
Regenerative Medicine Therapies for Rare Diseases
Abstract
The field of regenerative medicine is growing rapidly with the introduction of new therapies that have the potential to treat and cure serious medical conditions, including rare diseases, for which there are no available treatments. In the United States, the development of novel medical products is regulated and guided by the Food and Drug Administration (FDA). As scientific and technological advances are discovered and adopted by the medical industrial enterprise, the FDA’s implementation of policies that create a climate conducive to safe development and rapid availability of novel medical products is one of the pillars which support the Agency’s mission of protecting and promoting the public health. With advancements in cell modifications and tissue engineering, innovative creation of biomaterials, adoption of three-dimensional bioprinting, and rapid development of human genome editing technologies, the need for Agency’s work in ensuring that its science-based policies remain relevant and helpful in facilitating the availability of new treatments to the most vulnerable populations of patients becomes more pressing than ever before.
In December 2016, Congress amended section 506 of the Food, Drug, and Cosmetic (FD&C) Act [21 U.S.C. 356] by adding a new section 506(g), which defines the categories of products considered to be regenerative medicine therapies. As further described by FDA [1], regenerative medicine therapies are considered to include cell therapies, therapeutic tissue engineering products, human cell and tissue products, and combination products using any such therapies, as well as gene therapies, including genetically modified cells that lead to a durable modification of cells or tissues. The development and approval of regenerative medicine therapies are regulated by FDA’s Office of Tissues and Advanced Therapies (OTAT) in the Center for Biologics Evaluation and Research (CBER).
In this review article, we present practical considerations for investigating regenerative medicine therapies intended for the treatment of rare diseases. The material presented may be useful to researchers who are undertaking the challenging task of finding and delivering new treatments for those in need.
1.Introduction
The Orphan Drug Act of 1983 [2], defines a rare disease by a prevalence of less than 200,000 individuals (∼6 per 10,000 based on ∼325 million population). There are approximately 7000 different rare diseases described which are estimated to collectively affect about 30 million people in the United States. Notwithstanding the considerable demand, pharmaceutical product development for the treatment of rare diseases is fraught with multiple challenges, some unique to individual product development programs and others common across different programs. These various issues include a range of problems that present specific challenges, for example: (1) lack of sufficient knowledge about the disease’s genotypes, phenotypes, and course of progression; (2) lack of understanding of the disease’s underlying molecular mechanisms and the resulting difficulties with finding the right pathways for therapeutic interventions; (3) scarce or absent clinical scientific, psychometric, and patient experience data to support the use of valid, reliable, and clinically meaningful outcome measures that are sensitive to change and could be employed as trial endpoints; (4) rarity of individual conditions and the resulting difficulties with finding and recruiting patients in trials throughout the world; (5) delayed diagnosis due to difficulties in recognizing a rare disease; (6) challenges with identifying clinical care specialists who can serve as qualified clinical investigators in product programs; (7) need to adjust product manufacturing throughout development and the resulting challenges with demonstrating product comparability, which may have disproportionate impact on small-size development programs; (8) lack of knowledge of the regulatory policies for product development on the part of some stakeholders; and (9) limited resources to support data acquisition, patient outreach, and the logistics of product development.
Along with the many challenges, the rare disease field holds unprecedented opportunities for the development of novel treatment technologies, advanced platforms for delivery of biological therapies, and a high potential for stimulation of innovation and flexibility in the application of regulatory frameworks to medical product development. Owing to much advancement in medical sciences, the rare disease field is quickly becoming a fruitful soil for new developments in precision medicine where stratification of therapies may be employed based on groupings of genetic and phenotypic characteristics and where individualized treatments may be developed through modifications of patient’s own (autologous) cells and tissues. A new technology initially targeted to treat a small population of patients with one disease can find other uses and stimulate innovations in other therapeutic areas. It is a field where product development and manufacturing are often planned with consideration for global marketing. As previously reported [3, 4], regulatory flexibility has been applied to rare diseases with continuous collaboration and support from major regulatory agencies around the world. In the United States, financial incentives in the form of tax credits for clinical research, waivers for marketing application user fees, and additional years of marketing exclusivity, along with other incentives, are available to those embarking on the journey of finding a new treatment for a rare disease. Thirty five years following the enactment of the Orphan Drug Act, medical product development for rare diseases continues to attract unprecedented support from a wide range of stakeholders.
2.Regenerative medicine therapies
Many rare diseases result from inherited or spontaneously occurring genetic mutations, which can occur in both germ-line and somatic cells. Hence, discovery and manufacture of cell-based therapies and gene therapy products may hold promise for developing effective treatments and discovering cures for many rare diseases.
The 21st Century Cures Act was signed into law in December 2016 [5] and included provisions which, building on the FDA’s previous work, spurred policies supporting and encouraging scientific and technological advances in medical product development and, specifically, stimulating the development of regenerative medicine therapies. This article discusses several of these provisions and programs, along with their application to therapies investigated for the treatment of rare diseases, including: the regenerative medicine advanced therapy designation program, the approach to regulation of devices used with regenerative medicine advanced therapies, the rare pediatric disease voucher program, the FDA’s research grant program for natural history studies, and the initiatives supporting incorporation of patient input into medical product development. The article also includes some noteworthy considerations for conducting investigations with regenerative medicine therapies.
3.Regenerative Medicine Advanced Therapy (RMAT) program
The RMAT designation program constitutes the latest addition to the armamentarium of expedited development and review programs [6] available to sponsors of promising products for the treatment of serious conditions with unmet medical needs. In contrast to the Fast Track (FT) and Breakthrough Therapy (BT) expedited programs, which apply to both small molecule-based drugs and biological products, the RMAT designation program applies only to products that are regenerative medicine therapies (see the definition above). By the same token, a regenerative medicine therapy that is developed for a serious condition can be eligible for any expedited program and be granted a respective designation (i.e., FT, BT, and RMAT), provided that the expedited program’s specific criteria are met. A detailed explanation of the expedited programs other than the RMAT designation program is beyond the scope of this article; and the readers are referred to the FDA’s guidance on this topic [6].
A product eligible for RMAT designation must meet the definition of a regenerative medicine therapy (see the definition above), be intended to treat, modify, reverse, or cure a serious or life-threatening disease or condition, and have preliminary clinical evidence indicating the product’s potential to address unmet medical needs for such disease or condition [5]. As described by FDA [1], the sources of clinical evidence to support RMAT designation may range from randomized controlled trials, to open-label studies, to well-designed retrospective studies, and, in some cases, to clinical case series that provide compelling data collected systematically. Further [1], factors that may be considered in the assessment of the clinical evidence often include the rigor of data collection, the quality of the study conduct, the number of investigational sites contributing to the evidence, and the severity, prevalence, and rarity of the condition. Sponsors of programs that receive RMAT designation have the opportunity for early and frequent interactions with OTAT to obtain advice on efficient product development, including early interactions to discuss any potential surrogate and intermediate trial endpoints to be used to support accelerated approval [5]. Meetings with FDA can be requested throughout all stages of product development [33] and, when requested early, can be beneficial in reducing regulatory uncertainty and making strategic decisions for research and resource allocation, particularly in the context of rare disease programs with novel regenerative medicine products. In addition, early non-binding, regulatory advice can be obtained from OTAT through the Initial Targeted Engagement for Regulatory Advice on CBER products (INTERACT) program [39], which can be used to discuss early preclinical, manufacturing, or clinical aspects of product development.
Regenerative medicine advanced therapies may be used with medical devices employed for product delivery, for example, devices used for infusion, injection, topical application, inhalation, or other types of delivery. In addition, devices may be used for recovery or isolation of cells or tissues during the manufacturing of a regenerative medicine therapy. As outlined by FDA [31], devices used for isolation, recovery, and delivery of RMAT products may be developed through different regulatory pathways [7], with application of the least burdensome approach [8] to the regulation of such devices. Devices of different types, ranging from simple to complex (e.g., intra-ventricular delivery systems) may be co-developed or used with various RMAT products. The device characteristics and the level of evidence needed to show the device’s safety and effectiveness are often based on the specific RMAT product’s characteristics and its conditions of manufacturing and administration [31].
Stakeholders developing regenerative medicine therapies for the treatment of rare diseases may consider the RMAT program as one potential tool to aid in product development.
4.Prescription Drug User Fee waiver and Rare Pediatric Disease Review Voucher program
The Prescription Drug User Fee Act (PDUFA) was created by Congress in 1992 and authorizes FDA to collect fees from companies that produce certain human drug and biological products [32]. Section 736(d) of the Food, Drug, and Cosmetic Act (FDCA) provides that FDA will grant a waiver of or reduction in one or more user fees where the Agency finds that an applicant meets the eligibility criteria [38]. To support the development of new medical products for rare diseases, under Section 736(a)(1)(F) of the FDCA, FDA will waive an application fee for an application for a human drug or biological product that has been designated as a drug for a rare disease or condition under section 526 of FDCA [27] (orphan drug designation), unless the application includes an indication for other than a rare disease or condition. In addition, FDA provides support to small businesses and grants user fee waivers to applicants who are small business entities, provided that certain criteria are met [32]. FDA guidance [38] describes how user fee waivers, reductions, and refunds may be requested.
The Rare Pediatric Disease Priority Review Voucher program is intended to encourage the development of safe and effective drug and biological products, including regenerative medicine therapies, for rare pediatric diseases. A rare pediatric disease meets the definition of a rare disease under Section 526 of the Food, Drug, and Cosmetic Act [27] and is “ … serious and life-threatening in which serious and life-threatening manifestations primarily affect individuals aged from birth to 18 years, including neonates, infants, children, and adolescents” [9]. Under the Rare Pediatric Disease Priority Review Voucher program, FDA can award a priority review voucher to a sponsor who receives marketing approval for a drug or biological product, including a regenerative medicine therapy, for a rare pediatric disease. Such a voucher can be redeemed to receive a priority review (i.e., on shortened timelines) of a subsequent marketing application for a different product [10], which may or may not be for treatment of a rare disease, a pediatric indication, or a regenerative medicine therapy. As of September 2018, FDA has issued 15 Rare Pediatric Disease Priority Review Vouchers, including for two products recently approved by OTAT for the treatment of rare pediatric diseases. These two products are Kymriah (tisagenlecleucel), CD19-directed genetically modified autologous chimeric antigen receptor (CAR) T-cell immunotherapy, approved for patients up to 25 years of age with B-cell precursor acute lymphoblastic leukemia that is refractory to treatment or in second or later relapse, and Luxturna (voretigene neparvovec), adeno-associated virus vector-based gene therapy, approved for the treatment of patients with confirmed biallelic RPE65 mutation-associated retinal dystrophy. Additional information about the voucher program and examples of products that have been granted vouchers can be further obtained from publicly available sources [10, 34].
5.Natural History studies
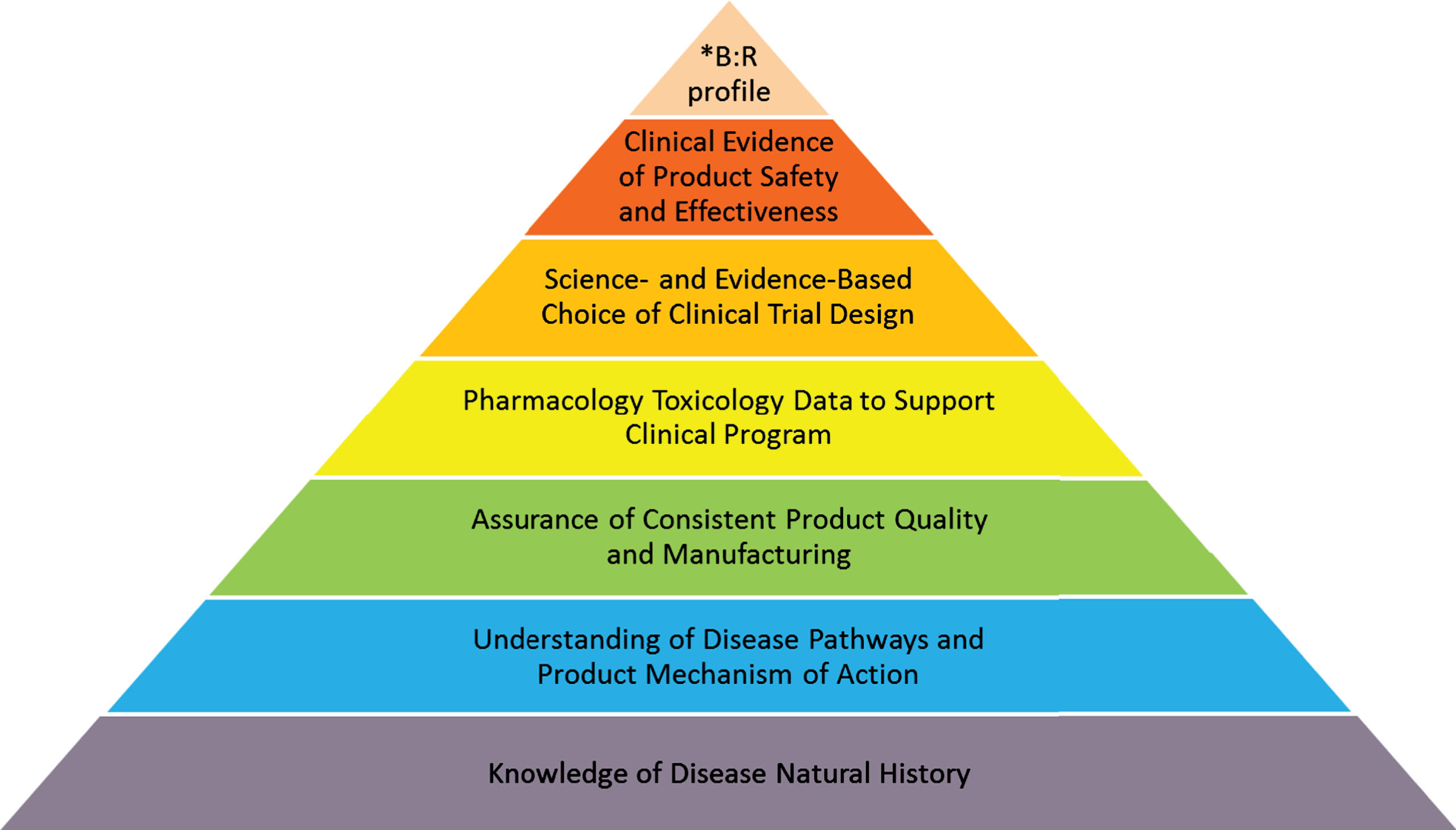
The natural history of a disease has been referred to as the progression of a disease process in an individual over time, in the absence of treatment [28]. It is not surprising that there is generally limited information about the natural history of most rare diseases. The geographic dispersion of disease incident cases, frequent lack of timely diagnosis, and scarcity of validated methods to assess specific conditions are just a few issues that frequently hinder collection of natural history data for many rare diseases. In small patient populations, piecemeal and anecdotal evidence of efficacy is often far more prevalent than adequately designed studies. Yet, a good understanding of a disease’s natural history is fundamentally important for any medical product development program (see Fig. 1).
Fig. 1.
Product Development Program: Building Blocks.
B: R – benefit: risk.
A well-designed natural history (NH) study is a great opportunity to collect credible data about the full spectrum of disease phenotypes and genotypes, intra- and interpatient variability in the disease course, the sequence of appearance of disease features, and the rate of disease progression. NH studies help identify disease prognostic factors, important serological, tissue, and imaging biomarkers, along with clinical endpoints reflective of disease outcomes, thus providing important information and tools utilizable in clinical trials. Furthermore, data collected from NH studies may help with learning the dynamics of laboratory and clinical changes that, in turn, may be useful in identifying the point of a meaningful therapeutic intervention in the disease pathway by targeting the upstream and downstream effects of the complex chemical, metabolic, and epigenetic interactions. In addition, knowledge of different disease stages and their rate of progression may help identify the optimal time for an intervention in order to have the largest impact on the reversibility of the physiological and functional changes.
A new NH study may be designed as a prospective longitudinal observational study in a single-center or multi-center setting run by a team of experts, including patients. A NH study can decrease many risks of product development, guide recruitment into future clinical trials, and increase opportunities for diversifying the portfolio of products in development for a rare disease. NH studies often include evolving protocols with study objectives incorporating plans to refine data collection to ensure flexibility in addressing all anticipated purposes of the study. In some cases, a prospectively designed NH study that collects data on the same variables that are intended for clinical development may provide an adequate comparison to serve as a control to assess efficacy in clinical trials. In addition, data from NH studies may be leveraged for development of trial endpoints, including clinical, surrogate, and patient-reported outcomes, while ensuring that the chosen endpoints have the desired characteristics of reliability, feasibility, resistance to bias, clinical meaningfulness, sensitivity to change, and sufficient discriminatory capacity. Similarly, observations of disease progression in NH studies are useful to assess the background risks associated with an untreated rare disease and, thus, provide context for assessment of potential risks from future treatments.
Regenerative medicine therapies may have prolonged effects on cells and tissues; therefore, observations of a disease’s natural course can be important for the interpretation of such prolonged treatment effects. However, in some cases, incorporating data from previously conducted natural history studies into development programs for regenerative medicine therapies may be challenging because of insufficient previous data collection, particularly for newly discovered cellular, tissue, and genetic parameters that may be affected by the prospective treatments. When developing a regenerative medicine therapy for a rare disease, it is helpful to consider use of data from NH studies and collect relevant information in the early stages of product development.
FDA offers a grant program intended to fund NH studies for rare diseases [11]. The goal of the program is to support studies that advance rare disease medical product development through characterization of the natural history of rare diseases, identification of genotypic and phenotypic subpopulations, and assessment and validation of clinical outcome measures, biomarkers, and companion diagnostics. For example, several such grants have been awarded by FDA to conduct NH studies with rare diseases including studies in Friedreich’s ataxia, sickle cell anemia, Angelman’s syndrome, and Myotonic Dystrophy Type 1; the latter two given in collaboration with the National Institutes of Health (NIH), National Center for Advancing Translational Sciences (NCATS) [35]. In addition, some research projects with gene and cell therapy products have recently received grants through the FDA’s Orphan Products Clinical Trials Grants Program [37]. Examples of such grants include funding for clinical trials investigating talimogene laherparepvec for the treatment of advanced pancreatic cancer, genetically modified autologous fibroblasts for the treatment of dystrophic epidermolysis bullosa, and CD-8 reduced T cells for the treatment of acute myeloid leukemia [36].
6.Patient engagement
Patients who live with a disease have a direct stake in development of new treatments, especially for rare diseases for which there are no available treatments. Engaging patients as partners in product development is important to better understand the disease and patient perspectives, while assessing effects of new treatments. Patients and caregivers are often the best advocates for raising awareness and improving treatment options for their disease, for describing the clinical meaningfulness and life- or activity- changing impacts related to new treatments, and for providing context for development of trial endpoints as measures of disease improvement. Patients with rare diseases and their caregivers can give valuable input when FDA evaluates risk tolerance in the context of a specific disease and its available treatments, as part of the benefit-risk profile for a novel intervention.
In recent years, much attention has been given to developing methodological approaches to collect patient experience data throughout the drug development process, to implement methodological considerations for collection, reporting, management, and analysis of patient experiences and preferences related to therapies, and to systematically incorporate patient input into clinical trials with subsequent application to regulatory decision-making. The Office of Tissues and Advanced Therapies actively supports the continued progress in developing new medicines through rigorous data collection and systematic inclusion of patient experiences and preferences into the development of new therapies [12]. Patients may find it useful to engage in the Patient Representative Program offered by FDA [13] and participate in Advisory Committee meetings on individual products.
In February 2018, the National Organization for Rare Disorders (NORD) and FDA signed a Memorandum of Understanding (MOU) which defined the framework for collaboration between the two organizations. This MOU delineated shared goals to improve the safety of the American public when using FDA-regulated medical products and to promote scientific progress in innovation, patient education, drug safety communications, training, research, and adverse event reporting [14]. Under this MOU, FDA and NORD initiated a series of listening sessions where FDA’s review staff has an opportunity to hear from patient communities about their disease burden, risk tolerance, and impacts of both approved and investigational treatments on their diseases.
7.Special considerations for developing regenerative medicine therapies
As mentioned earlier, regenerative medicine therapies include cell therapies, therapeutic tissue engineering products, human cell and tissue products, and combination products using any such therapies or products, as well as gene therapies, including genetically modified cells, that lead to a durable modification of cells or tissues [1]. Cell therapies include stem cell products that may be derived, for example, from adult, perinatal, fetal, embryonic, or induced pluripotent cells; somatic functionally differentiated cells; and various types of other cell lineages such as antigen-presenting cells and lymphocytes. Depending on the source of the product and its potential recipient(s), regenerative medicine therapies that are cell or tissue-based may be autologous or allogeneic. Regenerative medicine therapies that involve genetic modifications made to cells and tissues either ex vivo (such as modifications made to cells outside of the body, as in the case of chimeric antigen receptor (CAR) T-cells) or in vivo (such as delivering a corrective gene to specific target cells, as in some products developed for the treatment of hemophilia) often require sophisticated product manufacturing and delivery with engineered viral vectors, plasmids, or other types of genetic materials.
The schematic representation of the process of product development for a rare disease is shown in Fig. 1 It emphasizes the foundational essence of understanding the natural history of the disease, augmented by building blocks of quality manufacturing, informative toxicology data sufficient to support safe administration of the investigational therapy to humans, evidence-based clinical trial design and selection of endpoints, and demonstration of the product’s effectiveness in the context of acceptable safety for the condition to be treated.
Development of regenerative medicine therapies for the treatment of rare diseases often presents unique challenges that are important to take into consideration early on. Like all other biologics, regenerative medicine therapies are expected to meet regulatory standards of quality, purity, safety, and potency [15, 16], be manufactured according to the Current Good Manufacturing Practice-CGMP [16] and conform to national and international guidelines for biologic product development [17]. Besides selecting an appropriate product source and measuring the product’s functional activity with a potency assay [21], it is important to delineate the different cell types present in the product. In addition, development and establishment of assays for product characterization, understanding and controlling product source-related variations in cellular products, and controlling impurities in the viral vectors can become essential for manufacturing quality products and reducing any potential for undesired adverse events. For tissue scaffolds and tissue-based constructs, product design aspects need to anticipate possible in vivo environmental conditions that can either help promote or impair product engraftment. To this end, establishing critical quality attributes for the product and critical process parameters for its manufacturing [22], while using appropriate specifications, are essential to ensure the quality of the final product. Under the CGMP, manufacturers of regenerative medicine therapies may choose to establish quality management systems that assure proper design, monitoring, and control of manufacturing processes and facilities [18] with robust operating procedures and product tracking systems that ensure adequate detection of quality deviations [19].
Prior to clinical testing, the characterization of a specific product’s mechanism of action and safety profile is often complex. Therefore, incorporation of appropriate in-vitro, in-silico, in-vivo, and other testing paradigms can contribute to product assessment [30]. Use of biologically relevant animal species, as well as animal models of disease, to support a reasonable expectation of efficacy and to determine product safety and distribution profiles, can provide insight into the product’s ability to treat a specific disease population. For cell therapy products, an understanding of in-vivo cell fate, including engraftment, migration, differentiation, integration, and persistence can help further characterize the product’s effects. For gene therapy products, assessment of in-vivo vector biodistribution and subsequent transgene expression can help characterize the product’s biology and mechanisms of action. In conjunction with these parameters, the resulting data from proof-of-concept studies and toxicology studies are most informative when these studies and their evaluation parameters closely mimic the anticipated clinical conditions of product administration [23].
While the overall development of investigational therapies may be somewhat delineated into different focus areas related to product quality, preclinical, and clinical studies, certain considerations related to regenerative medicine therapies are common to all focus areas and need to be addressed throughout all stages of development. These considerations include concerns related to a product’s potential for immunogenicity and tumorigenicity, and the general recognition that, for many regenerative medicine therapy products, their safe and effective delivery to the target tissue is as important as their therapeutic effect.
Concerns with immunogenicity [24] result from observations of an altered response of the host’s immune system to the introduced biological therapies, and include a wide range of potential immune reactions. Such reactions can range from administration site reactions to systemic inflammatory responses, including graft-versus-host disease, various autoimmune phenomena, and cytokine release syndrome. Appropriate anticipation and investigation of these risks at the product design stage and the preclinical testing stage, followed by adequate clinical monitoring and management of these reactions, are paramount to the development and subsequent clinical use of safe and effective regenerative medicine therapies.
The risk of tumorigenicity stems from challenges with predicting and preventing the potential for unexpected cellular differentiation and proliferation, leading to ectopic tissue or tumor formation attributable to the product. In some development programs, this risk has been addressed by various measures, including minimizing residual non-target cell types in the final product, ensuring cytogenetic stability of the cell lines, and assuring quality control testing of the product at different stages, as well as the consistency of the manufacturing process. Assessment of the potential for tumorigenicity is also evaluated in animal studies, for example, by using models susceptible to tumor formation (such as athymic nude mice). Some viral vectors used for delivery of gene therapy products, while given with the intent to induce therapeutic effects, might on rare occasions integrate at inappropriate locations into the recipient’s genome and activate oncogenes introducing a potential for tumor formation and other unanticipated clinical outcomes. Use of various in vitro, in silico, and other methods, including extensive sequence homology searches through gene libraries and knowledge-based vector designing strategies help predict product safety profile, where feasible.
Source material, product intermediates, and final products can have short storage time, require specific conditions for storage and transfer, including cryopreservation, and may have short shelf lives. To this end, appropriate process parameters are established to ensure adequate restrictions on the manufacturing and shipping conditions, which may impact subsequent clinical use of the product and the timing and conditions of its administration. Safe and effective product administration is also generally of great importance for regenerative medicine products. Administration of regenerative medicine therapies often requires invasive procedures ranging from intravenous infusion to targeted tissue-specific delivery (e.g., intramuscular, intra-cardiac, or intra-cranial) or to operative surgical implantation (e.g., cartilage replacement or blood vessel insertion). If a device is used for tissue-targeted product delivery, it is important to assure product-device compatibility and adequate flow of product material through the device with no or minimal interference with cellular or tissue product activity and viability [29]. In addition, the effective and safe treatment administration is generally highly dependent on the skills and training of the personnel responsible for the treatment delivery. The criticality of careful planning for product manufacturing, storage, and clinical administration cannot be over-emphasized in rare disease patient populations, where the limited number of patients, seriousness of the disease, and frequent need for urgent treatment may severely limit the number of attempts for product manufacturing and administration.
Regenerative medicine therapies often produce long-lasting effects, and patients who receive these therapies may need to be clinically monitored for extended periods. This need for long-term follow-up is frequently compounded by the reality that most development programs investigating products for rare diseases include pediatric patients, patients who may have a high baseline risk for development of both disease-related poor outcomes and treatment-related adverse events, and individuals with little or no physiological reserve left due to progression of their disease. To this end, clinical investigations testing regenerative medicine therapies must be informative from the very first clinical study designed and should be supported by solid and diligent data collection to ensure both the possibility of therapeutic benefits for the participating subjects and the operational efficiency for the overall development program [25]. Safe product delivery and administration are paramount to any treatment, and are often critical issues for novel regenerative medicine therapies. Therefore, clinical protocols for first-in-human studies with novel treatments frequently specify staggered product dosing and sufficient monitoring after the initial product administration, before subsequent trial participants are dosed with the new therapy. Furthermore, for patients with rare pediatric diseases, clinical and regulatory considerations include the ethical and safe conduct of studies in children [26]. For rare genetic disorders, development of genetic tests or companion diagnostics can aid the overall management of individual conditions and improve selection of study subjects who would be most likely to respond to novel treatments.
8.A snapshot of OTAT experience
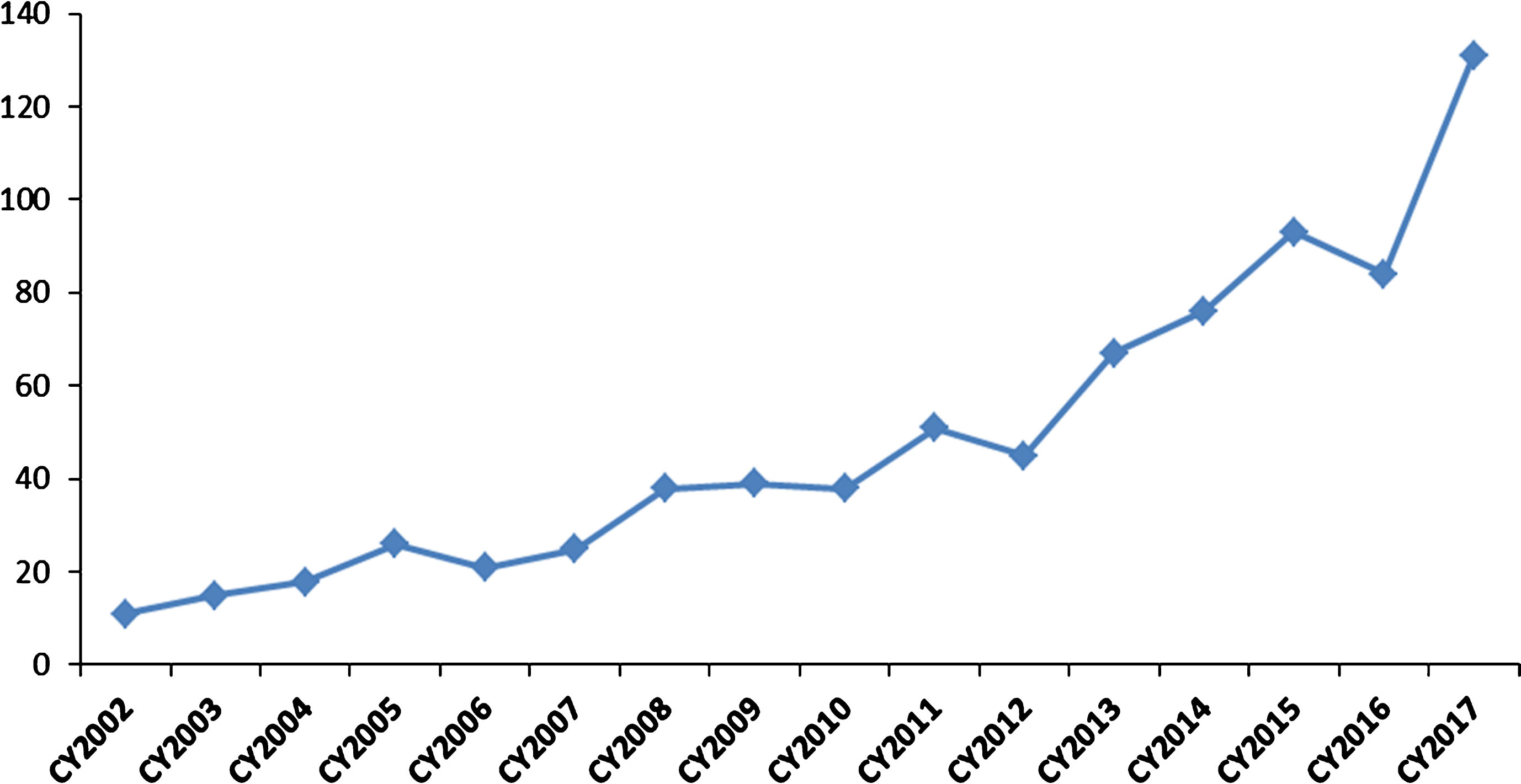
The Office of Tissues and Advanced Therapies (OTAT) in the Center for Biologics Evaluation and Research oversees the regulation of certain biological products developed for a wide range of therapeutic indications. These include products derived from stem cells (e.g., adult, hematopoietic, neural, mesenchymal, and induced pluripotent), somatic cells (e.g., retinal pigment epithelial cells, pancreatic islet cells, chondrocytes), gene therapy products (e.g., viral and non-viral vectors, genetically-modified cells, and genome-editing product technologies), therapeutic vaccines and other antigen-specific active immunotherapies, plasma-derived products, tissue-based and engineered tissue-based products, and devices used with certain biologics. Over the past 15 years, the Office has had a growing portfolio of programs with products that have the potential to impact the treatment of rare diseases (see Fig. 2).
Fig. 2.
Investigational New Drug (IND) applications with development programs that included study populations with rare diseases. The graph depicts the rising numbers of new IND applications with rare disease populations for cell, gene, tissue-based, and other biological products submitted to OTAT (formerly Office of Cellular, Tissues, and Gene Therapies – OCTGT) in calendar years 2002–2017.
As a snapshot demonstration of implementation of the RMAT designation program, between December 13, 2016 and October 1, 2018, OTAT granted 27 RMAT designation requests (see Table 1). Of those, 17/27 (63%) programs were for products developed for the treatment of rare, including rare genetic, diseases. Fourteen development programs (52%) were for products that required tissue-specific product delivery.
Table 1
Programs with products granted Regenerative Medicine Advanced Therapy designation between December 13, 2016 and October 1, 2018
| RMAT | Cell Therapy Products | Gene or Genetically Modified Cell Therapy Products | Total n (%) |
| All RMAT programs | 18 | 9 | 27 (100%) |
| RMAT programs designed for the treatment of rare diseases | 10 | 7 | 17 (63%) |
| RMAT programs with products requiring tissue-specific delivery via device or surgical procedure | 11 | 3 | 14 (52%) |
9.Conclusion
Regenerative medicine is a field that involves replacing, engineering, or regenerating human cells, tissues, or organs to establish, restore, or enhance normal function [20]. Discovery, manufacture, and development of such therapies, with individualization and precision executed on genetic, molecular, and cellular levels, hold great promise for patients and families with rare diseases. The pioneering efforts of rare disease stakeholders will continue to have a great impact on innovation in medicine creating a tremendous potential for finding new curative treatments and treatments that rapidly and substantially reduce disease burdens. Maturing molecular technologies are giving rise to new ways of treatment delivery where targeted biological therapies can be offered to patients with intent for restoration of their cell, tissue, and organ function to normal. Consistent with the technological advances supporting product quality assurance in the early stages of manufacturing, the traditional landscape of pharmaceutical product development will continue to be reshaped by the introduction of effective regenerative medicine therapies supported by fewer and more targeted clinical trials, safe treatment delivery, and the meaningful integration of the continued monitoring for long-term treatment effects into the evidence-based clinical practice.
Disclaimer
The opinions presented in this article are those of the authors and do not necessarily represent the views or policies of FDA. This article does not include all programs, policies, and incentives available for development of novel therapies for rare diseases; the topics discussed in this article represent recent advancements relevant to regenerative medicine therapies.
Acknowledgments
The authors would like to thank Drs. Mercedes Serabian, Xiaofei Wang, Rachael Anatol, Julia Tierney, and Wilson Bryan for their contributions to this article.
References
[1] | Draft Guidance for Industry: Expedited Programs for Regenerative Medicine Therapies for Serious Conditions, November 2017 https://www.fda.gov/downloads/BiologicsBloodVaccines/GuidanceComplianceRegulatoryInformation/Guidances/CellularandGeneTherapy/UCM585414.pdf |
[2] | Orphan Drug Act, Pub L.97-414. 96. Stat. 2049 (1983) Amended in 1984 by Pub L. 98-551 to add a numeric prevalence threshold to the definition of rare diseases. Government Publishing Office website. January 4, 1983. https://www.gpo.gov/fdsys/pkg/STATUTE-96/pdf/STATUTE-96-Pg2049.pdf |
[3] | F.J. Sasinowski and A.J. Varond , FDA’s Flexibility in Subpart H Approvals: Assessing Quantum of Effectiveness Evidence, Food Drug Law J 71: (1) ((2016) ), 135–157. |
[4] | F.J. Sasinowski , Quantum of effectiveness evidence in FDA’s approval of orphan drugs: Cataloging FDA’s flexibility in regulating therapies for persons with rare disorders, Drug Information Journal 46: (2) ((2012) ), 238–263. |
[5] | 21st Century Cures Act Pub L.114-255; 130 Stat 1033. https://www.congress.gov/114/plaws/publ255/PLAW-114publ255.pdf |
[6] | Guidance for Industry: Expedited Programs for Serious Conditions - Drugs and Biologics, May 2014 https://www.fda.gov/downloads/drugs/guidancecomplianceregulatoryinformation/guidances/ucm358301.pdf |
[7] | Medical Device Guidance Documents https://www.fda.gov/MedicalDevices/DeviceRegulationandGuidance/GuidanceDocuments/ucm418448.htm |
[8] | Draft Guidance for Industry: The Least Burdensome Provisions: Concept and Principles, December 2017 https://www.fda.gov/downloads/medicaldevices/deviceregulationandguidance/guidancedocuments/ucm588914.pdf |
[9] | Advancing Hope Act of 2016. Pub. L114-229, Stat.1878 https://www.gpo.gov/fdsys/pkg/BILLS-114s1878enr/pdf/BILLS-114s1878enr.pdf |
[10] | FDA’s Rare Pediatric Disease Priority Review Voucher Program https://www.fda.gov/ForIndustry/DevelopingProductsforRareDiseasesConditions/RarePediatricDiseasePriorityVoucherProgram/default.htm |
[11] | Orphan Products Natural History Grants Program https://www.fda.gov/ForIndustry/DevelopingProductsforRareDiseasesConditions/OrphanProductsNaturalHistoryGrantsProgram/default.htm |
[12] | FDA’s Plan for Issuance of Patient-Focused Drug Development Guidance under 21st Century Cures Act, Title III Section 3002. https://www.fda.gov/downloads/ForIndustry/UserFees/PrescriptionDrugUserFee/UCM563618.pdf |
[13] | FDA’s Patient Representative Program https://www.fda.gov/ForPatients/PatientEngagement/ucm412709.htm |
[14] | Memorandum of Understanding between the U.S.Department of Health and Human Services, Food and Drug Administration, and the National Organization for Rare Disorders. https://www.fda.gov/aboutfda/partnershipscollaborations/memorandaofunderstandingmous/othermous/ucm597454.htm |
[15] | Current Good Manufacturing Practice, 21 CFR ć210-211, ć600-610. |
[16] | International Conference on Harmonisation Guidance for Industry: Q7 Good Manufacturing Practice Guide for Active Pharmaceutical Ingredients. International Conference on Harmonisation, ed.2000. http://academy.gmp-compliance.org/guidemgr/files/3-1-18.PDF |
[17] | International Conference on Harmonisation of Technical Requirements for Pharmaceuticals for Human Use. http://www.ich.org/about/mission.html |
[18] | E. Dollins , P. Lynch , P. Varadkar and K. Wonnacott , The FDA Perspective on the Manufacturing, Production, and Processing of Regulated Cellular Therapies. In: Areman EM , Loper K , Cellular Therapy: Principles, Methods, and Regulations. Bethesda, MD: AABB; (2014) . |
[19] | ICH.Guidance for Industry: Q10 Pharmaceutical Quality System. http://www.ich.org/products/guidelines/quality/quality-single/article/pharmaceutical-quality-system.html |
[20] | C. Mason and P. Dunnill , A brief definition of regenerative medicine, Regen Med 3: ((2008) ), 1–5. |
[21] | Guidance for Industry: Potency Tests for Cellular and Gene Therapy Products, January 2011. https://www.fda.gov/downloads/BiologicsBloodVaccines/GuidanceComplianceRegulatorylnformation/Guidances/CellularandGeneTherapy/UCM243392.pdf |
[22] | Guidance for Industry: Process Validation: General Principles and Practices, January 2011. https://www.fda.gov/downloads/drugs/guidances/ucm070336.pdf |
[23] | Guidance for Industry: Preclinical Assessment of Investigational Cellular and Gene Therapy Products, November 2013. https://www.fda.gov/downloads/BiologicsBloodVaccines/GuidanceComplianceRegulatoryInformation/Guidances/CellularandGeneTherapy/UCM376521.pdf |
[24] | Guidance for Industry: Immunogenicity Assessment for Therapeutic Protein Products, August 2014. https://www.fda.gov/downloads/drugs/guidances/ucm338856.pdf |
[25] | Guidance for Industry: Considerations for the Design of Early-Phase Clinical Trials of Cellular and Gene Therapy Products, June 2015. https://www.fda.gov/downloads/BiologicsBloodVaccines/GuidanceComplianceRegulatoryInformation/Guidances/CellularandGeneTherapy/UCM564952.pdf |
[26] | Additional Safeguards for Children in Clinical Investigations, 21 CFR ć50-53, ć55. |
[27] | Food, Drug, and Cosmetic Act, Section 526 [21 USC 360bb] https://www.fda.gov/forindustry/developingproductsforrarediseasesconditions/howtoapplyfororphanproductdesignation/ucm364750.htm |
[28] | Center for Disease Control: Principles of Epidemiology in Public Health Practice, 3rd Edition; an introduction to applied epidemiology and biostatistics. Section 9 https://www.cdc.gov/ophss/csels/dsepd/ss1978/lesson1/section9.html |
[29] | Guidance for Industry: Cellular Therapy for Cardiac Disease, October 2010. https://www.fda.gov/downloads/BiologicsBloodVaccines/GuidanceComplianceRegulatoryInformation/Guidances/CellularandGeneTherapy/UCM164345.pdf |
[30] | R. Robinson , 3Rs of Animal Testing for Regenerative Medicine Products, Science Translational Medicine 3: (112) ((2011) ), 112fs11. |
[31] | Draft Guidance for Industry: Evaluation of Devices Used with Regenerative Medicine Advanced Therapies, November 2017 https://www.fda.gov/downloads/biologicsbloodvaccines/guidancecomplianceregulatoryinformation/guidances/cellularandgenetherapy/ucm585417.pdf |
[32] | Prescription Drug User Fee Amendments https://www.fda.gov/forindustry/userfees/prescriptiondruguserfee/ |
[33] | Draft Guidance for Industry: Formal Meetings Between the FDA and Sponsors or Applicants of PDUFA Products, December 2017 https://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM590547.pdf |
[34] | Priority Review Vouchers. http://www.priorityreviewvoucher.org |
[35] | FDA awards six grants for natural history studies in rare diseases. https://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm579375.htm |
[36] | FDA awards 12 grants to fund new clinical trials to advance the development of medical products for the treatment of rare diseases. https://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm621490.htm |
[37] | Orphan Products Clinical Trials Grants Program. https://www.fda.gov/forindustry/developingproductsforrarediseasesconditions/whomtocontactaboutorphanproductdevelopment/default.htm |
[38] | Guidance for Industry: User Fee Waivers, Reductions, and Refunds for Drug and Biological Products. https://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/ucm079298.pdf |
[39] | INTERACT Meetings (Initial Targeted Engagement for Regulatory Advice on CBER products). https://www.fda.gov/BiologicsBloodVaccines/ResourcesforYou/Industry/ucm611501.htm |


