
Conducting an investigator-initiated randomized double-blinded intervention trial in acute decompensation of inborn errors of metabolism: Lessons from the N-Carbamylglutamate Consortium
Abstract
Organic acidemias and urea cycle disorders are ultra-rare inborn errors of metabolism characterized by episodic acute decompensation, often associated with hyperammonemia, resulting in brain edema and encephalopathy. Retrospective reports and translational studies suggest that N-carbamylglutamate (NCG) may be effective in reducing ammonia levels during acute decompensation in two organic acidemias, propionic and methylmalonic acidemia (PA and MMA), and in two urea cycle disorders, carbamylphosphate synthetase 1 and ornithine transcarbamylase deficiency (CPSD and OTCD). We established the 9-site N-carbamylglutamate Consortium (NCGC) in order to conduct two randomized double-blind, placebo-controlled trials of NCG in acute hyperammonemia of PA, MMA, CPSD and OTCD. Conducting clinical trials is challenging in any disease, but poses unique barriers and risks in the ultra-rare disorders. As the number of clinical trials in orphan diseases increases, evaluating the successes and opportunities for improvement in such trials is essential. We summarize herein the design, methods, experiences, challenges and lessons from the NCGC-conducted trials.
1Introduction
Inborn errors of metabolism (IEMs) are rare genetic disorders caused by defects in enzymes, transporters, or other functional proteins, resulting in blocks in a biochemical pathway. The pathophysiology of these disorders typically derives from the accumulation of toxic substrates upstream of the block or deficiency of essential products beyond the block. Although the collective incidence of all IEMs is estimated to be as high as 1 in 800 births [1] virtually all individual IEMs are rare diseases, defined in the US Code of Federal Regulations (CFR) as affecting fewer than 200,000 people (approximately 1 in 1600 Americans) [2] or in Europe, fewer than 1 in 2,000 [3]. Although prevalence varies by population, the frequency of most IEMs is so low that they meet the definition of ‘ultra-rare’, a concept which has been proposed for diseases with a prevalence of less than 1:50,000 [4]. Ultra-rare disorders present unique challenges for the conduct of randomized clinical trials.
Propionic acidemia (PA), methylmalonic acidemia (MMA), ornithine transcarbamylase deficiency (OTCD), and carbamyl phosphate synthetase 1 deficiency (CPSD) are among many ultra-rare disorders associated with elevated blood ammonia levels, or hyperammonemia (HA) [5], which may result in brain edema causing encephalopathy during acute events and if untreated, could cause death. Even with current therapies for acute hyperammonemia, long-term sequelae often include developmental delays leading to intellectual and developmental disability [5].
In mammals, including humans, toxic ammonia is converted into urea via the urea cycle, a biochemical pathway of which carbamyl phosphate synthetase 1 (CPS1) catalyzes the first and rate-limiting step and ornithine transcarbamylase (OTC) catalyzes the second step [6]. For enzymatic activity, CPS1 requires an allosteric activator, N-acetylglutamate (NAG), which is produced by N-acetylglutamate synthetase (NAGS). In the absence of NAG, CPS1 is catalytically inactive. Thus, a deficiency of the NAGS, CPS1 or OTC enzyme results in reduced nitrogen flux through the urea cycle, causing HA [6]. In PA and MMA, the accumulated toxic substrates competitively inhibit the NAGS enzyme [7], thus also causing HA.
N-carbamylglutamate (NCG), a stable analogue of N-acetylglutamate, is currently approved in both the United States (US) and Europe for the treatment of NAGS deficiency. There have been retrospective reports [8–11], pre-clinical experiments [7, 12], and translational studies [13], that suggest that NCG may be effective in reducing ammonia levels during acute decompensation in PA, MMA, CPS1D, and OTCD. However, none of these reports were based on randomized, controlled trials.
Six years ago, we initiated two studies to evaluate whether the addition of NCG to standard therapy during acute HA 1) would result in improved neurocognitive outcomes; and 2) would more rapidly reduce ammonia levels, improve function and shorten hospitalization relative to standard therapy alone. The first study randomly assigned participants (all events per participant) to NCG or placebo; in the second study, each separate HA event experienced by participants is randomized to NCG or placebo. Both studies were investigator-initiated trials (IND 68,185), with no input from industry in their design or conduct and were developed to primarily answer scientific questions. The study sites included nine pediatric academic institutions in the US (Fig. 1), referred to collectively as the N-carbamylglutamate Consortium (NCGC).
Fig.1
The N-Carbamylglutamate Consortium. Date each site joined shown in parentheses.
These studies were primarily funded through an R01 award from the NIH National Institute of Child Health and Human Development (HD058567), and cost-shared with philanthropic funds. Orphan Europe, the only pharmaceutical distributor of NCG (Carbaglu) provided drug and placebo free-of-charge. Once NIH funding ended, Orphan Europe committed to funding the study to completion.
Herein, we summarize the design, methods, experiences, challenges and lessons from the NCGC-conducted trials, one of which is still in progress.
2Methods
Two randomized double-blind, placebo-controlled studies with NCG were designed and launched in 2012.
The first, “long-term neurocognitive outcome study” or LTO (Clinicaltrials.gov registry number: NCT01597440), focused on enrolling newly-diagnosed infants with PA and MMA, presenting with HA at or less than 4 weeks of age. Enrollees were randomly assigned to receive NCG or placebo, in addition to standard therapy, during all their hospitalizations for episodes of HA. Follow-up assessments were scheduled for 9, 15, 21, and 30 months of age. Each of these assessments included the Bayley Scales of Infant Development-Third Edition (all ages), Vineland Adaptive Behavior Scales (all ages), and the MacArthur Inventory of Communicative Development, Second edition (all ages), and the Brief Infant/Toddler Social Emotional Assessment at ages 15, 21, and 30 months.
The second, “short-term outcome” (STO) study (NCT01599286) focused on enrolling persons of all ages with PA, MMA, CPSD or OTCD not eligible for the LTO and randomizing them to NCG vs placebo added to standard care at hospitalizations for each separate HA event (Fig. 2). The primary endpoint of the STO study was the difference in average trajectory of ammonia decline during events receiving NCG vs. placebo. The secondary endpoints were based on change in clinical status during the hospital course and time to hospital discharge (Fig. 2). The Functional Status Scale (FSS) [14], ascertained pre-NCG and once daily thereafter, rated the patient’s functional status in 6 domains: mental status, sensory, communication, motor, feeding, and respiratory functions. Each domain was scored from normal (1) to very severe dysfunction (5) for potential score range from 6 to 30.
Fig.2
Schema of the Short-Term Outcome trial.
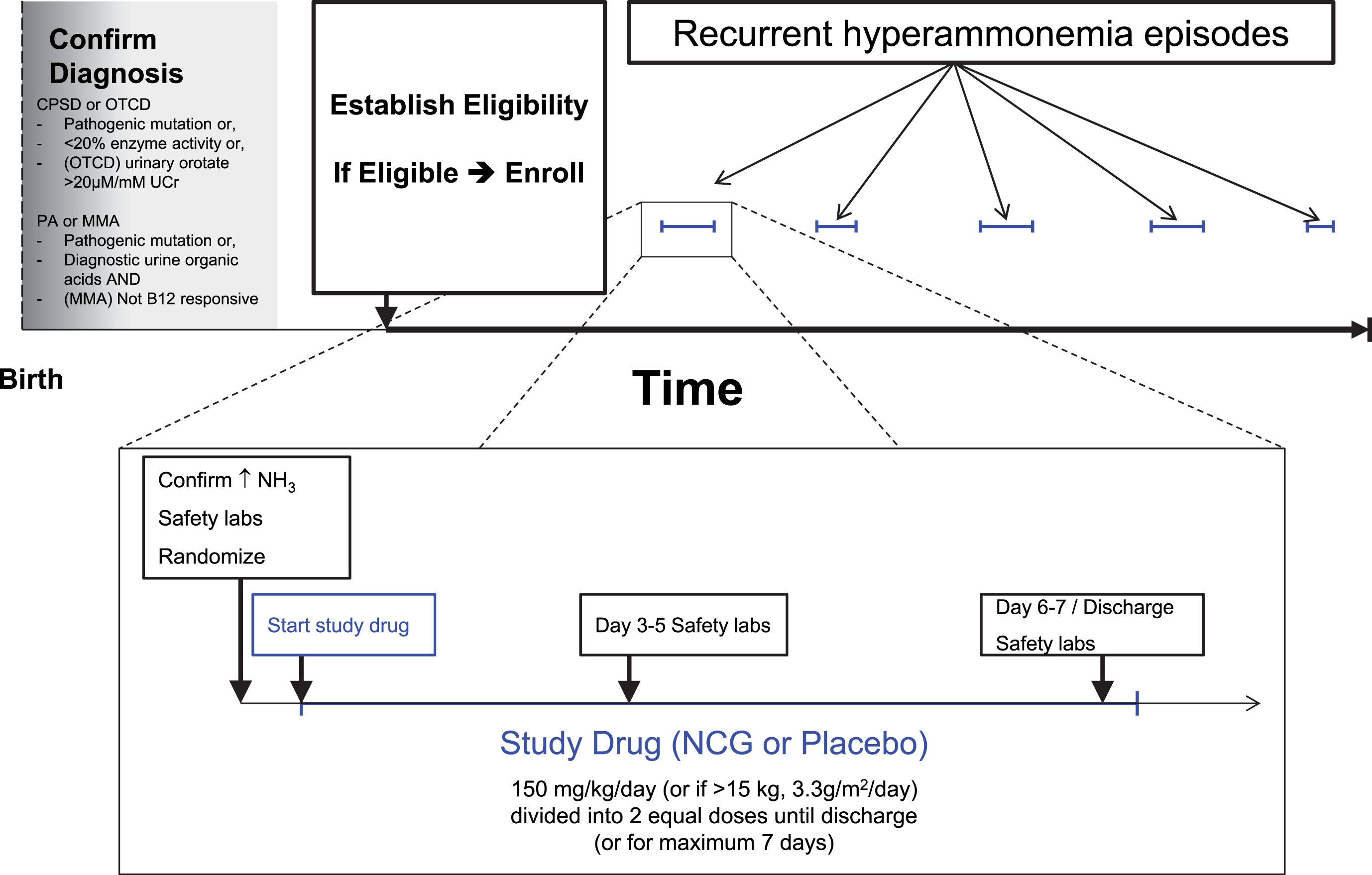
Drug/placebo assignments were block-randomized within study site but also controlled allocation balance within each participant, so that participants would receive approximately an equal number of assignments to either NCG or placebo over the course of multiple hospitalizations. This somewhat complex randomization allowed both balancing allocations to treatment within the site as a whole and balancing participant contributions to each treatment arm and resulting in multiple crossovers in patients with multiple episodes of hyperammonemia. A defined wash-out period prevented otherwise eligible participants from being randomized during any repeat hospitalizations falling within 7 days of the last dose of a prior episode.
For both studies, the prescribed dose was 150 mg/kg/d or 3.3 g/m2/d if >15 kg, which was continued for the earlier of 7 days or discharge. Per protocol, plasma ammonia levels were measured prior to NCG administration, and at least every 12 hours during the first 48 hours and daily thereafter. In addition, clinically-ordered ad hoc ammonia measurements were also captured, as well as any ad hoc plasma amino acid profiles. Safety investigations included a complete blood count, serum liver enzymes, and creatinine, and were obtained pre-NCG dosing and 2–4 days after the start of NCG. Electrocardiograms (ECG) were added to the safety investigations following a recommendation by the FDA 3 years after the studies had been opened for enrollment.
Children’s National was the lead site and coordinating center for the studies. Seven inaugural sites in the N-carbamylglutamate Consortium (NCGC) were chosen mostly based on their ongoing collaboration as members of the Urea Cycle Disorders Consortium (UCDC) [15]. These included Boston Children’s Hospital, Rainbow Babies & Children’s Hospital in Cleveland, Children’s Hospital Colorado, University of California Los Angeles Medical Center, Children’s Hospital of Philadelphia, Lucille Packard’s Children’s Hospital Stanford, and Children’s National Health System in Washington, DC. This approach ensured that sites had the requisite expertise, experience, patient base and established working relationship with the coordinating center at Children’s National. During study design, each site provided information based on a review of records from the recent past, of numbers of putatively eligible new patients and currently active patients as well as the expected frequency of admissions with HA of the four study disorders. Based on these data, each participant was expected to have an average of 4 HA admissions over the study period, which was used to estimate the study sample size for both trials. Two additional sites were later added to increase enrollment: Mount Sinai Health System in New York, and Children’s Hospital of Pittsburgh. Monthly calls with the principal investigators and staff at each site provided an opportunity to discuss updates, changes to the protocol and challenges in the conduct of the studies. Additionally, internal meetings of the study leadership and members of the coordinating center to discuss study progress were scheduled initially weekly, and then twice monthly, once the lead site was open for enrollment.
These trials have made use of the Children’s Hospital Research Information System (CHRIS) [16]. CHRIS supported screening, enrollment, random allocation of episodes to treatment arms, protocol management and form-based data collection. Challenges encountered with this platform forced a switch to REDCap (Research Electronic Data Capture) [17] within the first year of opening the study to enrollment.
All sites received IRB approval from their local IRB. The initial protocol submission as well as all modifications were first approved at the lead site, followed by approval at all other sites. A Data and Safety Monitoring Board (DSMB) was established that included a neurologist chair, an ethicist, two neonatologists, and a biostatistician. Per protocol, meetings of the DSMB were scheduled every six months to review participant safety as well as study progress and data completeness. Also per protocol, an interim analysis was scheduled at the projected halfway point of the study (72 episodes in the STO).
Sample and effect sizes were estimated in PASS (Hintze, J. PASS 11. NCSS, LLC. Kaysville, Utah, USA. www.ncss.com, 2011). Sample size projections were based on preliminary site reports, 2-tailed testing, 80% power, and assuming a type 1 error of 5% comparing timewise differences in the trajectory of neuropsychological test scores in the LTO trial and in ammonia levels in the STO trial. For the LTO study, site reports indicated the ability to collectively recruit and retain at least 34 PA and MMA participants. Evaluating Bayley scores assessed at 2-3 times per participant (assumed to be correlated at r = 0.8), we estimated that randomly assigning 17 participants to each arm would allow us to detect an effect size difference based on differences in the score trajectories between groups of at least 0.9 standard deviations (SD). This minimum detectable effect size was considered realistic given the severity of the projected insult in young participants and thus the potential for protection afforded by NCG. For the STO study, we considered the projected 4-episode experience within participant with random assignment to NCG+standard treatment (NCG-STD) vs. placebo+standard treatment (PLBO-STD) per episode to be most closely equivalent to a crossover design and thus developed our estimates based on this design and assumed a linear slope for the decline in ammonia. Accounting for gains in efficiency associated with an average of 4 episodes per enrollee, a sample size of 36 participants (144 episodes, 72 per group) was estimated to provide 80% power to detect a difference in ammonia level trajectories between groups of at least 0.6 SD. Based on recent experience at participating sites and on preliminary results regarding the magnitude of the NCG effect, the sample size was considered feasible and the projected effect was considered attainable if NCG proved to be effective. These numbers were also considered adequate to assess secondary outcomes of safety and functional improvement leading to earlier discharge. Sample and effect size estimates for the STO trial were also adjusted to include error spending to accommodate a single interim analysis targeted for the halfway point of that study (72 randomized episodes).
3Results
From December, 2012 through April, 2015, we enrolled only 1 participant into the Long-Term Outcome study. The study was therefore closed in May 2015, as it was determined that it was unlikely to recruit sufficient participants to reach enrollment targets and produce a meaningful conclusion.
In the Short-Term Outcome study, among all sites, median time (range) from initial submission to IRB approval was 68 days (range 25–164); from non-personnel amendment submission to approval was 27 days (range 11.8–39.1) and from submission of continuing review to approval was 24.5 days (range 16–54) (Table 1).
Table 1
Days to IRB approval by site
| Site 1 | Site 2 | Site 3 | Site 4 | Site 5 | Site 6 | Site 7 | Site 8 | Site 9 | Median | |
| Initial IRB | 42 | 68 | 68 | 25 | 131 | 164 | 70 | 63 | 56 | 68 |
| Amendment | 39 | 25 | 31 | 12 | 32 | 19 | N/A | 18 | 29 | 27 |
| Continuing Review | 33 | 16 | 16 | 16 | 54 | 16 | N/A | 45 | 41 | 25 |
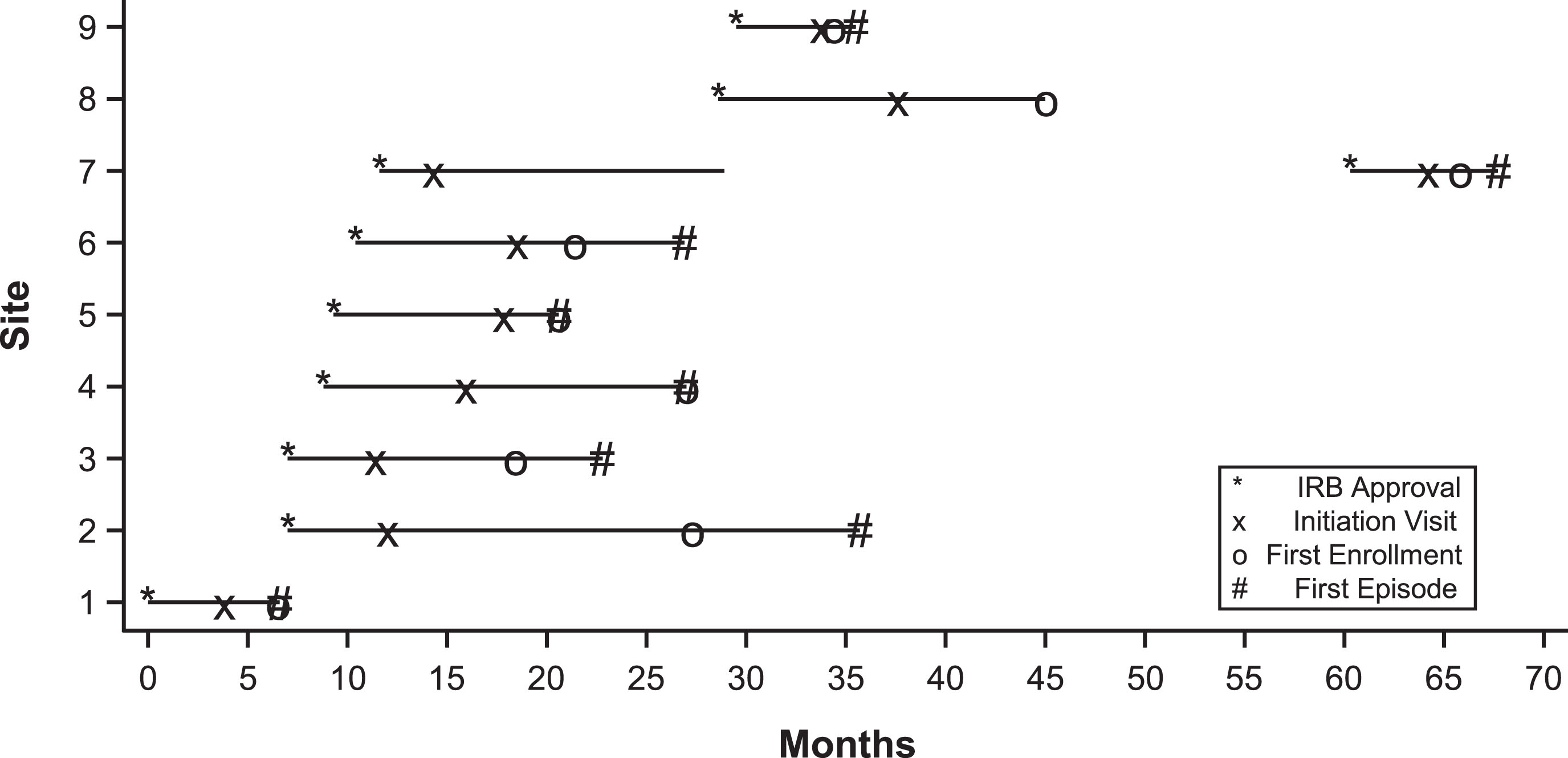
A timeline of the STO study is shown in Fig. 3. IRB protocol applications at all other sites began only after approval was obtained at the lead site. One site (Site 4) obtained IRB approval, but due to insufficient screening and enrollment efforts, was first placed on probation and after lack of progress, subsequently discontinued from the study. Reactivation was considered several years later, along with a change in site-PI, and the site was ultimately reactivated approximately 5 years into the study.
Fig.3
Study time line of the Short-Term Outcome trial. Initial IRB approval, study initiation visit, first enrollment and first episode is shown for each site. Site 7 was initially discontinued from the study for lack of progress, but subsequently re-activated.
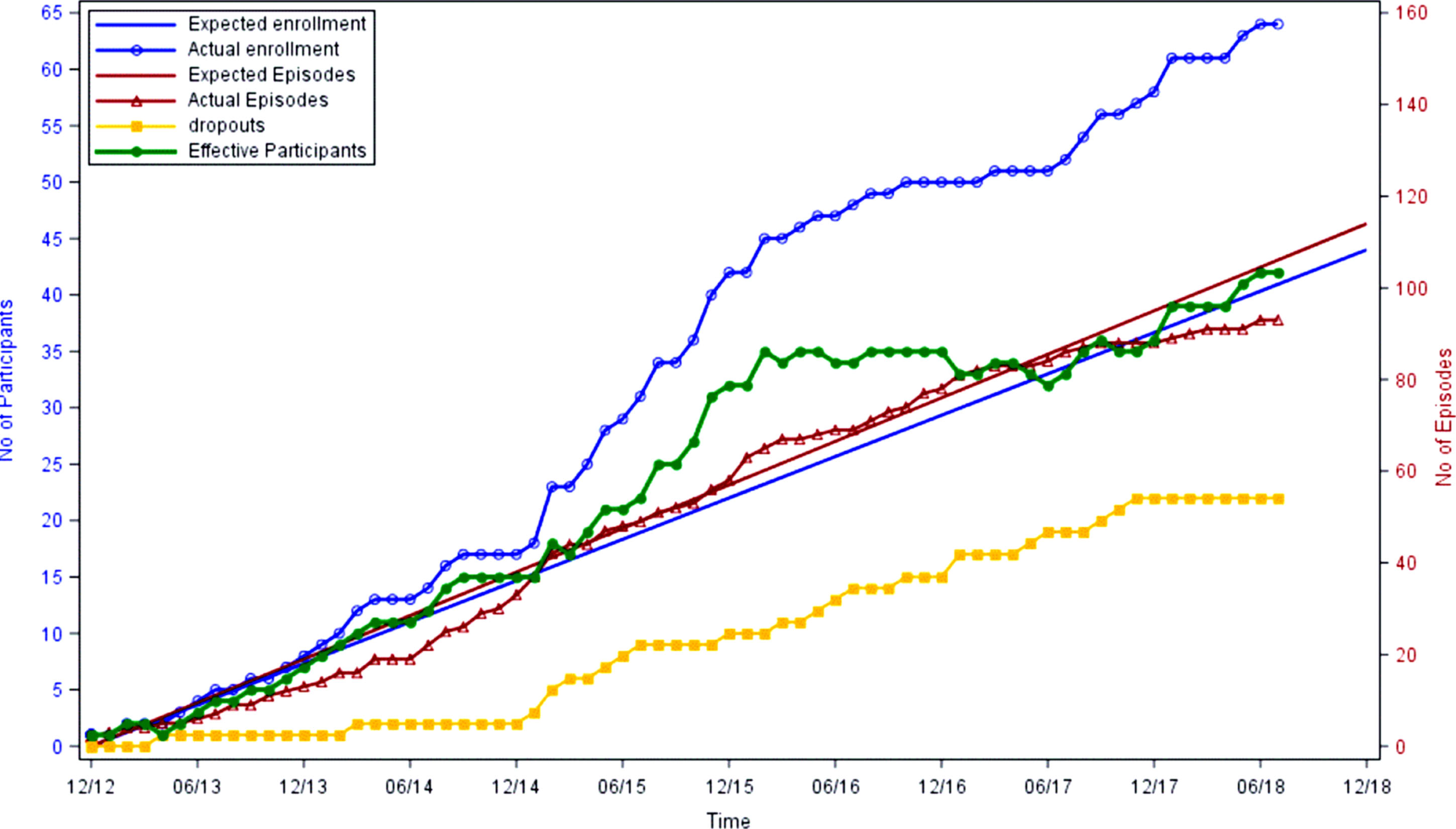
As of July, 2018, 61 out of 123 patients who met full eligibility criteria were enrolled. 22 (36%) of the 61 participants have dropped from the study, primarily due to liver transplantation. Figure 4 shows cumulative enrollment of participants as well as randomized HA hospitalization episodes. Although actual enrollment has exceeded projections, the active number of participants has plateaued since 2015 due to patient dropout. Accrual of study episodes has been relatively linear, with a slight increase during winter months.
Fig.4
Cumulative Enrollment, Episodes and Dropouts of the Short-Term Outcome trial.
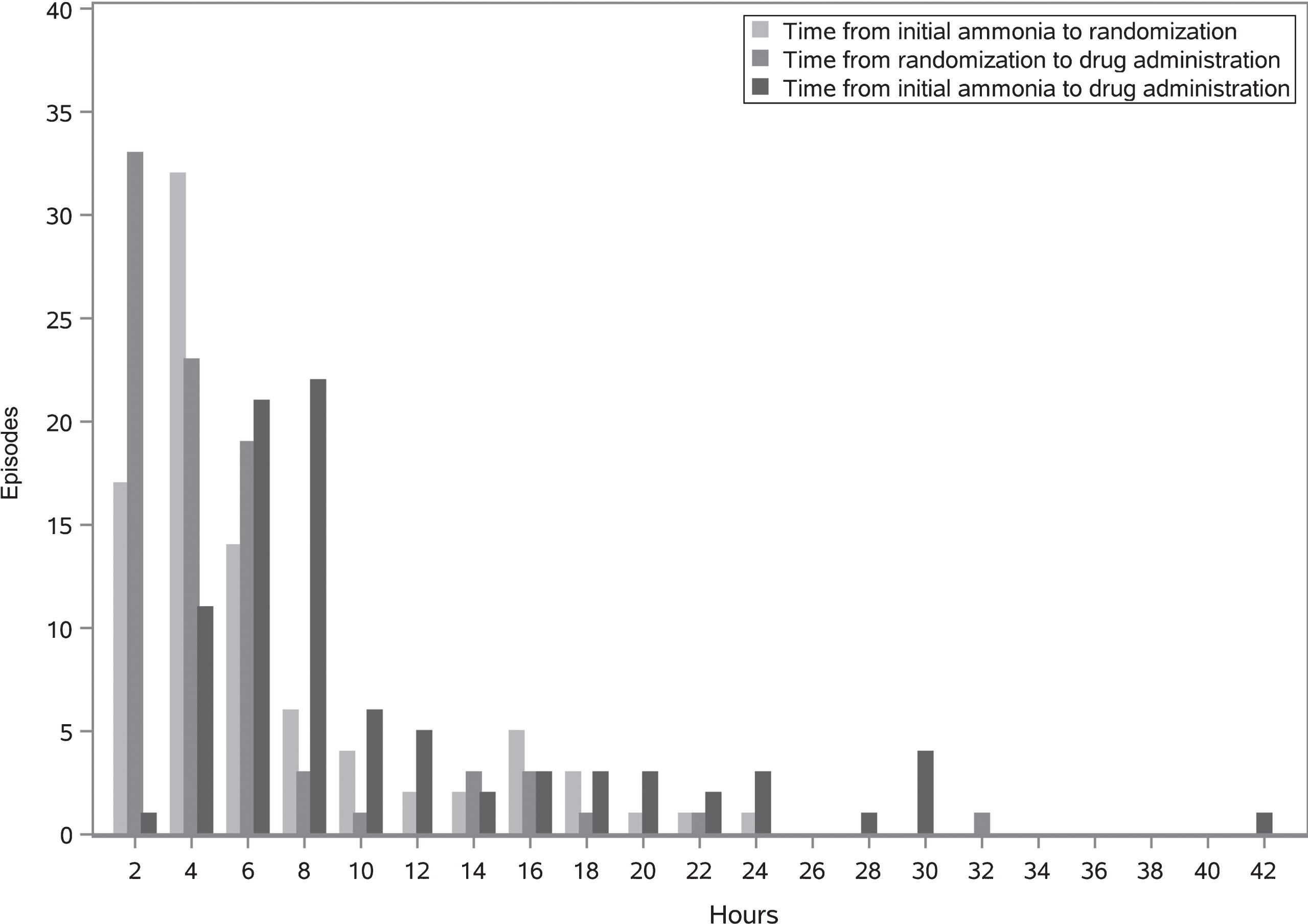
Time delay from initial qualifying ammonia level drawing until drug administration is shown in Fig. 5. Median time from ammonia level until randomization was 2.4 hours (range 0 –22.6). Median time from randomization to drug administration was 3 hours (range: 0.2 –31.2) (Fig. 5).
Fig.5
Time between initial ammonia draw, randomization and first administration of study drug for all episodes.
Plots of overall ammonia trajectory revealed that the decline was not linear. Instead, rates of ammonia decline varied at least by day, tending to plateau by day 3 following randomization, creating the need for piecewise regression models, which may vary by treatment group, to evaluate the hypothesized difference in the ammonia decline by treatment group. This unanticipated level of complexity made us reconsider the viability of the primary outcome.
The high variability in ammonia decline by day prompted us to choose an alternative composite primary outcome and modeling strategy., The composite outcome was time to reach an ammonia level of 50μmol/L or time to discharge, whichever occurred first and therefore the analysis approach used was a Cox regression model, which was simpler and required fewer assumptions. This change unburdens the primary outcome of serious modeling complexity while using a well-defined and more clinically-relevant outcome for comparison of treatment effects. The choice of 50μmol/L as the target for ammonia decline was based on the extensive experience of metabolic physicians that a patient reaching this level of ammonia is no longer at risk of acute hyperammonemic injury and would be a candidate for discharge in the absence of other complications. The original primary outcome of difference in ammonia trajectory was retained as a secondary outcome.
This change required re-evaluation of the adequacy of the study sample size to ensure at least 80% power to test the hazard ratio (HR) reflecting the difference by treatment group. Using the Cox Regression procedure in PASS we found that total sample sizes of 146, 114, and 93 episodes were sufficient to detect HR estimates of 1.6, 1.7, 1.8 and 1.9, respectively. The change allowed us to detect realistic and clinically meaningful differences at sample sizes smaller than our original projection resulting in a new target of 114 episodes after accounting for error spending associated with one interim analysis.
4Discussion
Studies of ultra-rare disorders face more challenges than those that merely meet the prevalence criteria for ‘rare disease’. The ultra-rare frequency of urea cycle disorders and organic acidemias impacted every aspect of the study, from recruitment, to regulatory compliance and protocol adherence, and importantly to the rate of accrual of eligible patient episodes.
In a hypothetical city with a population of 1 million, there would be approximately 650 affected with a disorder meeting the minimum CFR prevalence definition of ‘rare’. In contrast, there would only be 20 or fewer affected with a disorder meeting the proposed definition of ‘ultra-rare’ (1:50,000). The prevalence for PA, MMA, CPS1, and OTCD each meet this criterion, as their estimated birth incidences are all less than 1:100,000 live births [18–21], and many die in childhood. Additional eligibility criteria further reduce the number of prospective participants, not all of whom are willing to participate in clinical research. Each large academic institution joining the study would be expected to only add a small number of eligible participants. As a result, in our study, in 2014 when cumulative episodes continued to lag behind projections (Fig. 4), there was no choice but to add additional sites to the study in the hope of increasing enrollment and episodes. Recruitment may in fact be so challenging in the ultra-rare environment that in some studies, the number of sites exceeds the target number of enrolled participants (e.g., NCT02991144).
The extremely low incidence of ultra-rare disorders can add significant variability resulting in the study unfortunately hitting a recruitment ‘drought’. In the 3 years during which the LTO study was open, only 1 infant meeting eligibility criteria was born at all study sites. This was far fewer than historical numbers had predicted, and far fewer than have been born following closure of the LTO study. In the STO protocol, between the time that sites reported their potential subject numbers and the time that regulatory approval allowed recruitment, many of the most severely affected patients (and thus most likely to have eligible episodes of HA) either died or underwent liver transplantation. This loss of potential subjects, along with the natural variability of birth of additional eligible patients, combined to greatly slow accrual of episodes. Although neither could be considered unexpected, the time lag between conceptualization and planning of the study, and the beginning of enrollment, created a significant barrier to accrual.
Regulatory requirements in rare and more common disorders are similar, thereby creating a much higher ratio of administrative “overhead” effort-per-enrollee in rare disorders, whereas studies of common disorders benefit from economies of scale. It was also difficult to benefit from prior experience with study procedures. Hyperammonemia episodes happened so infrequently that study personnel would essentially have to re-learn the protocol details with every new episode.
4.1Site selection
Capitalizing on an existing network infrastructure can facilitate the establishment of a new clinical trial. In this study, 8 of 9 sites are members of the UCDC within the NIH Rare Disorders Clinical Research Network. These sites not only had an established working relationship with the lead site, Children’s National, but also had the requisite experienced personnel and facilities to conduct this acute intervention trial. Similarly, the Children’s Oncology Group (COG) maintains numerous active sites in the US and internationally, providing a source of centers equipped to launch trials of novel cancer therapies. In retrospect, the need for tighter timelines and the fact that most centers could enroll no more than a few subjects with any ultra-rare disorder, suggest that the reliance on a, geographically broad, consortium of referral centers for ultra-rare disease clinical trial centers would facilitate timely deployment of new protocols. Given their existing interests in rare disease, the consortia of the Rare Diseases Clinical Research Network may be ideally suited to serving as such clinical trial centers.
4.2Protocol development and regulatory issues
We aware that it was essential to start protocol development early and thus drew on prior experience based on establishing the Longitudinal Study of the UCDC, which took over 2 years to enroll its first participant. Protocol development for the studies described herein started nearly a year before NIH funding was secured and an initial draft was completed 8 months before NIH funding was started. This allowed us to enroll our first participant within 9 months of the funding start date.
With activation prior to the NIH-issued regulations regarding IRB reliance agreements, the studies were did not use a central IRB; instead, each site required individual IRB approval. As a result, site initiation was set back nearly a year due to the need to first have the final protocol approved at the lead site delaying application and approval at all other sites. Each site was also responsible for individually processing amendments and managing regulatory training of study staff. All protocol amendments were first drafted and approved at the Children’s National Coordinating Center before being distributed to other sites. In practice, this two-step process meant that sites were not always operating on the same protocol version.
The use of a central IRB of record would likely have expedited activation of all sites, and eliminated the complexities of having various sites operating under different versions of the protocol. The recent National Institutes of Health Policy, mandating the use of a single IRB for multi-site NIH-funded studies, has now made the use of a central IRB common. Harmonization of this policy across other sources of federal funding, such as the Patient-Centered Outcomes Research Institute (PCORI) and the FDA may help to further reduce regulatory burden and decrease delays in activation of multi-site studies.
Median time from initial IRB application until final IRB approval (68 days; Table 1) was in fact longer than prior published mean times in interventional trials (38-42 days [22–24]). Although in some cases, this length of time was increased due to personnel turnover, it may also highlight the difficulties that Institutional Review Boards have with understanding rare genetic disorders which they may not have previously encountered.
4.3Study Design
In rare disorders, scientific evidence-based practice is weak. Although treatment guidelines have been published, there is no agreed-upon standard of care for acute management of these disorders [25–27], and management varies greatly between metabolic centers. Although perhaps semantic, the term ‘standard care’ was instead employed, and essentially describes that each center was to utilize its routine management approach to treat acute hyperammonemia in addition to the trial’s random drug assignment. The multiple balanced crossover design will greatly reduce any bias introduced by differences in management between centers.
One issue of debate was the use of ammonia scavenging medications to treat acute hyperammonemia in PA and MMA. These medications were approved for use in UCDs, but would be considered ‘off-label’ investigational treatment of organic acidemias. In the organic acidemias, these medications may even have adverse effects. Phenylacetate or its derivatives are the ammonia scavengers of choice in the US. These medications can deplete the glutamine/glutamate pool in patients who have low glutamine levels and who may rely on these stores for anaplerosis of the TCA cycle [25]. Additionally, use of ammonia scavengers may deplete the already diminished pool of free Coenzyme-A, potentially worsening the mitochondrial dysfunction and the acidosis in the organic acidemias [28]. Inconsistent use of ammonia scavengers between sites or even within a site would also introduce a serious statistical and regulatory confounder. Therefore, ammonia scavenging medications were not permitted to be used in the protocol for PA and MMA, and all participating sites agreed to avoid ammonia scavenger use in patients with organic acidemia enrolled in these trials.
It is important that industry has had no role in study design or oversight. Orphan Europe committed to provide study drug and placebo for the study, and neither Recordati, nor its subsidiaries Recordati Rare Diseases, or Orphan Europe, the distributor of N-carbamylglutamate (Carbaglu), had any other role. The company also has committed to provide funding to complete the study when NIH funding ended. Nevertheless, the company remains at arm’s length from study conduct and direction and will have no role in the analysis or interpretation of results. Instead, the study team intends to publish our results in the public peer-reviewed literature, which Orphan Europe may then use as additional supportive data if they seek expansion of Carbaglu’s current indication.
As the primary purpose of our clinical trials was scientific rather than regulatory, the study team did not meet with the FDA review divisions prior to initiation of the study. However, 3 years after subjects had been enrolled into the study, the study team was made aware that cardiac safety data might be important for regulatory approval of any new drug or indication. Given that it is exceedingly unlikely that a separate placebo-controlled RCT would be initiated for the regulatory approval of Carbaglu in acute hyperammonemia, we opted to include pre- and post-drug EKGs as a safety measure for this study. In retrospect, even in the absence of primary regulatory objectives for this study, consultation with the FDA during the planning phase of the study may have obviated the need for a late change to the study safety measures.
There are few validated measures of clinical benefit, and none which apply to children with intoxication-type inherited metabolic disorders. We therefore pioneered the use of the Functional Status Score (FSS) in this study, which was originally designed for the critical-care patient population, and thus not ideally tailored to our patients. For instance, in general, affected participants do not have lung parenchymal involvement; therefore, participants are virtually always on room air, unless they require intubation for decreased mental status. This study, therefore typically only utilizes a narrow range of FSS. Additionally, in other categories such as mental status or feeding, the category seems insufficiently detailed to provide a refined categorization of their status.
4.4Patient advocacy organizations
The support of patient advocacy organizations is critical to the success of any clinical trial in rare diseases. The National Urea Cycle Disorders Foundation, Organic Acidemia Foundation, and Propionic Acidemia Association are among the most active rare disease patient advocacy organizations. Our study team was invited to present and recruit at each of their annual meetings and successful recruitment and conduct of this study would not be possible without the strong support of these organizations.
4.5Participant enrollment
As the number of affected patients with these particular ultra-rare diseases is low, any reduction in enrollment or missed episodes has profound and serious impact on the study. For instance, liver transplant performed in participants greatly impacted our active participant numbers since participants become ineligible once transplanted (Fig. 4).
Concurrent studies which compete for the same limited pool of participants may potentially result in recruitment failures for all studies. It also creates logistic and ethical issues regarding which studies are initially offered to patients. Each institution needed to determine a policy for resolving such issues, and in some situations individual investigators in the same institution might have different approaches. At Children’s National, for example, there was an industry-sponsored study and an investigator-initiated study, both offering a novel therapeutic. This created a conflict regarding study choice, as there was some overlap in participant eligibility. To alleviate this conflict, we ultimately developed a policy to first offer enrollment in the NIH-funded NCGC study, given the institutional commitment based on receiving federal funding.
Off-label prescription of the NCG also reduced availability of participants. Based only on anecdotal evidence of NCG efficacy in study disorders, some otherwise eligible patients were already prescribed chronic daily doses of the drug, thereby making them ineligible for our study. Desire in the medical community to offer new treatments when the list of approved options is limited, pressure from patients [29] to receive new therapies, coupled with seeking out physicians willing to prescribe novel therapeutics ‘off-label’, all contribute to this problem.
Enrollment was typically performed during routine outpatient clinic visits, which required the site physicians to be aware of the study and to remember to refer their patients to the site PI. Despite prioritizing enrollment in eligible patients not in crisis, occasionally, the opportunity was missed owing primarily to the focus on clinical care during long outpatient clinic visits in patients with complex multi-system disorders. Enrollment during non-crisis situations was preferred over approaching patients in crisis during a hyperammonemic episode, which would not only prolong the time to randomization, but also made recruitment more difficult because of the perceived coercive component when the prospective participant was critically ill. These issues contributed to the problem that anticipated episode numbers were higher than actual. In addition, reduced incidence of HA as patients age and become more stable over time, as well as increased use of liver transplantation, notably decreased the number of eligible participants.
4.6Episode capture
These disorders are rare, and the occurrence of hospitalizations for hyperammonemia is infrequent. Consequently, establishing a commonly accessible working document which outlines the workflow was helpful to facilitate the capturing of an episode. Our experience suggests that the most hospitalizations for hyperammonemia occur on evenings or weekends, rather than during typical work-week hours. This introduces an additional logistical challenge as the study personnel (e.g., physicians, coordinators, pharmacists) must be available and ready at any time for a very low-likelihood, yet highly stressful, urgent, and time-consuming event. We also observed that even the largest metabolic centers have limited investigational pharmacy coverage during off-hours.
Most episodes were identified when a participant arrived to the emergency department (ED). Families were instructed to inform hospital staff that they were enrolled in a research study. Additionally, participants were provided with wallet-sized ‘notification cards’ during enrollment, which indicated how to reach study personnel. However, this strategy rarely resulted in the alerting of study staff, as participants either forgot to bring their notification card, or failed to alert ER staff. Some, but not all, study sites have alerts embedded within their electronic health record systems that notify personnel that a presenting patient is enrolled in a research study. Nevertheless, no system is perfect and thus the study relies heavily upon a sequence of actions when an enrolled study participant presents with criteria for an eligible episode. The ED alerts the on-call metabolic physician. If the on-call metabolic physician is not a study investigator, he/she alerts study personnel either before or after a qualifying ammonia level has been determined. Unless the patient is already enrolled, study personnel must consent and enroll the participant at that time. The study investigator must verify that the participant’s episode meets eligibility criteria and randomize the participant using the online portal (REDCap). The investigational pharmacy must then be informed to prepare study drug based on the random assignment. Off-hours, this may require the on-call unblinded investigational pharmacist to instruct an on-site inpatient pharmacist how to interpret the coded random assignment and access the appropriate study drug. Each site has its own process for ensuring this is done in a timely manner. Once the study drug is prepared, it must be brought to the participant’s bedside maintaining chain-of-custody and validated by signature (i.e., cannot be sent via pneumatic tube). This often requires the participant’s ED nurse or investigator to retrieve the medication from the inpatient pharmacy.
This sequence of events not only impacts whether the participant is enrolled and how quickly, but also time to the participant receiving study drug (Fig. 5). Each person involved in this sequence potentially adds delays to the process. Episode eligibility is dependent on a clinical laboratory ammonia result, which is highly prone to pre-analytical error, frequently questioned, requiring a request for a repeat sample.
4.7Statistical challenges and considerations
Because of the lag in episodes, the interim analysis was delayed well beyond the scheduled (calendar) halfway point of the study. Concern mounted that delaying the interim analysis until 72 episodes had been randomized posed a fairness problem for enrollees, especially if the study needed to be stopped due to futility. It was decided therefore, to conduct the interim analysis at the point when 60 episodes were accrued. After the review of the report and the interim analysis, the DSMB voted to continue the trial, which remains in process.
4.8Financial considerations
This study was funded primarily through an NIH R01 grant award; however, it would not have been possible without cost sharing involving supplementary philanthropic funds. Even then, for this study to reach completion, we have received additional funding from Recordati Rare Disease. Although this phase II/III study will be completed at a fraction of the cost of an equivalent industry-sponsored trial, these costs are still considerable and probably prohibitive for investigator-initiated trials for ultra-rare disorders. Given the fixed start-up costs at each site, clinical trials in ultra-rare diseases bear a very high cost per enrolled participant. Additionally, because the actual yearly enrollment numbers for such ultra-rare disease studies are small, the start-up effort much exceeds the efforts needed to maintain the study, and therefore this was reflected in the yearly effort compensation. This point suggests strongly that meaningful evidence based medicine in ultra-rare diseases will require the development of pre-existing clinical research networks, with robust data collection mechanisms and integrated regulatory management systems, and with tight control of overhead cost. Such a structure would greatly benefit patients and patient care, healthcare payers, and industry partners, as has been seen in the field of childhood cancer.
4.9Single points of failure
This study has had multiple potential points of failure. As a result, it is extremely important to have (preferably multiple) motivated study coordinators and investigators, who are committed to overcoming inevitable breakdowns. Ideally, having someone ‘on call’ for the study (i.e., reachable at any time) allows for the highest chance of successful capturing of episodes. A member of the Coordinating Center carries a beeper and is available to assist sites when questions or challenges arise.
The utilization of a home-grown study information system, the Children’s Hospital Research Information System (CHRIS), to support randomization and data capture was tailored to this study. It was easy to make changes in and had customizable functionality. However, when the lead programmer for this system was incapacitated, we quickly needed to find another solution. REDCap, a commonly used research data capture system, was then implemented, which while customizable, was not perfectly tailored to this study. It required custom changes to support the multiple crossover function, as well as, to address other challenges imposed by this unique application. REDCap is still not infallible: when there is a network failure or update, there needs to be a back-up system in place to cover enrollment during off-hours. In addition, we assigned an on-call person and later back-up on-call persons with access to randomization charts to provide the coded drug/placebo assignment by telephone.
4.10Motivation of study members
In addition to providing ample effort compensation for both primary investigator and study coordinator, this study offered generous compensation for capturing episodes. However, this remuneration was insufficient to ensure capturing of all possible episodes, as retrospective review identified a number of episodes that were missed. The threat of probation also did not improve site performance by those sites unable to capture episodes. Instead, face-to-face engagement proved to be the single most important motivating factor. There was a distinct uptick in study enrollment and episode capture immediately following annual in-person meetings of the NCGC that was far greater than the limited impact of monthly phone calls with sites.
Acknowledgments
This work was funded by R01-HD058567 grant (Mendel Tuchman, PI) from the Eunice Kennedy Shriver National Institute of Child Health and Human Development. This work was also generously supported by the O’Malley Family Fund. NAM’s work was supported by the Rashid Family Fund. N-Carbamylglutamate and placebo was provided by Orphan Europe.
We thank the NCGC study coordinators, Angela Leshinski, Arang Kim, Audrey Lynn, Genevieve Nesom, Jennafer Dotson, Kimberly Wallis, Kyla Almeida, Leslie Atley, Luca Fierro, Rebecca Signer, Sandra Braden, Sandra Yang, Thu Quan, and Vera Anastasoaie, for their expert support.
References
[1] | Mak C.M. et al., Inborn errors of metabolism and expanded newborn screening: Review and update, Crit Rev Clin Lab Sci 50: (6) ((2013) ), 142–162. |
[2] | ORPHAN DRUGS, 21C.F.R. §316.10, ((1993) ) https://www.ecfr.gov/cgi-bin/text-idx?c=ecfr&SID=51cf70689d51f0ea4147c0a8ac649321&rgn=div5&view=text&node=21:5.0.1.1.6&idno=21. |
[3] | REGULATION (EC) No 141/OF THE EUROPEAN PARLIAMENT AND OF THE COUNCIL of of 16 December 1999 on orphan medicinal products. https://ec.europa.eu/health/sites/health/files/files/eudralex/vol-1/reg_2000_141_cons-2009-07/reg_2000_141_cons-2009-07_en.pdf. |
[4] | Hughes D.A. , Tunnage B. , Yeo S.T. Drugs for exceptionally rare diseases: Do they deserve special status for funding? QJM 98: (11) ((2005) ), 829–836. |
[5] | Kolker S. et al., The phenotypic spectrum of organic acidurias and urea cycle disorders. Part 1: The initial presentation, J Inherit Metab Dis 38: (6) ((2015) ), 1041–1057. |
[6] | Ah Mew N. , et al. Urea Cycle Disorders Overview, in GeneReviews((R)), M.P. Adam , et al., Editors. (2017) : Seattle (WA). |
[7] | Dercksen M. et al., Inhibition of N-acetylglutamate synthase by various monocarboxylic and dicarboxylic short-chain coenzyme A esters and the production of alternative glutamate esters, Biochim Biophys Acta (2013) . |
[8] | Valayannopoulos V. et al., Carglumic acid enhances rapid ammonia detoxification in classical organic acidurias with a favourable risk-benefit profile: A retrospective observational study, Orphanet J Rare Dis 11: ((2016) ), 32. |
[9] | Abacan M. , Boneh A. Use of carglumic acid in the treatment of hyperammonaemia during metabolic decompensation of patients with propionic acidaemia, Mol Genet Metab 109: (4) ((2013) ), 397–401. |
[10] | Gebhardt B. et al., N-carbamylglutamate enhances ammonia detoxification in a patient with decompensated methylmalonic aciduria, Mol Genet Metab 79: (4) ((2003) ), 303–304. |
[11] | Yap S. et al., N-Carbamylglutamate Is an Effective Treatment for Acute Neonatal Hyperammonaemia in a Patient with Methylmalonic Aciduria, Neonatology 109: (4) ((2016) ), 303–307. |
[12] | Coude F.X. , Sweetman L. , Nyhan W.L. Inhibition by propionyl-coenzyme A of N-acetylglutamate synthetase in rat liver mitochondria, A possible explanation for hyperammonemia in propionic and methylmalonic academia, J Clin Invest 64: (6) ((1979) ), 1544–1551. |
[13] | Ah Mew N. , et al. N-carbamylglutamate augments ureagenesis and reduces ammonia and glutamine in propionic academia, Pediatrics 126: (1) ((2010) ), e208–e14. |
[14] | Pollack M.M. et al., Functional Status Scale: New pediatric outcome measure, Pediatrics 124: (1) ((2009) ), 18–28. |
[15] | Seminara J. et al., Establishing a consortium for the study of rare diseases: The Urea Cycle Disorders Consortium, Mol Genet Metab 100: (Suppl 1) ((2010) ), S97–105. |
[16] | McCarter R. , et al. An Epidemiological Research Information Management System, in 16th Annual International Conference of the IEEE Engineering in Medicine and Biology Society (1994) . Baltimore, MD. |
[17] | Harris P.A. et al., Research electronic data capture (REDCap)–a metadata-driven methodology and workflow process for providing translational research informatics support, J Biomed Inform ((2009) ) 42: (2), 377–381. |
[18] | Lichter-Konecki U. et al., Ornithine Transcarbamylase Deficiency, in GeneReviews((R)), M.P. Adam , et al., Editors. (2016) : Seattle (WA). |
[19] | Manoli I. , Sloan J.L. , Venditti C.P. Isolated Methylmalonic Acidemia, in GeneReviews((R)), M.P. Adam , et al., Editors. (2016) : Seattle (WA). |
[20] | Shchelochkov O.A. , Carrillo N. , Venditti C. Propionic Acidemia, in GeneReviews((R)), M.P. Adam , et al., Editors. (2016) : Seattle (WA). |
[21] | Summar M.L. et al., The incidence of urea cycle disorders, Mol Genet Metab 110: (1–2) ((2013) ), 179–180. |
[22] | Kastner B. et al., Clinical Research in Vulnerable Populations: Variability and Focus of Institutional Review Boards’ Responses, PLoS One 10: (8) ((2015) ), e0135997. |
[23] | Stair T.O. et al., Variation in institutional review board responses to a standard protocol for a multicenter clinical trial, Acad Emerg Med 8: (6) ((2001) ), 636–641. |
[24] | Mansbach J. et al., Variation in institutional review board responses to a standard, observational, pediatric research protocol, Acad Emerg Med 14: (4) ((2007) ), 377–380. |
[25] | Baumgartner M.R. et al., Proposed guidelines for the diagnosis and management of methylmalonic and propionic academia, Orphanet J Rare Dis 9: ((2014) ), 130. |
[26] | Chapman K.A. et al., Acute management of propionic academia, Mol Genet Metab 105: (1) ((2012) ), 16–25. |
[27] | Haberle J. et al., Suggested guidelines for the diagnosis and management of urea cycle disorders, Orphanet J Rare Dis 7: ((2012) ), 32. |
[28] | Al-Hassnan Z.N. et al., The relationship of plasma glutamine to ammonium and of glycine to acid-base balance in propionic acidaemia, J Inherit Metab Dis 26: (1) ((2003) ), 89–91. |
[29] | Pontes M.C. , Pontes N.M. To treat or not to treat: Evidence for “treatment bias” when consumers evaluate physicians, Health Mark Q 20: (4) ((2003) ), 43–61. |


