
Neuronal ceroid-lipofuscinoses
Abstract
The neuronal ceroid-lipofuscinoses (NCLs) are a group of recessively inherited diseases characterized by progressive neuronal loss, accumulation of intracellular lipofuscin-like autofluorescent storage material with distinctive ultrastructural features, and clinical signs and symptoms of progressive neurodegeneration. Initially classified by the age of onset of clinical signs and symptoms as well as the ultrastructural morphology of the storage material, the growing family of NCLs can now also be biologically classified by their underlying genetic defects. For a few NCLs, we now understand the functions of the proteins encoded by the NCL genes, which has enabled refining of the diagnostic algorithms and ignited hopes for finding effective treatments in the future. Ongoing research into understanding the functions of the remainder of the NCL proteins as well as continuing advancements in technology (such as massively-parallel gene sequencing) has and will continue to inform the clinical approach, diagnostic algorithms, and treatment strategies.
1Introduction
The neuronal ceroid-lipofuscinoses (NCLs) are a group of autosomal recessive inherited, neurodegenerative diseases that have the following features in common:
1. progressive neuronal loss;
2. accumulation, in the cytoplasm of neurons and other cells, of lipofuscin-like autofluorescent storage material that has characteristic ultrastructural appearance;
3. clinical signs and symptoms including progressive loss of vision, myoclonic seizures, loss of cognitive function, pyramidal and extrapyramidal motor dysfunction, and early demise [1].
2Biological/functional classification
NCLs were initially classified by the age of onset and the ultrastructural morphology of the storage material. Since then, the discovery of their molecular attributes has helped to create a biologically rational classification of the NCLs [2, 3]. Table 1 shows the current classification that is likely to be updated in the near future as we better understand the pathophysiology and as more biologically distinct genetic entities are added to the growing list of NCLs. The table broadly classifies NCLs into two categories: those caused by deficiencies of proteins (all of which happen to be enzymes) with defined functions and those caused by deficiencies of proteins with currently debated or unknown functions. The implication of such a classification is that the diagnosis of the first group of NCLs may be confirmed by functional enzymatic assays.
Table 1
Current biological classification of NCLs
| Name | Gene |
| NCLs caused by deficiencies of proteins (enzymes) with known function | |
| CLN1 (INCL, Santavuori-Haltia disease) | PPT1 on 1p32 |
| CLN2 (LINCL, Jansky-Bielchowsky disease) | TPP1 on 11p15.5 |
| CLN10 (CNCL) | CTSD on 11p15.5 |
| CLN13 | CTSF on 11q13.2 |
| NCLs caused by deficiencies of proteins without unequivocally characterized function | |
| CLN3 (JNCL, Batten/Spielmeyer-Vogt-Sjogren disease) | CLN3 on 16p12.1 |
| CLN4A (Kufs disease, ANCL, autosomal recessive) | CLN6 on 15q21-q23 |
| CLN4B (Parry disease, ANCL, autosomal dominant) | DNAJC5 on 20q13.33 |
| CLN5 (LINCL, Finnish variant) | CLN5 on 13q21.1-q32 |
| CLN6 (LINCL, Costa Rican/Turkish/Indian/Roma Gypsy/Portugal variants) | |
| CLN6 on 15q21-q23 | |
| CLN7 (INCL, Turkish/Indian variants) | MFSD8 on 4q28.1-q28.2 |
| CLN8 (EPMR and LINCL, Turkish variant) | CLN8 on 8pter-8p22 |
| CLN9 | ? |
| CLN11 | GRN on 17q21.31 |
| CLN12 | ? ATP13A2 on 1p36.13 |
| CLN14 | KCTD7 on 7q11.21 |
CLN –Ceroid-lipofuscinosis, Neuronal. INCL –Infantile Neuronal Ceroid-Lipofuscinosis. LINCL –Late Infantile Neuronal Ceroid-Lipofuscinosis. CNCL –Congenital Neuronal Ceroid-Lipofuscinosis. JNCL –Juvenile Neuronal Ceroid-Lipofuscinosis. ANCL –Adult Neuronal Ceroid-Lipofuscinosis. EPMR –Progressive epilepsy with mental retardation.
As shown in Table 1, four of the characterized human NCLs are caused by deficiencies of soluble lysosomal proteins with now known enzymatic functions. CLN1 (the traditional Infantile Neuronal Ceroid-Lipofuscinosis, INCL, Santavuori-Haltia disease) is caused by a deficiency of the soluble lysosomal enzyme, palmitoyl-protein thioesterase 1 (PPT1), that cleaves the fatty acid moiety from cysteine residues of S-palmitolyated proteins [4, 5]. CLN2 (the traditional Late Infantile Neuronal Ceroid-Lipofuscinosis, LINCL, Jansky-Bielchowsky disease) is caused by a deficiency of a lysosomal serine protease, tripeptidyl peptidase (TPP1) [6, 7]. CLN10 (Congenital Neuronal Ceroid-Lipofuscinosis, CNCL) is caused by a deficiency of cathepsin D, a soluble lysosomal cysteine protease [8–10]; and CLN13, a rare late onset form of NCL, has been recently shown to be caused by mutations in the gene encoding cathepsin F, also a soluble lysosomal protease [11]. Animal studies suggest that other proteins of known function may be candidates for yet unclassified human NCLs. For instance, mice deficient in cathepsin F had previously been shown to develop late-onset neuronal ceroid-lipofuscinosis thus predicting CLN13 [12], and deficiencies of chloride channel proteins CLC-3, CLC-6, and CLC-7 produce neuronal ceroid-lipofuscinosis phenotypes in mice [13–15]. A truncating mutation in ATP13A2 that encodes an ATPase transporter of inorganic cations has been described in Tibetan terriers with adult onset NCL [16]. This animal model predicted the human CLN12 disorder which has so far been seen in only one family with ATP13A2 mutations [17]. Interestingly, the ATP13A2 gene is also associated with human Parkinsonism [18].
Other neuronal ceroid-lipofuscinoses are caused by mutations in genes that encode proteins whose functions are either not known or are yet to be unequivocally established. All but one of these proteins have been predicted to have a transmembrane localization. CLN3 (the traditional Juvenile Neuronal Ceroid-Lipofuscinosis, JNCL, Batten disease, Spielmeyer-Vogt-Sjogren disease) is caused by mutations in CLN3. Although JNCL is the most common of all neuronal ceroid-lipofuscinoses and CLN3 was identified approximately 20 years ago, the highly hydrophobic nature of the CLN3 protein (CLN3P) has precluded its purification and therefore hindered attempts at direct functional studies. While the primary function of CLN3P may be debated, its absence affects numerous cellular functions including pH regulation, arginine transport, organelle or membrane trafficking, and apoptosis [2, 20]. We have suggested that the unifying primary function of CLN3P may be in a novel palmitoyl-protein Δ-9 desaturase (PPD) activity, which is present at intermediate levels in heterozygotes. This enzyme activity could explain all of the various functional abnormalities seen in the JNCL cells [21–23]. Another group of researchers has shown a correlation between the CLN3P expression and the synthesis of bis (monoacylglycerol) phosphate (BMP) and suggested that CLN3P may play a role in the biosynthesis of BMP [24]. CLN4A (Kufs disease, autosomal recessive) has been shown to be caused by mutations in the CLN6 gene [25]. CLN4B (Kufs disease, autosomal dominant) has been shown to be caused by mutations in DNAJC5 that encodes cysteine-string protein alpha (CSPα) [26]. CLN5 (LINCL, Finnish variant) is caused by mutations in CLN5 that encodes a lysosomal protein; it is a subject of debate whether this protein is membrane-bound or soluble [27, 28]. CLN6 (LINCL, Costa Rican/Turkish/Indian/Roma Gypsy/Portugal variants) is caused by mutations in CLN6 [29–31]. CLN7 (INCL, Turkish/Indian variants) has been associated with a novel gene, MFSD8, which encodes a membrane protein belonging to the major facilitator superfamily of transporter proteins [32]. CLN8, which includes Northern epilepsy or progressive epilepsy with mental retardation (EPMR) and LINCL, Turkish variant, is caused by mutations in CLN8 [33–35]. CLN9 has not yet been mapped to a gene locus but the defect has been shown to be one of ceramide metabolism [36]. CLN11 arises from mutations in the progranulin gene GRN, which is also associated with frontotemporal lobar degeneration [37]. CLN14, a rare infantile NCL, results from mutations in KCTD7 that encodes the potassium channel tetramerization domain-containing protein 7, which is related to the ubiquitin-proteasome system [38].
3Ultrastructural patterns
As noted earlier, NCLs are characterized by storage, in the cytoplasm of neurons as well as non-neuronal cells such as endothelial cells and skin adnexal epithelial cells, of autofluorescent lipopigment that has features similar to the wear-and-tear lipofuscin and ceroid pigments that accumulate with aging in normal cells. The NCL pigment stains bright red with the Periodic acid-Schiff stain, blue with the Luxol-fast blue stain, is fuschinophilic, and stains with Sudan black-B. The lipopigment shows a bright yellow autofluorescence under ultraviolet light. Histochemically, lysosomal acid phosphatase activity is associated with the storage material. Immunohistochemcially, a few extremely hydrophobic proteins are detected in the lipopigment storage material; these include subunit c of the mitochondrial ATP synthase and sphingolipid activator proteins (SAPs or saposins) A and D [39–45].
Ultrastructural examination has been a diagnostic cornerstone for NCLs. The storage inclusions occur not only in central nervous system neurons but have been seen in the peripheral nervous system neurons and Schwann cells, conjunctival epithelial cells, fibroblasts, endothelial cells, smooth muscle cells, skeletal muscle cells, trophoblastic cells, eccrine sweat gland epithelial cells, and peripheral blood lymphocytes. The inclusions in the latter two cell types are especially significant because they allow for ultrastructural examination of easily accessible skin biopsy and peripheral blood buffy coat preparations. Other commonly biopsied tissues include skeletal muscle, conjunctiva, and rectal mucosa. The presence of storage material in trophoblasts allows for examination of first trimester biopsies of chorionic villi for prenatal diagnosis [46–54].
When examined by transmission electron microscopy, the storage material is seen as cytoplasmic storage bodies or cytosomes that may be membrane-enclosed suggesting lysosomal accumulation. Pertinently, as mentioned earlier, lysosomal acid phosphatase activity is associated with the storage material. Four ultrastructural morphologic patterns are seen, which correlate with the NCL types (Table 2) [3, 55]. In particular, the traditional INCL or CLN1 is characterized by granular osmiophilic deposits (GRODs), the traditional LINCL or CLN2 is characterized by curvilinear profiles (CLPs), and the traditional JNCL or CLN3 is characterized by fingerprint profiles (FPPs). The fourth ultrastructural pattern is rectilinear profiles (RLPs). Biochemically, GRODs are associated with an accumulation of sphingolipid activator proteins or saposins A. On the other hand, CLPs, FPPs, and RLPs are associated with an accumulation of subunit c of the mitochondrial ATP synthase [56]. GRODs are membrane-enclosed, homogeneous or finely granular rounded bodies that measure 0.5 micron in diameter and may form aggregates measuring up to 5 micron in diameter (Fig. 1). CLPs are membrane-enclosed or appear free in the cytoplasmic matrix as small electron-dense, C- or S-shaped alternating dark and light lines measuring 1.9–2.4 nm in thickness (Fig. 2). RLPs are stacks of straight dark and light bands measuring 2.8–3.8 nm in thickness. FPPs, usually membrane-enclosed, are compactly bound stacks of paired parallel dark lines that resemble fingerprints. Each pair of dark lines measures 7.6–9.6 nm in thickness and is separated from the adjacent pair by 3-4 nm space. Each dark line within a pair may be separated from each other by 1–3 nm lucent zone (Fig. 3). Mixed patterns of FPPs with CLPs or RLPs may occur in the same cytosome [57].
Fig.1
Granular osmiophilic deposits (GRODs) in an endothelial cell. 20,000x magnification.
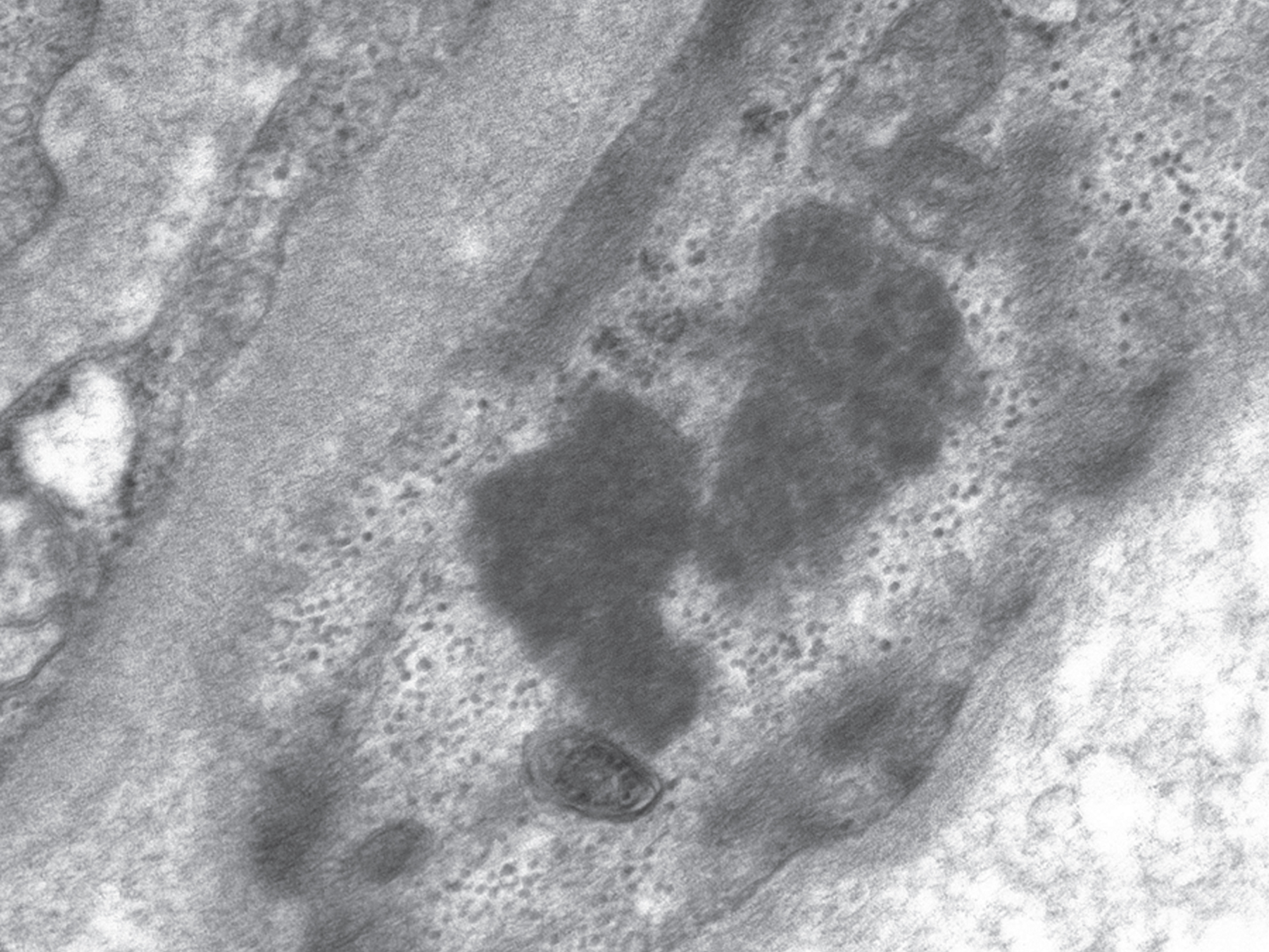
Fig.2
Curvilinear profiles (CLPs) in an endothelial cell. 20,000x magnification.
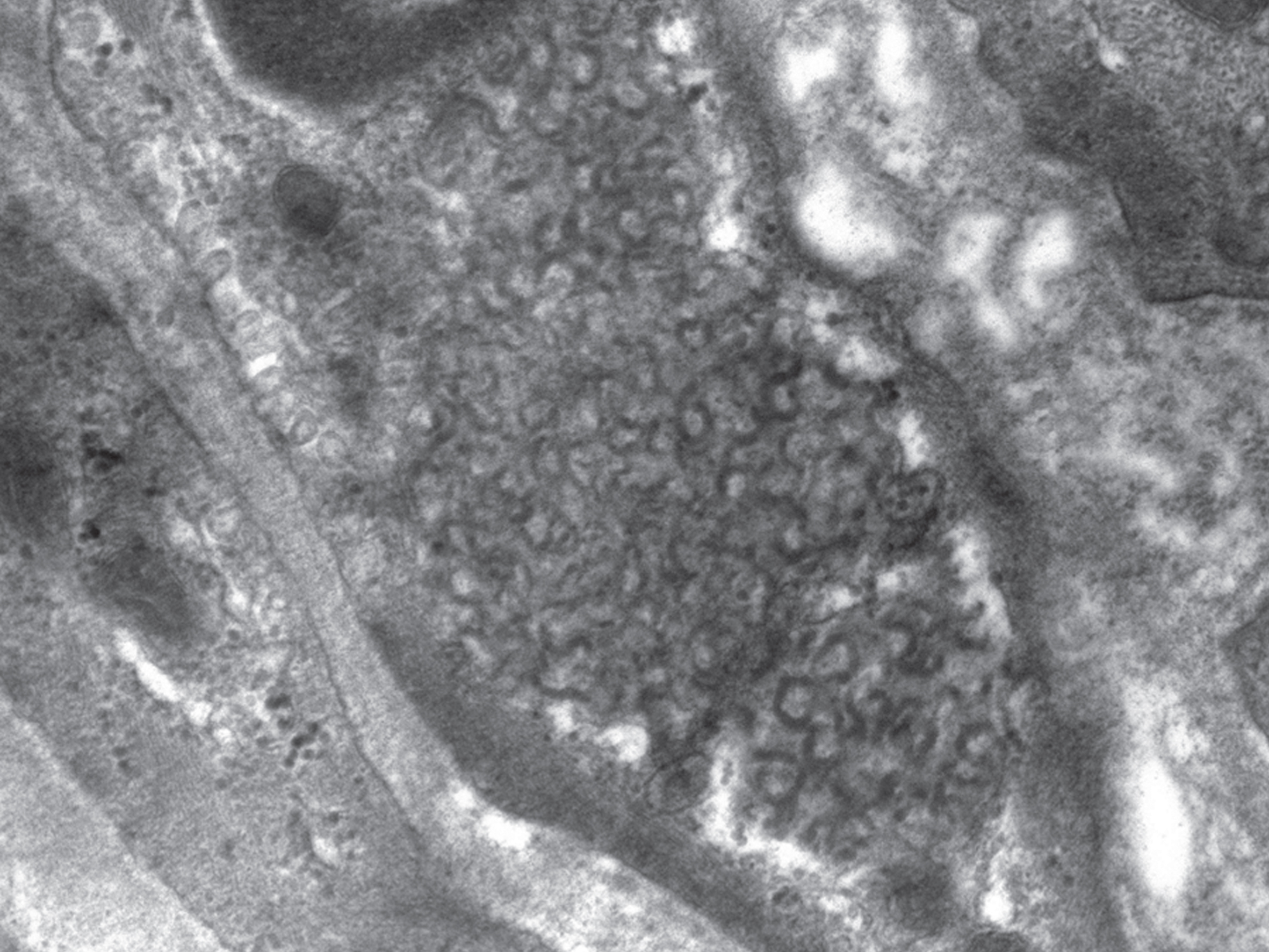
Fig.3
Fingerprint profiles (FPPs) in a brain neuron. 80,000x magnification.
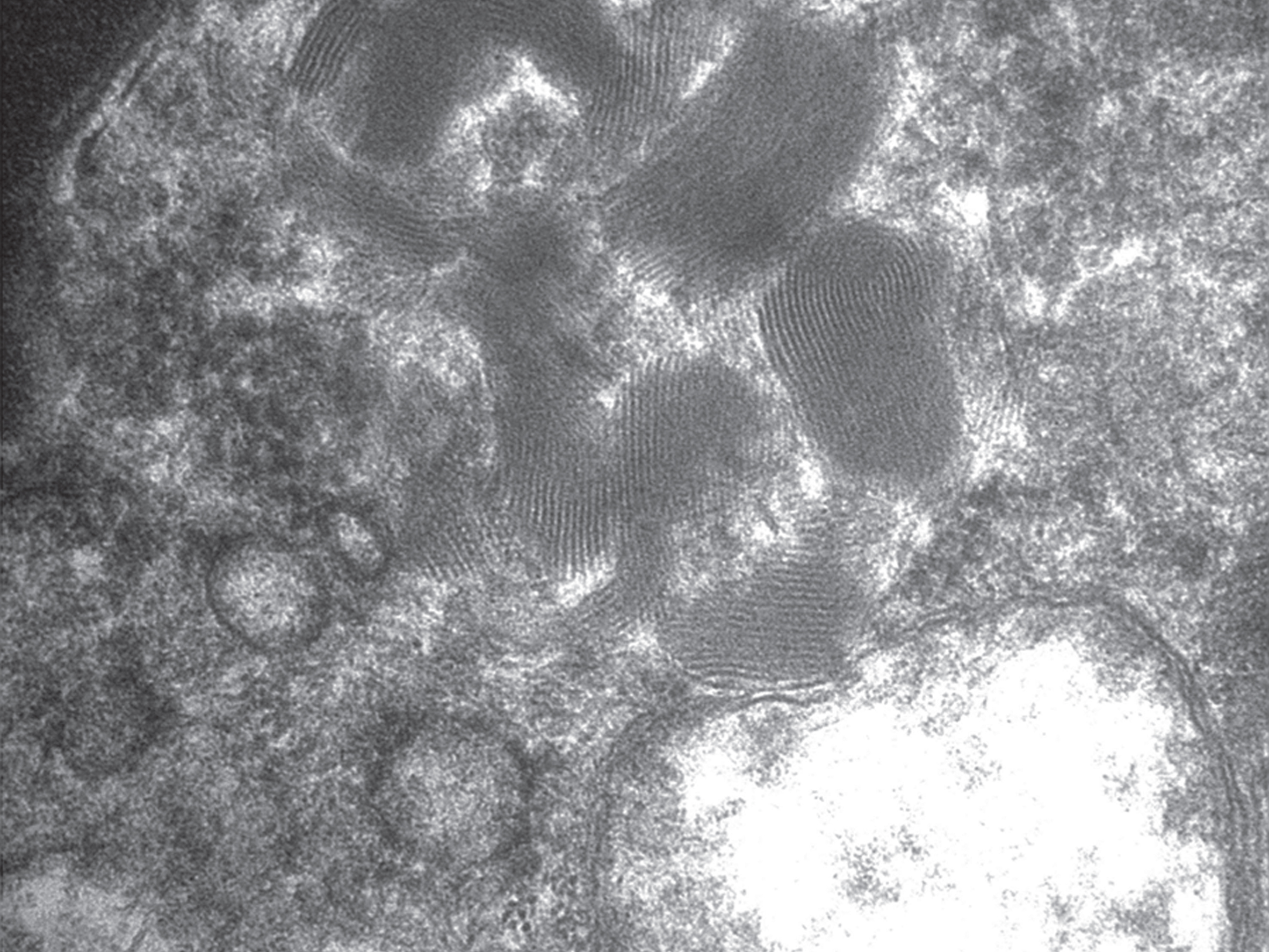
Table 2
Ultrastructural morphology of the storage material seen in NCLs
| Ultrastructural morphology | NCL type |
| Granular osmiophilic deposit (GROD) | CLN1, CLN4B, CLN9, CL10 |
| Curvilinear profiles (CLP) | CLN2, CLN5, CLN6, CLN7, CLN8, |
| CLN9 | |
| Fingerprint profiles (FPP) | CLN3, CLN4A, CLN5, CLN6, |
| CLN7, CLN9 | |
| Rectilinear profiles (RLP) | CLN5, CLN6, CLN7 |
4Clinical presentation
While the biological classification of NCLs is more rational and provides a pathophysiologic basis for the disease process, it is still useful for the laboratorian to know the clinical course of the disease, such as the age at onset of the clinical manifestations, to help narrow down the differential diagnosis (Table 3) [3, 55]. The traditional Infantile, Late Infantile, Juvenile, and Adult forms of NCL correspond to CLN1, CLN2, CLN3, and CLN4, respectively. However, as is evident from Table 3, there is an overlap in the clinical presentations. Furthermore, the discovery of newer types of NCLs has also contributed to the clinical overlap of biologically distinct NCLs.
Table 3
Classification of NCLs based on the age at the onset of clinical manifestations
| Age at onset of clinical disease | Chronological name | Biological name |
| At birth | CNCL | CLN10 |
| Within 1st year | INCL | CLN1, CLN14 |
| 2–4 years | LINCL | CLN2, CLN1, CLN5, CLN6, |
| CLN7, CLN8, CLN10 | ||
| School age | JNCL | CLN3, CLN1, CLN2, CLN8, |
| CLN10, CLN9 | ||
| Adulthood | ANCL | CLN4, CLN6, CLN1, |
| CLN10, CLN5, CLN13, | ||
| CLN11 |
Congenital or neonatal onset disease is the earliest and the most aggressive presentation of NCL. The affected neonates may present with respiratory insufficiency and seizures. Status epilepticus may occur and the patients die within hours to weeks. Typically, the affected neonate is microcephalic with a remarkably small brain weighing 15–50% of the expected weight for the gestational age. Neuropathologic examination shows diffuse gliosis and a subtotal loss of neurons in the cerebral cortex. The cerebellum is atrophic. Ultrastructural examination of the brain shows GRODs in most cells [58–64]. CLN10, caused by a deficiency of cathepsin D, is currently the only NCL known to present in this manner [10].
Infantile onset disease typically manifests in the second half of the first year of life following normal development up to the age of 6 to 12 months. The clinical pattern is that of mental slowing followed by arrest of motor development, hypotonia, and ataxia. Visual deterioration becomes manifest around the first birthday and progresses to complete vision loss by 2 years. Around this time, myoclonic jerks appear with generalized convulsions in a minority. Flexion contractures develop after 5 years and the most severely affected children die by the seventh year. Microcephaly is universal, and at autopsy, there is diffuse cerebral cortical and cerebellar atrophy. The cortical neuronal loss is accompanied by gliosis and macrophage accumulation. The cells in the brain, retina, skin, rectum, and the peripheral blood lymphocytes show GRODs on ultrastructual examination [39, 65–72]. CLN1 (the traditional Infantile Neuronal Ceroid-Lipofuscinosis, INCL, Santavuori-Haltia disease), caused by a deficiency of the soluble lysosomal enzyme, palmitoyl-protein thioesterase 1 (PPT1), that cleaves the palmitate moiety from cysteine residues of S-palmitolyated proteins, classically presents in this manner [4, 5]. CLN14 is a rare infantile NCL.
Late infantile onset disease classically manifests between the ages of 2 and 4 years. The children first come to medical attention because of seizures that may be myoclonic or generalized tonic-clonic. This is followed by progressive mental retardation, ataxia, and blindness, with death usually occurring by the first half of the second decade. Autopsy examination shows cerebral and cerebellar atrophy with neuronal loss and gliosis. Ultrastructural examination shows CLPs in the cells of the central nervous system as well as the spleen, colon, skeletal muscle, peripheral nerves, and skin [39, 73–78]. CLN2, the initially described traditional Late Infantile Neuronal Ceroid-Lipofuscinosis (LINCL, Jansky-Bielchowsky disease), is caused by a deficiency of a lysosomal serine protease, tripeptidyl peptidase (TPP1) [6, 7]. Milder mutations of PPT1 (CLN1) and CTSD (CLN10) may also give rise to late infantile presentation. In addition, variant LINCL (vLINC) occurs with slightly later disease onset at 5 to 7 years with death by late second to third decades [39]. Similar clinical presentations may also occur in patients with CLN5, CLN6, CLN7, or CLN8, none of which are caused by proteins with currently known functions.
Juvenile onset disease usually manifests in the first decade of life, typically between the ages of 4 and 8 years. The first symptom is usually a deterioration of visual acuity. However, subtle psychological disturbances may be noticed before problems with vision become manifest. The visual failure progresses to blindness by the time the patient reaches adolescence. Fundoscopic examination reveals macular degeneration, optic atrophy, and retinal degeneration with pigment aggregations. Non-corneal electroretinogram is abolished early. The flash-evoked visual potential is abolished by the second decade. Cognitive dysfunction is usually discovered when the child starts going to school and gradually worsens to severe dementia by the second decade. Epileptic seizures of generalized, complex-partial, or myoclonic type may occur by this time. Motor dysfunction is a combination of extrapyramidal, pyramidal, and cerebellar disturbances. Compulsive speaking begins around the mid-second decade and progressively worsens to severe dysarthria. Sleep disturbances may occur. Psychiatric disturbances may include social, thought, attention problems, somatic complaints, and aggressive behavior. Electroencephalogram shows nonspecific background disturbance and increased paroxysmal activity. Magnetic resonance imaging scans detect cerebral and cerebellar atrophy and low signal intensity in thalami and high signal intensity in periventricular white matter on T2-weighted images. By late second to early third decades, the patients are nonambulatory. This is followed by death, usually within a year [39, 40, 79–89]. At autopsy, the brain is small with a moderate to severe decrease in weight and grey and white matter atrophy. The skull bones are thickened because of the gradual brain atrophy. There is diffuse loss of neurons, with the few remnant neurons showing distension of the neuronal cytoplasm by granular lipopigment, such that the neuronal cell bodies become rounded. The accumulated cytoplasmic lipopigment pushes the nucleus to the periphery; the lipopigment also accumulates in the axon hillock and may extend down the axons and dendrites. This so-called Shaffer-Spielmeyer process is most prominent in the cortical neurons of the cerebellum and cerebrum, with the small neurons of layer II and the pyramidal neurons of layer V being the most affected in the early stages. There may be loss of neuronal melanin in the substantia nigra, correlating with the Parkinsonian-like motor disturbance. There is gliosis, and the microglia contain the granular lipopigment, which is also present in the endothelial cells lining the neighboring small blood vessels. Similar lipopigment is also present systemically in cells other than neurons, most commonly in myocardium, liver, spleen, intestinal wall, and kidneys. In addition, vacuolated lymphocytes may be seen in the peripheral blood smear preparations. In the eyes, the retinal neuroepithelium, especially in the central portion of the retina, shows degenerative changes the earliest. Subsequently, there is extensive loss of retinal neurons in all layers. On the other hand, lipopigment-containing neurons, gliotic astrocytes, and melanin-containing macrophages abound [39, 40]. Ultrastructural examination shows FPPs that may occur alone or in association with CLPs and/or RLPs and rarely GRODs [46–53]. CLN3 (the traditional Juvenile Neuronal Ceroid-Lipofuscinosis, JNCL, Batten disease, Spielmeyer-Vogt-Sjogren disease) classically manifests as juvenile onset disease, but other NCLs that may present in this manner include CLN1, CLN2, CLN8, CLN10, and CLN9.
Adult onset NCL is rare and characterized by sparing of vision, in contrast to all other clinical presentations of NCL. The traditional adult onset NCL, called Kufs disease, is divided into two overlapping clinical presentations. The patients may first come to clinical attention in the second decade to as late as the sixth decade, but most typically present around the age of 30 years. Kufs disease type A (CLN4A) is inherited in an autosomal recessive manner and presents with progressive myoclonus. Kufs disease type B (CLN4B), which presents with dementia and various motor-system signs, is the only NCL with presumed autosomal dominant inheritance pattern [90–94]. Ultrastructural examination may show GRODs or FPPs. The putative gene loci CLN4A and CLN4B were only recently mapped. The autosomal recessive Kufs disease, CLN4A, has been shown to be caused by mutations in the CLN6 gene [25], while the autosomal dominant Kufs disease, CLN4B, has been shown to be caused by mutations in DNAJC5 that encodes cysteine-string protein alpha (CSPα) [26]. Other NCLs that may rarely present with adult onset disease include CLN1, CLN10, CLN5, CLN13, and CLN11.
5Diagnostic approach
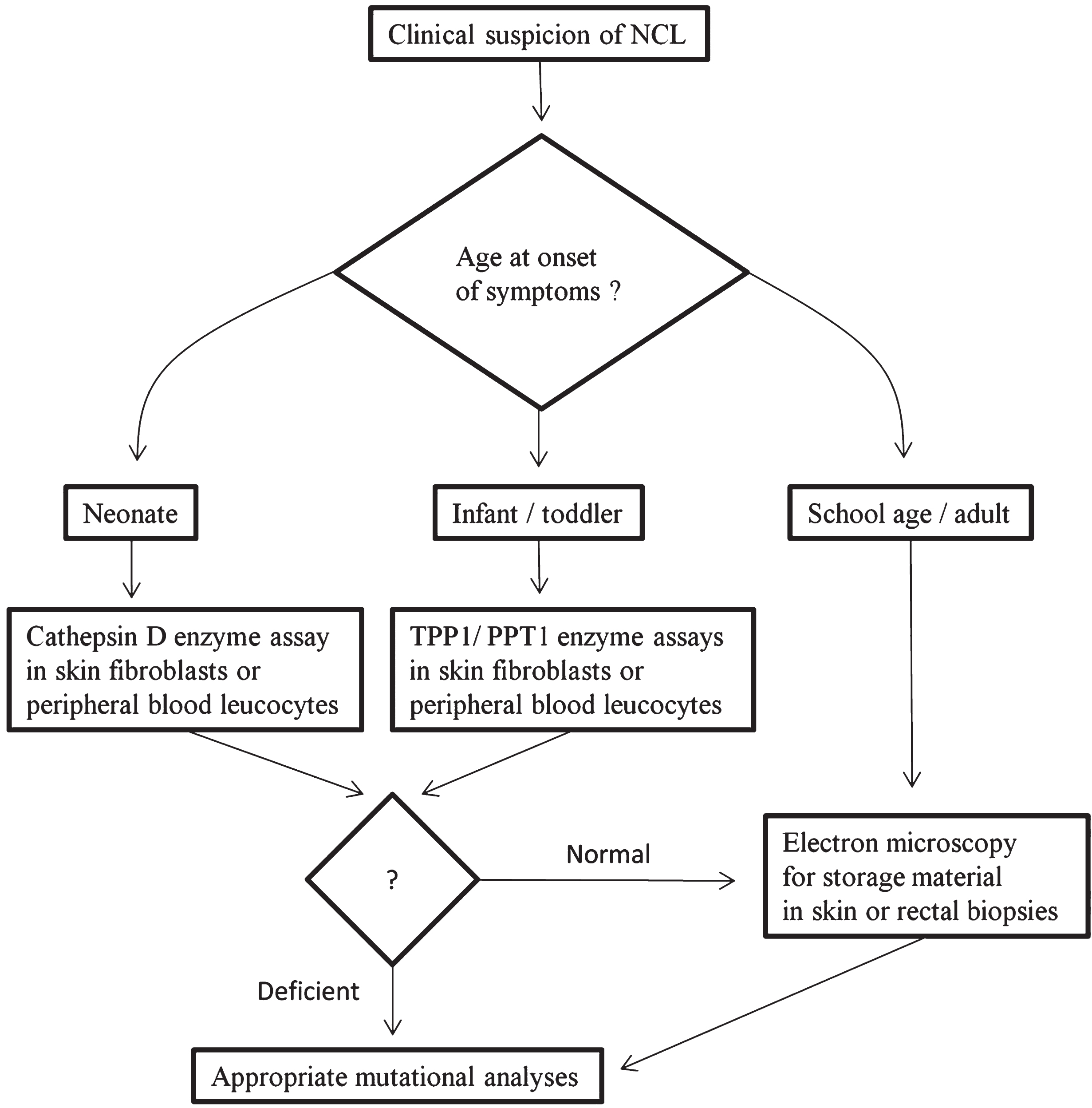
We suggest a step-wise, rather than a shot-gun, logical diagnostic approach based on the current knowledge of the NCL types (Fig. 4) [55]. The first factor that should be considered when making a diagnosis is the clinical phenotype including the age at onset of clinical presentation. For example, CLN10 should be considered in a neonate with microcephaly and seizures. In this scenario, measurement of reduced cathepsin D activity in skin fibroblasts or white blood cells would clinch the diagnosis. Similarly, CLN1 and CLN2 should be considered in infants with motor delay and visual deterioration. In this situation, reduced PPT1 activity in white blood cells or skin fibroblasts would be diagnostic of CLN1 and reduced TPP1 activity in white blood cells or skin fibroblasts would be diagnostic of CLN2. The enzyme assays can also be performed on chorionic villous cells for prenatal diagnosis [95]. If the enzyme activities are normal and in older children or adults with suspected NCL, a skin or rectal biopsy may be performed to look for the storage material by ultrastructural examination. Peripheral blood smears may be examined for cytoplasmic vacuolations in lymphocytes. Mutational analysis for the genes involved may then be performed based on the clinical and ultrastructural phenotype and the results of enzyme assays. Given the increasing number of genes associated with the NCLs and the decreasing cost of massively-parallel (next generation) sequencing, targeted panels or even whole exome/genome sequencing are becoming routinely available. Already, a few laboratories (for example, the genetics laboratories at Children’s Health of Dallas, University of Chicago, and Greenwood Genetic Center) offer “NCL panels” or cover NCL genes as part of an “epilepsy panel”. Functional enzyme testing when available will remain an important component of the diagnostic pathway especially when variants of unknown significance are identified in patients using molecular testing.
Fig.4
Suggested diagnostic algorithm for Neuronal Ceroid-Lipofuscinoses (NCLs).
66. Treatment
Currently, none of the NCLs is curable and supportive therapy is the only way to improve the quality of life for the patients. For NCLs caused by deficiencies of soluble lysosomal enzymes, enzyme replacement therapy, gene therapy, and stem cell therapy approaches to treatment are under evaluation but so far without demonstrative success [96]. Recently, enzyme replacement therapy with a recombinant human tripeptidyl peptidase-1 cerliponase alpha (Brineura®) has been approved for CLN2 patients three years and older by the Food and Drug Administration in the USA and for CLN2 patients of all ages in the European Union [97]. The only other approved therapies at the present time are to control signs and symptoms such as seizures and psychiatric problems.
References
[1] | Haltia M. , The neuronal ceroid-lipofuscinoses, Journal ofNeuropathology and Experimental Neurology 62: ((2003) ), 1–13. |
[2] | Rakheja D. , Narayan S.B. , Bennett M.J. , The function of CLN3P,the Batten disease protein, Mol Genet Metab 93: ((2008) ), 269–274. |
[3] | Jalanko A. , Braulke T. , Neuronal ceroid lipofuscinoses, Biochim Biophys Acta 1793: ((2009) ), 697–709. |
[4] | Hofmann S.L. , Lee L.A. , Lu J.Y. , Verkruyse L.A. , Palmitoyl-protein thioesterase and the molecular pathogenesis of infantile neuronal ceroid lipofuscinosis, Neuropediatrics 28: ((1997) ), 27–30. |
[5] | Verkruyse L.A. , Hofmann S.L. , Lysosomal targeting ofpalmitoyl-protein thioesterase, J Biol Chem 271: ((1996) ), 15831–15836. |
[6] | Sleat D.E. , Donnelly R.J. , Lackland H. , Liu C.G. , Sohar I. , Pullarkat R.K. , et al., Association of mutations in a lysosomal proteinwith classical late-infantile neuronal ceroid lipofuscinosis, Science 277: ((1997) ), 1802–1805. |
[7] | Sohar I. , Sleat D.E. , Jadot M. , Lobel P. , Biochemicalcharacterization of a lysosomal protease deficient in classical lateinfantile neuronal ceroid lipofuscinosis (LINCL) and development ofan enzyme-based assay for diagnosis and exclusion of LINCL in humanspecimens and animal models, J Neurochem 73: ((1999) ), 700–711. |
[8] | Tyynela J. , Sohar I. , Sleat D.E. , Gin R.M. , Donnelly R.J. , Baumann M. , et al., A mutation in the ovine cathepsin D gene causes acongenital lysosomal storage disease with profoundneurodegeneration, Embo J 19: ((2000) ), 2786–2792. |
[9] | Tyynela J. , Sohar I. , Sleat D.E. , Gin R.M. , Donnelly R.J. , Baumann M. , et al., Congenital ovine neuronal ceroid lipofuscinosis–acathepsin D deficiency with increased levels of the inactive enzyme, Eur J Paediatr Neurol 5: (Suppl A) ((2001) ), 43–45. |
[10] | Siintola E. , Partanen S. , Stromme P. , Haapanen A. , Haltia M. , Maehlen J. , et al., Cathepsin D deficiency underlies congenital humanneuronal ceroid-lipofuscinosis, Brain 129: ((2006) ), 1438–1445. |
[11] | Smith K.R. , Dahl H.H. , Canafoglia L. , Andermann E. , Damiano J. , Morbin M. , et al., Cathepsin F mutations cause Type B Kufs disease, anadult-onset neuronal ceroid lipofuscinosis, Hum Mol Genet 22: ((2013) ), 1417–1423. |
[12] | Tang C.H. , Lee J.W. , Galvez M.G. , Robillard L. , Mole S.E. and Chapman H.A. , Murine cathepsin F deficiency causes neuronallipofuscinosis and late-onset neurological disease, Mol CellBiol 26: ((2006) ), 2309–2316. |
[13] | Kasper D. , Planells-Cases R. , Fuhrmann J.C. , Scheel O. , Zeitz O. , Ruether K. , et al., Loss of the chloride channel ClC-7 leads tolysosomal storage disease and neurodegeneration, Embo J 24: ((2005) ), 1079–1091. |
[14] | Yoshikawa M. , Uchida S. , Ezaki J. , Rai T. , Hayama A. , Kobayashiet K. , et al., CLC-3 deficiency leads to phenotypes similar to humanneuronal ceroid lipofuscinosis, Genes Cells 7: ((2002) ), 597–605. |
[15] | Poet M. , Kornak U. , Schweizer M. , Zdebik A.A. , Scheel O. , Hoelter S. , et al., Lysosomal storage disease upon disruption of theneuronal chloride transport protein ClC-6, Proc Natl Acad Sci US A 103: ((2006) ), 13854–13859. |
[16] | Farias F.H. , Zeng R. , Johnson G.S. , Wininger F.A. , Taylor J.F. , Schnabel R.D. , et al., A truncating mutation in ATP13A2 is responsiblefor adult-onset neuronal ceroid lipofuscinosis in Tibetan terriers, Neurobiol Dis 42: ((2011) ), 468–474. |
[17] | Bras J. , Verloes A. , Schneider S.A. , Mole S.E. , Guerreiro R.J. , Mutation of the parkinsonism gene ATP13A2 causes neuronalceroid-lipofuscinosis, Hum Mol Genet 21: ((2012) ), 2646–2650. |
[18] | Yang X. , Xu Y. , Mutations in the ATP13A2 gene and Parkinsonism: Apreliminary review, Biomed Res Int 2014: ((2014) ), 371256. |
[19] | Lerner T.J. , Boustany R.N. , Anderson J.W. , D’Arigo K.L. , Schlumpf K.S. , Buckler A.J. , Gusella J.F. , Haines Less J.L. , et al., Isolation of a novel gene underlying Batten disease, CLN3. The International Batten Disease Consortium, Cell 82: ((1995) ), 949–957. |
[20] | Rakheja D. , Narayan S.B. , Bennett M.J. , Juvenile neuronalceroid-lipofuscinosis (Batten disease): A brief review and update, Curr Mol Med 7: ((2007) ), 603–608. |
[21] | Narayan S.B. , Rakheja D. , Tan L. , Pastor J.V. , BennettCLN3P M.J. , the Batten’s disease protein, is a novel palmitoyl-proteinDelta-9 desaturase, Ann Neurol 60: ((2006) ), 570–577. |
[22] | Narayan S.B. , Tan L. , Bennett M.J. , Intermediate levels of neuronal palmitoyl-protein Delta-9 desaturase in heterozygotes formurine Batten disease, Mol Genet Metab 93: ((2008) ), 89–91. |
[23] | Rakheja D. , Narayan S.B. , Pastor J.V. , Bennett M.J. , CLN3P, theBatten disease protein, localizes to membrane lipid rafts (detergent-resistant membranes), Biochem Biophys Res Commun 317: ((2004) ), 988–991. |
[24] | Hobert J.A. , Dawson G. , A novel role of the Batten disease gene CLN3: Association with BMP synthesis, Biochem Biophys ResCommun 358: ((2007) ), 111–116. |
[25] | Arsov T. , Smith K.R. , Damiano J. , Franceschetti S. , Canafoglia L. , Bromhead C.J. , et al., Kufs disease, the major adult form of neuronal ceroid lipofuscinosis, caused by mutations in CLN6, AmJ Hum Genet 88: ((2011) ), 566–573. |
[26] | Noskova L. , Stranecky V. , Hartmannova H. , Pristoupilova A. , Baresova V. , Ivanek R. , et al., Mutations in DNAJC5, encodingcysteine-string protein alpha, cause autosomal-dominant adult-onsetneuronal ceroid lipofuscinosis, Am J Hum Genet 89: ((2011) ), 241–252. |
[27] | Isosomppi J. , Vesa J. , Jalanko A. , Peltonen L. , Lysosomallocalization of the neuronal ceroid lipofuscinosis CLN5 protein, Hum Mol Genet 11: ((2002) ), 885–891. |
[28] | Savukoski M. , Klockars T. , Holmberg V. , Santavuori P. , Landerand E.S. , Peltonen L. , CLN5, a novel gene encoding a putativetransmembrane protein mutated in Finnish variant late infantile neuronal ceroid lipofuscinosis, Nat Genet 19: ((1998) ), 286–288. |
[29] | Gao H. , Boustany R.M. , Espinola J.A. , Cotman S.L. , Srinidhi L. , Antonellis K.A. , et al., Mutations in a novel CLN6-encoded transmembrane protein cause variant neuronal ceroid lipofuscinosis in man andmouse, Am J Hum Genet 70: ((2002) ), 324–335. |
[30] | Teixeira C.A. , Espinola J. , Huo L. , Kohlschutter J. , PersaudSawin D.A. , Minassian B. , et al., Novel mutations in the CLN6 genecausing a variant late infantile neuronal ceroid lipofuscinosis, Hum Mutat 21: ((2003) ), 502–508. |
[31] | Sharp J.D. , Wheeler R.B. , Parker K.A. , Gardiner R.M. , Williamsand R.E. , Mole S.E. , Spectrum of CLN6 mutations in variant late infantileneuronal ceroid lipofuscinosis, Hum Mutat 22: ((2003) ), 35–42. |
[32] | Siintola E. , Topcu M. , Aula N. , Lohi H. , Minassian B.A. , Paterson A.D. , et al., The Novel neuronal ceroid lipofuscinosis geneMFSD8 encodes a putative lysosomal transporter, Am J Hum Genet 81: ((2007) ), 136–146. |
[33] | Mitchell W.A. , Wheeler R.B. , Sharp J.D. , Bate S.L. , Gardiner R.M. , Ranta U.S. , et al., Turkish variant late infantile neuronal ceroidlipofuscinosis (CLN7) may be allelic to CLN8, Eur J PaediatrNeurol 5: (Suppl A) ((2001) ), 21–27. |
[34] | Ranta S. , Topcu M. , Tegelberg S. , Tan H. , Ustubutun A. , Saatciet I. , et al., Variant late infantile neuronal ceroid lipofuscinosis in asubset of Turkish patients is allelic to Northern epilepsy, HumMutat 23: ((2004) ), 300–305. |
[35] | Ranta S. , Zhang Y. , Ross B. , Lonka L. , Takkunen E. , Messer A. , et al., The neuronal ceroid lipofuscinoses in human EPMR and mnd mutantmice are associated with mutations in CLN8, Nat Genet 23: ((1999) ), 233–236. |
[36] | Schulz A. , Mousallem T. , Venkataramani M. , Persaud-Sawin D.A. , Zucker A.Z , Luberto C. , et al., The CLN9 protein, a regulator ofdihydroceramide synthase, J Biol Chem 281: ((2006) ), 2784–2794. |
[37] | Smith K.R. , Damiano J. , Franceschetti S. , Carpenter S. , Canafoglia L. , Morbin M. , et al., Strikingly differentclinicopathological phenotypes determined by progranulin-mutationdosage, Am J Hum Genet 90: ((2012) ), 1102–1107. |
[38] | Staropoli J.F. , Karaa A. , Lim E.T. , Kirby A. , Elbalalesy N. , Romansky S.G. , et al., A homozygous mutation in KCTD7 links neuronalceroid lipofuscinosis to the ubiquitin-proteasome system, Am JHum Genet 91: ((2012) ), 202–208. |
[39] | Hofmann S.L. , Peltonen L. , The neuronal ceroid lipofuscinoses, in The Metabolic And Molecular Bases Of Inherited Disease. III: , Scriver C. R. , Beaudet A. L. , Sly W. S. and Valle D. , Eds., 8th New York: McGraw-Hill, (2001) , pp. 3877–3894. |
[40] | Williams R.E. , Aberg L. , Autti T. , Goebel H.H. , Kohlschutter A. , Lonnqvist T. , Diagnosis of the neuronal ceroid lipofuscinoses: Anupdate, Biochim Biophys Acta 1762: ((2006) ), 865–872. |
[41] | Hall N.A. , Lake B.D. , Dewji N.N. , Patrick A.D. , Lysosomal storageof subunit c of mitochondrial ATP synthase in Batten’s disease(ceroid-lipofuscinosis), Biochem J 275: (Pt 1) ((1991) ), 269–272. |
[42] | Palmer D.N. , Fearnley I.M. , Walker J.E. , Hall N.A. , Lake B.D. , Wolfe L.S. , et al., Mitochondrial ATP synthase subunit c storage in theceroid-lipofuscinoses (Batten disease), Am J Med Genet ((1992) ) 42: , 561–567. |
[43] | Palmer D.N. , Jolly R.D. , van Mil H.C. , Tyynela J. and Westlake V.J. , Different patterns of hydrophobic protein storage in different formsof neuronal ceroid lipofuscinosis (NCL, Batten disease), Neuropediatrics 28: ((1997) ), 45–48. |
[44] | Palmer D.N. , Tyynela J. , van Mil H.C. , Westlake V.J. and Jolly R.D. , Accumulation of sphingolipid activator proteins (SAPs) A and D ingranular osmiophilic deposits in miniature Schnauzer dogs withceroid-lipofuscinosis, J Inherit Metab Dis 20: ((1997) ), 74–84. |
[45] | Tyynela J. , Palmer D.N. , Baumann M. , Haltia M. , Storage ofsaposins A and D in infantile neuronal ceroid-lipofuscinosis, FEBS Lett 330: ((1993) ), 8–12. |
[46] | Mole S.E. , Williams R.E. , Goebel H.H. , Correlations betweengenotype, ultrastructural morphology and clinical phenotype in theneuronal ceroid lipofuscinoses, Neurogenetics 6: ((2005) ), 107–126. |
[47] | Aberg L. , Jarvela I. , Rapola J. , Autti T. , Kirveskari E. , Lappiet M. , et al., Atypical juvenile neuronal ceroid lipofuscinosis withgranular osmiophilic deposit-like inclusions in the autonomic nervecells of the gut wall, Acta Neuropathol (Berl) 95: ((1998) ), 306–312. |
[48] | Anderson G.W. , Smith V.V. , Brooke I. , Malone M. , Sebire N.J. , Diagnosis of neuronal ceroid lipofuscinosis (Batten disease) byelectron microscopy in peripheral blood specimens, UltrastructPathol 30: ((2006) ), 373–378. |
[49] | Carlen B. , Englund E. , Diagnostic value of electron microscopy ina case of juvenile neuronal ceroid lipofuscinosis, UltrastructPathol 25: ((2001) ), 285–288. |
[50] | Conradi N.G. , Uvebrant P. , Hokegard K.H. , Wahlstrom J. and Mellqvist L. , First-trimester diagnosis of juvenile neuronal ceroidlipofuscinosis by demonstration of fingerprint inclusions inchorionic villi, Prenat Diagn 9: ((1989) ), 283–287. |
[51] | Boldrini R. , Biselli R. , Santorelli F.M. , Bosman C. , Neuronalceroid lipofuscinosis: An ultrastructural, genetic, and clinicalstudy report, Ultrastruct Pathol 25: ((2001) ), 51–58. |
[52] | Farrell D.F. , Sumi S.M. , Skin punch biopsy in the diagnosis ofjuvenile neuronal ceroid-lipofuscinosis. A comparison with leukocyteperoxidase assay, Arch Neurol 34: ((1977) ), 39–44. |
[53] | Ikeda K. , Goebel H.H. , Ultrastructural pathology of lymphocytesin neuronal ceroid-lipofuscinoses, Brain Dev 1: ((1979) ), 285–292. |
[54] | Goebel H.H. , Morphological aspects of the neuronal ceroidlipofuscinoses, Neurol Sci 21: ((2000) ), S27–S33. |
[55] | Kohlschutter A. , Schulz A. , Towards understanding the neuronalceroid lipofuscinoses, Brain Dev 31: ((2009) ), 499–502. |
[56] | Haltia M. , The neuronal ceroid-lipofuscinoses: From past to present, Biochim Biophys Acta 1762: ((2006) ), 850–856. |
[57] | Anderson G. , Elleder M. , Goebel H.H. , Morphological diagnosticand pathological considerations, in The Neuronal CeroidLipofuscinoses (Batten Disease), Mole S. E. , Williams R. E. and Goebel H.H. , Eds., 2nd New York: Oxford University Press, (2011) , pp. 35–49. |
[58] | Wisniewski K.E. , Kida E. , Congenital ceroid-lipofuscinosis, Pediatr Neurol 8: ((1992) ), 315. |
[59] | Barohn R.J. , Dowd D.C. , Kagan-Hallet K.S. , Congenitalceroid-lipofuscinosis, Pediatr Neurol 8: ((1992) ), 54–59. |
[60] | Garborg I. , Torvik A. , Hals J. , Tangsrud S.E. , LindemannCongenital R. , neuronal ceroid lipofuscinosis. A case report, ActaPathol Microbiol Immunol Scand A 95: ((1987) ), 119–125. |
[61] | Norman R.M. , Wood N. , A congenital form of amaurotic familyidiocy, J Neurol Psychiatry 4: ((1941) ), 175–190. |
[62] | Brown N.J. , Corner B.D. , Dodgson M.C. , A second case in the samefamily of congenital familial cerebral lipoidosis resembling amaurotic family idiocy, Arch Dis Child 29: ((1954) ), 48–54. |
[63] | Humphreys S. , Lake B.D. , Scholtz C.L. , Congenital amauroticidiocy–a pathological, histochemical, biochemical and ultrastructural study, Neuropathol Appl Neurobiol 11: ((1985) ), 475–484. |
[64] | Sandbank U. , Congenital amaurotic idiocy, Pathol Eur 3: ((1968) ), 226–229. |
[65] | Haltia M. , Rapola J. , Santavuori P. , Infantile type of so-calledneuronal ceroid-lipofuscinosis. Histological and electron microscopic studies, Acta Neuropathol 26: ((1973) ), 157–170. |
[66] | Haltia M. , Rapola J. , Santavuori P. , Keranen A. , Infantile typeof so-called neuronal ceroid-lipofuscinosis. 2. Morphological andbiochemical studies, J Neurol Sci 18: ((1973) ), 269–285. |
[67] | Santavuori P. , Haltia M. , Rapola J. , Infantile type of so-called neuronal ceroid-lipofuscinosis, Dev Med Child Neurol 16: ((1974) ), 644–653. |
[68] | Santavuori P. , Haltia M. , Rapola J. , Raitta C. , Infantile type of so-called neuronal ceroid-lipofuscinosis. 1. A clinical study of 15 patients, J Neurol Sci 18: ((1973) ), 257–267. |
[69] | Santavuori P. , Vanhanen S.L. , Sainio K. , Nieminen M. , Wallden T. , Launes J. , et al., Infantile neuronal ceroid-lipofuscinosis (INCL):Diagnostic criteria, J Inherit Metab Dis 16: ((1993) ), 227–229. |
[70] | Goebel H.H. , Klein H. , Santavuori P. , Sainio K. , Ultra structural studies of the retina in infantile neuronal ceroid-lipofuscinosis, Retina 8: ((1988) ), 59–66. |
[71] | Haynes M.E. , Manson J.I. , Carter R.F. , Robertson E. , Electronmicroscopy of skin and peripheral blood lymphocytes in infantile(Santavuori) neuronal ceroid lipofuscinosis, Neuropadiatrie 10: ((1979) ), 245–263. |
[72] | Rapola J. , Santavuori P. , Savilahti E. , Suction biopsy of rectalmucosa in the diagnosis of infantile and juvenile types of neuronalceroid lipofuscinoses, Hum Pathol 15: ((1984) ), 352–360. |
[73] | Wheeler R.B. , Schlie M. , Kominami E. , Gerhard L. , GoebelNeuronal H.H. , ceroid lipofuscinosis: Late infantile or Jansky Bielschowsky type–re-revisited, Acta Neuropathol 102: ((2001) ), 485–488. |
[74] | Andrews J.M. , Sorenson V. , Cancilla P.A. , Price H.M. and Menkes J.H. , Late infantile neurovisceral storage disease withcurvilinear bodies, Neurology 21: ((1971) ), 207–217. |
[75] | Buhl L. , Muirhead D. , Litthander J. , Raj G. , Late infantileneuronal ceroid lipofuscinosis: An ultrastructural investigation, Pediatr Pathol 14: ((1994) ), 397–404. |
[76] | Goebel H.H. , Gerhard L. , Kominami E. , Haltia M. , Neuronalceroid-lipofuscinosis–late-infantile or Jansky-Bielschowskytype–revisited, Brain Pathol 6: ((1996) ), 225–228. |
[77] | Kurata K. , Hayashi M. , Satoh J. , Kojima H. , Nagata J. , Tamagawaet K. , et al., Pathological study on sibling autopsy cases of the lateinfantile form of neuronal ceroid lipofuscinosis, Brain Dev 21: ((1999) ), 63–67. |
[78] | Markesbery W.R. , Shield L.K. , Egel R.T. , Jameson H.D. , Late-infantile neuronal ceroid-lipofuscinosis. An ultrastructuralstudy of lymphocyte inclusions, Arch Neurol 33: ((1976) ), 630–635. |
[79] | Aberg L.E. , Backman M. , Kirveskari E. , Santavuori P. , Epilepsyand antiepileptic drug therapy in juvenile neuronal ceroidlipofuscinosis, Epilepsia 41: ((2000) ), 1296–1302. |
[80] | Backman M.L. , Santavuori P.R. , Aberg L.E. , AronenPsychiatric E.T. , symptoms of children and adolescents with juvenileneuronal ceroid lipofuscinosis, J Intellect Disabil Res 49: ((2005) ), 25–32. |
[81] | Jarvela I. , Autti T. , Lamminranta S. , Aberg L. , Raininko R. and Santavuori P. , Clinical and magnetic resonance imaging findings inBatten disease: Analysis of the major mutation (1.02-kb deletion), Ann Neurol 42: ((1997) ), 799–802. |
[82] | Lamminranta S. , Aberg L.E. , Autti T. , Moren R. , Laine T. , Kaukoranta J. , et al., Neuropsychological test battery in the follow-upof patients with juvenile neuronal ceroid lipofuscinosis, JIntellect Disabil Res 45: ((2001) ), 8–17. |
[83] | Santavuori P. , Neuronal ceroid-lipofuscinoses in childhood, Brain Dev 10: ((1988) ), 80–83. |
[84] | Santavuori P. , Linnankivi T. , Jaeken J. , Vanhanen S.L. , Telakiviand T. , Heiskala H. , Psychological symptoms and sleep disturbances inneuronal ceroid-lipofuscinoses (NCL), J Inherit Metab Dis 16: ((1993) ), 245–248. |
[85] | Weleber R.G. , The dystrophic retina in multisystem disorders: The electroretinogram in neuronal ceroid lipofuscinoses, Eye 12: (Pt 3b) ((1998) ), 580–590. |
[86] | Eksandh L.B. , Ponjavic V.B. , Munroe P.B. , Eiberg H.E. , Uvebrant P.E. , Ehinger B.E. , et al., Full-field ERG in patients withBatten/Spielmeyer-Vogt disease caused by mutations in the CLN3 gene, Ophthalmic Genet 21: ((2000) ), 69–77. |
[87] | Autti T. , Raininko R. , Santavuori P. , Vanhanen S.L. , Poutanenand V.P. , Haltia M. , MRI of neuronal ceroid lipofuscinosis. II. PostmortemMRI and histopathological study of the brain in 16 cases of neuronalceroid lipofuscinosis of juvenile or late infantile type, Neuroradiology 39: ((1997) ), 371–377. |
[88] | Autti T. , Raininko R. , Vanhanen S.L. , Santavuori P. , MRI ofneuronal ceroid lipofuscinosis. I. Cranial MRI of 30 patients withjuvenile neuronal ceroid lipofuscinosis, Neuroradiology 38: ((1996) ), 476–482. |
[89] | Autti T. , Hamalainen J. , Aberg L. , Lauronen L. , Tyynela J. and Van Leemput K. , Thalami and corona radiata in juvenile NCL (CLN3): Avoxel-based morphometric study, Eur J Neurol 14: ((2007) ), 447–450. |
[90] | Burneo J.G. , Arnold T. , Palmer C.A. , Kuzniecky R.I. , Oh S.J. and Faught E. , Adult-onset neuronal ceroid lipofuscinosis (Kufs disease)with autosomal dominant inheritance in Alabama, Epilepsia 44: ((2003) ), 841–846. |
[91] | Sadzot B. , Reznik M. , Arrese-Estrada J.E. , Franck G. , FamilialKufs’ disease presenting as a progressive myoclonic epilepsy, JNeurol 247: ((2000) ), 447–454. |
[92] | Josephson S.A. , Schmidt R.E. , Millsap P. , McManus D.Q. and Morris J.C. , Autosomal dominant Kufs’ disease: A cause of early onsetdementia, J Neurol Sci 188: ((2001) ), 51–60. |
[93] | Vital A. , Vital C. , Orgogozo J.M. , Mazeaux J.M. , Pautrizel B. , Lariviere J.M. , Adult dementia due to intraneuronal accumulation ofceroidlipofuscinosis (Kufs’ disease): Ultrastructural study of twocases, J Geriatr Psychiatry Neurol 4: ((1991) ), 110–115. |
[94] | Berkovic S.F. , Carpenter S. , Andermann F. , Andermann E. and Wolfe L.S. , Kufs’ disease: A critical reappraisal, Brain 111: (Pt 1) ((1988) ), 27–62. |
[95] | Young E.P. , Worthington V.C. , Jackson M. , Winchester B.G. , Pre-and postnatal diagnosis of patients with CLN1 and CLN2 by assay ofpalmitoyl-protein thioesterase and tripeptidyl-peptidase Iactivities, Eur J Paediatr Neurol 5: (Suppl A) 193-6 ((2001) ), 80–83. |
[96] | Wong A.M. , Rahim A.A. , Waddington S.N. , Cooper J.D. , Currenttherapies for the soluble lysosomal forms of neuronal ceroidlipofuscinosis, Biochem Soc Trans 38: ((2010) ), 1484–1488. |
[97] | Markham A. , Cerliponase Alfa: First Global Approval, Drugs 77: ((2017) ), 1247–1249. |


