
Novel SLC16A2 mutations in Chinese patients with Allan-Herndon-Dudley Syndrome
Abstract
We aimed to delineate the clinical profiles of Chinese patients with Allan-Herndon-Dudley Syndrome (AHDS), an X-linked intellectual disability caused by SLC16A2 mutation and affecting males only. Clinical features of five males from four Chinese families, who manifested with severe cognition impairment, developmental delay, hypotonia accompanied by dystonia, were described and summarized. All of them displayed a distinctive thyroid hormone change, with increased FT3, reduced FT4, and normal TSH. Three of them had neuroimaging tests and all presented with hypomyelination. Two of them demonstrated some facial anomalies, including long face, narrow forehead and tent mouth. Targeted next-generation sequencing associated with intellectual disability was performed. Four SLC16A2 mutations (c.916C>T, p.Gln306*; c.61G>T, p.Glu21*; c.695_699delATGGT, p.Asn232Sfs*7; c.42delC, p.Trp15Glyfs*69) were identified, of which three SLC16A2 mutations were novel. Our findings expand mutational spectrumof SLC16A2 and provide support for delineating the clinical features of Chinese patients with AHDS. In addition, this is the first report of AHDS in Chinese cohort. We recommend that male patients with intellectual disability, developmental delay and severe hypotonia, especially those with distinctive thyroid hormone change should be tested for SLC16A2 mutations.
1Introduction
It is generally known that thyroid hormone (TH) is important for the growth and development of different tissues, especially the brain [1, 2]. It is established that TH is involved in the regulation of expression of hundreds of genes in primary astrocytes [2]. Thyroid hormone has two forms: T4 (3,3’,5,5’-tetraiodothyronine) and T3 (3,3’,5-triodothyronine). TH activity is mediated through the binding of T3 to nuclear thyroid hormone receptors (TRs). The active sites of the TRs are located intracellularly. Consequently, TH has to cross multiple membranes in order to reach the nuclear receptors. It was originally assumed that TH enters target cells by passive diffusion because of its lipophilic structures. However, it has become increasingly clear that cellular influx and efflux of TH are facilitated by transmembrane protein transporters [3, 4]. Several transporter families have been identified, of which monocarboxylate transporter 8 (MCT8) deserves special attention because of its role in disease [5].
MCT8 protein is encoded by the SLC16A2 gene, which is located on chromosome Xq13.2. The protein has a wide tissue distribution, such as liver, kidney, pituitary and thyroid gland, as well as brain [5]. SLC16A2 mutations have been associated with Allan-Herndon-Dudley syndrome (AHDS) (OMIM #300523), an X-linked diseaseand affecting males only, characterized by abnormal thyroid hormone level and severe neurological deficits, with axial hypotonia, spastic quadriplegia, impaired or absent speech, and muscle hypoplasia [5]. Here we describe five males from four Chinese families with SLC16A2 gene mutations.
2Case presentation
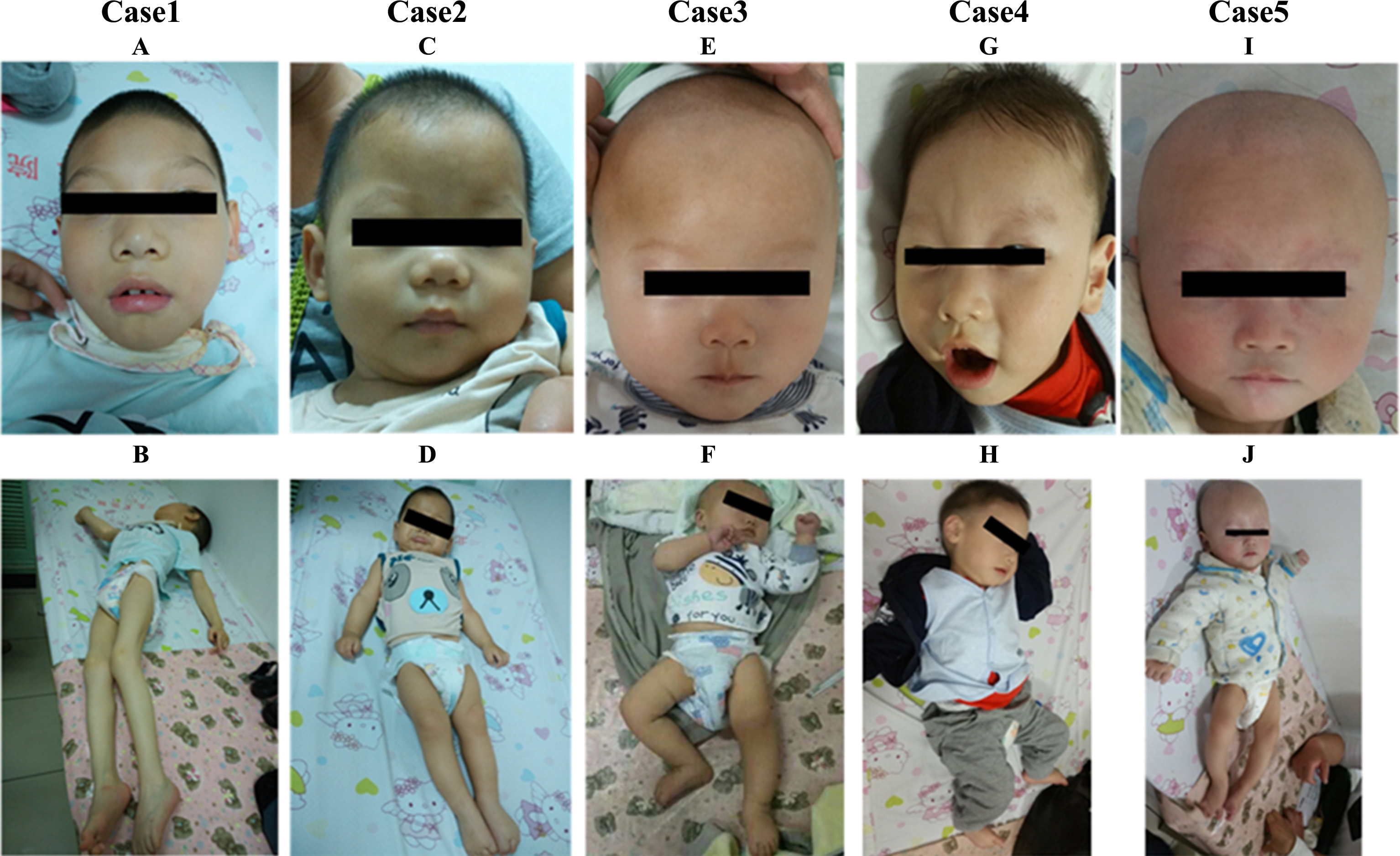
All five patients were male, of whom the age ranged from 8 months and 19 days to 8 years and 9 months. Case 1 and Case 2 were siblings from one Chinese family. The pregnancy was uneventful for all of them. They all presented with severe psychomotor retardation, and never achieved head control, sitting, crawling, standing, or verbal communication. They lacked autonomic activities, had no speech and showed poor response to the outside. Hypotonia accompanied by dystonia was another marked feature observed in all patients. The parents described that when excited or crying, they always became rigid like a stick or developed a twisted posture, but became floppy when relaxed or sleeping. Two of them displayed long face, narrow forehead and cheeks, opened mouth, thick lips, tented upper lips and large ears (Fig. 1). There was no significant regression during the course of the disease in our patients.
Fig.1
Pictures of 5 AHDS males in our study. A and G show that patients (Case 1 and 4) had facial appearance, including elongated and myopathic face, prominent eyes, narrow forehead, narrow cheeks, bigger ears, tented upper lip. C, E and I show that the patients (Case 2, 3 and 5) had normal appearance. B and J showed patients (Case 1 and 4) displaying twisted posture. Muscle hypoplasia was also obvious in Case 1.
Laboratory tests including liver and renal function, lactic acid, creatine kinase, and ammonia were normal. Metabolic screening including plasma amino acids, acylcarnitine profile and urine organic acids were unremarkable. Thyroid hormone test revealed a distinct set of changes in all 5 patients, including increased FT3, decreased FT4, and normal TSH (Table 1). Thyroid hormone level was normal in two female carriers in this study. All three patients who had neuroimaging demonstrated delayed myelination (Fig. 2). Three out of five patients had sinus tachycardia on electrocardiography (ECG).
Fig.2
MRI of Case 1, 4 and 5. The red arrow points to delayed myelination of white matter.
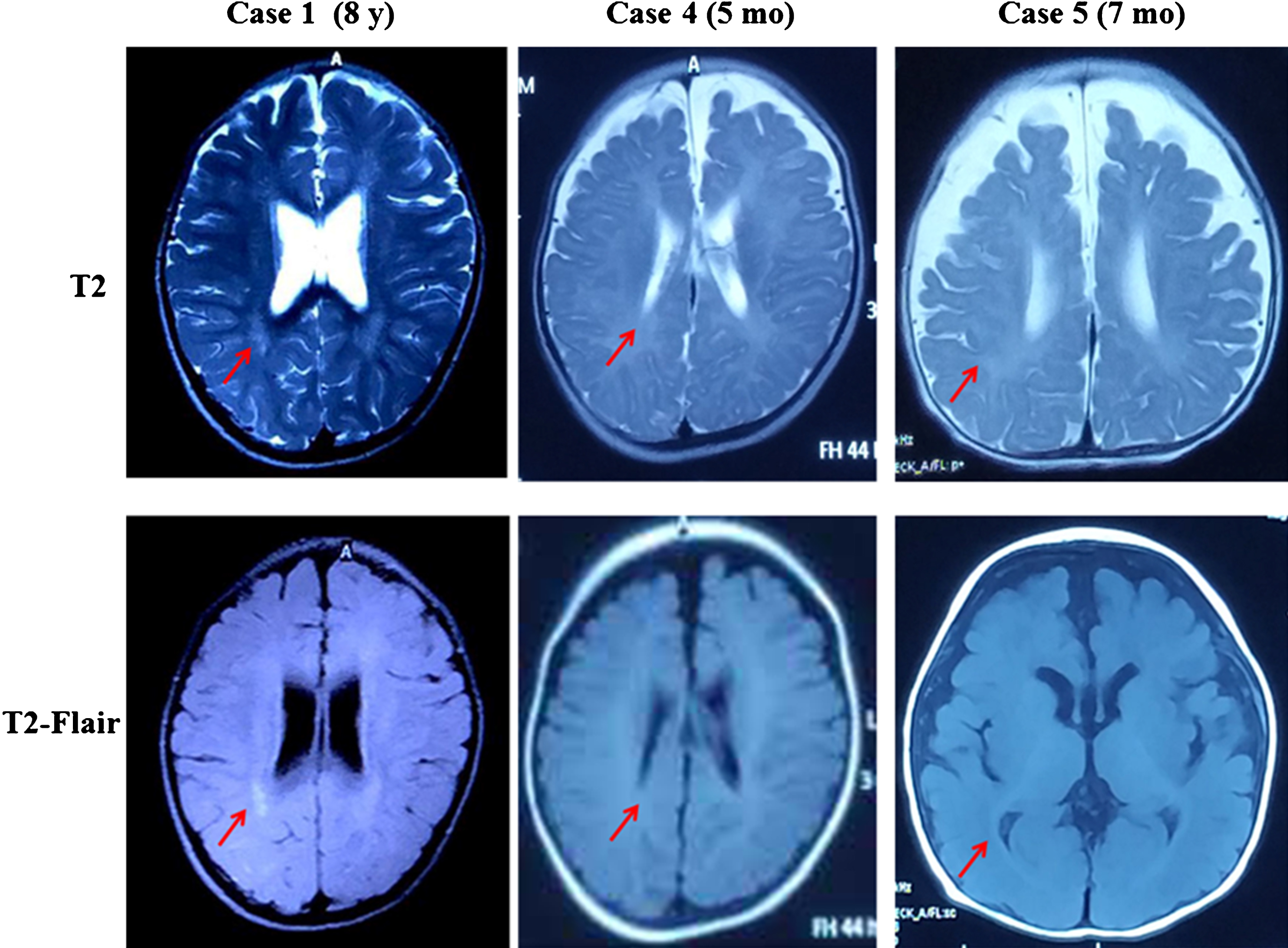
Table 1
Thyroid hormone levels of five patients and two heterozygous carrier in our study
| Patient | Case 1 (8 y, 9 mo) | Case 2 (9 mo) | Case 3 (8 mo) | Case 4 (8 mo) | Case 5 (7 mo) | Het 1 | Het 2 | Reference range |
| TT3 (nmol/L) | 3.32 | 3.32 | 4.64↑ | 5.34↑ | 5.17↑ | 2.10 | 1.71 | 1.6–4.3 |
| FT3 (pmol/L) | 7.77 ↑ | 8.61↑ | 7.01↑ | 13.75↑ | 10.31↑ | 5.13 | 5.76 | 3.50–6.50 |
| TT4 (nmol/L) | 54.20↓ | 31.30↓ | 58.80↓ | 43.52↓ | 48.6↓ | 101.6 | 84.9 | 82.4–171.2 |
| FT4 (pmol/l) | 10.67↓ | 8.45↓ | 10.13↓ | 7.12↓ | 7.99↓ | 13.68 | 14.99 | 11.48–22.70 |
| TSH (μIU/ml) | 3.56 | 3.38 | 3.38 | 4.96 | 4.86 | 2.79 | 1.27 | 0.64–6.27 |
y, years; mo, months. Het 1, mother of Case 1 and Case 2. Het 2, mother of Case 5.
Through targeted next-generation gene sequencing panel consisting 264 genes (supplemental material 1) associated with intellectual disability, four mutations were identified in SLC16A2 gene in four families; three of these mutations were novel. These SLC162A mutations (c.916C>T, p.Gln306*; c.61G>T, p.Glu21*; c.695_699delATGGT, p.Asn232Sfs*7; c.42delC, p.Trp15Glyfs*69) included two nonsense mutations and two micro-deletions. Two of these four SLC16A2 mutations were de novo, and the other two were of maternal origin. A schematic representation of the MCT8 protein, including the mutations identified in this study is shown in Fig. 3.
The detailed clinical information and mutations of five patients were summarized in Table 2.
Fig.3
Schematic representation of the MCT8 protein and the locations of mutations found in this study. Red dot: novel mutations. Black dot: previously reported mutations.
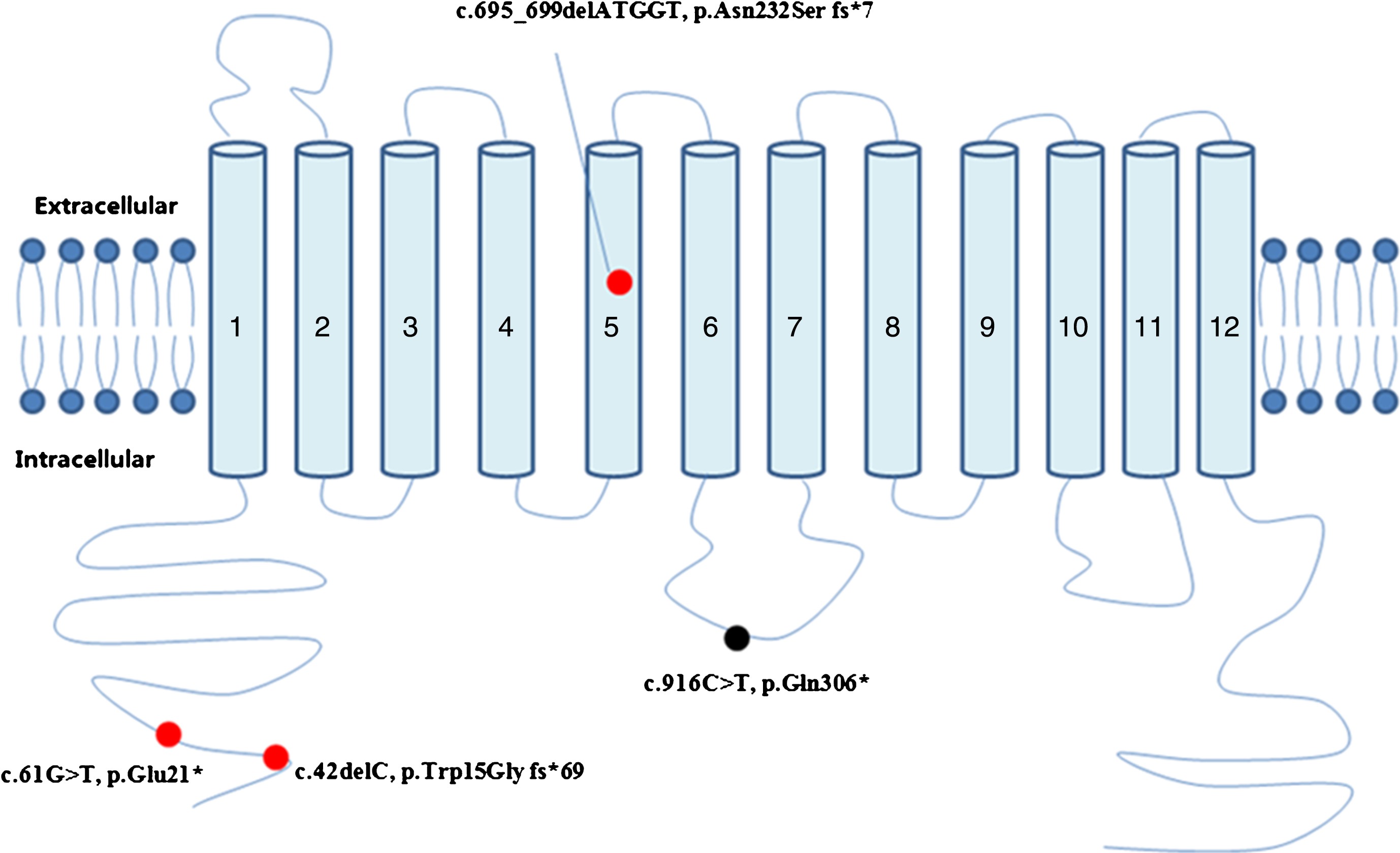
Table 2
Clinical information of 5 patients with AHDS
| Patients | Case 1 | Case 2 | Case 3 | Case 4 | Case 5 |
| Age | 8 y, 9 mo | 9 mo | 8 mo, 19 d | 1 y, 3 mo | 9 mo |
| Hypotonia | + | + | + | + | + |
| Dystonia | + | + | + | + | + |
| Motor development | Severe delayed | Severe delayed | Severe delayed | Severe delayed | Severe delayed |
| Cognition | Severe delayed | Severe delayed | Severe delayed | Severe delayed | Severe delayed |
| Speech | No speech | No speech | No speech | No speech | No speech |
| Weight | 17.2 kg (< –3SD) | 7.6 kg (–1SD∼–2SD) | 9 kg (Normal) | 9.5 kg (–1SD) | 8 kg (–1SD∼–2SD) |
| MRI | demyelination (8 y, 9 mo) | – | – | demyelination (5 mo) | demyelination (7 mo) |
| ECG | sinus tachycardia (161//min, Ref: 80–100/min) | sinus tachycardia, (142/min, Ref: 110–130/min) | Normal (115/min, Ref: 110–130/min) | Normal (106/min, Ref: 110–130/min) | sinus tachycardia (164/min, Ref: 110–130/min) |
| BMD | Normal | Normal | – | Normal | – |
| Hyperreflexia | + | + | + | + | + |
| Babinski | + | + | + | + | + |
| SLC16A2 mutation | c.916C>T, p.Gln306* | c.916C>T, p.Gln306* | c.61G>T, p.Glu21* | c.695_699delATGGT, p.Asn232Ser fs*7 | c.42delC, p.Trp15Gly fs*69 |
| Origin | Maternal | Maternal | De novo | De novo | Maternal |
| Reported/Novel | R | R | N | N | N |
y, years; mo, months; SD, standard deviation; MRI, magnetic resonance imaging; Ref, reference; ECG, Electrocardiograph; BMD, bone mineral density; N, novel; R, reported.
3Discussion
In this study, four SLC16A2 mutations were identified, including three novel mutations, which provided support for delineating the clinical features of Chinese patients with SLC16A2 mutations. In addition, this is the first report of AHDS in Chinese cohort.
Due to the deficiency of MCT8, caused by SLC16A2 mutations, T3 cannot enter the neurons. The reduced uptake of T3 into neurons results in T3 accumulation in serum. TH has two forms: T3 and T4. T3 is the active form of TH. In liver, T4 is converted to T3 by type 1 iodothyronine deiodinase (D1) [6]. The elevated serum T3 level causes liver hyperthyroidism and positively regulates DIO1 gene expression, which consequently increases D1 activity. This results in increased production of T3 at the expense of T4, which further enhances the serum T3 accumulation and causes the consumption of serum T4 [7]. In addition, reduced TH entry into the paraventricular nucleus of hypothalamus and the pituitary gland impairs the negative feedback loop of the hypothalamic-pituitary axis [8]. As a result, AHDS patients display a characteristic thyroid hormone profile with increased FT3, decreased FT4 and normal TSH. Thyroid hormone changes in our patients were in accordance with this characterization. As AHDS is an X-linked disorder, which affects males only, female carriers are usually asymptomatic, but increased serum FT3 was described in 25% of female carriers [8]. In our study, two female carriers had normal thyroid hormone level.
Thyroid hormone is vital for normal brain development, especially within the first years of life. MCT8 deficiencyresults in thyroid hormone deprivation in the brain. Hypothyroidism in the central nervous system leads to severe neurological symptoms, including psychomotor retardation, hypotonia, dystonia, and lack of speech [5]. However, other tissues such as liver, kidney, and muscle, which do not depend exclusively on MCT8, are exposed to toxic levels of T3 . The hyperthyroidism in the peripheral tissue leads to low body weight, tachycardia, sleeplessness, and reduced muscle mass [5, 9]. All five patients of our study presented with severe psychomotor stagnation, axial hypotonia and dystonia. Hyperthyroidism as well as muscle weakness, muscle hypoplasia and low weight were more obvious in the older patients (Case 1 and Case 4). Sinus tachycardia, which is attributed to the peripheral hypermetabolism was detected in three of five patients.
Besides, it has been reported that affected males usually have distinct facial features including elongated and myopathic face with prominence of the eyes, synophrys and prominence of the lower lip [5]. In our study, Case 1 and Case 4 (aged 8 years and 9 months, 1 year and 3 months, respectively) had the similar facial characteristics, while the facial features ofthe other three younger patients were unremarkable, suggesting facial features change as patients get older.
Hypomyelination on brain MRI is common in AHDS. Markedly delayed myelination had been reported in some patients [10–12]. Three patients in our study displayed hypomyelination on MRI. Therefore, AHDS should be considered in males with severe psychomotor retardation, increased T3 and decreased T4 and hypomyelinationon brain MRI.
To date, more than 80 pathogenic mutations in SLC16A2 have been registered in the Human Gene Mutation Database (HGMD) and some genotype-phenotype correlations are recognized [13]. Patients with Ser194Phe, Lys568Pro, Lys434Trp mutations had milder psychomotor retardation in comparison to patients with other mutations. They had fairly well-preserved fine motor skills and were able to walk with some support. Some of them even developed some dysarthric speech. In vitro function analysis demonstrated that the mutated protein possessed residual function of T3 uptake [14]. Other mutations (for example, Val235Met, Ser448*, Phe229del, Lys471Proand Lys512Pro) have previously been reported to result in complete loss of MCT8 function. In vitro functional analysis showed that these mutations almost had no functions, they either lost the capability of trafficking to the plasma membrane, or lost the affinity to substrate. Meanwhile, the expression of mutant protein was profoundly reduced. Therefore, compared with mutations with some residual functions, these mutations lead to more severe clinical outcome [13]. The four SLC16A2 mutations found in our study lead to a premature termination of MCT8 protein, losing the last several TMDs and the cytoplasmic region at the carboxy end of the protein. The truncated MCT8 protein might lose most of its functions, leading to the severe phenotype of our patients.
So far, there are no effective therapies for AHDS. Some patients were treated with propylthiouracil (PTU) in combination with Levothyroxine (LT4). The thyrotoxic condition in peripheral organs could be improved. However, there was no benefit on the severe cognition impairment [15–17]. The key in the treatment of AHDS is to supply appropriate amount of intracellular thyroid hormone, bypassing MCT8 during the brain developmental period. Some patients were treated with Diiodothyropropionic Acid (DITPA), a TH analog, which could enter the neurons independent of MCT8 transporter activity. However, no intellectual improvement was seen in these patients [18]. A recent study found that 3,3’,5-triiodothyroacetic acid (TRIAC) can restore neuronal differentiation in the hypothyroid brain of Mct8/Oatp1c1-double knockout mice, which replicates the clinical picture of human MCT8 deficiency [19]. TRIAC has been used in patients with thyroid cancer and with resistance to TH [20]. In Erasmus Medical Center in Rotterdam, Netherlands there is an ongoing clinical trial treating AHDS patients with TRIAC (https://clinicaltrials.gov/ct2/results?cond=ALLAN-HERNDON-DUDLEY% 20SYNDROME). No study results have been reported so far. The clinical use of TRIAC in AHDS patients needs further studies.
Acknowledgments
We would like to thank the patients and their family for their cooperation in this study. The study was funded by 985 Peking University and Clinical Hospital Cooperation Project (2013-1-06), clinical research special fund of Wu Jieping Medical Foundation (320.6750.17091), and Technology innovation talents special fund of Harbin Science and Technology Bureau (2016RAXYJ089).
References
[1] | Morreale de Escobar G. , Obregon M.J. and Escobar del Rey F. , Role ofthyroid hormone during early brain development, Eur JEndocrinol 151: (Suppl 3) ((2004) ), U25–U37. |
[2] | Morte B. , Gil-Ibanez P. , Bernal J. , Regulation of gene expressionby thyroid hormone in primary astrocytes: Factors influencing thegenomic response, Endocrinology 159: (5) ((2018) ), 2083–2092. |
[3] | Wirth E.K. , Schweizer U. , Kohrle J. , Transport of thyroid hormonein brain, Front Endocrinol (Lausanne) 5: ((2014) ), 98. |
[4] | Visser W.E. , et al., Thyroid hormone transport by monocarboxylatetransporters, Best Pract Res Clin Endocrinol Metab 21: (2) ((2007) ), 223–236. |
[5] | Schwartz C.E. , Stevenson R.E. , The MCT8 thyroid hormonetransporter and Allan-Herndon-Dudley syndrome, Best Pract ResClin Endocrinol Metab 21: (2) ((2007) ), 307–321. |
[6] | Gereben B. , et al., Cellular and molecular basis ofdeiodinase-regulated thyroid hormone signaling, Endocr Rev 29: (7) ((2008) ), 898–938. |
[7] | Trajkovic M. , et al., Abnormal thyroid hormone metabolism in micelacking the monocarboxylate transporter 8, J Clin Invest 117: (3) ((2007) ), 627–635. |
[8] | Dumitrescu A.M. , et al., Tissue-specific thyroid hormone deprivationand excess in monocarboxylate transporter (mct) 8-deficient mice, Endocrinology 147: (9) ((2006) ), 4036–4043. |
[9] | Schwartz C.E. , et al., Allan-Herndon-Dudley syndrome and themonocarboxylate transporter 8 (MCT8) gene, Am J Hum Genet 77: (1) ((2005) ), 41–53. |
[10] | Kobayashi S. , et al., Clinical course and images of four familialcases of Allan-Herndon-Dudley syndrome with a novel monocarboxylatetransporter 8 gene mutation, Pediatr Neurol 51: (3) ((2014) ), 414–416. |
[11] | Gika A.D. , et al., White matter abnormalities and dystonic motordisorder associated with mutations in the SLC16A2 gene, Dev MedChild Neurol 52: (5) ((2010) ), 475–482. |
[12] | Tonduti D.V.A. , Berardinelli A. , Schmidt J.L. , Collins C.D. , Novara F. , Genni A.D. , Mita A. , Triulzi F. , Brunstrom-Hernandez J.E. , Zuffardi O. , Balottin U. and Orcesi S. , MCT8 deficiency:Extrapyramidal symptoms and delayed myelination as prominentfeatures, J Child Neurol 28: (6) ((2013) ). |
[13] | Jansen J. , et al., Genotype-phenotype relationship in patients withmutations in thyroid hormone transporter MCT8, Endocrinology 149: (5) ((2008) ), 2184–2190. |
[14] | Visser W.E. , et al., Novel pathogenic mechanism suggested by ex vivoanalysis of MCT8 (SLC16A2) mutations, Hum Mutat 30: (1) ((2009) ), 29–38. |
[15] | Wemeau J.L. , et al., Beneficial effects of propylthiouracil plusL-thyroxine treatment in a patient with a mutation in MCT8, JClin Endocrinol Metab 93: (6) ((2008) ), 2084–2088. |
[16] | Dumitrescu A.M.L.X. , Best T.B. , Brockmann K. and Refetoff S. , Anovel syndrome combining thyroid and neurological abnormalities isassociated with mutations in a monocarboxylate transporter gene, Am J Hum Genet 74: (1) ((2004) ), 168–175. |
[17] | Anik A. , et al., Psychomotor retardation caused by a defectivethyroid hormone transporter: Report of two families with differentMCT8 mutations, Horm Res Paediatr 82: (4) ((2014) ), 261–271. |
[18] | Verge C.F. , et al., Diiodothyropropionic acid (DITPA) in thetreatment of MCT8 deficiency, J Clin Endocrinol Metab 97: (12) ((2012) ), 4515–4123. |
[19] | Kersseboom S. , et al., In vitro and mouse studies supporttherapeutic utility of triiodothyroacetic acid in MCT8 deficiency, Mol Endocrinol ((2015) ) me00009999. |
[20] | Anzai R. , et al., Long-term 3,5,3’-triiodothyroacetic acid therapyin a child with hyperthyroidism caused by thyroid hormoneresistance: Pharmacological study and therapeutic recommendations, Thyroid 22: (10) ((2012) ), 1069–1075. |


