease, J Am Soc Nephrol 27: (3) ((2016) ), 722–729. |
[38] | Everson G.T. , Helmke S.M. and Doctor B. , Advances in management of polycystic liver disease, Expert Rev Gastroenterol Hepatol 2: (4) ((2008) ), 563–576. |
[39] | Everson G.T. and Taylor M.R. , Management of polycystic liver disease, Curr Gastroenterol Rep 7: (1) ((2005) ), 19–25. |
[40] | O’Brien K. , Font-Montgomery E. , Lukose L. , Bryant J. , Piwnica-Worms K. , Edwards H. , et al., Congenital hepatic fibrosis and portal hypertension in autosomal dominant polycystic kidney disease, J Pediatr Gastroenterol Nutr 54: (1) ((2012) ), 83–89. |
[41] | Pirson Y. , Chauveau D. and Torres V. , Management of cerebral aneurysms in autosomal dominant polycystic kidney disease, J Am Soc Nephrol 13: (1) ((2002) ), 269–276. |
[42] | International Study of Unruptured Intracranial Aneurysms I. Unruptured intracranial aneurysms–risk of rupture and risks of surgical intervention, N Engl J Med 339: (24) ((1998) ), 1725–1733. |
[43] | Lumiaho A. , Ikaheimo R. , Miettinen R. , Niemitukia L. , Laitinen T. , Rantala A. , et al., Mitral valve prolapse and mitral regurgitation are common in patients with polycystic kidney disease type 1, Am J Kidney Dis 38: (6) ((2001) ), 1208–1216. |
[44] | Parisi M , Glass I. Joubert Syndrome. Jul 9 [Updated Jun 29]. In: Adam MP , Ardinger HH , Pagon RA , et al., editors. GeneReviews®[Internet]. Seattle (WA): University of Washington, Seattle; (1993) -(2019) . Available from: https://www.ncbi.nlm.nih.gov/books/NBK1325/. |
[45] | Poretti A. , Snow J. , Summers A.C. , Tekes A. , Huisman T. , Aygun N. , et al., Joubert syndrome: Neuroimaging findings in 110 patients in correlation with cognitive function and genetic cause, J Med Genet 54: (8) ((2017) ), 521–529. |
[46] | Vilboux T. , Doherty D.A. , Glass I.A. , Parisi M.A. , Phelps I.G. , Cullinane A.R. , et al., Molecular genetic findings and clinical correlations in 100 patients with Joubert syndrome and related disorders prospectively evaluated at a single center. Genet Med 19: (8) ((2017) ), 875–882. |
[47] | Brooks B.P. , Zein W.M. , Thompson A.H. , Mokhtarzadeh M. , Doherty D.A. , Parisi M. , et al., Joubert syndrome: Ophthalmological findings in correlation with genotype and hepatorenal disease in 99 patients prospectively evaluated at a single center, Ophthalmology 125: (12) ((2018) ), 1937–1952. |
[48] | Bulgheroni S. , D’Arrigo S. , Signorini S. , Briguglio M. , Di Sabato M.L. , Casarano M. , et al., Cognitive, adaptive, and behavioral features in Joubert syndrome, Am J Med Genet A 170: (12) ((2016) ), 3115–3124. |
[49] | Summers A.C. , Snow J. , Wiggs E. , Liu A.G. , Toro C. , Poretti A. , et al. Neuropsychological phenotypes of 76 individuals with Joubert syndrome evaluated at a single center, Am J Med Genet A ((2017) ). |
[50] | Bachmann-Gagescu R. , Dempsey J.C. , Phelps I.G. , O’Roak B.J. , Knutzen D.M. , Rue T.C. , et al., Joubert syndrome: A model for untangling recessive disorders with extreme genetic heterogeneity, Journal of Medical Genetics 52: (8) ((2015) ), 514–522. |
[51] | Fleming L.R. , Doherty D.A. , Parisi M.A. , Glass I.A. , Bryant J. , Fischer R. , et al., Prospective evaluation of kidney disease in joubert syndrome, Clin J Am Soc Nephrol 12: (12) ((2017) ), 1962–1973. |
[52] | Strongin A. , Heller T. , Doherty D. , Glass I.A. , Parisi M.A. , Bryant J. , et al., Characteristics of liver disease in 100 individuals with joubert syndrome prospectively evaluated at a single center, J Pediatr Gastroenterol Nutr 66: (3) ((2018) ), 428–435. |
[53] | Bachmann-Gagescu R. , Ishak G.E. , Dempsey J.C. , Adkins J. , O’Day D. , Phelps I.G. , et al., Genotype-phenotype correlation in CC2D2A-related Joubert syndrome reveals an association with ventriculomegaly and seizures, J Med Genet 49: (2) ((2012) ), 126–137. |
[54] | Hartill V. , Szymanska K. , Sharif S.M. , Wheway G. and Johnson C.A. , Meckel-gruber syndrome: An update on diagnosis, clinical management, and research advances, Front Pediatr 5: ((2017) ), 244. |
[55] | Salonen R. , The meckel syndrome: Clinicopathological findings in 67 patients, Am J Med Genet 18: (4) ((1984) ), 671–689. |
[56] | Sergi C. , Adam S. , Kahl P. and Otto H.F. , Study of the malformation of ductal plate of the liver in Meckel syndrome and review of other syndromes presenting with this anomaly, Pediatr Dev Pathol 3: (6) ((2000) ), 568–583. |
[57] | Paetau A. , Salonen R. and Haltia M. , Brain pathology in the Meckel syndrome: A study of 59 cases, Clin Neuropathol 4: (2) ((1985) ), 56–62. |
[58] | Ahdab-Barmada M. and Claassen D. , A distinctive triad of malformations of the central nervous system in the Meckel-Gruber syndrome, J Neuropathol Exp Neurol 49: (6) ((1990) ), 610–620. |
[59] | Bisceglia M. , Galliani C.A. , Senger C. , Stallone C. and Sessa A. , Renal cystic diseases: A review, Adv Anat Pathol 13: (1) ((2006) ), 26–56. |
[60] | Collin G.B. , Won J. , Hicks W.L. , Cook S.A. , Nishina P.M. and Naggert J.K. , Meckelin is necessary for photoreceptor intraciliary transport and outer segment morphogenesis, Invest Ophthalmol Vis Sci 53: (2) ((2012) ), 967–974. |
[61] | Forsythe E. and Beales P.L. , Bardet-Biedl Syndrome. In: Adam M.P. , Ardinger H.H. , Pagon R.A. , Wallace S.E. , Bean L.J.H. , Stephens K. , et al., editors. GeneReviews((R)). Seattle WA. |
[62] | Forsythe E. and Beales P.L. , Bardet-Biedl syndrome, European Journal of Human Genetics: EJHG 21: (1) ((2013) ), 8–13. |
[63] | Moore S.J. , Green J.S. , Fan Y. , Bhogal A.K. , Dicks E. , Fernandez B.A. , et al., Clinical and genetic epidemiology of Bardet-Biedl syndrome in Newfoundland: A 22-year prospective, population-based, cohort study, Am J Med Genet A 132A: (4) ((2005) ), 352–360. |
[64] | Kerr E.N. , Bhan A. and Heon E. , Exploration of the cognitive, adaptive and behavioral functioning of patients affected with Bardet-Biedl syndrome, Clin Genet 89: (4) ((2016) ), 426–433. |
[65] | Barnett S. , Reilly S. , Carr L. , Ojo I. , Beales P.L. and Charman T. , Behavioural phenotype of Bardet-Biedl syndrome, J Med Genet 39: (12) ((2002) ), e76. |
[66] | Imhoff O. , Marion V. , Stoetzel C. , Durand M. , Holder M. , Sigaudy S. , et al., Bardet-Biedl syndrome: A study of the renal and cardiovascular phenotypes in a French cohort, Clin J Am Soc Nephrol 6: (1) ((2011) ), 22–29. |
[67] | Forsythe E. , Sparks K. , Best S. , Borrows S. , Hoskins B. , Sabir A. , et al., Risk factors for severe renal disease in bardet-biedl syndrome, J Am Soc Nephrol 28: (3) ((2017) ), 963–970. |
[68] | Linne T. , Wikstad I. and Zetterstrom R. , Renal involvement in the Laurence-Moon-Biedl syndrome, Functional and radiological studies. Acta Paediatr Scand 75: (2) ((1986) ), 240–244. |
[69] | Beales P.L. , Elcioglu N. , Woolf A.S. , Parker D. and Flinter F.A. , New criteria for improved diagnosis of Bardet-Biedl syndrome: Results of a population survey, J Med Genet 36: (6) ((1999) ), 437–446. |
[70] | Weihbrecht K. , Goar W.A. , Pak T. , Garrison J.E. , DeLuca A.P. , Stone E.M. , et al., Keeping an eye on bardet-biedl syndrome: A comprehensive review of the role of bardet-biedl syndrome genes in the eye, Med Res Arch 5: (9) ((2017) ). |
[71] | Hamel C.P. , Cone rod dystrophies, Orphanet J Rare Dis 2: ((2007) ), 7. |
[72] | Mujahid S. , Hunt K.F. , Cheah Y.S. , Forsythe E. , Hazlehurst J.M. , Sparks K. , et al., The endocrine and metabolic characteristics of a large bardet-biedl syndrome clinic population, J Clin Endocrinol Metab 103: (5) ((2018) ), 1834–1841. |
[73] | Feuillan P.P. , Ng D. , Han J.C. , Sapp J.C. , Wetsch K. , Spaulding E. , et al., Patients with Bardet-Biedl syndrome have hyperleptinemia suggestive of leptin resistance, J Clin Endocrinol Metab 96: (3) ((2011) ), E528–E535. |
[74] | Grace C. , Beales P. , Summerbell C. , Jebb S.A. , Wright A. , Parker D. , et al., Energy metabolism in Bardet-Biedl syndrome, Int J Obes Relat Metab Disord 27: (11) ((2003) ), 1319–1324. |
[75] | Vaisse C. , Reiter J.F. and Berbari N.F. , Cilia and obesity, Cold Spring Harb Perspect Biol 9: (7) ((2017) ). |
[76] | Nakamura F. , Sasaki H. , Kajihara H. and Yamanoue M. , Laurence-Moon-Biedl syndrome accompanied by congenital hepatic fibrosis, J Gastroenterol Hepatol 5: (2) ((1990) ), 206–210. |
[77] | Baker K. and Beales P.L. , Making sense of cilia in disease: The human ciliopathies, Am J Med Genet C Semin Med Genet 151C: (4) ((2009) ), 281–295. |
[78] | Branfield Day L. , Quammie C. , Heon E. , Bhan A. , Batmanabane V. , Dai T. , et al., Liver anomalies as a phenotype parameter of Bardet-Biedl syndrome, Clin Genet 89: (4) ((2016) ), 507–509. |
[79] | Elbedour K. , Zucker N. , Zalzstein E. , Barki Y. and Carmi R. , Cardiac abnormalities in the Bardet-Biedl syndrome: Echocardiographic studies of 22 patients, Am J Med Genet 52: (2) ((1994) ), 164–169. |
[80] | Olson A.J. , Krentz A.D. , Finta K.M. , Okorie U.C. and Haws R.M. , Thoraco-abdominal abnormalities in bardet-biedl syndrome: Situs inversus and heterotaxy, J Pediatr 204: ((2019) ), 31–37. |
[81] | Paisey R.B. , Carey C. and Macdermott S. , Alstrom Syndrome. In: Adam M.P. , Ardinger H.H. , Pagon R.A. , Wallace S.E. , Bean L.J.H. , Stephens K. , et al., editors. GeneReviews((R)). Seattle WA, (1993) . |
[82] | Marshall J.D. , Bronson R.T. , Collin G.B. , Nordstrom A.D. , Maffei P. , Paisey R.B. , et al., New Alstrom syndrome phenotypes based on the evaluation of 182 cases, Arch Intern Med 165: (6) ((2005) ), 675–683. |
[83] | Collin G.B. , Cyr E. , Bronson R. , Marshall J.D. , Gifford E.J. , Hicks W. , et al., Alms1-disrupted mice recapitulate human Alstrom syndrome, Human Molecular Genetics 14: (16) ((2005) ), 2323–2333. |
[84] | Nasser F. , Weisschuh N. , Maffei P. , Milan G. , Heller C. , Zrenner E. , et al., Ophthalmic features of cone-rod dystrophy caused by pathogenic variants in the ALMS1 gene, Acta Ophthalmol 96: (4) ((2018) ), e445–e54. |
[85] | Marshall J.D. , Paisey R.B. , Carey C. and Macdermott S. , Alstrom Syndrome. In: Pagon R.A. , Adam M.P. , Ardinger H.H. , Wallace S.E. , Amemiya A. , Bean L.J.H. , et al., editors. GeneReviews(R). Seattle WA, (1993) . |
[86] | Malm E. , Ponjavic V. , Nishina P.M. , Naggert J.K. , Hinman E.G. , Andreasson S. , et al., Full-field electroretinography and marked variability in clinical phenotype of Alstrom syndrome, Archives of Ophthalmology 126: (1) ((2008) ), 51–57. |
[87] | Brofferio A. , Sachdev V. , Hannoush H. , Marshall J.D. , Naggert J.K. , Sidenko S. , et al., Characteristics of cardiomyopathy in Alstrom syndrome: Prospective single-center data on 38 patients, Mol Genet Metab 121: (4) ((2017) ), 336–343. |
[88] | Louw J.J. , Corveleyn A. , Jia Y. , Iqbal S. , Boshoff D. , Gewillig M. , et al., Homozygous loss-of-function mutation in ALMS1 causes the lethal disorder mitogenic cardiomyopathy in two siblings, European Journal of Medical Genetics 57: (9) ((2014) ), 532–535. |
[89] | Shenje L.T. , Andersen P. , Halushka M.K. , Lui C. , Fernandez L. , Collin G.B. , et al., Mutations in Alstrom protein impair terminal differentiation of cardiomyocytes, Nature Communications 5: ((2014) ), 3416. |
[90] | Edwards N.C. , Moody W.E. , Yuan M. , Warfield A.T. , Cramb R. , Paisey R.B. , et al., Diffuse left ventricular interstitial fibrosis is associated with sub-clinical myocardial dysfunction in Alstrom Syndrome: An observational study, Orphanet Journal of Rare Diseases 10: ((2015) ). |
[91] | Corbetti F. , Razzolini R. , Bettini V. , Marshall J.D. , Naggert J. , Tona F. , et al., Alstrom syndrome: Cardiac magnetic resonance findings, International Journal of Cardiology 167: (4) ((2013) ), 1257–1263. |
[92] | Toulany A. , Shea S. and Warren A.E. , Doppler tissue, strain, and strain rate imaging in pediatric patients with Alstrom syndrome: Are there regional functional abnormalities? Journal of the American Society of Echocardiography: Official Publication of the American Society of Echocardiography 19: (1) ((2006) ), 14–20. |
[93] | Han J.C. , Reyes-Capo D.P. , Liu C.Y. , Reynolds J.C. , Turkbey E. , Turkbey I.B. , et al., Comprehensive endocrine-metabolic evaluation of patients with alstrom syndrome compared With BMI-matched controls, J Clin Endocrinol Metab 103: (7) ((2018) ), 2707–2719. |
[94] | Gathercole L.L. , Hazlehurst J.M. , Armstrong M.J. , Crowley R. , Boocock S. , O’Reilly M.W. , et al., Advanced non-alcoholic fatty liver disease and adipose tissue fibrosis in patients with Alstrom syndrome, Liver Int 36: (11) ((2016) ), 1704–1712. |
[95] | Marshall J.D. , Maffei P. , Collin G.B. and Naggert J.K. , Alstrom syndrome: Genetics and clinical overview, Curr Genomics 12: (3) ((2011) ), 225–235. |
[96] | Favaretto F. , Milan G. , Collin G.B. , Marshall J.D. , Stasi F. , Maffei P. , et al., GLUT4 defects in adipose tissue are early signs of metabolic alterations in Alms1GT/GT, a mouse model for obesity and insulin resistance, PloS one 9: (10) ((2014) ), e109540. |
[97] | Jaykumar A.B. , Caceres P.S. , King-Medina K.N. , Liao T.D. , Datta I. , Maskey D. , et al., Role of Alstrom syndrome 1 in the regulation of blood pressure and renal function, JCI Insight 3: (21) ((2018) ). |
[98] | Baig S. , Paisey R. , Dawson C. , Barrett T. , Maffei P. , Hodson J. , et al., Defining renal phenotype in Alstrom syndrome, Nephrol Dial Transplant ((2018) ). |
[99] | Waldman M. , Han J.C. , Reyes-Capo D.P. , Bryant J. , Carson K.A. , Turkbey B. , et al., Alstrom syndrome: Renal findings in correlation with obesity, insulin resistance, dyslipidemia and cardiomyopathy in 38 patients prospectively evaluated at the NIH clinical center, Mol Genet Metab 125: (1-2) ((2018) ), 181–191. |
[100] | Izzi C. , Maffei P. , Milan G. , Tardanico R. , Foini P. , Marshall J. , et al., The Case mid R: Familial occurrence of retinitis pigmentosa, deafness, and nephropathy, Kidney Int 79: (6) ((2011) ), 691–692. |
[101] | Koc E. , Bayrak G. , Suher M. , Ensari C. , Aktas D. and Ensari A. , Rare case of Alstrom syndrome without obesity and with short stature, diagnosed in adulthood, Nephrology 11: (2) ((2006) ), 81–84. |
[102] | Lindsey S. , Brewer C. , Stakhovskaya O. , Kim H.J. , Zalewski C. , Bryant J. , et al., Auditory and otologic profile of Alstrom syndrome: Comprehensive single center data on 38 patients, American Journal of Medical Genetics Part A 173: (8) ((2017) ), 2210–2218. |
[103] | Boerwinkle C. , Marshall J.D. , Bryant J. , Gahl W.A. , Olivier K.N. and Gunay-Aygun M. , Respiratory manifestations in 38 patients with Alstrom syndrome, Pediatr Pulmonol 52: (4) ((2017) ), 487–493. |
[104] | Franco B. and Thauvin-Robinet C. , Update on oral-facial-digital syndromes (OFDS), Cilia 5: ((2016) ), 12. |
[105] | Ferrante M.I. , Zullo A. , Barra A. , Bimonte S. , Messaddeq N. , Studer M. , et al., Oral-facial-digital type I protein is required for primary cilia formation and left-right axis specification, Nat Genet 38: (1) ((2006) ), 112–117. |
[106] | Prattichizzo C. , Macca M. , Novelli V. , Giorgio G. , Barra A. , Franco B. , et al., Mutational of the oral-facial-digital type I syndrome: A study on a large collection of patients, Human mutation 29: (10) ((2008) ), 1237–1246 spectrum. |
[107] | Saal S. , Faivre L. , Aral B. , Gigot N. , Toutain A. , Van Maldergem L. , et al., Renal insufficiency, a frequent complication with age in oral-facial-digital syndrome type I, Clinical Genetics 77: (3) ((2010) ), 258–265. |
[108] | Chetty-John S. , Piwnica-Worms K. , Bryant J. , Bernardini I. , Fischer R.E. , Heller T. , et al., Fibrocystic disease of liver and pancreas; under-recognized features of the X-linked ciliopathy oral-facial-digital syndrome type 1 (OFD I), Am J Med Genet A 152A: (10) ((2010) ), 2640–2645. |
[109] | Zhang W. , Taylor S.P. , Ennis H.A. , Forlenza K.N. , Duran I. , Li B. , et al., Expanding the genetic architecture and phenotypic spectrum in the skeletal ciliopathies, Human Mutation 39: (1) ((2018) ), 152–166. |
[110] | Arts H. and Knoers N. , Cranioectodermal Dysplasia. In: Adam M.P. , Ardinger H.H. , Pagon R.A. , Wallace S.E. , Bean L.J.H. , Stephens K. , et al., editors. GeneReviews((R)). Seattle WA, (1993) . |
[111] | Mookherjee S. , Chen H.Y. , Isgrig K. , Yu W. , Hiriyanna S. , Levron R. , et al., A CEP290 C-terminal domain complements the mutant CEP290 of Rd16 mice in trans and rescues retinal degeneration, Cell Rep 25: (3) ((2018) ), 611–623 e6. |
[112] | Deng W.L. , Gao M.L. , Lei X.L. , Lv J.N. , Zhao H. , He K.W. , et al., Gene correction reverses ciliopathy and photoreceptor loss in iPSC-derived retinal organoids from retinitis pigmentosa patients, Stem Cell Reports 10: (4) ((2018) ), 1267–1281. |
[113] | Cornec-Le Gall E. , Alam A. and Perrone R.D. , Autosomal dominant polycystic kidney disease, Lancet 393: (10174) ((2019) ), 919–935. |
[114] | Ramsbottom S.A. , Molinari E. , Srivastava S. , Silberman F. , Henry C. , Alkanderi S. , et al., Targeted exon skipping of a CEP290 mutation rescues Joubert syndrome phenotypes in vitro and in a murine model, Proc Natl Acad Sci U S A 115: (49) ((2018) ), 12489–12494. |
[115] | Forsythe E. , Kenny J. , Bacchelli C. and Beales P.L. , Managing bardet-biedl syndrome-now and in the future, Front Pediatr 6: ((2018) ), 23. |


