
Japanese pathogenic variant database: DPV
Abstract
Databases of pathogenic variants form the basis for clinical genomic diagnosis using next-generation sequencers. ClinVar and the Human Gene Mutation Database (HGMD) are two major databases for pathogenic variants. However, ethnic diversity in the distributions of pathogenic variants in these databases has been observed; thus, geographic region-specific variant databases are required. We established a Japanese pathogenic variant database in 2016 and began registering pathogenic variants from published articles written by Japanese researchers. The criteria for registration are as follows: 1) the variant has been validated using Sanger sequencing, 2) the minor allele frequency of the variant is lower than 0.03 in the normal Japanese population, and 3) the clinical features of the patient have been critically evaluated by multiple clinicians and geneticists. All registered variants can be downloaded as an aggregated single VCF file to use in laboratories. We are presently curating variants from more than 2000 articles and estimate the registration of >10,000 pathogenic variants derived from the Japanese population. Although the number of registered variants remains relatively small, the clinical genetics community in Japan has begun to work on this initiative in a concerted manner.
1Background
Databases of pathogenic variants form the basis for clinical genomic diagnosis using next-generation sequencing (NGS). The Human Gene Mutation Database (HGMD) [12] and ClinVar [4] are two major resources. ClinVar is publicly available and serves the international rare disease community. ClinVar has accumulated over 582,190 variants, including 116,473 pathogenic or likely-pathogenic variants. However, ethnic diversity in the distributions of pathogenic variants has been observed, and nation-specific variant databases, such as FinDis [http://www.findis.org], have been established in some countries [10].
At present, Japan does not have a nationwide pathogenic variant database. Unfortunately, the coverage of Japanese variants in ClinVar is far from complete. For example, among the 60+ pathogenic variants of the phenylalanine hydroxylase [PAH] locus that have been described by a Japanese biochemical geneticist [7], only 20 have been registered in the ClinVar database. We have designed a pathogenic variant database dedicated to rare genetic diseases in Japan and have accumulated more than 2,000 variants so far.
2Methods
The present project was approved by the institutional review board of Keio University School of Medicine. No identifiable personal information were gathered or registered. Data were obtained from various resources including published literature, locus-specific databases, output from a Japanese program for undiagnosed diseases(Initiative on Rare and Undiagnosed Disease, IRUD [1]), and reports published by the Japanese government (Nam-byo study).
Published articles that were potentially relevant to our project were collected in a semi-automatic manner using the software programs tmVar for the selection of variants and GNormPlus for the selection of gene symbols [14]. The entire PubMed database was searched using EDirect software and the keywords “case reports” and “address equals Japan” (https://dataguide.nlm.nih.gov/edirect/overview.html).
We selected articles for which the last names of the principal investigators were Japanese. The database was designed so that the minimal dataset required for ClinVar submission would be included. The database was mainly built using MySQL. Some scripts for annotation and data processing were built using Python and Java. The programmed script for the database displays the variants. The annotation was performed using relatively accepted public programs including TransVar for the coordinate conversion [18], vcfanno for the table annotation [8], Variant Effect Predictor to convert the g.HGVS format to the VCF format [16], CrossMap for the lift-over between hg10 and GRCh38 [17], and InterVar for the semi-automatic variant scoring [5].
Positional information on the variants at the cDNA sequence or protein sequence levels were converted to genome coordinates (GRCh37 or GRCh38) using TransVar software [18]. Once the genome coordinates were determined by the curators, the variant information per the genome coordinate was again converted to the cDNA sequence or protein sequence level to ensure that the entire conversion process was performed in an appropriate manner.
Only those variants that met the following criteria were registered: 1) the variant had been validated using Sanger sequencing 2) the minor allele frequency of the variant had been evaluated using a database of 3554 normal Japanese subjects (Integrative Japanese Genome Variation Database [iJGVD], developed by the Tohoku Medical Megabank Organization [15]) and, for genes that cause autosomal recessive disorders, had been found to be lower than 3% (genes that cause severe pediatric autosomal dominant diseases, as defined by the Clinical Genomics Database [11] were discarded), and 3) the clinical features of the patient from whom the variant was derived had been critically evaluated by two independent pediatricians and two medical geneticists.
As a criterion for validation, we basically followed the rule suggested by the 2015 guideline, Standards and Guidelines for the Interpretation of Sequence Variants: A Joint Consensus Recommendation of the American College of Medical Genetics and Genomics [ACMG] and the Association for Molecular Pathology. Our policy for evaluating the pathogenicity of variants is posted on the website that hosts the database. Software that implements the ACMG rule, InterVar, was used to assist with the manual curation of the variants.
After a manual curation process, variants that met the above criteria for “pathogenic” and “likely pathogenic” were made available to the public. These variants can be viewed according to the name of the genes or the OMIM disease name.
The distributions of the Combined Annotation Dependent Depletion [CADD] scores [6] for the registration of non-synonymous variants were evaluated and compared with those of the CADD scores for non-synonymous variants obtained from the iJGVD. The CADD scores were evaluated according to standard methods. The distribution curves were drawn using R, an open source software environment for statistical computing and graphics [13].
3Results
A total of 93,351 articles written by Japanese researcher were searched in PubMed in a semi-automatic manner, and 2126 abstracts for case reports describing a genetic analysis performed using tmVar were selected. In addition, articles provided by the Nan-byo Study group and the IRUD were also collected, and about 300 articles have been manually curated. Variants from three locus-specific databases (Japanese Familial Alzheimer’s Disease, Resource of Asian Primary Immunodeficiency Diseases, and Fabry-database.org) were added after confirming the genome coordinates of the variants [2, 3, 9]. The database has been made public at http://dpv.cmg.med.keio.ac.jp/dpv-pub/variants. Currently, 1474 variants of 224 genes that cause 213 rare diseases have been made available. These variants can be downloaded as a single VCF file for inclusion in next-generation sequencing pipelines. The content of the entire database is provided in a variant call format file (VCF). The VCF contains various information on annotation that has been used for curation.
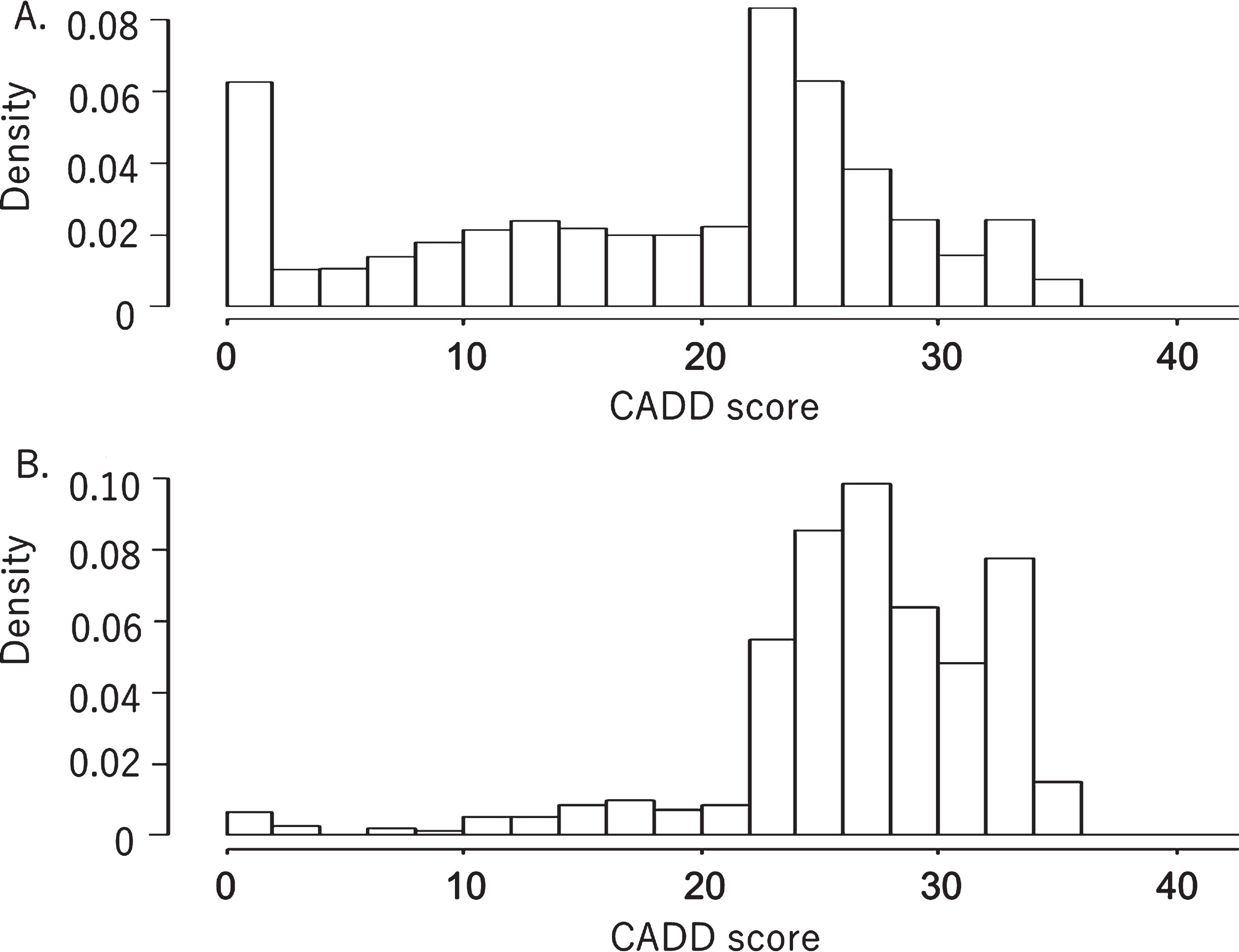
Fig. 1 shows the distribution of the CADD scores of the 1474 registered non-synonymous variants compared with the CADD scores of the 288621 non-synonymous variants obtained from 3554 normal Japanese subjects (iJGVD). Most of the CADD scores of the variants registered in the DPV were above 25, with a very small number of variants having CADD scores below 25. In contrast, many of the non-synonymous variants from the iJGVD had CADD scores below 25. Nevertheless, quite a large number of variants in the iJGVD had CADD scores of above 25.
Fig. 1
Distributions of CADD scores for non-synonymous variants in a normal Japanese population database (A) and a database of pathogenic variants (B). Both graphs have been standardized to show the same integral value of frequency.
4Discussion
We have successfully developed a prototypic database for pathogenic variants enriched in the Japanese population. With the help of the iJGVD database of normal population variants, we have systematically removed common variants. The distribution of CADD scores among the collected pathogenic variants and the distribution within the normal population database support the overall validity of our curation process.
This database should facilitate exome sequencing/medical exome sequencing for clinical diagnostic purposes, since publicly accessible databases of human genetic variants can serve as sources of valid scientific evidence to support the clinical validity of genotype-phenotype relationships when performing NGS-based tests (FDA’s guidance, “Use of Public Human Genetic Variant Databases to Support Clinical Validity for Next Generation Sequencing [NGS]-Based In Vitro Diagnostics”).
The aggregation, annotation and evaluation of the variants were performed by three independent curators. We plan to continue curating the list of variants that have been collected in a semi-automatic manner. We are also encouraging researchers investigating individual rare genetic disorders to register the variants that they have collected to date. The proof-of-concept demonstrated herein is applicable to any other country that has yet to establish a pathogenic variant database.
5Conclusion
We have established a pathogenic variant database that registers mainly variants obtained from Japanese patients with rare diseases. Although the number of registered variants remains relatively small, the clinical genetics community in Japan has begun to work on this initiative in a concerted manner.
Acknowledgments
The authors thank T. Ikeuchi (Department of Molecular Genetics, Brain Research Institute, Niigata University, Japan), S. Usami (Department of Otorhinolaryngology, Shinshu University School of Medicine, Japan), S. Nishio (Department of Otorhinolaryngology, Shinshu University School of Medicine, Japan), and their laboratory members for sharing data. This research was supported by AMED under “Program for an Integrated Database of Clinical and Genomic Information” (Grant Number: JP18kk0205002 and JP18kk0205014) and “Program for Evaluation methods of NGS based in-vitro diagnostics” (Grant Number: JP18mk0102093).
References
[1] | Adachi T. , Kawamura K. , Furusawa Y. , Nishizaki Y. , Imanishi N. , Umehara S. , Izumi K. , Suematsu M. , Japan’s initiative on rare and undiagnosed diseases (IRUD): Towards an end to the diagnostic odyssey, Eur J Hum Genet 25: ((2017) ), 1025–1028. |
[2] | Kasuga K. , Kikuchi M. , Tokutake T. , Nakaya A. , Tezuka T. , Tsukie T. , Hara N. , Miyashita A. , Kuwano R. , Ikeuchi T. , Systematic review and meta-analysis of Japanese familial Alzheimer’s disease and FTDP-17, J Hum Genet 60: ((2015) ), 281–283. |
[3] | Keerthikumar S. , Raju R. , Kandasamy K. , Hijikata A. , Ramabadran S. , Balakrishnan L. , Ahmed M. , Rani S. , Selvan L.D. , Somanathan D.S. , Ray S. , Bhattacharjee M. , Gollapudi S. , Ramachandra Y.L. , Bhadra S. , Bhattacharyya C. , Imai K. , Nonoyama S. , Kanegane H. , Miyawaki T. , Pandey A. , Ohara O. , Mohan S. , RAPID: Resource of Asian Primary Immunodeficiency Diseases, Nucleic Acids Res 37: ((2009) ), D863–867. |
[4] | Landrum M.J. , Lee J.M. , Riley G.R. , Jang W. , Rubinstein W.S. , Church D.M. , Maglott D.R. , ClinVar: Public archive of relationships among sequence variation and human phenotype, Nucleic Acids Res 42: ((2014) ), D980–985. |
[5] | Li Q. , Wang K. , InterVar: Clinical Interpretation of Genetic Variants by the 2015 ACMG-AMP Guidelines, Am J Hum Genet 100: ((2017) ), 267–280. |
[6] | Mather C.A. , Mooney S.D. , Salipante S.J. , Scroggins S. , Wu D. , Pritchard C.C. , Shirts B.H. , CADD score has limited clinical validity for the identification of pathogenic variants in noncoding regions in a hereditary cancer panel, Genet Med 18: ((2016) ), 1269–1275. |
[7] | Okano Y. , Kudo S. , Nishi Y. , Sakaguchi T. , Aso K. , Molecular characterization of phenylketonuria and tetrahydrobiopterin-responsive phenylalanine hydroxylase deficiency in Japan, J Hum Genet 56: ((2011) ), 306–312. |
[8] | Pedersen B.S. , Layer R.M. , Quinlan A.R. , Vcfanno: Fast, flexible annotation of genetic variants, Genome Biol 17: ((2016) ), 118. |
[9] | Saito S. , Ohno K. , Sakuraba H. , Fabry-database.org: Database of the clinical phenotypes, genotypes and mutant alpha-galactosidase A structures in Fabry disease, J Hum Genet 56: ((2011) ), 467–468. |
[10] | Sipila K. , Aula P. , Database for the mutations of the Finnish disease heritage, Hum Mutat 19: ((2002) ), 16–22. |
[11] | Solomon B.D. , Nguyen A.D. , Bear K.A. , Wolfsberg T.G. , Clinical genomic database, Proc Natl Acad Sci U S A 110: ((2013) ), 9851–9855. |
[12] | Stenson P.D. , Ball E.V. , Mort M. , Phillips A.D. , Shiel J.A. , Thomas N.S. , Abeysinghe S. , Krawczak M. , Cooper D.N. , Human Gene Mutation Database (HGMD): 2003 update, Hum Mutat 21: ((2003) ), 577–581. |
[13] | R Development Core Team ((2008) ). R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. ISBN 3-900051-07-0, URL http://www.R-project.org |
[14] | Wei C.H. , Kao H.Y. , Lu Z. , GNormPlus: An Integrative Approach for Tagging Genes, Gene Families and Protein Domains, Biomed Res Int 2015: ((2015) ), 918710. |
[15] | Yamaguchi-Kabata Y. , Nariai N. , Kawai Y. , Sato Y. , Kojima K. , Tateno M. , Katsuoka F. , Yasuda J. , Yamamoto M. , Nagasaki M. , iJGVD: An integrative Japanese genome variation database based on whole-genome sequencing, Hum Genome Var 2: ((2015) ), 15050. |
[16] | Yourshaw M. , Taylor S.P. , Rao A.R. , Martin M.G. , Nelson S.F. , Rich annotation of DNA sequencing variants by leveraging the Enbl Variant Effect Predictor with plugins, Brief Bioinform 16: ((2015) ), 255–264. |
[17] | Zhao H. , Sun Z. , Wang J. , Huang H. , Kocher J.P. , Wang L. , CrossMap: A versatile tool for coordinate conversion between genome asblies, Bioinformatics 30: ((2014) ), 1006–1007. |
[18] | Zhou W. , Chen T. , Chong Z. , Rohrdanz M.A. , Melott J.M. , Wakefield C. , Zeng J. , Weinstein J.N. , Meric-Bernstam F. , Mills G.B. , Chen K. , TransVar: A multilevel variant annotator for precision genomics, Nat Methods 12: ((2015) ), 1002–1003. |

