
Translational research advances a new era of prenatal diagnosis and newborn screening
Abstract
This article provides a review of selected metabolic disorders resulting from genetic mutations and the methods used to identify them prenatally or facilitate diagnosis in the early neonatal period. Prenatal and neonatal diagnostic technologies have expanded and improved dramatically in the 21st century, as is their application in population-based screening and/or targeted assessment of at-risk couples. For instance, preimplantation genetic diagnosis has been a major advance. Emphasis herein has been placed on prototype diseases such as phenylketonuria, cystic fibrosis, and Tay-Sachs that have stimulated seminal efforts to improve medical practices in these fields. As more molecular strategies evolve, future developments in prenatal screening and diagnosis, along with newborn screening expansion, seem likely to continue rapid translation to the bedside.
Metabolic disorders resulting from genetic mutations have challenged obstetricians and pediatricians to develop diagnostic methods to identify them prenatally or in the early neonatal period [1]. The strategy of translational research has been ideal to close this gap as biotechnological advances are applied from the stage of novel laboratory developments to the practice of maternal-fetal medicine and neonatology. Although prenatal and neonatal diagnostic technologies have evolved gradually over four decades, they are expanding and changing dramatically in the 21st century, as is their application in population-based screening and/or targeted assessment of at-risk couples [1]. These changes have resulted in part from the Human Genome Project, a multicenter project supported by public and private funding geared to the sequencing of the human genome, which was completed in 2003. Clearly, the demand for prenatal genetic screening testing will continue as a result of individual and organizational advocacy efforts. In addition, advances in analytical technologies applying sophisticated cellular and molecular biology methods have increased the number and quality of tests available. Therefore, we are providing an overview with emphasis on prenatal diagnosis and suggest more comprehensive references in the bibliography [2–5]. We also recommend that the reader consult practice guidelines publications of The American College of Obstetricians and Gynecologists (ACOG) [6, 7], the American Academy of Pediatrics (AAP) [8], and the American College of Medical Genetics (ACMG) [9, 10]. For a variety of reasons, our review has focused almost exclusively on clinical care in the United States, where the variety and complexity of medical, legal, social, and ethical issues are generally greater than in other countries.
We have also taken the liberty of emphasizing certain disorders such as Tay Sachs disease and cystic fibrosis because they are prototype genetic disorders in prenatal and neonatal screening, respectively.
1Evolution of prenatal and neonatal genetic diagnostic methods and screening
Prenatal and perinatal medicine advanced dramatically, though incrementally, during the second half of the twentieth century because of combination of research discoveries and technological developments applicable to the fetus and newborn. The important discoveries and their translation into patient care depended on interdisciplinary science and teamwork among health care providers. Perhaps no other area of medical practice has been so immediately and profoundly influenced by the genetics revolution as represented by the completion of the Human Genome Project in 2003, as cellular and molecular biology techniques developed in research laboratories to sequence the human genome became widely available in clinical practice. Thus, recent years have witnessed an extraordinary improvement in our ability to diagnose genetic conditions either prenatally or in the early neonatal period; almost all of these depend upon collaboration with clinical laboratories.
Prenatal genetics emerged in the 1960’s and 1970’s with amniocentesis used to assess fetal conditions. During the 1980’s, chorionic villus sampling technology with cell culture emerged as an important step forward. This was followed during the next decade by ultrasound-based assessment of fetal morphology, as originally applied by radiologists and subsequently by maternal-fetal specialists and reproductive geneticists. As novel genomic and proteomic technologies were applied, the precision of diagnostic technologies has improved dramatically and non-invasive screening methods now predominate in prenatal genetics practice. The interval from laboratory discovery to bedside application is no longer measured in years, as previously has been the case in medicine, but in months or even weeks. In recent years, the most significant advances have come about by applying both biochemical and molecular technologies. The burgeoning field of prenatal diagnosis has fostered a host of specialty journals, most notably Prenatal Diagnosis, books, and internet sites. Thus, driven by a combination of excellent diagnostic technologies and patient demand, and also augmented by growing proportion of first pregnancies in women beyond thirty years of age, a new era in prenatal diagnosis of genetic disease has been well established worldwide [11].
In parallel, the diagnosis of genetic disorders in newborn infants has improved for a wide variety of inborn errors of metabolism due to single gene defects. The advent of newborn screening is historically interesting because it depended primarily on development of a neonatal blood sampling technique rather than analytical biochemistry advances [12]. Specifically, to promote early diagnosis of phenylketonuria (PKU) in newborn populations, Robert Guthrie devised a technique for routine procurement of blood using a “heel stick” to obtain about 0.4 ml of blood on filter paper. During the early 1960’s, the potential opportunity to prevent mental retardation through early diagnosis aroused great interest, and Guthrie was motivated to focus his attention on PKU using blood phenylalanine levels determined microbiologically to ensure early diagnosis through population-based screening of all newborns. Driven by parent advocacy groups, mandatory statewide screening for PKU was first instituted in Massachusetts and Oregon during 1963, and two years later 28 more states had statutory requirements for PKU screening of all newborns. Next, galactosemia screening was implemented on the same dried blood specimens using microbiologic determinations and in the early 1970’s congenital hypothyroidism followed. Many other genetic disorders classified as inborn errors of metabolism were added in the 1980’s and 1990’s. With the recent advent of tandem mass spectrometry, more than 50 inborn errors of metabolism can be readily diagnosed using dried blood specimens. The analytical methods extended in the 1990’s to DNA analyses for carrier screening and diagnosis of cystic fibrosis (CF). Soon after nationwide newborn screening for CF began in the USA, another DNA-based test was introduced for Severe Combined Immunodeficiency (SCID). Because congenital metabolic and endocrine disorders have a latent period from onset to irreversible pathology, the opportunity exists for intervention with preventive therapies, and these have also been developing.
2Diagnostic strategies
Despite the incorporation of new and novel laboratory techniques, conclusively establishing the diagnosis of disease still typically depends on history, physical examination, and laboratory studies confirming preexisting suspicion. In the case of the fetus and asymptomatic neonate, laboratory tests are likely to increase in importance compared to other factors applicable to the newborn or infant. Screening for early detection of disease is especially valuable in prenatal and neonatal practice. According to Hennekens and Buring [13], “screening refers to the application of a test to people who are as yet asymptomatic for the purpose of classifying them with the respect to their likelihood of having a particular disease…the screening procedure itself does not diagnose illness…those who test positive are sent on for further evaluation by a subsequent diagnostic test or procedure to determine whether they do, in fact, have the disease.” As modern laboratory methods improve, however, some screening tests can actually become diagnostic tests as in the case of CF when DNA analyses on neonatal blood specimens reveal two F508del mutations of the cystic fibrosis transmembrane conductance regulator (CFTR) gene [14].
There are important differences between prenatal diagnostic strategies practiced by obstetricians and the early diagnosis of genetic disorders in newborns through population-based screening. In general, the former has relied on the traditional medical model of intervention in individuals (pregnant women), although the interventions are usually triggered by risk factors rather than illnesses. Newer approaches to population-based screening applied to pregnant women are represented by the use of massive parallel shotgun sequencing of cell-free fetal nucleic acids obtained from maternal blood to noninvasively screen pregnant women for fetal trisomy 21 and will likely be applicable to other fetal chromosomal abnormalities and Mendelian disorders [15, 16].
Important screening test characteristics include two measures of validity, namely sensitivity and specificity, and also measures of probability. Sensitivity and specificity are inherent characteristics of screening tests with a defined cut-off value and are not affected by disease prevalence, whereas the predictive values (probability measures) of the tests are influenced by prevalence. The most important consideration in prenatal and neonatal genetics practice is that screening tests achieve close to 100% sensitivity, i.e. avoidance of false negative results. Unfortunately, it is impossible to achieve 100% sensitivity over a long duration because even when there are no biologic false negatives, the possibility always exists for laboratory or human error. Therefore, health care providers should always assume that no screening test is perfect, just as no laboratory test is infallible. Test specificity, or the true negative rate, indicates probability of the person without the disease will have a negative test result. As with sensitivity, specificity should ideally approach 100% but this is difficult to achieve while optimizing sensitivity. Finally, the positive predictive value of (PPV) should be regarded as extremely important because it provides a measure of the post-test probability or likelihood of disease following a positive screening test. PPV indicates the proportion of persons with positive test results who have the disease.
3Methods of prenatal diagnosis
A brief overview of the well-known conventional techniques is provided below and is followed by comments about newer preimplantation techniques.
A. Sampling Techniques
1. Chorionic villus sampling
Since amniocentesis (discussed below) is most commonly performed in the mid-second trimester (15 to 16 weeks), fetal diagnosis is not usually established prior to 16 weeks gestation. A technique that could be performed during the first trimester is most desirable to reduce the psychological stress of awaiting results until mid-pregnancy and to allow a safer method of pregnancy termination. Chorionic villus sampling (CVS) is such a technique. Following fertilization, the zygote differentiates first into the blastocyst, which contains an inner cell mass that develops into the fetus and an outer trophoblastic layer that develops into nonfetal structures such as amnion, chorion, and placenta. The genetic complement of the outer cell mass nearly always reflects the genetic constitution of the inner cell mass (i.e., the fetus) because both are derived from the same zygote. It follows that cytogenetic, DNA, or biochemical analysis on trophoblastic cells should provide information comparable with that obtained from cultured amniotic fluid cells. The one major exception is that assays requiring amniotic fluid, specifically alpha fetoprotein (AFP), require amniocentesis. The procedure is usually performed between 10 and 14 weeks’ gestation. Under ultrasound guidance, chorionic villi are aspirated mostly by passing a catheter transcervically into the placenta or passing a needle through the maternal abdomen and uterus into the placenta [17, 18]. Recent assessments suggest that the risk for CVS-related loss among skilled and experienced clinicians is not different than for amniocentesis, with an increased risk for loss at approximately 1 in 1,000 [19] although earlier studies on the safety of CVS suggested an approximate 1% increased loss of pregnancy [20]. The risk for fetal limb defects following CVS was initially believed to be increased but is now considered to be no greater among women undergoing CVS than those not undergoing invasive prenatal diagnosis [20, 21].
2. Amniocentesis
Biological specimens obtainable include amniotic fluid and cells. The procedure is performed from 14 weeks’ gestation onward. The technique involves passing a needle transabdominally into the amniotic cavity under continuous ultrasound monitoring.
Analyses are the same as for CVS. AFP can also be evaluated in amniotic fluid, as can additional metabolites (e.g., proteins, hormones). The procedure was previously associated with an increased risk of loss of 0.5% above the baseline risk of spontaneous loss or stillbirth, depending on the expertise of the physician performing the procedure [3, 22]. However, more recent assessments of the safety of amniocentesis now indicate a far lower risk of loss, with a large, prospective study indicating no significant increased risk of pregnancy loss among women undergoing second-trimester amniocentesis [23].
3. Percutaneous umbilical blood sampling
Access to the fetal circulation was initially accomplished by fetoscopy, a method of directly visualizing the fetus, umbilical cord, and chorionic surface of the placenta, using endoscopic instruments [24]. Fetoscopy for this purpose has now been replaced by ultrasound-directed percutaneous umbilical blood sampling (PUBS), also termed cordocentesis or funipuncture. Fetal blood chromosome analysis has been used to help clarify purported chromosome mosaicism detected in cultured amniotic fluid cells [25] or chorionic villi. More recently, fluorescent in situ hybridization (FISH) with chromosome-specific DNA probes has also been used for rapid prenatal diagnosis of aneuploidy using nucleated fetal blood cells from umbilical cord blood, as well as chorionic villus and amniotic fluid cells [26]. Fetal blood sampling appears to be a relatively safe procedure when performed by experienced physicians, albeit carrying more risk than CVS or amniocentesis. Collaborative data from 14 North American centers, sampling 1600 patients at varying gestational ages and for a variety of indications, revealed an uncorrected fetal loss rate of 1.6% [27].
B. Preimplantation Genetic Diagnosis
Preimplantation genetic diagnosis (PGD) is a prenatal diagnostic approach allowing for the detection of embryonic abnormalities that can prevent the implantation of an abnormal or affected conceptus [28, 29]. Many couples choose PGD rather than traditional prenatal genetic diagnostic approaches for assessing Mendelian disorders, aneuploidy, or chromosomal imbalance; however, such couples must undergo in vitro fertilization (IVF) in order to avail themselves of PGD. Unlike early amniocentesis, chorionic villus sampling, and other invasive techniques, PGD is not just an earlier option for prenatal diagnosis. It allows genetic diagnosis prior to the establishment of an in utero pregnancy, a preconceptional approach that has unique advantages. Moreover, potential application of PGD extends beyond traditional prenatal genetic diagnosis and improved pregnancy rates in assisted reproductive technology (ART).
A successful preimplantation genetics diagnostic program requires 1) high quality ART, 2) micromanipulation skills sufficient to obtain a specimen (polar body or blastomere) for analysis, and 3) molecular technology more sophisticated than that required for traditional prenatal diagnosis. Preimplantation genetic diagnosis requires access to gametes (oocyte) or embryos before 6 days post conception, the time when implantation occurs. There are three potential approaches: 1) blastomere biopsy or aspiration of 1 to 2 blastomeres from the 6 to 8 cell embryos (2 to 3 days), 2) polar body biopsy, and 3) trophectoderm biopsy from the 5- to 6-day blastocyst.
Blastomere (6 – 8 cells) biopsy
Biopsy at this stage was the first technique developed. The approach is to aspirate 1-2 of the 6–8 cells contained within the zona pellucida. This can be accomplished by mechanical (razor) or chemical (pronase, EDTA) dissociation of the zona pellucida, followed by aspiration with a second pipette.
Polar Body Biopsy
A second general approach is to remove either the first or second polar body, or both. Polar body biopsy (PBB) is technically similar to blastomere biopsy, but the embryo is not entered per se. Literally this constitutes preconceptional diagnosis, as opposed to preimplantation diagnosis per se. Suppose a woman and her mate were both heterozygous for the same autosomal recessive disorder. A polar body having the mutant allele should be complemented by a primary oocyte presumed to have the normal allele. The normal oocyte could thus be allowed to fertilize in vitro and then transferred for potential implantation. Conversely, if the polar body were normal, fertilization would not be allowed to proceed because the oocyte would presumably contain the mutant allele. The same reasoning would apply for detecting aneuploidy. A polar body containing only one chromosome 21 (as determined by fluorescent in situ hybridization with a chromosome-specific probe) should be complemented by a primary oocyte also containing a single number 21 chromosome. If the first polar body failed to show a 21, the oocyte can be presumed to have two number 21 chromosomes, which would lead to a trisomic zygote.
Disadvantages include inability to assess the paternal genotype, precluding application if a father has an autosomal dominant disorder. Lacking information about paternal transmission also results in reproductive inefficiencies. Presence of a mutant autosomal recessive allele in a polar body precludes allowing that oocyte to be fertilized, whereas in reality the embryo resulting from fertilization of that oocyte would be affected only if the sperm also contained the father’s mutant allele. If it were known that the father’s normal allele were transmitted, the embryo would be heterozygous and phenotypically normal.
Another disadvantage is recombination, the meiotic phenomenon that occurs routinely between homologous chromosomes. Diagnostic hazards resulting from recombination are a greater problem for genes located nearer the telomeres because these genes display recombination frequencies approximating 50%.
Blastocyst (Trophectoderm) Biopsy
An underlying difficulty when aspirating either the polar body or a blastomere from the 6–8 cell embryo is that diagnosis must depend upon only a single cell, or occasionally two. Many more cells would be available if one biopsied the 5- to 6- day blastocyst, which contains hundreds of cells. The strategy is to biopsy the trophectoderm overlying the anembryonic pole. One major problem is that blastocysts are less readily obtainable as 6–8 cell embryos. Blastocysts can be obtained by culture in vitro, but this process has traditionally proved very inefficient. After 3 to 4 days in vitro, embryonic development is not sustained efficiently to the blastocyst stage.
In PGD, diagnosis may depend upon information from a single cell. Indeed, this unique characteristic of PGD is likely responsible for much of the diagnostic inaccuracy attributed to PGD, given the likelihood that one of these early cells may be characterized by a cytogenetic or genomic alteration not found in the rest of the zygote. Metaphase analysis is possible for only a single cell, but this is not optimal. Other approaches to determine chromosomal status include comparative genome hybridization (CGH) [30]. However, none of these approaches can prevent diagnostic inaccuracies resulting from inherent cellular mosaicism. The preferable approach for chromosomal analysis in PGD involves fluorescence in situ hybridization (FISH) with chromosome-specific probes. FISH is a molecular genetic technique based on analysis of DNA sequences that are chromosome-specific. Chromosome-specific probes can hybridize to denatured DNA, in either metaphase or interphase. In interphase cells the number of FISH signals should equal the number of a given chromosome. Using a chromosome 21-specific probe would detect trisomy 21 if three signals were visualized in the nucleus. This approach is also well suited for sex determination, X and Y probes being utilized concurrently. A diagnostic dilemma unique to PGD is the very high frequency of mosaicism in the early embryos.
Molecular analysis of a single cell requires techniques more sensitive than those necessary for analyzing chorionic villi or amniotic fluid cells. The small amount of DNA in one cell (6 pg), is insufficient for conventional multi-cell molecular diagnostic approaches. In traditional polymerase chain reaction (PCR) methods, a targeted DNA sequence flanked by specific primers is amplified to increase numbers of copies, usually 105 to 106 fold. Amplification from a single cell using a single set of primers is successful if a repetitive DNA sequence is being evaluated (e.g., Y-chromosome). However, technical modifications are necessary when analyzing unique sequence mutations because sensitivity for detecting unique sequence DNA cannot be readily achieved below approximately 100 pg DNA. Rather than a single round of PCR (20 to 30 cycles), nested primers are required. The second set of primers is constructed internal to the first. Following approximately 20 cycles of PCR with the outer primers, the product is subjected to PCR with a second (internal) set of primers. Using nested PCR, as little as 1 pg of DNA suffices for diagnosis.
The list of Mendelian disorders for which PGD has been clinically used has grown rapidly in recent years. These include cystic fibrosis (F508del homozygosity), Tay-Sachs disease, Rh (D), sickle cell anemia, certain β-thalassemias, phenylketonuria, spinal muscular atrophy and myotonic dystrophy, and adenosis polyposis coli and Li-Fraumeni syndrome. Of special note is that PGD has been accomplished for a variety of adult onset autosomal dominant disorders, including Huntington disease and Marfan syndrome. Any Mendelian disorder whose gene is localized or whose molecular basis is known is potentially detectable by linkage analysis if not otherwise.
4Genetic conditions of special concern and their prenatal identification
Within the context of this chapter, prenatal diagnosis is defined as a process that rules out or detects and manages embryonic and fetal abnormalities or disease and that provides information to parents and physicians to optimize both pregnancy outcome and family well-being. Although prenatally diagnosable disorders may be categorized in many ways, it is simplest to consider only two groups of couples or families who may benefit from prenatal testing. Theoretically the two groups encompass all types of embryonic or fetal disorders that may be amenable to prenatal testing or management.
The first are those patients who themselves, or whose physicians, are (or theoretically can be) aware before conception of a known risk for fetal abnormality or unfavorable pregnancy outcome. This is the group of families in whose medical, family, or personal history there are known factors that might result in prenatally detectable or even preventable fetal disorders. They might include families of increased parental age, those with previous infants with chromosomal abnormalities or other genetically determined disorders, or families of specific ethnic origin that might increase their risk for abnormal pregnancy outcome.
The second are those who during pregnancy, without previously recognizable known risk factors, have an unexpected risk of fetal abnormality or potential for unfavorable outcome identified. Examples include those in whom a routine blood test or prenatal ultrasound reveals an unexpected abnormal finding.
1. Metabolic Single Gene Disorders Detectable by Biochemical Analysis
In considering pregnancy in association with biochemical disorders, most of which are inherited in autosomal recessive fashion, it is important to take into account the type of disorder, whether it is a parent who is affected or the fetus which is at risk, and whether and by what method the disorder is prenatally detectable. Clinical consideration of biochemical disorders can be divided into two categories: 1) carrier (heterozygote) screening to identify couples at risk for having an affected fetus and thereby candidates for prenatal diagnosis; and 2) diagnosis and management, of a first affected child.
Important technological developments in recent years have facilitated prenatal diagnosis of metabolic disorders. In fact, a number of metabolic disorders can now be diagnosed in the fetus prenatally or others through newborn screening [1] as listed in Table 1. Diagnosis relies on either the demonstration of a block in a metabolic pathway by examination of amniotic fluid cells, a sampling of chorionic villus biopsy, preimplantation diagnosis via blastomere of the 6- to 10-cell embryo, or through DNA studies that document the presence of a mutant gene or a marker associated with the mutant gene. Demonstration of a metabolic block requires either the measurement of the enzyme activity with natural or artificial substrates or the ability to measure flux through a metabolic pathway. Flux refers to the depletion rate of a labeled substrate that may feed into a metabolic pathway distant from the actual metabolic block. Alternatively, one can measure the metabolites that accumulate secondary to the metabolic block. The tissue used for prenatal diagnosis depends on the level of expression of the metabolic pathway, that is, tissue specificity, which may be influenced by the developmental age of the conceptus [2].
Table 1
Genetic disorders recommended for routine newborn screening
| Disorders detected by MS/MS (“expanded newborn screening”) |
| Amino acid disorders Phenylketonuria (PKU) |
| Maple syrup urine disease (MSUD) |
| Classic homocystinuria (HCY) |
| Classic homocystinuria (HCY) |
| Tyrosinemia type I (TYR-I) |
| Citrullinemia (CIT) |
| Argininosuccinic academia (ASA) |
| Organic acid disorders |
| Glutaric academia type I (GA-I) |
| Propionic academia (PA) |
| Methylmalonic academia (mutase) (MUT) |
| Methylmalonic academia (Cb1 A,B) (CBL, A/CBL B) |
| Isovaleric academia (IvA) |
| 3-Methylcrotonyl-CoA carboxylase deficiency (3MCC) |
| 3-hydroxy 3-methylglutaric aciduria (HMG) |
| β-Ketothiolase deficiency (BKT) |
| Multiple carboxylase deficiency (MCD) |
| Mitochondrial fatty acid oxidation disorders |
| Medium-chain acyl-CoA dehydrogenase deficiency (MCAD) |
| Long-chain 3-OH acyl-CoA dehydrogenase deficiency (LCHAD) |
| Trifunctional protein deficiency (TFP) |
| Very long-chian acyl-CoA dehydrogenase deficiency (VLCAD) |
| Carnitine uptake defect (CUD) |
| Disorders not detected by MS/MS |
| Hemoglobinopathies |
| Hb SS disease (Sickle cell anemia) |
| Hb S/β-Thalassemia |
| Hb S/C disease |
| Other disorders |
| Congenital hypothyroidism |
| Biotimidase deficiency |
| Congenital adrenal hyperplasia |
| Cystic fibrosis |
| Severe Combined Immunodeficiency |
| Hearing loss |
CoA indicates coenzyme A; Hb-hemoglobin; MS/MS-tandem mass spectrometry; SA-succinylacetone
For disorders in which the specific enzyme defect can be demonstrated in cultured skin fibroblasts, prenatal diagnosis can be accomplished using cultured amniotic fluid cells or cultured or directly analyzed chorionic villus tissue. This approach requires determination of normal values of the specific enzymes, under identical conditions. Care must also be taken to ensure that heterozygote values do not fall within the range expected for affected individuals. In addition, in chorionic villus analysis, one must avoid maternal contamination by removing decidual tissue before culture or direct enzyme assay [3].
With our increasing knowledge and experience with the human genome, there are only a few diseases in which the enzyme is not normally expressed within chorionic villi or amniotic fluid cells and for which DNA assays have not yet been developed. Prenatal diagnosis of these disorders may rely on either detection of an abnormal metabolite in the cell-free amniotic fluid or measurement of the enzyme in fetal tissue known to express the enzyme. The latter approach requires a biopsy of fetal organs or collection of fetal blood by cordocentesis [3].
There are inherited metabolic diseases, such as neuronal ceroid lipofuscinosis (NCL), for which there are no specific biochemical tests. In these instances, morphologic diagnoses may be possible; for example, electron microscopy (EM) of uncultured amniotic fluid cells or chorionic villi has been diagnostically valuable for NCL. Amniotic fluid cells obtained at 16 weeks’ gestation in NCL contain curvilinear bodies. Curvilinear cytosomes are present in the placenta of the affected fetus [4]. Similarly, fingerprint cytosomes, characteristic of juvenile NCL, are present in the syncytiotrophoblast, making prenatal diagnosis possible by CVS.
Prenatal diagnosis of mucolipid storage diseases is possible by biochemical studies as well as by EM of uncultured and cultured amniotic fluid cells that contain numerous membranous and granular cytosomes [5, 6]. By using histochemistry, immunofluorescence, and EM to identify the peroxisomes in chorionic villi or cultured villus tissue, peroxisomal deficiency disease may also be diagnosed prenatally [7, 8].
The following discussion of selected disorders illustrates the principles and practices applied to prenatal diagnosis of metabolic disorders. More information is available in the references cited.
Tay-Sachs disease, a lysosomal sphingolipid storage disorder, is a typical example of a disorder that is detectable in the first pregnancy of a couple at risk. Its clinical manifestations, occurring more frequently in the Ashkenazi Jewish, Cajun, and some French Canadian populations, are caused by the absence of the alpha subunit of the enzyme hexosaminidase A (Hex A), which is a normal gene product. The clinical course of the infantile form is relentlessly fatal, usually within 4 to 6 years after birth. It is characterized by loss of developmental skills and milestones in an initially healthy-appearing infant; a typical cherry-red spot on the ocular macula; gradual deterioration (associated with intracellular accumulation of the abnormal metabolite, GM2 ganglioside) of visual, auditory, and motor function; and frequent seizures. Hex A levels in the serum of heterozygotes are decreased by approximately 50% in comparison to those in unaffected homozygotes and are easily detectable in populations at risk. In women who are pregnant or on oral contraceptives, measurement of serum Hex A levels may lead to false positive results. Therefore, a leukocyte Hex A assay is used for these women.
The International Tay-Sachs Disease Network, through the participation of multiple centers around the world with well-organized, targeted, and accurate screening surveys, has been indirectly responsible for the dramatic reduction of this fatal disorder from about 60 newly diagnosed patients per year in the United States around 1970 to 3 per year in 1992 [31]. Most cases were prevented through prenatal diagnosis and pregnancy termination of affected fetuses of couples found through screening to be at 25% risk. The fetal diagnosis was initially established by quantitative Hex A determination in amniocytes or chorionic villus tissue; more recently it has become molecularly based and several mutations, as well as multiple forms of disease in addition to the infantile one, are known to exist. Thus, Tay-Sachs disease has become not only a preventable (and in its severe, infantile form, now almost extinct) disorder. In addition to Tay-Sachs disease, it is recommended that Ashkenazi Jewish individuals be offered screening for Canavan disease, familial dysautonomia, and cystic fibrosis. Additional tests that may be added to a screening panel include Fanconi anemia group C, mucolipidosis IV, Bloom syndrome, and Gaucher disease [32].
For most inborn errors of metabolism, carrier screening is not available. After the birth of an affected child the parents are identified as heterozygotes for a particular disease and prenatal diagnosis may be an option in subsequent pregnancies.
Table 2
Examples of disorders utilizing DNA analysis for prenatal diagnosis
| Disorder | Locus | DNA Analysis | Direct | Linkage |
| Achondroplasia | 4p16.3 | + | + | Ultrasound |
| Batten disease | 16P12 | + | ||
| Breast cancer – early | 17q21 | + | ||
| Cystic fibrosis | 7q31.3 | + | ||
| DiGeorge syndrome | 22q11 | + | + | Ultrasound |
| Duchenne/Becker muscular dystrophy | Xp21.2 | + | + | |
| Fragile X syndrome | Xp27.3 | + | ||
| Huntington disease | 4p16.3 | + | ||
| Miller-Dieker syndrome | 17p13.3 | + | + | Cytogenetic |
| Myotonic dystrophy | 19q13.3 | + | Ultrasound rule out congenital | |
| Neurofibromatosis I | 17q11.2 | + | ||
| Neurofibromatosis II | 22q | + | ||
| Sickle cell disease | 11p15.5 | + | ||
| Tay-Sachs disease | 15q23.3 | + | Enzyme assay | |
| Tuberous sclerosis | 12q23.3 | + | Other types, other loci | |
| Waardenburg syndrome | 2q37.3 | + | ||
| X-linked mental retardation | X | + | Many types |
From: Gilbert-Barness E. (Ed). Potter?s Pathology of the Fetus, Infant and Child, 2nd ed., Mosby Elsevier, 2007, pg. 633.
5Single gene disorders detectable by direct molecular analysis
There is an ever increasing list of disorders detectable by molecular analysis, which permits identification of the mutation directly in fetal (or embryonic) tissue. However, this should not be offered to any couple unless the couple is known to be at risk for the disorder in question, and unless testing is preceded by extensive, supportive, and informative counseling of all aspects and implications of potential results, as well as complications and ambiguities. Table 2 provides a selective list of disorders prenatally diagnosable by direct identification of the mutation, using a variety of DNA analytical methods, as well as the chromosomal location and mechanism of inheritance. Prototypes of more commonly occurring disorders within this group include cystic fibrosis (CF), sickle-cell disease, Duchenne muscular dystrophy.
Cystic fibrosis
CF is a multi-system disease inherited in autosomal recessive fashion. In the white population approximately 1 in 4000 individuals in North American and northern Europe are affected). The frequency of heterozygous carriers is about 1 in 29 individuals in Caucasians (Northern European origin) and Ashkenazi Jews. The heterozygote frequency is lower in Hispanic, Asians and African Americans. CF is a disease of exocrine glands that results in viscid secretions which most commonly and most severely affects the respiratory and gastrointestinal systems. In 1989, the CF gene was mapped to chromosome 7 [33]. Its gene product, a transport protein called cystic fibrosis transmembrane conductance regulator (CFTR), functions as a chloride channel activated by cAMP-dependent protein kinase A phosphorylation [33]. More than 2,000 different mutations have been discovered that can potentially result in an abnormal CFTR, but only about 10–15% of these actually cause the disease CF.
The most common disease-associated variant is F508del (originally referred to as ΔF508 and now more properly referred to as p.Phe508del), which is a deletion of 3 base pairs that code for phenylalanine. The only other mutation present in high frequency in any ethnic group in W1282X, the most common mutant allele in Ashkenazi Jews.
The pathogenesis in cystic fibrosis involves a blocked or closed chloride channel in the epithelial cell membrane. As result, chloride ions are trapped inside the cell. This draws sodium ions and water into the cell resulting in dehydration of mucus secretions. In the lungs, this can lead to airway obstruction and predisposition to infection, dyspnea, bronchiectasis and pulmonary fibrosis. The major cause of morbidity and mortality in these individuals is progressive, chronic bronchopulmonary disease. In the pancreas, the dehydrated secretions are also insufficiently alkalinized, hence causing not only duct obstruction but also a reduction in digestive enzymes and fibrosis. The fibrosis can involve the endocrine cells and may lead to not only pancreatic insufficiency but also diabetes mellitus. In the intestinal tract, the lack of digestive enzymes leads to malabsorption of fat and protein and the altered mucus can lead to bowel obstruction. Infants born with meconium ileus have a significantly higher risk of subsequent malnutrition. Sweat electrolytes are also abnormal with elevated concentrations of sodium and chloride. This in turn can lead to heat intolerance, hyponatremia, and a hypochloremic metabolic alkalosis.
The clinical presentation and course of individuals affected with CF is highly variable. The median age at diagnosis by traditional methods is 6–8 months. Some individuals have severe pulmonary and/or gastrointestinal disease, whereas others have mild expression with presentation in adolescence and young adulthood. There is a close correlation of genotype with phenotype in relation to pancreatic function; however, identification of the specific CFTR mutation has not proved highly predictive of the severity of pulmonary disease. Modifier genes and environmental factors undoubtedly play roles in this varied expressivity. Recently, a project known as CFTR2 has provided very helpful descriptions of the clinical signs and symptoms that can be expected with individual mutations [34].
With advances in medical treatment, the prognosis for individuals affected with CF has improved. In the United States, median survival now exceeds 40 years. Data from the Cystic Fibrosis Foundation indicate that about half of affected individuals are 18 years or older, and survival into the fifth decade is not uncommon (https://www.cff.org/2013_CFF_Annual_Data_ Report_to_the_Center_Directors.pdf).
Women with CF have compromised fertility. They may show delayed onset of menarche; those with malnutrition and advanced lung disease often have anovulatory cycles with secondary amenorrhea. Dehydrated and thickened cervical secretions also may pose a physical barrier to sperm penetration of the cervical os. Nonetheless, as more women with CF enter reproductive age, they should be appropriately counseled about contraception, pregnancy and the potential increased risks associated with CF. Pregnancy rates among CF women have increased. During pregnancy these women are at risk for respiratory and cardiac compromise because their underlying lung disease has the potential to be further exacerbated by gestationally related hypoxemia and pulmonary hypertension. The changes in ventilatory drive, cardiac function, blood volume and increased nutritional requirements all pose potential problems for the woman with CF [35]. Most women with CF can complete a successful pregnancy. Careful monitoring of lung function and ensuring adequate energy intake is recommended. Affected women should be counseled about the symptoms and signs of preterm labor and routine cervical exams should be considered.
As mentioned above, over 2,000 mutations have been identified in the CFTR gene, but only about 10% of them are CF disease-causing [34]. The F508del mutation is represented in almost all populations, although its relative frequency varies among different geographic locations. The highest frequency for F508del is observed in white populations, especially of Northern European origin, where it accounts for 70–75% of the CF alleles. F508del is present in 48% of CF alleles in African Americans, 46% in Hispanics, 30% in Asian Americans, and 30% in Ashkenazi Jews. About 15 to 20 other mutations account for 2–15% of the other CF alleles, depending on the ethnic group studied. Most remaining mutations are rare. A panel of 23 mutations will detect 87% of cases in Caucasians and differing frequencies in other ethnic groups. In several populations, the combination of F508del with other ethnic-specific mutations allows for a 90% to 95% detection rate: Ashkenazi Jews, Celtic Britons, French Canadians from Quebec, and some Native Americans [36]. In Ashkenazi Jews, F508del and W1282X alone account for 85% of CF alleles.
An important implication of the various mutation detection rates among different populations is the number of couples in whom both partners can actually be identified as CF carriers thereby enabling unequivocal prenatal diagnosis. For example, if the mutation detection rate is 75% and one partner is determined to be a carrier and the other not, the risk of having an affected offspring is 1 in 400. By contrast, if the mutation detection rate is 90% and one partner is determined to be a carrier and the other not, the risk for an affected offspring is 1 in 1000. If both partners are screen negative, the risk is 1 in 240,000 [37]. Experience during the period of 2005-11 when CF newborn screening was being implemented nationwide demonstrated the practical ramifications of a sensitivity as low as 75%, i.e., the usual situation in which the mother is a carrier who tests positive for one CFTR mutation and the father/partner is negative. In that circumstance, after the prospective parents have been reassured, no matter how cautiously this communication occurs, countless pregnancies have been continued and numerous infants have been born with a positive newborn screening test and then a confirmatory sweat test, thus diagnosing CF. The implications of such outcomes need to be explored with regard to both prenatal and neonatal screening.
If amniotic fluid analysis were possible for the cystic fibrosis gene product (i.e., mutant protein), the inability to detect all mutations would not matter so much in situations where one parent carries an identified CF mutation and the other is screen negative, but still has a residual risk of carrying an unidentified CF mutation. Unfortunately, no such assay exists, meaning that prenatal diagnosis is not possible except to exclude the fetus who inherited the mutation from the known heterozygous parent. Measuring microvillus intestinal enzymes in amniotic fluid is specifically not helpful, for a low value is more likely to connote a false-positive than a true-positive value.
Another issue with carrier screening is genotype-phenotype correlations. Predictions of the severity and course of pulmonary disease are variable for specific mutations, especially for fetuses detected to be compound heterozygotes; however, prediction for pancreatic function is generally reliable [38]. The issue of phenotype being different from CF with pancreatic and pulmonary insufficiency becomes most relevant when one is confronted with a male having two CF alleles that results in congenital bilateral absence of the vas deferens (CBAVD).
Males with classic CF are virtually always sterile due to CBAVD. There is an increased frequency of CF mutations in otherwise healthy men with CBAVD. About 25% of these men with CBAVD are compound heterozygotes for two mutant alleles (one being a “mild” mutation); half have one identifiable mutation, and the remainder have no identifiable mutation [39]. Eighty-four percent with no identifiable mutation carry this splice site variant for a CF mutation called the 5T allele and 25% with no identifiable mutation carry a splice site variant. The 5T refers to the number of thymines present at the end of intron 8, which alters the likelihood of proper splicing of exon 9. The 7T allele is usually associated with normal splicing. The 5T allele is present in men with CBAVD at a four-fold higher frequency than the general population [40–42].
5.3Recommendations for cystic fibrosis carrier screening
Population-based programs offering CF carrier screening in the U.S. were introduced routinely in the U.S. in 2001 [43]. The following guidelines and recommendations have been provided [44]:
• CF carrier screening should be offered to all couples with a positive family history of CF. All partners of individuals with CF and all Caucasian couples of European or Ashkenazi Jewish descent planning a pregnancy or seeking prenatal care. Ideally screening is performed prior to conception or during the first or early second trimester. Information about CF screening should also be provided to patients in other ethnic and racial groups. Counseling and screening should be provided and made available to individuals in the lower risk groups upon their request.
• The clinician should identify couples to whom screening should be offered based on family history and ascertainment of the ethnicity and race of the partners during the initial history. Counseling and standardized written educational material or other formats such as videos, and/or interactive computer programs should be used to inform the woman and whenever possible, her partner. In the event that her partner does not accompany the woman to a prenatal or preconception visit, written educational material should be provided for her to give to her partner. Women and their providers may elect to use either a simultaneous or sequential carrier screening strategy for CF. Simultaneous testing may be particularly important when there are time constraints for making a decision regarding prenatal diagnosis or on the availability of termination of affected pregnancies.
• The obstetrical care provider should offer CF screening when appropriate and may choose to provide or refer for pre-test counseling. Post-test counseling for couples with positive/negative, positive/untested or positive/positive screening results, requires special knowledge of CF with regard to range of severity, prognosis, treatment options, etc. and calculation of genetic risk. Referral to a geneticist or a provider with special expertise with CF testing may be considered in these situations. Referral to a geneticist or individual with special expertise with CF testing should also be considered when: there is a family history of CF; when carriers are identified with CF mutations that may be associated with congenital absence of the vas deferens in the male offspring; or when an affected adult or an affected fetus is identified.
To implement these guidelines a minimum panel of 23 mutations to be included in CF screening was recommended. Provider educational materials, patient pamphlets and model informed consent forms are available. More recent attempts to expand the number of mutations incorporated into the screening algorithm have been undertaken by several private laboratories, and have been associated with some acceptance in the medical community. However, it is unclear whether expanding the CF screening panel will improve or make more complicated the screening process for CF. Regardless, the increasing use of new DNA multi-mutation analytical methods will assuredly further expand the number of mutations used to screen the high-risk and general populations for CF mutation carriers.
5.4Sickle-cell disease
Sickle-cell disease, is an autosomal recessive disorder occurring commonly (1 in 400 to 500) in the African-American population, with heterozygote frequency of about 1 in 10. Heterozygotes are detected by hemoglobin electrophoresis. Sickle cell disease is attributable to the SS hemoglobin molecule’s instability, which leads to anemia, blood flow reduction, and increased susceptibility to pneumococcal infections that can be prevented with penicillin prophylaxis [45]. Historically, the discovery of the prenatal diagnosis of sickle-cell disease with restriction endonucleases [46] was one of the more dramatic breakthroughs in prenatal diagnosis. Direct detection of the mutation at the specific site within the beta-globin gene on chromosome 11, where an exchange occurs from glutamine to valine, is one of the simplest and most accurate tests available. It utilizes polymerase chain reaction, and needs no adjuvant family studies, apart from the identification of (a) parent(s) at risk.
5.5Fragile X syndrome
Fragile X syndrome is thought to be one of the most common causes of an inherited form of mental retardation, rivaled only by the incidence of Down syndrome, and possibly by the effects of intrauterine exposure to maternal alcohol ingestion. It is inherited in X-linked dominant fashion, affecting about 1 in 1000 and 1 in 2000 males and females, respectively, with no male-to-male transmission. The syndrome is defined by a combination of a characteristic phenotype and behaviors, cognitive impairment, the presence of a fragile site (gap) detectable cytogenetically in folate-free culture medium on Xq27.3, called FRA-X-A. Molecularly, it is caused by transcriptional inhibition, through overmethylation, of a messenger ribonucleic acid (mRNA) protein binding gene called FMR-1 (fragile X mental retardation-1).
The characteristic but subtle phenotype includes elongated face and mandible, large ears, macrocephaly with bizygomatic pinching, soft skin, inconsistent mitral valve prolapse, macro-orchidism, mildly shortened stature in adulthood, and occasional seizures. Characteristic behaviors may resemble autism and attention deficit disorders. Intellectual impairment in affected individuals varies from mild to severe with most affected males within the moderate range of cognitive disability. However, 20% of males who have the fragile X mutation are phenotypically and intellectually unaffected. They are called transmitting males. Female heterozygotes may be indistinguishable from the general population or may have subtle physical signs of both physical and intellectual impairment.
The mutation is characterized by varying lengths of DNA fragments, consisting of varying numbers of repeats of the trinucleotide CGG, located 5’ to the gene (FMR-1) itself. The trinucleotide is repeated approximately 6 to 50 times in the normal population, approximately 51 to 200 times in unaffected individuals with a so-called premutation, who are at risk for expansion and transmission to offspring in expanded form, and over 200 times in individuals who are (usually) affected and said to have a full mutation. These CGG repeats are called unstable (their numbers change) and tandem (they are next to one another) repeats, and there are several disorders the molecular basis of which includes such unstable tandem repeats [47].
Prenatal diagnosis of fragile X syndrome should be contemplated only in families with (a) member(s) in whom the diagnosis has been unequivocally confirmed by DNA analysis. The next step is the determination, first from the family history, then by DNA analysis, whether one of the parents of the pregnancy is at risk. If the mother is a heterozygote with a premutation (∼50 to 200 repeats), she could be at risk for transmitting it in similar (i.e., premutational) or expanded (i.e., mutational) form, or not at all, by transmitting her normal X. Expansion occurs only during oogenesis, not spermatogenesis; therefore if the father is a transmitting male (i.e., unaffected, symptomless, with a premutation), it is unlikely that his daughters would be affected. They would all have premutations also, and it would be their offspring (more frequently sons, less frequently daughters) who would be at risk for having a full mutation and therefore clinical manifestations of the syndrome.
Direct DNA analysis of fetal tissue either by CVS or amniocentesis can differentiate between a normal, a premutational, and a mutational number of CGG repeats. It should be preceded by cytogenetic fetal sex determination. Although males are affected more frequently than females, carrier mothers can have affected offspring of both sexes, and awareness of fetal sex can sometimes be helpful in attempts to provide the parents with prognostic information about the severity of the disorder, if present in their future child.
5.6Muscular dystrophies
Duchenne muscular dystrophy (DMD) and Becker muscular dystrophy (BMD) are allelic X-linked recessive (XLR) disorders, caused by a gene on Xp21, the malfunction of which results in a deficiency of the muscle protein dystrophin. The two disorders can occur within members of the same family and differ in severity. While boys with DMD are usually wheelchair bound by age 8 to 10 years, those with BMD are sometimes ambulatory into their twenties and thirties. Their life expectancy also differs significantly, and may not exceed 20 years in patients with DMD, whereas patients with BMD frequently live into middle age or longer. In contrast to fragile X syndrome, the genetic transmission of DMD and BMD is straightforward XLR. Thus, with very few exceptions, only males are affected within the family and in most cases they are sons of carrier females. The exceptions may include females with X autosomal chromosomal translocations involving the crucial segment of Xp21, females with a 45, X (Turner syndrome) karyotype, or, rarely, those in whom the Active X is the abnormal one, enhancing the expression of the gene.
The prenatal diagnosis of DMD and BMD begins, as in all other instances, with an accurately confirmed diagnosis, often including a muscle biopsy in an affected family member. Wherever possible, the identification of carrier status in the mother should also be established. Localization of the dystrophin gene to Xp21 now allows prenatal diagnosis for DMD and BMD. Many affected boys with the disorder have a deletion, at the molecular level, of the dystrophin gene [48], as do their carrier mothers and other female relatives. In the simplest instance, then, prenatal diagnosis in such families is based on the detection, by direct DNA analysis, of a deletion in a male fetus with a known carrier mother. Such a result is accurate and reliable. Occasionally an affected infant with a deletion is born to an apparent non-carrier mother (she does not have a detectable gene deletion). If the birth of her affected son were the result of a new mutation (the remainder of the family history is negative), there should be no increased risk for recurrence, and prenatal diagnosis would ordinarily not be indicated for a subsequent pregnancy. However, the possibility of maternal gonadal mosaicism cannot be ruled out, and has indeed been confirmed in several instances [49] in which two affected boys have been born to mothers whose carrier status has been undetectable by currently available techniques. Since it is not possible to rule out gonadal mosaicism, it is advisable to offer prenatal diagnosis to all couples who have had an affected child with DMD, even if the family history is negative and the mother appears not to be a carrier.
A third scenario is even more confusing. Not all affected boys have a molecularly detectable deletion, even though they are unequivocally affected. The technique of choice in such families is then by linkage analysis – that is, the tracking of the crucial chromosomal segment with molecular markers and comparison among known affected and unaffected individuals. If a mother and her male fetus have inherited the same X chromosome as her affected brothers, sons, or maternal uncles, the probability that the fetus is affected is high, but never certain. It is inversely proportionate to the probability of recombination (crossing over) between homologous chromosomal segments. The closer the compared markers are, the lower the probability of crossing over, and the higher the probability of the presence of the gene in question.
Myotonic dystrophy, or dystrophia myotonica (DM), is thought to be the most common form of muscular dystrophy that affects adults. It is a systemic disease inherited in an autosomal dominant manner, but the well-known congenital form that is most severe occurs only in children of affected mothers.
Like all pleiotropic dominant disorders, adult-onset DM varies in phenotype, age of onset, and degree of involvement of different organ systems. It can be so mild that its presence in an elderly grandfather may not be recognized until the birth of a grandchild with the severe congenital form. The disorder is characterized by progressive muscle weakness, myotonia (inability of the muscles to relax after contraction), cataracts, frontal alopecia, cardiac conduction defects, endocrine failure with subsequent infertility in males, dilatation of the gastrointestinal tract with absence of peristalsis, progressive dementia, and respiratory failure. The congenital form may be suspected prenatally by reports – and ultrasonic evidence – of lack of fetal movement and polyhydramnios. At birth the infant is frequently immobile, with a characteristic myopathic open-mouthed face and inability to suck or swallow. Gradually these signs tend to improve and the child begins to move and develop gross motor skills, though very slowly. Such children are almost always not only physically but also psychometrically delayed. Historically, anticipation (namely, increasing severity of the disease in successive generations) was recognized as early as 1947 [50] but was attributed mostly to ascertainment bias [51]. The discovery of heritable unstable trinucleotide repeats (CTG) finally provided a molecular explanation not only for the phenomenon of anticipation but also for some of the variable expression of the disease. [52] The gene is now known to be located on chromosome 19q; before that it was found to be linked to the Lutheran blood group and secretor status genes, and this was used in the early 1970s for prenatal diagnosis but the recombination rate was high and many families were uninformative. As more markers became available, linkage was the prenatal diagnostic method of choice, using family parental and fetal (amniotic) material.
The breakthrough in 1992 by Aslanidis and colleagues [53] resulted in the possibility of direct molecular prenatal diagnosis without the need for extensive family studies. It consists of the detection of a CTG triplet located in the 3’ untranslated region of an mRNA-encoded polypeptide of the protein kinase family. The triplet is repeated between about 5 to 27 times in the general (unaffected) population; mildly affected patients with DM may have 50 or more, while severely affected ones have over 1000 repeats.
It seems therefore that the size of the expansion is correlated with the severity of the disease and age of onset, but contraction (or mosaicism) has been observed in male somatic cells as well as in sperm [54]. Thus patients with DM may have gonosomal mosaicism and contraction in sperm may account for the presence of congenital DM only in infants of affected mothers. This explanation is probably incomplete.
Hecht and colleagues [52] investigated directly several fetal tissues and chorionic villi (after CVS and pregnancy termination). They found no heterogeneity or mosaicism in lengths of repeats among tissues or villi thus evaluated. However, expansion did occur during culture of CVS cells. The authors therefore recommend that prenatal diagnosis for DM be performed on direct rather than cultured cells. More experience is needed before these findings enable the provision of unequivocal and unambiguous information to parents who request not only prenatal diagnosis for DM but who, in the case of maternal transmission, may wish to know early during the pregnancy whether or not their fetus has the congenital or adult-onset form. The clinical severity and progression of the disease in the affected parent might also be considered by the couple, as they plan their future and that of their family.
6Newborn screening: Principles and practices
Newborn screening may be defined [5] as “a population based public health program applying preventive medicine in defined regions to reduce newborn morbidly and mortality from certain biomedical and genetic disorders by using presymptomatic detection/diagnosis with dried blood specimens analyzed in central laboratories and employing automated procedures and linked to clinical follow-up systems.” It should be emphasized that each aspect of the sequence from blood collection to follow-up care is essential for quality assurance. The importance of consistent processes and procedures has been emphasized in a series of guideline publications of the Clinical and Laboratory Standards Institute (CLSI) that describe best practices [55–60].
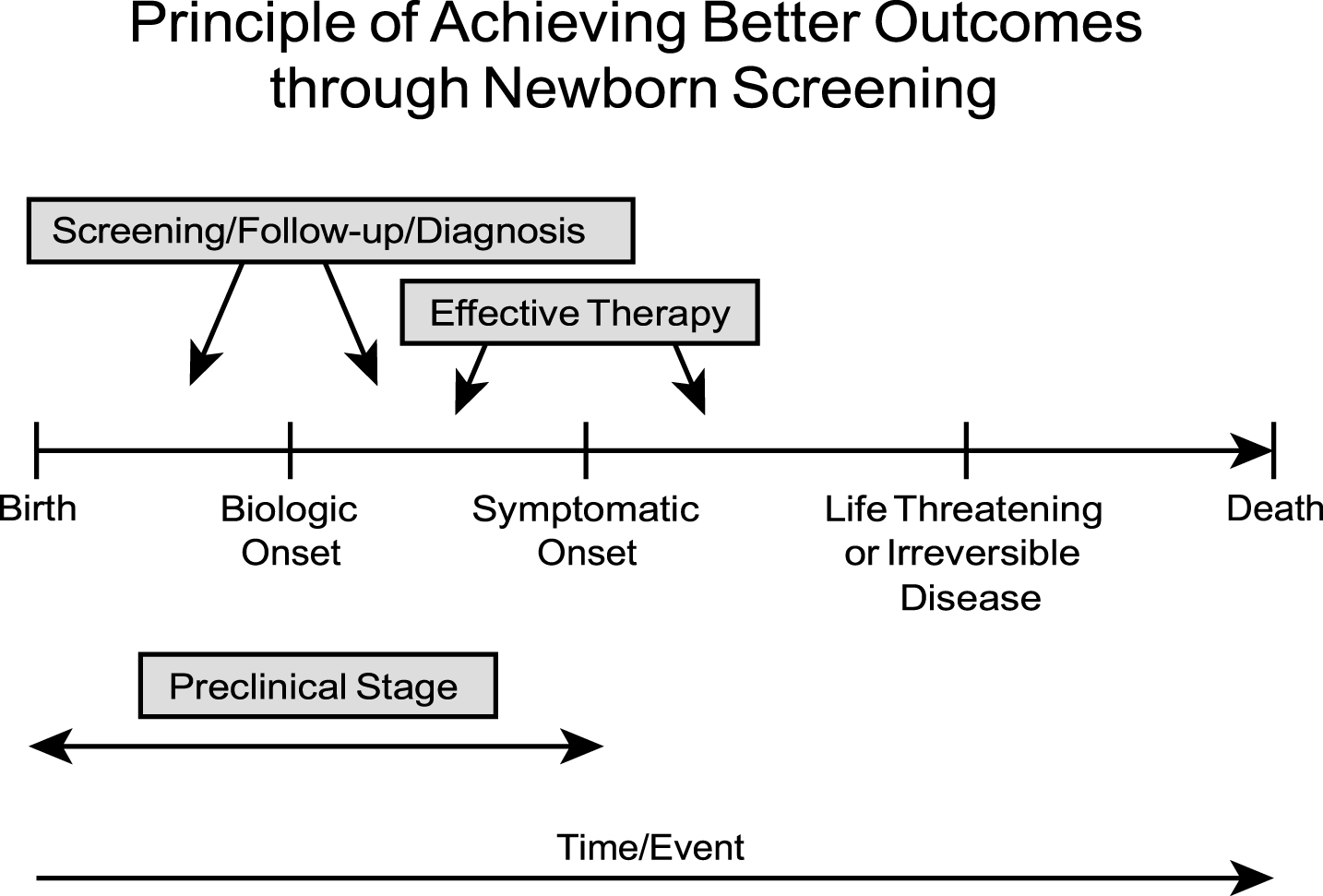
The fundamental principle for achieving benefits through newborn screening is illustrated in Fig. 1. In essence, early diagnosis through newborn screening provides children with an opportunity for better outcomes when there is a detectable preclinical stage of a congenital disorder, ideally soon after birth, and effective, accessible therapy exists that prevents irreversible abnormalities such as severe morbidity (e.g., mental retardation) or death. Often the biologic onset and symptomatic onset are chronologically close, and only limited time (days to weeks) exists from that stage to a “point of no return” where irreversible pathology has supervened. The goals of newborn screening are summarized in Table 3. For most genetic diseases included in newborn screening programs, prevention of mental retardation and developmental disabilities has been the primary objective and indeed was the driving force for the advent of this model public health initiative. Also, most of these disorders are autosomal recessive and very difficult to diagnose using the traditional history/physical exam strategy; even the very best pediatricians will fail to diagnose “accurately and promptly” enough to prevent long term adverse consequences [1].
Table 3
Goals of neonatal screening
| PKU* and hypothyroidism | Prevent MR/DD |
| Biotinidase | Prevent DD and possibly deaths |
| Congenital adrenal hyperplasia | Prevent neonatal deaths in males at risk for salt-wasting crisis and prevent incorrect gender assignment |
| Galactosemia | Prevent MR/DD and infant deaths |
| Sickle cell disease | Prevent deaths |
| Cystic fibrosis† | Prevent malnutrition, hyponatremia, deaths and early progressive respiratory disease |
| Maple syrup urine disease† | Prevent infant deaths and DD |
| Homocystinuria† | Prevent MR/DD, thromboembolic episodes, and death |
| Severe combined immunodeficiency | Prevent severe, life-threatening infections |
*PKU = phenylketonuria; MR = mental retardation; DD = developmental disabilities. †Goals may not be fully achievable. Modified from: Gilbert-Barness E. (Ed). Potter’s Pathology of the Fetus, Infant, and Child, 2nd ed., Mosby Elsevier, 2007, pg. 636.
Fig.1
Principles of achieving benefits for infants through early diagnosis and therapy.
As described previously, the first newborn screening programs were implemented in the early 1960’s for PKU [12], a disorder associated with phenylalanine hydroxylase deficiency. Initially voluntary, the PKU test became mandatory by legislative actions throughout the nation, such that by 1973 a total of 43 states had formal statutes. In general, state health departments, particularly their maternal and child health programs funded by Title V of the Social Security Act, assumed responsibility for implementing the new laws and ensuring neonatal blood collection and analysis. Thus, PKU became the prototype [61, 62] genetic disorder for newborn screening because it is very difficult to diagnose early in primary care practice, has a relatively short latent (preclinical) stage, a high risk for serious, irreversible pathology (mental retardation), and can be effectively treated using dietary phenylalanine restriction [63]. Although early implementation efforts were fraught with problems, attention to the entire newborn screening system led to quality improvement as centralized laboratories became responsible for the lab tests and a network of centers assumed the essential follow-up role. All states screen every newborn for PKU, but the analytical tests have varied considerably from the original bacterial inhibition assay of Guthrie [5] to automated methods using sophisticated tandem mass spectrometry (MS/MS) [4, 58]. As described subsequently under “The Future of Newborn Screening” the antiquated methods from the 1960’s are being replaced by MS/MS [58].
All states also screen their newborn populations for congenital hypothyroidism using the same dried blood specimen and a variety of radioimmunoassay methods [4]. The testing protocols have varied and included simple measurement of blood thyroxin, a combination of thryopin and thyroid stimulating hormone (TSH), or TSH alone. There has been a trend to use primary TSH screening in recent years because it can be both effective and cost-effective [5]. As shown in Table 4, congenital hypothyroidism is five times more common than PKU, and thus was very attractive in the 1970’s to add to newborn screening panels, especially in view of the curative therapy available (thyroxin) [64]. In the U.S., about 1200 confirmed cases per year are identified with an assumed sensitivity of 100%, specificity of 98.5%, and PPV of about 2%, i.e., 50 false positives for every diagnosis [65]. A variety of problems were encountered as implementation occurred nationwide and quite rapidly [5, 66, 67] (e.g., imperfect sensitivity of laboratory protocols and follow-up problems) but congenital hypothyroidism screening has been a great success. In retrospect, the lessons learned from flawed implementation of PKU screening promoted an expeditious, constructive response of leaders that was very effective. AAP guidelines include the recommendations provided below [68].
Table 4
Incidence of genetic diseases included most commonly in U.S. newborn screening programs
| Condition* | Prevalence (Approximate) | |
| Congenital hypothyroidism | 1:3,000 | |
| Sickle cell disease | 1:3,000 | |
| Cystic fibrosis | 1:4,000 | |
| MCAD deficiency+ | 1:10,000 | |
| Congenital adrenal hyperplasia | 1:15,000 | |
| Phenylketonuria | 1:20,000 | |
| Galactosemia | 1:70,000 | |
| Biotinidase deficiency | ||
| Severe combined immunodeficiency | 1:70,000 | |
| 1:70,000 | ||
| Homocytinuria | 1:225,000 | |
| Maple syrup urine disease (branched-chain ketoaciduria) | 1:250,000 |
*All autosomal recessive, except for some varieties of congenital hypothyroidism. +MCAD = Medium chain acyl-CoA dehydrogenase. Modified from: Gilbert-Barness E. (Ed). Potter’s Pathology of the Fetus, Infant, and Child, 2nd ed., Mosby Elsevier, 2007, pg. 637.
• Every infant should be tested before discharge from the nursery.
• In instances such as home births, or in the case of a critically ill or premature neonate, blood should be obtained within 7 days after birth.
• The responsibilities for transmission of screening test results back to the physician or hospital identified on the screening filter paper card should rest with the authority or agency that performed the test.
• Infants with low T4 and elevated TSH concentrations have congenital hypothyroidism until proved otherwise.
• Administration of l-thyroxine is the treatment of choice.
• Routine clinical examination, including assessment of growth and development, should be performed at regular intervals, approximately every few months during the first 3 years.
Another hereditary endocrinopathy, congenital adrenal hyperplasia (CAH), is now screened for in about half the states as part of newborn screening programs. The goal of early diagnosis through neonatal screening is to prevent deaths associated with the salt wasting form of the disease. This disorder is identified by radioimmunoassay of 17-hydropyprogesterone and is readily treated with cortisol replacement [5, 69]. The major problem with CAH screening is the relatively low PPV of 0.5%, which leads to large proportion of false positive tests, especially in premature infants [5], i.e., up to 200 for each diagnosis [65].
Most regions also screen newborns for galactosemia in an effort to prevent mental retardation. A variety of methods are used [4]. Classical galactosemia is caused by a deficiency of galactose-1-phosphate uridyl-transferase. A growing number are now screening for biotinidase deficiency, which develops because of an ability to recycle biotin. Both of these disorders occur with an incidence of 1:70,000 and thus are somewhat infrequently diagnosed, i.e., about 50 cases of each disorder per year among the approximate 4 million newborns in the USA. Delays in diagnosis, however, can be catastrophic for those 50 infants.
Other hereditary metabolic disorders are being included in newborn screening panels, although the value of early diagnosis is debatable for some conditions. For instance both homocystinuria, which is caused by defective metabolism of sulfur – containing amino acids, and Maple Syrup Urine Disease (branchedchain ketoaciduria) screening were temporarily discontinued in Wisconsin because of their very low incidence (about 1:225,000) and uncertain benefits. Indeed, these two disorders do not meet the criteria used in Wisconsin [70, 71, 72] (listed below) or the World Health Organization (WHO) criteria [72]. With their low incidence only about 18 cases of each disorder are likely to be diagnosed per year in the United States. Unfortunately, preventing mental retardation may not be feasible through screening newborns. On the other hand, these disorders will progress and be life threatening or fatal with long delays in diagnosis and their detection may be more feasible with new analytical, automated technology.
The implementation of tandem mass spectrometry has promoted screening for numerous disorders of amino acid and organic acid metabolism as well as disturbances in fatty acid oxidation (Table 5). Tandem mass spectrometry is arguably the most significant procedural advance in newborn screening since the “Guthrie card” method of obtaining dried blood specimens was introduced in the 1960’s. Consequently, we have included a detailed review of this technological advance and recommend reading the CLSI guidelines [58] for more information on laboratory aspects such as quality control. Prior to MS/MS, the laboratory strategy was to use a separate biochemical test or even two tests for each disorder. Automated, computer-assisted MS/MS transformed that paradigm into a modern strategy of using that method to screen for multiple hereditary metabolic disorders. Nearly a quarter century of evolutionary advances, however, was needed to reach the current status of routine MS/MS in newborn screening laboratories. The early phase depended on a combination of liquid chromatography and mass spectrometry detection of metabolite derivatives (analytes). By the 1990’s, instrumentation advances with techniques such as fast atom bombardment and fast ion bombardment coupled to a series of units controlling separation, fragmentation and detection set the stage for creating a high throughput system employing electrospray ionization as the optimal sample delivery interface. With the addition of computer-assisted data monitoring, it became possible for a newborn screening laboratory to analyze up 500 samples per day and screen for 50 or more disorders (Table 5).
Table 5
Expanded panel of newborn screening tests
| 1. Amino Acid Disorders |
| Argininemia |
| Arginosuccinic Aciduria (ASA Lyase Deficiency) |
| 5-Oxoprolinuria (Pyroglutamic Aciduria) |
| Carbamoylphosphate Synthetase Deficiency |
| Citrulinemia |
| Galactosemia |
| Homocystinuria |
| Hypermethioninemia |
| Hyperammonemia, Hyperomithinemmia, Homocitrullinemia Syndrome (HHH) |
| Hyperomithinemia with Gyral Atrophy |
| Maple Syrup Urine Disease (MSUD) |
| Phenylketonuria (PKU) |
| Tyrosinemia |
| 2. Biotinidase Deficiency |
| 3. Congenital Endocrinopathies |
| Congenital Adrenal Hyperplasia |
| Congenital Hypothyroidism |
| 4. Cystic Fibrosis |
| 5. Fatty acid oxidation disorders |
| Carnitine/Acylcarnitine Translocase Deficiency (CAT) |
| Carnitine Palmitayl Transferase Deficiency Type |
| 3-Hydroxy Long Chain Acy-CoA Dehydrogenase Deficiency (LCHAD) |
| 2, 4-Dienoyl-CoA Reductase Deficiency |
| Multiple Acyl-CoA Dehydrogenase Deficiency (MADD or Glutaric Acidemia II) |
| Medium Chain Acyl-CoA-Dehydrogenase Deficiency (MCAD) |
| Short Chain Acyl-CoA Dehydrogenase Deficiency (SCHAD) |
| Trifunctional Protein Deficiency (TFP) |
| Long Chain Acyl-CoA Dehydrogenase Deficiency (LCAD) |
| Very Long Chain Acyl-CoA Dehydrogenase Deficiency (VLCAD) |
| 6. Hemoglobinopathies: Sickle Cell Disease |
| 7. Organic Acid Disorders |
| 3-Hydroxy 3-Methylgutary CoA Lyase Deficiency (HMG) |
| Glutaric Acidemia Type I (GA-I) |
| Isobutyryl-Co-A Dehydrogenase Deficiency |
| Isovoleric Acidemia (IVA) |
| 2-Methylbutyryl-CoA Dehydrogenase Deficiency |
| 3-Methylcrotonyl-CoA Carboxylase Deficiency (3MCC) |
| 3-Methylglutacony-CoA Hydratase Deficiency |
| Methylmalonic Acidemia: Methylmalony-CoA Mutase |
| Deficiency (MUT) |
| Mitochondrial Acetocetyl-CoA Thiolase Deficiency |
| Propionic Acidemia (PA) |
| Multiple CoA Carboxylase Deficiency Malonic Aciduria |
| 8. Severe combined immunodeficiency |
Modified from: Gilbert-Barness E. (Ed). Potter’s Pathology of the Fetus, Infant, and Child, 2nd ed., Mosby Elsevier, 2007, pg. 638.
MS/MS-detectable hereditary metabolic disorders include aminoacidopathies, fatty acid oxidation deficiencies, organic academia disturbances, and urea cycle defects. The detection of aminoacidopathies and urea cycle disorders relies on determination of elevated amino acids such as phenylalanine for PKU and lencine/isoleucine/valine for MSUD. The abnormalities in fatty acid oxidation and organic acid metabolism are identified by measuring acylcarnitines, an intermediate metabolite consisting of a fatty acid or organic acid and carnitine, which is required for transfer across the mitochondrial membrane for oxidation. Each disorder shown in Table 5 has a distinct profile of abnormal levels of specific acylcarnitines. Although these disorders are individually uncommon, when MS/MS as a single test is used to facilitate their early diagnosis, the cumulative incidence among newborns is approximately 1:4000 and thus is similar to congenital hypothyroidism and CF (Table 4). Decisions to add MS/MS and screen newborns with an expanded panel have been made in different regions based on a variety of criteria, including technical capability and financial capacity. When Wisconsin decided to proceed in the 2000, it was clear that predetermined criteria [70] summarized below were met [73]. Since then most states have implemented MS/MS and excellent guidelines for its application published [58].
• In general, screening should not take place unless the disorder is recognized as an important public health problem for which there will be a commitment of financial support. Benefits to identified infants should outweigh the costs of the screening program.
• Incidence: The identification of 1 case per 100,000 births should be the minimum number of any specific disorder. With an annual birth rate of 70,000–75,000, Wisconsin should expect to identify approximately 1 case per year.
• Morbidity and Mortality: Assessment of scientific evidence should support the expectation that benefits of identification in the neonatal period (treatment effective only when started before the age at which clinical diagnosis is usually made) will outweigh adverse consequences such as psychological effects on false-positive population and the costs of screening.
• Potential for Successful Treatment: There should be evidence that effective management can be implemented to benefit infants and their families; diagnosis without effective treatment is inappropriate.
• Cost: Laboratory costs of any test to be added should be comparable to the costs of well established tests such as PKU.
• Laboratory Feasibility: Test must be adaptable to a mass screening program. The health care system and laboratory technology limits tests to those that use blood specimens at this time.
Medium chain acyl-CoA dehydrogenase deficiency (MACD) is the prototype disorder in this category and causes potentially fatal vomiting, lethargy, encephalopathy, hepatomegaly, seizures and coma [74]. The approximate incidence is 1:10,000. Effective treatment to prevent a metabolic crisis depends on a low fat, high carbohydrate diet. Early results of screening are promising.
When it became clear that penicillin prophylaxis could be life saving for children with sickle cell disease (SCD) [45], hemoglobinopathy screening was incorporated into many programs and now is offered almost universally in the USA. Originally depending on protein electrophoresis, after using cellular acetate, programs now use other molecular diagnostic methods such as isoelectric focusing and HPLC [4]. Prevention of deaths associated with pneumococcal infections can be readily achieved in children with SCD [4, 53] and thus it seems surprising that some regions don’t include hemoglobinopathy screening in their panels. Although a lower incidence (e.g., 1:40,000) of SCD associated with regional demographic differences (i.e., a lower proportion of African-Americans) is offered as an explanation, some of these states screen for hereditary metabolic disorders that are even less prevalent [8]. This situation is typical of the regional disparities currently evident in the newborn screening programs of North America and suggests the need for national policy development [8, 71, 72, 73].
In the case of cystic fibrosis (CF), a quarter century of extensive, well-organized research in the US, Western Europe, New Zealand, and Australia [14] led to national recommendations [75] in 2004 by leaders of the Centers for Disease Control and Prevention (CDC) following their earlier conclusion that more research was needed [76]. After CDC recommended more CF newborn screening, all states began planning efforts and by 2011 the entire USA and many European countries had implemented such programs. The methods that have been used commonly are described in Table 6 and more detail is available in the CLSI Guideline document [60]. The role of the CDC in reviewing evidence and issuing policy recommendations for CF newborn screening was unique. In addition, the long term emphasis on prospective research related to CF newborn screening is also noteworthy because in general only limited systematic outcomes research has occurred regarding other genetic conditions included in newborn screening panels. Other features are also special such as the screening test used most commonly which involves DNA analysis. Specifically a two tiered method employing immunoreactive trysinogen (IRT) is used first and this is followed by DNA analysis for CFTR mutations when the IRT level exceeds a cutoff selected to maximize sensitivity. The IRT/DNA test was the first molecular genetics method applied to complete populations in community-based screening [70]. Because there are more than 2000 CFTR mutations, the DNA component has been challenging but fortunately about 70% of CF chromosomes have the principal and first mutation discovered [33], namely F508del (deletion of 3 bases at codon 508 and loss of phenylalanine in the mutant chloride channel protein). Furthermore, about 90% of CF patients in North America, Europe, and Australasia have at least one F508del mutation, so that simple IRT/DNA (F508del) testing detects most of them and presumptively diagnoses half the CF population who are homozygous for F508del [1, 30, 77]. However, the recent CF Founation consensus guidelines for diagnosis emphasizes that even these infants need to have a diagnostic sweat test performed [78]. The IRT/DNA (F508del) screening test has a high PPV (17%) compared to other disorders on routine newborn screening panels [79]. In addition, there are advantages to using IRT/DNA (CFTR) multi-mutation analysis, and development of such tests has proceeding rapidly [1, 31]. New analytical technologies such as next generation sequencing [80] provide an opportunity to detect more than 100 CFTR mutations simultaneously within a few hours. As shown in Table 4, CF is relatively common among disorders included in newborn screening programs. In fact, CF is the most common, life-threatening autosomal recessive disorder among Caucasians, and over 1000 patients per year are diagnosed in the US.
Table 6
Newborn screening methods for cystic fibrosis
| Testing method | Description | Follow-up sweat testing |
| 1. IRT* | IRT analysis of the initial specimen obtained at 24–72 hours of life | Required |
| 2. IRT/IRT | IRT analysis of the initial specimen obtained at 24–72 hours of life plus repeat analysis (to reduce false positive results) on a routine recall specimen | Required |
| 3.IRT/DNA(F508del) | IRT analysis of the initial specimen, with positive specimens subsequently tested for the F508del mutation | Required for confirming diagnosis, even with F508del/F508del is deleted |
| 4. IRT/DNA (CFTR)† | IRT analysis of the initial specimen, with positive specimens subsequently tested for a panel of the most common mutations in the area. There is also a IRT/IRT/DNA (CFTR) method in which the IRT test is repeated to demonstrate persistent elevation before DNA analysis is performed | Required for confirming diagnosis, even if detection of two CF-causing mutations has led to a presumptive diagnosis |
*IRT: immunoreactive trypsinogen. †CFTR: multimutation analysis for mutations in the cystic fibrosis transmembrane conductance regulator gene. Modified from: Gilbert-Barness E. (Ed). Potter’s Pathology of the Fetus, Infant, and Child, 2nd ed., Mosby Elsevier, 2007, pg. 638.
Early diagnosis through IRT/DNA screening provides an opportunity to prevent the potentially fatal salt depletion with sweating, and the severe malnutrition that can cause long term problems such as growth retardation and cognitive dysfunction [14]. In addition, early diagnosis facilitates therapy of CF lung disease, although currently it can’t be prevented [14]. On the other hand, the development of CFTR modulator therapy [81] offers the potential to ameliorate lung disease through enhanced chloride channel functioning.
CF illustrates the challenges that need to be addressed to enhance the quality of newborn screening programs, including: 1) improved testing protocols to optimize both sensitivity and positive predictive value; 2) enhanced quality assurance with consistent policies; 3) better education of families and providers, especially in the pre-screening, prenatal period; and 4) improved risk communication for both true and false positive families, i.e., better genetic counseling. Specific recommendations for improvements include:
• It is likely that multiplex methods of newborn screening using methods such as IRT/DNA for CF and tandem mass spectrometry for numerous metabolic disorders will usher in a new era of expansion featuring molecular methods and more accountability. The CDC has recognized the dynamic state of newborn screening with the CF-related recommendations summarized below [75]. In addition, CLSI has published recent comprehensive guidelines for CF newborn screening that recommend using as a “core panel” the 23 CF-causing mutations specified by ACMG [82]. The magnitude of the health benefits from screening for CF is sufficient that regions should consider including routine newborn screening for CF in conjunction with networked systems to ensure access to high-quality care.
• In reaching a decision as to whether to add newborn screening for CF, regions should consider available resources and priorities as well as available national guidelines regarding CF screening, diagnosis, and treatment.
• Regions that implement newborn screening for CF should collect follow-up data in collaboration with CF care centers and analyze this information to monitor and improve the quality of CF newborn screening. In particular, all regions should collect, share, and analyze data by using standard protocols to evaluate and optimize laboratory algorithms used to screen for CF and refer for diagnosis. Regions seeking guidance on optimal laboratory protocols might wish to consult with other programs having more experience in conducting CF screening of newborns.
• Newborn screening for CF should be accompanied by rigorous infection control practices to minimize the risk to children with CF detected at an early age of acquiring infectious organisms associated with lung disease from older patients. Further research is needed to evaluate and optimize these practices.
• Newborn screening systems should ensure parental and provider education and communication of screening results to primary-care providers in a manner that will ensure prompt referral to diagnostic centers. For CF, these should be centers skilled in providing both sweat tests to young, presymptomatic children with CF and accurate and effective counseling to families, including those with infants identified as carriers. Regions are recommended to work with each other and with professional organizations and federal agencies to develop approaches to provide newborn screening information to parents during the prenatal and perinatal periods on all conditions, including CF, to facilitate informed choices and appropriate responses to positive screening results.
7. The future of newborn screening
Although the time frame for future evolution of newborn screening programs is difficult to predict, it is clear that more and better tests will be applied to newborn populations to ensure early diagnosis and hopefully reduce health disparities further. More precise and technologically advanced, though not necessarily more expensive, methods are readily available such as tandem mass spectrometry, next generation sequencing for DNA analysis, and possibly whole genome sequencing (WGS); these procedures are likely to be implemented during the next decade.
Clearly tandem mass spectrometry is an evolution that is already causing a revolution. Not only are more regions implementing MS/MS [58], but it is causing an expansion of testing panels to literally quadruple overnight the number of congenital disorders being screened for routinely. Completion of the Human Genome Project has given us molecular genetics analytical biotechnology capability far beyond what is currently being used in newborn screening programs, and thus more DNA-based tests and possibly whole genome sequencing will emerge over time. Indeed, the CDC [76] referred to newborn screening for CF using IRT/DNA analysis as “a paradigm for public health genetics policy development.”
It is safe to predict that as DNA-based assessment of newborns for CF proliferates, other DNA-linked screening tests will be implemented. For instance, CAH screening programs, because of their low positive predictive value (up to 200 false positives for every diagnosis) [64], could be improved quite significantly with a two-tier strategy using 17-hydroxyprogesterone coupled to detection of 21-hydroxylase mutations [5]. SCID screening provides an excellent example of how public health laboratories can quickly adapt to new, DNA-based technologies [85–88].
In addition to more and better tests, the glaring lack of consistencies among regions needs to be addressed effectively and eliminated through quality improvement methods that address all six components of the newborn screening system (http://mchb.hrsa.gov/screening/) namely: 1) education of professionals and parents; 2) screening, i.e. specimen collection, submission, and testing; 3) follow-up of abnormal and unsatisfactory test results; 4) confirmatory testing and diagnosis; 5) medical management and periodic outcome evaluation; and 6) system evaluation and quality assurance.
The variations relate to policies, financing, testing panels and protocols, follow-up efforts, and education. Consequently, the American Academy of Pediatrics Newborn Screening Task Force has issued “a call for a national agenda on state newborn screening programs.” Some of their most important recommendations follow.
• Federal agencies must take action to strengthen the public health infrastructure for newborn screening.
• State public health agencies should direct their newborn screening program to be consistent with professional guidelines and recommendations.
• States and state public health agencies should implement mechanisms to inform and involve health professionals and the public.
• States and state public health agencies should provide support for coordination and integration of program activities, including information and services.
• A federally-funded newborn screening research agenda should be outlined that aims to: develop better tests (more sensitive, more specific, and less costly); assess the validity and utility of new technologies (e.g., tandem mass spectrometry, DNA-based testing, and other evolving technologies); and define appropriate uses of residual biologic samples for a population-based research and surveillance.
• Pilot studies should be undertaken to demonstrate the safety, effectiveness, validity, and clinical utility of tests for additional conditions and new testing modalities.
• States should assure adequate financing of all parts of the newborn screening system: screening, short-term follow-up, diagnostic testing, comprehensive medical care/treatment, and evaluation of the system.
The AAP ‘call for a national agenda’ was followed by a landmark effort under the leadership of the ACMG to address some of the critical issues. Commissioned by the Maternal and Child Health Bureau of the Health Resources and Services Administration (US Department of Health and Human Services), a consensus-developing group of multidisciplinary experts published their analyses, conclusions, and recommendations recently in a document entitled “Newborn Screening: Toward a Uniform screening Panel and System” (http://mchb.hrsa.gov/screening/). This group recommended a core panel of 29 genetic conditions based on predetermined criteria “as primary targets for screening that warrant the full breadth of the newborn screening system” (see Table 1). In addition to congenital hearing impairment, the list includes biotinidase deficiency, classical galactosemia, congenital adrenal hyperplasia, congenital hypothyroidism, cystic fibrosis, three hemoglobinopathies associated with the hemoglobin S allele, five disorders of fatty acid oxidation, six amino acidurias, and nine organic acidurias. Subsequently, screening for severe combined immunodeficiency (SCID) with molecular methods [86–88] was added to the list, so that it now the core group recommended consists of 30 genetic disorders, as shown in Table 1. These new methods for SCID involve quantitating T-cell receptor excision circles (TRECs) using real-time quantitative polymerase chain reaction on DNA extracted from dried blood spots to detect infants with T-cell lymphopenia. Multiplex technologies such as MS/MS will detect blood analyte abnormalities found in 23 of the 30 conditions of the core panel. The group also emphasized that newborn screening “programs are obligated to establish a diagnosis and communicate the result to the health care provider and family” and furthermore to make available the information “when newborn screening laboratory results definitely establish carrier status.”
This AAP list of recommendations describes a wide range of issues that need to be addressed in quality improvement efforts. Enhancing quality assurance is difficult because although newborn screening is a system, its application varies markedly in different regions. In addition, as advances in biotechnology occur, the rate and effectiveness of implementation also vary. Furthermore, the ability of providers and parents to understand the laboratory results, whether they be diagnoses or false positives, is variable at best. Currently, about 5% of newborn screening tests are associated with a reportable result that needs to be explained to the parents. This amounts to 200,000 families per year in the United States or about 4,000 per week (ironically, the same number as the total diagnosed through newborn screening on an annual basis). The communication challenges faced by providers are daunting. The content of the message, how and when to deliver the stress-promoting news to parents, and the kind of follow-up required are all difficult issues. It is ironic that the biotechnology has advanced far beyond the capability of the system to meet the most fundamental of human interactions – communication. Clearly, more research is needed to improve risk communication methods and to evaluate their impact.
Despite the need for multidimensional improvement, it should be emphasized that newborn screening programs have been one of the great public health success stories. Thousands of children have benefited with reductions in mortality and morbidity. Even greater success will undoubtedly be achieved in the future.
References
[1] | Farrell P.M. Neonatal Screening: Toward earlier diagnosis and improved outcome, Doershuk C. Cystic Fibrosis in the 20th Century AM Publishing, Cleveland, 120–137, (2001) . |
[2] | Simpson J.L. , Elias S. , Genetics in Obstetrics and Gynecology, ed 2 Philadelphia, (2003) . Saunders. |
[3] | Elias S. , Simpson J.L. , Amniocentesis and fetal blood sampling, In Milunsky A ed. Genetic Disorders and the Fetus: Diagnosis,revention, and Treatment. Baltimore: The Johns Hopkins University Press, (2004) , 66–99. |
[4] | Therrell B.L. Jr , Laboratory methods for neonatal screening, American Public Health Association, Washington, DC, (1993) . |
[5] | Allen D.B. and Farrell P.M. , Newborn screening, Adv Pediatr 43: ((1996) ), 231–270. |
[6] | American College of Gynecology Practice Bullet, renatal diagnosis of fetal chromosomal abnormalities. Number 27, (2001) . |
[7] | American College of Gynecology Practice Bullet, Neural tube defects. Number 44, (2003) . |
[8] | Newborn Screening Task Force. Newborn Screening: A Blueprint for the Future, Pediatr 106: ((2000) ), 386–388. |
[9] | Driscoll D.A. and Gross SJ . Screening for fetal aneuploidy and neural tube defects, Genet Med 11: ((2009) ), 818–821. |
[10] | Manning M. and Hudgins L. , Array-based technology and recommendations for utilization in medical genetics practice for detection of chromosomal abnormalities, Genet Med 12: ((2010) ), 742–745. |
[11] | Resta R.G. , Changing demographics of advanced maternal age (AMA) and the impact on the predicted incidence of Down syndrome in the United States. Implications for prenatal screening and genetic counselling, Am J Med Genet 133A: ((2005) ), 31–36. |
[12] | Guthrie R. , The origin of newborn screening, Screening 1: ((1992) ), 5–15. |
[13] | Hennekens C. , Buring J. , Epidemiology in Medicine Boston/Toronto: Little, Brown and Company, (1987) . |
[14] | Farrell M.H. and Farrell P.M. , Newborn screening for cystic fibrosis: Ensuring more good than harm, J Pediatr 143: ((2003) ), 707–711. |
[15] | Ehrich M. , Deciu C. , Zwiefelhofer T. , et al., Noninvasive detection of fetal trisomy 21 by sequencing of DNA in maternal blood: A study in a clinical setting,205e, Am J Obstet Gynecol 204: ((2011) ), 1–211. |
[16] | Palomaki G.E. , Kloza E.M. , Lambert-Masserlian G.M. , et al., DNA sequencing of maternal plasma to detect Down syndrome: An international clinical validation study, Genet Med 13: ((2011) ), 913–920. |
[17] | Elias S. , Simpson J.L. , Martin A.O. , et al., Chorionic villus sampling for first trimester prenatal diagnosis: Northwestern University Program, Am J Obstet Gynecol 152: ((1985) ), 204–210. |
[18] | Elias S. , Simpson J.L. , Shulman L.P. , et al., Transabdominal chorionic villus sampling for first-trimester prenatal diagnosis, Am J Obstet Gynecol 320: ((1989) ), 609–617. |
[19] | Caughey A.B. , Hopkins L.M. and Norton M.E. , Chorionic villus sampling compared with amniocentesis and the difference in the rate of pregnancy loss, Obstet Gynecol 108: ((2006) ), 612–616. |
[20] | D’Alton M.E. and DeCherney A.H. , Prenatal diagnosis, N Engl J Med 328: ((1993) ), 114. |
[21] | Evans M.I. and Wapner R.J. , Invasive prenatal diagnostic procedures, Semin Perinatol 29: ((2005) ), 215–218. |
[22] | Seeds J.W. , Diagnostic mid trimester amniocentesis: How safe? Am J Obstet Gynecol 191: ((2004) ), 608–616. |
[23] | Eddleman K.A. , Malone F.D. , Sullivan L. , et al., Pregnancy loss after midtrimester amniocentesis, Obstet Gynecol 108: ((2006) ), 1067–1072. |
[24] | Elias S. , The role of fetoscopy in antenatal diagnosis, Clin Obstet Gynaecol 7: ((1980) ), 73. |
[25] | Gosden C. , Nicolaides K.H. and Rodeck C.H. , Fetal blood sampling in investigation of chromosome mosaicism in amniotic fluid culture, Lancet 2: ((1988) ), 613–617. |
[26] | Bryndorf T. , Lundsteen C. , Lamb A. , et al., Rapid prenatal diagnosis of chromosome aneuploidies by interphase in situ hybridization: A one-year clinical experience with high-risk and urgent fetal and postnatal samples, Acta Obstet Gynecol Scand 79: ((2000) ), 8–14. |
[27] | Nicolaides K.H. , Thorpe-Beeston J.G. , Noble P. , Cordocentesis in assessment and care of the fetus. In Eden RD, Boehm FH (eds). Assessment and Care of the Fetus. & Lange, Norwalk, CT, (1990) , p. 291. |
[28] | Verlinsky Y. , Cohen J. , Munne S. , et al., Over a decade of experience with Preimplantation genetic diagnosis, Fertil Steril 82: ((2004) ), 302–303. |
[29] | Grace J. , Toukhy T. and Braude P. , Pre-implantation genetic testing, Brit J Ob Gyn 111: ((2004) ), 1165–1173. |
[30] | Elias S. , Preimplantation genetic diagnosis by comparative genomic hybridisation, N Engl J Med 345: ((2001) ), 1569–1571. |
[31] | Kaback M. , Lim-Steele J. , Dabholkar D. , et al., Tay-Sachs disease – carrier screening, prenatal diagnosis, and the molecular era: An international perspective, -, JAMA 270: ((1993) ), 2307–2315. |
[32] | ACOG Committee Opinion No. 298, Prenatal and preconceptional carrier screening for genetic diseases in individuals of Eastern European Jewish descent. American College of Obstetricians and Gynecologists, Washington, DC. |
[33] | Kerem B.S. , Rommens J.M. , Buchanan J.A. , et al., Identification of the cystic fibrosis gene: Genetic analysis, Science 245: ((1989) ), 1073–1080. |
[34] | Sosnay P.R. , Siklosi K.R. , Van Goor F. , , et al., Defining the disease liability of variants in the cystic fibrosis transmembrane conductance regulator gene, Nat Genet 45: ((2013) ), 1160–1167. |
[35] | Frangolias D.D. , Nakielna E.M. and Wilcox P.G. , Pregnancy and cystic fibrosis: A case-controlled study, Chest 111: ((1997) ), 963–969. |
[36] | Genetic Testing for Cystic Fibrosis, NIH Consensus Statement, (1997) . |
[37] | Lemna W.K. , Feldman G.L. , Kerem B. , et al., Mutation analysis for heterozygote detection and the prenatal diagnosis of cystic fibrosis, N Engl J Med 322: ((1990) ), 291–296. |
[38] | Genetic Testing for Cystic Fibrosis, NIH Consensus Statement, (1997) . |
[39] | Mercier B. , Verlingue C. , Lissens W. , et al., Is congenital bilateral absence of the vas deferens a primary form of cystic fibrosis? Analyses of CFTR in 67 patients, Am J Hum Genet 56: ((1995) ), 272–277. |
[40] | Costes B. , Girodon E. , Ghanem N. , et al., Frequent occurrence of CFTR intron 8 (TG)n 5T allele in men with congenital bilateral absence of the vas deferens, Eur J Hum Genet 3: ((1995) ), 285–293. |
[41] | Kiesewetter S. , Macek M. , Davis C. , et al., A mutation in CFTR produces different phenotypes depending on chromosomal background, Nat Genet 5: ((1993) ), 274–278. |
[42] | Chillon M. , Casals T. , Mercier B. , et al., Mutations in cystic fibrosis gene in patients with congenital absence of the vas deferens, N Engl J Med 332: ((1995) ), 1475–1480. |
[43] | Grody W.W. , Cutting G.R. , Klinger K.W. , et al., Laboratory standards and guidelines for populationbased cystic fibrosis carrier screening, Med 3: ((2001) ), 149–154. |
[44] | The American College of Obstetricians and Gynecologist and The American College of Medicine Genetics. Preconception and Prenatal Carrier Screening for Cystic Fibrosis. October The American College of Obstetricians and Gynecologists, 409 12th Street, 2001, Washington, DC. |
[45] | Gaston M.H. , Verter J.I. , Woods G. , et al., Prophylaxis with oral penicillin in children with sickle cell anemia, N Engl J Med 314: ((1986) ), 1593–1599. |
[46] | Kan Y.W. , Trecartin R.F. , Golbus M.S. , et al., Prenatal diagnosis of beta-thalassemia and sickle cell anemia: Experience with 24 cases, Lancet 1: ((1971) ), 269–271. |
[47] | Hagerman R.J. , Silverman A.C. , Fragile X syndrome: Diagnosis, treatment and research, Baltimore: The, Johns Hopkins University Press, (1991) . |
[48] | Hoffman E.P. , Brown R.H. and Kunkel L.M. , Dystrophin: The protein product of the Duchenne muscular dystrophy locus, Cell 51: ((1987) ), 919–928. |
[49] | Bakker E. , Veenema H. , den J.T. , et al., Dunnen, Germinal mosaicism increases the recurrence risk for “new” Duchenne muscular dystrophy mutations, J Med Genet 26: ((1989) ), 553–559. |
[50] | Bell J. , Dystrophia myotonica and allied diseases, Treas Hum Inherit 4: ((1947) ), 343. |
[51] | Penrose L.S. , The problem of anticipation in pedigrees of dystrophia myotonica, Ann Eugen Lond 14: ((1948) ), 125–132. |
[52] | Hecht B.K. , Donnelly A. , Gedeon A.K. , et al., Direct molecular diagnosis of myotonic dystrophy, Clin Genet 43: ((1993) ), 276–285. |
[53] | Aslanidis C. , Jansen G. , Amemiya C. , et al., Cloning of the essential myotonic dystrophy region and mapping of the putative defect, Nature 355: ((1992) ), 548–551. |
[54] | Jansen G. , Willems P. , Coerwinkel M. , et al., Gonosomal mosaicism in myotonic dystrophy patients: Involvement of mitotic events in (CTG)n repeat variation and selection against extreme expansion in sperm, Am J Hum Genet 54: ((1994) ), 575–585. |
[55] | CLSI. Blood Collection on Filter Paper for Newborn Screening Programs Approved Standard – Fifth Edition. CLSI document LA04-A5. Wayne, PA: Clinical and Laboratory Standards Institute; (2007) . |
[56] | CLSI Newborn Screening Follow-up; Approved Guideline. CLSI document I/LA27-A. Wayne, PA: Clinical and Laboratory Standards Institute, (2006) . |
[57] | CLSI Newborn Screening Follow-up; Approved Guideline. CLSI document I/LA27-A. Wayne, PA: Clinical and Laboratory Standards Institute, (2009) . |
[58] | CLSI Newborn Screening Follow-up; Approved Guideline. CLSI document I/LA27-A. Wayne, PA: Clinical and Laboratory Standards Institute, (2010) . |
[59] | CLSI. Molecular Diagnostic Methods for Genetic Diseases; Approved Guideline – Second Edition. CLSI document MM01-A2. Wayne, PA: Clinical and Laboratory Standards Institute; (2006) . |
[60] | CLSI. Newborn Screening for Cystic Fibrosis; Approved Guideline. CLSI document I/LA35-A. Wayne, PA: Clinical and Laboratory Standards Institute; (2011) . |
[61] | Scriver C.R. and Clow C.L. , Phenylketonuria: Epitome of human biochemical genetics I, N Engl J Med 303: ((1980) ), 1336–1342. |
[62] | Scriver C.R. and Clow C.L. , Phenylketonuria: Epitome of human biochemical genetics II, N Engl J Med 303: ((1980) ), 1394–1400. |
[63] | Bickel H. , Gerrard J. and Hickmans E.M. , Influence of phenylalanine intake on phenylketonuria, Lancet 2: ((1953) ), 812–813. |
[64] | Dussault J.H. , Coulombe P. , Laberge C. , et al., Preliminary report on a mass screening program for neonatal hypothyroidism, J Pediatr 86: ((1975) ), 670–674. |
[65] | Kwon C. and Farrell P.M. , The magnitude and challenge of false-positive newborn screening test results, Arch Pediatr Adoles Med 154: ((2000) ), 714–718. |
[66] | Fisher D.A. , Dussault J.H. and Foley T.P. , Screening for congenital hypothyroidism: Results of screening one million North American infants, J Pediatr 94: ((1979) ), 700–705. |
[67] | Allen D.B. , Hendricks S.A. and Sieger J.E. , Screening programs for congenital hypothyroidism: How can they be improved? Am J Dis Child 142: ((1989) ), 232–236. |
[68] | AAP Section on Endocrinology and Committee on Genetics, American Thyroid Association Committee on Public Health: Newborn screening for congenital hypothyroidism: Recommended guidelines, Pediatrics 91: ((1993) ), 1203–1209. |
[69] | Pang S. , Wallace M.A. , Hofman L. , et al., Worldwide experience in newborn screening for classical congenital adrenal hyperplasia due to 21-hydroxylase deficiency, Pediatrics 81: ((1988) ), 866–874. |
[70] | Farrell P.M. , Aronson R.A. , Hoffman G. , et al., Newborn screening for cystic fibrosis in Wiscons First alication of population-based molecular genetics testing Wisc Med J 93: ((1994) ), 415–421. |
[71] | Stoddard J.J. and Farrell P.M. , State-to-state variations in newborn screening policies, Arch Pediatr Adolesc Med 151: ((1997) ), 561–564. |
[72] | Wilson J.M.G. , Junger G. (1968) Principles and practice of screening for disease, World Health Organization.ublic Health Papers, No 34. Geneva: WHO. |
[73] | Ciske J.B. , Hoffman G. , Hanson K. , Annable K.M. , Wolff J. , Litscheim T. , Lessig R. and Aronson R. , Newborn screening in Wiscons, rogram overview and test addition. Wisc Med J 99: ((2000) ), 38–42. |
[74] | Roe C.R. , Coates P.M. , Mitochondrial fatty acid disorders, In: The Metabolic Basis of Inherited Disease , 7th edition, Scriver et al, (eds), McGraw Hill, New York, pp. 1501–1634, (1995) . |
[75] | Centers for Disease Control and Prevention. Newborn screening for cystic fibrosis: Evaluation of benefits and risks and recommendations for state newborn screening programs, MMWR 53: (No. RR-13) ((2004) ). |
[76] | Centers for Disease Control and Prevention: Newborn screening for cystic fibrosis: A paradigm for public health genetics policy development-proceedings of a workshop, MMWR ((1997) ) 456: (No. RR-16), 11–12. |
[77] | Gregg R.G. , Simantel A. , Farrell P.M. , Koscik R.E. , Kosorok M.R. , Laxova A. , Laessig R.H. , Hoffman G.L. , Hassemer D.J. , Mischler E.H. and Splaingard M. , Newborn screening for cystic fibrosis in Wiscons, paris Comon of biochemical and molecular methods. Pediatrics 99: ((1997) ), 819–824. |
[78] | Farrell P.M. , White T.B. , Ren C.L. , Hempstead S.E. , Accurso F. , Derichs N. , Howenstine M. , McColley S.A. , Rock M. , Rosenfeld M. , Sermet-Gaudelus I. , Southern K.W. , Marshall B.C. and Sosnay P.R. , Diagnosis of cystic fibrosis: Consensus guidelines from the cystic fibrosis foundation, J Pediatr 181S: ((2017) ), S4–S18. |
[79] | Bobadilla J.L. , Farrell M.H. and Farrell P.M. , Applying CFTR molecular genetics to facilitate the diagnosis of cystic fibrosis through neonatal screening, Pediatr 49: ((2002) ), 131–190. |
[80] | Baker M.W. , Atkins A.E. , Cordovado S.K. , Hendrix M. , Earley M.C. and Farrell P.M. , Improving newborn screening for cystic fibrosis using next-generation sequencing technology: A technical feasibility study, Genet Med 18: ((2016) ), 231–238. |
[81] | Amaral M.D. , Novel personalized therapies for cystic fibrosis: Treating the basic defect in all patients, J Intern Med 277: ((2015) ), 155–166. |
[82] | Koscik R.L. , Farrell P.M. , Kosorok M.R. , Zaremba K.M. , Laxova A. , Lai H.C. , et al., Cognitive function of children with cystic fibrosis: Deleterious effect of malnutrition, Pediatr 113: ((2004) ), 1549–1558. |
[83] | Farrell P.M. , Li Z. , Kosorok M.R. , Laxova A. , Green C.G. , Collins J. , Lai H.-C. , Rock M.J. and Splaingard M.L. , Bronchopulmonary disease in children with cystic fi after early or delayed diagnosis, J Resp Crit Care Med 168: ((2003) ), 1100–1108. |
[84] | Grody W.W. , Cutting G.R. , Klinger K.W. , Richards C.S. , Watson M.S. and Desnick R.J. , Subcommittee on Cystic Fibrosis Screening, Accreditation of Genetic Services Committee, American College of Medical Genetics. Laboratory standards and guidelines for population-based cystic fibrosis carrier screening, Genet Med 3: (2) ((2001) ), 149–154. |
[85] | Watson M.S. , Cutting G.R. , Desnick R.J. , et al., Cystic fibrosis population carrier screening: revision of American College of Medical Genetics mutation panel, Genet Med 6: (5) ((2004) ), 387–391. |
[86] | The Working Group for Newborn Screening for SCID (Baker JC, Baker MW, et al) Puck JM. Development of population-based newborn screening for SCID, J Allergy Clin Immunol 120: (4) ((2007) ), 760–768. |
[87] | Baker M.W. , Grossman W.J. , Laessig R.H. , Hoffman G.L. , Brokopp C.D. , Kurtycz D.F. , Cogley M.F. , Litsheim T.J. , Katcher M.L. and Routes J.M. , Development of a routine newborn screening protocol for severe combined Immunodeficiency, J Allergy Clin Immunol 124: (3) ((2009) ), 522–527. |
[88] | Routes J.M. , Grossman W.J. , Verbsky J. , Laessig R.H. , Hoffman G.L. , Brokopp C.D. and Baker M.W. , Statewide newborn screening for severe T-cell lymphopenia, JAMA 302: (22) ((2009) ), 2466–2471. |


