
Cerebro-facio-thoracic dysplasia (Pascual-Castroviejo syndrome): Identification of a novel mutation, use of facial recognition analysis, and review of the literature
Abstract
BACKGROUND:
Cerebro-facio-thoracic dysplasia (CFTD) is a rare, autosomal recessive disorder characterized by facial dysmorphism, cognitive impairment and distinct skeletal anomalies and has been linked to the TMCO1 defect syndrome.
OBJECTIVE:
To describe two siblings with features consistent with CFTD with a novel homozygous p.Arg114* pathogenic variant in the TMCO1 gene.
METHODS:
We conducted a literature review and summarized the clinical features and laboratory results of two siblings with a novel pathogenic variant in the TMCO1 gene. Facial recognition analysis was utilized to assess the specificity of facial traits.
CONCLUSION:
The novel homozygous p.Arg114* pathogenic variant in the TMCO1 gene is responsible for the clinical features of CFTD in two siblings. Facial recognition analysis allows unambiguous distinction of this syndrome against controls.
1Background
Cerebro-facio-thoracic dysplasia (CFTD) was first described by Pascual-Castroviejo et al. in 1975 in three children with characteristic findings of cognitive impairment, narrow forehead, bushy eyebrows, synophrys, hypertelorism, broad nose, wide philtrum, triangular-shaped mouth, short neck, maxillary hypoplasia, low hairline, brachycephaly and multiple bony abnormalities in the upper thoracic vertebrae and ribs [1]. The authors postulated that CFTD is an autosomal recessive disorder given parental consanguinity in two of the unrelated children. Subsequent authors described additional features of CFTD including: hypoplasia of the corpus callosum and cerebellar vermis [2], cleft lip and palate and hypothyroidism [3], Chiari I malformation [4], colobomas of the optic nerve, ptosis, small conical teeth, talipes, hypermobility and hypodensity of the grey matter [5].
With the advent of whole genome sequencing, researchers have now linked CFTD to pathogenic variants in the human transmembrane and coiled-coil domains protein 1 (TMCO1) gene and have concluded that it is synonymous to the TMCO1 defect syndrome [6]. The TMCO1 gene encodes a transcription factor, which is expressed ubiquitously in human and fetal tissue and plays a crucial role in human development. Xin et al. identified a 2 bp homozygous frameshift mutation in exon 2 of TMCO1 (c.139_140delAG/p.Ser47*) as the cause of the TMCO1 defect syndrome in 11 Amish patients in Ohio [7]. These patients’ features included craniofacial dysmorphism (brachycephaly, high arched bushy eyebrows, synophrys, long eyelashes, low-set ears, microdontia and gingival fibromatosis), skeletal anomalies of the chest (spinal fusion, rib abnormalities and Sprengel deformity of the scapula) and neurologic findings (intellectual disability, depressed deep tendon reflexes, unstable gait, and a loud, hoarse voice in verbal patients). They also experienced a high incidence of anatomical abnormalities of the genitourinary system, otitis media, sinusitis, and strabismus. Caglayan et al. reported another case of TMCO1 defect syndrome in a male patient, with similar features to the Amish patients and used whole-exome sequencing to identify a homozygous nonsense mutation p.Arg87* in exon 5 of TMCO1 [8]. Likewise, Alanay et al. identified the same homozygous nonsense founder mutation, p.Arg87*, in the TMCO1 in four out of five Turkish families with CFTD [9].
Here, we report two new patients with cerebro-facio-thoracic dysplasia, identify a novel mutation, use facial recognition analysis to assess the specificity of facial traits for disease recognition and review the literature based on the 19 patients whom up to this date have been confirmed to have mutations in the TMCO1 gene.
2Clinical report
Two siblings (currently a 19-year-old male and a 12-year-old female) with similar dysmorphic features have been followed by clinical geneticists at Children’s National Health System since age two years and 15 months, respectively. Both patients share low insertion of anterior hairline, optic nerve hypoplasia/atrophy with exotropia, central and axial hypotonia, dysphagia when young, gingival hyperplasia, intellectual disability and autism spectrum disorder with motor stereotypies including frequent rocking and repetitive hand movements. The male sibling also has cleft lip and palate and right clubfoot (repaired), self-mutilation (biting hands and scratching) when young, chronic otitis media and sinusitis, hypertelorism, left hip subluxation, dysmorphic spine and multiple rib abnormalities (fusion of 3rd and 4th ribs, see Fig. 1). He is more cognitively impaired than his sister, is non-verbal but can walk. The brother’s MRI, completed at 2 years of age, showed moderate atrophy and a normal corpus callosum. The sister has bilateral pes planus, synophrys and can speak in simple sentences, write part of her name and count to 5. The sister’s MRI at 13 months showed hypogenesis of the corpus callosum, mild volume loss, small optic nerves, small left olfactory bulb, dysplasia of the hippocampal formations, advanced myelination with bands of relative decreased myelination in the deep white matter.
Fig.1
Fusion of the posterior aspect of the 3rd and 4th right ribs.
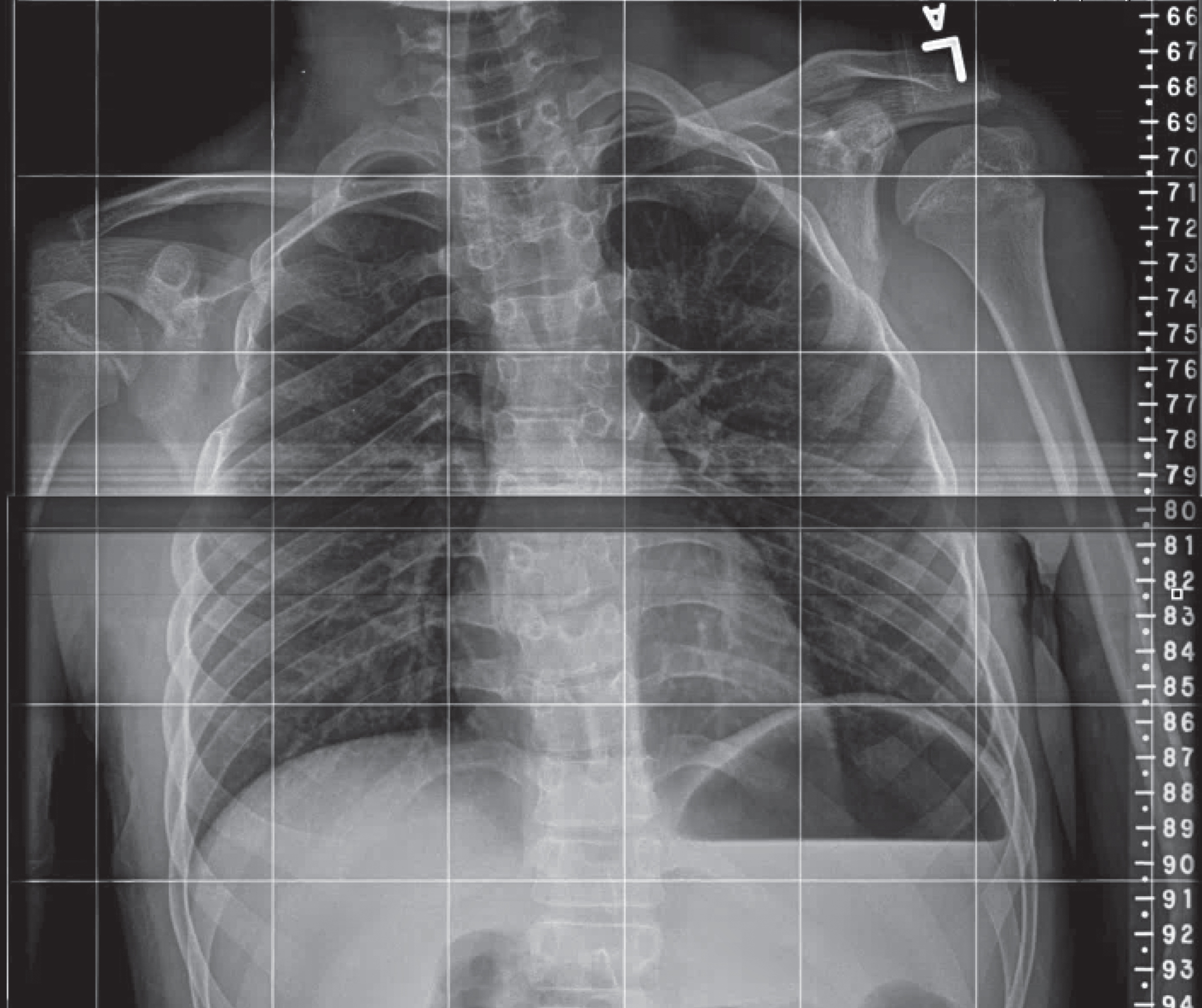
The parents were born in a small Guatemalan village, but denied consanguinity. The siblings’ cousin reportedly has similar phenotypic features and cognitive impairment and is the product of the maternal sister and paternal cousin.
3Methods
The siblings have been evaluated by clinical geneticists and multiple other specialists including neurology, orthopedics, physical medicine, ophthalmology, audiology, otolaryngology, craniofacial and cardiology. They underwent extensive metabolic and genetic work-up including karyotype and fragile X testing, comparative genomic hybridization array, MECP2 sequencing, plasma amino acids, urine organic acids, ammonia, lactate, beta-hydroxybutyrate, serum copper and ceruloplasmin, urine qualitative and quantitative glycosaminoglycans, urine oligosaccharide and free glycan analysis, acylcarnitine profile and uric acid levels. After obtaining signed consent from the mother, whole exome sequencing was performed on a commercial basis. Briefly, genomic DNA was extracted and exonic regions plus flanking splice junctions of the genome were sequenced on an Illumina sequencing system with 100bp or greater paired-end reads. These reads were aligned to the human genome build GRCh37/hg19, and analyzed for sequence variants using a custom-developed analysis tool. Sanger sequencing was used to confirm the pathogenic variants identified in the siblings.
We performed facial recognition analysis using the Facial Dysmorphology Novel Analysis (FDNA, Boston MA, USA) technology, which allows for assessment of subtle craniofacial dysmorphic features and facial pattern recognition. The input consisted of 53 facial frontal photos from 50 unaffected controls, as well as photos from one of our patients, and those obtained from prior publications (specifically, 14 patients with confirmed TMCO1 mutations).
A binary comparison was then performed between both groups, and the capacity to distinguish them was assessed by measuring the Area Under the Curve (AUC) of the Receiver Operating Characteristic (ROC) curve, which plots the true positive rate as function of the true negative rate. An AUC of 1 indicates perfect accuracy, while an AUC of 0.5 is the performance obtained by a totally random system (as for example a coin toss). Cross validation was performed by recurrently and randomly splitting the data into training sets, each set contained half of the photos. This random split was repeated 10 times [10].
4Results
The initial metabolic and genetic tests were normal except for mildly elevated uric acid in the sister and the brother with a balanced translocation 46,XY,t(11;15)(p15.1;q22.1). This translocation did not explain his condition because his unaffected mother carried the same translocation while his affected sister did not.
Whole exome sequencing demonstrated a homozygous pathogenic variant in the TMCO1 gene (c.340C>T/p.Arg.114* in exon 3, rs765824628) in the siblings, found in the heterozygous state in the mother. The father is deceased and therefore could not be tested. Data on the coordinates, allele frequency and in silico prediction of this novel variant are found on Table 1.
Table 1
Information regarding novel mutation
| Chromosomal location (GRCh37/hg19) | cDNA change (NM_019026.4) | Protein change (NP_061899.2) | Frequency (gnomAD) | In silico prediction CADD [11] |
| chr1:165728783 | c.340C>T | p.Arg114* | 8/276642 | 38 |
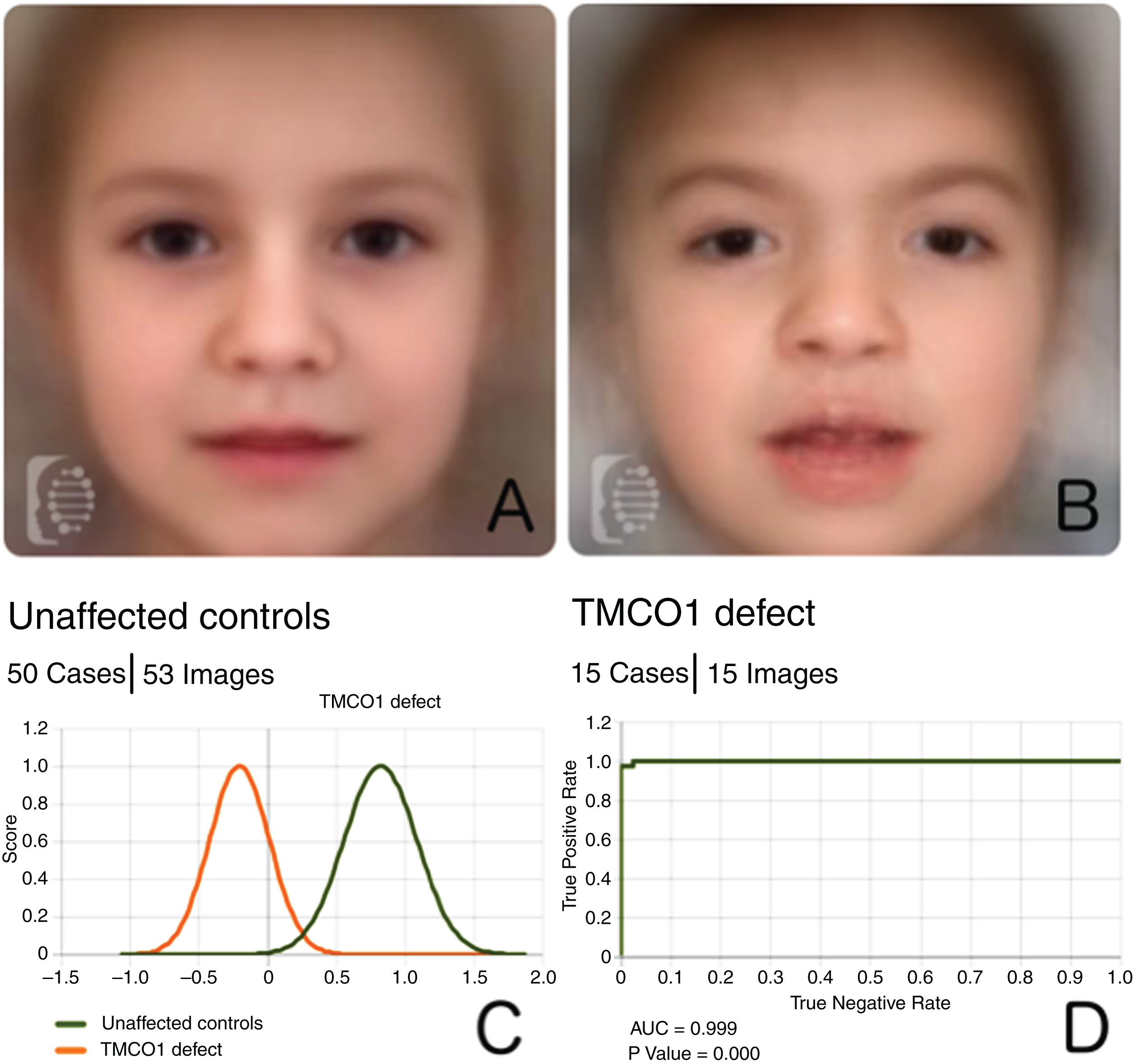
A composite image of unaffected controls versus that of patients can be found in Fig. 2A, B. Facial recognition analysis allowed for the discrimination of patients with TMCO1 vs controls, with an AUC of 0.999 (100% accuracy, p value 0.000, see Fig. 2C, D).
Fig.2
A: Composite photo created by averaging the extracted mathematical information of the photos in the control cohort. B: Composite photo obtained from images of patients with TMCO1 mutations. C: Score distribution of the binary comparison between unaffected controls and patients with TMCO1 mutations. D: ROC curve with pertinent statistics obtained after conducting 10 random splits.
5Discussion
With advancements in genetics, CFTD has now been linked to TMCO1 defect syndrome. Two siblings with similar phenotypic features and consistent with CFTD were found by whole genome sequencing to have a novel homozygous p.Arg114* pathogenic variant in the TMCO1 gene.
These siblings shared features previously described in other CFTD/TMCO1 defect syndrome patients. Both have severe cognitive impairment, low insertion of anterior hairline, exotropia, feeding difficulties, gingival hyperplasia, central and axial hypotonia, optic nerve atrophy and exotropia. The sister also has synophrys, hypogenesis of the corpus callosum while the brother has rib fusion, cleft lip/palate, self-mutilation and club foot. Both patients have autism spectrum disorder (ASD) with poor eye contact and motor stereotypies. Although ASD has not been previously described in CFTD patients, Pascual-Castroviejo did comment that one of his patients at 7.5 years “showed little interest in toys or in playing with other children. Her speech was still limited to a few words.” This patient, in retrospect, may have had ASD.
We performed a review of the literature, focusing on the clinical features of the 17 previous patients reported in the literature that were confirmed to have mutations in the TMCO1 gene, in addition to our two patients (see Table 2). As can be seen, the clinical and radiological findings caused by mutations in this gene are quite uniform, and thus the prior practice of separating the ‘craniofacial dysmorphism, skeletal anomalies, and mental retardation syndrome” from the “cerebro-facio-thoracic syndrome” is no longer warranted. In fact, the original patient reports of CFTD by Pascual-Castroviejo in 1975 clearly had the same condition, despite the lack of molecular confirmation.
Table 2
Review of the literature of 19 patients with molecular confirmation
| Feature | Xin et al. [7] | Caglayan et al. [8] | Alanay et al. [9] | Pehlivan et al. [6] | Present cases | Total |
| Polyhydramnios | 4/11 | – | 1/4 | 1/1 | 0/2 | 6/19 |
| Brachycephaly | 11/11 | – | 4/4 | 1/1 | 2/2 | 19/19 |
| Flat face | 11/11 | – | 4/4 | 1/1 | 2/2 | 19/19 |
| Low-set ears | 11/11 | 1/1 | 4/4 | 1/1 | 2/2 | 19/19 |
| Low hairline | 11/11 | 1/1 | 4/4 | 1/1 | 2/2 | 19/19 |
| Widely spaced eyes | 11/11 | 1/1 | 3/4 | 1/1 | 2/2 | 18/19 |
| Synophrys | 11/11 | 1/1 | 4/4 | 1/1 | 2/2 | 19/19 |
| Thick eyebrows | 11/11 | – | 4/4 | 1/1 | 2/2 | 19/19 |
| Short nose | 11/11 | 1/1 | 4/4 | 1/1 | 0/2 | 17/19 |
| Cleft lip/palate | 3/11 | 0/1 | 4/4 | 1/1 | 1/2 | 9/19 |
| Microdontia | 9/9 | – | 3/3 | nd | 2/2 | 14/14 |
| Gingival hypertrophy | 8/8 | – | 2/2 | nd | 2/2 | 12/12 |
| Short neck | 6/11 | 1/1 | 4/4 | 1/1 | 2/2 | 14/19 |
| DD/ID | 11/11 | 1/1 | 4/4 | 1/1 | 2/2 | 19/19 |
| Vertebral anomalies | 6/11 | – | 4/4 | 1/1 | 1/2 | 12/18 |
| Rib anomalies | 6/11 | 1/1 | 4/4 | 1/1 | 1/2 | 13/19 |
| Genitonurinary anomalies | 5/11 | – | 1/4 | 1/1 | 0/2 | 7/17 |
| Mutation | c.139_140delAG/ | c.259 | c.259 | c.323+3G>C/- | c. 340 | |
| p.Ser47* | C>T/ | C>T/ | C>T/ | |||
| p.Arg87* | p.Arg87* | p.Arg.114* |
Abbreviations: DD/ID: Developmental Delay/Intellectual Disability; nd: not documented.
Regarding the molecular characterization of the condition, there have been 3 variants so far reported in this gene, and our novel variant represents only the fourth mutation. It is important to note that all four mutations so far described in the TMCO1 gene have been truncating mutations. Two of the four reported variants represent founder mutations, one in the Amish population, and one in the Turkish population.
6Conclusion
We describe a novel homozygous p.Arg114* pathogenic variant in the TMCO1 gene–only the fourth variant reported in this gene–in two siblings with features consistent with CFTD.
URLs
genome aggregation Database (gnomAD browser): http://gnomad.broadinstitute.org
UCSC genome browser: https://genome.ucsc.edu
Face2Gene RESEARCH: http://www.Face2Gene.com
Conflict of interest
The authors report no conflict of interest.
Acknowledgments
The authors would like to thank the patients and their families for their kind cooperation as well clinical input from Anupama Tate, DMD, MPH (dentistry) and Laura Tosi, MD (orthopedics).
References
[1] | Pascual-Castroviejo I. , Santolaya J.M. , Martin V.L. , Rodriguez-Costa T. , Tendero A. and Mulas F. , Cerebro-facio-thoracic dysplasia: Report of three cases, Dev Med Child Neurol 17: ((1975) ), 343–351. |
[2] | Rufo-Campos M. , Riveros-Huckstadt P. , Rodríguez-Criado G. and Hernández-Soto R. , Another case of cerebro-facio-thoracic dysplasia (Pascual-Castroviejo syndrome), Brain Dev 26: ((2004) ), 209–212. |
[3] | Smigiel R. , Barg E. , Gabrysz M. , Szpich E. , Sasiadek M. and Sasiadek M. , A new case of cerebro-facio-thoracic dysplasia in a 3-year-old girl with short stature and hypothyroidism, Clin Dysmorphol 21: ((2012) ), 167. |
[4] | Cortesi A. , Rossi M. , Mazzi M. , Marseglia G. , Pescucci C. , Palchetti S. , Torricelli F. and Orrico A. , An additional case of cerebrofaciothoracic dysplasia associated with Chiari type I malformation, Clin Dysmorphol 22: ((2013) ), 115–117. |
[5] | Cilliers D. , Alanay Y. , Boduroglu K. , Utine E. , Tunçbilek E. and Clayton-Smith J., Cerebro-facio-thoracic dysplasia: Expanding the phenotype, Clin Dysmorphol 16: ((2007) ), 121–125. |
[6] | Pehlivan D. , Karaca E. , Aydin H. , Beck C.R. , Gambin T. , Muzny D.M. and Bilge B. , Geckinli, A. Karaman and S.N. Jhangiani, Centers for Mendelian Genomics, R.A. Gibbs, and J.R. Lupski, Whole-exome sequencing links TMCO1 defect syndrome with cerebro-facio-thoracic dysplasia, Eur J Hum Genet EJHG 22: ((2014) ), 1145–1148. |
[7] | Xin B. , Puffenberger E.G. , Turben S. , Tan H. , Zhou A. and Wang H. , Homozygous frameshift mutation in TMCO1 causes a syndrome with craniofacial dysmorphism, skeletal anomalies, and mental retardation, Proc Natl Acad Sci U S A 107: ((2010) ), 258–263. |
[8] | Caglayan A.O. , Per H. , Akgumus G. , Gumus H. , Baranoski J. , Canpolat M. , Calik M. , Yikilmaz A. , Bilguvar K. , Kumandas S. and Gunel M. , Whole-exome sequencing identified a patient with TMCO1 defect syndrome and expands the phenotic, Clin Genet 84: ((2013) ), 394–395 spectrum. |
[9] | Alanay Y. , Ergüner B. , Utine E. , Haçariz O. , Kiper P.O.S., Taşkiran E.Z., Perçin F., Uz E., Sağiroğlu Mş, Yuksel B., Boduroglu K. and Akarsu N.A., TMCO1 deficiency causes autosomal recessive cerebrofaciothoracic dysplasia, Am J Med Genet A 164A: ((2014) ), 291–304. |
[10] | Lumaka A. , Cosemans N. , Lulebo Mampasi A. , Mubungu G., Mvuama N., Lubala T., Mbuyi-Musanzayi S., Breckpot J., Holvoet M., de Ravel T., Van Buggenhout G., Peeters H., Donnai D., Mutesa L., Verloes A., Lukusa Tshilobo P. and Devriendt K., Facial dysmorphism is influenced by ethnic background of the patient and of the evaluator, Clin Genet 92: ((2017) ), 166–171. |
[11] | Kircher M. , Witten D.M. , Jain P. , O’Roak B.J. , Cooper G.M. and Shendure J. , A general framework for estimating the relative pathogenicity of human genetic variants, Nat Genet 46: ((2014) ), 310–315. |


