
Meeting report from the Mitochondrial Medicine Southeast Regional Symposium — understanding mitochondrial disease and mitochondrial dysfunction: Opportunities and impacts in the clinic and laboratory
Abstract
On April 7th, 2017, the Foundation for Mitochondrial Medicine and the United Mitochondrial Disease Foundation held a Southeast Regional Symposium in Birmingham, Alabama to explore mitochondrial dysfunction, diseases and conditions related to mitochondrial dysfunction, their clinical management, and new therapies and tools to address those conditions. This article provides a report of the proceedings.
1Introduction
On April 7th, 2017, the Foundation for Mitochondrial Medicine and the United Mitochondrial Disease Foundation held a Southeast Regional Symposium in Birmingham, Alabama to explore mitochondrial dysfunction, diseases and conditions related to mitochondrial dysfunction, their clinical management, and new therapies and tools to address those conditions.
Thought leaders and presenters included:
• Bruce H. Cohen, MD, Course Co-Chair, Akron Children’s Hospital, Akron, OH—Potential Therapies and Treatments for Mitochondrial Disease
• Leon S. Dure, MD, Course Co-Chair, Children’s Hospital of Alabama, Birmingham, AL-Pediatric Neurology and Mitochondrial Medicine at UAB
• Victor Darley-Usmar, PhD, University of Alabama at Birmingham School of Medicine, Birmingham, AL—Bioenergetics and Mitochondrial Medicine
• David Ferrick, Agilent Technologies—Bioenergetics and Mitochondrial Medicine
• Matthew Goldberg, PhD, University of Alabama at Birmingham School of Medicine, Birmingham, AL—Parkinson’s and Mitochondrial Dysfunction
• Gene Kelly, Stealth Biotherapeutics, Clinical Trials Targeting Mitochondrial Dysfunction
• Shilpa Iyer, PhD, University of Arkansas, Fayetteville, AR—DOD Funding for Study of Leigh’s Disease
• Amel Karaa, MD, Massachusetts General Hospital, Boston, MA—Mitochondrial Disease Primer
• Matthew Klein, MD, Bioelectron Technology Corporation—Clinical Trials Targeting Mitochondrial Dysfunction
• Robin Morris, PhD, Georgia State University, Neuropsychology, Atlanta, GA—Autism and Mitochondrial Dysfunction
• Nell Rebowe, Anavex Pharmaceuticals, Clinical Trials Targeting Mitochondrial Dysfunction
• Alex Sherman, PhD, Massachusetts General Hospital, Boston, MA—ALS and Mitochondrial Dysfunction
• Diana Shineman, PhD, Alzheimer’s Drug Discovery Foundation—Alzheimer’s and Mitochondrial Dysfunction
• Keshav Singh, PhD, University of Alabama at Birmingham, AL—Cancer and Mitochondrial Dysfunction
• Phil Yeske, PhD, United Mitochondrial Disease Foundation
2Basic science of mitochondrial dysfunction and disease
Amel Karaa, MD, Instructor in Pediatrics at Massachusetts General Hospital, provided an overview of mitochondria and their functions within cells.
Subcellular organelles, the cigar-shaped mitochondria are approximately 1 micron in length. Mitochondria, which play many roles in the cell, reside in the cytoplasm of all cells except red blood cells, and there can be thousands per cell. They are mobile within the cell and can make relationships with other organelles, and they look histologically different from cell to cell in response to the energy needs of that cell or tissue.
In addition to the main function of generating adenosine triphosphate (ATP), mitochondria perform many functions, such as being a critical component of apoptosis and maintaining calcium homeostasis. Because of their myriad roles within cells, mitochondrial dysfunction plays a role in many neurodegenerative diseases and in some more common disorders such as cancers, heart disease, and diabetes.
Mitochondria have their own DNA (mtDNA), which is inherited from the mother and contains 37 genes. However, because nuclear DNA (nDNA), inherited from the mother, father, or both, has input into the normal function of mtDNA, other inheritance patterns can be observed (autosomal dominant, recessive, X-linked). Overall, the maternal pattern of inheritance is due to the greater number of mitochondria in the oocyte, which is rich in mitochondria, compared to the sperm, which contains only a few. The process of fertilization further dilutes the number of sperm mtDNA as most sperm are destroyed. Image 1 shows mitochondrial inheritance.
Image.1
Mitochondrial inheritance. Source: Greenwood Genetic Center.
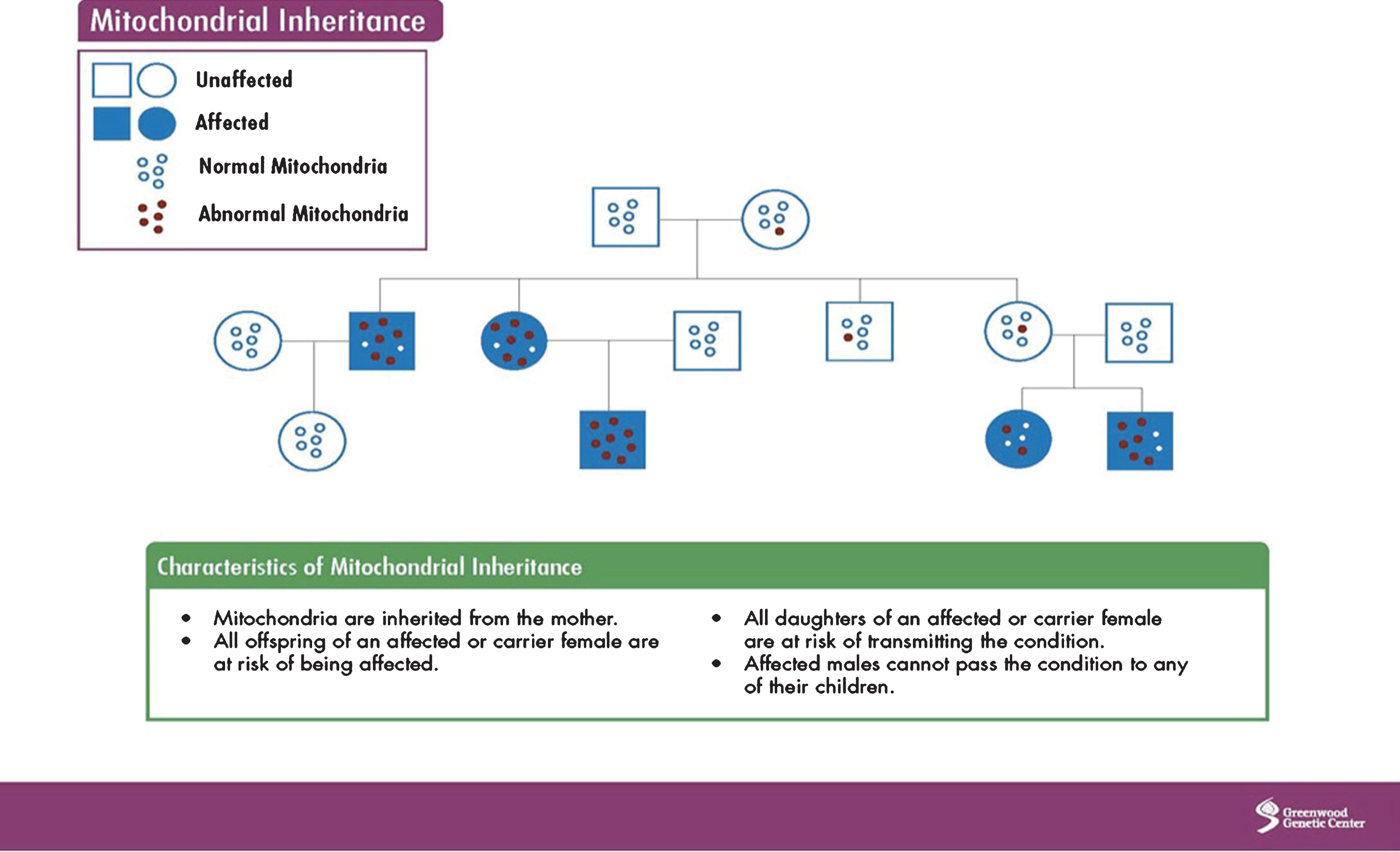
MtDNA can undergo point, deletion, and duplication mutations, as well as depletion, which occurs when mtDNA become so low in numbers that they cannot carry out normal mitochondrial function.
It is estimated that 1 in 200 people have an mtDNA mutation. So why do patients, even related ones, have such different signs and symptoms? A few concepts help explain this situation:
1. Random segregation: The egg is rich in mitochondria, but its normal and abnormal mitochondria are randomly segregated every time it divides. This produces an uneven distribution of normal and abnormal mitochondria within each cell.
2. Threshold expression: A cell can function normally if it has enough normal mtDNA. But over time, the number of mutant mtDNA increases, so at some point (the threshold) the cell cannot sustain normal function. This threshold is different for each person and can make mitochondrial disease look very different from one person to another. Heteroplasmy is used to describe and measure the number of normal versus abnormal mitochondria in a cell. Ten percent heteroplasmy means 10% of mitochondria carry an abnormality.
The tissues that are the primary targets for mitochondrial disease include those with high-energy needs that cannot handle oxidative damage. Tissues mostly affected include the brain, retina, heart, and over time, the kidneys, liver, pancreas, hearing, and peripheral nervous system. Mitochondrial dysfunction can cause complex, heterogeneous diseases that can be challenging to treat. For example, two well-characterized mitochondrial diseases are MELAS (mitochondrial encephalomyopathy, lactic acidosis, and stroke-like episodes) and MERRF (myoclonic epilepsy with ragged red fibers). The primary symptoms of the diseases represent wide-ranging clinical features.
Mitochondrial disease impairs energy function, often by creating errors in the proteins involved in the electron transport (ET) chain that produces ATP. This dynamic system can be rearranged to make more energy when it is required, and the various complexes can combine and re-arrange depending on energy requirements. The 5 complexes of the oxidative phosphorylation system are coded by mtDNA and nDNA, which explains why these 2 genomes need to both be intact for optimal mitochondrial function and that abnormalities in one or the other can lead to mitochondrial disease.
MtDNA and nDNA are closely intertwined in areas outside of the ET chain, too. There are about 1500 nuclear genes that can affect mitochondrial function. NDNA can affect mtDNA through single base change, deletions, or duplications. For example, polymerase gamma (POLG) is one of the most common nDNA mutations that affect how mtDNA is regulated. POLG has 3 domains where changes occur and that can lead to mtDNA changes and disease. Therefore, one specific gene can lead to a tremendous amount of heterogeneity in outcomes with diseases that can occur in early childhood to later in life. For example, all of the patients below have a mutation in POLG:
• An 8-month-old female with jaundice, liver failure, and seizures;
• A 3-year-old male with developmental regression, failure to thrive, and lactic acidosis;
• A 15-year-old female with migraines, severe constipation, and fainting;
• A 24-year-old male with speech and balance problems;
• A 55-year-old male with weakness, muscle pain, and droopy eyelids.
Overall, children with mitochondrial disease usually present with liver, kidney, and brain issues and their diseases are often more devastating. Adult-onset disease is often musculoskeletal, initially noted by exercise intolerance. But, the course of mitochondrial diseases is unpredictable. A child may grow up and his or her condition may improve, or an adult’s condition may progress quickly. Further, the diseases can also be episodic and flare due to metabolic stressors, which could be anything that puts extra demands on the system, such as infections, surgeries, toxins, or drugs.
There is also a distinction between primary and secondary mitochondrial disease. Primary mitochondria disease has an inborn genetic error as the cause of the mitochondrial disease. Secondary mitochondrial diseases, such as Parkinson’s disease or Alzheimer’s disease, are due to acquired mitochondrial abnormalities that happen after birth and require other factors, such as lifestyle choices, drugs, or toxins that can damage the mitochondria and result in disease states.
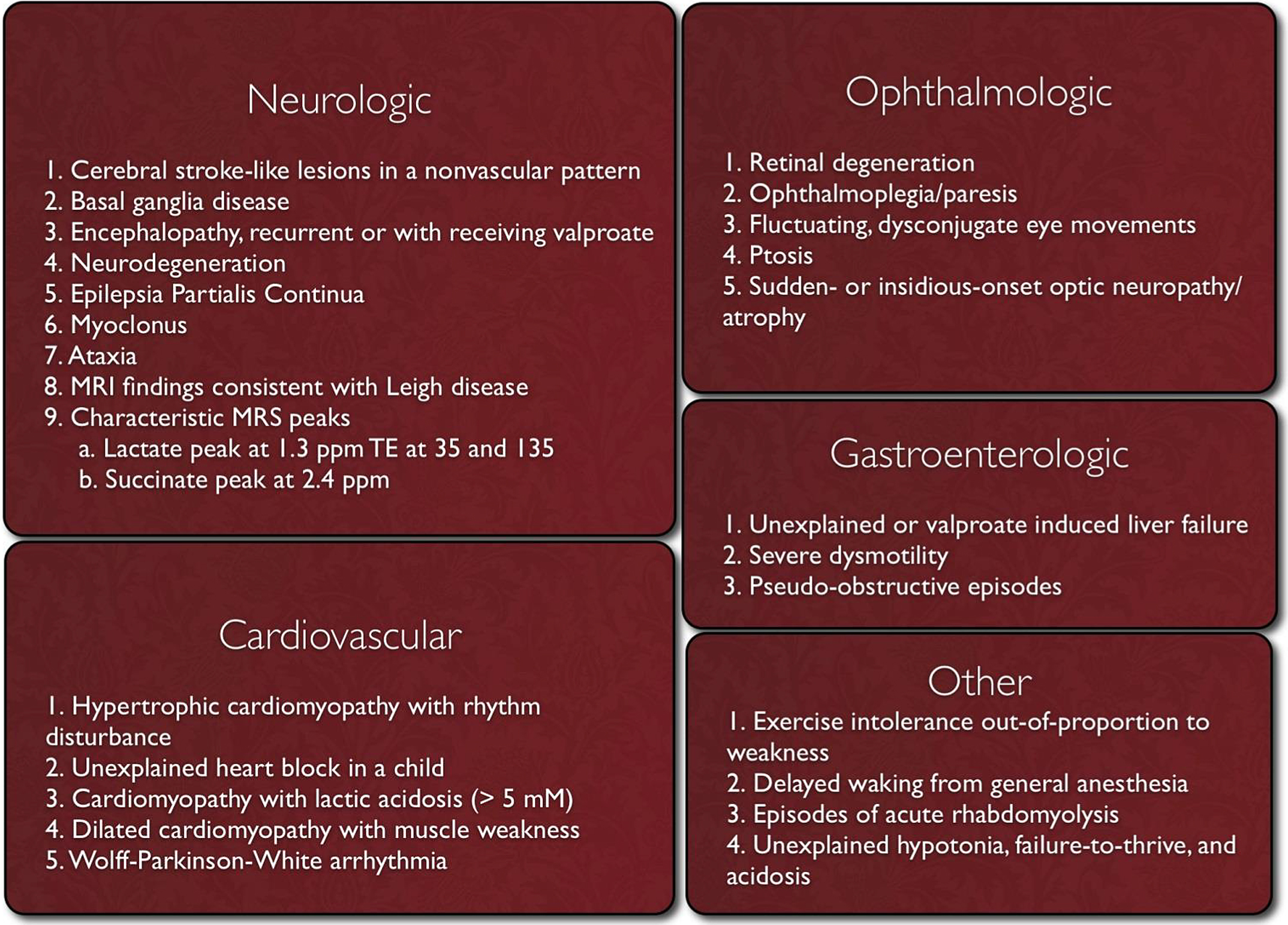
Clinicians should suspect mitochondrial disease if patients present with the red flag symptoms listed in Image 2.
Image.2
Red Flag Symptoms for Mitochondrial Disease. Source: Mitochondrial Medicine Society http://www.mitosoc.org/toolkit/.
Even with the defining of red flag symptoms, there is a great need for a diagnostic algorithm for mitochondrial disease. Many symptoms are nonspecific, and muscle testing, which used to be the gold standard of diagnosis, has high rates of false positives and false negatives. One such effort at creating a diagnostic process for mitochondrial disease suggests the following [1]:
• The presence of red flag symptoms;
• Basic screening tests (liver enzymes, blood tests, urine tests etc.), imaging to assess organ involvement;
• Molecular testing – the new gold standard. Can test mtDNA and nDNA for mutations.
The Mitochondrial Medicine Society maintains publications providing specific details on diagnostics for mitochondrial diseases at http://www.mitosoc.org/toolkit/.
The clinical management for mitochondrial disease is mostly supportive and focuses on symptom management and the prevention of complications. Treatment is often individualized, and there is no single treatment due to the variable course of disease among patients. Patients are counseled on balancing energy needs – avoiding stressors, getting restorative sleep, practicing good nutrition, and exercising in order to minimize energy loss and to optimize the creation and maintenance of energy.
Clinical advancements for mitochondrial disease are in the near future: multiple clinical trials, new diagnostic tools, and mitochondrial replacement therapy has been approved in the United Kingdom, [2] making it possible for women with defective mtDNA to have children without passing along the mutations due to the insertion of healthy mitochondria from a donor.
3Management of patients with mitochondrial dysfunction
Bruce Cohen, MD, Director of The NeuroDevelopmental Science Center and Center for Mitochondrial Research at Akron Children’s Hospital, provided profiles of patients he treated over the years to exemplify the range of signs and symptoms exhibited by mitochondrial dysfunction and how his patient-centered treatments have evolved [3].
Patient profiles included several patients that fit the clinical criteria for mitochondrial disease but have eluded a genetic diagnosis:
• A 6-year-old, who had early developmental delays, intellectual disability, recurrent high fevers without viral illness, low-grade neural crest tumor, lactic acidosis, thalamic changes as seen on an MRI, RRF changes as seen on muscle biopsy, sleeping only a few hours per day. Patient died in her sleep during a febrile event.
• A high school track-and-field star who repeatedly became encephalopathic and hemiparetic during races, especially on hot days. He had mild analyte abnormalities and ANT defect on polarography. He now works in a high-powered job in New York City.
• A 4-year-old girl with developmental delays, ataxia, intellectual disability, autism spectrum disorder (ASD), hemiparesis with headache and encephalopathy, lactic acidosis, high alanine. MRI showed corpocephaly and a muscle biopsy showed ET complex I deficiency. The patient fell asleep under a tree on a hot day and was not able to be aroused, but she returned to baseline in a month. Overall, her diagnosis was a loose MELAS phenotype.
• A young adult, who was ambulatory but ataxic with dystonia and chorea, dysarthria, ASD.
• 1-year-old boy with mild motor delays, sudden onset cardiomyopathy, carnitine deficiency, lactic acidosis, heart transplant candidate, muscle biopsy, complex I defect, very long chain acyl-CoA dehydrogenase (VLCAD) defect.
• 61-year-old man, with many classic mitochondrial signs, was referred to cardiology for pacemaker. He became anxious in the waiting room, then lapsed into deep stupor. CT showed bilateral calcified basal ganglia. Medical history included 8 abdominal surgeries for “acute abdomen,” progressive sarcopenia and lipodystrophy, ptosis without further progressive external opthalmoplegia (PEO), neuropathy without DM, hepatic fibrosis, loss of intellectual prowess, family history (mother with similar illness, son with early signs of illness), and persistent lactic acidosis.
From these few patient descriptions, it is easy to see how wide-ranging the signs and symptoms of mitochondrial disease can be. Further, signs and symptoms may vary depending on whether the patient has primary or secondary mitochondrial disease or disorders (see Image 3). Finally, some patients, even those having extensive genetic evaluations, may escape a complete diagnosis.
Image.3
Primary mitochondrial disease versus secondary mitochondrial disorders. Source: Bruce Cohen, MD, Akron Children’s Hospital. Modified from Mechanism of Disease: Monogenic Mitochondrial Disorders. N Engl J Med 2012; 366:1132-1141. Available at http://www.nejm.org/doi/full/10.1056/NEJMra1012478 [4].
![Primary mitochondrial disease versus secondary mitochondrial disorders. Source: Bruce Cohen, MD, Akron Children’s Hospital. Modified from Mechanism of Disease: Monogenic Mitochondrial Disorders. N Engl J Med 2012; 366:1132-1141. Available at http://www.nejm.org/doi/full/10.1056/NEJMra1012478 [4].](https://content.iospress.com:443/media/trd/2017/2-3-4/trd-2-3-4-trd014/trd-2-trd014-g003.jpg)
The classic mitochondrial phenotype includes:
• Step-wise central nervous system (CNS) deterioration (movement disorder ataxia);
• Ptosis and PEO;
• Progressive optic atrophy or retinitis pigmentosa;
• High frequency hearing loss;
• Cardiac conduction defects;
• Myopathy or cardiomyopathy;
• Hepatopathy;
• Neuropathy (large fiber);
• Systemic lipomatosis or lipodystrophy;
• Classic MRI findings (symmetric, deep gray);
• Biochemical: true lactic acidosis (LA), classic amino acid pattern, classic organic acid patterns.
There are no FDA-approved therapies for the specific treatment of mitochondrial diseases. Therapy should be directed at symptomatic control. Although the use of supplements remains unproven for groups of patients with mitochondrial disease, some patients have responses that warrant continued treatment. Thus, vitamin and cofactor therapy has become part of most patients’ treatment plans. Because patients exhibit such a range of signs and symptoms, the therapies need to be individualized to address each patient’s needs.
Overall, the goals of vitamin and cofactor therapy and the basis for the rational to try them include:
• Stimulate poorly functioning enzymes (example, using thiamine for pyruvate dehydrogenase deficiency);
• Increase antioxidant activity to reduce oxidative stress and effects (CoQ10, N-acetyl cysteine);
• Provide alternative energy sources (succinate, medium-chain triglyceride oil);
• Improve muscle bulk (creatine monohydrate);
• Scavenge free-fatty acids and poisonous organic acids (levocarnitine);
• Bypass blocked components of the electron transport chain (vitamins K and C);
• Address vascular effects (L-arginine, L-citrulline);
• Replace deficient vitamins and cofactors (Co-Q10, folinic acid);
• Offer a medical therapy; even if not a proven therapy;
• Offer the chance for increased partnership between clinician and patients/families;
• Offer hope.
Over the years, Dr. Cohen’s treatment has evolved from using 17 vitamins and cofactors to using about 6 today. The reasons for the evolution were the recognition that the shotgun approach of using all the vitamins and cofactors does not always work, the availability of evolving clinical trial evidence showing that some therapies do not work at all, and the expense involved in taking many vitamins and cofactors [5].
Dr. Cohen’s typical treatment for mitochondrial disorder or disease includes:
• Maximal exercise, including endurance and resistance training, and a combination of physical, occupational, or speech therapies;
• Sleep hygiene, including polysomnograms for all patients to evaluate brain activity, oxygen levels in blood, heart rate, breathing, and eye and leg movements during sleep;
• Hydration, including early IV hydration during viral illnesses;
• Basic supplements:
• CoEnzyme Q10 5–20 mg per kg per day;
• Carnitor only if carnitine deficient;
• B2 100–600 mg per day;
• Creatine monohydrate 0.1 grams per kg per day; max 5 grams a day;
• Alpha-Lipoic Acid 300 mg bid for an adult;
• Folinic Acid for CSF folate-deficient patients: 5–25 mg tid;
• L-arginine 0.15-0.3 gram per kg per day; 4–24 grams a day for an adult;
• L-citrulline 0.1 grams per kg per day;
• Antioxidants (gamma form of vitamin E, vitamin C, selenium, zinc);
• Miralax polyethylene glycol for constipation;
• Avoidance and prompt treatment of stressors, such as illness, fever, malnutrition, and lack of sleep.
Nutraceuticals are a relatively recent addition to the portfolio of mitochondrial treatments. They can including alpha lipoic acid, L-arginine or L-citrulline (for MELAS patients, especially), and folinic acid
One of the reasons that treatments for patients with mitochondrial disorders are so individualized is because there are few randomized, controlled clinical trials (RCTs) that study treatments for these disorders. The individualized and often-fluctuating nature of mitochondrial disease contributes to the problem of creating RCTs. For example
• There are many different disease manifestations of mitochondrial disease – different pathways are affected by different genes.
• Defining the outcomes of treatments is difficult due to the heterogeneous nature of patient’s illnesses.
• Mitochondrial disease is not static – it often has a long and unpredictable course.
• Patients with mitochondrial disease often experience swings in symptoms, making it difficult to know when to measure function.
• It can be difficult to get patients to meet criteria.
Additionally, statistical considerations, cost, travel, and the quality of data further impede the development of clinical trials for mitochondrial dysfunction.
The differences between primary and secondary mitochondrial diseases and differences in what insurance companies will pay for contribute to an individualized approach to managing mitochondrial diseases. Therefore, physicians who treat patients with mitochondrial disease often pursue an individualized approach based on what works best in their patients.
4Panel discussion: Highlights from ongoing clinical trials in therapies targeting mitochondrial dysfunction: BioElectron Technology Corporation (formerly, Edison Pharmaceuticals), Stealth BioTherapeutics, Reata Pharmaceuticals, and Anavex Pharmaceuticals
Philip Yeske, PhD, Science and Alliance Officer for UMDF, moderated a panel on new drug development that included:
Matthew Klein, MD, Chief Medical Officer for BioEelectron Technology Corporation, who described his company’s research into two compounds: EPI-743 and EPI-589.
• EPI-743 is a drug candidate in late-stage clinical development for inherited mitochondrial diseases. EPI-743 is administered orally, passes into the brain, and works by regulating key enzymes involved in the synthesis and regulation of energy metabolism. EPI-743 has been studied in patients with Leigh syndrome, Friedriech’s ataxia, Rett syndrome, Cobalamin C defect, NIH undiagnosed diseases of redox and metabolism, MELAS, and Parkinson’s disease.
• EPI-589 is a drug candidate in phase II development for adult neurodegenerative diseases, including amyotrophic lateral sclerosis (ALS) and Parkinson’s disease.
About Edison Pharmaceuticals/BioElectron Technology Corporation: BioElectron is the first hybrid biotech/tech platform company with a focus on dynamic energy parameters within all living systems. The mission of BioElectron is to learn how biological energy works to engineer solutions to complex dynamic problems in biology. BioElectron has built the first biological energy learning engine, combining biotech and tech tools to develop robust and predictive models of complex biological systems using unique data derived from electron transfer chemical reactions in annotated biological systems.
Gene Kelly, Senior Director of Commercial & Scientific Liaison at Stealth BioTherapeutics explained how Stealth’s compound, elamipretide, targets the inner mitochondrial membrane to restore electron transport, which helps improve cellular energy production and reduce oxidative stress created by the over-production of reactive oxygen species (ROS), which are important signaling molecules in the mitochondria.
Elamipretide works by penetrating the cellular and outer mitochondrial membranes, and targets cardiolipin, which is found exclusively in the inner mitochondrial membrane. Elamipretide has been shown in preclinical studies to positively affect dysfunctional mitochondria, with no effect on healthy mitochondria. Image 4 shows a schematic of elamipretide’s penetration of mitochondrial membranes.
Image.4
Schematic of Elamipretide’s penetration of mitochondrial membrane. Source: Stealth BioTherapeutics.
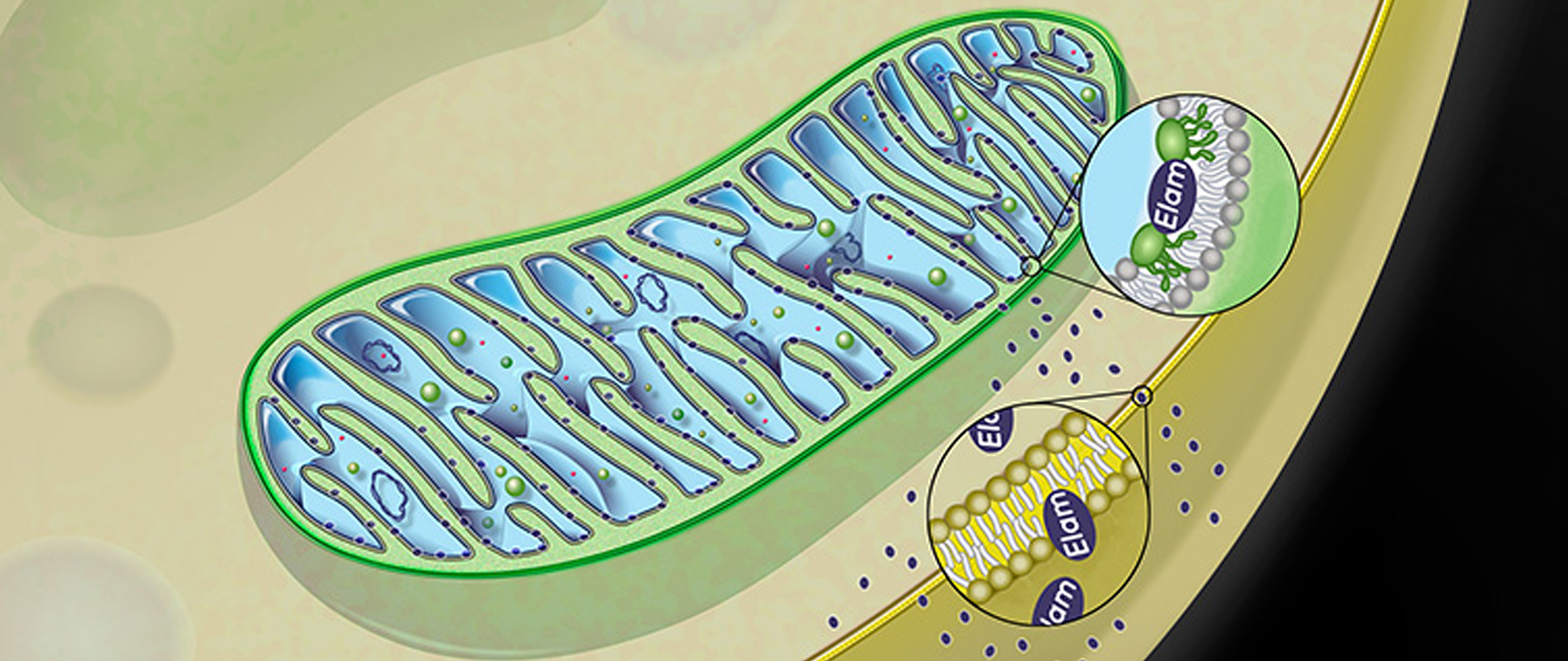
The history of elamipretide is one of serendipity and vision – vision because researchers were looking for a therapeutic opportunity for mitochondrial treatment and serendipity because the licensed compound they ended up with, which they thought initially to be a new opioid, caused mice to be more active and playful. Eventually, researchers learned of elamipretide’s effects on bioenergetics- it increases production of ATP, decreases ROS, and causes an increase in mitochondrial crista structure. Stealth is focused on treating both rare and common diseases associated with mitochondrial dysfunction.
Elamipretide is currently in multiple phase I and II clinical studies for a variety of diseases, both rare and common. Data from these clinical trials suggest that elamipretide is well tolerated in those patients and conditions studied.
About Stealth BioTherapeutics: Stealth BioTherapeutics is an innovative biopharmaceutical company, working to develop therapies to treat mitochondrial dysfunction associated with genetic mitochondrial diseases and other common diseases of aging. Founded in 2007, Stealth BioTherapeutics is a clinical stage biopharmaceutical company developing investigational drugs for the treatment of diseases involving mitochondrial dysfunction, with a scientific focus on the mitochondria and the ways in which mitochondrial therapy can positively impact the lives of patients.
Bruce Cohen, MD, Akron Children’s Hospital/Reata Pharmaceuticals, who presented on the compound RTA 408, which, in clinical trials, has been shown to upregulate mitochondrial function in a safety and efficacy study.
RTA 408 binds to Keap-1 activating transcription factor Nrf2 and inhibits transcription factor NF-kB (nuclear factor kappa-light-chain enhancer of activated B cells) to control transcription of DNA, cytokine production, and cell survival. Image 5 is a graphic representation of RTA 408’s method of action.
Image.5
RTA 408 method of action. Source: Reata Pharmaceuticals.
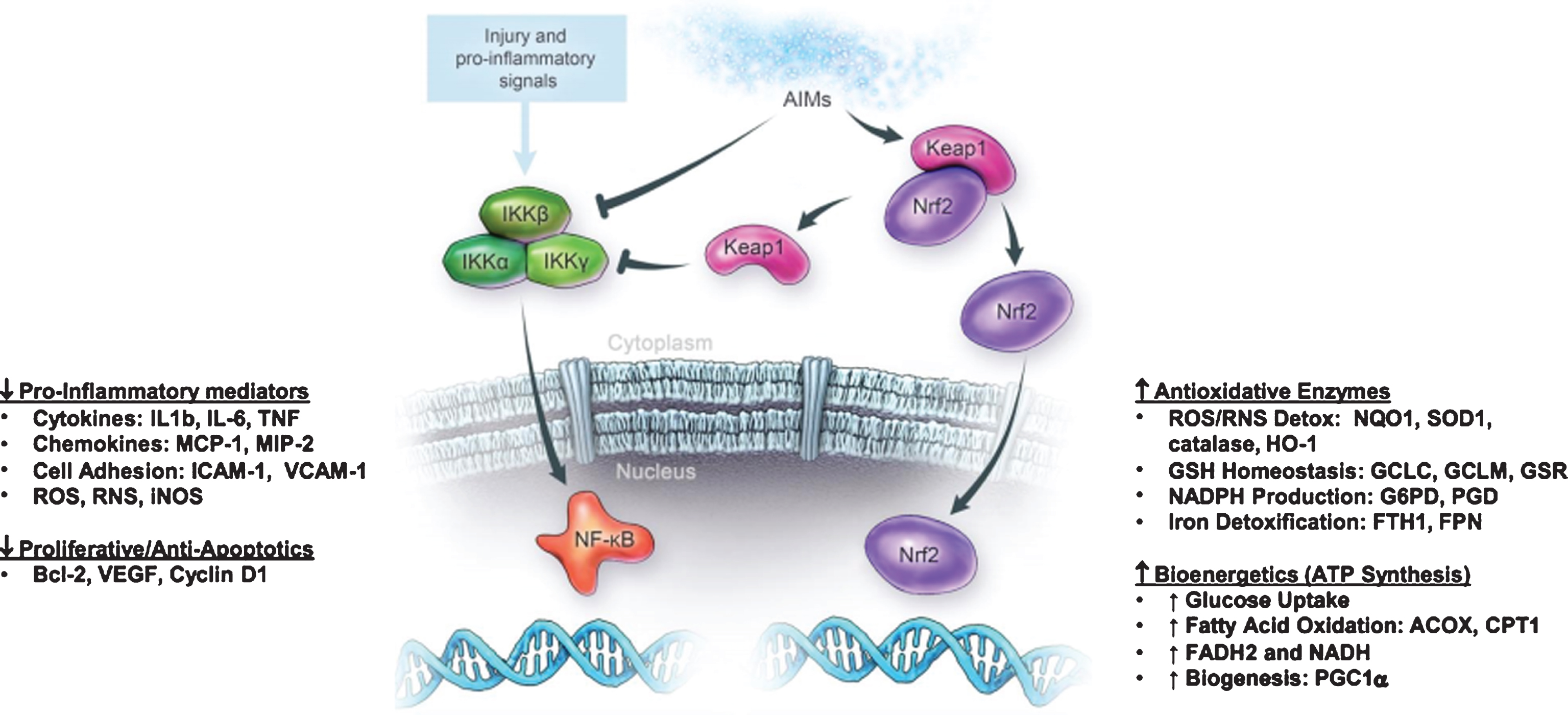
RTA 408 has been tested in Parkinson’s disease, ALS, and Fanconi anemia patient fibroblasts and has shown to restore mitochondrial function, as assessed by transmembrane potential and ATP levels. It increases the glutathione and the nicotinamide adenine dinucleotide hydrate (NADH) pool (reducing equivalents for electron transport chain). Work is ongoing in models of epilepsy, dementia, and others.
About Reata Pharmaceuticals: Reata Pharmaceutical’s mission is to develop novel therapeutics for patients with rare and life-threatening diseases by targeting molecular pathways that regulate cellular metabolism and inflammation.
Nell Rebowe, Director of Business Development and Investor Relations, Anavex Life Sciences Corporation who presented on Anavex 2–73, the lead compound being developed by Anavex Life Sciences Corporation.
Anavex 2–73 is a sigma 1 receptor agonist. Sigma receptors may be targets for therapeutics to combat many human diseases, including Alzheimer’s disease, Rett syndrome, and Parkinson’s disease. When bound by the appropriate ligands, sigma receptors influence the functioning of multiple biochemical signals that are involved in the pathogenesis of disease.
Anavex completed a Phase IIa trial with Anavex 2–73 in November 2016. It plans to initiate a Phase II/III trial, expected to include up to 300 mild-to-moderate Alzheimer’s patients. In a Phase I trial, Anavex 2–73 showed reversal of memory loss and neuroprotection in several models of Alzheimer’s disease. Anavex plans to begin studying the compound in patients with Rett syndrome this year, in conjunction with Dr. Alan Percy at the University of Alabama, Birmingham.
About Anavex Life Sciences Corporation: Anavex Life Sciences Corporation is dedicated to the development of differentiated therapeutics for the treatment of neurodegenerative and neurodevelopmental diseases, including Alzheimer’s diseases, other central nervous system diseases, pain, and various types of cancer.
5Bioenergetics and mitochondrial medicine
Victor Darley-Usmar, PhD, Professor of Mitochondrial Medicine and Pathology at the University of Alabama, Birmingham, presented on bioenergetic health, a new concept to establish the relationship between mitochondrial function and metabolism with the evolution of precision medicine and pathology in the context of mitochondrial diseases arising from direct genetic lesions or as a secondary consequence of disease processes.
Mitochondrial function is integrated with metabolism and services the specific demands of varied cell types. Importantly, this extends far beyond the canonical function of providing ATP for cellular energetics. The key concept is the integrated measures of bioenergetics health can serve as an early warning system for the bioenergetics crises and can reflect the impact of aging, genetics, lifestyle and the environment. Based on this concept and using an established cellular test of mitochondrial function, the Bioenergetic Health Index (BHI) [6] has been developed to monitor changes in patients’ bioenergetics, which can help determine severity and progression of complex diseases and their responses to treatment. The test can be run on patient platelets and leukocytes, which act as a systemic biomarker of mitochondrial function in the brain, kidney, skeletal muscle and other organs.
Image 6 shows the progressive deterioration of bioenergetic health related to mitochondrial quality. Healthy subjects have a high reserve capacity, high ATP-linked respiration, and low proton leak. In response to pathological or environmental stress, damage to mitochondria accumulates and bioenergetic health deteriorates. Subjects with mitochondrial issues display low ATP-linked respiration, low reserve capacity, and high non-mitochondrial respiration. These mitochondria show increased proton leak and the need for higher levels of ATP. As chronic stress continues, further deterioration in mitochondrial function occurs, which can be measured as a decreasing BHI.
Image.6
BHI as a dynamic measure of the response of the body to stress. Source: Chacko BK, Kramer PA, Ravi S, et al. The Bioenergetic Health Index: A new concept in mitochondrial translational research. Clin Sci (Lond). 2014;127(pt 6):367-373. Available at http://www.clinsci.org/content/127/6/367/.
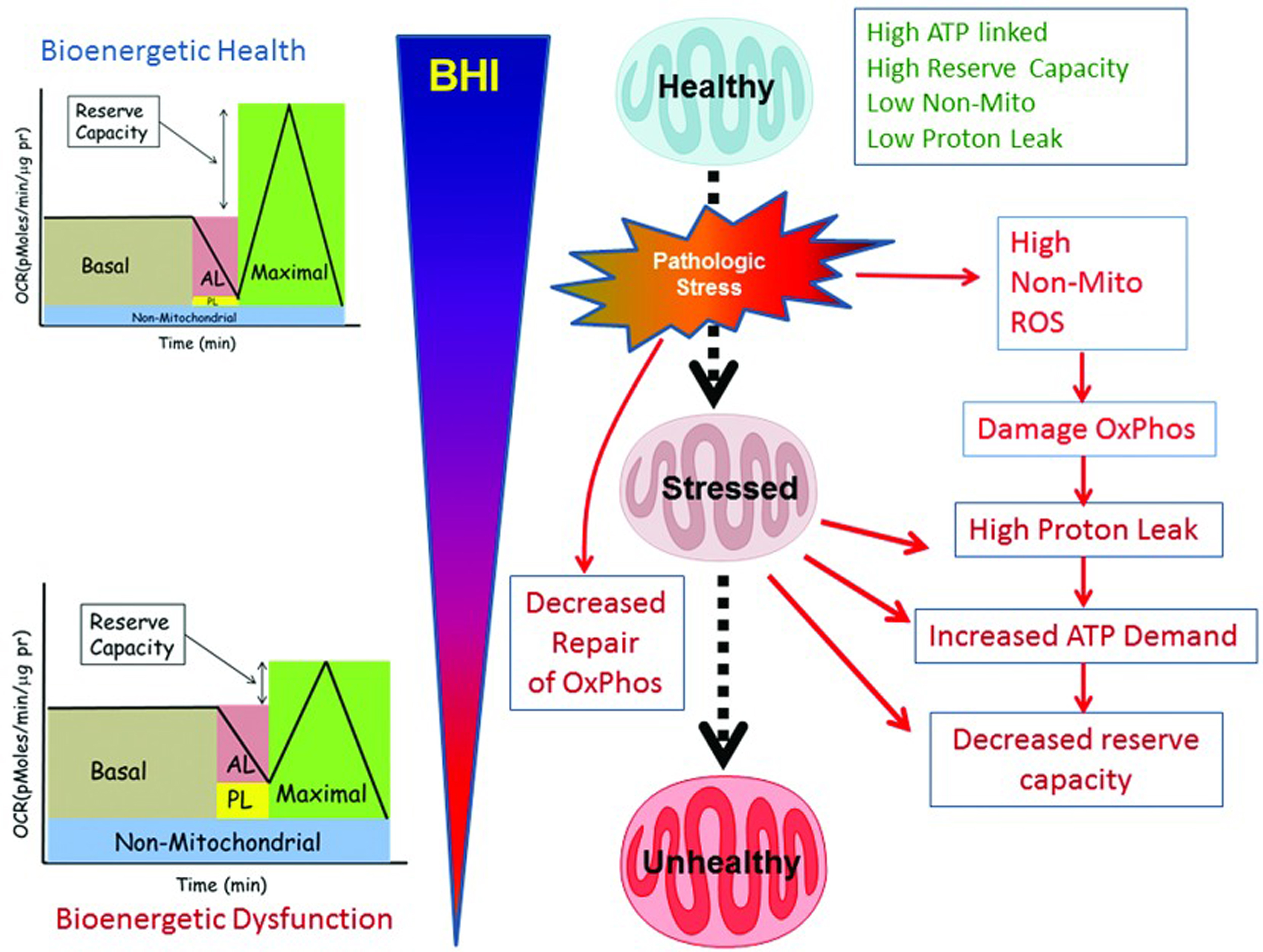
The general process of obtaining a BHI is based upon an established mitochondrial stress test used widely in the field, which has been specifically adapted for platelets and cells isolated from blood. While these concepts are in their early stages, several independent research groups have now confirmed validation of the general approach.
The BHI could have many potential uses. For example, BHI could be used to
• Measure the efficacy of an intervention;
• Pinpoint treatment that could be useful for certain conditions;
• Predict the outcome of a treatment;
• Monitor the differing bioenergy health of different cells in the body;
• Provide an objective measure of fatigue and replace muscle biopsies;
• Differentiate between those with fatigue due to mitochondrial dysfunction and those whose fatigue is due to other issues.
A measurement such as the BHI has potential as a standardized clinical test as a measure of target engagement in clinical trials with novel metabolically targeted therapeutics.
6Department of Defense funding for the study of origins of Leigh’s disease
Shilpa Iyer, PhD, Assistant Professor in the J. William Fulbright College of Arts & Sciences at the University of Arkansas, presented on two areas: the education of the general public, doctors, schools, and others about bioenergetics; and basic research into the personalized medicine of turning a patient’s stem cells into viable therapies for mitochondrial dysfunction.
6.1Educating about bioenergetics
Dr. Iyer’s group created the Bio-E program, which consists of a walk-through exhibit that combines art and science to explain the science of mitochondria and bioenergetics in way that is accessible to consumers [7]. Through the use of images, dance, poetry, and videos, the exhibit conveys the science of bioenergetics and the reality of living with mitochondria dysfunction that affects the bioenergetics. Over 80,000 individuals and 39,726 people in groups, such as school groups, have viewed the exhibit. Outcome measures taken after individuals have viewed the exhibit showed an 8% increase in knowledge of bioenergetics. Their website, https://bioenergetics.uark.edu, includes all patient videos, so visitors can view these videos by accessing the website.
6.2Research
Because the symptoms that patients with mitochondrial disease face are so disparate depending on which tissues in the body are affected, Dr. Iyer’s group focuses on creating individual therapies based on a patient’s stem cells, which can become one of 220 different cells types in the body, such as brain, nerve, or heart cells. Dr. Iyer’s lab’s approach is to
• Create specific precursor stem cell models for drug discovery and testing studies;
• Create patient-specific induced pluripotent stem cell models for understanding energetics and tissues specific variability due to mitochondrial DNA mutations [8].
Dr. Iyer’s group is genetically reprogramming skin cells from patients diagnosed with mitochondrial and other energy-deficiency diseases into stem cells, and then transforming those stem cells into neurons and muscle cells. This groundbreaking work developed over a series of steps.
Initially, Dr. Iyer’s group showed that pathogenic mtDNA could be introduced and expressed into hNPs (human pluripotent stem cell-derived neural progenitor cells) without loss of phenotype or neuronal differentiation potential. This mitochondrial gene replacement technology allows for creation of in vitro stem cell-based models useful for understanding neuronal development and treatment of neurodegenerative disorders [9].
The next approach was to create precursor stem cells for personalized medicine purposes. The researchers chose Leigh’s Syndrome, a well-characterized syndrome resulting from a genetic mutation in mtDNA. They successfully created a clinical grade induced pluripotent cell line for this mitochondrial disorder. The mutation is stably expressed over time. The next steps are to
• Create human pluripotent stem cells (hPSCs) from multiple patient cell lines;
• Study mitochondrial DNA transmission and its role in self-renewal and differentiation;
• Study how mitochondrial DNA transmission affects disease severity;
• Study how mitochondrial DNA transmission affects stem cell energetics;
• Apply cell-based models in our models for drug discovery and testing studies.
While Dr. Iyer’s work is still preliminary, it offers great hope to provide individualized treatment for mitochondrial-based disorders.
7Panel: Mitochondria and related diseases: Alzheimer’s disease, cancer, Parkinson’s disease, Amyotrophic lateral sclerosis (ALS), and autism
7.1Targeting Mitochondrial function in Alzheimer’s disease
Diana Shineman, PhD, Senior Director of Scientific Affairs for the Alzheimer’s Drug Discovery Foundation (ADDF), provided an overview of the brain changes that occur with Alzheimer’s disease and noted that aging is the primary risk factor for developing it. The ADDF is investing in the areas of genetics, oxidation, neuronal energy failure, inflammation, and damaged proteins that lead to the main pathologic hallmarks – the tangles and plaques – associated with Alzheimer’s. Many of those basics of Alzheimer’s are associated with mitochondrial dysfunction. For example:
• Genes involved in oxidative phosphorylation are downregulated in the brains of people with mild cognitive impairment.
• Recessive mutations in nuclear and mitochondrial oxidative phosphorylation genes are associated with dementia.
• Mitochondrial gene expression is altered in blood early in Alzheimer’s.
The ADDF has three programs exploring different targets that may be useful in preventing or treating Alzheimer’s disease:
1. Targeting mTOT, which is a complex located on the inner mitochondrial membrane that is a binding site for thiazolidinediones. Research has shown that mTOT can affect oxidative metabolism, including mTOR, which coordinates mitochondrial activity and proliferation (see Image 7).
2. Targeting Complex I of the ET system (see Image 8). These compounds mildly inhibit Complex I, which leads to an increase in AMPK and a decrease in cognitive decline.
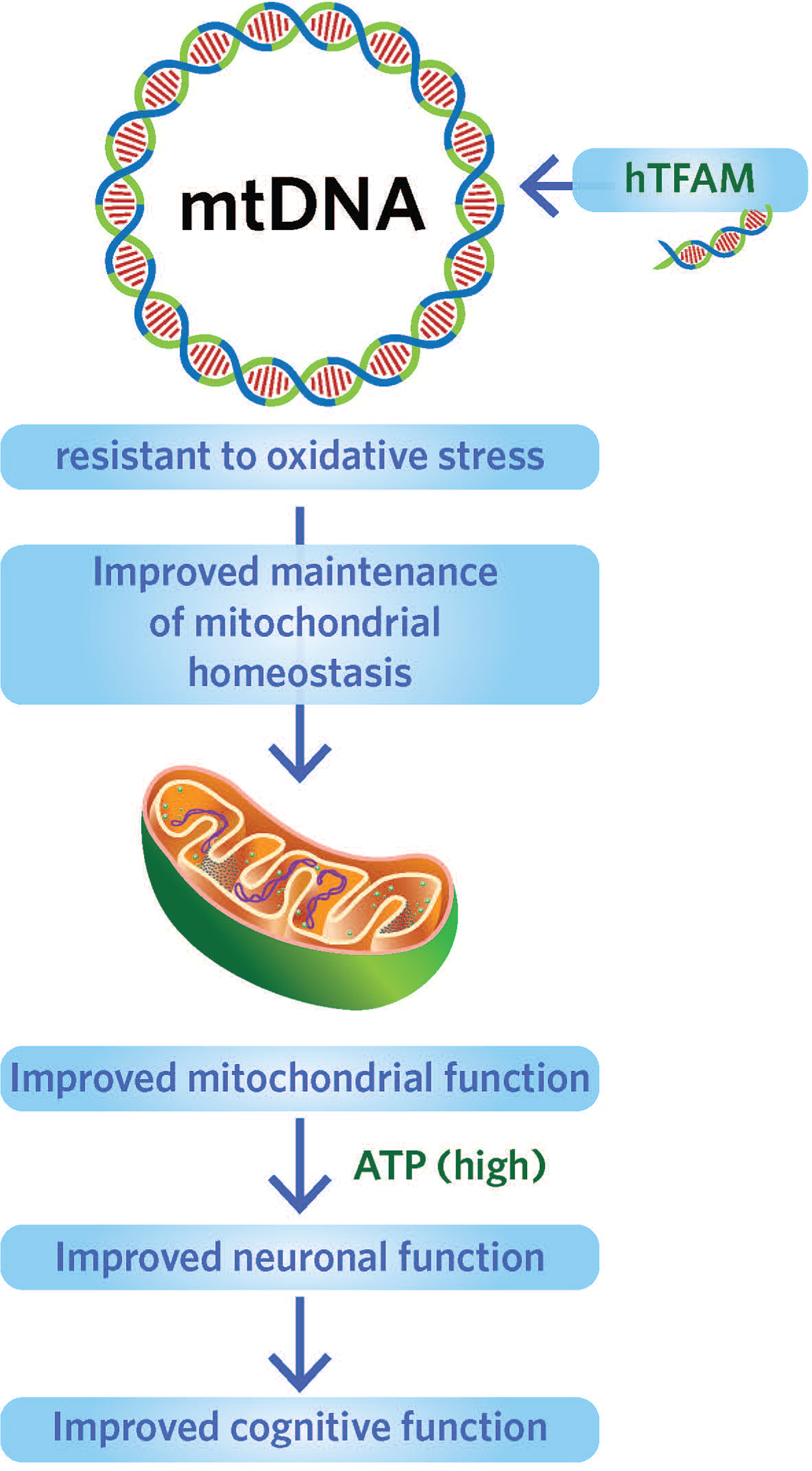
3. Targeting human mitochondrial transcription factor (hTFAM)(see Image 9). [10] hTFAM protects mtDNA from oxidative stress. Overexpression of hTFAM can lead to increases in cognitive function and decrease the accumulation of deleterious substances.
Image.7
Targeting mTOT to affect mitochondrial activity and proliferation. Source: Metabolic Solutions Development Company.
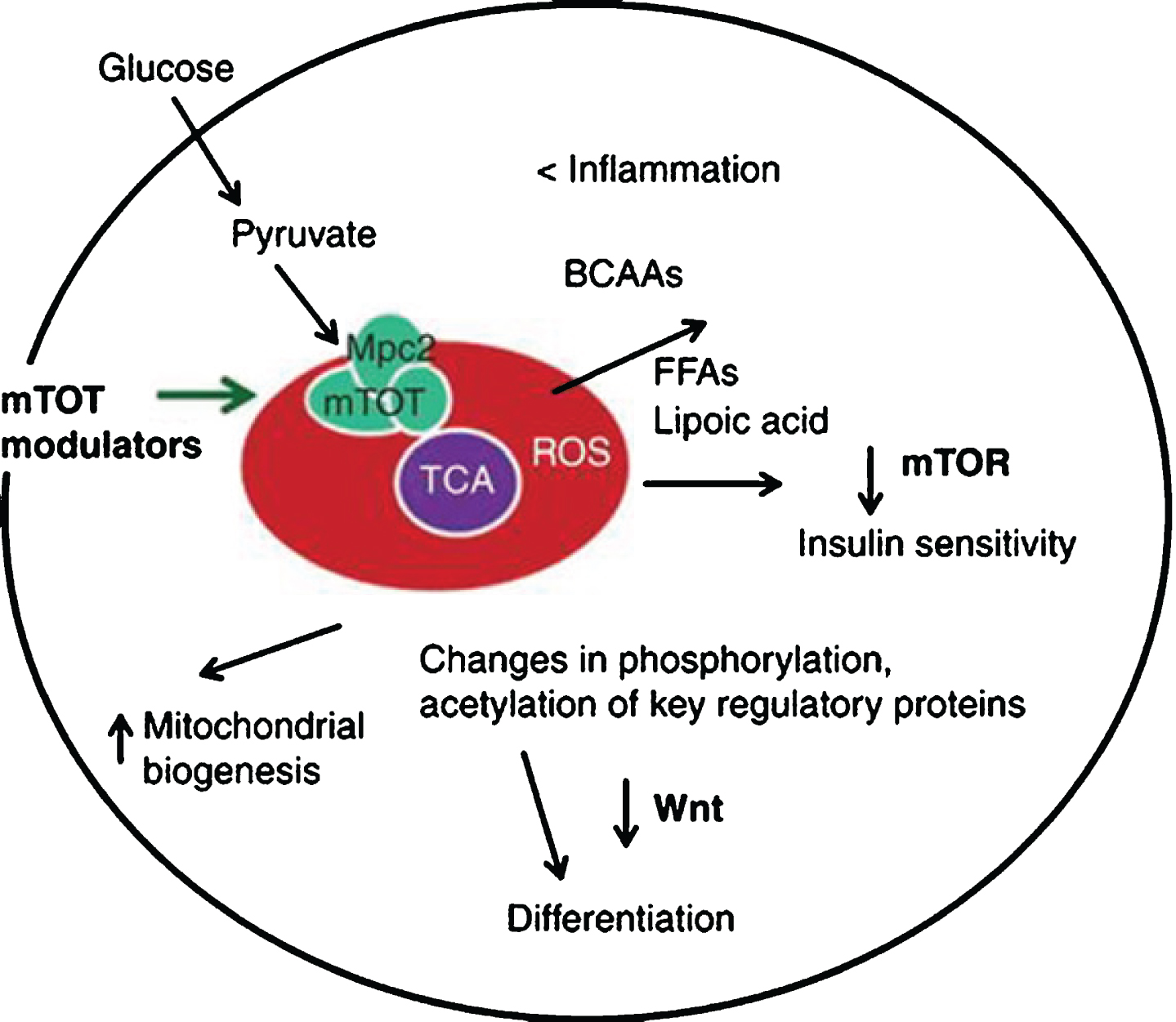
Image.8
Targeting Complex I to decrease cognitive decline. Source: Eugenia Trushina, PhD, Mayo Clinic.
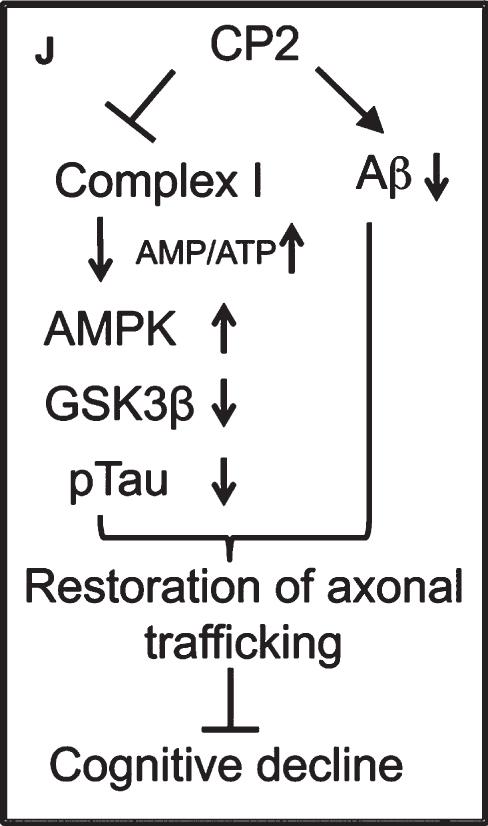
Image.9
Targeting human mitochondrial transcription factor to protect mtDNA from oxidative stress. Source: Foundation for Mitochondrial Medicine.
Dr. Shineman concluded by suggesting ways different groups can work together to create effective mitochondria-targeted therapies:
• Share resources;
• Share experimental tools, research models, patient samples;
• Share knowledge;
• Foster cross-disciplinary opportunities and training for early career researchers;
• Share funds;
• Collective funding of projects/resources can advance research for all research areas related to mitochondrial dysfunction.
7.2Mitochondrial function and cancer
Keshav Singh, PhD, Senior Scientist in the Cancer Cell Biology Program, Director of the Cancer Genetics Program, Joy and Bill Harbert Endowed Chair in Cancer Genetics at the University of Alabama, Birmingham Comprehensive Cancer Center presented on mitochondrial function and cancer.
Numtogenesis is the term for fragments of mtDNA that are transferred into a cell’s nucleus. These fragments are called nuclear mtDNA, or NUMTs. It is relevant for cancer and other issues because this DNA can also mutate and be inherited by other cells [11].
It has been documented that patients with mitochondrial disease have an increased prevalence of cancer [12, 13]. Researchers have found that mitochondrial disease can lead to genomic instability, and have measured the distribution of NUMTs in various cancers as compared to NUMTs in normal genomes.
Normally, there are “mitocheckpoints” that monitor the functional state of mitochondria. The checkpoints turn on an intergenomic response that can lead to cell death in the event the mitochondria are not working properly. However, if the checkpoint is not working as it should, the dysfunctional mitochondrial may result in transfer of mitochondria and/or mtDNA-induced numtogenesis and tumor development can occur.
YME1L1 has been identified as a protein of interest due to its proposed function in mitochondria pathologies. In normal situations, the YME1L1 gene serves as a suppressor gene in controlling cell growth and division. What is known about YME1L1 functions in mitochondria:
• It maintains the integrity of the mitochondria.
• It is a member of AAA (ATPase Associated with diverse cellular Activities) protease family.
• It is a catalytic subunit of the mitochondrial inner membrane AAA protease super-complex required for mitochondrial inner membrane protein turnover.
• It binds unfolded proteins in an ATPase-independent manner.
• It is required both for the degradation of unassembled subunit2 of cytochrome C oxidase (COX2) and for efficient assembly of mitochondrial respiratory chain.
• It ensures cell proliferation, maintains normal cristae morphology and complex I respiration activity, promotes antiapoptotic activity and protects mitochondria from the accumulation of oxidatively damaged membrane proteins.
• It is an orthologue of yeast YME1 in mammals.
Dr. Singh presented what is known about NUMTs and human pathologies:
• Tumor genomes contain more somatic NUMTs than matched normal genomes.
• Tumors in females contained more NUMT than tumors in men.
• NUMT abundance in tumor correlate with abundance in blood.
• Increased numtogenesis is associated with higher mortality.
• YME1L1 is frequently mutated (20% ) in colorectal tumors demonstrating increased NUMTs abundance.
• YME1L1 is mutated in tumors derived from other tissues.
• Inactivation of human YME1L1results in increased migration of mtDNA in the nucleus.
• YME1L1 is the first NUMT suppressor gene identified in human cells.
Further research into this area could help better understand how the process of numtogenesis is involved in various malignancies, as well as other diseases, and the role NUMTs play in human health.
7.3Mitochondrial dysfunction and Parkinson’s disease
Matthew Goldberg, PhD, at the University of Alabama, Birmingham presented on the evidence linking mitochondrial dysfunction and Parkinson’s disease. For example
• Environmental exposures to mitochondrial inhibitors such as MPTP cause acute Parkinsonism.
• Mitochondrial dysfunction has been noted in the brain and blood cells of Parkinson’s patients.
• Genetic mutations in mitochondrial proteins cause inherited forms of Parkinson’s disease.
With aging, environmental assaults, or a genetic predisposition, all cellular components, including DNA, proteins, and lipids, accumulate damage over time. Further, the population of mitochondria in each cell ranges from healthy to damaged to dysfunctional. To keep cells healthy, dysfunctional mitochondria are normally removed from cells. But when these mechanisms for removal fail, the dysfunctional mitochondria can affect the function of the cell and create diseases, such as Parkinson’s. Focusing on the mechanisms to remove dysfunctional mitochondria is an important area of Parkinson’s research.
By studying inherited Parkinson’s disease, researchers have been able to define some processes of how dysfunction in mitochondria can lead to Parkinson’s. Two genes, in particular, are implicated as playing a significant role in this process: PINK1 and Parkin, both of which, in normal conditions, work together to remove dysfunctional mitochondria. Researchers have shown that deletion mutations in PINK1 and Parkin cause early-onset Parkinson’s disease. Image 10 shows how Parkin and PINK1 work together to remove dysfunctional mitochondria.
Image.10
Parkin and PINK1 work together to remove dysfunctional mitochondria. Source: Matthew Goldberg, PhD, University of Alabama, Birmingham [14].
![Parkin and PINK1 work together to remove dysfunctional mitochondria. Source: Matthew Goldberg, PhD, University of Alabama, Birmingham [14].](https://content.iospress.com:443/media/trd/2017/2-3-4/trd-2-3-4-trd014/trd-2-trd014-g010.jpg)
The researchers have engineered mice with activated Parkin, but do not have answers yet to key questions, such as
• Does activated Parkin affect mitochondrial health?
• Can it protect against mitochondrial stress?
• Does activated Parkin have any adverse effects?
• Could small molecule Parkin activators be therapeutically beneficial?
By focusing on these genes, in addition to others, the researchers hope to create therapies that will benefit patients with Parkinson’s disease.
7.4Mitochondria and amyotrophic lateral sclerosis (ALS)
Alex Sherman, Director of Strategic Development and Systems at the Neurological Clinical Research Institute at Massachusetts General Hospital and Harvard Medical School, presented on collaborating and big data in prognosis and treatment of ALS.
ALS, also known as Lou Gehrig’s disease, is a progressive neurodegenerative disorder affecting upper and lower motor neurons and characterized by progressive loss of motor neurons in the brainstem and spinal cord. Symptoms include muscle weakness, paralysis and eventually death, usually within 3 to 5 years from disease onset. Currently, there is no cure or effective treatment for ALS and the cause of disease is unknown in most cases.
Neuronal mitochondrial dysfunction is one of the earliest features of ALS. Mitochondria are highly dynamic organelles that undergo continuous fission, fusion, trafficking and turnover, all of which contribute to the maintenance of mitochondrial function. Abnormal mitochondrial dynamics have been repeatedly reported in ALS and increasing evidence suggests altered mitochondrial dynamics as possible pathomechanisms underlying mitochondrial dysfunction in ALS.
One substantial obstacle to understanding and developing an effective treatment for ALS is the heterogeneity of the disease course, ranging from under a year to over 10 years. It is challenging to predict how a given patient’s disease will progress and thereby to demonstrate the effect of a potential therapy, making clinical trials especially challenging. Understandably, the uncertainty surrounding prognosis is an enormous burden for patients and their families.
Identification and development of new sources and approaches for large datasets are critical for creation of disease models and potential therapies development. Individual medical centers may not see enough patients with ALS to develop their own meaningful datasets, and competition for research funds is fierce and long-term collaborations are challenging. Large data sets created by pulling together data from clinical trials and individual patients’ clinical visits could provide information for clinicians.
Dr. Sherman discussed an implemented approach and platforms for patient-centered clinical and research information aggregation, collaboration, and sharing. These platforms allow the rare neurological diseases clinical/research community to enter the age of “Big Data”. A tremendous amount of disease-specific information already exists and is being created daily in platforms such as electronic medical records, clinical trials, patient registries, and biobanks. Sharing collected information could help define disease progression and create therapies.
One of the first collaborative efforts in ALS was the PRO-ACT (Pooled Resource Open Access ALS Clinical Trials) project to combine data from multiple clinical trials, allowing researchers to access thousands of patient records to find similarities, clues for disease progression, and areas for future clinical trials. Currently, the PRO-ACT database comprises de-identified data from 10,700 patient records derived from 23 Phase II/III clinical trials. It includes demographic information, outcomes, safety, laboratory, medical and family histories; overall, it contains more than 10 million longitudinally collected data points. The entire dataset is available for download at www.alsdatabase.org.
NeuroBANK™ is another example of a novel and already successful approach in international clinical research collaboration. It is a patient-centric clinical research platform that enables the capture and aggregation of clinical and clinical research data from simultaneously running research projects and links these data with biorepositories, image banks, genetic information, and patient-reported outcomes (PROs). Embedded Neurological Global Unique Identifier (NeuroGUID™) technology generates a patient-specific 11-character string that uniquely and securely identifies a patient without risk of exposure of that patient’s identity. It allows aggregation of information from the same person from multiple sources, regardless of where and when the data were collected. The character string prevents a patient from being created multiple times in a research database if he or she participated in a study at different sites or in multiple studies at the same time.
Harmonization of clinical and research common data elements, best practices, and standards for information collection during patient clinical and research visits is paramount. The NeuroBANK™ ecosystem aims to share PROs, scales, questionnaires and trackers, standard nomenclature for bio-specimen collection, image banks, and genetic information; provides tools to all stakeholders, and creates efficiencies in research and care. The centralized search facility allows researchers to query clinical and phenotypical data and find corresponding biospecimens, images, and genetic files.
More than twenty simultaneously executed clinical research studies utilize the NeuroBANK™ platform, with enrollments ranging from 50 to 1,500 patients and more than 80 participating sites from 17 countries. All these projects capture and are committed to sharing not just longitudinal clinical data but also tens of thousands of vials with biofluids, thousands of files resulting from whole-genome sequencing, cell lines, and results of genomics analyses. All these resources are available to the global ALS research community.
7.5Mitochondrial dysfunction and autism
Robin Morris, PhD, Regents Professor of Psychology and Executive Officer for Entrepreneurship at Georgia State University, presented on the subgroup of people with mitochondrial dysfunction, some with autism and some without autism.
While autism may be caused by different combinations of genetic and environmental influences, mitochondrial defects and markers appear in a subset of people with autism.
The brain has one of the highest energy expenditures/ATP requirements of any organ. It is particularly vulnerable to mitochondrial dysfunction, and high metabolic areas within the brain are most sensitive to such problems, potentially disrupting normal development and functioning of the brain.
For example, researchers have shown that the ratio of lactate to pyruvate can be related to the brain’s processing speed, with very high levels of lactate/pyruvate leading to a decrease in mental processing ability and speed. The related concept of cognitive fatigue, defined as a “time-related deterioration in the ability to perform certain mental tasks” has also been suggested because the reserves of energy are not enough to sustain the high-energy needs of the brain.
The aim of Dr. Morris’s research is to determine if children with oxidative phosphorylation (OXPHOS) defects in mitochondrial disease/dysfunction show indices of such brain “fatigue” through testing by functional magnetic resonance imaging (fMRI), diffusion tensor imaging (DTI), and neuropsychological testing.
The hypothesis is that the inability to utilize delivered O2 during high-demand mental processes consistently causes a decrease in cerebral metabolic rate of oxygen (CMRO2), resulting in an up-regulation of local blood flow. The increased blood flow and decreased CMRO2 result in a greater blood oxygenation level dependent signal (BOLD) response, suggesting some evidence of cognitive fatigue.
Identifying reliable measures of cognitive fatigue and identifying relevant treatments could expand the patient’s ability to focus and would benefit the patient greatly. For example, the patient could attend school more regularly or could adapt to the school day.
While the study is ongoing, early trends note a very diverse pattern of developmental outcomes of the children who were diagnosed with mitochondrial disease with autism or ASD when they were much younger, but who now do not fully meet the diagnostic criteria for these disorders that are standard (Autism Diagnostic Interview-Revised [ADI-R] or Autism Diagnostic Observation Schedule [ADOS] or both). The research team continues to focus on the identification and measurement of cognitive fatigue in these children and hopes to have fMRI results that will provide a more quantitative method for documenting that factor and its impact on these children’s functioning in the near future.
8About the Foundation for Mitochondrial Medicine
The Foundation for Mitochondrial Medicine’s (FMM) mission is to support the development of the most promising research and treatments for the many forms of mitochondrial disease. Treatments for mitochondrial disease could impact cures for Autism, Alzheimer’s, Parkinson’s, muscular dystrophy and more. FMM partners with related disease organizations and others to raise awareness for mitochondrial disease, fuel connections, and to co-fund treatments where mitochondrial dysfunction is a core element. For more information on FMM funded research such as functional MRI studies on cognitive fatigue and testing of new drug compounds, visit www.mitochondrialdiseases.org
9About the United Mitochondrial Disease Foundation
Founded in 1996, the United Mitochondrial Disease Foundation (UMDF) works to promote research and education for the diagnosis, treatment and cure of mitochondrial diseases, and to provide support for affected individuals and families. Since its inception, the UMDF has funded nearly $13 million in research, making it the leading non-governmental contributor of grants focused solely on mitochondrial disease. The UMDF, based in Pittsburgh, PA, is a national organization, represented around the world by thousands of members. For more information about mitochondrial disease or the UMDF, visit www.umdf.org.
10Glossary of Abbreviations
• AAA | ATPases associated with various cellular activities |
• ADI-R | Autism diagnostic interview – revised |
• ADOS | Autism diagnostic observation |
• ATP | Adenosine triphosphate |
• ALS | Amyotrophic lateral sclerosis |
• AmPK | AMP-activated protein kinase |
• ASD | Autism spectrum disorder |
• BHI | Bioenergetic health index |
• BOLD | Blood oxygenation level dependent signal |
• CMR02 | Cerebral metabolic rate of oxygen |
• CT | Computerized tomography |
• CNS | Central nervous system |
• DTI | Diffusion tensor imaging |
• ET | Electron transport |
• FA | Fanconi anemia |
• fMRI | Functional magnetic resonance imaging |
• hNP | Human pluripotent stem cell-derived neural progenitor cells |
• hPSC | Human pluripotent stem cells |
• hTFAM | Human mitochondrial transcription factor |
• LA | Lactic acidosis |
• mDNA | mitochondrial DNA |
• MELAS | mitochondrial encephalomyopathy, lactic acidosis, and stroke-like episodes |
• MERRF | Myoclonic epilepsy with ragged red fibers |
• MRI | Magnetic resonance imaging |
• mTOR | Mechanistic target of rapamycin |
• mTOT | Mitochondrial target of thiazolidinedione |
• NADH | Nicotinamide adenine dinucleotide hydrate |
• nDNA | nuclear DNA |
• NUMTS | Nuclear mitochondrial DNA |
• OXPHOS | Oxidative phosphorylation |
• PEO | Progressive external opthalmoplegia |
• PINK1 | PTEN-induced putative kinase 1 |
• POLG | Polymerase gamma |
• PRO | Patient-reported outcome |
• Pro-ACT | Pooled resource open access ALS clinical trials |
• ROS | Reactive oxygen species |
• RRF | Ragged red fibers |
• VLCAD | Very long chain acyl-CoA dehydrogenase |
References
[1] | Morava E. and van den Heuvel L. , Hol F., et al., Mitochondrial disease criteria: Diagnostic application in children, Neurology 67: ((2006) ), 1823–1826. https://www.ncbi.nlm.nih.gov/pubmed/17130416 |
[2] | Castro R.J. , Mitochondrial replacement therapy: The UK and US regulatory landscapes, J Law Bio Sci 3: ((2016) ), 726–735. https://academic.oup.com/jlb/article/3/3/726/2566730/Mitochondrial-replacement-therapy-the-UK-and-US |
[3] | Cohen B., Interventions in Primary Mitochondrial Disorders: Developing an Evidence Base ((2017) ). Available at: http://mitochondrialdiseases.org/wp-content/uploads/Therapies-for-Mitochondrial-Disease-Dr.-Bruce-Cohen.pdf |
[4] | Koopman W.J.H. , Willems O.H.G.M. and Smeitink J.A.M. , Monogenic mitochondrial disorders, N Engl J Med 366: ((2012) ), 1132–1141. Available at http://www.nejm.org/doi/full/10.1056/NEJMra1012478 |
[5] | Kerr D.S. , Review of clinical trials for mitochondrial disorders: 1997–2012, Neurotherapeutics 10: (2) ((2013) ), 307. https://www.ncbi.nlm.nih.gov/pubmed/23361264 |
[6] | Chacko B.K. , Kramer P.A. , Ravi S. et al., The Bioenergetic Health Index: A new concept in mitochondrial translational research, Clin Sci (Lond) 127: (pt 6) ((2014) ), 367–373. Available at http://www.clinsci.org/content/127/6/367/ |
[7] | Iyer S., Bioenergetics: Art meets Gentle Science in Sickness and in Health: https://wp.vcu.edu/bioenergetics/ |
[8] | Rao R.R. and Iyer S. , Stem cells, neural progenitors, and engineered stem cells, Methods Mol Bio 1254: ((2015) ), 255–267. https://www.ncbi.nlm.nih.gov/pubmed/1071 |
[9] | Iyer S. , Xiao E. , Alsayegh K. et al., Mitochondrial gene replacement in human pluripontent stem cell-derived neural progenitors, Gene Ther 19: ((2012) ), 469–475. https://www.ncbi.nlm.nih.gov/pubmed/21918550 |
[10] | Oka S., Leon J., Sakumi K., et al., Human Mitochondrial Transcriptional Factor A breaks the Mitochondria-Mediated Vicious Cycle in Alzheimer’s Disease. Sci Reports 6: ((2016) ), https://www.nature.com/articles/srep37889 |
[11] | Singh K.K. , Choudhurry A.R. and Tiwari H.K. , Numtogenesis as a mechanism for development of cancer, Semin Cancer Biol ((2017) ), pii: S1044-579X(17)30126-8. doi: 10.1016/j.semcancer.2017.05.003. https://www.ncbi.nlm.nih.gov/pubmed/28511886 [Epub ahead of print] |
[12] | Finsterer J. and Frank M. , Prevalence of neoplasms in definite and probable mitochondrial disorders, Mitochondrion 29: ((2016) ), 31–34. https://www.ncbi.nlm.nih.gov/pubmed/27181047 |
[13] | Finsterer J. and Krexner E. , Increased prevalence of malignancy in adult mitochondrial disorders, J Med Life 6: ((2013) ), 477–481. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4034300/ |
[14] | Barodia S.K., Creed R.B., and Goldberg M.S., Parkin and PINK1 functions in oxidative stress and neurodegeneration, Brain Research Bulletin 133: ((2017) ). http://www.sciencedirect.com/science/article/pii/S0361923016304683?via%3Dihub. |


