
Disorders of metal metabolism
Abstract
Trace elements are chemical elements needed in minute amounts for normal physiology. Some of the physiologically relevant trace elements include iodine, copper, iron, manganese, zinc, selenium, cobalt and molybdenum. Of these, some are metals, and in particular, transition metals. The different electron shells of an atom carry different energy levels, with those closest to the nucleus being lowest in energy. The number of electrons in the outermost shell determines the reactivity of such an atom. The electron shells are divided in sub-shells, and in particular the third shell has s, p and d sub-shells. Transition metals are strictly defined as elements whose atom has an incomplete d sub-shell. This incomplete d sub-shell makes them prone to chemical reactions, particularly redox reactions. Transition metals of biologic importance include copper, iron, manganese, cobalt and molybdenum. Zinc is not a transition metal, since it has a complete d sub-shell. Selenium, on the other hand, is strictly speaking a nonmetal, although given its chemical properties between those of metals and nonmetals, it is sometimes considered a metalloid. In this review, we summarize the current knowledge on the inborn errors of metal and metalloid metabolism.
1Copper homeostasis
The human body contains approximately 100 mg of copper, of which 35 mg is in muscle, 20 mg in brain, 10 mg in connective tissue, 5 mg in kidney, and 10 mg in blood [1, 2]. Copper absorption and excretion are normally 1 to 5 mg daily (Table 1), with a net balance of zero. The alimentary system generally achieves this balance through intestinal absorption and biliary excretion.
Table 1
Copper balance and copper and ceruloplasmin values in health and disease
| Normal | Wilson Disease | Menkes Disease | |
| Copper dietary intake (mg/day) | 5 | 5 | 5 |
| Copper intestinal absorption (mg/day) | 2 | 2 | 0.1–0.2 |
| Copper biliary excretion (mg/day) | 2 | 0.2–0.4 | Not known |
| Copper urinary excretion (μg/day) | 15–60 | 100–1000 | Increased |
| Copper balance | 0 | Positive | Negative |
| Serum copper (mg/L) | 0.75–1.45 | 0.19–0.63 | <0.70 |
| Serum ceruloplasmin (mg/L) | 180–360 | 0–200 | <50 |
| Liver copper (μg/g dry weight) | 70–140 | 200–3000 | 10–20 |
| Duodenal copper (μg/g dry weight) | 7–29 | ? | 50–80 |
| Adapted from Danks DM [1] and Cullotta VC &Gitlin JD [2]. | |||
Between 80 and 95% of plasma copper is bound to ceruloplasmin, an alpha-glycoprotein with amine oxidase and ferroxidase activities; the process of ceruloplasmin binding copper occurs in the liver shortly after hepatic uptake of copper. Other copper-containing enzymes are listed in Table 2 and include tyrosine hydroxylase and dopamine hydroxylase, which produce neurotransmitters in the central nervous system, and cytochrome c oxidase, critical for mitochondrial electron transport and energy production. The major copper storage protein is metallothionein, which binds a variety of heavy metals extremely tightly and whose promoter is very responsive to induction by heavy metals [3].
Table 2
Copper-containing enzymes
| Enzyme | Function | Result of Deficiency |
| Ceruloplasmin | Ferroxidase | Anemia |
| Diamine oxidases | Degrade histamine | Increased histamine response |
| Superoxide dismutase | Free radical detoxification | Decreased protection against oxygen free radicals |
| Cytochrome c oxidase | Electron transport chain | Decreased energy in muscle, neurons; impaired myelination |
| Tyrosinase | Melanin production in skin | Reduced pigmentation |
| Dopamine-β-hydroxylase | Dopamine production in neurons | Reduced neurotransmitters |
| Catecholamine production | Neurologic effects; temperature instability pupillary constriction | |
| Lysyl oxidase | Cross-linking of collagen and elastin | Arterial abnormalities; bladder diverticulae; lax skin and joints |
| Peptidylglycine α-aminating monoxygenase | Removes carboxy-terminal glycine to activate neuroendocrine peptides | Reduced activity of gastrin, cholecystokinin, VIP, CRH, TRH, calcitonin, vasopressin, MSH |
| Sulfhydryl oxidase | Cross-linking of keratin | Pili torti |
| Ascorbate oxidase | Dehydroascorbate production | Skeletal demineralization |
| Adapted from Danks DM [1] and Kaler SG [22]. | ||
1.1Wilson disease
Wilson disease (hepatolenticular degeneration) is an inherited disorder that was first described by the neurologist Samuel Alexander Kinnier Wilson [4]. The clinical manifestations of this disorder result from an excessive accumulation of copper, first in the liver and then in the brain [5]. The disease has an autosomal recessive mode of inheritance with a prevalence of 1 in 30000 live births and a prevalence of the heterozygote-carrier state slightly higher than 1 in 100. With increased rates of consanguinity, as occurs in certain isolated communities in Japan, Sardinia, and the Middle East, the frequency of the gene for Wilson disease can be as high as 2% to 3%. Heterozygotes do not have any clinical manifestations, although approximately 20% have low levels of ceruloplasmin and serum copper[6, 7].
The Wilson disease gene, ATP7B on chromosome 13q14.3, encodes a cation-transporting, P-type adenosine triphosphatase (ATPase) [8–10]. The gene contains 21 exons. Its mutations number over 700 [11]; more than half of the disease-causing mutations are missense changes, the most common being H1069Q [12]. In addition, the mutation R778L occurs in 30% of Asians with Wilson disease [13–15]. There appears to be some genotype-phenotype correlation [16–20].
The ATP7B protein functions in the trans-Golgi region of hepatocytes, where it is responsible for transporting copper into the secretory system. The Long Evans Cinnamon rat serves as a bone fide model for human Wilson disease, since its mutations occur in the rat orthologue of ATP7B.
The basic defect in Wilson disease involves failure to excrete copper in the bile for elimination, and failure to incorporate copper into its main circulating binding protein, ceruloplasmin. As a consequence, copper builds up in the liver, exceeds the binding capacity of metallothionein, overflows into the circulation bound to amino acids (especially cysteine), and deposits in susceptible tissues. Hence, levels of hepatic copper are elevated and serum ceruloplasmin and copper levels are decreased.
1.1.1Clinical characteristics
Although the biochemical defect that leads to the accumulation of copper in Wilson disease is present at birth, clinical symptoms typically appear for the first time in older children, adolescents, and young adults. Most patients present with hepatic [23, 24] or neurologic dysfunction; tremors and ataxia reflect basal ganglia involvement, which can be seen as metal accumulation on imaging of the brain [25, 26]. The brain MRI and MRS findings in Wilson disease have been reported [27, 28]. Electroencephalographic (EEG) changes are rarely helpful in diagnosis of neurologic cases.
Psychiatric, hematologic, renal, musculoskeletal, cardiac, ophthalmologic, endocrinologic, and dermatologic presentations have all been described in Wilson disease, including acute hemolytic crisis, joint symptoms, renal stones, renal tubular acidosis, pancreatic disease, cardiomyopathy, and hypoparathyroidism [5]. Renal manifestations include elements of renal tubular malfunction; some have the full picture of the Fanconi syndrome, including aminoaciduria, glycosuria, acidemia, rickets, and poor growth. Copper overload has deleterious effects on the heart; many patients with Wilson disease have electrocardiographic abnormalities, including conduction disturbances [29]. Anemia (normochromic) is not common except when there is hemolysis, but thrombocytopenia (50% of cases) and neutropenia (30% of cases) are found frequently [30].
Up to 30% of patients with Wilson disease present with clinical, biochemical, and histologic features similar to those of other forms of chronic hepatitis [23, 31]. Therefore, biochemical screening for Wilson disease should be performed in all patients under the age of 40 years in whom the clinical or histologic findings are consistent with chronic hepatitis and in whom autoimmune and viral disorders have been eliminated as diagnoses. In some cases, the coagulopathy associated with the hepatic failure of Wilson disease can interfere with obtaining a liver biopsy.
Patients with fulminant Wilson disease can be clinically indistinguishable from those with virus-induced or drug-induced hepatic necrosis [24]. Viral illnesses may occur immediately preceding the onset of acute hepatitis [32], raising the possibility that a patient’s hepatic decompensation may be triggered by a viral infection. The sudden release of copper from the liver into the blood can cause massive hemolysis, as copper is subsequently taken up by erythrocytes, causing oxidative damage to cellular membranes. Acute hemolysis in Wilson disease is associated with hepatic damage, renal insufficiency, and death [33].
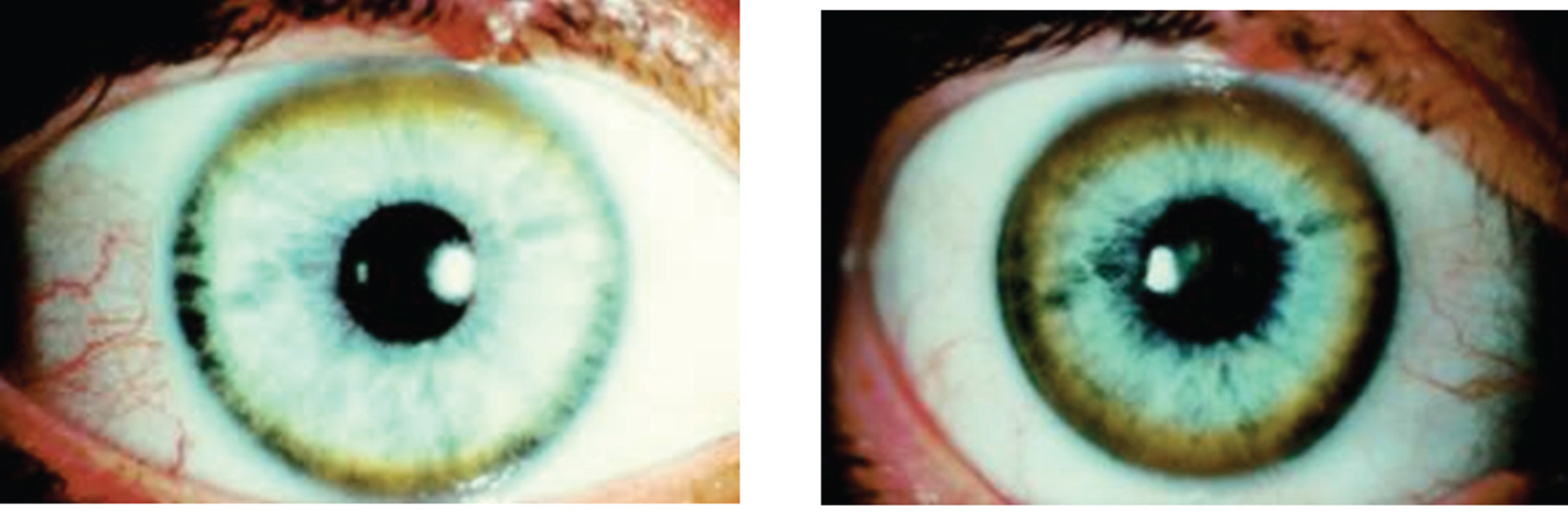
The Kayser-Fleischer ring (Fig. 1) reflects the corneal accumulation of copper that has entered the circulation from the liver. Kayser-Fleischer rings are golden-brown or greenish discolorations of the limbic region of the cornea caused by copper deposition in the Descemet membrane; they occur in patients with symptomatic Wilson disease, particularly those with neurologic manifestations [34]. The presence of these rings, however, is not pathognomonic of Wilson disease, since they are occasionally observed in patients with other cholestatic hepatic diseases. Conversely, the rings are absent in many patients with Wilson disease, even those with fulminant hepatitis [24]. Sunflower cataracts occur in a smaller proportion of cases (15% –20%); both of these ocular signs improve with effective chelation therapy [1].
Fig.1
Appearance of Kayser-Fleischer ring in Wilson disease. Dark pigment at periphery of the cornea is due to deposition of copper.
1.1.2Diagnosis
The diagnosis of Wilson disease requires a clinical suspicion combined with laboratory evidence [35]. Clinical findings may include hepatic, neurological, or renal abnormalities, and possibly Kayser-Fleischer rings. Laboratory findings generally include elevated urinary copper levels [24] and low serum copper and ceruloplasmin. However, in 5–15% of Wilson disease patients, the serum ceruloplasmin values may be normal. In these cases, the ceruloplasmin, measured as the apoprotein, may not bind copper appropriately, or may be increased because of hepatic inflammation, since ceruloplasmin is an acute phase reactant [23, 31]. Liver copper is usually elevated in Wilson disease, but hepatic copper values may be normal because of the massive release of hepatic copper stores.
One noninvasive and reasonably definitive test is measurement of urinary copper after a penicillamine challenge [36]. Urinary excretion of 1000 to 2000 mg copper within 24 hours after oral administration of 1 g penicillamine is diagnostic [6]. Alternatively, molecular diagnostics can be performed for mutations in the ATP7B gene [5].
Wilson disease cannot be ruled out on the basis of any single laboratory test.
1.1.3Pathology
The pathologic effects on the liver, kidneys, and brain are directly related to the accumulation of copper ions. Four pathophysiologic phases are acute hepatitis, fulminant hepatitis, chronic active hepatitis, and cirrhosis.
In the precirrhotic stage, the changes resemble a chronic, active hepatitis with focal necrosis, scattered acidophilic bodies, and moderate to marked steatosis [37]. Glycogenated nuclei in periportal hepatocytes are a typical finding. Kupffer cells are hypertrophied and may contain hemosiderin. Submassive or massive hepatocellular necrosis may occur; the clinical and biochemical findings in such cases may resemble those of chronic active hepatitis [4, 23, 37]. In later stages, periportal fibrosis, portal inflammation (Fig. 2), and finally cirrhosis occur. The presence of copper may not be cytochemically demonstrable in the precirrhotic stage. In young, asymptomatic patients, the copper is diffusely distributed in the cytoplasm but later accumulates in lysosomes [37]. Rhodamine and rubeanic acid stains are specific to detect the presence of copper (Fig. 3) [23]. Copper-associated protein can be stained with orcein or aldehyde fuchsin [38, 39]. Cirrhosis may be micronodular or macronodular (Fig. 4), or it may be mixed. Variable degrees of bile duct proliferation and inflammation have been reported [40]. Mallory bodies are frequently present in periportal hepatocytes [40, 41]. Single-cell necrosis and submassive or massive coagulative necrosis [40] may be seen. Tissue copper deposition, when present, is generally confined to the periportal zone in early stages of the disease but may be diffusely distributed by the time cirrhosis has developed [32, 40, 42].
Fig.2
Wilson disease. (A) Microscopic section of liver in inflammatory reaction in the portal area. The hepatocytes contain copper pigment. (B) High-power view showing Mallory body (arrow).
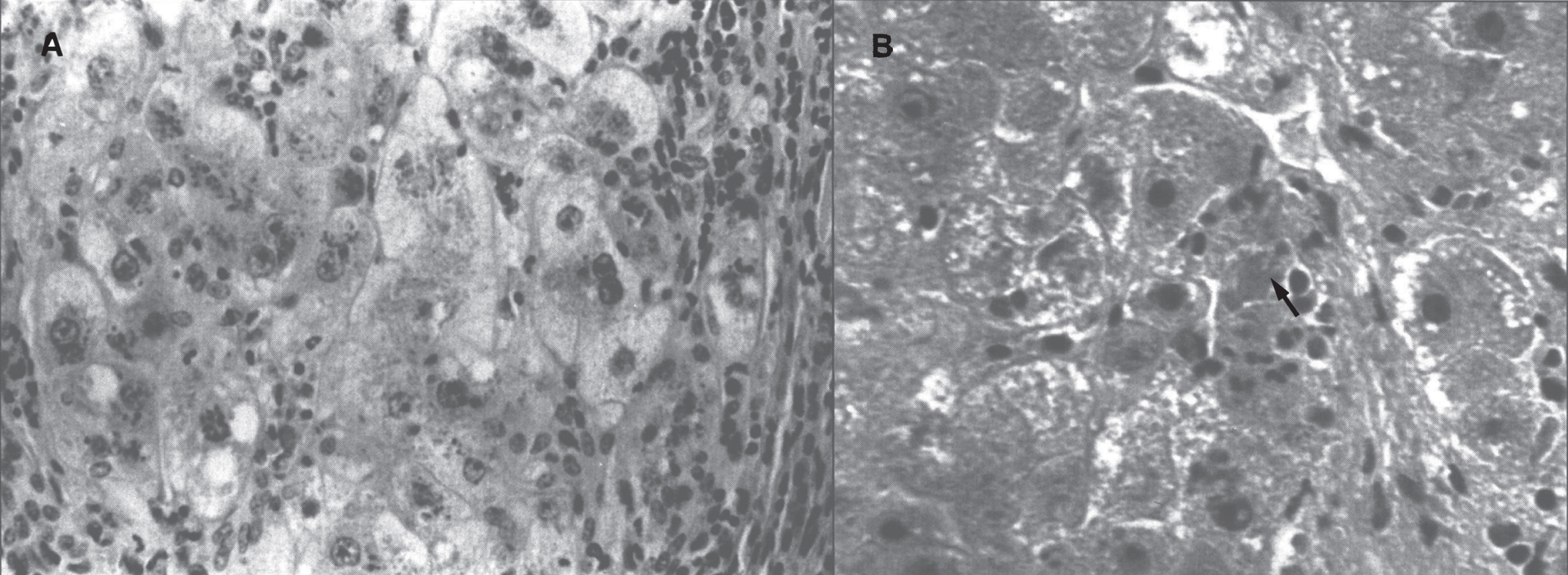
Fig.3
Microscopic section of liver in Wilson disease showing copper pigmentation of hepatocytes stained with Rhodamine stain.

Fig.4
Gross appearance of cirrhotic liver in Wilson disease.

Liver copper, typically measured by Inductively Coupled Plasma Optical Emission Spectroscopy (ICP-OES) or ICP-Mass Spectrometry (ICP-MS) may detect 10 to 35μg of copper per gram of dry liver weight in normal individuals. More than 250μg/g is diagnostic of Wilson disease, although lower cut-off values have been proposed [43–45].
Extensive sampling may be necessary to confirm the diagnosis of Wilson disease. Furthermore, increased hepatic copper, with levels as high as 250μg/g of dry weight, is seen in diseases other than Wilson disease, including chronic cholestatic diseases such as primary biliary cirrhosis and primary sclerosing cholangitis, Indian childhood cirrhosis, and copper-associated cirrhosis [42, 44].
On histologic examination, a negative copper stain of the liver does not rule out a diagnosis of Wilson disease [42], particularly during the initial stages of copper accumulation when the metal is distributed diffusely throughout the cytoplasm of the hepatocyte and may be undetectable with routine staining techniques [32, 38, 46, 47]. It is imperative to establish the correct diagnosis, since the morbidity and mortality rates are very high among untreated persons [23], and chelation therapy or liver transplantation frequently results in long-term survival.
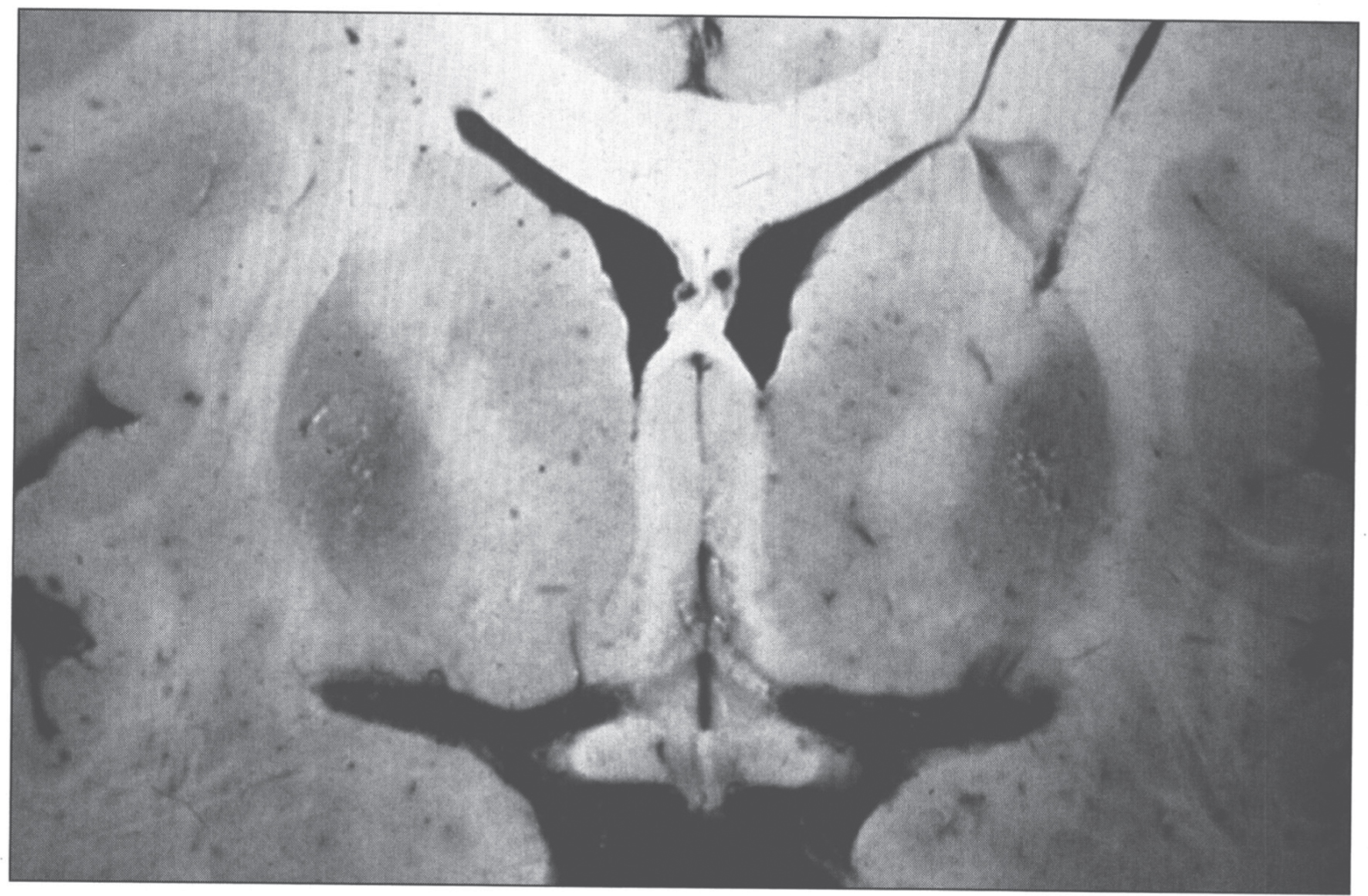
The pathologic changes in the brain (Fig. 5) are characterized by rarefactive spongy change progressing to cavitary necrosis with microcysts primarily in the putamen and rarely in neocortical layers V and VI. Severe white matter degeneration and neuronal loss may be seen. Pathological types of astroglia can be seen in the form of Alzheimer type I cells (mainly in the putamen) and Alzheimer type II cells (distributed diffusely). Opalski cells, round, large cells (>35μm) with a small nucleus and finely granular, eosinophilic cytoplasm, are characteristic of Wilson disease and are mainly found in the putamen [48, 49]. The brain has high concentrations of copper.
Fig.5
Wilson disease. Coronal section of brain showing small cystic cavitation and light brown discoloration in the basal ganglia, particularly in the putamen.
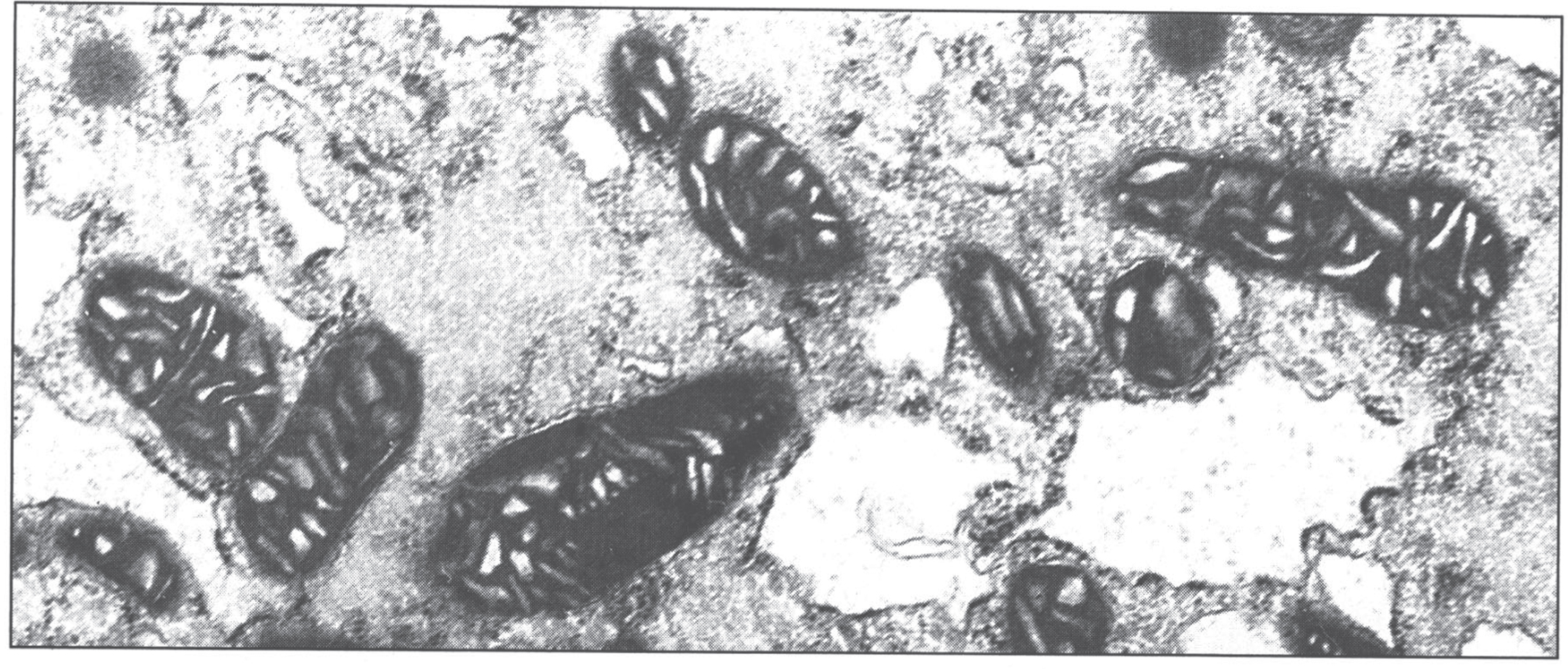
The ultrastructural changes are pathognomonic (Fig. 6) [50]. The mitochondria show marked pleomorphism: the cristae are wide and microcysts form at the tips of the cristae. Copper deposits are extremely electron-dense.
Fig.6
Electron micrograph of hepatocyte in Wilson disease. The mitochondria vary in size and shape, with dilated and microcystic cristae. (Courtesy Dr. Roma Chandra.)
The histologic differential diagnosis of Wilson disease includes diabetes mellitus, ethanol abuse, viral or autoimmune hepatitis, α1-antitrypsin deficiency, and rare idiosyncratic drug reactions, all of which can produce findings similar to those of Wilson disease. Overdoses of drugs such as acetaminophen [40] and ingestion of toxins such as carbon tetrachloride and tannic acid [36] may cause submassive necrosis with steatosis, as in Wilson disease.
1.1.4Treatment
Patients with acute hepatic failure from Wilson disease tend to be young and have a fulminant clinical course, with survival requiring acute management followed rapidly by liver transplantation. A prognosis index for Wilson disease patients with fulminant hepatitis is based on the serum bilirubin, aspartate aminotransferase, and prothrombin time [50, 51]. Hemolysis and renal insufficiency are grave prognostic signs [24]. Hepatic transplantation for Wilson disease is associated with a survival rate of approximately 72 to 79% at 5 years [52–54]. All transplant recipients have a complete reversal of the underlying defects in copper metabolism, and many have improvement in symptoms and signs [25].
Treatment of chronic or subacute Wilson disease involves lifelong chelation therapy with penicillamine [55] or trientine hydrochloride [56, 57]. Chelators may decrease pyridoxine, which should be added. An alternative approach to chelation is the administration of zinc, which induces metallothionein in the gastrointestinal mucosa. Dietary copper displaces the zinc, and subsequent sloughing of mucosal cells results in decreased copper absorption. This therapy is not as effective as chelation [58], and should be considered complementary to chelation therapy, except in children with mild liver disease [59].
During these medical therapies, serum copper levels should be monitored, since therapy may be inadequate or may result in copper deficiency [60–62]. If treatment regimens prove inadequate, liver transplantation should be considered [63, 64]. Gene therapy has been discussed for Wilson disease [65].
1.2Menkes disease
In 1962, John Menkes and colleagues from Columbia University in New York described five males from a single family with progressive neurologic deterioration after a few months of life, failure to thrive, and peculiar hair [66]. David Danks in Melbourne noticed that the hair of children with this condition was similar to the wool produced by sheep that grazed on grass from copper-depleted soil in Australia, where wool remained an important industry [67]. This led him to evaluate copper metabolism in 7 patients with Menkes disease, all of whom were found to have decreased serum copper and ceruplasmin levels [68]. Inherited as an X-linked recessive disorder of copper metabolism, Menkes disease is characterized by a defect in the transport and incorporation of copper into copper-containing enzymes [22, 66, 67, 69]. As a result, serum levels of copper and ceruloplasmin are low in affected male infants [67, 69], and all copper-containing enzymes (Table 2) have impaired activity. Copper levels are low in most tissues except the kidney and intestinal mucosa, with levels in liver remarkably decreased (Table 1) [67].
The incidence of Menkes disease is approximately 1 in 250,000 [69]. The Menkes disease gene, ATP7A, resides on chromosome Xq13.3 and contains 23 exons [10]. A wide variety of mutations have been described; most are private, with some genotype-phenotype correlation [70]. As for most X-linked disorders, one-third of cases are predicted to be new mutations. Full manifestation of the disease has been described in 23 girls [71–81].
The Menkes disease protein, ATP7A, is 55% identical to the Wilson disease protein, ATP7B [2]. ATP7A resides in the trans-Golgi network [82] and transports copper into the secretory vesicle system for incorporation into copper-containing enzymes [2]. The basic defect in Menkes disease prevents the transport of copper out of gastrointestinal mucosal cells and into the circulation; consequently, intestinal mucosal cells store large amounts of copper. Copper egress from cultured fibroblasts is impaired in Menkes disease [1, 2]. The mottled mouse is a model for Menkes disease [83].
1.2.1Clinical characteristics
The severity of Menkes disease is quite variable [84]; the clinical manifestations result from deficiency of different copper enzymes [67, 69]. Tyrosinase deficiency causes hypopigmentation of hair and skin pallor. Lysyl oxidase deficiency results in frayed and split arterial intima due to defective elastin and collagen cross-linking. Deficiency of cytochrome c oxidase causes hypothermia and ascorbate oxidase deficiency causes skeletal demineralization. Dopamine β-hydroxylase deficiency results in decreased catecholamine synthesis.
Infants with Menkes disease have a sparse, steel wool appearance to their hair, which is coarse and brittle; this phenomenon is called pili torti (Fig. 7). The hair becomes tangled, lusterless, and grayish or ivory-colored, with a stubble of broken hairs palpable over the occiput and temporal regions where the hair rubs on sheets. Microscopic examination of the hair will reveal the pili torti [1]. Menkes disease infants have pudgy cheeks, skeletal changes [85] including wormian bones, metaphyseal widening -particularly of the ribs and femora- and lateral spurs, progressive cerebral deterioration with seizures, and widespread arterial elongation and tortuosity [67, 69]. There can be metaphyseal fractures mimicking the classic metaphyseal lesion from child abuse. This, in combination with subdural hematomas, can lead to confusion with non-accidental trauma [86, 87].
Fig.7
Menkes syndrome child with typical coarse kinky hair.
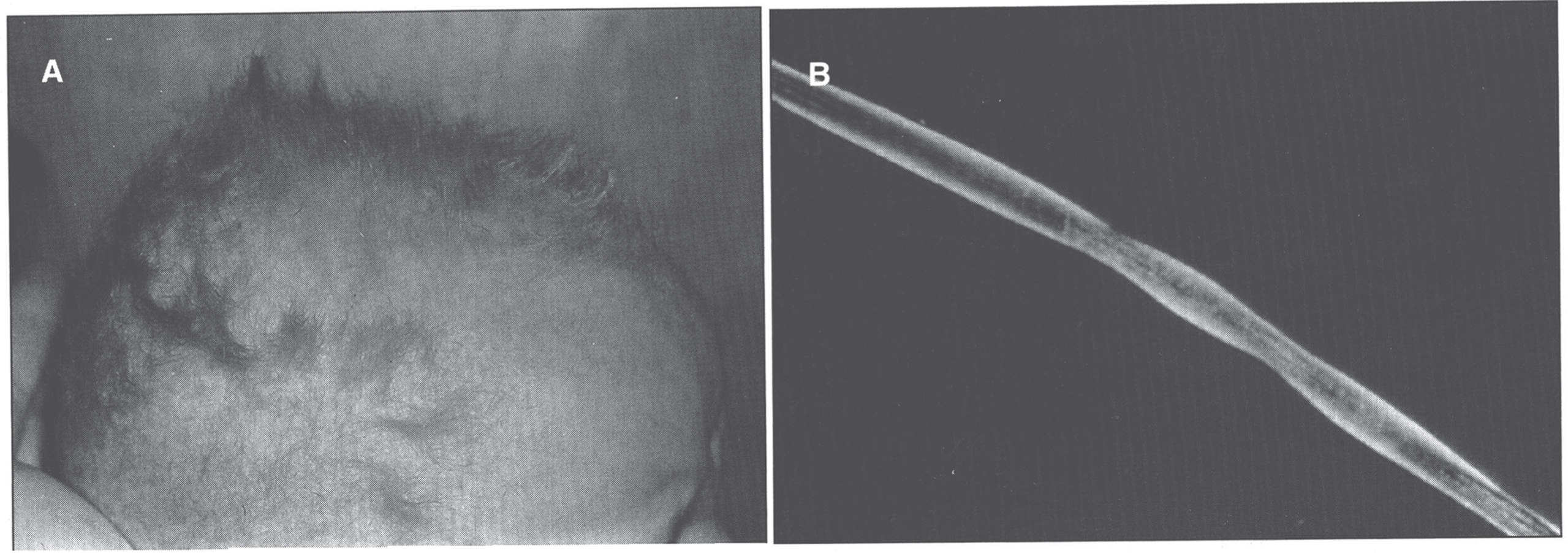
Many of the effects of Menkes disease are established in utero. Premature delivery is very frequent, as are neonatal hypothermia and hyperbilirubinemia. Hypothermia may also occur in older babies. In the neonatal period, the child’s growth and appearance can be normal, with fine hair and sometimes an unusual facies. Some Menkes disease babies, however, show early symptoms. By about 3 months of age, most affected infants develop flagrant symptoms of developmental delay, loss of early developmental skills, and seizures. Cerebral degeneration then dominates the clinical picture, with vascular complications, particularly subdural hematomas [1, 69]. Progressive neurologic degeneration leads to death in infancy. The clinical and laboratory manifestations of classic Menkes disease are shown in Table 3.
Table 3
Major clinicopathologic findings in menkes disease and related phenotypes
| Craniofacial | Coarse hair, with pili torti |
| Wormian bones, especially within lambdoidal sutures | |
| Occipital exostoses (in the occipital horn variant and some mild phenotypes) | |
| Brain | Cerebral and cerebellar neuronal cell loss (marked, in classic severe Menkes) |
| Demyelination | |
| Subdural hematoma | |
| Thorax | Pectus excavatum/carinatum |
| Rib fractures (typical in classic Menkes) | |
| Urinary tract | Massive bladder diverticula |
| Skin and joints | Generalized laxity |
| Vasculature | Generalized tortuosity/ectasia |
| Adapted from Kaler SG [22]. | |
About half of obligate heterozygotes for Menkes disease can be identified by studies of copper incorporation into fibroblasts obtained from a skin biopsy [88] or by examination of hair for pili torti [89]. Patchy suntanning can occur in white heterozygotes. Finding elevated placental copper levels seems to be a particularly good method of diagnosing heterozygotes when females are born into affected families [90].
1.2.2Diagnosis
As for Wilson disease, the diagnosis of Menkes disease relies upon a combination of clinical and laboratory findings [69, 91]. Clinical diagnostic tests are often characteristic [67]. Electroencephalograms are usually moderately to severely abnormal [92] although normal tracings have been recorded in some classically affected individuals [93]. Brain MRI findings typically include white matter abnormalities reflecting impaired myelination, tortuosity of cerebral blood vessels, and/or diffuse atrophy with ventriculomegaly [94]. Subdural hematomas are common in infants, and cerebrovascular accidents can occur in patients who survive longer. Cystography or pelvic ultrasound reveals diverticula of the urinary bladder in nearly every patient. Radiographs often disclose abnormalities of bone formation in the skull (wormian bones), long bones (metaphyseal spurring), and ribs (anterior flaring, multiple fractures) [95, 96].
Laboratory findings include low levels of serum copper and ceruloplasmin (Table 1). Serum copper and ceruloplasmin are unreliable as diagnostic markers during the first several weeks of life, since children under age 6 months normally have low serum levels of both markers [69]. Instead, one should rely upon concentrations of neurochemicals. Partial deficiency of dopamine hydroxylase, a critical enzyme in the catecholamine biosynthetic pathway, is responsible for a distinctively abnormal plasma and cerebrospinal fluid neurochemical pattern in Menkes patients [97, 98]. The ratio of a proximal compound in the pathway, dihydroxyphenylalanine (DOPA) –or dopamine metabolite dihydroxyphenylacetic acid (DOPAC)- to a distal metabolite dihydroxyphenylglycol (DHPG), provides a better index of DBH deficiency in Menkes patients than norepinephrine levels alone, as does the ratio of dopamine to norepinephrine [22, 99]. Definitive diagnosis can also be made based upon mutation analysis of ATP7A.
1.2.3Pathology
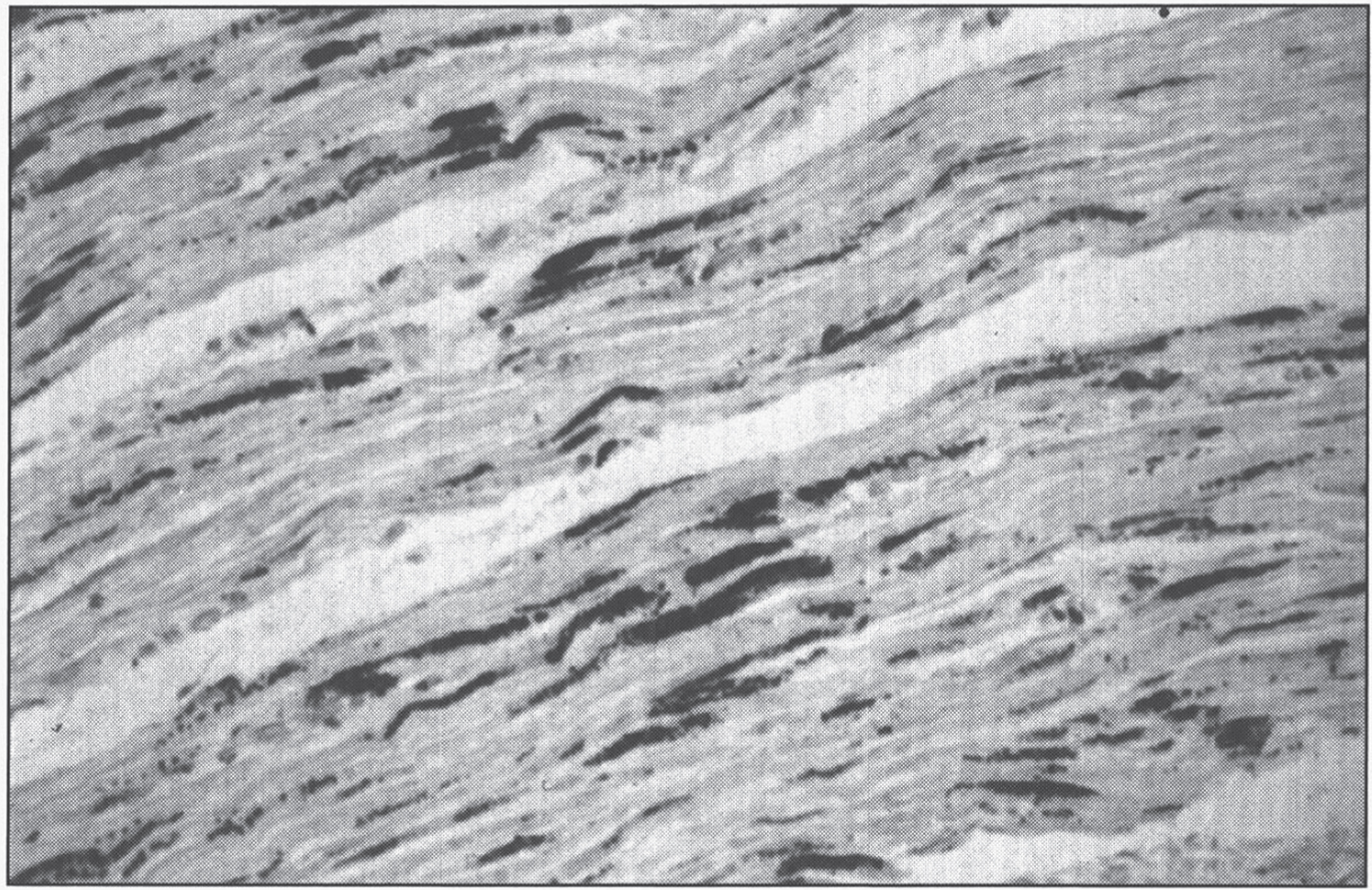
Pathologic changes involve the cardiovascular system. Developmental and degenerative changes are seen in the arteries with elongation, aneurysmal dilations, telangiectasia (Fig. 8), rupture, stenoses, and areas of intimal proliferation and thrombosis. Extreme fragmentation, disruption, and reduplication of the internal elastic lamina are seen microscopically (Fig. 9), especially in the large arteries [100]. Intimal changes are also seen. Aorta and skin ultrastructural changes suggest defective formation rather than disruption of tissue elastic fibers [101].
Fig.8
Section of cerebral white matter in Menkes disease showing telangiectatic capillary.
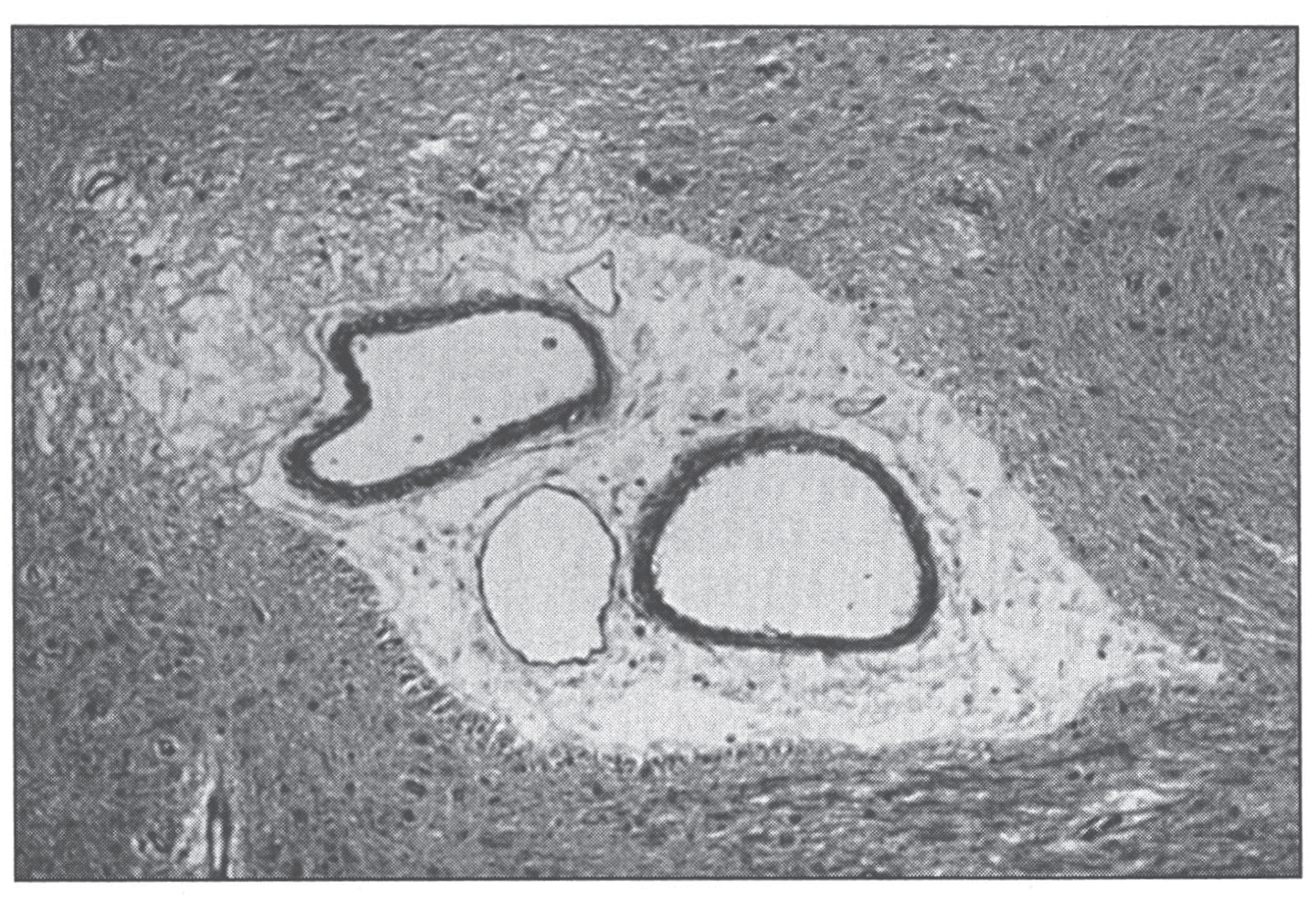
Fig.9
Menkes disease. (A) Microscopic section of the aorta showing disruption and piling up of elastic fibrils of internal elastic lamella. (B) Fluorescence photomicrograph of longitudinal section of peripheral nerve. Note torpedo-like swelling of intense fluorescence-positive axon. Inset shows cross section of peripheral nerve with nonspecific fluorescence of myelin sheath (formaldehyde-induced fluorescence). (Courtesy of Dr. Hideo Uno.)
There is widespread neuronal destruction in the cerebral cortex and the cerebellum, as well as associated gliosis [102–104]. Cerebellum changes are severe with neuronal loss in the granular cell layer and molecular layer. There is proliferation of the Bergmann glia [71, 105]. Many Purkinje cells are lost, and those that remain show several abnormalities: 1) heterotopia, or displacement into the granular layer; 2) somal sprouts, or short, radial dendrites emanating directly from the soma of the Purkinje cell; 3) cacti, or enlarged, swollen dendrites; 4) abnormal arborization of dendrites, with a weeping willow configuration (downward extension), or a Medusa head formation; 5) focal axonal swellings, either fusiform (torpedoes) or round (retraction bulbs); 6) confronting cisternae complexes, seen ultrastructurally as laminated structures with alternating 5–83 nm clear zones and 30 nm dense zones; enlarged, swollen mitochondria with tubule-vesiculated cristae and occasional glycogen accumulation, also seen on electron microscopy [66, 71,105–110].
Changes in the skeletal muscle (glycogen accumulation, mitochondrial disorganization, “ragged red fibers”) and in the iris and retina have been described, including progressive degeneration of ganglion cells, leading to loss of nerve fibers and optic atrophy, abnormal retinal pigment with small and irregular melanin granules, and abnormal elastica in Bruch’s membrane [111].
By formaldehyde-induced fluorescence for the identification of catecholamines, peculiar torpedo-like swellings of catecholamine-containing axons are seen in the peripheral nerve tracts, as are reduced numbers of adrenogenic fibers in the mid-forebrain [100, 112]. Deficient cytochrome c oxidase activity seems likely to be a major factor in the devastating brain damage associated with classic Menkes disease. Neuropathologic changes in Menkes patients are similar to those found in individuals with Leigh disease (subacute necrotizing encephalomyelopathy) in whom cytochrome c oxidase deficiency is caused by complex IV respiratory chain defects[113].
1.2.4Treatment
There is no effective treatment for Menkes disease, although some benefit may accrue following treatment with copper histidinate started soon after birth, particularly if there is residual ATP7A activity [91, 114].
1.3Occipital horn syndrome
Occipital horn syndrome (OHS) is a mild allelic variant of Menkes disease [115, 116] in which connective tissue features (lax skin, loose joints, hernias, bladder diverticula, abnormally shaped bones, and vascular tortuosity) predominate [115]. Survival into adult life is usual [1]. OHS patients have mutations in ATP7A. The disease is named for the pathognomonic wedge-shaped calcification that forms within the trapezium and sternocleidomastoid muscles at their attachment to the occipital bone in affected individuals. Intellect is in the low-normal to mildly impaired range. Additionally, some patients have signs of autonomic dysfunction, such as syncope and episodic diarrhea that are highly suggestive of DBH deficiency [117]. In typical OHS, neurologic findings are otherwise essentially normal, whereas other clinical variants have been described that fall between the classic Menkes and occipital horn syndromes with respect to neurologic phenotype [118, 119]. Definitive diagnosis can be made based upon mutation analysis of ATP7A. Plasma copper and ceruloplasmin levels are in the low-normal range. Copper egress in cultured fibroblasts is impaired to the same degree as in classic Menkes disease. Treatment with copper histidinate gives results similar to those for Menkesdisease [91].
1.4X-linked distal hereditary motor neuropathy
This is the third, and mildest, condition caused by mutations in ATP7A. It is characterized by an adult-onset, progressive distal muscle weakness of hands and feet, with absent or minimal sensory involvement, and with normal serum copper, ceruloplasmin and neurochemical ratios [69, 120]. Sural nerve biopsies in one patient revealed no abnormalities [121] Two unique mutations, p.T994I and p.P1386S, have been associated with this phenotype [122]. Normally, ATP7A recycles continuously between the Golgi and the plasma membrane, while the two aforementioned mutations result in preferential accumulation in the plasma membrane [120].
1.5MEDNIK syndrome
The name MEDNIK syndrome is an acronym for some of the main clinical features of the disease, including Mental retardation, Enteropathy (diarrhea), Deafness (in the form of sensorineural hearing loss), peripheral Neuropathy, Ichthyosis, and Keratodermia [123]. Other features include hepatopathy with elevated transminases and cholestasis, decreased serum ceruplasmin and copper although with increased non-ceruloplasmin bound copper, increased urine copper excretion, and increased liver copper concentration [123]. There is also a mild increase in plasma very long-chain fatty acid levels, as well as bilateral T2-weighted hyperintensities of the basal ganglia. Most reported patients have been from the Kamouraska municipality of Quebec [124]. It is caused by mutations in AP1S1, encoding the σ1A subunit of the adaptor protein complex AP-1 [125]. Normally, the copper ATPases recycle from the trans-Golgi network to the plasma membrane, depending on the intracellular concentrations of copper. At basal concentration, the ATPases transfer copper to cuproenzymes in the Golgi, while at increased copper concentrations, they translocate to the plasma membrane in order to contribute to copper excess efflux. This intracellular trafficking is dependant on adaptor proteins, and thus mutations in AP1S1 disrupt the cycle [123]. Treatment with zinc acetate might improve cholestasis and intellectual development [123]. Liver biopsies revealed intrahepatic cholestasis in one patient, and fibrosis and/or cirrhosis in other two [123]. A skin biopsy from what was likely the first patient reported with the condition revealed prominent epidermal hyperkeratosis with mild inflammatory infiltrate around dermal blood vessels. A muscle biopsy from the same patient showed focal infiltration of lymphocytes around isolated muscle fibers [126].
1.6Huppke-Brendel syndrome
Huppke, Brendel and colleagues described five patients from four independent families with developmental delay, congenital cataracts, nystagmus, hearing loss, white matter hypomyelination, cerebral and cerebellar atrophy, early lethality, and low serum copper and ceruloplasmin levels [127]. Urine copper was normal. Horváth et al. had previously reported one of those patients, whose muscle biopsies revealed a dilated sarcoplasmic reticulum as the most conspicuous finding [128]. Huppke-Brendel syndrome is caused by mutations in the SLC33A1 gene, encoding for a transporter (AT-1) that translocates acetyl-CoA into the ER lumen [127]. It is postulated that decreased acetylation of ceruloplasmin leads to its decreased secretion, and the low serum copper is just a secondary phenomenon, as ceruloplasmin carries 95% of serum copper [127].
2Neurodegeneration with brain iron accumulation
Neurodegeneration with brain iron accumulation (NBIA) comprises a group of various inherited neurodegenerative disorders characterized by the excessive accumulation of iron in the basal ganglia, and sometimes elsewhere, within the central nervous system. The neuropathology of NBIA is characterized by iron accumulation, axonal spheroids, and variable findings of tau-positive neurofibrillary tangles (NFTs) or α-synuclein-positive Lewy bodies [129]. Ferrous iron is stained by Turnbull’s blue, while ferric iron is stained by Prussian blue (also known as Berlin blue, or Perls stain). A summary of the different subtypes of NBIA can be found in Table 4.
Table 4
Subtypes of neurodegeneration with brain iron accumulation
| Disease | Synonyms | Gene | Locus | Inheritance | MRI finding | Iron accumulation | Neuropathology |
| Pantothenate kinase-associated neurodegeneration (PKAN) | NBIA1 | PANK2 | 20p13 | AR | “Eye-of-the-tiger” | GP >SN | NFTs, spheroids. |
| PLA2G6-associated neurodegeneration (PLAN) | NBIA2, PARK14 | PLA2G6 | 22q13.1 | AR | Cerebellar atrophy | GP >SN | Lewy bodies, NFTs, spheroids |
| Mitochondrial membrane protein-associated neurodegeneration (MPAN) | NBIA4, SPG43 | C19orf12 | 19q12 | AR | T2 hyperintensity of medial medullary lamina | GP, SN | Lewy bodies, spheroids, tau |
| Beta-propeller protein-associated neurodegeneration (BPAN) | NBIA5, SENDA | WRD45 | Xp11.23 | XLD | T1 hyperintensity with central hypointensity in SN | SN >GP | Lewy bodies, NFTs, spheroids |
| COASY protein-associated neurodegeneration (CoPAN) | NBIA6 | COASY | 17q21.2 | AR | GP calcification in one case (can mimic the “eye-of-the-tiger”) | GP, SN | N/A |
| Fatty acid hydroxylase-associated neurodegeneration (FAHN) | SPG35 | FA2H | 16q23 | AR | Pontocerebellar atrophy. White matter changes. Thin CC | GP, SN | N/A |
| Kufor-Rakeb syndrome | PARK9, CLN12 | ATP13A2 | 1p36.13 | AR | Cerebral, cerebellar and brainstem atrophy | CN, P | Lipofuscin? |
| Woodhouse-Sakati syndrome | – | DCAF17 | 2q31.1 | AR | White matter changes | GP, SN | N/A |
| Neuroferritinopathy | NBIA3 | FTL | 19q13.33 | AD | Cystic cavitation in basal ganglia | P, CN, GP, Th, DN | Distorted, iron-laden nuclei |
| Aceruloplasminemia | – | CP | 3q25.1 | AR | Cerebellar atrophy. White matter changes | P, CN, GP, Th, DN | Bizarre astrocytes; glomerular structures |
| Adapted from Kruer [129], Gregory and Hayflick [130], Venco et al. [131] and Wiethoff et al. [132]. | |||||||
AR, autosomal recessive; AD, autosomal dominant; CC, corpus callosum; CN, caudate nucleus; DN, dentate nucleus; GP, globus pallidus; N/A, not available; NFT, neurofibrillary tangles; P, putamen; SN, substantia nigra; Th, thalamus; XLD, X-linked dominant.
2.1Pantothenate kinase-associated neurodegeneration (PKAN)
The first case of NBIA was described by neuropathologists Julius Hallervorden and Hugo Spatz in 1922 [133]. Five sisters (including a pair of twins) presented with dystonia, dysarthria and progressive cognitive impairment starting between the ages of 7 and 9 years, ending with death between 16 to 27 years old. The neuropathology revealed abnormal accumulation of iron in the globus pallidus and substantia nigra [134]. For a while the disease was known as Hallervorden-Spatz syndrome, but given their involvement with the Nazi regime, the use of this eponym has decreased significantly over the last couple of decades [135]. PKAN represents 35 to 50% of all cases of NBIA [130]. Classic PKAN presents in the first decade of life (at a mean age of 3.4 years) with progressive gait disturbances due to the presence of dystonia and rigidity [136]. Loss of ambulation occurs 10 to 15 years after the onset of the disease. The rate of cognitive impairment might be overestimated given the severity of motor disability over time [136]. Two thirds of patients have pigmentary retinal degeneration. One quarter of patients have atypical PKAN, presenting after the age of 10 years (at a mean age of 13.6 years), with loss of ambulation 15 to 40 years later [136]. HARP syndrome (Hypoprebetalipoproteinemia, Acanthocytosis, Retinitis pigmentosa, and Pallidal degeneration) is part of the disease spectrum. Iron is primarily deposited in the form of coarse hemosiderin granules around blood vessels, in the cytoplasm of astrocytes (not in microglia or oligodendroglia), and less prominently so in the cytoplasm of neurons, and in the neuropil (“iron dust”) [137]. Neuroaxonal spheroids can be seen in the globus pallidus, corpus callosum and subcortical white matter. Degenerating neurons stain strongly for ubiquitin, and weakly for tau [137]. Pantothenate kinase is the first enzyme in the biosynthesis of coenzyme A from pantothenate (vitamin B5). It phosphorylates pantothenate into 4-phosphopantothenate. Pantethine supplementation has been proposed as a treatment for PKAN, since it bypasses the metabolic defect [138].
2.2PLA2G6-associated neurodegeneration (PLAN)
This disease accounts for 20% of all NBIA cases [130]. It can be classified into three overlapping phenotypes, from more to less severe: infantile neuroaxonal dystrophy (INAD, presenting before age 3 years), atypical neuroaxonal dystrophy (with onset before age 20 years), and PLA2G6-related dystonia-parkinsonism [139]. INAD was first described by Seitelberger in 1952 [140], thus its old nomenclature of Seitelberger disease. What was originally described as Karak syndrome [141] is now known to be atypical NAD. In neuroaxonal dystrophy, developmental regression is the rule. Neuropathologically, the disease is characterized by pronounced demyelination associated with the presence of spheroids in the axons of the white matter of the cerebral hemispheres (Fig. 10) [142]. Ultrastructure shows amorphous electron-dense material in unmyelinated axons (Fig. 11). Diagnosis is made by pathologic examination of brain, peripheral nerve, conjunctiva, skin, or rectal biopsy. The characteristic axonal swellings are detected by histochemical and ultrastructural techniques [143–148]. Macroscopically, the brain of affected infants shows atrophy of the cerebral hemispheres with ventricular dilation and cerebellar atrophy. Histologically, axonal spheroids are seen in the grey matter of the nervous system with degeneration of corticospinal and spinobulbar tracts associated with reactive astrocytosis. An infant with cerebellar localization has been described [149]. Alpha-synuclein-positive Lewy bodies and Lewy neurites have been described in all forms of the disease, mainly in the substantia nigra and neocortex [129, 150], while early-onset cases tend to have tau-positive neurofibrillary tangles and neuropil threads [150]. Iron deposition is inconsistent, seen in less then 50% of patients [151].
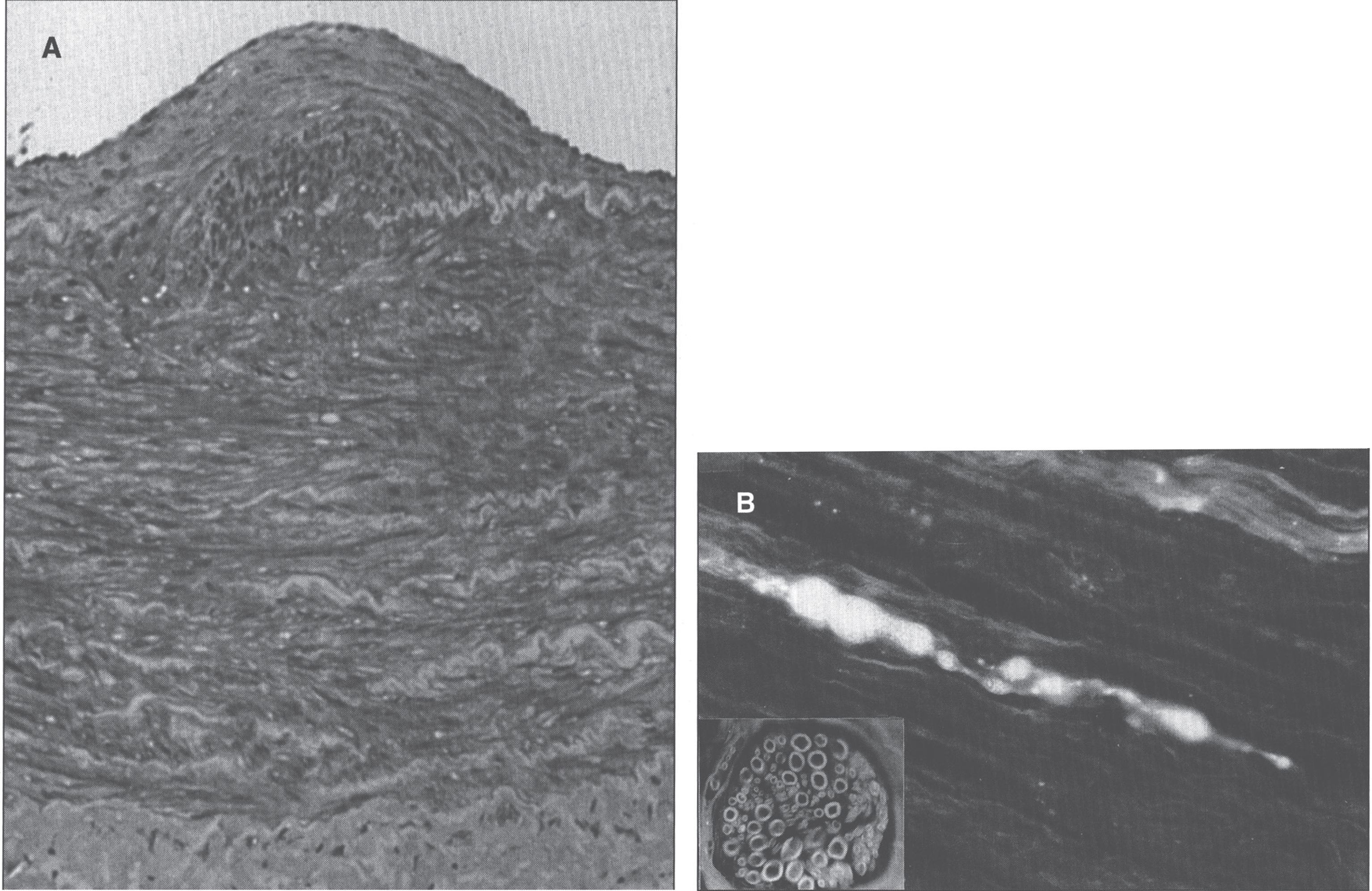
Fig.10
Neuroaxonal leukodystrophy. There are axonal spheroids in white matter associated with myelin degeneration.
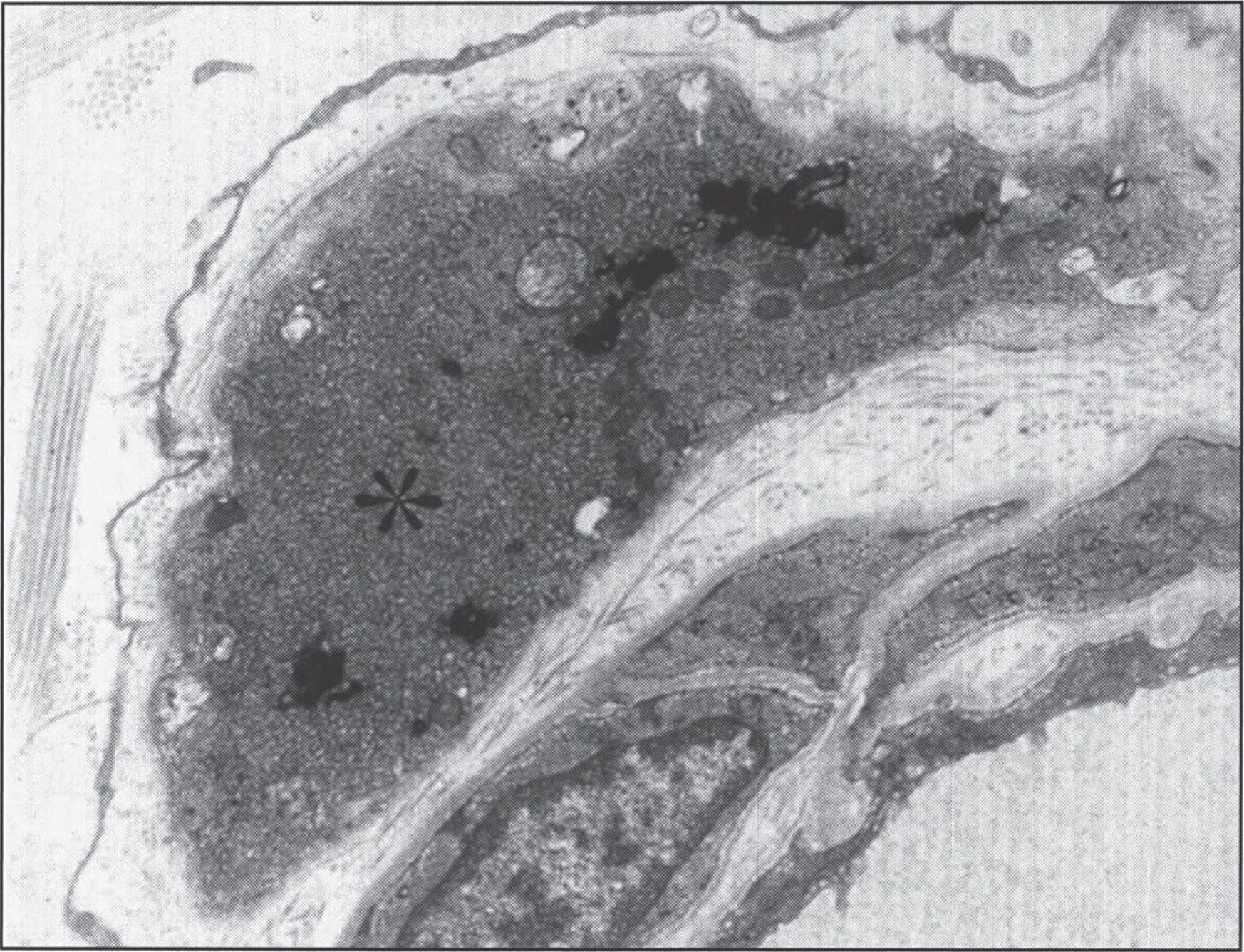
Fig.11
Infantile neuroaxonal dystrophy. A portion of one of the unmyelinated axons (asterisk) in this conjuctival perivascular nerve is distended by an accumulation of tubulo-membranous structures, degenerate organelles, and amorphous electron-dense materials. With these, distinctive axonal “spheroids” become more condensed and display characteristic clefts. (Courtesy of Dr. Gary Mireau.)
2.3Mitochondrial membrane protein-associated neurodegeneration (MPAN)
The disease accounts for 6–10% of all cases of NBIA [130]. Gait impairment is the most common presenting feature, dystonia and dysarthria are common during the course of the disease, and progressive cognitive deterioration is the rule [152]. Disease progression is typically slow, with survival well into adulthood, distinguishing it from other types of NBIA [152]. Widespread Lewy bodies and Lewy neurites are found in basal ganglia, substantia nigra and neocortex, with Lewy bodies being about 6 times more abundant in MPAN than in PLAN [129, 153]. Spheroids can be found both in the central and peripheral nervous systems, just as in PLAN [153]. One patient showed decreased estimated iron content in the substantia nigra, with stable content in the globus pallidus after months of treatment with deferiprone [154].
2.4Beta-propeller protein-associated neurodegeneration (BPAN)
This is the only X-linked NBIA, accounting for 1–2% of all cases [130]. BPAN was previously referred to as “Static Encephalopathy of childhood with NeuroDegeneration in Adulthood”, or SENDA, since it presents with static developmental delay during childhood, with progressive parkinsonism, dystonia and dementia in early adulthood [155]. Neuropathology reveals numerous large axonal spheroids primarily in the globus pallidum and substantia nigra, axonal torpedoes in the cerebellar granular cell layer [156], and extensive deposition of tau-positive neurofibrillary tangles, pre-tangles and neuropil threads [157].
The rest of the causes of NBIA account for less than 1% of cases each [158].
2.5COASY protein-associated neurodegeneration (CoPAN)
Coenzyme A synthase (COASY) is a bifunctional enzyme that performs the last two steps in coenzyme A synthesis. These include the conversion of 4’-phosphopantetheine into dephospho-coenzyme A by phosphopantetheine adenylyltransferase (PPAT), and the dephosphorylation of dephospho-coenzyme A into the final product, coenzyme A, by dephospho-CoA kinase (DPCK) [159]. This is the second inborn error of CoA biosynthesis to be associated with NBIA, although the mechanism by which coenzyme A deficiency leads to altered iron metabolism remains unknown. There are two other enzymes participating in the biosynthesis of CoA that have not yet been associated with human disease. The disease has been reported in two patients presenting with spasticity before the age of 3 years, followed by progressive dystonia, dysarthria and parkinsonism [160]. Neuropathology is not available.
2.6Fatty acid hydroxylase-associated neurodegeneration (FAHN)
The deficient enzyme normally α-hydroxylates myelin galactolipids, galactosylceramide, and sulfatides [161]. The disease has been reported in less than 30 individuals from different ancestry. It can present as a dysmyelinating leukodystrophy and spasticity with or without dystonia, or as spastic paraplegia 35 [162]. Spasticity is a universal finding, dystonia, ataxia, cognitive decline and optic atrophy are seen in the majority of patients, and seizures are common [162]. The disease typically presents between the ages of 3 and 11 years [162]. The finding of a thin corpus callosum in the setting of NBIA should raise concern for FAHN [132]. Neuropathology of the central nervous system is not available. Sural nerve biopsy in one patient showed mild decrease in myelinated fibers [161], while sural nerve, skin and muscle biopsies in another patient were normal [163], and muscle biopsy in one more patient showed signs of denervation and reinnervation [164]. Although not required for the diagnosis, a bone marrow biopsy may reveal granular histiocytes [162].
2.7Kufor-Rakeb syndrome
The disease was initially described in the village of Kufor-Rakeb, in Jordan, in 1994 [165]. It is characterized by parkinsonism starting in adolescence of early adulthood, oculomotor abnormalities such as supranuclear gaze palsy or oculogyric crisis, cognitive involvement including dementia and visual hallucinations, and a characteristic facial-faucial-finger mini-myoclonus [166]. As in PLAN, iron accumulation is inconsistent [151]. Membrane-bound electron-dense lamellated inclusions have been found in fibrocytes, endothelial cells, and macrophages from skin biopsies, and intermyofibrillar, subsarcolemmal and endomysial regions from muscle biopsies [167]. Another patient had a sural nerve biopsy showing reduced myelinated fiber density, axonal degeneration, and numerous cytoplasmic inclusions in endoneurium, perineurium and vascular smooth muscle cells. Ultrastructurally, these inclusions are membrane-bound, electron dense, occasionally folded, and irregular [168]. Neuropathology in one patient with mutations in ATP13A2 revealed extensive neuronal and glial liposfuscinosis, with sparing of the white matter [169].
2.8Woodhouse-Sakati syndrome
The disease, first described by Woodhouse and Sakati in 1983, is characterized by diabetes mellitus, alopecia, hypogonadism, deafness, dystonia, and intellectual disability [170]. There is often no iron accumulation, as in PLAN and Kufor-Rakeb syndrome [151]. Neuropathology is not available.
2.9Neuroferritinopathy
It is the only NBIA with autosomal dominant inheritance. This adult-onset condition presents with chorea in half of patients and dystonia in 42.5%, while over the course of the disease dysarthria develops in 77.5% of patients and orolingual dyskinesia in 65% [171]. Serum ferritin is low (<20μg/L) in most males and post-menopausal women, but it is normal in pre-menopausal females [172]. The characteristic finding is that of swollen, distorted, vacuolated nuclei in neurons and glia, primarily in the putamen [129, 173]. Hepatocytes also contain vacuolated nuclei that stain positively for iron [173]. Ubiquitin- and tau-positive neuroaxonal spheroids can be seen in the putamen, although they are not prominent [129].
2.10Aceruloplasminemia
The disease was first described by Miyajima in 1987 [174]. Ceruloplasmin is synthesized in hepatocytes and secreted into the serum with 6 atoms of copper incorporated during biosynthesis [175]. Ceruloplasmin is an acute-phase reactant, and the serum concentration increases during infection, inflammation, and trauma largely due to effects on hepatic ceruloplasmin gene expression [176]. The ceruloplasmin gene is on chromosome 3q23-25, and abundant, tissue-specific expression of this gene is found in the liver [177].
Aceruloplasminemia is an extremely rare autosomal recessive disorder of iron metabolism characterized by diabetes, retinal degeneration, and neurologic symptoms [178]. Affected patients have marked parenchymal iron accumulation in conjunction with an absence of circulating serum ceruloplasmin, and molecular genetic analysis reveals inherited mutations in the ceruloplasmin gene [2, 177].
Ceruloplasmin is essential for the release of iron from reticuloendothelial storage sites. Specifically, the ferroxidase activity of ceruloplasmin is required to oxidize ferrous to ferric iron for incorporation into transferrin and subsequent transport out of cells [179]. Hence, in aceruloplasminemia, ferrous iron accumulates in the plasma and is rapidly removed from the circulation by the liver and other tissues [180]. There is a marked increase in iron in both hepatocytes and reticuloendothelial cells [181]. The serum ferritin concentration is elevated (850–4000 ng/mL; normal 30–200), the serum iron concentration is decreased (<45μg/dL; normal 60–180), and most patients have a mild anemia (178]. Autopsy findings reveal iron in the pancreas with accumulation in the islets of Langerhans in association with a selective loss of insulin-producing cells [181]. All patients exhibit abnormal glucose metabolism and most develop insulin-dependent diabetes. The presence of iron overload in the liver, insulin-dependent diabetes mellitus, and hyperferritinemia can mimic hemochromatosis, and in fact in at least one occasion had led to its misdiagnosis [182]. The iron content in the liver is greater than in the brain [183].
The most significant pathology, however, involves progressive degeneration of the basal ganglia due to iron deposition. Patients present with dysarthria, dystonia, and dementia in the fourth decade of life. Brain tissue at autopsy shows abundant iron deposition, more prominent in astrocytes than in neurons, with neuronal loss [181, 183]. The main site of iron accumulation is the globus pallidus, followed by the putamen >thalami >cerebellar cortex >hippocampus [183]. There is marked loss of Purkinje cells in the cerebellum [183]. The characteristic features are abnormal, deformed astrocytes and globular structures [184]. Astrocytes in the basal ganglia are enlarged, up to 50μm in size, with abundant cytoplasm and prominent nuclei, some with lobulated nuclei or multinucleated [183–185]. These are similar to Alzheimer type 1 astrocytes [185]. Globular structures are grumose or foamy spheroid bodies, 10 to 60μm in diameter, seen mainly in the basal ganglia and superficial layers of the frontal cortex [183–185]. These globular structures stain strongly with anti-GFAP antibodies, but only faintly with antibodies for neuronal markers such as neurofilment and synaptophysin [185]. This attests to their glial origin, and distinguishes them from spheroids, which are axonal swellings from neurons [183]. Ultrastructurally, many electron-dense bodies can be seen inside the globular structures [183]. With increased duration of the disease, the aforementioned pathologic processes extend from the basal ganglia into the cerebral cortex [186]. Ophthalmologic examination reveals iron deposition and photoreceptor loss in the peripheral regions of the retina. Involvement of the central nervous system indicates that ceruloplasmin plays an essential role in brain iron metabolism.
Although all patients with aceruloplasminemia have low serum copper (<10μg/dL) owing to the absence of this protein in the serum, copper metabolism in such patients remains normal [93]. Patients with aceruloplasminemia have normal liver histology on biopsy with no evidence of hepatic copper accumulation [181].
Iron chelators such as desferrioxamine and deferasirox reduce body iron stores and can ameliorate the neurologic symptoms of patients with aceruloplasminemia if initiated early enough in the course of the disease [187]. In one case, however, only hepatic and not brain iron was chelated [188].
3Iron homeostasis
Humans absorb iron through the gastrointestinal tract and excrete iron only through the bile and urine. The total body iron pool ranges from 2 to 6 g in normal adults; about 0.5 g is stored in the liver, 98% of which is in hepatocytes. The normal flux is 1 mg/day for men and 1.5–2.0 for women [189]. Iron absorption requires transporters at the apical and basolateral membranes of gastrointestinal mucosal cells. The basolateral transporter, ferroportin, can bind a protein called hepcidin that inactivates ferroportin and causes iron to remain within mucosal cells, which eventually slough. Hepcidin, a hormone produced in the liver in response to high transferrrin saturation [102], also inhibits iron release from macrophages. Iron overload occurs whenever net absorption of iron is excessive.
3.1Hereditary hemochromatosis
Hemochromatosis is the excessive accumulation of body iron, most of which is deposited in the parenchymal cells of various organs, particularly liver and pancreas [190]. In hemochromatosis, total iron accumulation may exceed 50 g. Iron accumulation is lifelong; symptoms usually first appear in the fifth to sixth decades of life.
Hereditary hemochromatosis is the most common autosomal recessive genetic disease in the Caucasian population [191–193]. With an incidence of 1 in 200 live births, it affects 1.5 million people in the United States, although most are asymptomatic. Biallelic mutations in one of five different genes cause hemochromatosis: HFE; HJV and HAMP, both causative of juvenile hemochromatosis -or hemochromatosis type 2- leading to severe iron overload before the age of 30 years); TFR2, causing hemochromatosis type 3; and FTH1, causing hemochromatosis type 5, or Japanese iron overload. All are autosomal recessive conditions, with the exception of hemochromatosis type 5, which is dominant. Their gene products function in the signaling pathway that produces hepcidin in hepatocytes. Lack of hepcidin allows for unregulated gastrointestinal absorption of iron. The primary hemochromatosis gene, HFE, is located on the short arm of chromosome 6, close to the HLA gene locus [194]. Two mutations, C282Y and H63D (new nomenclature: p.Cys282Tyr and p.His63Asp), account for the vast majority of hemochromatosis [191–193]. Affected patients are either homozygous for C282Y or compound heterozygous for C282Y and H63D. Hemochromatosis can also be caused by ferroportin resistance to hepcidin (see below) [195]. Iron overload is also seen in congenital atransferrinemia due to mutations in the TF gene, characterized by early-onset: 1) severe microcytic, hypochromic anemia refractory to iron therapy; 2) liver iron overload with significant iron deposition in both hepatocytes and Kupffer cells; 3) absent transferrin, leading to a decreased β-globulin fraction on serum protein electrophoresis [196]. Finally, yet another hereditary condition that can lead to early-onset severe hepatic iron overload with microcytic, hypochromic anemia is caused by mutations in the gene SLC11A2, encoding for the divalent metal transporter DMT1 [197, 198].
3.1.1Clinical characteristics
In hemochromatosis patients, excessive intestinal iron absorption leads to net iron accumulation of 0.5 to 1.0 g per year [191]. Symptoms typically develop after 20 g of storage iron have accumulated. Males predominate (5–7 : 1) with slightly earlier clinical presentation, partly because physiologic iron loss (menstruation, pregnancy) delays iron accumulation in women. The proposed mechanisms for iron toxicity is through lipid peroxidation via iron-catalyzed free radical reaction (Fenton reaction), iron stimulation, and collagen formation. Direct interaction of iron with DNA leads to alterations in hepatic morphology and predisposes to hepatocellular carcinoma. It is reversible in cells not injured by iron accumulation, and removal of excess iron during therapy promotes recovery of tissue function.
Patients with hemochromatosis manifest hepatomegaly, abdominal pain, skin pigmentation (particularly in sun-exposed areas), deranged glucose homeostasis or frank diabetes mellitus, cardiac dysfunction (arrhythmias, cardiomyopathy), and atypical arthritis [193, 199] (Table 5). In some patients, the presenting complaint is hypogonadism, with amenorrhea in the female and loss of libido and impotence in the male. The classic triad of pigment cirrhosis with hepatomegaly, skin pigmentation, and diabetes mellitus does not always develop. Death may result from cirrhosis or cardiac disease. Hemochromatosis patients carry a risk for hepatocellular carcinoma that is 200-fold greater than that of the general population [200]. Depletion of iron, and even reversal of cirrhosis, does not totally prevent the occurrence of this fatal neoplasm [200].
Table 5
Hereditary hemochromatosis
| Homozygote frequency | 1 in 200 |
| Carrier frequency (N. Europeans) | Approx. 1 in 10 |
| Disease expression: males to females | Approx. 5 : 1 (juvenile cases show equal sex ratio) |
| Other modifying factors | Alcohol (approx. 80% of established cases) underlying red blood cell disorders, such as spherocytosis, β-thalassemia |
| Age of onset | >40-year-long presymptomatic period |
| Clinical features | Nonspecific fatigue; asthenia; joint pains; loss of libido and impotence; diabetes mellitus; liver enlargement; increased skin pigmentation; cardiac disease; increased risk of hepatocellular carcinoma. |
| Treatment | Phlebotomy and/or iron chelators. |
| Prognosis | Untreated disease shortens life; treatment before cirrhosis develops compatible with normal life expectancy. |
The severity of myocardial involvement varies widely and does not correlate with the severity in other organs. Cardiac involvement, whose severity is proportional to the quantity of myocardial iron deposition, leads to a mixed dilated-restrictive cardiomyopathy, with both systolic and diastolic dysfunction [201]. The possibility of cardiac hemochromatosis should be considered in any patient with unexplained heart failure.
3.1.2Diagnosis
Screening for HHC is accomplished by measuring serum iron, total iron-binding capacity, and ferritin levels, followed by liver biopsy, if indicated. Most important for identification of patients with asymptomatic HHC is screening of family members. Heterozygotes for HHC accumulate excessive iron, but not to the degree required to cause significant tissue damage. A diagnosis of hemochromatosis can be suspected based upon a serum ferritin concentration of more than 400μg per liter or a transferrin saturation of more than 45%. The serum ferritin concentration correlates well with the level of hepatic iron stores; the transferrin saturation test is believed to be sufficient as a screening test. A bone marrow biopsy is not helpful in diagnosis since the bone marrow does not store iron in HHC. Hepatic iron content can be estimated non-invasively through magnetic resonance imaging [202, 203].
3.1.3Pathology
Although iron overload occurs via excessive intestinal absorption, iron accumulates as ferritin and hemosiderin in the parenchymal tissues of the body and in the linings of joints, without significant deposition in the bone marrow. In the liver [204], iron becomes evident first as golden yellow hemosiderin granules in the cytoplasm of periportal hepatocytes, particularly in lysosomes (siderosomes) within the pericanalicular cytoplasm [205]. With increasing iron load, there is progressive involvement of the lobule –extending from zone 1 toward zone 3- and bile duct epithelium. Kupffer cell pigmentation is less marked, in distinction from secondary causes of iron overload such as hemoglobinopathies or blood transfusions [206]. Because iron is a direct hepatotoxin, inflammation is characteristically absent. At this stage, the liver is typically slightly larger than normal, dense, and brown. Fibrous septa develop slowly, leading ultimately to a micronodular pattern of cirrhosis (Fig. 12) [41]. The overall reduction in liver size is rarely marked.
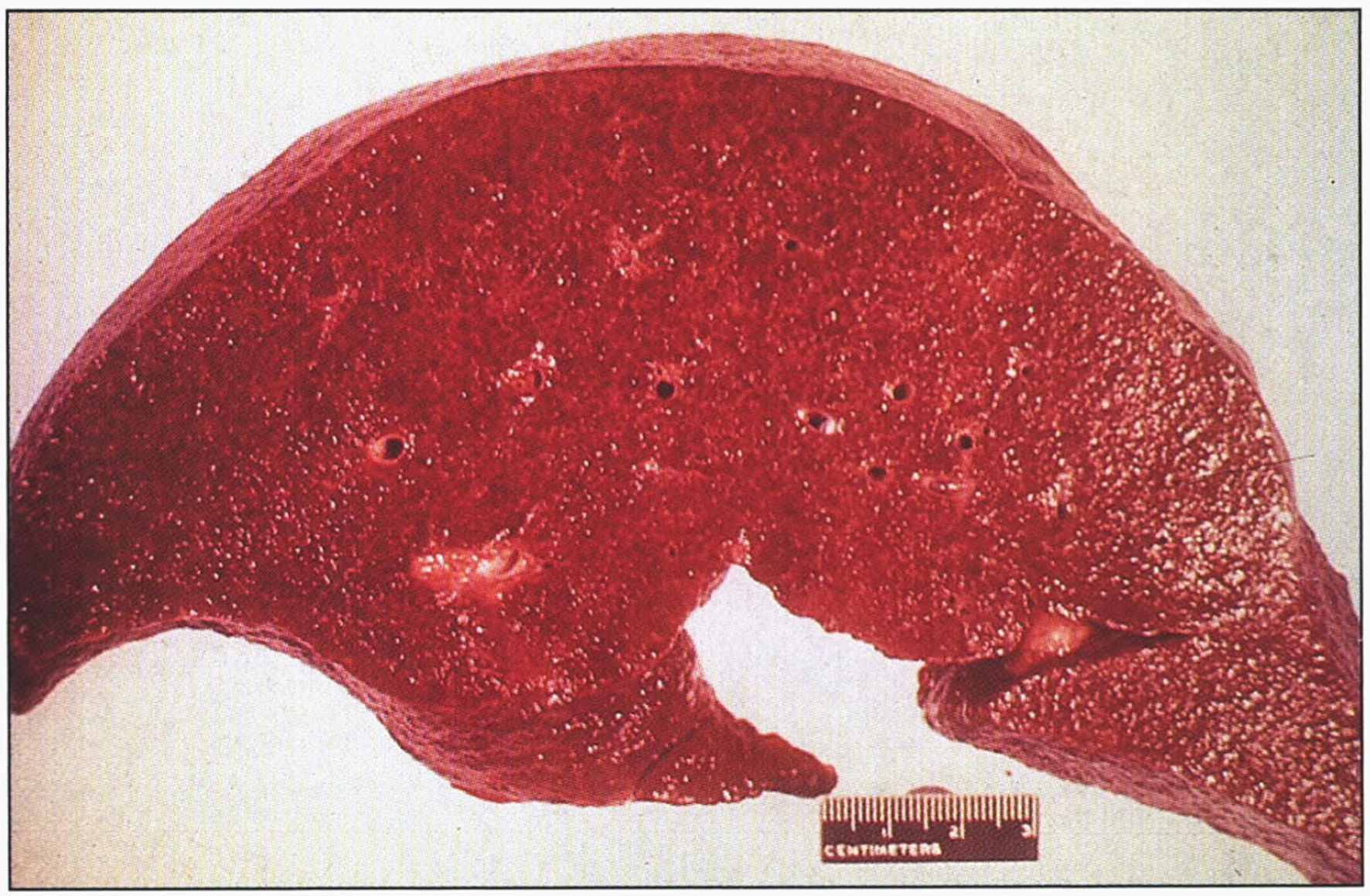
Fig.12
Hereditary hemochromatosis. Gross appearance of micronodular cirrhotic liver.
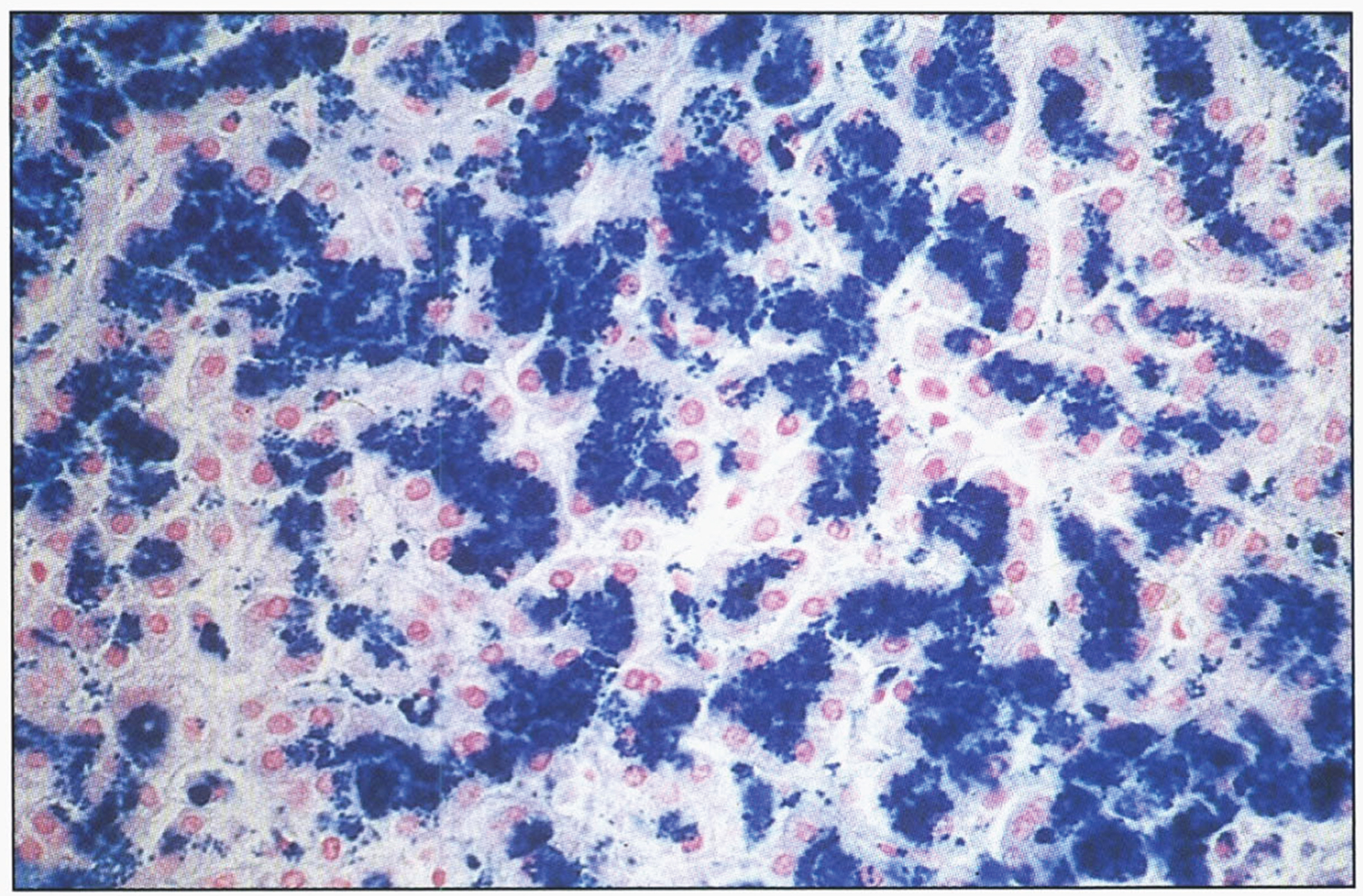
The Prussian blue reaction (potassium ferrocyanide and hydrochloric acid) is used to stain the hemosiderin granules blue-black (Fig. 13). Because this stain does not stain ferritin-bound iron, biochemical determination of hepatic iron concentration in unfixed tissue is the standard for quantitating iron content. In normal individuals, it is less than 1000μg iron/g dry weight; hepatic iron concentrations in excess of 22,000μg/g dry weight (300μmol/g) carry a risk for the development of fibrosis and cirrhosis [207].
Fig.13
Microscopic appearance of liver in hereditary hemochromatosis showing iron in hepatocytes. Prussian blue stain.
The heart is often enlarged and has hemosiderin granules within the myocardial fibers, inducing a striking brown coloration to the myocardium (Fig. 15). A delicate interstitial fibrosis may appear. Myocardial iron deposition (Fig. 16) results in an increase in the thickness of the left ventricular wall and reduces left ventricular compliance, with an eventual decline in the left ventricular systolic function and ventricular dilation. Myocardial iron deposits are sarcoplasmic and are most common in the subepicardial region, followed by the subendocardial region; they are least common in the midmyocardial wall. Iron deposition is more extensive in the ventricles than in the atria, and the cardiac conduction system is frequently involved.
Fig.14
Microscopic appearance of the pancreas in hereditary hemochromatosis showing iron pigment in pancreatic acinar and islet cells. Prussian blue stain.
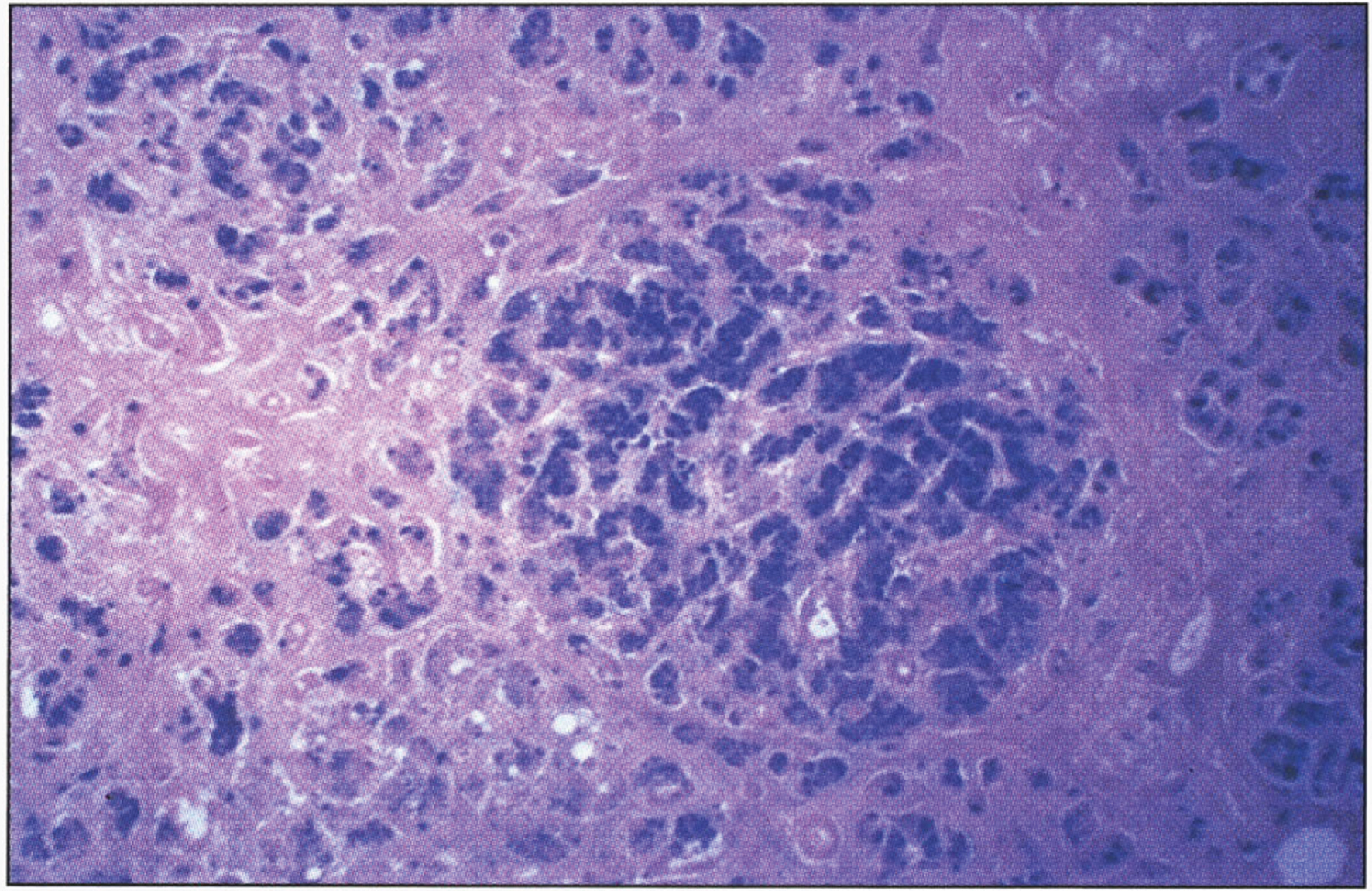
The pancreas becomes intensely pigmented (Fig. 15), has diffuse interstitial fibrosis, and may exhibit some parenchymal atrophy. Hemosiderin is found in both the acinar and the islet cells and sometimes in the interstitial fibrous stroma. The intensity of iron staining in the pancreatic islets correlates with the occurrence and severity of the diabetes.
Fig.15
Gross appearance of the heart in hereditary hemochromatosis showing iron pigmentation.
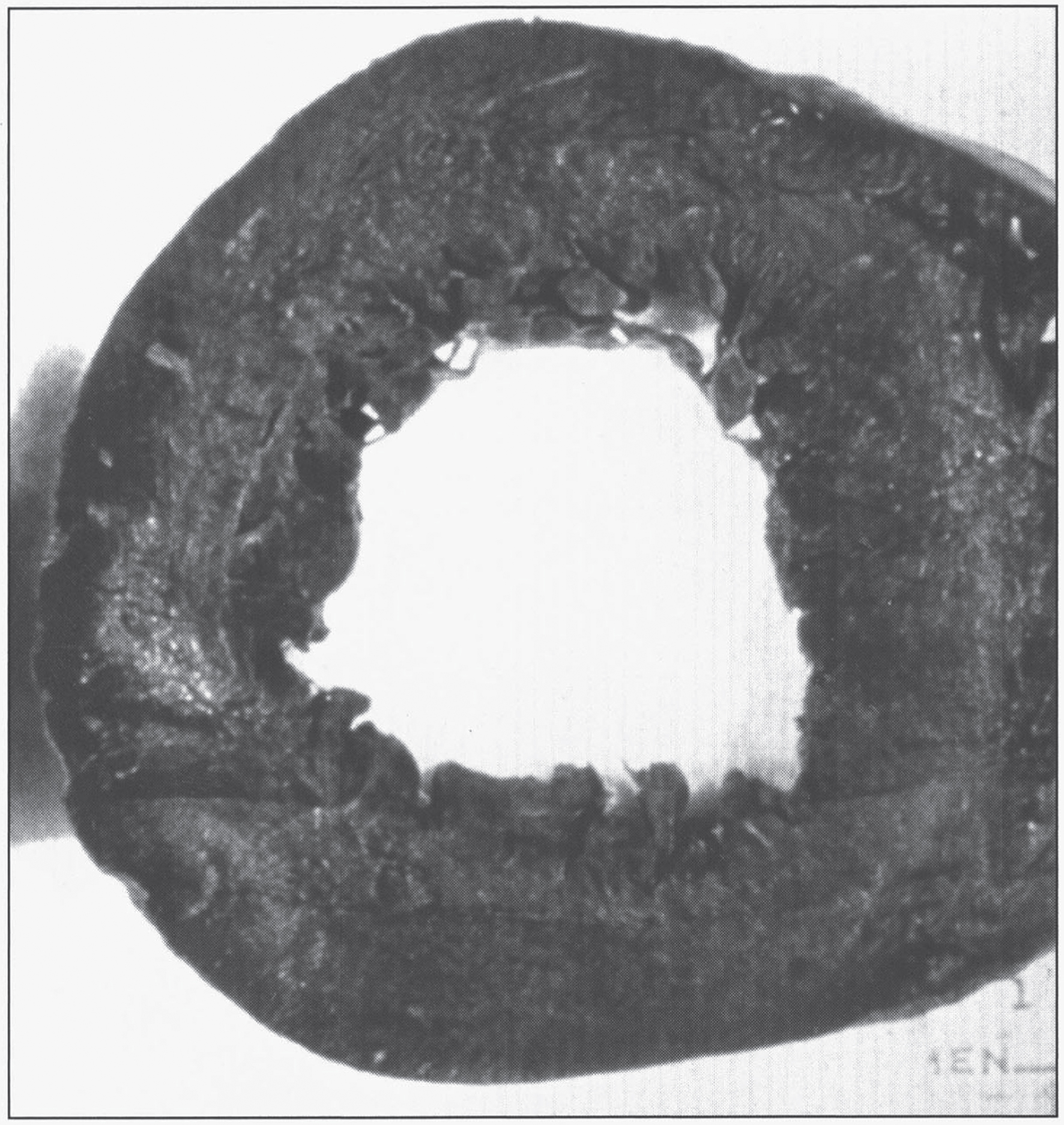
Fig.16
Microscopic section of the heart in hereditary hemochromatosis showing iron pigment in the myocardial cells.
The skin usually has increased pigmentation, mainly owing to increased epithelial melanin (seen also with other forms of cirrhosis). A distinctive metallo-slate-gray pigmentation is related to accumulation of hemosiderin in dermal macrophages and fibroblasts.
With hemosiderin deposition in the joint synovium, an acute synovitis may develop. Excessive deposition of calcium pyrophosphate damages the articular cartilage, producing disabling arthritis referred to as pseudogout.
The testes may be small and atrophic but are not usually significantly pigmented. It is thought that the atrophy is secondary to a derangement in the hypothalamic-pituitary axis. Other parenchymal organs, particularly endocrine (e.g., adrenals, thyroid), may also accumulate iron.
3.1.4Treatment
The treatment of choice for hemochromatosis is regular phlebotomy, although iron chelators can also be used. The rate of phlebotomy is usually 3-4 units per year after the initial removal of 500 mL of blood once or twice each week upon diagnosis [191]. Treatment goal is a serum ferritin of around 50μg/L, since lower levels -of around 20μg/L- are associated with a rebound increase in nonheme iron absorption [208]. The serum ferritin concentration usually becomes normal before the transferrin saturation, and in fact transferrin saturation can still remain elevated under conditions of iron deficiency. Thus, the best marker for monitoring goals of therapy is serum transferrin [208]. Patients with hemochromatosis diagnosed in the subclinical, precirrhotic stage and treated by regular phlebotomy have a normal life expectancy [200].
3.1.5Ferroportin disease
SLC40A1 encodes ferroportin, the only known mammalian iron exporter, present in macrophages and in the basolateral membrane of hepatocytes and enterocytes. Loss-of-function mutations lead to classical–or type A–ferroportin disease, characterized by iron accumulation predominantly in macrophages, with increased ferritin but low-to-normal transferrin saturation until late in the disease. Both features distinguish it from hemochromatosis, although ferroportin disease has also been called hemochromatosis 4. However, non-classical –or type B- ferroportin disease, caused by mutations leading to hepcidin resistance, is associated with iron overload within hepatocytes and increased transferrin saturation and ferritin, thus mimicking HHC [209]. However, ferroportin disease is inherited in an autosomal dominant manner.
4Zinc homeostasis
An average-sized adult male has 1.4 to 2.3 g of zinc, about 40% of the total body iron content, and 10–20 times that of copper. Mean consumption of zinc for an adult is about 10–15 mg daily, absorbed predominantly in the duodenum and proximal jejunum, and excreted mainly in the feces [210]. Zinc is a cofactor for several enzymes, including ornithine transcarbamylase, alkaline phosphatase, carbonic anhydrase, superoxide dismutase, DNA and RNA polymerases, lactate dehydrogenase, alcohol dehydrogenase, insulin-degrading enzyme, matrix metalloproteinases, and N-acyl D-aspartate acylase. All major metabolic pathways are regulated by zinc metalloenzymes. The functions of these enzymes include catalytic, structural, and regulatory roles [211].
4.1Acrodermatitis enteropathica
This disturbance of zinc homeostasis results from a partial block in the intestinal absorption of zinc, resulting in a severe zinc deficiency state with impaired function of zinc metalloenzymes. It is inherited in an autosomal recessive fashion, and the responsible gene is SLC39A4, encoding for ZIP4 [212–214], a zinc transport protein.
4.1.1Clinical characteristics
The most dramatic clinical feature of acrodermatitis enteropathica is a skin rash that has a characteristic symmetrical, circumoral, retroauricular, and acral distribution. The skin lesions are erythematous in the acute stages, then vesicobullous and pustular, and later hyperkeratotic. Secondary infection is common, usually with candida or staphylococci, which may lead to misdiagnosis. Mucosal lesions include gingivitis, stomatitis, and glossitis. Symptoms are usually present in infancy. The onset is delayed in breast-fed infants until after weaning, whereas babies fed infant formula develop the disorder as early as the first 2–4 weeks of life. During early infancy, frequent passage of watery stools, anorexia, and failure to thrive often precede the skin lesions. Total alopecia occurs frequently. Nail deformities and ophthalmologic problems including blepharitis, conjunctivitis, photophobia, and impaired dark adaption may occur. Mood changes, irritability, lethargy, or depressed mental status are also early features of zinc deficiency, as are recurrent infections. Symptoms are aggravated during infections and physiologic stress, at growth spurts in early childhood, and in puberty. After puberty, men are less vulnerable to zinc deficiency than women. One-third of pregnancies of untreated patients result in spontaneous abortion or in congenital defects of the skeletal or central nervous system [215]. Fluctuations occur, but the clinical course is usually progressive without zinc therapy.
4.1.2Diagnosis
The diagnosis in patients with the characteristic clinical picture can be suspected if markedly reduced plasma zinc values are found. In most patients, plasma zinc is reduced to 3 to 6μmol/L (normal range, 9 to 20μmol/L). The diagnosis is established if, after successful zinc therapy and clinical remission, a withdrawal of zinc leads to a relapse.
Blood samples for zinc analysis should be taken in the morning in the fasting state and the patient should preferably be without infection. By analyzing plasma zinc, the diagnosis of AE can never be proven beyond doubt. In some plasma, zinc may be normal because of zinc released from catabolized tissues. On the other hand, plasma zinc may be low because of acquired zinc deficiency or redistribution of zinc to other body pools during stress and infection. Neutrophils, lymphocytes, or red blood cells are unsuitable for zinc determination. Usually urinary zinc excretion is decreased if plasma zinc is low, and sometimes plasma copper is at the upper range of normal. In general, plasma alkaline phosphatase parallels plasma zinc during severe zinc deficiency. Hair zinc is unreliable in acrodermatitis enteropathica because hair growth is often impaired. Elevated blood ammonia (due to ornithine transcarbamylase deficiency), hypobetalipoproteinemia, and an altered fatty acid pattern also occur. In many patients, impaired immune responses are associated with zinc deficiency states, pointing to depressed humoral and cell-mediated immunity.
4.1.3Pathology
Skin biopsy shows epidermal vacuolation with clear epidermal cells (Fig. 17) and parakeratosis. The dermis may show a sparse lymphohistiocytic inflammatory infiltrate. Ultrastructurally, Paneth cells of the intestinal mucosa show inclusions, as well as pleomorphic granules, sometimes becoming confluent, giving rise to giant granules [216–218]. Some enterocytes contain large lysosomal inclusion bodies [216]. These changes regress with therapy.
Fig.17
Microscopic appearance of skin in acrodermatitis enteropathica showing epidermal vacuolar degeneration.

4.1.4Treatment
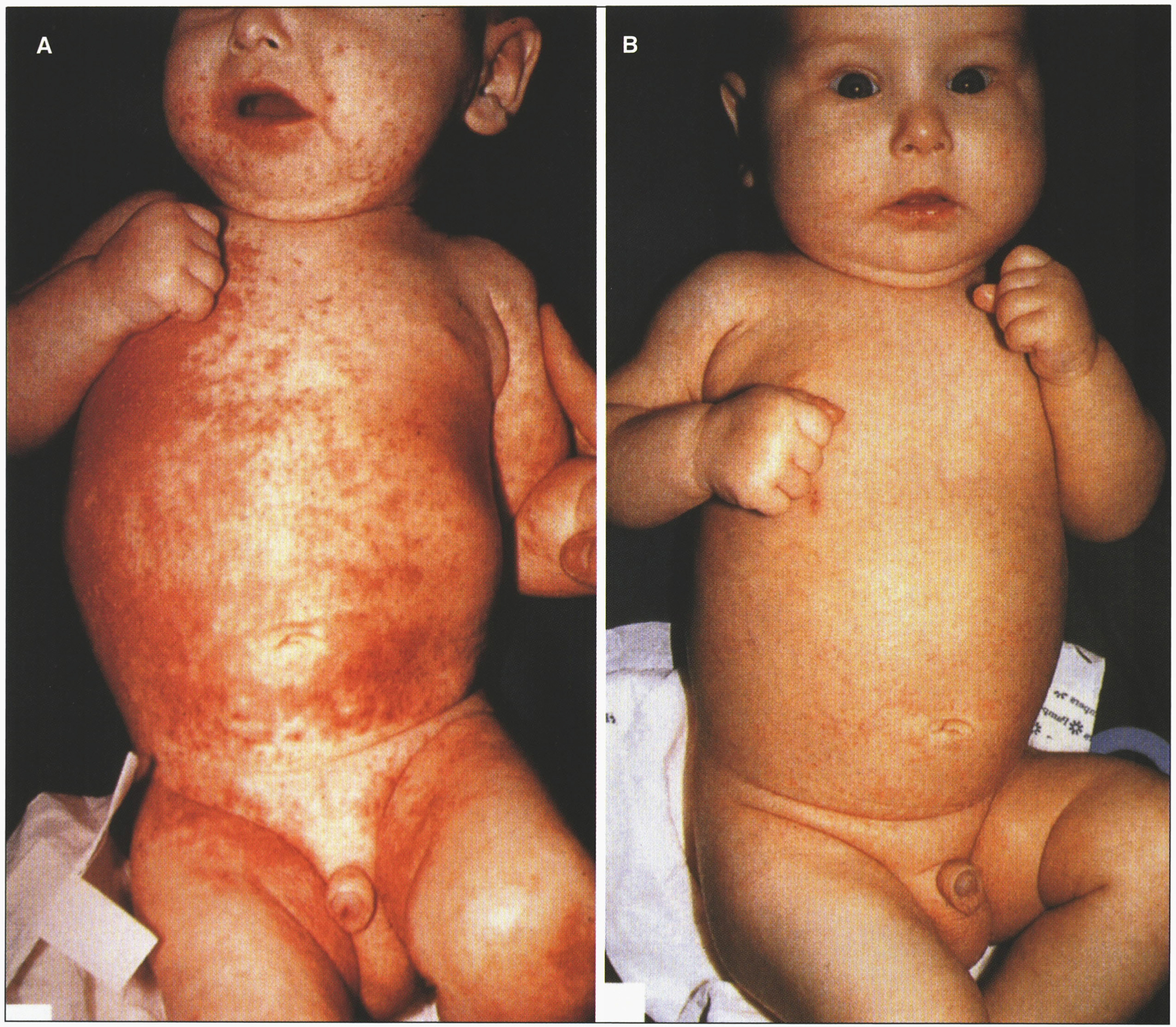
The bioavailability of zinc from human milk is higher than from other dietary sources, so breast-fed babies may not develop symptoms until breast-feeding is discontinued. Zinc supplementation results in the disappearance of skin lesions within a week (Fig. 18), as plasma and urinary excretion of zinc, as well as serum alkaline phosphatase values, increase to normal levels. The usual therapeutic dose is between 30 and 50 mg Zn/day (10–30μmol Zn/kg/day). Zinc is not very toxic, thus higher amounts of 50 to 200 mg do not cause side effects. Plasma copper should be monitored to avoid hypocupremia. The zinc salts used are gluconate, acetate, and, more often, sulfate, administered in 3 divided doses daily if gastric problems occur. In many patients the total dose per day remains constant during their entire childhood.
Fig.18
Acrodermatitis enteropathica showing skin rash. (A) Before treatment. (B) After zinc treatment. (Courtesy of Dr. Bernard Cohen.)
4.2Transient neonatal zinc deficiency
SLC30A2 encodes ZnT2, a zinc transporter in mammary epithelial cells. Transient neonatal zinc deficiency is caused by maternal heterozygous mutations in SLC30A2, leading to low maternal milk zinc concentrations. Infants have a clinical phenotype similar to acrodermatitis enteropathica while receiving breastmilk, but the symptoms resolve after weaning [217–219].
4.3Spondylocheirodysplastic Ehlers-Danlos syndrome (SCD-EDS)
This disorder is caused by biallelic mutations in SLC39A13, which encodes the zinc transporter ZIP13. It is characterized by EDS-like features and radiological findings of a skeletal dysplasia [220]. The EDS-like features include hyperelastic, thin, translucent skin with easy bruising, atrophic scars, wrinkled palms, flat feet, joint hypermobility with subsequent contractures of small joints, down-slanting palpebral fissures, bluish sclerae and protuberant eyes. Short stature is a common feature. Radiological findings include platyspondyly, osteopenia, irregular endplates of the vertebral bodies, metaphyseal widening and epiphyseal flattening [220]. The urinary deoxypyridinoline-to-pyridinoline ratio is about 5 times the normal mean, but 6 times less than in the kyphoscoliotic form of EDS. Serum zinc levels are normal, but the intracellular distribution of zinc is abnormal, leading to abnormal nuclear translocation of SMADs, which in turn alters TGF-β and BMP signaling in collagen-producing cells [221]. Skin biopsies evaluated by electron microscopy revealed normal elastin amount and structure, and normal shape and diameter of the collagen fibrils [220].
4.4Birk-Landau-Perez syndrome
This novel condition is associated with biallelic mutations in SLC30A9, encoding ZnT9 [222]. Clinically, it manifests as a cerebro-renal syndrome with early-onset intellectual disability and tubulointerstitial nephropathy. Patients exhibit normal development for the first 1-2 years of life, but subsequently show progressive neurodeterioration. Brain MRI does not show any specific structural anomalies. ZnT9 is located in the endoplasmic reticulum, and its deficiency leads to disrupted zinc homeostasis, with decreased cytosolic zinc concentrations [222].
5Manganese homeostasis
The total body content of manganese in an adult human is 12–20 mg. The daily turnover of manganese is estimated at 20μg, while the dietary intake varies from 2–22 mg/day, with 2–10% being absorbed throughout the length of the small intestine [223]. Thus, the dietary intake of manganese greatly exceeds its daily need. There is an inverse correlation between iron intake and manganese absorption, as they both compete for the same divalent metal transporter, DMT1 [224]. Once absorbed, excess manganese is excreted by the liver into the bile [224]. Examples of manganese-binding enzymes can be found in Table 6.
Table 6
Manganese-dependent enzymes
| Enzyme | Function |
| Glycosyl tranferases | Glycosaminoglycan synthesis |
| Prolidase | Collagen recycling |
| Arginase | Urea synthesis |
| Glutamine synthetase | Glutamine synthesis |
| Superoxide dismutase (SOD)* | Antioxidant |
| Pyruvate carboxylase | Gluconeogenesis |
| Phosphoenolpyruvate carboxykinase | |
| Isocitrate dehydrogenase | Krebs cycle |
*The mitochondrial SOD uses manganese as a cofactor, while the cytoplasmic SOD uses copper and zinc.
5.1Hypermanganesemia with dystonia, polycythemia and cirrhosis (HMDPC)
This disorder, also known as hypermanganesemia with dystonia type 1, is caused by biallelic mutations in SLC30A10, which encodes for a manganese exporter [227] mainly expressed in the small intestine, liver and brain [228]. The disease has been described in 20 individuals from 10 families, and is characterized by childhood-onset dystonia or adult-onset parkinsonism, chronic liver disease, polycythemia, and hypermanganesemia with levels often above 2,000 nmol/L (reference: <320 nmol/L) [229]. Four-limb dystonia leads to a characteristic “cock-walk” gait [230]. Liver disease is present in most patients, with hepatomegaly, increased transaminases and indirect hyperbilirubinemia. Mean hemoglobin concentration is 18.6 mg/dL, and the polycythemia is thought to be related to induction of erythropoietin gene expression by manganese [229]. Brain MRI reveals T1-weighted hyperintensity of the basal ganglia. There is depletion of iron stores with low serum ferritin and iron levels. Chelation therapy with intravenous disodium calcium edetate infusion leads to amelioration of symptoms and decrease in manganese levels. Iron supplementation is also helpful, as it competes for the same transporter as manganese. Mutations causing this disease are known to alter intracellular trafficking and efflux activity of the exporter [227]. Liver pathology in one patient showed micronodular cirrhosis, scattered macrovesicular steatosis, and brown-orange pigment that stains with rhodanine within the cytoplasm of hepatocytes [231]. The patient also showed hypertrophic cardiomyopathy with rhodanine-positive aggregates in rare cardiomyocytes. Gross brain pathology revealed yellow-gray discoloration of the basal ganglia, predominantly in the globus pallidus. Microscopically, the basal ganglia showed severe neuronal loss with reactive astrocytosis. Rhodanine-positive pigment was seen in the remaining neurons, astrocytes, neuropil and scattered macrophages in the globus pallidus. The substantia nigra had poorly pigmented neurons, although regular in number. Ultrastructurally, membrane-bound, electron-dense multigranular deposits could be found in the remaining pallidal neurons and in hepatocytes [231].
5.2Hypermanganesemia with dystonia type 2
This new syndrome is associated with biallelic mutations in SLC39A14, encoding ZIP14 [232]. Patients typically present between 6 months and 3 years of life with delay or loss of motor milestones, followed by dystonia, spasticity, and Parkinsonism [233]. Brain MRI reveals T1-weighted hyperintensities of the basal ganglia with sparing of the thalami, and of the white matter of the cerebellum, spinal cord, and dorsal pons, with ventral pons sparing [233]. There is decreased visceral uptake of manganese, so the liver is spared, but there is consequent elevation of whole blood manganese concentrations, leading to brain deposition. Chelation therapy with disodium calcium edetate leads to dramatic clinical improvement. Neuropathology at autopsy in one individual revealed marked neuronal loss and gliosis in the globus pallidus and dentate nucleus of the cerebellum, the latter also with a few rounded eosinophilic spheroid structures. A patchy vacuolated myelinopathy was observed in the white matter of the cerebrum and cerebellum. Iron deposition, axonal spheroids, tau or alpha-synuclein pathology were not observed [232, 233].
5.3SLC39A8 deficiency
A deficiency of this transporter leads to variable short stature and neurologic symptoms, such as intellectual disability, infantile spasms, hypotonia and cerebellar atrophy [234, 235]. There is a founder mutation in the Hutterite population [234]. Aside from cerebral and cerebellar atrophy, the brain MRI can also reveal restricted diffusion and T2-weighted hyperintensities of the basal ganglia [236]. Variably low concentrations of manganese and zinc are found in blood, with elevated concentrations in the urine, indicating renal wasting [234]. The low availability of manganese leads to a deficient activity of manganese-dependent enzymes, such as for example beta-1,4-galactosyltransferase. The deficiency of the latter leads to a type II glycosylation pattern on transferrin analysis; supplementation with galactose and uridine led a normalization of glycosylation [235]. More recently, treatment with manganese supplementation was attempted in two patients, with improvement of neurologic symptoms, although it should be noted that patients under this treatment should be carefully monitored for findings of manganism [237].
6Selenium homeostasis
The total body content of selenium for an adult living in the US varies from 13.0 to 20.3 mg [238], although this value is lower in people living in countries with low selenium intake. Selenium is mainly found in foods in its organic form -selenomethionine in plant sources, selenocysteine in animal sources- while the inorganic forms of selenium –selenite and selenite- are mainly found in dietary supplements. More than 80% of the ingested selenium is absorbed through the small intestines. There are 25 human selenoproteins, including 5 forms of glutathione peroxidase, 3 forms of thioredoxin reductase, and 3 deiodinases, the latter participating in thyroid hormone metabolism [239, 240]. Selenoproteins contain selenocysteine, the 21st proteinogenic amino acid. In selenocysteine, the oxygen molecule from serine or the sulfur molecule from cysteine is replaced by selenium. Selenocysteine lacks its own tRNA synthetase, and is instead synthesized on its cognate tRNA, called tRNA[Ser] Sec. tRNA[Ser] Sec is first loaded with serine by seryl-tRNA synthetase (SARS). The newly-formed ser-tRNA[Ser] Sec is then phosphorylated by O-phosphoseryl-tRNA kinase (PSTK). The phosphate group from the resulting O-phosphoseryl-tRNA is then replaced by selenium –using selenophosphate as a donor- by the action of the O-phosphoseryl-tRNA:selenocysteinyl-tRNA synthase (SepSecS) [241]. A selenocysteinyl-tRNA-specific eukaryotic elongation factor (EEFSEC) then carries the Sec- tRNA[Ser] Sec to the ribosome [242].
Selenocysteine is not encoded by an independent codon, but rather by the UGA stop codon. The UGA codon is interpreted as a Sec codon, as opposed to a stop codon, by the presence of a stem-loop structure -the Sec-insertion sequence (SECIS) element- in the 3’-untranslated region of the selenoprotein mRNA. SECIS-binding protein 2 (SBP2) binds to the SECIS element and recruits the Sec-specific elongation factor carrying the Sec- tRNA[Ser] Sec to the mRNA of the selenoprotein [242].
6.1SBP2 deficiency
The disease is caused by biallelic mutations in SECISBP2, encoding SBP2. In 10 patients from 8 different families, all had abnormal thyroid metabolism, with elevated T4 and rT3, high/normal TSH, and low T3 levels. Thyroid testing was performed due to delayed growth and delayed bone age [243]. Developmental motor and/or speech delays were seen in 5 patients, sensorineural hearing loss in 4, and progressive congenital myopathy in 2 [243]. Serum selenium concentration is low, indicating a global selenoprotein synthesis deficiency. Thyroid function abnormalities do not normalize after organic or inorganic selenium supplementation [244]. Muscle biopsy in one patient showed marked type 1 fiber predominance, with occasional minicores [245].
6.2Progressive cerebello-cerebral atrophy
Caused by biallelic mutations in SEPSECS, the disease is characterized by profound intellectual disability, progressive postnatal microcephaly and spasticity, progressive atrophy of the cerebrum and cerebellum, and variable myoclonic or generalized tonic-clonic seizures [246, 247]. Neuropathology reveals subtotal laminar necrosis of the neocortex, mainly involving the parietooccipital regions, with relative sparing of the hippocampus [248]. There is severe olivopontocerebellar atrophy, involving the basis pontis, inferior olives, tegmentum of the medulla oblongata, and cerebellar cortex, the latter with thin molecular and granular layers, and subtotal loss of Purkinje cells [248]. There is also spinal cord atrophy, predominantly involving the posterior columns [248].
REFERENCES
[1] | Danks D.M. , Disorders of copper transport. In: Scriver CR , Beaudet AL , Sly WS , Valle DL , editors. Metabolic and Molecular Bases of Inherited Diseases. 7th ed. New York: McGraw Hill; (1995) |
[2] | Culotta V.C. and Gitlin J.D. , Disorders of Copper Transport. In: Valle DL , Beaudet AL , Vogelstein B , Kinzler KW , Antonarakis SE , Ballabio A , et al., editors. The Online Metabolic and MolecularBases of Inherited Disease [Internet]. New York NY: The McGraw-Hill Comanies, Inc.; (2014) [cited 2015 Jul 27]. Available from: http://mhmedical.com/content.aspx?aid=1102891541 |
[3] | Palmiter R.D. , The elusive function of metallothioneins, Proc Natl Acad Sci U S A 95: (15) ((1998) ), 8428–8430. |
[4] | Wilson S. , Progressive lenticular degeneration: A familial nervous disease associated with cirrhosis of the liver, Brain ((1912) ) 34: , 295–509. |
[5] | Weiss K.H. , Wilson Disease. In: Pagon RA , Adam MP , Ardinger HH , Wallace SE , Amemiya A , Bean LJ , et al., editors. GeneReviews(®) [Internet]. Seattle (WA): University of Washington, Seattle; (1993) [cited 2015 Aug 6]. Available from: http://www.ncbi.nlm.nih.gov/books/NBK1512/ |
[6] | Scheinberg I.H. and Sternlieb I. , Wilson’s disease. Vol. 23: . WB Saunders Company; (1984) . |
[7] | Cousins R.J. , Absorption, transport, and hepatic metabolism of copper and zinc: Special reference to metallothionein and ceruloplasmin, Physiol Rev 65: (2) ((1985) ), 238–309. |
[8] | Bull P.C. , Thomas G.R. , Rommens J.M. , Forbes J.R. and Cox D.W. , The Wilson disease gene is a putative copper transporting P-type ATPase similar to the Menkes gene, Nat Genet 5: (4) ((1993) ), 327–337. |
[9] | Petrukhin K. , Fischer S.G. , Pirastu M. , Tanzi R.E. , Chernov I. , Devoto M. , et al., Mapping, cloning and genetic characterization of the region containing the Wilson disease gene, Nat Genet 5: (4) ((1993) ), 338–343. |
[10] | Chelly J. and Monaco A.P. , Cloning the Wilson disease gene, Nat Genet 5: (4) ((1993) ), 317–318. |
[11] | Stenson P.D. , Mort M. , Ball E.V. , Shaw K. , Phillips A. and Cooper D.N. , The human gene mutation database: Building a comprehensive mutation repository for clinical and molecular genetics, diagnostic testing and personalized genomic medicine, Hum Genet 133: (1) ((2014) ), 1–9. |
[12] | Stapelbroek J.M. , Bollen C.W. , van Amstel J.K.P. , van Erpecum K.J. , van Hattum J. , van den Berg L.H. , et al., The HQ mutation in ATP7B is associated with late and neurologic presentation in Wilson disease: Results of a meta-analysis, J Hepatol 41: (5) ((2004) ), 758–763. |
[13] | Chuang L.M. , Wu H.P. , Jang M.H. , Wang T.R. , Sue W.C. , Lin B.J. , et al., High frequency of two mutations in codon 778 in exon 8 of the ATP7B gene in Taiwanese families with Wilson disease, J Med Genet 33: (6) ((1996) ), 521–523. |
[14] | Kim E.K. , Yoo O.J. , Song K.Y. , Yoo H.W. , Choi S.Y. , Cho S.W. , et al., Identification of three novel mutations and a high frequency of the Arg778Leu mutation in Korean patients with Wilson disease, Hum Mutat 11: (4) ((1998) ), 275. |
[15] | Yoo H.-W. , Identification of novel mutations and the three mostcommon mutations in the human ATP7B gene of Korean patients withWilson disease, Genet Med Off J Am Coll Med Genet 4: (Suppl 6) ((2002) ), 43S–48S. |
[16] | Horslen S. and Hahn S.H. , Genotype-phenotype correlation in wilsondisease, J Clin Gastroenterol [Internet] 44: (6) ((2010) ), Available from: http://journals.lww.com/jcge/Fulltext/2010/07000/Genotype_Phenotype_Correlation_in_Wilson_Disease.6.aspx |
[17] | Leggio L. , Addolorato G. , Loudianos G. , Abenavoli L. and Gasbarrini G. , Genotype–phenotype correlation of the Wilson disease ATP7B gene, Am J Med Genet A 140: (8) ((2006) ), 933–933. |
[18] | Panagiotakaki E. , Tzetis M. , Manolaki N. , Loudianos G. , Papatheodorou A. , Manesis E. , et al., Genotype-phenotype correlations for a wide spectrum of mutations in the Wilson disease gene (ATP7B), Am J Med Genet A 131: (2) ((2004) ), 168–173. |
[19] | Usta J. , Wehbeh A. , Rida K. , El-Rifai O. and Estiphan T.A. , T.Majarian, et al., Phenotype-genotype correlation in wilson diseasein a large lebanese family: Association of c.2299insC with hepaticand of p. Ala1003Thr with neurologic phenotype, PLoS One 9: (11) ((2014) ), e109727. |
[20] | Vrabelova S. , Letocha O. , Borsky M. and Kozak L. , Mutation analysis of the ATP7B gene and genotype/phenotype correlation in 227 patients with Wilson disease, Mol Genet Metab 86: (1-2) ((2005) ), 277–285. |
[21] | Wu J. , Forbes J.R. , Chen H.S. and Cox D.W. , The LEC rat has a deletion in the copper transporting ATPase gene homologous to the Wilson disease gene, Nat Genet 7: (4) ((1994) ), 541–545. |
[22] | Kaler S.G. , Metabolic and molecular bases of Menkes disease and occipital horn syndrome, Pediatr Dev Pathol Off J Soc Pediatr Pathol Paediatr Pathol Soc 1: (1) ((1998) ), 85–98. |
[23] | Scott J. , Gollan J. , Samourian S. and Sherlock S. , Wilson’s disease, presenting as chronic active hepatitis, Gastroenterology 74: (4) ((1978) ), 645–651. |
[24] | McCullough A.J. , Fleming C.R. , Thistle J.L. , Baldus W.P. , Ludwig J. , McCall J.T. , et al., Diagnosis of Wilson’s disease presenting as fulminant hepatic failure, Gastroenterology 84: (1) ((1983) ), 161–167. |
[25] | Williams F.J. and Walshe J.M. , Wilson’s disease. An analysis of the cranial computerized tomographic appearances found in 60 patients and the changes in response to treatment with chelating agents, Brain J Neurol 104: (Pt 4) ((1981) ), 735–752. |
[26] | Das M. , Misra U.K. and Kalita J. , A study of clinical, MRI and multimodality evoked potentials in neurologic Wilson disease, Eur J Neurol Off J Eur Fed Neurol Soc 14: (5) ((2007) ), 498–504. |
[27] | Hedera P. , Brewer G.J. and Fink J.K. , White matter changes inWilson disease, Arch Neurol 59: (5) ((2002) ), 866–867. |
[28] | Page R.A. , Davie C.A. , MacManus D. , Miszkiel K.A. , Walshe J.M. , Miller D.H. , et al., Clinical correlation of brain MRI and MRS abnormalities in patients with Wilson disease, Neurology 63: (4) ((2004) ), 638–643. |
[29] | Kuan P. , Cardiac Wilson’s disease, Chest 91: (4) ((1987) ), 579–583. |
[30] | Hogland H.C. and Goldstein N.P. , Hematologic (cytopenic) manifestations of Wilson’s disease (hepatolenticular degeneration), Mayo Clin Proc 53: (8) ((1978) ), 498–500. |
[31] | Schilsky M.L. , Scheinberg I.H. and Sternlieb I. , Prognosis of Wilsonian chronic active hepatitis, Gastroenterology 100: (3) ((1991) ), 762–767. |
[32] | Zandman-Goddard G. , Weiss P. , Avidan B. , Bar-Meir S. and Shoenfeld Y. , Acute varicella infection heralding Wilsonian crisis, J Clin Gastroenterol 18: (3) ((1994) ), 265–266. |
[33] | Roche-Sicot J. and Benhamou J.P. , Acute intravascular hemolysis and acute liver failure associated as a first manifestation of Wilson’s disease, Ann Intern Med 86: (3) ((1977) ), 301–303. |
[34] | Wiebers D.O. , Hollenhorst R.W. and Goldstein N.P. , The ophthalmologic manifestations of Wilson’s disease, Mayo Clin Proc 52: (7) ((1977) ), 409–416. |
[35] | Roberts E.A. and Schilsky M.L. , American Association for Study of Liver Diseases (AASLD). Diagnosis and treatment of Wilson disease: An update, Hepatol Baltim Md 47: (6) ((2008) ), 2089–2111. |
[36] | Schilsky M.L. , Non-invasive testing for Wilson disease: Revisiting the d-penicillamine challenge test, J Hepatol 47: (2) ((2007) ), 172–173. |
[37] | Archer G.J. and Monie R.D. , Wilson’s disease and chronic active hepatitis, Lancet Lond Engl 1: (8009) ((1977) ), 486–487. |
[38] | Goldfischer S. and Sternlieb I. , Changes in the distribution of hepatic copper in relation to the progression of Wilson’s disease (hepatolenticular degeneration), Am J Pathol 53: (6) ((1968) ), 883–901. |
[39] | Irons R.D. , Schenk E.A. and Lee J.C. , Cytochemical methods for copper. Semiquantitative screening procedure for identification of abnormal copper levels in liver, Arch Pathol Lab Med 101: (6) ((1977) ), 298–301. |
[40] | Davies S. , Williams R. and Portmann B. , Hepatic morphology and histochemistry of Wilson’s disease presenting as fulminant hepatic failure: A study of 11 cases, Histopathology 15: (4) ((1989) ), 385–394. |
[41] | Hubscher S.G. , Burt A.D. , Portmann B.C. and Ferrell L.D. , MacSween’s pathology of the liver. Elsevier Health Sciences; (2011) . |
[42] | Salaspuro M. and Sipponen P. , Demonstration of an intracellular copper-binding protein by orcein staining in long-standing cholestatic liver diseases, Gut 17: (10) ((1976) ), 787–790. |
[43] | Ferenci P. , Steindl-Munda P. , Vogel W. , Jessner W. , Gschwantler M. , Stauber R. , et al., Diagnostic value of quantitative hepatic copper determination in patients with Wilson’s Disease, Clin Gastroenterol Hepatol Off Clin Pract J Am Gastroenterol Assoc 3: (8) ((2005) ), 811–818. |
[44] | Yang X. , Tang X.P. , Zhang Y.H. , Luo K.Z. , Jiang Y.F. , Luo H.Y. , et al., Prospective evaluation of the diagnostic accuracy of hepatic copper content, as determined using the entire core of a liver biopsy sample, Hepatol Baltim Md 62: (6) ((2015) ), 1731–1741. |
[45] | Stremmel W. and Merle U. , Liver: A new copper cut-off value for diagnosis of Wilson disease? Nat Rev Gastroenterol Hepatol 12: (9) ((2015) ), 493–494. |
[46] | Faa G. , Nurchi V. , Demelia L. , Ambu R. , Parodo G. , Congiu T. , et al., Uneven hepatic copper distribution in Wilson’s disease, J Hepatol 22: (3) ((1995) ), 303–308. |
[47] | Goldfischer S. , Popper H. and Sternlieb I. , The significance of variations in the distribution of copper in liver disease, Am J Pathol 99: (3) ((1980) ), 715–730. |
[48] | Bertrand E. , Lechowicz W. , Szpak G.M. , Lewandowska E. , Członkowska A. and Dymecki J. , Quantitative study of pathological forms of astroglia in Wilson’s disease, Folia Neuropathol Assoc Pol Neuropathol Med Res Cent Pol Acad Sci 35: (4) ((1997) ), 227–232. |
[49] | Bertrand E. , Lewandowska E. , Szpak G.M. , Hoogenraad T. , Blaauwgers H.G. , Członkowska A. , et al., Neuropathological analysis of pathological forms of astroglia in Wilson’s disease, Folia Neuropathol Assoc Pol Neuropathol Med Res Cent Pol Acad Sci 39: (2) ((2001) ), 73–79. |
[50] | Ludwig J. , Moyer T.P. and Rakela J. , The liver biopsy diagnosis of Wilson’s disease. Methods in pathology, Am J Clin Pathol 102: (4) ((1994) ), 443–446. |
[51] | Fischer R.T. , Soltys K.A. , Squires R.H. , Jaffe R. , Mazariegosand G.V. and Shneider B.L. , Prognostic scoring indices in Wilson disease: Acase series and cautionary tale, J Pediatr Gastroenterol Nutr 52: (4) ((2011) ), 466–469. |
[52] | Schilsky M.L. , Scheinberg I.H. and Sternlieb I. , Liver transplantation for Wilson’s disease: Indications and outcome, Hepatol Baltim Md 19: (3) ((1994) ), 583–587. |
[53] | Medici V. , Mirante V.G. , Fassati L.R. , Pompili M. , Forti D. , Del Gaudio M. , et al., Liver transplantation for Wilson’s disease: The burden of neurological and psychiatric disorders, Liver Transplant Off Publ Am Assoc Study Liver Dis Int Liver Transplant Soc 11: (9) ((2005) ), 1056–1063. |
[54] | Sevmis S. , Karakayali H. , Aliosmanoglu I. , Yilmaz U. , Ozcay F. , Torgay A. , et al., Liver transplantation for Wilson’s disease, Transplant Proc 40: (1) ((2008) ), 228–230. |
[55] | Weiss K.H. , Thurik F. , Gotthardt D.N. , Schäfer M. , Teufel U. , Wiegand F. , et al., Efficacy and safety of oral chelators in treatment of patients with Wilson disease,–, Clin Gastroenterol Hepatol Off Clin Pract J Am Gastroenterol Assoc 11: (8) ((1028) ), 1035–2. |
[56] | Fox A.N. and Schilsky M. , Once daily trientine for maintenance therapy of Wilson disease, Am J Gastroenterol 103: (2) ((2008) ), 494–495. |
[57] | Ala A. , Aliu E. and Schilsky M.L. , Prospective pilot study of a single daily dosage of trientine for the treatment of Wilson disease, Dig Dis Sci 60: (5) ((2015) ), 1433–1439. |
[58] | Weiss K.H. , Gotthardt D.N. , Klemm D. , Merle U. , Ferenci-Foerster D. , Schaefer M. , et al., Zinc monotherapy is not aseffective as chelating agents in treatment of Wilson disease, Gastroenterology 140: (4) ((2011) ), 1189–1198.e1. |
[59] | Ranucci G. , Dato F.D. , Spagnuolo M.I. , Vajro P. and Iorio R. , Zincmonotherapy is effective in Wilson’s disease patients with mildliver disease diagnosed in childhood: A retrospective study, Orphanet J Rare Dis 9: (1) ((2014) ), 41. |
[60] | Horvath J. , Beris P. , Giostra E. , Martin P-Y and Burkhard P.R. , Zinc-induced copper deficiency in Wilson disease, J Neurol Neurosurg Psychiatry 81: (12) ((2010) ), 1410–1411. |
[61] | Cortese A. , Zangaglia R. , Lozza A. , Piccolo G. and Pacchetti C. , Copper deficiency in Wilson’s disease: Peripheral neuropathy and myelodysplastic syndrome complicating zinc treatment, Mov Disord Off J Mov Disord Soc 26: (7) ((2011) ), 1361–1362. |
[62] | Foubert-Samier A. , Kazadi A. , Rouanet M. , Vital A. , Lagueny A. , Tison F. , et al., Axonal sensory motor neuropathy in copper-deficient Wilson’s disease, Muscle Nerve 40: (2) ((2009) ), 294–296. |
[63] | Yoshitoshi E.Y. , Takada Y. , Oike F. , Sakamoto S. , Ogawa K. , Kanazawa H. , et al., Long-term outcomes for 32 cases of Wilson’s disease after living-donor liver transplantation, Transplantation 87: (2) ((2009) ), 261–267. |
[64] | Arnon R. , Annunziato R. , Schilsky M. , Miloh T. , Willis A. , Sturdevant M. , et al., Liver transplantation for children with Wilson disease: Comparison of outcomes between children and adults, Clin Transplant 25: (1) ((2011) ), E52–E60. |
[65] | Merle U. , Stremmel W. and Encke J. , Perspectives for gene therapy of Wilson disease, Curr Gene Ther 7: (3) ((2007) ), 217–220. |
[66] | Menkes J.H. , Alter M. , Steigleder G.K. , Weakley D.R. and Sung J.H. , A sex-linked recessive disorder with retardation of growth,peculiar hair, and focal cerebral and cerebellar degeneration, Pediatrics 29: (5) ((1962) ), 764–779. |
[67] | Kaler S.G. , Menkes disease, Adv Pediatr ((1994) ) 41: , 263–304. |
[68] | Danks D.M. , Campbell P.E. , Stevens B.J. , Mayne V. and Cartwright E. , Menkes’s kinky hair syndrome. An inherited defect in copper absorption with widespread effects, Pediatrics 50: (2) ((1972) ), 188–201. |
[69] | Kaler S.G. , ATP7A-Related Copper Transport Disorders. In: Pagon RA , Adam MP , Ardinger HH , Wallace SE , Amemiya A , Bean LJ , et al., editors. GeneReviews(®) [Internet]. Seattle (WA): University of Washington, Seattle; (1993) [cited 2015 Aug 8]. Available from: http://www.ncbi.nlm.nih.gov/books/NBK1413/ |
[70] | Møller L.B. , Mogensen M. and Horn N. , Molecular diagnosis of Menkes disease: Genotype-phenotype correlation, Biochimie 91: (10) ((2009) ), 1273–1277. |
[71] | Matsubara O. , Takaoka H. , Nasu M. , Iwakawa Y. and Okeda R. , An autopsy case of Menkes kinky hair disease, Acta Pathol Jpn 28: (4) ((1978) ), 585–594. |
[72] | Kapur S. , Higgins J.V. , Delp K. and Rogers B. , Menkes syndrome in a girl with X-autosome translocation, Am J Med Genet 26: (2) ((1987) ), 503–510. |
[73] | Gerdes A.M. and Tønnesen T. , Horn N. , Grisar T. , Marg W. , MüllerA. , et al., Clinical expression of Menkes syndrome in females, Clin Genet 38: (6) ((1990) ), 452–459. |
[74] | Beck J. , Enders H. , Schliephacke M. , Buchwald-Saal M. and Tümer Z. , X;1 translocation in a female Menkes patient: Characterization by fluorescence in situ hybridization, Clin Genet 46: (4) ((1994) ), 295–298. |
[75] | Abusaad I. , Mohammed S.N. , Ogilvie C.M. , Ritchie J. , Pohl K.R. and Docherty Z. , Clinical expression of Menkes disease in a girl with X;13 translocation, Am J Med Genet 87: (4) ((1999) ), 354–359. |
[76] | Sugio Y. , Sugio Y. , Kuwano A. , Miyoshi O. , Yamada K. , Niikawa N. , et al., Translocation t(X;21)(q13.3; p11.1) in a girl with Menkes disease, Am J Med Genet 79: (3) ((1998) ), 191–194. |
[77] | Sirleto P. , Surace C. , Santos H. , Bertini E. , Tomaiuolo A.C. , Lombardo A. , et al., Lyonization effects of the t(X;16) translocation on the phenotypic expression in a rare female with Menkes disease, Pediatr Res 65: (3) ((2009) ), 347–351. |
[78] | Møller L.B. , Lenartowicz M. , Zabot M-T , Josiane A. , Burglen L. , Bennett C. , et al., Clinical expression of Menkes disease in females with normal karyotype, Orphanet J Rare Dis ((2012) ) 7: , 6. |
[79] | Smpokou P. , Samanta M. , Berry G.T. , Hecht L. , Engle E.C. and Lichter-Konecki U. , Menkes disease in affected females: The clinical disease spectrum, Am J Med Genet A 167A: (2) ((2015) ), 417–420. |
[80] | Favier A. , Boujet C. and Joannard A. , Possibility of a Menkes-like disorder of copper metabolism in a girl, J Inherit Metab Dis 6: (2) ((1983) ), 89–89. |
[81] | Barton N.W. , Dambrosia J.M. and Barranger J.A. , Menkes’ Kinky-HairSyndrome: Report of a Case in a Female Infant: 10:45 AM: 6.[Abstract], Neurology 33: (4 Suppl 2) ((1983) ), 154. |
[82] | Yamaguchi Y. , Heiny M.E. , Suzuki M. and Gitlin J.D. , Biochemical characterization and intracellular localization of the Menkes disease protein, Proc Natl Acad Sci U S A 93: (24) ((1996) ), 14030–14035. |
[83] | Camakaris J. , Mann J.R. and Danks D.M. , Copper metabolism in mottled mouse mutants: Copper concentrations in tissues during development, Biochem J 180: (3) ((1979) ), 597–604. |
[84] | Gerdes A.M. , Tønnesen T. , Pergament E. , Sander C. , Baerlocher K.E. , Wartha R. , et al., Variability in clinical expression of Menkes syndrome, Eur J Pediatr 148: (2) ((1988) ), 132–135. |
[85] | Amador E. , Domene R. and Fuentes C. , Carreño J-C and Enríquez G. , Long-term skeletal findings in Menkes disease, Pediatr Radiol 40: (8) ((2010) ), 1426–1429. |
[86] | Arita J.H. , Faria E.C. , Peruchi M.M. , Lin J. , Rodrigues Masruha M. and Vilanova L.C.P. , Menkes disease as a differential diagnosis of child abuse, Arq Neuropsiquiatr 67: (2B) ((2009) ), 507–509. |
[87] | Cronin H. , Fussell J.N. , Pride H. and Bellino P. , Menkes syndrome presenting as possible child abuse, Cutis 90: (4) ((2012) ), 170–172. |
[88] | Horn N. , Menkes X linked disease: Heterozygous phenotype in uncloned fibroblast cultures, J Med Genet 17: (4) ((1980) ), 257–261. |
[89] | Moore C.M. and Howell R.R. , Ectodermal manifestations in Menkes disease, Clin Genet 28: (6) ((1985) ), 532–540. |
[90] | Horn N. , Menkes X-linked disease: Prenatal diagnosis of hemizygous males and heterozygous females, Prenat Diagn 1: (2) ((1981) ), 107–120. |
[91] | Kaler S.G. , Holmes C.S. , Goldstein D.S. , Tang J. , Godwin S.C. , Donsante A. , et al., Neonatal diagnosis and treatment of Menkes disease, N Engl J Med 358: (6) ((2008) ), 605–614. |
[92] | Friedman E. , Harden A. , Koivikko M. and Pampiglione G. , Menkes’ disease: Neurophysiological aspects, J Neurol Neurosurg Psychiatry 41: (6) ((1978) ), 505–510. |
[93] | White S.R. , Reese K. , Sato S. and Kaler S.G. , Spectrum of EEG findings in Menkes disease, Electroencephalogr Clin Neurophysiol 87: (1) ((1993) ), 57–61. |
[94] | Johnsen D.E. , Coleman L. and Poe L. , MR of progressive neurodegenerative change in treated Menkes’ kinky hair disease, Neuroradiology 33: (2) ((1991) ), 181–182. |
[95] | Stanley P. , Gwinn J. and Sutcliffe J. , The osseous abnormalities in Menke’s syndrome In (1976) . p. 167. |
[96] | Kozlowski K. and McCrossin R. , Early osseous abnormalities in Menkes’ kinky hair syndrome, Pediatr Radiol 8: (3) ((1979) ), 191–194. |
[97] | Kaler S.G. , Goldstein D.S. , Holmes C. , Salerno J.A. and Gahl W.A. , Plasma and cerebrospinal fluid neurochemical pattern in Menkes disease, Ann Neurol 33: (2) ((1993) ), 171–175. |
[98] | Kaler S.G. , Gahl W.A. , Berry S.A. , Holmes C.S. and Goldstein D.S. , Predictive value of plasma catecholamine levels in neonatal detection of Menkes disease, J Inherit Metab Dis 16: (5) ((1993) ), 907–908. |
[99] | Goldstein D.S. , Holmes C.S. and Kaler S.G. , Relative efficiencies of plasma catechol levels and ratios for neonatal diagnosis of menkes disease, Neurochem Res 34: (8) ((2009) ), 1464–1468. |
[100] | Uno H. and Arya S. , Neuronal and vascular disorders of the brain and spinal cord in Menkes kinky hair disease. Opitz JM , Bernstein J , editors, Am J Med Genet 28: (S3) ((1987) ), 367–377. |
[101] | Oakes B.W. , Danks D.M. and Campbell P.E. , Human copper deficiency: Ultrastructural studies of the aorta and skin in a child with Menkes’ syndrome, Exp Mol Pathol 25: (1) ((1976) ), 82–98. |
[102] | Aguilar M.J. , Chadwick D.L. , Okuyama K. and Kamoshita S. , Kinky hair disease. I. Clinical and pathological features, J Neuropathol Exp Neurol 25: (4) ((1966) ), 507–522. |
[103] | Goto S. , Hirano A. and Rojas-Corona R.R. , A comparative immunocytochemical study of human cerebellar cortex in X-chromosome-linked copper malabsorption (Menkes’ kinky hair disease) and granule cell type cerebellar degeneration, Neuropathol Appl Neurobiol 15: (5) ((1989) ), 419–431. |
[104] | Vuia O. and Heye D. , Neuropathologic aspects in Menkes’ Kinky hair disease (trichopoliodystrophy). Menkes’ Kinky hair disease, Neuropädiatrie 5: (3) ((1974) ), 329–339. |
[105] | Troost D. , van Rossum A. , Straks W. and Willemse J. , Menkes’ kinky hair disease. II. A clinicopathological report of three cases, Brain Dev 4: (2) ((1982) ), 115–126. |
[106] | Martin J.J. , Flament-Durand J. , Farriaux J.P. , Buyssens N. , Ketelbant-Balasse P. and Jansen C. , Menkes kinky-hair disease. A report on its pathology, Acta Neuropathol (Berl) 42: (1) ((1978) ), 25–32. |
[107] | Kato S. , Ito M. , Ohama E. , Mikoshiba K. , Maeda N. , Yen S-H , et al., Immunohistochemical Studies on Cerebellar Purkinje Cells of Patients with Menkes’ Kinky Hair Disease, Neuropathology 13: (2) ((1993) ), 159–166. |
[108] | Okeda R. , Gei S. , Chen I. , Okaniwa M. , Shinomiya M. and Matsubara O. , Menkes’ kinky hair disease: Morphological and immunohistochemical comparison of two autopsied patients, Acta Neuropathol (Berl) 81: (4) ((1991) ), 450–457. |
[109] | Robain O. , Aubourg P. , Routon M.C. , Dulac O. and Ponsot G. , Menkes disease: A Golgi and electron microscopic study of the cerebellar cortex, Clin Neuropathol 7: (2) ((1988) ), 47–52. |
[110] | Yoshimura N. and Kudo H. , Mitochondrial abnormalities in Menkes’kinky hair disease (MKHD), Electron-microscopic study of thebrain from an autopsy case. Acta Neuropathol (Berl) 59: (4) ((1983) ), 295–303. |
[111] | Wray S.H. , Kuwabara T. and Sanderson P. , Menkes’ kinky hair disease: A light and electron microscopic study of the eye, Invest Ophthalmol 15: (2) ((1976) ), 128–138. |
[112] | Uno H. , Arya S. , Laxova R. and Gilbert E.F. , Menkes’ syndrome with vascular and adrenergic nerve abnormalities, Arch Pathol Lab Med 107: (6) ((1983) ), 286–289. |
[113] | Robinson B.H. , De Meirleir L. , Glerum M. , Sherwood G. and Becker L. , Clinical presentation of mitochondrial respiratory chain defects in NADH-coenzyme Q reductase and cytochrome oxidase: Clues to pathogenesis of Leigh disease, J Pediatr 110: (2) ((1987) ), 216–222. |
[114] | Kaler S.G. , Liew C.J. , Donsante A. , Hicks J.D. , Sato S. and Greenfield J.C. , Molecular correlates of epilepsy in early diagnosedand treated Menkes disease, J Inherit Metab Dis 33: (5) ((2010) ), 583–589. |
[115] | Donsante A. , Tang J. , Godwin S.C. , Holmes C.S. , Goldstein D.S. , Bassuk A. , et al., Differences in ATP7A gene expression underlie intrafamilial variability in Menkes disease/occipital horn syndrome, J Med Genet 44: (8) ((2007) ), 492–497. |
[116] | Kaler S.G. , Gallo L.K. , Proud V.K. , Percy A.K. , Mark Y. , Segal N.A. , et al., Occipital horn syndrome and a mild Menkes phenotype associated with splice site mutations at the MNK locus, Nat Genet 8: (2) ((1994) ), 195–202. |
[117] | Biaggioni I. , Goldstein D.S. , Atkinson T. and Robertson D. , Dopamine-beta-hydroxylase deficiency in humans, Neurology 40: (2) ((1990) ), 370–373. |
[118] | Das S. , Levinson B. , Vulpe C. , Whitney S. , Gitschier J. and Packman S. , Similar splicing mutations of the Menkes/mottled copper-transporting ATPase gene in occipital horn syndrome and the blotchy mouse, Am J Hum Genet 56: (3) ((1995) ), 570–576. |
[119] | Proud V.K. , Mussell H.G. , Kaler S.G. , Young D.W. and PercyDistinctive A.K. , Menkes disease variant with occipital horns:Delineation of natural history and clinical phenotype, Am JMed Genet 65: (1) ((1996) ), 44–51. |
[120] | Yi L. and Kaler S. , ATP7A trafficking and mechanisms underlying the distal motor neuropathy induced by mutations in ATP7A, Ann N Y Acad Sci ((2014) ) 1314: , 49–54. |
[121] | Takata R.I. , Speck Martins C.E. , Passosbueno M.R. , Abe K.T. , Nishimura A.L. , Da Silva M.D. , et al., A new locus for recessive distal spinal muscular atrophy at Xq13.1-q21, J Med Genet 41: (3) ((2004) ), 224–229. |
[122] | Kennerson M.L. , Nicholson G.A. , Kaler S.G. , Kowalski B. , Mercer J.F.B. , Tang J. , et al., Missense mutations in the copper transporter gene ATP7A cause X-linked distal hereditary motor neuropathy, Am J Hum Genet 86: (3) ((2010) ), 343–352. |
[123] | Martinelli D. , Travaglini L. , Drouin C.A. , Ceballos-Picot I. , Rizza T. , Bertini E. , et al., MEDNIK syndrome: A novel defect of copper metabolism treatable by zinc acetate therapy, Brain J Neurol 136: (Pt 3) ((2013) ), 872–881. |
[124] | Saba T.G. , Montpetit A. , Verner A. , Rioux P. , Hudson T.J. , Drouin R. , et al., An atypical form of erythrokeratodermia variabilis maps to chromosome 7q22, Hum Genet 116: (3) ((2005) ), 167–171. |
[125] | Montpetit A. , Côté S. , Brustein E. , Drouin C.A. , Lapointe L. , Boudreau M. , et al., Disruption of AP1S1, causing a novel neurocutaneous syndrome, perturbs development of the skin and spinal cord, PLoS Genet 4: (12) ((2008) ), e1000296. |
[126] | Beare J.M. , Nevin N.C. , Froggatt P. , Kernohan D.C. and Allen I.V. , Atypical erythrokeratoderma with deafness, physical retardation and peripheral neuropathy, Br J Dermatol 87: (4) ((1972) ), 308–314. |
[127] | Huppke P. , Brendel C. , Kalscheuer V. , Korenke G.C. , Marquardt I. , Freisinger P. , et al., Mutations in SLC33A1 cause a lethal autosomal-recessive disorder with congenital cataracts, hearing loss, and low serum copper and ceruloplasmin, Am J Hum Genet 90: (1) ((2012) ), 61–68. |
[128] | Horváth R. , Freisinger P. , Rubio R. , Merl T. , Bax R. , Mayr J.A. , et al., Congenital cataract, muscular hypotonia, developmental delay and sensorineural hearing loss associated with a defect in copper metabolism, J Inherit Metab Dis 28: (4) ((2005) ), 479–492. |
[129] | Kruer M.C. , The neuropathology of neurodegeneration with brain iron accumulation, Int Rev Neurobiol ((2013) ) 110: , 165–194. |
[130] | Gregory A. , Hayflick S. , Neurodegeneration with Brain IronAccumulation Disorders Overview. In: Pagon RA , Adam MP , Ardinger HH , Wallace SE , Amemiya A , Bean LJ , et al., editors. GeneReviews(®) [Internet]. Seattle (WA): University of Washington, Seattle; (1993) [cited 2015 Aug 26]. Available from: http://www.ncbi.nlm.nih.gov/books/NBK121988/. |
[131] | Venco P. , Dusi S. , Valletta L. and Tiranti V. , Alteration of the coenzyme A biosynthetic pathway in neurodegeneration with brain iron accumulation syndromes, Biochem Soc Trans 42: (4) ((2014) ), 1069–1074. |
[132] | Wiethoff S. , Bhatia K.P. , Houlden H. , Genetics of NBIA Disorders, In: Movement Disorder Genetics. Sringer; (2015) . pp. 263–291. |
[133] | Hallervorden J. and Spatz H. , Eigenartige Erkrankung imextrapyramidalen System mit besonderer Beteiligung des Globuspallidus und der Substantia nigra, Z Für Gesamte Neurol Psychiatr 79: (1) ((1922) ), 254–302. |
[134] | Donaldson I. , Marsden C.D. , Schneider S.A. , Bhatia K.P. , editors, Chapter 11: Pantothenate kinase-associated neurodegeneration (PKAN), previously also known as Hallervorden–Spatz disease and related disorders of neurodegeneration with brain iron accumulation, In: Marsden’s Book of Movement Disorders [Internet]. Oxford University Press; (2012) . pp. 521–536. Available from: http//oxfordmedicine.com/10.1093/med/9780192619112.001.0001/med-9780192619112-chapter-011 |
[135] | Zeidman L.A. and Pandey D.K. , Declining use of theHallervorden-Spatz disease eponym in the last two decades, J Child Neurol 27: (10) ((2012) ), 1310–1315. |
[136] | Gregory A. , Hayflick S.J. , Pantothenate Kinase-AssociatedNeurodegeneration, In: Pagon RA , Adam MP , Ardinger HH , Wallace SE , Amemiya A , Bean LJ , et al., editors. GeneReviews(®)[Internet]. Seattle (WA): University of Washington, Seattle; (1993) [cited 2015 Aug 26]. Available from: http://www.ncbi.nlm.nih.gov/books/NBK1490/. |
[137] | Kruer M.C. , Hiken M. , Gregory A. , Malandrini A. , Clark D. , Hogarth P. , et al., Novel histopathologic findings in molecularly-confirmed pantothenate kinase-associated neurodegeneration, Brain J Neurol 134: (Pt 4) ((2011) ), 947–958. |
[138] | Brunetti D. , Dusi S. , Giordano C. , Lamperti C. , Morbin M. , Fugnanesi V. , et al., Pantethine treatment is effective in recoveringthe disease phenotype induced by ketogenic diet in a pantothenatekinase-associated neurodegeneration mouse model, Brain[Internet] ((2013) ); Available from: http://brain.oxfordjournals.org/content/early/2013/12/05/brain.awt325.abstract |
[139] | Gregory A. , Kurian M.A. , Maher E.R. , Hogarth P. and Hayflick S.J. PLA2G6-Associated Neurodegeneration. In: Pagon RA , Adam MP , Ardinger HH , Wallace SE , Amemiya A , Bean LJ , et al., editors. GeneReviews(®) [Internet]. Seattle (WA): University of Washington, Seattle; (1993) [cited 2015 Aug 26]. Available from: http://www.ncbi.nlm.nih.gov/books/NBK1675/ |
[140] | Seitelberger F. , Eine unbekannte Form von infantiler Lipoid-speicher-Krankheit des Gehirns. In Rosenberg and Sellier Turin; (1952) . pp. 323–333. |
[141] | Mubaidin A. , Roberts E. , Hampshire D. , Dehyyat M. , Shurbaji A. , Mubaidien M. , et al., Karak syndrome: A novel degenerative disorder of the basal ganglia and cerebellum, J Med Genet 40: (7) ((2003) ), 543–546. |
[142] | Minagawa M. , Maeshiro H. , Kato K. and Shioda K. , A rare case of leucodystrophy–neuroaxonal leucodystrophy (Seitelberger) (author’s transl), Seishin Shinkeigaku Zasshi 82: (8) ((1980) ), 488–503. |
[143] | Ramaekers V.T. , Lake B.D. , Harding B. , Boyd S. , Harden A. , Brett E.M. , et al., Diagnostic difficulties in infantile neuroaxonal dystrophy. A clinicopathological study of eight cases, Neuropediatrics 18: (3) ((1987) ), 170–175. |
[144] | Goebel H.H. , Kohlschütter A. and Schulte F.J. , Rectal biopsy findings in infantile neuroaxonal dystrophy, Neuropediatrics 11: (4) ((1980) ), 388–392. |
[145] | Kimura S. , Terminal pathology in infantile neuroaxonal dystrophy, Pediatr Neurol 7: (2) ((1991) ), 116–120. |
[146] | Miike T. , Ohtani Y. , Nishiyama S. and Matsuda I. , Pathology of skeletal muscle and intramuscular nerves in infantile neuroaxonal dystrophy, Acta Neuropathol (Berl) 69: (1-2) ((1986) ), 117–123. |
[147] | Wisniewski K. and Wisniewski H.M. , Diagnosis of infantile neuroaxonal dystrophy by skin biopsy, Ann Neurol 7: (4) ((1980) ), 377–379. |
[148] | Ferreira R.C. , Mierau G.W. and Bateman J.B. , Conjunctival biopsy in infantile neuroaxonal dystrophy, Am J Ophthalmol 123: (2) ((1997) ), 264–266. |
[149] | Tachibana H. , Hayashi T. , Kajii T. , Takashima S. and Sasaki K. , Neuroaxonal dystrophy of neonatal onset with unusual clinicopathological findings, Brain Dev 8: (6) ((1986) ), 605–609. |
[150] | Paisán-Ruiz C. , Li A. , Schneider S.A. , Holton J.L. , Johnson R. , Kidd D. , et al., Widespread Lewy body and tau accumulation in childhood and adult onset dystonia-parkinsonism cases with PLA2G6 mutations, Neurobiol Aging 33: (4) ((2012) ), 814–823. |
[151] | Rouault T.A. , Iron metabolism in the CNS: Implications for neurodegenerative diseases, Nat Rev Neurosci 14: (8) ((2013) ), 551–564. |
[152] | Gregory A. , Hartig M. , Prokisch H. , Kmiec T. , Hogarth P. and Hayflick S.J. , Mitochondrial. Membrane Protein-AssociatedNeurodegeneration. In: Pagon RA , Adam MP , Ardinger HH , Wallace SE , Amemiya A , Bean LJ , et al., editors. GeneReviews(®)[Internet]. Seattle (WA): University of Washington, Seattle; (1993) [cited 2015 Aug 26]. Available from: http://www.ncbi.nlm.nih.gov/books/NBK1675/. |
[153] | Hogarth P. , Gregory A. , Kruer M.C. , Sanford L. , Wagoner W. , Natowicz M.R. , et al., New NBIA subtype: Genetic, clinical, pathologic, and radiographic features of MPAN, Neurology 80: (3) ((2013) ), 268–275. |
[154] | Löbel U. , Schweser F. , Nickel M. , Deistung A. , Grosse R. , Hagel C. , et al., Brain iron quantification by MRI in mitochondrial membrane protein-associated neurodegeneration under iron-chelating therapy, Ann Clin Transl Neurol 1: (12) ((2014) ), 1041–1046. |
[155] | Saitsu H. , Nishimura T. , Muramatsu K. , Kodera H. , Kumada S. , Sugai K. , et al., De novo mutations in the autophagy gene WDR45 cause static encephalopathy of childhood with neurodegeneration in adulthood, Nat Genet 45: (4) ((2013) ), 445–449, 449e1. |
[156] | Hayflick S.J. , Kruer M.C. , Gregory A. , Haack T.B. , Kurian M.A. , Houlden H.H. , et al., β-Propeller protein-associated neurodegeneration: A new X-linked dominant disorder with brain iron accumulation, Brain J Neurol 136: (Pt 6) ((2013) ), 1708–1717. |
[157] | Paudel R. , Li A. , Wiethoff S. , Bandopadhyay R. , Bhatia K. , de Silva R. , et al., Neuropathology of Beta-propeller protein associated neurodegeneration (BPAN): A new tauopathy, Acta Neuropathol Commun 3: (1) ((2015) ), 39. |
[158] | Arber C.E. , Li A. , Houlden H. and Wray S. , Insights into molecular mechanisms of disease in neurodegeneration with brain iron accumulation: Unifying theories, Neuropathol Appl Neurobiol 42: (3) ((2015) ), 220–241. |
[159] | Daugherty M. , Polanuyer B. , Farrell M. , Scholle M. , Lykidis A. , de Crécy-Lagard V. ,et al., Complete reconstitution of the human coenzyme A biosynthetic pathway via comparative genomics, J Biol Chem 277: (24) ((2002) ), 21431–21439. |
[160] | Dusi S. , Valletta L. , Haack T.B. , Tsuchiya Y. , Venco P. , Pasqualato S. , et al., Exome sequence reveals mutations in CoA synthase as a cause of neurodegeneration with brain iron accumulation, Am J Hum Genet 94: (1) ((2014) ), 11–22. |
[161] | Edvardson S. , Hama H. , Shaag A. , Gomori J.M. , Berger I. , Soffer D. , et al., Mutations in the fatty acid 2-hydroxylase gene are associated with leukodystrophy with spastic paraparesis and dystonia, Am J Hum Genet 83: (5) ((2008) ), 643–648. |
[162] | Kruer M.C. , Gregory A. , Hayflick S.J. , Fatty AcidHydroxylase-Associated Neurodegeneration. In: Pagon RA , Adam MP , Ardinger HH , Wallace SE , Amemiya A , Bean LJ , et al., editors. GeneReviews(®) [Internet]. Seattle (WA): University ofWashington, Seattle; (1993) [cited 2015 Aug 26]. Available from: http://www.ncbi.nlm.nih.gov/books/NBK56080/ |
[163] | Pierson T.M. , Simeonov D.R. , Sincan M. , Adams D.A. , Markello T. , Golas G. , et al., Exome sequencing and SNP analysis detect novel compound heterozygosity in fatty acid hydroxylase-associated neurodegeneration, Eur J Hum Genet EJHG 20: (4) ((2012) ), 476–479. |
[164] | Dick K.J. , Eckhardt M. , Paisán-Ruiz C. , Alshehhi A.A. , Proukakis C. , Sibtain N.A. , et al., Mutation of FA2H underlies a complicated form of hereditary spastic paraplegia (SPG35), Hum Mutat 31: (4) ((2010) ), E1251–E1260. |
[165] | Najim al-Din A.S. , Wriekat A. , Mubaidin A. , Dasouki M. and Hiari M. , Pallido-pyramidal degeneration, supranuclear upgaze paresisand dementia: Kufor-Rakeb syndrome, Acta Neurol Scand 89: (5) ((1994) ), 347–352. |
[166] | Williams D.R. , Hadeed A. , al-Din A.S.N. , Wreikat A-L and Lees A.J. , Kufor Rakeb disease: Autosomal recessive, levodopa-responsive parkinsonism with pyramidal degeneration, supranuclear gaze palsy, and dementia, Mov Disord Off J Mov Disord Soc 20: (10) ((2005) ), 1264–1271. |
[167] | Malandrini A. , Rubegni A. , Battisti C. , Berti G. and Federico A. , Electron-dense lamellated inclusions in 2 siblings with Kufor-Rakeb syndrome, Mov Disord Off J Mov Disord Soc 28: (12) ((2013) ), 1751–1752. |
[168] | Paisán-Ruiz C. , Guevara R. , Federoff M. , Hanagasi H. , Sina F. , Elahi E. , et al., Early-onset L-dopa-responsive parkinsonism with pyramidal signs due to ATP13A2, PLA2G6, FBXO7 and spatacsin mutations, Mov Disord Off J Mov Disord Soc 25: (12) ((2010) ), 1791–1800. |
[169] | Bras J. , Verloes A. , Schneider S.A. , Mole S.E. and Guerreiro R.J. , Mutation of the parkinsonism gene ATP13A2 causes neuronal ceroid-lipofuscinosis, Hum Mol Genet 21: (12) ((2012) ), 2646–2650. |
[170] | Woodhouse N.J. and Sakati N.A. , A syndrome of hypogonadism, alopecia, diabetes mellitus, mental retardation, deafness, and ECG abnormalities, J Med Genet 20: (3) ((1983) ), 216–219. |
[171] | Chinnery P.F. , Neuroferritinopathy. In: Pagon RA , Adam MP , Ardinger HH , Wallace SE Amemiya A , Bean LJ , et al., editors. GeneReviews(regd) [Internet]. Seattle (WA): University of Washington, Seattle; (1993) [cited 2015Aug 1205 26]. Available from: http://www.ncbi.nlm.nih.gov/books/NBK1141/ |
[172] | Chinnery P.F. , Crompton D.E. , Birchall D. , Jackson M.J. , Coulthard A. , Lombès A. , et al., Clinical features and natural history of neuroferritinopathy caused by the FTL1 460InsA mutation, Brain J Neurol 130: (Pt 1) ((2007) ), 110–119. |
[173] | Mancuso M. , Davidzon G. , Kurlan R.M. , Tawil R. , Bonilla E. , Di Mauro S. , et al., Hereditary ferritinopathy: A novel mutation, its cellular pathology, and pathogenetic insights, J Neuropathol Exp Neurol 64: (4) ((2005) ), 280–294. |
[174] | Miyajima H. , Nishimura Y. , Mizoguchi K. , Sakamoto M. , Shimizu T. and Honda N. , Familial apoceruloplasmin deficiency associated with blepharospasm and retinal degeneration, Neurology 37: (5) ((1987) ), 761–767. |
[175] | Ortel T.L. , Takahashi N. and Putnam F.W. , Structural model of human ceruloplasmin based on internal triplication, hydrophilic/hydrophobic character, and secondary structure of domains, Proc Natl Acad Sci U S A 81: (15) ((1984) ), 4761–4765. |
[176] | Gitlin J.D. , Transcriptional regulation of ceruloplasmin gene expression during inflammation, J Biol Chem 263: (13) ((1988) ), 6281–6287. |
[177] | Gitlin J.D. , Aceruloplasminemia, Pediatr Res 44: (3) ((1998) ), 271–276. |
[178] | Miyajima H. , Aceruloplasminemia. In: Pagon RA , Adam MP , Ardinger HH , Wallace SE , Amemiya A , Bean LJ , et al., editors. GeneReviews(®) [Internet]. Seattle (WA): University ofWashington, Seattle; (1993) [cited 2015 Aug 9]. Available from: http://www.ncbi.nlm.nih.gov/books/NBK1493/ |
[179] | Roeser H.P. , Lee G.R. , Nacht S. and Cartwright G.E. , The role of ceruloplasmin in iron metabolism, J Clin Invest 49: (12) ((1970) ), 2408–2417. |
[180] | Craven C.M. , Alexander J. , Eldridge M. , Kushner J.P. , Bernstein S. and Kaplan J. , Tissue distribution and clearance kinetics of non-transferrin-bound iron in the hypotransferrinemic mouse: A rodent model for hemochromatosis, Proc Natl Acad Sci U S A 84: (10) ((1987) ), 3457–3461. |
[181] | Morita H. , Ikeda S. , Yamamoto K. , Morita S. , Yoshida K. , Nomoto S. , et al., Hereditary ceruloplasmin deficiency with hemosiderosis: A clinicopathological study of a Japanese family, Ann Neurol 37: (5) ((1995) ), 646–656. |
[182] | Fasano A. , Bentivoglio A.R. and Colosimo C. , Movement disorder due to aceruloplasminemia and incorrect diagnosis of hereditary hemochromatosis, J Neurol 254: (1) ((2007) ), 113–114. |
[183] | Miyajima H. , Aceruloplasminemia, an iron metabolic disorder, Neuropathol Off J Jpn Soc Neuropathol 23: (4) ((2003) ), 345–350. |
[184] | Miyajima H. , Aceruloplasminemia, Neuropathology 35: (1) ((2015) ), 83–90. |
[185] | Kaneko K. , Yoshida K. , Arima K. , Ohara S. , Miyajima H. , Kato T. , et al., Astrocytic deformity and globular structures are characteristic of the brains of patients with aceruloplasminemia, J Neuropathol Exp Neurol 61: (12) ((2002) ), 1069–1077. |
[186] | Kaneko K. , Hineno A. , Yoshida K. , Ohara S. , Morita H. and Ikeda S. , Extensive brain pathology in a patient with aceruloplasminemia with a prolonged duration of illness, Hum Pathol 43: (3) ((2012) ), 451–456. |
[187] | Miyajima H. , Takahashi Y. , Kamata T. , Shimizu H. , Sakai N. and Gitlin J.D. , Use of desferrioxamine in the treatment of aceruloplasminemia, Ann Neurol 41: (3) ((1997) ), 404–407. |
[188] | Finkenstedt A. , Wolf E. , Höfner E. , Gasser B.I. , Bösch S. , Bakry R. , et al., Hepatic but not brain iron is rapidly chelated by deferasirox in aceruloplasminemia due to a novel gene mutation, J Hepatol 53: (6) ((2010) ), 1101–1107. |
[189] | Alexander J. and Kowdley K.V. , HFE-associated hereditary hemochromatosis, Genet Med 11: (5) ((2009) ), 307–313. |
[190] | Gehrke S.G. , Kulaksiz H. , Herrmann T. , Riedel H-D , Bents K. , Veltkamp C. , et al., Expression of hepcidin in hereditary hemochromatosis: Evidence for a regulation in response to the serum transferrin saturation and to non-transferrin-bound iron, Blood 102: (1) ((2003) ), 371–376. |
[191] | Nichols G.M. and Bacon B.R. , Hereditary hemochromatosis: Pathogenesis and clinical features of a common disease, Am J Gastroenterol 84: (8) ((1989) ), 851–862. |
[192] | Beutler E. , Bothwell T.H. , Charlton R.W. , Motulsky A.G. , Hereditary Hemochromatosis. In; Valle DL , Beaudet AL , Vogelstein B , Kinzler KW , Antonarakis SE , Ballabio A , et al., editors. The Online Metabolic and Molecular Bases of Inherited Disease [Internet]. New York, NY: The McGraw-Hill Comanies, Inc.; (2014) [cited 2015 Jul 27]. Available from: http://mhmedical.com/content.aspx?aid=1102891675 |
[193] | Kowdley K.V. , Bennett R.L. , Motulsky A.G. HFE-Associated Hereditary Hemochromatosis. In: Pagon RA , Adam MP , Ardinger HH , Wallace SE , Amemiya A , Bean LJ , et al., editors. GeneReviews(®) [Internet]. Seattle (WA): University of Washington, Seattle; (1993) [cited 2015 Aug 9]. Available from: http://www.ncbi.nlm.nih.gov/books/[NBK1440/] |
[194] | Jazwinska E.C. , Lee S.C. , Webb S.I. , Halliday J.W. and Powell L.W. , Localization of the hemochromatosis gene close to D6S105, Am J Hum Genet 53: (2) ((1993) ), 347–352. |
[195] | Sham R.L. , Phatak P.D. , Nemeth E. , Ganz T. , Hereditary hemochromatosis due to resistance to hepcidin: High hecidin concentrations in a family with C326S ferroportin mutation, Blood 114: (2) ((2009) ), 493–494. |
[196] | Hamill R.L. , Woods J.C. and Cook B.A. , Congenital atransferrinemia. A case report and review of the literature, Am J Clin Pathol 96: (2) ((1991) ), 215–218. |
[197] | Mims M.P. , Guan Y. , Pospisilova D. , Priwitzerova M. , Indrak K. , Ponka P. , et al., Identification of a human mutation of DMT1 in a patient with microcytic anemia and iron overload, Blood 105: (3) ((2005) ), 1337–1342. |
[198] | Beaumont C. , Delaunay J , Hetet G , Grandchamp B , de Montalembert M , Tchernia G . Two new human DMT1 gene mutations in a patient with microcytic anemia, low ferritinemia, and liver iron overload, Blood 107: (10) ((2006) ), 4168–4170. |
[199] | Carroll G.J. , Breidahl W.H. , Bulsara M.K. and Olynyk J.K. , Hereditary hemochromatosis is characterized by a clinically definable arthropathy that correlates with iron load, Arthritis Rheum 63: (1) ((2011) ), 286–294. |
[200] | Niederau C. , Fischer R. , Sonnenberg A. , Stremmel W. , Trampisch H.J. and Strohmeyer G. , Survival and causes of death in cirrhotic and in noncirrhotic patients with primary hemochromatosis, N Engl J Med 313: (20) ((1985) ), 1256–1262. |
[201] | Olson L.J. , Edwards W.D. , Holmes D.R. , Miller F.A. , Nordstrom L.A. and Baldus W.P. , Endomyocardial biopsy in hemochromatosis: Clinicopathologic correlates in six cases, J Am Coll Cardiol 13: (1) ((1989) ), 116–120. |
[202] | Gandon Y. , Olivié D. , Guyader D. , Aubé C. , Oberti F. , Sebille V. , et al., Non-invasive assessment of hepatic iron stores by MRI, Lancet Lond Engl 363: (9406) ((2004) ), 357–362. |
[203] | Tziomalos K. and Perifanis V. , Liver iron content determination by magnetic resonance imaging, World J Gastroenterol WJG 16: (13) ((2010) ), 1587–1597. |
[204] | Theise N.D., Chapter 18: Liver and Gallbladder, in: Kumar V., Abbas A., Aster J. (Eds.), Robbins and Cotran pathologic basis of disease, Elsevier Health Sciences ((2014) ), 821–881. |
[205] | Iancu T.C. , Deugnier Y. , Halliday J.W. , Powell L.W. and Brissot P. , Ultrastructural sequences during liver iron overload in genetic hemochromatosis, J Hepatol 27: (4) ((1997) ), 628–638. |
[206] | Batts K.P. , Iron overload syndromes and the liver, Mod Pathol 20: (1s), S31–S39. |
[207] | Tavill A.S. and Adams P.C. , A diagnostic approach to hemochromatosis, Can J Gastroenterol 20: (8) ((2006) ), 535–540. |
[208] | Adams P.C. and Barton J.C. , How I treat hemochromatosis, Blood 116: (3) ((2010) ), 317–325. |
[209] | Mayr R. , Janecke A.R. , Schranz M. , Griffiths W.J.H. , Vogel W. , Pietrangelo A. , et al., Ferroportin disease: A systematic meta-analysis of clinical and molecular findings, J Hepatol 53: (5) ((2010) ), 941–949. |
[210] | Aggett P.J. and Harries J.T. , Current status of zinc in health and disease states, Arch Dis Child 54: (12) ((1979) ), 909–917. |
[211] | Maverakis E. , Fung M.A. , Lynch P.J. , Draznin M. , Michael D.J. , Ruben B. , et al., Acrodermatitis enteropathica and an overview of zinc metabolism, J Am Acad Dermatol 56: (1) ((2007) ), 116–124. |
[212] | Küry S. , Dréno B. , Bézieau S. , Giraudet S. , Kharfi M. , Kamoun R. , et al., Identification of SLC39A4, a gene involved in acrodermatitis enteropathica, Nat Genet 31: (3) ((2002) ), 239–240. |
[213] | Andrews G.K. , Regulation and function of Zip4, the acrodermatitis enteropathica gene, Biochem Soc Trans 36: (Pt 6) ((2008) ), 1242–1246. |
[214] | Schmitt S. , Küry S. , Giraud M. , Dréno B. , Kharfi M. and Bézieau S. , An update on mutations of the SLC39A4 gene in acrodermatitis enteropathica, Hum Mutat 30: (6) ((2009) ), 926–933. |
[215] | Mambidge K.M. , Neldner K.H. and Walravens P.A. , Letter: Zinc,acrodermatitis enteropathica, and congenital malformations, Lancet Lond Engl 1: (7906) ((1975) ), 577–578. |
[216] | Jones J.G. , Elmes M.E. , Aggett P.J. and Harries J.T. , The effect of zinc therapy on lysosomal inclusion bodies in intestinal epithelial cells in acrodermatitis enteropathica, Pediatr Res 17: (5) ((1983) ), 354–357. |
[217] | Chowanadisai W. , Lönnerdal B. and Kelleher S.L. , Identification of a mutation in SLC30A2 (ZnT-2) in women with low milk zinc concentration that results in transient neonatal zinc deficiency, J Biol Chem 281: (51) ((2006) ), 39699–39707. |
[218] | Lasry I. , Seo Y.A. , Ityel H. , Shalva N. , Pode-Shakked B. , Glaser F. , et al., A dominant negative heterozygous G87R mutation in the zinc transporter, ZnT-2 (SLC30A2), results in transient neonatal zinc deficiency, J Biol Chem 287: (35) ((2012) ), 29348–29361. |
[219] | Itsumura N. , Inamo Y. , Okazaki F. , Teranishi F. , Narita H. , Kambe T. , et al., Compound heterozygous mutations in SLC30A2/ZnT2results in low milk zinc concentrations: A novel mechanism forzinc deficiency in a breast-fed infant, PLoS One 8: (5) ((2013) ), e64045. |
[220] | Giunta C. , Elçioglu N.H. , Albrecht B. , Eich G. , Chambaz C. , Janecke A.R. , et al., Spondylocheiro dysplastic form of the Ehlers-Danlos syndrome–an autosomal-recessive entity caused by mutations in the zinc transporter gene SLC39A13, Am J Hum Genet 82: (6) ((2008) ), 1290–1305. |
[221] | Fukada T. , Civic N. , Furuichi T. , Shimoda S. , Mishima K. , Higashiyama H. , et al., The zinc transporter SLC39A13/ZIP13 isrequired for connective tissue development; its involvement inBMP/TGF-beta signaling pathways, PLoS One 3: (11) ((2008) ), e3642. |
[222] | Perez Y. , Shorer Z. , Liani-Leibson K. , Chabosseau P. , Kadir R. , Volodarsky M. , et al., SLC30A9 mutation affecting intracellular zinc homeostasis causes a novel cerebro-renal syndrome, Brain J Neurol 140: (4) ((2017) ), 928–939. |
[223] | Brandt M. , Schramm V.L. Chapter 1 - Mammalian manganese metabolism and manganese uptake and distribution in rat hepatocytes. In: Wedler VLSC , editor. Manganese in Metabolism and Enzyme Function [Internet]. Academic Press; (1986) . pp. 3–16. Available from: http://www.sciencedirect.com/science/article/pii/B9780126290509500053 |
[224] | Tuschl K. , Mills P.B. and Clayton P.T. , Manganese and the brain, Int Rev Neurobiol ((2013) ) 110: , 277–312. |
[225] | Tuschl K. , Clayton P.T. , Gospe S.M. , Gulab S. , Ibrahim S. , Singhi P. , et al., Syndrome of hepatic cirrhosis, dystonia, polycythemia, and hypermanganesemia caused by mutations in SLC30A10, a manganese transporter in man, Am J Hum Genet 90: (3) ((2012) ), 457–466. |
[226] | Quadri M. , Federico A. , Zhao T. , Breedveld G.J. , Battisti C. , Delnooz C. , et al., Mutations in SLC30A10 cause parkinsonism and dystonia with hypermanganesemia, polycythemia, and chronic liver disease, Am J Hum Genet 90: (3) ((2012) ), 467–477. |
[227] | Leyva-Illades D. , Chen P. , Zogzas C.E. , Hutchens S. , Mercado J.M. , Swaim C.D. , et al., SLC30A10 Is a Cell Surface-Localized Manganese Efflux Transporter, and Parkinsonism-Causing Mutations Block Its Intracellular Trafficking and Efflux Activity, J Neurosci 34: (42) ((2014) ), 14079–14095. |
[228] | Bosomworth H.J. , Thornton J.K. , Coneyworth L.J. , Ford D. and Valentine R.A. , Efflux function, tissue-specific expression andintracellular trafficking of the Zn transporter ZnT10 indicateroles in adult Zn homeostasis, Metallomics 4: (8) ((2012) ), 771–779. |
[229] | Tuschl K. , Clayton P.T. , Gospe S.M. , Mills P.B. Dystonia/Parkinsonism, Hypermanganesemia, Polycythemia, andChronic Liver Disease. In: Pagon RA , Adam MP , Ardinger HH , Wallace SE , Amemiya A , Bean LJ , et al., editors. GeneReviews(®)[Internet]. Seattle (WA): University of Washington, Seattle; (1993) [cited 2015 Aug 29]. Available from: http://www.ncbi.nlm.nih.gov/books/NBK100241/ |
[230] | Avelino M.A. , Fusão E.F. , Pedroso J.L. , Arita J.H. , Ribeiro R.T. , Pinho R.S. , et al., Inherited manganism: The “cock-walk” gait and typical neuroimaging features, J Neurol Sci 341: (1-2) ((2014) ), 150–152. |
[231] | Lechpammer M. , Clegg M.S. , Muzar Z. , Huebner P.A. , Jin L-W and Gospe S.M. , Pathology of inherited manganese transporter deficiency, Ann Neurol 75: (4) ((2014) ), 608–612. |
[232] | Tuschl K. , Meyer E. , Valdivia L.E. , Zhao N. , Dadswell C. , Abdul-Sada A. , et al., Mutations in SLC39A14 disrupt manganese homeostasis and cause childhood-onset parkinsonism-dystonia, Nat Commun ((2016) ) 7: , 11601. |
[233] | Tuschl K. , Gregory A. , Meyer E. , Clayton P.T. , Hayflick S.J. , Mills P.B. , et al., SLC39A14 Deficiency. In: Pagon RA , Adam MP , Ardinger HH , Wallace SE , Amemiya A , Bean LJ , et al., editors. GeneReviews(®) [Internet]. Seattle (WA): University of Washington, Seattle; (1993) [cited 2017 Sep 25]. Available from: http://www.ncbi.nlm.nih.gov/books/NBK431123/. |
[234] | Boycott K.M. , Beaulieu C.L. , Kernohan K.D. , Gebril O.H. , Mhanni A. , Chudley A.E. , et al., Autosomal-Recessive IntellectualDisability with Cerebellar Atrophy Syndrome Caused by Mutation ofthe Manganese and Zinc Transporter Gene SLC39A8, Am J HumGenet 97: (6) ((2015) ), 886–893. |
[235] | Park J.H. , Hogrebe M. , Grüneberg M. , DuChesne I. , von der Heiden A.L. , Reunert J. , et al., SLC39A8 Deficiency: A Disorder of Manganese Transport and Glycosylation, Am J Hum Genet 97: (6) ((2015) ), 894–903. |
[236] | Riley L.G. , Cowley M.J. , Gayevskiy V. , Roscioli T. , Thorburn D.R. , Prelog K. , et al., A SLC39A8 variant causes manganese deficiency,and glycosylation and mitochondrial disorders, J Inherit Metab Dis 40: (2) ((2017) ), 261–269. |
[237] | Park J.H. , Hogrebe M. , Fobker M. , Brackmann R. , Fiedler B. , Reunert J. , et al., SLC39A8 deficiency: Biochemical correction and major clinical improvement by manganese therapy, Genet Med Off J Am Coll Med Genet ((2017) ), Epub ahead of print. |
[238] | Schroeder H.A. , Frost D.V. and Balassa J.J. , Essential trace metals in man: Selenium, J Chronic Dis 23: (4) ((1970) ), 227–243. |
[239] | Kryukov G.V. , Castellano S. , Novoselov S.V. , Lobanov A.V. , Zehtab O. , Guigó R. , et al., Characterization of mammalian selenoproteomes, Science 300: (5624) ((2003) ), 1439–1443. |
[240] | Reeves M.A. and Hoffmann P.R. , The human selenoproteome: Recent insights into functions and regulation, Cell Mol Life Sci CMLS 66: (15) ((2009) ), 2457–2478. |
[241] | Roman M. , Jitaru P. and Barbante C. , Selenium biochemistry and its role for human health, Met Integr Biometal Sci 6: (1) ((2014) ), 25–54. |
[242] | Schweizer U. , Dehina N. and Schomburg L. , Disorders of selenium metabolism and selenoprotein function, Curr Opin Pediatr 23: (4) ((2011) ), 429–435. |
[243] | Fu J. and Dumitrescu A.M. , Inherited defects in thyroid hormone cell-membrane transport and metabolism, Best Pract Res Clin Endocrinol Metab 28: (2) ((2014) ), 189–201. |
[244] | Schomburg L. , Dumitrescu A.M. , Liao X-H , Bin-Abbas B. , Hoeflich J. , Köhrle J. , et al., Selenium supplementation fails to correct the selenoprotein synthesis defect in subjects with SBP2 gene mutations, Thyroid Off J Am Thyroid Assoc 19: (3) ((2009) ), 277–281. |
[245] | Schoenmakers E. , Agostini M. , Mitchell C. , Schoenmakers N. , Papp L. , Rajanayagam O. , et al., Mutations in the selenocysteine insertion sequence-binding protein 2 gene lead to a multisystem selenoprotein deficiency disorder in humans, J Clin Invest 120: (12) ((2010) ), 4220–4235. |
[246] | Ben-Zeev B. , Hoffman C. , Lev D. , Watemberg N. , Malinger G. , Brand N. , et al., Progressive cerebellocerebral atrophy: A new syndrome with microcephaly, mental retardation, and spastic quadriplegia, J Med Genet 40: (8) ((2003) ), e96. |
[247] | Agamy O. , Ben Zeev B. , Lev D. , Marcus B. , Fine D. , Su D. , et al., Mutations disrupting selenocysteine formation cause progressive cerebello-cerebral atrophy, Am J Hum Genet 87: (4) ((2010) ), 538–544. |
[248] | Anttonen A.-K. , Hilander T. , Linnankivi T. , Isohanni P. , French R.L. , Liu Y. , et al., Selenoprotein biosynthesis defect causes progressive encephalopathy with elevated lactate, Neurology 85: (4) ((2015) ), 306–315. |


