
KCNMA1 mutation in children with paroxysmal dyskinesia and epilepsy: Case report and literature review
Abstract
Patients with KCNMA1 gene mutation present with paroxysmal dyskinesia and/or epilepsy. We describe a male with heterozygous mutation c.3158A>G, (p.N1053S) in KCNMA1 gene, displaying paroxysmal dyskinesia and moderate mental retardation. We also review 20 reported cases with KCNMA1 mutation. We summarize that there is clinical heterogeneity in these patients. The onset age of episodic events ranges from 20 days to 15 years old. 6/21 (29%) patients merely had epilepsy, 10/21(48%) patients had paroxysmal dyskinesia only, and 5/21 (24%) had both epilepsy and paroxysmal dyskinesia. Seizure types were various, including absence, generalized tonic–clonic seizures, and myoclonic seizures. Paroxysmal dyskinesia was nonkinesigenic, but can be induced by alcohol, fatigue or stress. Most patients had variable degrees of mental retardation. The clinical outlook for this condition is in general not good. Epilepsy or non-epileptic events were resistant in most patients. Most patients presented with mild to severe intellectual disability and developmental delay.
1Introduction
KCNMA1 gene encodes α-subunit of the large conductance calcium-sensitive potassium channel (Kca1.1) [1]. Kca1.1 has a wide distribution in central nervous system, especially in excitatory neurons of cortex and hippocampus. It plays important roles in regulating neuronal excitability [1–3]. In 2005, KCNMA1 gene was first reported as a pathogenic gene in a large family with autosomal dominant paroxysmal nonkinesigenic dyskinesia and generalized epilepsy [4]. Since then, several KCNMA1 gene mutations in twenty patients have been described [5, 6]. Here, to delineate the clinical characteristics of the disease caused by KCNMA1 gene mutation further, one patient with a de novo KCNMA1 gene mutation is described. Besides, the reports associated with diseases caused by KCNMA1 gene mutations are summarized.
2Case report
The 3.5-year-old boy is the first child of nonconsanguineous Chinese parents. Pregnancy was uneventful. He has a normal birth and an early development. Head circumference was 33 cm at birth. At 16 months old, he developed episodes of sudden weakness of lower limbs, occasionally accompanied by rolling his eyes, lasting one to ten seconds. It occurred 10–20 times per day, with higher frequency with fatigue or excitement. Symptoms were not trigged by starvation. Before coming to our hospital, he was treated with valproate (VPA) at 16 months old with no response. When oxcarbazepine (OXC) was added to his therapy, the frequency of episodic events increased. Consequently, OXC was stopped. At 3 years of age, lamotrigine (LTG) was added, but there was still no response. Then clonazepam (CLZ) was administrated, and the frequency of paroxysmal dyskinesia was reduced to 3–5 times per day, and the longest interval could be up to three weeks.
Development is moderately delayed. He could control his head at 3 months, sit independently at 6 months, walk alone at 2 years old and say single words at 3 years old. Head circumference was 48 cm at his age of 2 years and 11 months. There was no family history of epilepsy or dyskinesia with him.
Electroencephalogram (EEG) at age of 2 years and 11 months showed generalized spike wave complexes. Episodic events presented during the EEG test; there were no epileptic discharges simultaneously. MRI at age of 16 months revealed no anomalies. Lumbar puncture was performed so as to exclude GLUT1-deficiency syndrome. Routine CSF test was unremarkable. Glucose level of CSF (2.66 mmol/L, ref 2.5∼4.5 mmol/L) and serum (6.28 mmol/L, ref 3.9∼6.9 mmol/L) were normal.
A gene panel consisting of 380 genes (Additional files 1) related with epilepsy and/orparoxysmal dyskinesia was performed on the proband. In total, 34 variants (Additional file 2) were discovered, of which 32 variants were reported polymorphisms. Pathogenicity of one heterozygous variant in ACY1 gene was ruled out, as it is inherited as autosomal recessive pattern. Consequently, the mutation (c.3158A>G, p.N1053S) in KCNMA1 gene deserved most attention. PCR-Sanger sequencing was used to confirm the mutation and parental origin, which revealed de novo occurrence (Fig. 1). It was a known pathogenic mutation, which had been reported previously in a patient with paroxysmal dyskinesia and developmental delay [6]. The clinical information of patients with KCNMA1 mutation is summarized in Table 1.
Fig.1
Sequence chromatogram showing one base pair substitution in KCNMA1 gene (A) and conservation of the altered amino acid shown in the ClustalW alignments (B).
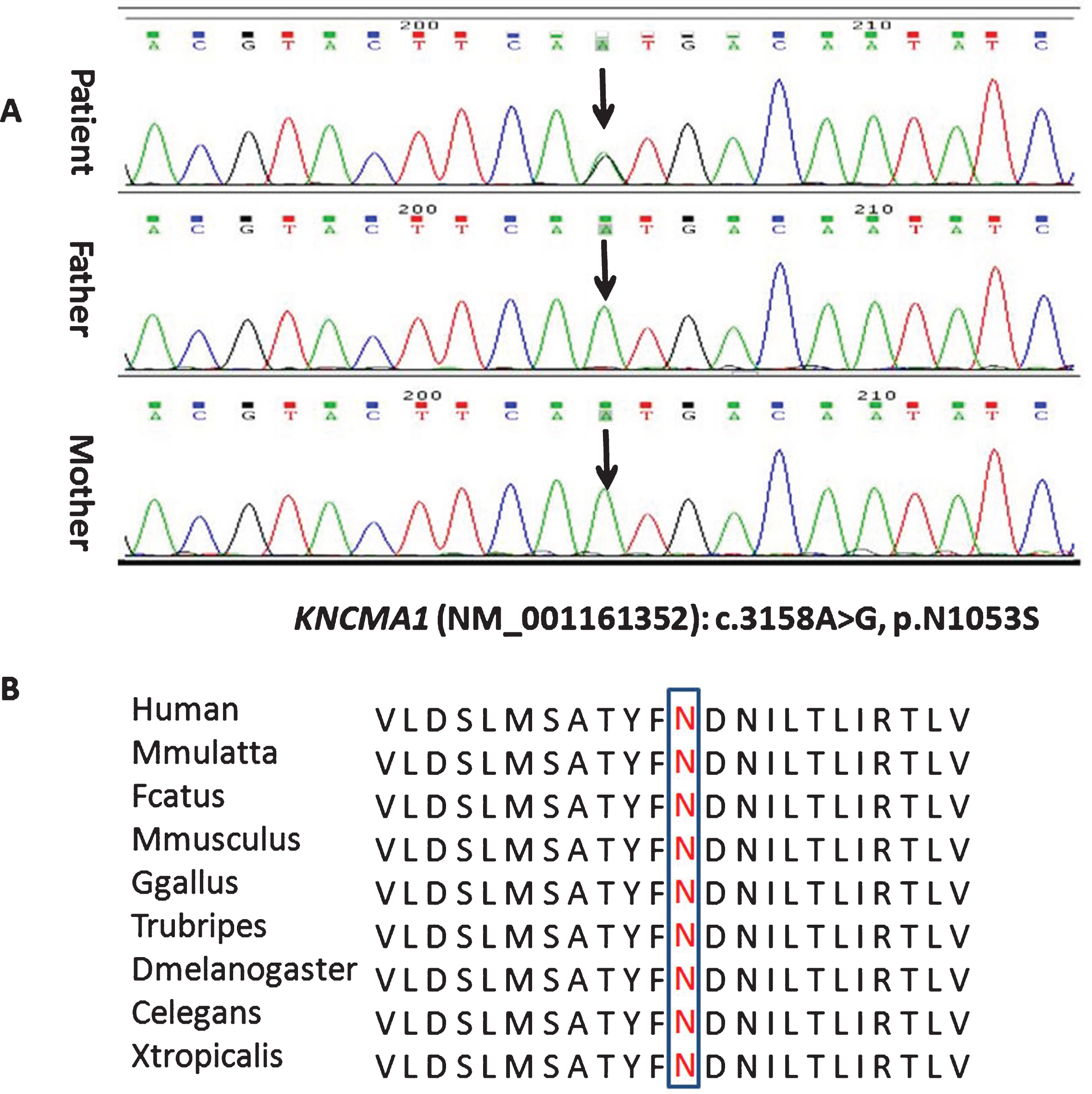
Table 1
Clinical features of patients with KCNMA1 gene mutation
| Patients | Du et al. [4] | Zhang et al. [6] | Tabarki et al. [5] | Our study | ||
| Sex | 10 M, 6 F | M | M | F | F | M |
| Age of onset | 6 mo- 15 y | 20 d | 7 mo | 8 mo | 8 mo | 16 mo |
| E and PD | 4 with E alone, 5 with E+PD, 7 with PD alone | PD | PD | E | E | PD |
| Seizure type | 4 with Ab, 2 with Ab and GTCS | No | No | Myoclonic seizures evolving to tonicand GTCS | Myoclonic seizures evolving to tonicand GTCS | No |
| PD | Involuntary dystonic or choreiform movements of the mouth, tongue and extremities | 1) Sudden onset of asymmetric limbdystonic posture, sometimes with nystagmus and strabismus, lasted several minutes to half an hour, and occurring once a week initially to 2–7 times per day after 1 year.2) Sudden decrease in voluntary movement of limbs, with hypotonia and occasional esotropia and yawning, lasting as long as 1 hour, and occurringonce to twice a day. | Paroxysmal dystonic postures, lasting several seconds to minutes, and occurring 3–5 times per day to once a week. | No | No | Sudden weakness of lower limbs, occasionally accompanied by rolling his eyes, and occurring 10–20 timesper day |
| Triggers | Alcohol, fatigue and stress | No | No | No | No | Fatigue or agitation |
| Development | NA | Severe delay | Mild delay | Severe delay | Severe delay | Moderate delay |
| EEG | Generalized spike wave complexes (the proband) | Normal | Normal | Lennox–Gastaut pattern | Mild backgroundslowing | Generalized spike wave complexes |
| MRI | NA | Normal | Normal | Cerebellar atrophy | Cerebellar atrophy | Normal |
| Treatment | Seizure frequency was reduced from daily to monthly with VPA and LTG in the proband; seizures and PD partially responsive to CLZ in other two patients | No response to OXC, VPA, LEV | Controlled by CLZ | Controlled by VPA | Controlled by VPA, LEV | Aggravated by OXC; VPA, LTG were not effective; frequency of episodic events was decreased after CLZ was added. |
| Mutation (NM_1161352) | c.1301A>G (heterozygous) | c.2650G>A (heterozygous) | c.3158A>G (heterozygous) | c.2026dupT (homozygous) | c.2026dupT (homozygous) | c. 3158A>G (heterozygous) |
| AA changed | D434G | E884K | N1053S | Y676Lfs*7 | Y676Lfs*7 | N1053S |
M, male; F, female; mo, months; y, year; E, epilepsy; PD, paroxysmal dyskinesia; Ab, absence; GTCS, generalized tonic clonic seizures; NA, not available; OXC, oxcarbazepine; VPA, valproate; LEV, levetiracetam; CLZ, clonazepam; LTG, lamotrigine.
3Discussion
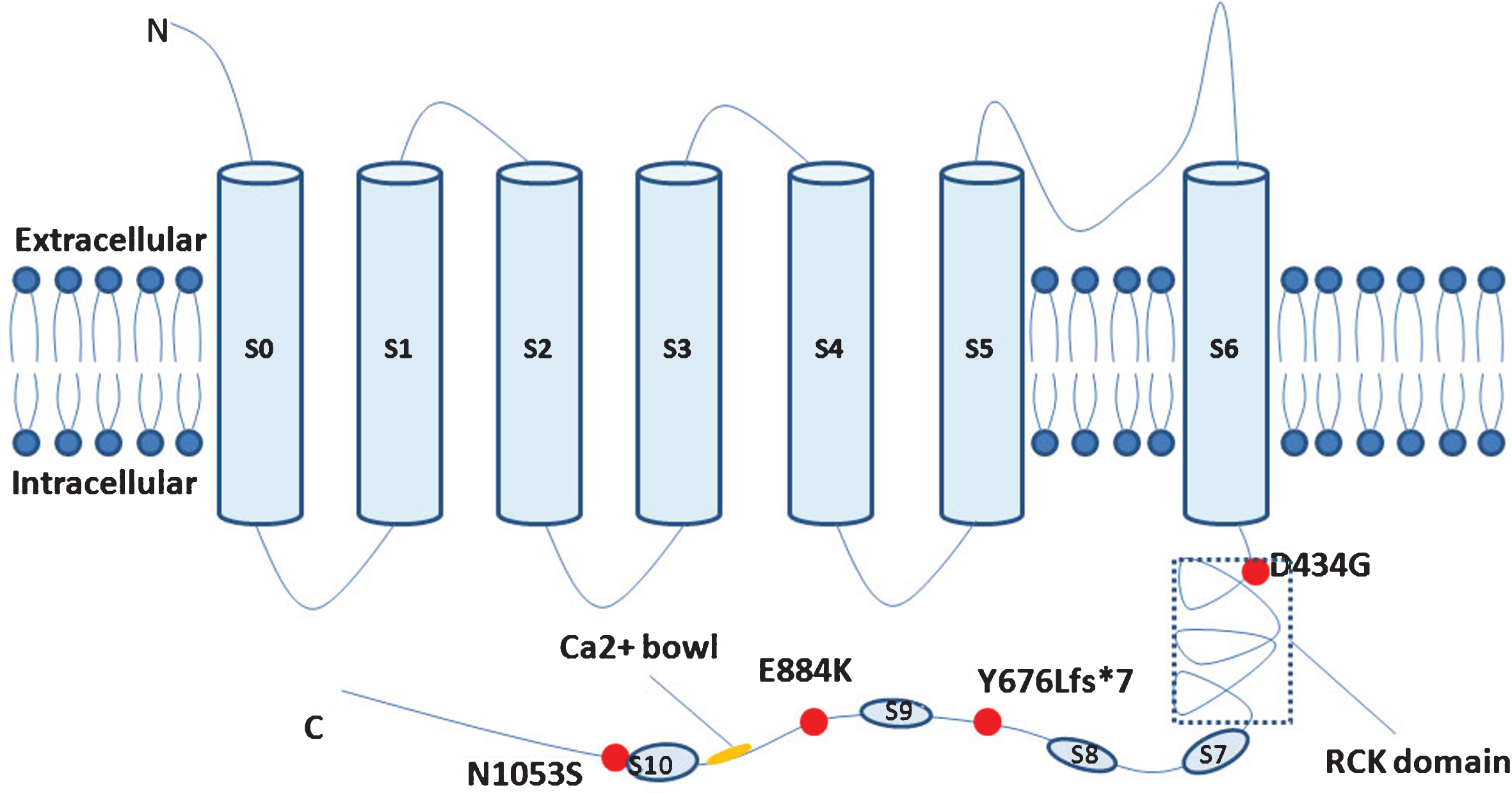
KCNMA1 gene, which is located at 10q22.3, encodes the alpha-subunit of the KCa1.1, consisting of seven transmembrane domains (S0–S6) at the N terminus, and an extensive C-terminal cytosolic domain which confers Ca2 + sensitivity to the channel. There are two putative high affinity Ca2 + binding sites, RCK domain and Ca2 + bowl, respectively [7, 8]. Kca1.1 has a wide distribution in central nervous system, and prominent expression is observed in excitatory neurons of cortex and hippocampus. It plays vital roles in driving action potential repolarization, mediating fast phase of AHP (after hyperpolarization potential), and regulating neurotransmitter release and dendritic excitability [1–3].
Since 2005, KCNMA1 gene has been associated with early onset epilepsy, paroxysmal dyskinesia and developmental delay [4]. To date, three publications with 21 patients (13 males and 8 females), have been found with KCNMA1 gene mutations, including two pedigrees (16 and 2 affected members, respectively) and three sporadic patients [4–6]. The age of onset ranged from 20 days after birth to 15 years old. Among the 18 patients with detailed clinical description, 33% (6/18) of patients had the onset of episodes within one year after birth, 55% (10/18) patients had symptoms between 2∼7 years old, and 17% (2/18) after age of 7 years.
The pathology associated with KCNMA1 mutations can manifest in patients as paroxysmal dyskinesia or epilepsy only, or both [4–6]. 10/21 (48%) had paroxysmal dyskinesia only, 6/21 (29%) had epilepsy only, 5/21 (24%) had both epilepsy and paroxysmal dyskinesia, including one patient who had paroxysmal dyskinesia within 6 months after birth and seizures attacks at age of 3 years, while the other four patients had epilepsy and paroxysmal dyskinesia simultaneously. Among 11 patients with epilepsy, 4 had absence seizure, 2 had absence seizures accompanied by GTCS (generalized tonic clonic seizures) occasionally, 2 had myoclonic seizures or myoclonic seizures evolving to tonicand GTCS, and 3 had epilepsy with no description about seizure types. Paroxysmal dyskinesia was nonkinesigenic, but in some patients it can be induced by alcohol, fatigue or stress. Patients presented different degrees of mental retardation. EEG abnormity was observed in patients with or without epilepsy, including generalized spike wave complexes, Lennox–Gastaut pattern, and mild background slowing.
There are not so many related reports on the treatments of patients with KCNMA1 mutations. In Zhang’s report, one patient with paroxysmal dyskinesia only was controlled by CLZ [6]. In Tabarki’s report, the seizure-free state was achieved in two patients with epilepsy only, by VPA and VPA combined with levetiracetam (LEV), respectively [5]. Three patients with both paroxysmal dyskinesia and epilepsy were partially responsive to antiepileptic drugs (Table 1) [4]. For our patient, OXC aggravated his paroxysmal events, while VPA and LTG had poor curative effective. He was partially responsive to CLZ, and the frequency of episodic events was reduced after CLZ was added. In vitro functional analysis revealed that gain of function of the BK channel leads to greater macroscopic potassium conductance, which results in more rapid repolarization of action potentials. Enhancing this repolarization leads to faster removal of inactivation of sodium channels, hence the neurons fire more frequently [5]. Consequently, considering previous reports and our study, sodium-channel blockers might be effective. Moreover, activation of inhibitory GABAB receptors by CLZ is effective as well.
The missense mutation (c.3158A>G, p.N1053S) identified in this study was previously mentioned by Zhang et al. [6]. Both patients merely presented with paroxysmal dyskinesia and developmental delay, while without epilepsy. But phenotype of patient in this study was a bit more severe (Table 1), which indicated the clinical heterogeneity of disorders caused by KCNMA1 mutations.
Including this study, four mutations of KCNMA1 were identified to be associated with epilepsy and/or paroxysmal dyskinesia. The majority of patients were heterozygous and the mutations were inherited as autosomal dominant. But patients with homozygous mutation in KCNMA1 gene were also reported, while the mutation was inherited from their heterozygous parents. Those parents were second cousins and had normal phenotype (Table 1) [5, 6]. All the mutations were located in the C-terminal of Kca1.1 (Fig. 2). D434G was located in the RCK domain, and the functional analysis revealed that the D434G speeds up channel activation and enhances Ca2 + sensitivity, suggesting a gain-of-function of Kca1.1 channel [4]. The functional impact of other mutations on BK channel activity remains unknown. The mutated N1053S identified in this study was located nearby S10, which might change the spatial conformation of the channel [6]. On the other hand, previous reports also indicated loss-of-function of KCNMA1 gene was pathogenic. Besides, Tabarki et al. described two siblings with homozygous truncated mutation in KCNMA1 gene, which presented with epilepsy and severe psychomotor retardation [5]. Kcnma1 homozygous knockout mice displayed severe motor dysfunction and cerebellar ataxia [9]. Taking all the above studies together, we could conclude that both gain-of-function and loss-of-function of Kca1.1 were responsible for epilepsy and movement disorders.
Fig.2
Simplified schematic of the large-conductance Ca2 +-activated K+ channel and KCNMA1mutations ever identified.
Our report summarized the mutation spectrum of KCNMA1and phenotypic profile of KCNMA1 gene related disorders. More mutations reports and function researches in the future might help to figure out the structure-function relationships of Kca1.1 and the mechanisms of its pathogenesis in neurological disorders.
Appendices
Additional file 1.
380 genes in the panel related with epilepsy accompanied with/without paroxysmal dyskinesia
| ABCC8 | CEP152 | EIF2B1 | HP | MTHFR | PPT1 | SLC9A6 |
| ACADSB | CHI3L1 | EIF2B2 | HRAS | NDN | PRICKLE1 | SLC9A9 |
| ACTB | CHRNA2 | EIF2B3 | HSD17B10 | NDUFA1 | PRICKLE2 | SNIP1 |
| ACY1 | CHRNA3 | EIF2B4 | HSD17B4 | NDUFA11 | PROC | SNRPN |
| ADK | CHRNA4 | EIF2B5 | HTR2A | NDUFAF1 | PRODH | SOBP |
| ADSL | CHRNA5 | ELP4 | HTT | NDUFAF2 | PRRT2 | SPAST |
| AFG3L2 | CHRNA7 | EMX2 | ICCA | NDUFAF3 | PTPN22 | SPTAN1 |
| AKT1 | CHRNB2 | EPB41L1 | IDH2 | NDUFAF4 | PUS1 | SPTLC2 |
| ALDH7A1 | CLCN2 | EPM2A | IDS | NDUFB3 | QDPR | SRPX2 |
| ALG1 | CLN3 | ERBB4 | IER3IP1 | NDUFS1 | RAB39B | STRADA |
| ALG11 | CLN5 | ERLIN2 | IFNG | NDUFS2 | RANBP2 | STS |
| ALG3 | CLN6 | EVC | IL6 | NDUFS4 | RELN | STXBP1 |
| AMACR | CLN8 | FADD | INS | NDUFS6 | ROGDI | SUOX |
| AMT | CNTNAP2 | FAM123B | KCNA1 | NDUFV1 | RPIA | SYN1 |
| APOL2 | COG7 | FASTKD2 | KCNJ10 | NDUFV2 | RTN4R | SYN2 |
| APOL4 | COH1 | FCGR2B | KCNJ11 | NEU1 | RYR1 | SYNGAP1 |
| APP | COMT | FKTN | KCNMA1 | NF1 | SCARB2 | SYP |
| ARG1 | COX6B1 | FLNA | KCNQ1 | NHLRC1 | SCN1A | TBC1D24 |
| ARHGAP31 | CPA6 | FOLR1 | KCNQ2 | NHS | SCN1B | TBP |
| ARHGEF9 | CPS1 | FOXG1 | KCNQ3 | NOTCH3 | SCN2A | TCF4 |
| ARSA | CSTB | FOXRED1 | KCTD7 | NR3C1 | SCN8A | TMEM165 |
| ARSE | CTSA | GABRA1 | KDM5C | NRXN1 | SCN9A | TPP1 |
| ARX | CTSD | GABRB3 | KIF11 | NTNG1 | SCZD1 | TREM2 |
| ASAH1 | CYB5R3 | GABRD | KIF1A | NUBPL | SCZD11 | TREX1 |
| ATIC | D2HGDH | GABRG2 | KRAS | OFD1 | SCZD12 | TSC1 |
| ATN1 | DAO | GAMT | L2HGDH | OPHN1 | SCZD2 | TSC2 |
| ATP1A2 | DAOA | GBA | LBR | PAFAH1B1 | SCZD3 | TSEN2 |
| ATP2A2 | DBH | GCK | LGI1 | PAH | SCZD5 | TSEN34 |
| ATP6AP2 | DCX | GCSH | LIAS | PAK3 | SCZD6 | TSEN54 |
| ATRX | DHFR | GLB1 | LMX1B | PANK2 | SCZD7 | TUBGCP6 |
| ATXN10 | DISC1 | GLDC | MAGI1 | PCDH19 | SCZD8 | TYROBP |
| BANK1 | DISC2 | GLRA1 | MAGI2 | PDHA1 | SERPINI1 | UBE3A |
| BOLA3 | DMPK | GOSR2 | MAN1B1 | PGK1 | SETBP1 | XK |
| BRP44L | DNAJC5 | GPHN | MANBA | PHF6 | SGCE | ZDHHC15 |
| C10orf2 | DNASE1 | GPR48 | MAPK10 | PHGDH | SHH | ZEB2 |
| C12orf62 | DOCK6 | GPR98 | MCCC2 | PIGL | SIAT9 | ZFYVE26 |
| C20orf7 | DPYD | GRIN1 | MCPH1 | PIGV | SIX3 | ZNF41 |
| C2orf64 | DRD2 | GRIN2A | MECP2 | PLA2G6 | SLC17A5 | STK11 |
| C4A | DRD3 | GSS | MEF2C | PLCB1 | SLC19A3 | HCN2 |
| CACNA1H | DTNBP1 | GYS1 | MFSD8 | PLP1 | SLC20A2 | SCN3A |
| CACNB4 | DXS423E | HAX1 | MLC1 | PNKP | SLC25A15 | GABRA6 |
| CACNG2 | EBP | HFE | MOCS1 | PNPO | SLC25A22 | CAPS |
| CASR | ECM1 | HLA-DQA1 | MOCS2 | POLG | SLC26A4 | SYNE1 |
| CCM1 | EFHC1 | HLA-DQB1 | MOCS3 | POMGNT1 | SLC2A1 | VLDLR |
| CDKL5 | EHMT1 | HNF1B | MR1 | PPOX | SLC46A1 | VPS13 |
| ABCB7 | ATP2B3 | AXK | FMR1 | NOP56 | POLG1 | TBP |
| AFG3L2 | ATPX | BEAN | FXN | NPHP1 | PPP2R2B | TDP1 |
| AHI1 | ATTP | C10orf2 | ITPR1 | PANR2 | PRKCG | TGM6 |
| APTX | ATXN1 | CA8 | JPH3 | PDYN | RPGRIP1L | TMEM216 |
| ARL13B | ATXN10 | CABC1 | KCNA1 | PEX1 | SACS | TTBK2 |
| ARX | ATXN2 | CACNA1A | KCNC3 | PEX2 | SETX | TTBK2 |
| ATCAY | ATXN3 | CACNB4 | KCNJ10 | PEX26 | SIL1 | TTPA |
| ATM | ATXN7 | CC2D2A | MPZ | PLEKHG4 | SLC1A3 | ADH3 |
| ATN1 | ATXN8OS | FGF14 | MRE11A | PMP22 | SPTBN2 | ATP13A2 |
| AAOPD | ADH1C |
Additional file 2.
Variants identified by NGS panel
| Gene | Transcript | Base change | AA change | Heterohomo | dbSNP | MAF | SIFT | PolyPhen-2 |
| ACY1 | NM_001198898.1 | c.584-9C>T | Hetero | Uncertain Significance | ||||
| APOL2 | NM_145637.2 | c.733A>G | p.V245V | Homo | rs132760 | 0 | Benign | |
| ATP1A2 | NM_000702.3 | c.1704C>T | p.F568F | Hetero | rs17846714 | 0.0278 | Benign | |
| CASR | NM_000388.3 | c.2244G>C | p.P758P | Homo | rs2036400 | 0.0272 | Benign | |
| CNTNAP2 | NM_014141.5 | c.3716-6C>G | Hetero | rs77025884 | Likely Benign | |||
| CPS1 | NM_001122633.2 | c.13_14insTCT | p.I5_K6insF | Hetero | rs3835047 | 0.477 | Benign | |
| CPS1 | NM_001122633.2 | c.204C>T | p.G68G | Hetero | rs529836556 | 0.0002 | Uncertain Significance | |
| CPS1 | NM_001122633.2 | c.1048A>G | p.T344A | Hetero | rs1047883 | Benign | Damaging | |
| DNAJC5 | NM_025219.2 | c.144C>T | p.P48P | Hetero | rs113987077 | 0.0278 | Benign | |
| DTNBP1 | NM_001271667.1 | c.268+7281C>A | Homo | rs6926401 | 0.0281 | Benign | ||
| HTT | NM_002111.7 | c.7182A>C | p.L2394L | Homo | rs2857790 | 0.0152 | Benign | |
| KCNA1 | NM_000217.2 | c.1296C>G | p.S432S | Hetero | rs76066681 | 0.025 | Benign | |
| KCNQ1 | NM_000218.2 | c.54C>T | p.I145I | Hetero | rs1800170 | 0.009 | Likely Benign | |
| KRAS | NM_004985.4 | c.451-5617G>A | p.R161R | Homo | rs4362222 | 0.0024 | Benign | |
| LGI1 | NM_001308275.1 | c.657T>C | p.F171F | Homo | rs1111820 | 0.0226 | Benign | |
| MCPH1 | NM_024596.3 | c.1175A>G | p.D344G | Homo | rs2515569 | 0.0056 | Benign | Tolerated |
| NDUFAF3 | NM_199070.1 | c.166+8G>A | Hetero | rs554862207 | 0.0002 | Uncertain Significance | ||
| NHS | NM_001291868.1 | c.566-12_566-11insT | Homo | rs5901624 | Benign | |||
| NR3C1 | NM_001018074.1 | c.1764C>T | p.H491H | Hetero | rs6194 | 0.0198 | Benign | |
| PDHA1 | NM_001173456.1 | c.958A>C | p.M251L | Homo | rs2229137 | 0.0495 | Benign | Tolerated |
| PRICKLE2 | NM_198859.3 | c.816T>C | p.D272D | Homo | rs27673 | 0.0162 | Benign | |
| PRRT2 | NM_145239.2 | c.751T>C | p.L251L | Homo | rs11150573 | 0.0082 | Benign | |
| RANBP2 | NM_006267.4 | c.8253G>A | p.E2751E | Homo | rs826580 | 0.0128 | Benign | |
| RELN | NM_173054.2 | c.3060C>T | p.D1020D | Hetero | rs115886170 | 0.0022 | Uncertain Significance | |
| RELN | NM_173054.2 | c.1888A>C | p.S630R | Hetero | rs115734214 | 0.0172 | Likely Benign | Damaging |
| SLC46A1 | NM_080669.5 | c.4417delA | Homo | rs5819844 | 0 | Benign | ||
| SPTAN1 | NM_001195532.1 | c.1330G>A | p.V444I | Hetero | rs77358650 | 0.0176 | Likely Benign | Tolerated |
| SPTAN1 | NM_001195532.1 | c.5085A>G | p.L1690L | Homo | rs1415568 | 0.0152 | Benign | |
| TCF4 | NM_001243226.2 | c.28G>C | p.P10P | Homo | rs611326 | 0.0032 | Benign | |
| TUBGCP6 | NM_020461.3 | c.4861G>C | p.L1621L | Homo | rs4838864 | 0.0012 | Benign | |
| TYROBP | NM_198125.2 | c.130G>T | p.V44L | Hetero | rs77782321 | 0.0232 | Benign | Activating |
| ZFYVE26 | NM_015346.3 | c.6405G>A | p.L2135L | Hetero | rs76327447 | 0.017 | Benign | |
| ZFYVE26 | NM_015346.3 | c.453C>T | p.S151S | Hetero | rs75391113 | 0.016 | Benign | |
| KCNMA1 | NM_1161352 | c.3158A>G | p.N1053S | Hetero | Pathogenic | Damaging |
Acknowledgments
We truly thank and appreciate the patients and their parents for their cooperation in this study. This study was financially supported by 985 Peking University and Clinical HospitalCooperation Project (2013-1-06), and Technology innovation talents special fund of Harbin Science and Technology Bureau (2016RAXYJ089).
References
[1] | Knaus H.G. , Schwarzer C. , Koch R.O. , Eberhart A. , Kaczorowski G.J. , Glossmann H. , Wunder F. , Pongs O. , Garcia M.L. , Sperk G. , Distribution of high-conductance Ca(2+)-activated K+ channels in ratbrain: Targeting to axons and nerve terminals, J Neurosci 16: (3) ((1996) ), 955–963. |
[2] | Misonou H. , Menegola M. , Buchwalder L. , Park E.W. , Meredith A. , Rhodes K.J. , Aldrich R.W. and Trimmer J.S. , Immunolocalization of the Ca2+-activated K+ channel Slo1 in axons and nerve terminals of mammalian brain and cultured neurons, J Comp Neurol 496: (3) ((2006) ), 289–302. |
[3] | Martire M. , Barrese V. , D’Amico M. , Iannotti F.A. , Pizzarelli R. , Samengo I. , Viggiano D. , Ruth P. , Cherubini E. and Taglialatela M. , Pre-synaptic BK channels selectively control glutamate versus GABArelease from cortical and hippocampal nerve terminals, J Neurochem 115: (2) ((2010) ), 411–422. |
[4] | Du W. , Bautista J.F. , Yang H. , Diez-Sampedro A. , You S. , Wang L. , Kotagal P. , Lüders H.O. , Shi J. and Cui J. , Calcium-sensitive potassium channelopathy in human epilepsy and paroxysmal movementdisorder, Nat Genet 37: (7) ((2005) ), 733–738. |
[5] | Tabarki B. , AlMajhad N. , AlHashem A. , Shaheen R. and Alkuraya F.S. , Homozygous KCNMA1mutation as a cause of cerebellar atrophy, developmental delay and seizures, Hum Genet 135: (11) ((2016) ), 1295–1298. |
[6] | Zhang Z. , Tian M. , Gao K. , Jiang Y. and Wu Y. , De novo KCNMA1 mutations in children withearly-onset paroxysmal dyskinesia and developmental delay, Movement Disord 30: (9) ((2015) ), 1290–1292. |
[7] | Xia X.M. , Fakler B. , Rivard A. , Wayman G. , Johnson-Pais T. , Keen J.E. , Ishii T. , Hirschberg B. , Bond C.T. and Lutsenko S. , Mechanism of calcium gating in small-conductance calcium-activated potassiumchannels, Nature 395: (6701) ((1998) ), 503–507. |
[8] | Xia X.M. , Zeng X. and Lingle C.J. , Multiple regulatory sites in large-conductance calcium-activatedpotassium channels, Nature 418: (6900) ((2002) ), 880–884. |
[9] | Sausbier M. , Hu H. , Arntz C. , Feil S. , Kamm S. , Adelsberger H. , Sausbier U. , Sailer C.A. , Feil R. and Hofmann F. , Cerebellar ataxiaand Purkinje cell dysfunction caused by Ca2+-activated K+channeldeficiency, Proc Natl Acad Sci U S A 101: (25) ((2004) ), 9474–9478. |


