
Extending the phenotypic spectrum of Sengers syndrome: Congenital lactic acidosis with synthetic liver dysfunction
Abstract
Sengers syndrome is a rare autosomal recessive mitochondrial disease characterized by lactic acidosis, hypertrophic cardiomyopathy and bilateral cataracts. We present here a case of neonatal demise, within the first day of life, who initially presented with severe lactic acidosis, with evidence of both chorioamnionitis and cardiogenic shock. Initial metabolic labs demonstrated a severe lactic acidosis prompting genetic testing which revealed a homozygous pathogenic variant for Sengers syndrome in AGK, c.979A > T; p.K327*. In addition to the canonical features of Sengers syndrome, our patient is the first reported case with liver dysfunction extending the phenotypic spectrum both in terms of severity and complications. This case also highlights the importance of maintaining a broad differential for congenital lactic acidosis.
1Introduction
Sengers syndrome was first identified in 1975 by the co-occurrence of the near pathognomonic triad of congenital cataracts, lactic acidosis and cardiomyopathy [1]. This disorder was subsequently shown to be caused by recessively inherited mitochondrial dysfunction and was noted to present in two distinct forms: a severe neonatal and more insidious later onset presentation [2, 3]. Sengers syndrome is caused by loss of function variants in AGK, the gene encoding acylglycerol kinase [4]. Acylglycerol kinase is a mitochondrial transmembrane enzyme that catalyzes the phosphorylation of monoacylglycerol and diacylglycerol to lysophosphatidic and phosphatidic acid, which are important secondary messengers and regulate mitochondrial ultrastructure. AGK was recently identified as a component of the mitochondrial TIM22 complex, and is essential for import and assembly of metabolite carrier proteins in a kinase-independent manner [5]. These kinase dependent and independent findings have helped elucidate the multiple functions of AGK, explaining the diverse metabolic findings present in Sengers syndrome.
To date there have been less than thirty molecularly diagnosed patients with Sengers syndrome, with the majority of these consisting of later onset disease as opposed to the neonatal form [3]. These distinct presentations differ in that neonatal forms are noted to have central nervous system involvement including cerebellar hypoplasia while adult onset forms are more likely to have pathogenic variants secondary to splicing defects suggesting possible low level gene expression [6]. We report here a case of neonatal demise within the first day of life, presenting with severe lactic acidosis and systemic hypotension, in the setting of both chorioamnionitis and cardiomyopathy, found to have Sengers syndrome.
2Materials and methods
A CMA SNP array (CytoScan® Dx) was performed showing areas of homozygosity over 5.8% of the array. Candidate genes within the areas of homozygosity were reviewed. A nuclear mitochondrial panel of 35 genes was done by Massively Paralleled Sequencing using TruSight One kit (v1.0). Plasma amino acids were run on a Biochrom 30 Amino Acid Analyzer.
3Case presentation
Our case is a 1-day-old full term boy born from a G1P0 31-year-old mother who had an uneventful pregnancy receiving routine prenatal care. He was the product of Iranian consanguineous parents, known to be second cousins, with reported history of infertility prior to this naturally conceived pregnancy. Rupture of membranes occurred 40 hours prior to delivery and meconium stained fluid was present. During labor, maternal fever was noted, and maternal administration of broad-spectrum antibiotics were started for clinically diagnosed chorioamnionitis. At birth, the newborn showed a weak cry and decreased tone. He was found to be tachycardic and febrile with APGAR scores of 7 and 8 and was started on broad spectrum antibiotics for presumed sepsis. Physical exam was notable for bilateral opacification of the corneas and hypospadias. At 5 hours of life, he began to have progressive encephalopathy and respiratory distress resulting in intubation. Initial laboratory findings were notable for a severe metabolic acidosis with insufficient respiratory compensation (pH = 6.94, pCO2 = 22.8, HCO3 = 6, Anion Gap = 49, Lactate of 354.6 mg/dl). Fluid resuscitation with normal saline and administration of intravenous sodium bicarbonate resulted in minimal improvement in acidosis. The infant became progressively hypotensive, despite initiation of continuous vasopressor infusion. A chest X-ray was notable for cardiomegaly and an echocardiogram revealed severe biventricular dysfunction and ventricular trabeculations suggestive of cardiomyopathy (Fig. 1A). Further studies showed evidence of synthetic liver dysfunction including low albumin, total protein, and fibrinogen, and prolonged PT, PTT with normal bilirubin and elevated transaminases. Hematologic labs were significant for leukocytosis with a predominance of immature bands, anemia, and thrombocytopenia which in combination with coagulation studies were concerning for disseminated intravascular coagulation. Bacterial cultures of blood, urine and cerebrospinal fluid were drawn and ultimately resulted as normal studies. Imaging included a head ultrasound showing “slit-like” lateral ventricles, decreased size of the extra-axial spaces and crowded posterior cranial fossa, suggestive of generalized brain edema. Abdominal ultrasound showed an edematous liver with increased periportal echogenicity, but no evidence of hepatitis. Initially, the combined clinical and laboratory findings were suggestive of both cardiogenic and septic shock. However, the degree of lactic acidosis seemed excessive relative to the infant’s hemodynamic status and was refractory to resuscitation, prompting a more detailed metabolic evaluation.
Fig.1
A) Portable CXR showing marked cardiomegaly. B) Plasma amino acid levels represented as ratio of the upper limit of normal. Dashed line is present at the upper level of normal, equal to one, with amino acids with asterisk denoting values more than double.
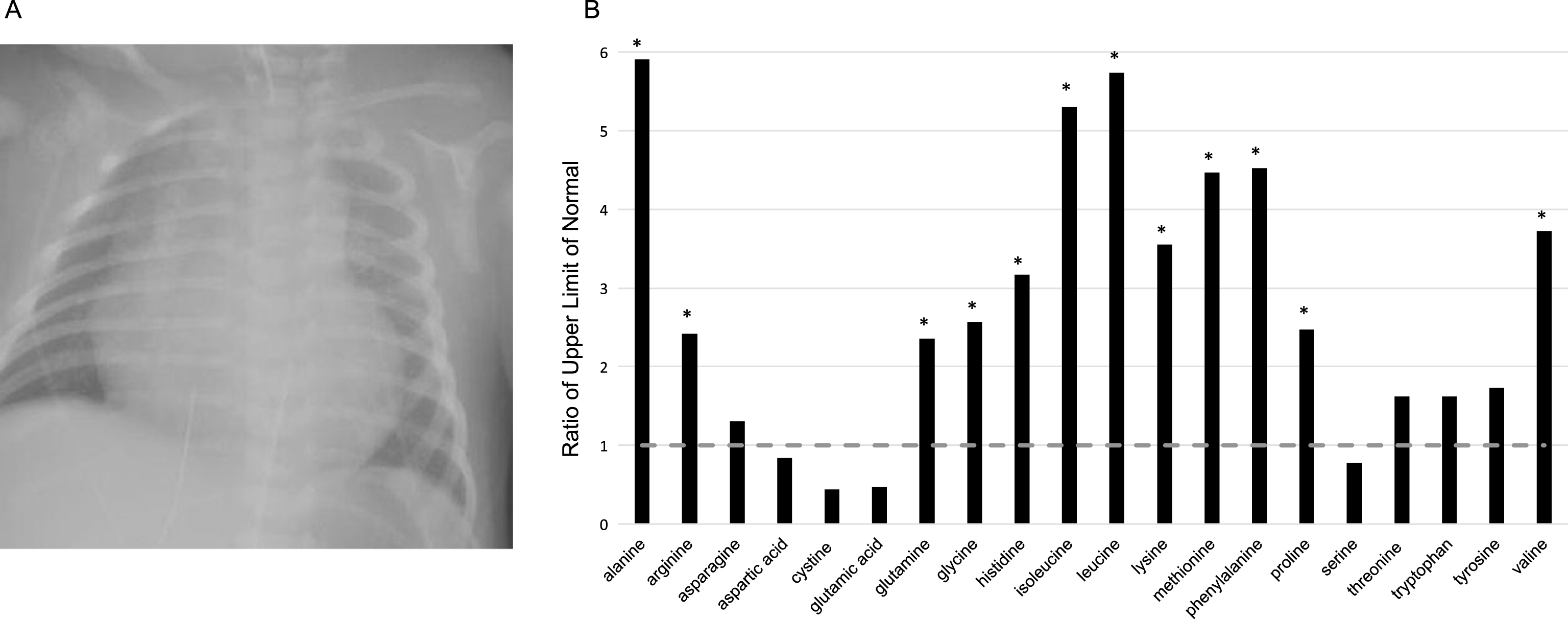
Serum lactate was elevated at 354.6 mg/dl, pyruvic acid 3.81 mg/dL, and the lactate to pyruvate ratio was 91, suggestive of mitochondrial disease. Additional genetic and metabolic testing included chromosomal microarray and plasma amino acids. Results from a nuclear mitochondrial gene panel revealed a homozygous pathogenic variant in AGK, c.979A > T p.K327*. This same genotype was present in a large consanguineous family with multiple neonatal Sengers syndrome cases [7, 8]. Plasma amino acid analysis revealed elevation of alanine, methionine, phenylalanine, and branched chain amino acids, indicating liver dysfunction and catabolism (Fig. 1B). These elevations were also consistent with in vitro experiments where loss of AGK leads to an accumulation of TCA cycle intermediates [5]. Despite aggressive medical management, the patient had a cardiac arrest likely due to worsening cardiac function secondary to refractory metabolic acidosis and multi-organ system dysfunction, dying on day of life one. An autopsy was declined per the family’s request.
The clinical course and genetic tests suggest that this infant had severe manifestation of Sengers syndrome, that may have been exacerbated by clinical chorioamnionitis and culture-negative sepsis. Although testing was unable to provide a diagnosis prior to our patient’s death, it allowed the family the option for pre-implantation genetic testing for future pregnancies.
4Conclusions
To our knowledge, we present the most severe case of Sengers syndrome reported to date. A novel finding in our patient was clinically-apparent synthetic liver dysfunction. While elevated transaminases and prolonged coagulation studies can result from septic shock, and/or disseminated intravascular coagulation, ultimately, infectious studies were negative. Rather, this may represent an additional manifestation of Sengers syndrome. Although a single patient is reported in the literature with primary liver involvement, characterized as cytosolic granules in liver cells found on autopsy, none has had clinically-evident liver impairment [8].
Alternatively, this patient may have had another autosomal recessive condition that could explain his synthetic liver dysfunction, especially considering the multiple areas of homozygosity noted by CMA; however, given the acute situation and rapid demise, whole exome sequencing was not able to be performed.
This case highlights the importance of maintaining a wide differential diagnosis for congenital lactic acidosis despite seemingly obvious etiologies [9]. This is particularly striking in our case given the evidence for septic and cardiogenic shock, yet these etiologies were unlikely to be the sole underlying cause of such profound, and disproportionate metabolic decompensation. Finally, despite recent advances in our understanding of AGK function, the phenotypic diversity attributed to other genes involved in the TIM22 complex, such as DDP1 in Mohr-Tranebjaerg Syndrome, suggest multiple mechanisms of action that may be better elucidated through further detailed genotype-phenotype correlations [10].
Acknowledgments
We thank the families for their generous contributions. The authors have no competing interests.
References
[1] | Sengers R.C. , et al., Congenital cataract and mitochondrial myopathy of skeletal and heart muscle associated with lactic acidosis after exercise, J Pediatr 86: (6) ((1975) ), 873–880. |
[2] | Haghighi A. , et al., Sengers syndrome: Six novel AGK mutations in seven new families and review of the phenotypic and mutational spectrum of 29 patients, Orphanet J Rare Dis 9: ((2014) ), 119. |
[3] | van Ekeren G.J. , et al., A retrospective study of patients with the hereditary syndrome of congenital cataract: Mitochondrial myopathy of heart and skeletal muscle and lactic acidosis, Eur J Pediatr 152: (3) ((1993) ), 255–259. |
[4] | Mayr J.A. , et al., Lack of the mitochondrial protein acylglycerol kinase causes Sengers syndrome, Am J Hum Genet 90: (2) ((2012) ), 314–320. |
[5] | Kang Y. , et al., Sengers Syndrome-Associated Mitochondrial Acylglycerol Kinase Is a Subunit of the Human TIM22 Protein Import Complex, Mol Cell 67: (3) ((2017) ), 457–470 e5. |
[6] | Perry M.S. and Sladky J.T. , Neuroradiologic findings in Sengers syndrome, Pediatr Neurol 39: (2) ((2008) ), 113–115. |
[7] | Pitkanen S. , et al., Familial cardiomyopathy with cataracts and lactic acidosis: A defect in complex I (NADH-dehydrogenase) of the mitochondria respiratory chain, Pediatr Res 39: (3) ((1996) ), 513–521. |
[8] | Siriwardena K. , et al., Mitochondrial citrate synthase crystals: Novel finding in Sengers syndrome caused by acylglycerol kinase (AGK) mutations, Mol Genet Metab 108: (1) ((2013) ), 40–50. |
[9] | Johns D.R. , Seminars in medicine of the Beth Israel Hospital: Boston, Mitochondrial DNA and disease, N Engl J Med 333: (10) ((1995) ), 638–644. |
[10] | Roesch K. , et al., Human deafness dystonia syndrome is caused by a defect in assembly of the DDP1/TIMM8a-TIMM13 complex, Hum Mol Genet 11: (5) ((2002) ), 477–486. |


