
Sengers syndrome in Asian Indians – two novel mutations and variant phenotype-genotype correlation
Abstract
Sengers syndrome is an autosomal recessive mitochondrial disease comprising a tetrad of congenital cataract, hypertrophic cardiomyopathy, skeletal myopathy and lactic acidosis. Mutations in the AGK gene cause Senger syndrome. We describe two unrelated Asian Indian families with two novel mutations, c.909 G>A (p.Trp303Ter) and c.982G>T (p.E328Ter), in the AGK gene. Similar nonsense mutations have previously been reported with a severe phenotype and early infantile death, while this patient is doing well at 3 years, suggesting a mild phenotype.
The second child tested positive for two previously reported mutations, c.841C>T (p.Arg281Ter) and c.424-3C>G. The presence of a splice site mutation typically predicts a milder phenotype with one exception reported to date. The second patient we report died at 9 months of age adding to the one previously reported exception.
Both these cases add onto the scant literature of genotype phenotype correlation in Sengers syndrome. We emphasize the importance of diagnosis of this clinically recognizable syndrome to counsel families of recurrence risks and option of prenatal diagnosis.
1Introduction
Sengers syndrome (MIM 212350) is a rare autosomal recessive mitochondrial disorder characterized by congenital cataracts, hypertrophic cardiomyopathy, skeletal myopathy, lactic acidosis and normal cognition [3, 8, 14, 21]. About 40 patients with Sengers syndrome have been reported worldwide [6, 11, 15, 18, 23, 24]. The clinical course is variable with early death at the severe end of the spectrum and a late onset, slowly progressive phenotype with survival to the fourth decade at the milder end [6]. AGK gene converts diacylglycerol (DAG) and monoacylglycerol (MAG) to phosphatidic and lysophosphatidic acid which helps in phospholipid synthesis. Excess accumulation of DAG and MAG leads to formation of reactive oxygen species (ROS) and oxidative damage of organs containing mitochondria [10]. Dysfunction of acylglycerol kinase (AGK) results in increased DAG levels with increased stimulation of ROS detoxification pathways. Less formation of phosphatidic acids will cause changes in the lipid composition of the inner mitochondrial membrane, affecting respiratory chain complexes, the adenine nucleotide translocator and protein import [8, 22]. Two simultaneous publications identified the molecular basis of the disorder [11, 16].
Here we present two families in whom Sengers syndrome was confirmed on molecular testing. Two novel mutations were observed, while the phenotype-genotype correlation varied from that reported in the literature in both the patients.
Case 1
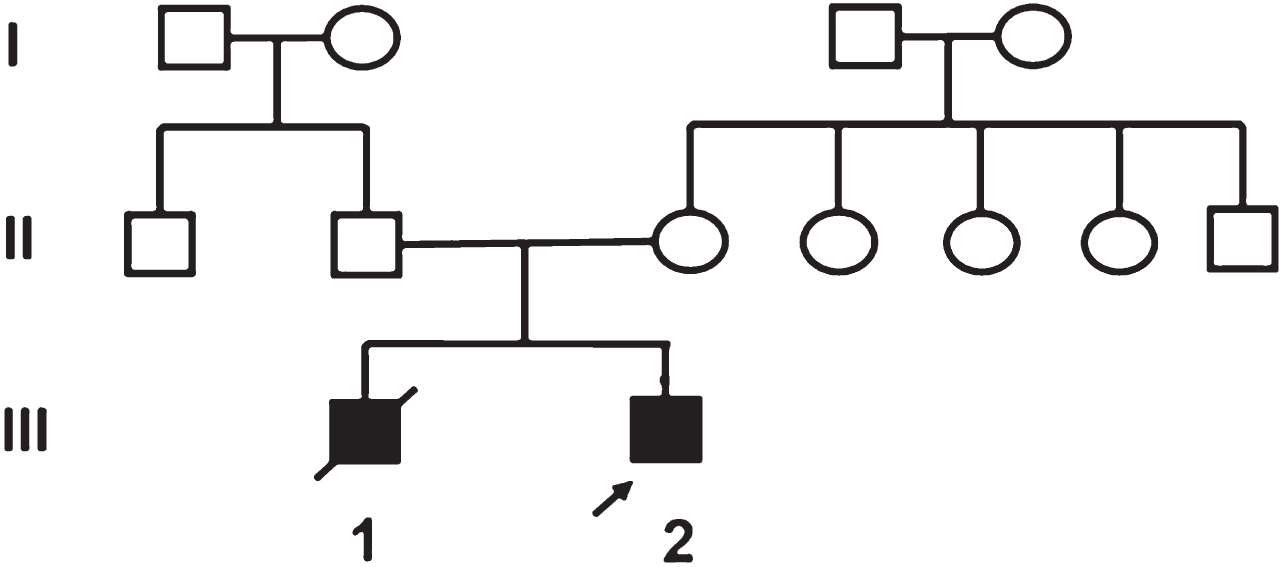
The first child (III 1) was born at term to non consanguineous parents. After a term, normal vaginal delivery, the neonate did not cry at birth. He was noted to have hydrocephalus and severe hypotonia. He remained in the neonatal intensive care unit till day 20 of life when he died without a definite diagnosis.
The proband (III 2) had an uneventful antenatal history. He was born at term with normal APGAR score. Bilateral cataracts were noted on day 3 of life. During evaluation at 2 months of age for cataract surgery he was incidentally detected to have left ventricular hypertrophy. He had mild motor developmental delay. At the age of 5 months he presented to the genetic clinic with history of feeding difficulty and respiratory distress for past few days.
On examination, his weight was 7.5 kg (75th centile), length 68 cm (90th centile) and occipitofrontal circumference 40 cm (3rd centile). There was no facial dysmorphism. There was cataract in one eye and the second was aphakic. Neck holding was partial, he followed light and social smile was present. There was mild tachypnea. The liver was palpable 3 cm below the right costal margin. Neurological examination was normal. Investigations revealed normal hematological, renal and thyroid profiles. The creatine kinase level was 121 IU/L Blood lactate was elevated (56.80 mg/dl) and ammonia was normal (78.0 μmol/l). Urine gas chromatography/mass spectrometry (GC/MS) and blood tandem mass spectrometry (TMS) did not reveal any abnormal metabolites. The blood FGF-21, a marker of mitochondrial disease [13], was 766 pg/ml (normal<13.5). A panel of nuclear encoded mitochondrial genes were analysed by next generation sequencing. Two novel variations in AGK gene, [c.982G>T (p.E328Ter) in exon 14 and c.909G>A (p.W303Ter) in exon 13] were identified, consistent with the diagnosis of Sengers syndrome. These variations are not reported in 1000Genome [1] and ExAC [17] and are predicted to be disease causing by MutationTaster [2]. These variations were present in a region of the gene that is conserved in mammals. Based on the standard guidelines for interpretation of sequence variants, they are predicted to be pathogenic [20].
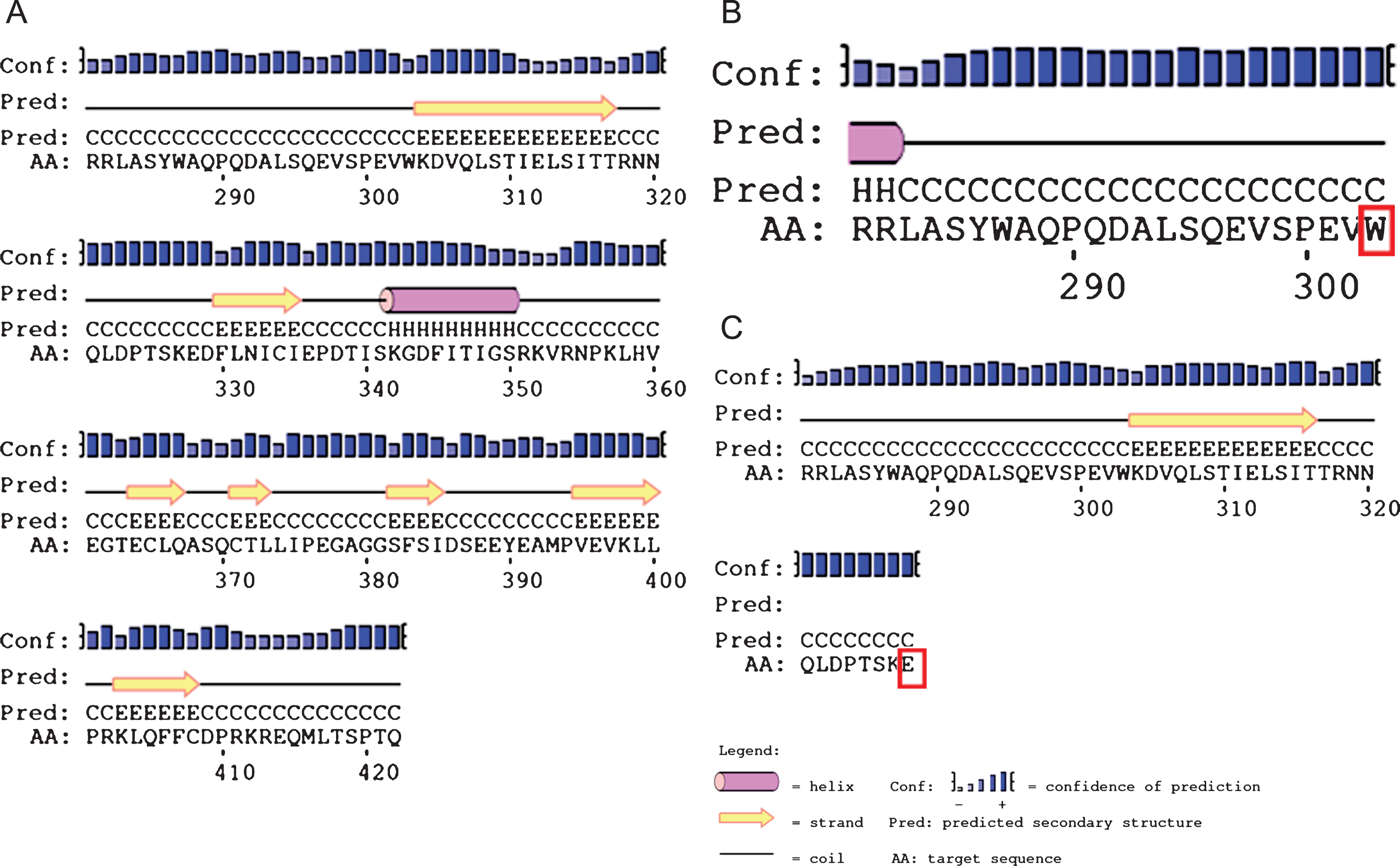
Protein structure of the AGK is still not identified and prediction of secondary structure of AGK protein sequence was done by using PSIpred software [12]. PSIpred predicted the loss of functional protein due to both truncating mutations at aminoacids 303 and 328. Results are shown inFig. 1.
Fig.1
Secondary structure prediction of AGK protein sequence by PSIpred in patient 1 with c.909G>A (p.Trp303Ter) and c.982G>T (p.E328Ter) mutations. The amino acids 281–422, 281–303, 281–328 have been shown in sections A, B and C respectively. Section A represents wild type 422 amino acid protein. Section B and C represents no further formation of protein due to non sense mutation at 303 and 328 amnioacid which are highlighted in red boxes. H, Helix; E, strand; C, coil.
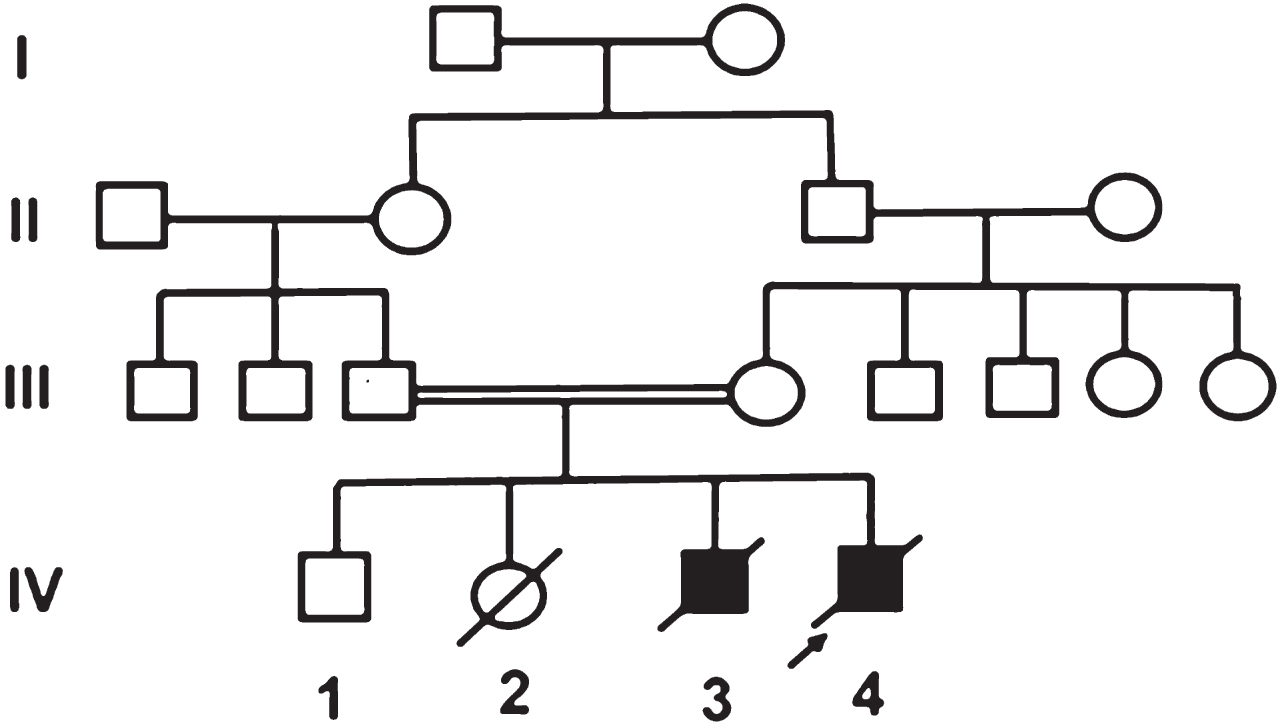
The first child is alive and healthy. The second baby was reported to have died on day 2 of life due to aspiration. The third child was detected to have cataracts at birth and subsequent developmental delay with failure to thrive. He died undiagnosed at 18 months of age.
The proband (IV 4), was a 5-month-old boy, born to consanguineous parents of Asian Indian origin, with bilateral cataracts, developmental delay and failure to thrive. On clinical examination the proband had failure to thrive with sunken eyes and growth parameters <3rd centile (head circumference 40 cm; weight 4865 g). Except for sparse scalp hair there was no other dysmorphism. Roving movements of eyes and bilateral cataract were present. The neurological examination was normal except for exaggerated deep tendon reflexes in all limbs. The liver was just palpable. Clinical examination of the cardiovascular and respiratory system was normal.
His complete blood count, liver function tests, arterial blood gas, creatine kinase, echocardiography, brain magnetic resonance imaging and magnetic resonance spectroscopy were normal. The urine metabolic screen, TMS and high performance liquid chromatography (HPLC) of urine for amino acids were normal as was screening for galactosemia, peroxisomal disorders and congenital disorders of glycosylation. A raised serum lactate of 46 mg/dl and elevated lactate, fumarate and ketones in the urine GC/MS suggested a mitochondrial pathology.
As analysis of respiratory chain enzyme is not available in India, exome sequencing was performed. This identified two previously reported pathogenic variations, c.424 -3C>G and c.841C>T(p.R281*) in the AGK gene shown in Fig. 2. This confirmed the diagnosis of Sengers syndrome.
Fig.2
Gene structure of AGK gene and localisation of the mutations in both the cases, novel mutations in case 1 are highlighted in blue and in case 2 are highlighted in red colour. Reference: Haghighi et al. [9].
![Gene structure of AGK gene and localisation of the mutations in both the cases, novel mutations in case 1 are highlighted in blue and in case 2 are highlighted in red colour. Reference: Haghighi et al. [9].](https://content.iospress.com:443/media/trd/2017/2-3-4/trd-2-3-4-trd017/trd-2-trd017-g002.jpg)
2Discussion
Molecular basis of Sengers disease was first identified in 2012 and since then, only around 40 families have been diagnosed with Sengers syndrome to the best of our knowledge [7, 11]. After the review of published literature related to Sengers syndrome, we found that milder phenotypes of the disease have at least one splice site or start codon mutation as compared to a severe presentation and early death in patients with biallelic nonsense mutations [11].
We report two patients, with affected siblings, from two unrelated families, with causative mutations in the AGK gene. The first child, presented with the phenotype of cardiomyopathy, cataracts and elevated lactate, he is currently 3 years old and stable inspite of having two nonsense mutations while the second child presented with bilateral cataract and failure to thrive and died in infancy and had one splice site mutation.
Both our cases are contrary to usual clinical phenotype severity and mutations.
Biallelic nonsense mutations in the AGK gene present as the severe, infantile phenotype. They manifest within the first few months of life and do not survive beyond their first birthday.
Table 1 compares phenotype of the proband in Family 1 and other similar patients reported in literature [11, 16]. Case 1 was compound heterozygous for two novel loss of function, nonsense mutations in the AGK gene. Growth failure, reported in these patients, within the spectrum of a mitochondriopathy typically occurs due to disruption of ATP generation with the OXPHOS pathway [3]. However, growth parameters were age appropriate in the index case 1 despite severe protein truncation mutations. The child is 3 years old, developing well with relatively age appropriate development and stable cardiac status. He has been operated for the bilateral cataract. This could partly be due to early recognition of the multisystem phenotype, appropriate referral and timely molecular diagnosis that has helped to plan appropriate symptomatic, multidisciplinary management. However, we assume some modifying factors for this mild phenotype inspite of having truncating mutations which needs to be explored.
Table 1
Comparison of clinical features in patients with biallelic nonsense mutations
| Cases | Sex | Mutation | Type | Status | CM | Cataract/Age | MD | LA | RD | Others |
| Case 1 | M | c.909G>A (p.W303*) c.982G>T (p.E328*) | Nonsense Nonsense | Alive 3 years | 2 months | Birth | – | + | + | – |
| PRC1 [16] | M | c.306C>T (p.Tyr102*), c.841C>T (p.Arg281*) | Nonsense Nonsense | Died 18 days | <1 yr | + | + | |||
| PRC2 [11] | F | c.409C>T (p.Arg137*) | Nonsense (Homozygous) | Died 3 months | Age | + | Esotropia, IUGR, feeding issues, Raised MMA and carnitines | |||
| PRC 3 [11] | M | c.409C>T (p.Arg137*) | Nonsense (Homozygous) | Died 6 months | Birth | Birth | + | + | + | Esotropia, nystagmus, floppy infant |
| PRC4 [22] | F | c.979A>T (p.Lys3278*) | Nonsense (Homozygous) | Died 5 months | Birth | Birth | Upper respiratory tract infection | |||
| PRC5 [22] | F | c.979A>T (p.Lys327*) | Nonsense (Homozygous) | Died 12 days | Birth | Birth | ||||
| PRC6 [22] | F | c.979A>T (p.Lys327*) | Nonsense (Homozygous) | Died 2 days | Birth | Birth | ||||
| PRC7 [22] | M | c.979A>T (p.Lys327*) | Nonsense (Homozygous) | Died 18 days | Birth | Birth | ||||
| PRC8 [7] | F | c.412C>T (p.R138*) | Died 3 months | Birth | Birth | + | + | + | Severe hypotonia |
Abbreviations: CM – Cardiomyopathy, F-Female, LA-Lactic acidosis, M – Male, MD-Motor delay, PRC – Previously reported cases, RD – Respiratory distress.
Table 2 compares the clinical features of reported patients and those in this study who harbour the one splice site and nonsense mutations. Both mutations (424-3C>G and c.841C>T) in the proband of family 2 were previously reported [4, 16].
Table 2
Comparison of clinical features in patients with splice site and nonsense mutations
| Cases | Sex | Mutation | Type | Status/age | CM | Cataract/age | MD | LA | RD | Others |
| Case 2 | M | c.424-3C>G c.841C>T (p.R281*) | Splice site Nonsense | Died/9 months | – | Day 8 | + | + | – | Thin sparse hair, nystagmus, failure to thrive |
| PRC 1 [16] | F | c.221+1G>A c.1213C>T (p.Gln405*) | Splice site Nonsense | Alive/12 yrs | + | Birth | + | + | – | – |
| PRC2 [5] | F | c.297+2T>C (p.Lys75Glnfs*12) c.1170T>A (p.Tyr390*) | Splice site Nonsense | Died/18 yrs | + | <1 yr | – | + | – | Recurrent headaches, osteopenia, premature ovarian failure |
| PRC3 [11] | F | c.297+2T>C (p.Lys75Glnfs*12), c.841C>T (p.Arg281*) | Splice site Nonsense | Alive 10 yrs | Birth | Birth | + | – | – | Cervical meningocele |
Abbreviations: CM-Cardiomyopathy, F-Female, LA-Lactic acidosis, M-Male, MD-Motor delay, PRC-Previously reported cases, RD-Respiratory distress.
Though both mutations predict a loss of function for the protein, the presence of one AGK splice site mutation usually favours a milder phenotype with longer survival [11]. This splice site mutation skips exon 8 resulting in a frameshift, premature truncation of the protein but a small amount of normally spliced transcript can still be formed. Aldahmesh et al. report three patients with homozygous 424-3C>G mutations in AGK gene with an isolated non syndromic congenital cataract, describing the mildest phenotype associated with mutations in AGK gene [4].
In contrast, our patient presented with a severe mitochondrial energy deficient phenotype and died within the first year of life. The absence of cardiomyopathy typically seen in the severe phenotype could be explained as a function of the previously reported mild phenotype with c.424-3C>G mutation due to presence of some amount of the normal protein [11]. There are only three previously reported patients with biallelic nonsense and splice site mutations [5, 11, 16]. They presented with a combination of cardiomyopathy, cataract and skeletal muscle weakness. However unlike the severe phenotype observed in our patient, all had milder presentations with one patient who died at 18 yrs and the other two are alive at 10 and 12 yrs. More literature is needed to explore the genotype phenotype in this combination of mutations. This child in addition was noted to have thin sparse hair and dry and rough skin that has not previously been reported. This could be related to the failure to thrive present in the child. The sibling (III 3) of family 2 had similar manifestations and died at 18 months. It is possible that the neonatal death in this family could be an undetected, severe lactic acidosis with cardiorespiratory collapse previously described in Sengers disease [7].
Sengers syndrome is an autosomal recessive mitochondrial disorder due to biallelic mutations in the nuclear-encoded mitochondrial gene. There is no definitive treatment [19] and families need to be counselled of the recurrence risk of 25% in each conception. Once the mutations are identified in the proband, prenatal testing at 11 weeks gestation in a chorionic villi sample is an option that can be discussed with the families. Recognition of the typical phenotype and molecular studies to confirm the diagnosis is imperative for appropriate management and genetic counselling [9].
References
[1] | A global reference for human genetic variation, The 1000 Genomes Project Consortium, Nature 526: ((2015) ), 68–74. 10.1038/nature15393 |
[2] | Adzhubei I.A. , Schmidt S. , Peshkin L. , et al., Nat Methods 7: ((2010) ), 248–249. |
[3] | Ohtake A. , Murayama K. , Mori M. , et al., Diagnosis and molecular basis of mitochondrial respiratory chain disorders: Exome sequencing for disease gene identification, Biochim Biophys Acta 1840: ((2014) ), 1355–1359. |
[4] | Aldahmesh M.A. , Khan A.O. , Mohamed J.Y. , Alghamdi M.H. and Alkuraya F.S. , Identification of a truncation mutation of acylglycerol kinase (AGK) gene in a novel autosomal recessive cataract locus, Hum Mutat 33: ((2012) ), 960–962. |
[5] | Calvo S.E. , Compton A.G. , Hershman S.G. , et al., Molecular diagnosis of infantile mitochondrial disease with targeted next-generation sequencing, Sci Transl Med 4: ((2012) ), 118ra10. |
[6] | Cruysberg J.R. , Sengers R.C. , Pinckers A. , Kubat K. and van Haelst U.J. , Features of a syndrome with congenital cataract and hypertrophic cardiomyopathy, Am J Ophthalmol 102: ((1986) ), 740–749. |
[7] | Kor D. , Yilmaz B.S. , Horoz O.O. , et al., Two novel mutations in the AGK gene: Two case reports with sengers syndrome, Gene Technol 5: ((2016) ), 1. |
[8] | Di Rosa G. , Deodato F. , Loupatty F.J. , Rizzo C. , Carrozzo R. , Santorelli F.M. , Boenzi S. , D’Amico A. , Tozzi G. and Bertini E. , Hypertrophic cardiomyopathy, cataract, developmental delay, lactic acidosis: A novel subtype of 3 methylglutaconic aciduria, J Inherit Metab Dis 29: ((2006) ), 546–550. |
[9] | Emery and Rimoin’s Principles and Practice of Medical Genetics, Emery A.E. , Korf B.R. , Rimoin D.L. , Connor J.M. and Pyeritz R.E. , Published by Churchill Livingstone, Rumford, ME, U.S.A, ((2002) ). |
[10] | Haack T.B. , Haberberger B. , Frisch E.M. , et al., Molecular diagnosis in mitochondrial complex I deficiency using exome sequencing, H J Med Genet 49: ((2012) ), 277–283. |
[11] | Haghighi, et al., Sengers syndrome: Six novelmutations in seven new families and review of the phenotypic and mutational spectrum of 29 patients, Orphanet Journal of Rare Diseases 9: ((2014) ), 119. |
[12] | Jones D.T. , Protein secondary structure prediction based on position-specific scoring matrices, J Mol Biol 292: ((1999) ), 195–202. |
[13] | Jia K. , Zhenga J. , Lva J. , et al., Skeletal muscle increases FGF21 expression in mitochondrial disorders to compensate for energy metabolic insufficiency by activating the mTOR-YY1-PGC1α pathway, Free Radic Biol Med 84: ((2015) ), 161–170. |
[14] | Lalived’Epinay S. , Rampini S. , Arbenz U. , Steinmann B. and Gitzelmann R. , Infantile cataract, hypertrophic cardiomyopathy and lactic acidosis following minor muscular exertion— a little known metabolic disease, Klin Monbl Augenheilkd 189: ((1986) ), 482–485. |
[15] | Atiq M. , Iqbal S. and Ibrahim S. , Sengers disease: A rare association of hypertrophic cardiomyopathy and congenital cataracts, Indian J Pediatrics 71: ((2004) ), 437–440. |
[16] | Mayr J.A. , Haack T.B. , Graf E. , et al., Lack of the mitochondrial protein acylglycerol kinase causes Sengers syndrome, Am J Hum Genet 90: ((2012) ), 314–320. |
[17] | Lek M. , Karczewski K.J. , Minikel E.V. , et al., Analysis of protein-coding genetic variation in 60,706 humans, Nature 536: ((2016) ), 285–291. |
[18] | Morava E. , Sengers R. and Ter Laak H. , et al., Congenital hypertrophic cardiomyopathy, cataract, mitochondrial myopathy and defective oxidative phosphorylation in two siblings with Sengers like syndrome, Eur J Pediatr 163: ((2004) ), 467–471. |
[19] | Parikh S. , Saneto R. , Falk M.J. , et al., A Modern approach to the treatment of mitochondrial disease, Current Treatment Options in Neurology 11: ((2009) ), 414–430. |
[20] | Richards S. , et al., Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the american college of medical genetics and genomics and the association for molecular pathology, Genetics in Medicine: Official Journal of the American College of Medical Genetics ((2015) ) 17: (5), 405–424. |
[21] | Sengers R.C. , Trijbels J.M. , Willems J.L. , Daniels O. and Stadhouders A.M. , Congenital cataract and mitochondrial myopathy of skeletal and heart muscle associated with lactic acidosis after exercise, J Pediatr 86: ((1975) ), 873–880. |
[22] | Siriwardena K. , Mackay N. , Levandovskiy V. , Blaser S. , Raiman J. and Kantor P.F. , Mitochondrial citrate synthase crystals: Novel finding in Sengers syndrome caused by acylglycerol kinase (AGK) mutations, Mol Genet Metab 108: ((2013) ), 40–50. |
[23] | Smeitink J.A. , Sengers R.C. , Trijbels J.M. , et al., Fatal neonatal cardiomyopathy associated with cancer and mitochondrial myopathy, Eur J Pediatr 148: ((1989) ), 656–659. |
[24] | Van Ekeren G.J. , Stadhouders A.M. , Smeitink J.A. and Sengers R.C. , A retrospective study of patients with the hereditary syndrome of congenital cataract, mitochondrial myopathy of heart and skeletal muscle and lactic acidosis, Eur J Pediatr 152: ((1993) ), 255–259. |


