
Ethical issues related to clinical research and rare diseases
Executive summary
The Alpha-1 Foundation (Foundation) has a long history of recognizing the importance of ethical, legal, and social issues (ELSI) in rare genetic disease research [1]. In April of last year, the Foundation assembled a distinguished panel of ethicists, social scientists, clinical investigators, and legal experts to explore ELSI issues related to research in rare diseases.
Marilyn Coors, PhD, Chair of the ELSI Working Group of the Foundation and of the meeting, explained the conference goal was to “discuss the ethics of rare disease research and, if possible, make recommendations that can inform future clinical research in rare diseases”. She continued “if we can’t accomplish those two charges with the people who are here today, I don’t know who can”.
Clinical research in rare diseases such as alpha-1 antitrypsin deficiency (AATD) encounters unique challenges including a small patient population for recruitment, limited research funding, and an urgent need for rapid drug development. Efforts have been under way to streamline research in rare diseases through flexible study protocols, while protecting the rights and interests of volunteer participants. The goal of this conference was to foster discussion about critical and emerging ethical issues in rare disease research that could contribute to the formulation of recommendations about future research.
Participants in the Conference included Foundation ELSI Working Group members, clinical scientists, patient representatives, and representatives from the Food and Drug Administration (FDA), National Institutes of Health (NIH), and Patient-Centered Outcomes Research Institute (PCORI).
Overall, the workshop successfully highlighted how the opportunities and challenges facing research on rare diseases are best addressed through strong partnerships among federal regulators, industry, patient communities, and clinical investigators. The workshop also provided the Foundation and its ELSI Working Group with a wealth of information that can be used to develop a clear research agenda.
The following summaries of the individual presentations were drafted by John Goodale, a science writer who attended the meeting, and rewritten or edited either by the presenters or Adam Wanner, Scientific Director of the Alpha-1 Foundation.
In 1966, the medical ethicist Henry Beecher’s article “Ethics and Clinical Research” in the New England Journal of Medicine, was instrumental in establishing federal rules on human experimentation. Beecher called for informed consent and a “conscientious, compassionate, and responsible investigator.” Today’s regulatory system reflects Beecher’s call for informed consent but adds an institutional review board (IRB) to it. It’s a system that minimizes potential biases and conflicts that investigators may bring to the table.
IRB review was initiated when the NIH Clinical Center opened in 1953. The review committee system was expanded and, by 1966, it became the policy for the entire Public Health Service. In 1974, the National Commission for the Protection of Subjects of Biomedical and Behavioral Research was established and the FDA codified international guidelines in 1981. The Common Rule established uniform regulations for federal agencies in 1991, and in 2011, the U.S. Department of Health and Human Services (DHHS) approved a series of changes to the Common Rule.
Today, the principles that contribute to ethical research include collaborative partnership with stakeholders, social value, scientific validity, fair subject evaluation, a balance of risk and benefit, independent review, informed consent, and respect for participants.
Independent review ensures that ethical requirements are fulfilled, biases are eliminated, ethical issues between investigators and participants are balanced, and that research does not exploit individuals or groups. Independent review is now an internationally recognized requirement with U.S. regulations codified for the DHHS and FDA. Criteria for IRB review include minimized risk balanced with anticipated benefits, fairly selected subjects who are fully informed and well monitored, and safeguards for vulnerable persons. Simply put, the ethical goal of clinical research is to promote responsible, beneficial research while protecting the rights and welfare of research participants.
One challenge to meeting these goals is the regulatory disparity between research endeavors that are funded by the federal government and covered under the Common Rule and those research projects that are not federally funded or subject to FDA approval. Another challenge involves recent changes in technology, the growth in spending for research, and a shift toward multi-site and multinational trials. There have also been changes in the responsibilities of IRBs, reflecting new research elements (stored samples, cell-based therapies, genomic sequencing, etc.) and an expanding role of IRBs in dealing with conflict of interest issues, regulatory compliance, and in the monitoring of clinical trials.
There is concern about a growing bureaucracy that has become overburdened, outmoded, and more concerned about protecting the institution than the individual participant. Data support some of these negative perceptions: inconsistent judgments, extended review time frames, variable levels of expertise, and an inability to measure effectiveness. Clearly, IRB processes need to be simplified and expedited. A 2012 article by Matt Hanson et al. in Arch Dis Child noted that two studies in the EU were under review for several years with no significant changes to study parameters. When a 2014 study dealing with Hirschsprung’s disease encountered similar obstacles, Dr. Aravinda Chakravarti of Johns Hopkins University noted that “...progress on collaborative research in this disease has been significantly hampered by the need for redundant IRB reviews...” This appears to be a common theme. Additional challenges specific to rare disease research include relatively few participants, the need for multi-site studies, innovative designs and active advocacy groups, and the need to protect participant privacy in a contained research environment.
In order to streamline the IRB process eight changes to the Common Rule, under the 2015 Notice of Proposed Rule Making (NPRM) have been proposed:
1. Improve informed consent.
2. Require informed consent for the use of stored biospecimens in secondary research, generally obtained by means of broad consent.
3. Exclude certain categories of activities that are not deemed to be research, are inherently low risk, or where protections similar to those usually provided by IRB review are separately mandated.
4. Add additional categories of exempt research to accommodate changes in the scientific landscape and to better calibrate the level of review to the level of risk.
5. Limit circumstances that allow waiver or alteration of consent for research involving biospecimens.
6. Mandate that U.S. institutions engaged in cooperative research rely on a single IRB.
7. Eliminate the continuing review requirement for studies that were expedited or are complete except for data analysis or follow-up.
8. Extend the scope of the policy to cover all clinical trials at U.S. institutions, regardless of the funding source.
Public comments on these proposed changes focused on the waiver of informed consent, alterations of consent for research involving biospecimens, and Notice of Proposed Rule Making (NPRM)-proposed changes involving a single IRB. Although some of these changes were not included in the Final Rule, single IRB review of multisite studies is required by the final changes to the Common Rule published in 2017.
The NIH is also requesting public comments regarding whether or not NIH should mandate single IRB review for multisite studies. Although there have been both positive and negative public comments about this proposed change in NIH policy, there is consensus that efficiency is a good idea. Carrie Wolinetz, Director of the Office of Science Policy, stated that “While the vast majority of comments (∼70%) support the use of a single IRB for multisite research studies, commenters across all demographics also provided very thoughtful recommendations, questions, and concerns. These comments were broad in scope and expressed differing, often conflicting, opinions for NIH to consider.”
Future changes in NIH policy will likely involve single IRB review of multisite studies, research with biospecimens, data sharing, and increased participant involvement.
References
1 | Grady C. Institutional Review Boards: Purpose and Challenges. Chest. (2015) ; 148: (5):1148–55. |
2 | Hansson M, Gattorno M, Stjernshcantz J, Feltelius N, Martini A, Ruperto N. Ethics bureaucracy: a significant hurdle for collaborative follow-up of drug effectiveness in rare childhood diseases Arch Dis Child (2012) ;97: :561–563. |
3 | DHS (Department of Homeland Security) et al. (2015) . Notice of proposed rulemaking (NPRM). Federal policy for the protection of human subjects. Federal Register 80: (173): 53933–54061. http://www.hhs.gov/ohrp/regulations-and-policy/regulations/nprm-home/. |
4 | Public Comments on the Draft NIH Policy on the Use of a Single Institutional Review Board for Multi-Site Research December 3, 2014 – January 29, 2015. Available at http://osp.od.nih.gov/sites/default/files/resources/sIRB%2007-21-2015.pdf. Comment #58 on page 31. |
5 | DHS et al. (2017) . Final rule. Federal policy for the protection of human subjects. Federal Register 82: (12): 7149–7274. https://www.gpo.gov/fdsys/pkg/FR-2017-01-19/pdf/2017-01058.pdf. |
The FDA became increasingly involved in the development of drugs designed to diagnose and treat rare diseases in 1983 with the passage of the Orphan Drug Act (ODA). The “Orphan” designation requires that a drug address a condition affecting fewer than 200,000 Americans and that there is a scientific rationale to establish a medically plausible basis for the use of the drug for the rare disease or condition. The Orphan designation provides financial incentives to pharmaceutical companies that undertake rare disease product development. Prescription Drug User Fee Act (PDUFA) fees are waived, and there is a 50 percent tax credit toward the cost of clinical trials. Companies that develop these drugs may also be eligible for FDA grants that support clinical research. In addition, they may receive seven years of marketing exclusivity for an approved Orphan product.
Orphan drug legislation has been successful. Prior to 1983, only 38 drugs were approved in the US to treat orphan diseases. In Congressional Year 2015, about 47 percent of the novel drugs approved at the FDA were approved to treat rare or “orphan” diseases. In 2015, the Office of Orphan Products Development (OOPD) designated a record 354 drugs as Orphans, a 22 percent increase over 2014. In addition to the incentives built into the legislation, patient advocates have been a driving force behind this surge in the development of Orphan products. Approximately 25 percent of products with the Orphan designation go on to receive marketing approval. To date, there have been 518 Orphan drug approvals.
The Rare Diseases Program (RDP) is part of the Center for Drug Evaluation and Research (CDER) and was established in the Office of New Drugs (OND) in 2010. Today, a team of five people and the Associate Director for Rare Diseases (Dr. Jonathan Goldsmith) report to the Director of the Office of New Drugs, Dr. John Jenkins. The RDP mission is to facilitate, support, and accelerate development of drug and biologic products for the treatment of patients with rare disorders. Although the RDP is not directly involved in the review of drugs, it does take on special projects, such as training FDA reviewers, reviewing rare pediatric disease voucher requests, working closely with the NIH, developing guidelines for industry, and meeting face-to-face with patient advocacy groups and sponsors. The RDP is also part of the FDA’s Rare Disease Council.
The OOPD is mainly responsible for administrating the Orphan Drug Act, reviewing Orphan designations, exclusivity, and Orphan grants. The RDP, on the other hand, advises various FDA review divisions working on rare disease applications and responds to rare disease-related congressional inquiries.
There are a number of challenges to the development of products for rare diseases. The natural history of these diseases is often poorly understood. Some rare diseases are well studied but further investigations have led to a better understanding of their pathogenesis and possible drug targets, while the disease mechanisms underlying other rare diseases are virtually unknown. Most rare diseases tend to be progressive, serious, and often life threatening. Yet they present in small populations, restricting study design and replication. Frequently, phenotypic diversity and genetic subsets are present within these disorders, which add to the complexity of research. Defined and validated outcome measures are often lacking, and there may be ethical considerations for enrolling children in certain clinical trials.
The FDA is supporting rare disease drug development through expedited programs, regulatory flexibility, multiple offices and departments, and patient-focused drug development. Expedited clinical development of rare disease products includes the following: Priority Review, Fast Track, Accelerated Approval, and Breakthrough Therapy. These programs are extremely useful in terms of getting these products approved. Eighty-six percent of all rare disease product approvals benefitted from at least one of these expedited programs, compared to only 34 percent of non-rare disease approvals.
Flexible clinical development programs are another effective way to expedite product approval. The law requires that a new drug establish efficacy through two “adequate and well-controlled” studies. However, we have the authority within the law to consider a product with only one well-controlled study if there is other supporting evidence. Eighty-one percent of rare drug approvals were made under these more flexible criteria versus 36 percent for non-rare drug approvals.
The FDA maintains a variety of offices focused on rare disease products. The Office of Health and Constituent Affairs (OHCA) serves as a liaison between the FDA and patient advocacy organizations, training patient representatives to serve on advisory committees and gathering patient input. The Professional Affairs and Stakeholder Engagement (PASE) group develops strategies to improve stakeholder relations, conducts research, and coordinates safe-medication-use projects across the FDA. The Clinical Outcome Assessments Staff program promotes the development and implementation of patient-focused endpoint measures in medical product development to describe clinical benefit in labeling. The FDA’s Patient-Focused Drug Development Program (PFDDP) brings patients and government officials together to gather patient input about specific clinically meaningful symptoms and treatment options available for a given condition. These meetings help the FDA conduct risk/benefit assessments and advise drug companies about patients’ perspectives on new products. The PFDDP has increased its number of patient meetings and also generates a “Voice of the Patient Report” to disseminate patient input received at these meetings.
Finally, the FDA has developed a Rare Disease Cluster to improve understanding of international approaches to rare disease drug development, specifically in Europe (European Medicines Agency/EMA). In summary, the FDA has introduced a host of programs and regulatory modifications that promote and facilitate clinical research in rare diseases. Knowledge of these procedural benefits should help clinical researchers, and biotech and pharmaceutical companies in the design and implementation of trials aimed at finding new therapeutic solutions in the rare disease space.
References
6 | Guidance for Industry Expedited Programs for Serious Conditions – Drugs and Biologics https://www.fda.gov/downloads/drugs/guidancecomplianceregulatoryinformation/guidances/ucm358301.pdf. |
7 | Patient-Focused Drug Development: Disease Area Meetings Planned for Fiscal Years 2013-2017 https://www.fda.gov/forindustry/userfees/prescriptiondruguserfee/ucm347317.htm |
8 | Developing Products for Rare Diseases & Conditions https://www.fda.gov/forindustry/DevelopingProduCTsforrareDiseasesConditions/default.htm |
The National Patient-Centered Clinical Research Network (PCORnet) seeks to improve the nation’s capacity to conduct clinical research by creating a large, highly representative, national patient-centered network that supports efficient clinical trials and observational studies.
PCORnet funds thirteen Clinical Data Research Networks (CDRNs) and twenty Patient-Powered Research Networks (PPRNs). The CDRNs consist of various integrated delivery systems, academic medical centers, and clinics—all collaborating to conduct research as a network. The PPRNs are operated and governed by groups of patients and their partners, focused on a particular condition, population of patients, or specific rare disease.
PCORnet’s approach to data collection is unique, combining electronic health records (EHR), administrative claims data from CMS and others, and patient-reported outcomes data collected via the NIH’s Patient Reported Measurement Information System (PROMIS) and other surveys. PCORnet’s Common Data model defines research criteria and ensures a streamlined, efficient way to address research-related questions and make data available quickly and in a widely acceptable format.
Networks are encouraged to collaborate with appropriate rare disease organizations to identify additional individuals with those conditions. In developing these cohorts, the CDRNs are expected to work to ensure that data standards support interoperability. Currently, thirteen CDRNs are funded as part of PCORnet. CDRN Rare Disease cohorts include the following:
• Alpha-1-antitrypsin deficiency
• Sickle cell disease
• Recurrent C. difficile colitis
• Amyotrophic lateral sclerosis (ALS)
• Rare cancers
• Osteogenesis imperfecta
• Cystic fibrosis
• Duchenne muscular dystrophy
• Idiopathic pulmonary fibrosis
• Hypoplastic left heart syndrome
• Severe congenital heart disease
• Kawasaki disease
• Pulmonary arterial hypertension
The ADVANCE CDRN, focused on AATD, has used electronic health records to define specific population characteristics for its 2.4 million safety-net patients. The ADVANCE CDRN and the COPD PPRN are collaborating with data collection. For example, EHR-based “computational phenotypes” are used to identify AATD patients and a Targeted Testing Population (TTP), and then a COPD PPRN survey is conducted among a sample of AATD patients in the ADVANCE cohort. All three ADVANCE ambulatory systems have identified patients who meet the criteria for inclusion in the cohort. The three ADVANCE CDRN data partners and the COPD PPRN are currently participating in data validation.
The PCORnet PPRNs patient cohorts are more than just a collection of patients. These cohorts are based on advisory groups that include patients, studies that address what patients want to learn, and proposals that are patient driven.
CDRN rare disease cohorts share a number of themes. Advisory groups, including patients, caregivers, clinicians, and researchers are established. There is institutional review board (IRB) oversight. Patient identification is done using computable phenotypes. Recruitment and consent are somewhat flexible. In rare disease populations, patients may respond favorably to face-to-face recruitment, but that takes more time and money. Novel approaches are needed, for example, where patients can opt out at the time of recruitment. Finally, data collection involves both EMR and patient surveys.
The future of PCORnet rare disease cohorts revolves around cohort identification and analysis using standardized queries against analysis-ready, standardized data. Moving from Phase 1 to Phase 2 will require evaluation of projected cohort status, available data elements, the ability to contact individuals to participate in research, and the establishment of research funding beyond PCORI.
References
9 | Gibson B, Butler J, Zirkle M, Hammond K, Weior C. Foraging for information in the EHR: The search for adherence related information by mental health clinicians. AMIA Annu Symp Proc (2017) ; 2016: : 600–608. |
10 | Ellenberg SS, Culbertson R, Gillen DL, Goodman S, Schrandt S, Zirkle M . Data monitoring committees for pragmatic clinical trials. Clin Trials (2015) ; 12: : 530–536. |
The mission of the National Center for Advancing Translational Sciences (NCATS) is to promote innovative methods and technologies to enhance the development of diagnostics and therapeutics that may be applied to many diseases. NCATS doesn’t focus on specific diseases, but on what is common among them and the translation science process. NCATS’ approach to this mission may be broadly divided into three domains: pre-clinical translational science, re-engineering translational science, and clinical translational science.
One example of preclinical translational science efforts is the NCATS Chemical Genomics Center. The center was founded as part of the Molecular Libraries Program and today is a collaboration of more than 200 investigators worldwide. The mission is to lead the way in developing assays, high-throughput screening, chemical informatics, and medicinal chemistry. The focus is on unprecedented targets and rare or neglected diseases. NCATS explores new ways to approach the drug-development process.
A second example of NCATS’s preclinical initiatives is the Assay Development and Screening Laboratory (ADST). The goal is to help disease foundations, such as the Alpha-1 Foundation, employ early-stage, translation drug discovery strategies. The program also conducts training, grant support, and outreach to strengthen the competencies of investigators involved in translational research.
Drs. Michael Iannotti and James Inglese of the Assay Development and Screening Technology (ADST) developed two cell-based models for alpha-1 antitrypsin deficiency-related liver disease. One looks at the intracellular accumulation of polymerized mutant alpha-1 antitrypsin, which has been implicated in liver toxicity. The other uses an assay for alpha-1 antitrypsin secretion that is impaired in alpha-1 antitrypsin deficiency. Thus, their model is well positioned to test new drugs and siRNA targeting the basic defect. This partnership with the Alpha-1 Foundation researchers is the type of collaborative effort that exemplifies NCATS’ mission.
Efficacy, safety, and toxicity of promising therapies traditionally has involved an animal model approach. NCATS, however, is shifting (re-engineering) this paradigm with our tissue chip program. The goal of this collaborative effort with the FDA and Defense Advanced Research Projects Agency (DARPA) is to develop an in vitro platform that uses human tissues to evaluate the efficacy, safety, and toxicity of promising therapies.
Tissue chips are fabricated from flexible plastic, with ports and channels that allow nutrients and oxygen to pass through them. Unlike cell-culture and animal models, the tissue chips replicate the complexity of the human body environment. The ultimate goal is to use these chips to advance precision medicine, test for environmental toxins, and develop disease-specific models—perhaps for cancer and rare diseases.
NCATS has two major initiatives in the clinical translational science space. First, the Rare Disease Clinical Research Network brings together dozens of patient advocacy groups (PAGs) and a number of NIH agencies to build a collaborative clinical research infrastructure that helps support rare disease clinical research.
The second clinical initiative, the Clinical and Translational Science Awards Program, is the largest program within NCATS. This program includes a network of over 50 medical centers seeking to improve clinical and translational research both locally and nationwide. It also provides innovative training for clinical and translational researchers.
A recent national focus of the Clinical and Translational Science Awards (CTSA) program is improving and streamlining the conduct of multi-site clinical trials. Causes of delayed startup for clinical trials include duplicative IRB reviews, subcontracting issues, and duplicative investigator site qualifications. To overcome these challenges, NCATS plans to set up centralized IRB review, utilize streamlined contracting templates, and establish Good Clinical Practice (GCP) training across the CTSA program sites. Future CTSA Trial Innovation Centers and Recruitment Innovation Centers will further implement these efforts and improve the planning, design, and execution of trials across the country.
The ultimate objective of NCATS is to facilitate all translational processes—from preclinical research to public health—while maintaining our patient-focused approach to the development of diagnostics and therapeutics. Ultimately, NCATS is about getting more treatments to more patients more quickly.
Reference
11 |
AlphaNet, a nonprofit organization founded in 1995, has developed an educational, patient-support program called the Alpha-1 Disease Management and Prevention Program (ADMAPP). The program is designed to provide patients, families, and healthcare providers with information about prophylactic measures that individuals with AATD can take to decrease the risk of further disease complications.
Recently, program administrators evaluated specific criteria to determine how the ADMAPP program may impact patients with AATD enrolled in the program. During this evaluation, data collection and data analysis were segregated to ensure protection of patients’ personal information.
The first criterion was life expectancy in patients who have severe AATD (ZZ genotype). The rationale was to determine whether life expectancy has actually improved in ZZ patients during recent years. Data were collected from a sample of 2,505 patients with AATD from 2002 through 2015. The average age of the total cohort at entry into the database was 53.7 + 10.2 years. Respondents were predominantly Caucasian (98.8 percent) and 51 percent male. To be included in the database, patients had to be on augmentation therapy (although some patients may not have been on therapy continuously).
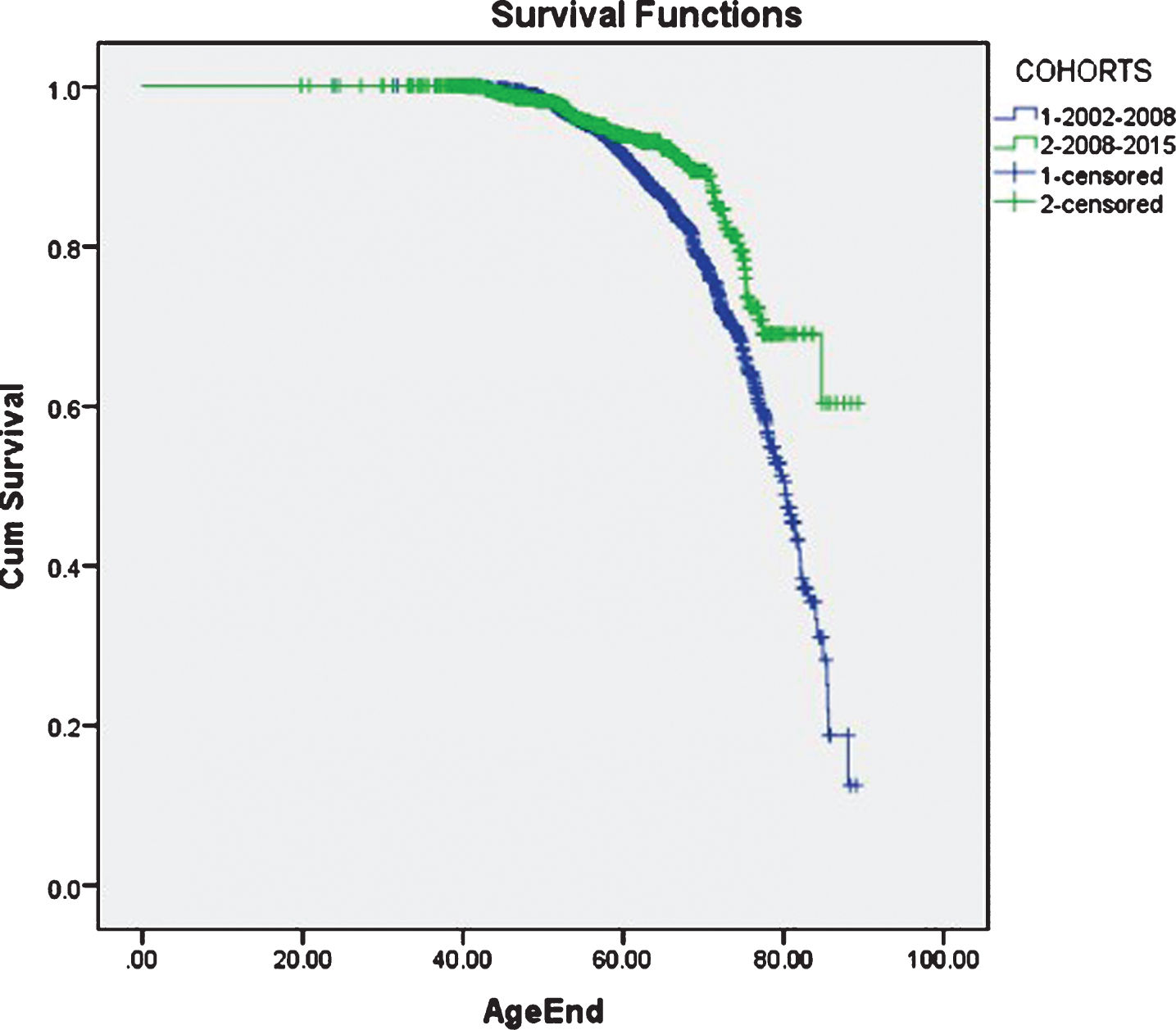
The study cohort was divided into two groups: patients enrolled in the program between 7/2002 and 6/2008 and patients enrolled in the program between 7/2008 and 3/2015.
Compared to the earlier cohort, patients in the more recent group were slightly older, less likely to use oxygen, and more likely to have been diagnosed with AATD because of emphysema.
Out of the total sample, 366 patients died and 2,139 were censored. Survival analysis was performed using the Kaplan-Meier method with a log-rank test to measure the differences in survival curves between the two cohorts. The more recent cohort had an improved life expectancy compared to the earlier cohort (see figure). The data may be biased because complete mortality data was only available after 2008. However, this life expectancy analysis of AATD patients in the ADMAP program could have an impact on augmentation therapy and other treatments for AATD-related COPD including pulmonary rehabilitation and transplantation policy.
The second criterion was patient compliance with program initiatives. The objective was to determine if there is an association between self-reported adherence to the ADMAP program components and health benefits. 3,526 ADMAPP patients with a higher self-reported level of compliance with the ADMAPP program initiatives (n = 1,605) were compared to 1,921 patients with a lower self-reported level of compliance. The results indicate that patients with higher adherence were:
- more comfortable with their knowledge of AATD
- more likely to read some part of a patient education booklet
- more likely to use augmentation therapy
- more likely to get vaccinations
- more likely to exercise
- less likely to smoke
These observations suggest that a disease management program such as the ADMAPP might be beneficial to patients with severe AATD.
Reference
12 | Perkins JT, Choate R, Mannino DM, Browning SR, Sandhaus RA. Benefits Among Patients with Alpha-1 Antitrypsin Deficiency Enrolled in a Disease Management and Prevention Program. J COPDF (2016) ; 4: : 56–64. |
The ethics of genetic research encompass study design, implementation, consent, and translation of results into clinical practice if the research leads to clinical benefit. Ethical considerations for study design address several questions. Are patients and advocacy groups along with researchers involved in framing the research? Are patient cohorts representative and free of potential bias? Are the privacy concerns of secondary subjects (family members) being considered along with individual participants? Finally, is there adequate human subject protection and ethics review?
These questions have particular relevance for the rare disease community. The families and advocacy groups may be eager to push research forward, and the consent process should therefore include families as well as individual patients.
Implementation of genetics research studies requires that researchers balance explanations of genetic information with patient recruitment and meaningful consent procedures. There needs to be a balance between risk/benefit considerations, privacy and confidentiality, voluntariness, and the notion of “therapeutic misconception”: Do participants understand the difference between clinical practice and this particular research study?
Patients with rare diseases may have few treatment options other than participating in a study, meaning that risk/benefit calculations must be carefully considered. Finally, genetic research among patients with rare diseases may entail a complex relationship with researchers. Many rare disease researchers are also clinicians, so a balance must be struck between the role of highly trusted “family” clinician and being an impartial researcher trying to recruit patients for a complicated study.
Looking ahead, a broader assessment must be done regarding the ethical implications of conducting studies with small cohorts of participants in multiple sites. There is a need for more dynamic consent models, including increased communication between researchers and patients about how samples and data are handled. Also, researchers need to move beyond simple consent and begin to assess patient values in an effort to promote patient and community engagement in genome research.
Genomic research also involves a balance between patient privacy and how results are—or are not—returned to individual patients and family members. If a study is anonymous, how can researchers make individual patients aware of the results? Incidental findings and unintended results could impact family members. Should results be entered into a patient’s medical record, and who decides what data are released—and when? Return of results requires additional consideration in the rare disease community where incidental findings and variants of unknown significance may lead to confusion among family members about potential causal genomic markers.
There are also issues surrounding the translation of study results, specifically, concerns about publication of open-source genetic information and stigmatization. The Genetic Information Nondiscrimination Act (GINA) prohibits discrimination based on genetic information, but there are limits. These translation concerns may be heightened in the rare disease community. Small sample sizes make these participants more identifiable than participants in larger studies. Yet, some rare disease study participants want to be identified and draw attention to the research and the disease itself. There is also potential in smaller genetic studies for a slower translation to clinical practice.
The future use of samples and data has implications for genetic research, particularly for studies involving rare diseases. What kind of limitations can patients place on the future use of their clinical samples? What about pediatric patients who grow up but whose samples are still stored? Can people limit the scope of their consent to restrict future use of those samples or even limit those samples for use in research on a particular disease? Potential changes to the Common Rule could impact this research, especially genetic research involving biospecimens. Patients with rare diseases may want to limit the scope of “future” consent differently from patients recruited to more traditional studies.
Quotes from patients are revealing. A patient who favored one-time consent reflects the views of many patients: “One-time consent creates an easier way for researchers to be able to actually utilize samples instead of waiting to get consent each time.” Some patients, however, feel that “I want to be contacted” if the parameters of the original study change. Other patients were unsure about the best approach: “I want to make sure it was completely de-identified. Although, you know, if they figure something out that could benefit me, I’d kind of want to know it.”
Future considerations for genomic research include translation of results for orphan genotypes, ethical obligations to reanalyze samples and re-contact participants, and the ramifications of future very large studies such as the Precision Medicine Initiative (PMI). In all these areas, the voices of rare disease communities must continue to be heard.
References
13 | Rivera SM, Goldenberg A, Rosenthal B, Augst H, Maschke KJ, Rothwell E, Anderson RA, Botkin J, Joffe S. Injvestigator experiences and attitudes about research with biospecimens. J Empir Res Hum Res Ethics (2015) ; 10: : 449–456. |
14 | Simpson EL, Goldenberg AJ, Culverhouse R, Daley D, Igo RP, Jarvik GP, Mandal DM, Mascalzoni D, Montgomkery CG, Pierce B, Plaetke R, Shete S, Goddard KA, Stein CM. Practical barriers and ethical challenges in genetic data sharing. Int J Environ Res Public Health (2014) ; 11: : 8383–8398. |
In recent years, there has been a shift in clinical research away from personalized patient engagement toward a more contractual approach. Unfortunately, a 50-page consent form or a detailed data-use agreement does not build trust. Researchers must rely on personal and institutional integrity because the relationships among researchers, patients, and organizations require trust, regardless of contractual obligations.
There are many reasons to trust someone: a common goal or shared interest, transparent and consistent rules, or perhaps a trusted mutual acquaintance. PCORnet recently hosted a conference on building trust between patients and researchers. One conclusion they reached is that the work is never done. Even when there is a longstanding trusted partnership, that relationship still requires attention. Also, feedback loops are critical. Many researchers focus on patient recruitment instead of providing feedback to patients about the research itself. This is unfortunate as it can erode the patient’s level of trust in the research. Too often researchers are not closing the feedback loop on their research practices.
The PCORnet conference also addressed the need to talk about mutual failures or shortfalls—in a compassionate way. The ability to recover from mistakes is an essential part of our work. Money, of course, is also often an obstacle to engagement: “My budget can only pay for data management and statisticians. We have to spend the money on the science.” For many researchers, the question remains: How can we best put real dollars behind actual engagement?
Mark Yarborough (University of California Davis), Richard Sharp (Mayo Clinic), and I examined how organizations rebuild public trust once it is lost. We found that investing in relationships was certainly important, but we also found that establishing accountability was critical. Having guidelines to track data, documenting what didn’t work, and implementing consistent decision-making were all critical accountability issues.
The reasons to engage with patients include speeding up translational research, learning new things, or meeting an organizational requirement. There are also ethical reasons to engage with patients. According to the 1978 Belmont report (Ethical Principles and Guidelines for the Protection of Human Subjects of Research, Report of the National Commission for the Protection of Human Subjects of Biomedical and Behavioral Research), these reasons might include enacting respect, assuring that the research is beneficial, and making sure that research proceeds fairly.
Timing is another important factor. When should researchers engage with patients? Engagement should be undertaken throughout the entire translational and research process—from conception to completion.
Many resources are available that deal with how to engage with patients. One partnership that’s often overlooked is the engagement among clinical groups, hospital organizations, and research enterprises.
I recently completed a project in collaboration with several disease-advocacy organizations. Our conclusions were the following:
- Remember why you are here—to help the community, find a cure, or alleviate symptoms.
- Be open to unexpected allies and cultures. Don’t presuppose that a given group isn’t going to work with you.
- Be nimble and flexible, so you can change as you go.
- Be humble. People matter and you need to know your biases.
Overall, researchers need to build trust, communicate respect, and personally engage patients throughout the research process. Importantly, researchers should not just enjoy the trust invested in them and the research but thrive to earn it. This requires integrity and transparency in the design and conduct of clinical research. It also requires patient involvement in the entire process, from trial conception to the sharing of study results with the participating patients.
Reference
15 | Edwards KT. Methods of legitimation: how ethics committees decide which reasons count in public policy decision making. Soc Sci Med (2014) ; 113: : 34–41. |
We care about conflicts of interest because they can impede our efforts to conduct good scientific research by requiring us to make “methodological choices that are biased by interests that are at odds with the established goals of science.”We also care about these conflicts because they can impede our efforts to do ethically responsible research, in which participants are not hurt, exploited, or put at unwarranted risk.
According to the Institute of Medicine report, Conflict of Interest in Medical Research, Education, and Practice, a “conflict of interest is a set of circumstances that creates a risk that professional judgment or actions regarding a primary interest will be unduly influenced by a secondary interest.” The definition is relatively simple, but recognizing and managing conflicts of interest may be more difficult than we like to admit.
The more mechanisms we have in place to manage these conflicts and the more we know about how well these mechanisms work, the better able we will be to routinely avoid and/or manage them. We would have a better sense of what these mechanisms are and how well they work in research related to rare diseases if we had a framework that helped us identify competing interests and the mechanisms for managing them. Such a framework could identify all the various competing interests—such as conducting good science, doing ethical research, making money, and managing personal prestige—in research and then label these interests as either primary or secondary.
That same framework could be used to identify the specific risks that can occur when we allow a secondary interest to trump a primary one. For example, there may be a risk of generating inaccurate or biased data when we place a secondary interest in saving money over a primary interest in conducting sound research.
Once the proposed framework identifies the potential secondary interests that might trump primary ones, it can also show the mechanisms that are currently available and used to minimize the risk that a secondary interest may supersede a primary one. For example, disclosure of financial relationships is the predominant risk management mechanism used to ensure that secondary financial interests don’t take precedence over primary scientific ones. Similarly, we use the mechanisms of IRB review and the threat of litigation to guard against the exploitation of research participants that can occur when secondary interests supersede primary ones.
By using the framework to identify primary and secondary interests and the risk management measures in place to prevent or manage the conflicts that can rise between them, we can better assess the adequacy of our efforts to address conflicts of interest in research. For example, to evaluate mechanisms to prevent the bias that can be introduced into research when secondary interests shove primary ones aside, we can ask the following questions:
• Are some types of bias more detrimental than others in terms of producing studies that improve healthcare?
• If so, are our risk management strategies weighted toward those that are most injurious?
• Or, are our prevention strategies weighted toward those that are most prevalent?
If we believe that all bias undermines the integrity of science, what difference should it make where that bias comes from? Bias introduced by poor study design or publication issues is more prevalent than bias introduced by financial relationships. Yet, the proposed framework reveals to us that the bulk of our energy currently is spent managing conflicts of interest involving the financial relationships that can introduce bias and thus it also reveals that additional tools are required.
Research related to rare diseases can raise novel conflicts of interest. For example, patient advocacy organizations in the rare disease community can create conflicts that can cloud judgment and interfere with sound or ethical research. Their secondary interest of recruiting sufficient numbers of people into clinical trials can possibly undermine the primary interest of assuring ethical research through a robust informed consent process if organizations use educational and recruitment activities that minimize the uncertainties and risks associated with clinical research.
Further magnifying the challenges of managing conflicts of interest in rare disease research are features that go hand-in-hand with rare diseases. Due to small numbers of affected individuals we often have only one chance to get the science right. Getting it right requires balancing a limited number of participants, a small number of animal models, and an inability to repeat studies. Improperly managed conflicts of interest can exacerbate all of these features.
Finally, several questions about current strategies for managing conflicts of interest in research need to be addressed. First, does a one-size-fits-all institutional approach to management help or hinder researchers in the rare disease community? Who is best qualified to ensure that the primary interests of doing good science and abiding by high ethical standards are respected in rare disease research? Also, do we recognize the extent to which compliance with federal regulations and institutional policies may provide a false sense of security that primary interests will be pursued over secondary ones?
In summary, we need improved frameworks for and approaches to managing conflicts of interest in the study of rare disease if we want to better assure scientifically sound and ethically appropriate research.
References
16 | Winsberg E, Huebner B, Kukla R. Accountability and values in radically collaborative research. Studies in History and Philosophy of Science Part A. (2014) ;46: : 16–23. |
17 | Tsilidis KK, Panagiotou OA, Sena ES, Aretouli E, Evangelou E, Howells DW, et al. Evaluation of excess significance bias in animal studies of neurological diseases. PLoS Biol. (2013) ;11: (7):e1001609. doi: 10.1371/journal.pbio.1001609. PubMed PMID: 23874156; PubMed Central PMCID: PMC3712913. |
18 | Sena ES, van der Worp HB, Bath PM, Howells DW, Macleod MR. Publication bias in reports of animal stroke studies leads to major overstatement of efficacy. PLoS Biol. (2010) ;8: (3):e1000344. doi: 10.1371/journal.pbio.1000344. PubMed PMID: 20361022; PubMed Central PMCID: PMCPMC2846857. |
19 | Landy DC, Brinich MA, Colten ME, Horn EJ, Terry SF, Sharp RR. How disease advocacy organizations participate in clinical research: a survey of genetic organizations. Genet Med. (2012) ;14: : 223–8. |
20 | Yarborough M. Openness in science is key to keeping public trust. Nature. (2014) ;515: : 313. |
The institutional and legal guidelines under which research is conducted are in part the result of past atrocities. The National Research Act of 1974, for example, was intended to prevent unethical research practices like those used in the 1932 Tuskegee Syphilis Study. In this infamous example, some 400 African American men were subjects in an interventional study without receiving information about the project and without providing valid consent. Contemporary regulatory guidelines were shaped by the recognition that credible oversight of human-subject research is essential.
Established a quarter-century ago, the Common Rule lays out the role and function of IRBs. It is intended to protect participants in biomedical research, and it has largely been successful. Success has, however, come at a price. IRB regulations are widely seen as onerous, inapplicable, and misguided impediments to the research mission they were designed to improve. These challenges provide insight into the human-subject protections needed in an era of big data, precision medicine, and learning healthcare systems.
Clinical data and patient information affect more than privacy. Researchers recognize the obligation to protect people’s data and the benefit of community-based, participatory research. Equally important, ordinary citizens must acknowledge the benefits they realize from research. Data from and about individuals benefits public health, improves clinical and hospital outcomes, and is the engine of biomedical discovery; this entails a collective duty to share appropriately de-identified data and information.
This may be put in the following way: If one wants, intends or expects to receive or enjoy a public benefit, it is perverse and selfish to be unwilling to contribute to the source of the benefit. Consider in this regard those who benefit from disease prevention efforts, but are unwilling to be vaccinated or have their children vaccinated; or one who would gladly accept an organ transplant but is unwilling to be an organ donor; or those who expect access to medical care but disdain the idea they should pay taxes to organizations that provide it. Indeed, there is growing reason to believe that most people are not such “free riders” and, when it comes to information, either assume or do not object to its use by trusted agents who are mindful of privacy and security.
This cannot be emphasized enough. Trust and privacy are closely intertwined. Any putative duty to share information entails a corresponding duty to safeguard that information. When such safeguards are absent or ineffective, the basis of trust is damaged or eroded. This means that investigators must make a substantial effort to identify and address potential sources of disquiet among those whose data and information are being sought. For instance, it is increasingly recognized that even de-identified data – stripped of all or most unique identifiers – can still be linked to individuals. But who would want to do such a thing, that is, re-identify a record? Longstanding tools include very strict penalties for inappropriately data use, security protections at least as strong as tools used to violate privacy, and education programs that teach or remind investigators of the values that underlie and motivate rules.
It would be hopeless to protect every research participant from all intrusions. This is quite similar to acceptance of some risk when driving or flying, receiving medical care, or shopping online. One assumes some risk in exchange for something else of value. In biomedical research, that risk must be kept as small as possible if investigators are to maintain the trust underlying people’s willingness to share data and information.
This is essential if we are to realize the promise of “learning healthcare systems,” or systems that more or less automatically acquire from all research and patient encounters evidence to improve health care. Suppose, for example, a pulmonologist learns something new about treating COPD and, a year later, another physician asks, “Will what worked for your patient also work for mine?” Does that second doctor need to go back to the original patient for permission to use that information? No, of course not. Such shared knowledge, and knowledge sharing, is at the foundation of learning healthcare systems.
Physicians have been doing this all along. It nevertheless remains information about an individual patient. So, where is the tipping point, that is, the juncture at which clinical data become research data the acquisition of which must be governed by traditional human-subject protection policies and laws? Among the most promising approaches to this challenge is the creation of “trusted broker” systems in which patient data and information flow through a process that includes expert committees and community representatives to filter the data and information for research. In the other direction, these “trusted brokers” are available to advise on the management of incidental findings, clinically relevant data, or information that surprise investigators and would be important to an individual patient.
What we seek might be termed “regulation without vexation.” To that end, we can:
- Do a better job of educating investigators about privacy and its importance to ordinary people.
- Work with institutional lawyers to help clarify legal risk and make the consent process more useful for patients and researchers.
- Improve the way we process requests to conduct research.
- Improve the Common Rule to adapt better to data science. In fact, the Common Rule is undergoing a major revision.
Improved health does not entail or require a decline in privacy. Smarter laws, policies, and regulations will reinforce recognition of researchers’ duty to communities and underscore the importance of trust. If our communities do not trust our scientists, then future claims to be helping these communities will seem hollow. Improved public health and biomedical research require trust—and trustworthiness.
References
21 | Goodman KW. Ethics, Medicine, and Information Technology: Intelligent Machines and the Transformation of Health Care. Cambridge: Cambridge University Press, (2016) . |
22 | Goodman KW. Ethics, information technology and public health: New challenges for the clinician-patient relationship, Journal of Law, Medicine and Ethics (2010) ;38: (1):58–63. |
23 | Goodman KW, Meslin EM. Ethics, information technology and public health: Duties and challenges in computational epidemiology. In Magnuson JA, Fu PC, eds., Public Health Informatics and Information Systems, Second Edition, London: Springer-Verlag, (2014) , 191–209. |
Patient engagement in clinical trials extends far beyond patients’ participation as subjects in the trial. Patient advocacy groups and the communities they serve have become far more educated about their condition, the experts in the field and the research taking place to study innovative therapies and cures. Engagement by way of trust, transparency and involving patients in clinical trial design, implementation and data reporting is a key endeavor for success in rare disease clinical research.
While AATD patients are largely motivated to make clinical research a success, they consider many variables while deciding whether or not to participate in clinical research. Their physical, financial, and geographical capacity are weighed, as are ethical and social issues. Those variables are sometimes dependent upon the status of disease and available treatments and may significantly alter a patient’s perspective of risk to benefit ratio, burden and ultimately their decision to participate in clinical research.
A survey of the AATD community was conducted to explore participants’ opinions about treatment considerations and possible participation in AATD-related clinical trials. The survey was administered as part of the FDA’s Patient-Focus Drug Development Initiative. Participants were broadly solicited and through the Alpha-1 Foundation support network, website and social media assets. Target audiences included self-identified parents of liver-affected children, caregivers of adult patients, lung-affected adults, liver-affected adults, and both lung- and liver-affected adults. A total of 1,655 patients responded to the survey. The target audience breakdown and age distribution of the Alpha-1 patients of focus in the survey follow.
Survey participants voiced three major concerns when asked what worried them most about AATD. First, nearly 90% of respondents reported that they were concerned about worsening and progression of AATD. This was followed closely by cost of care and then possible disease progression in the alternate organ—being a lung patient and getting liver disease, or vice versa. Not surprisingly, all respondents felt the ideal future treatment was a cure.
When asked to rank the ideal treatment option for AATD, adults with lung disease felt that ease of administration was the most important factor, followed by expense, duration therapy lasts, potential gene therapy, and home administration of alpha-1 antitrypsin augmentation therapy. When considering factors in future treatments, lung-affected patients ranked cost first, followed by safety/risk, location of administration, duration, and comfort.
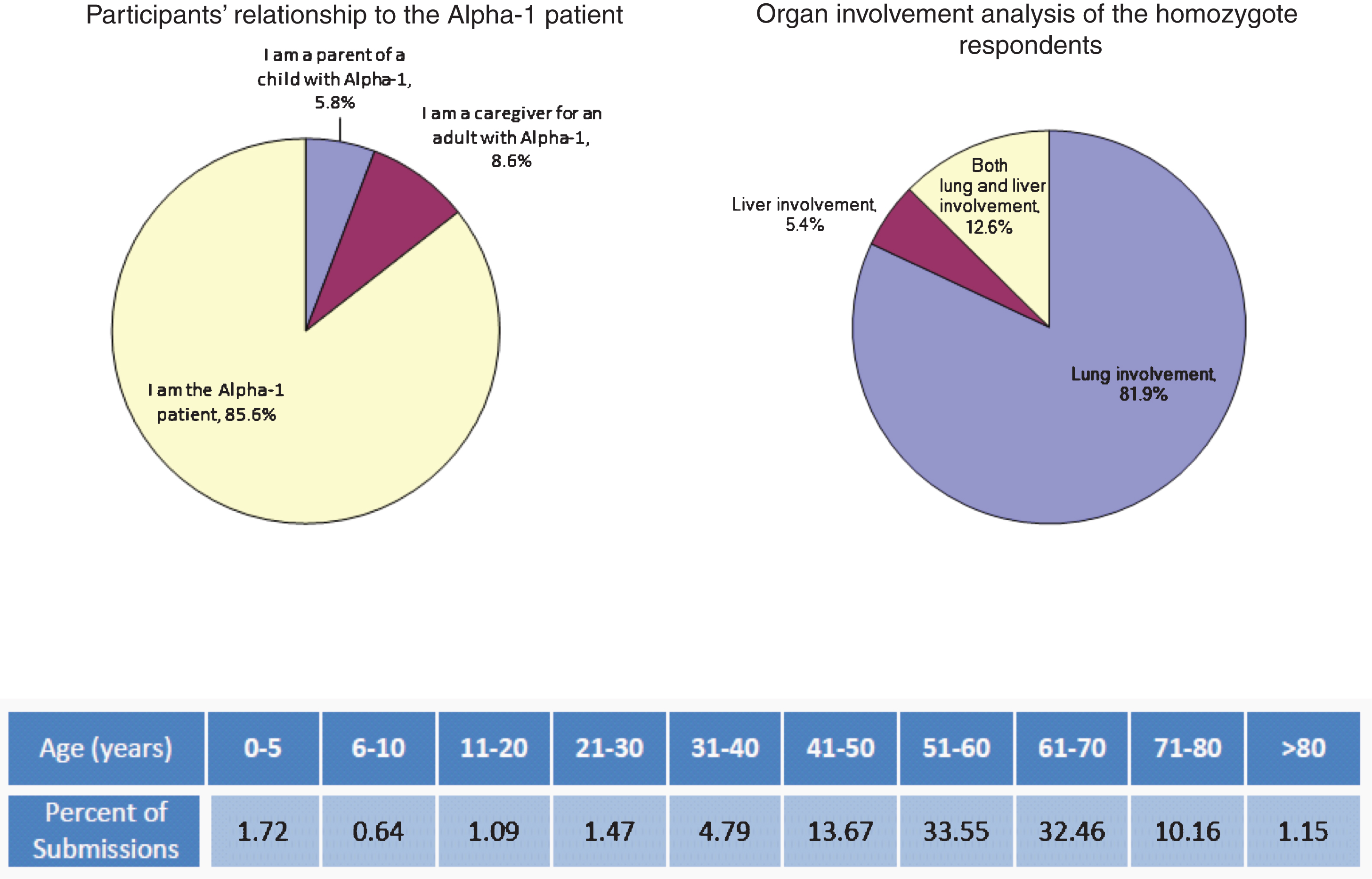
Parents of children with liver disease felt overwhelmingly that gene therapy was the ideal treatment, followed by treatment that was less expensive, easy to administer, administered at home, and administered with low frequency. With respect to future therapies, parents of liver-affected children ranked safety first, and then risk and comfort. Adult patients with liver disease expressed similar feelings about the ideal treatment, with more emphasis on potential curative options and symptom severity.
Asked about reasons that might affect a patient’s willingness to participate in a clinical trial, lung-affected patients listed safety/risk first, followed by location and cost (in terms of current health, finances, and ability to participate). This target population largely associates safety/risk concerns with clinical trial criteria to discontinue alpha-1 proteinase inhibitor augmentation for any portion or the duration of any given trial (placebo control). This is illustrated in the following table of top words, their prevalence and association/context from the survey analysis:
| Word/Context | Prevalence (% comments) | Associated with |
| Risk | 11.77 | Placebo, benefit, infection, requirement to stop medications, invasive treatments |
| Safety | 11.51 | Surgical, environment, level of disease, |
| Location | 11.34 | Expenses, time away, missed work, away from family |
| Travel | 10.65 | Hard for me, expenses, difficult, time, how often |
| Treatment | 9.62 | Placebo, Frequency, route of treatment, discontinuing mine, safety, cost, |
| Participate | 8.33 | Not with placebo, participate in many, every trial 1 qualify for, within 100 miles |
| Trial | 8.25 | Location, safety, placebo, distance |
| Study | 7.13 | Goal of study, Same as above (trial) |
| Placebo | 5.58 | Would not do, loss of lung capacity, stopping would kill me, no placebo |
| Health | 3.78 | Risk, nothing to lose, effect on job, long term |
| Side Effects | 3.26 | Severity, disease progression, impact on function |
Similarly, parents of liver-affected children and liver-affected adults ranked safety/risk as the most important consideration, followed by location and cost (time away from work and family, reimbursement). In their cases however, safety and risk were associated with comfort, long-term side effects, how invasive the trial therapy is and disease progression.
Patients had a variety of comments about the benefits of participating in a clinical trial:
It is important that the study benefits future patients with AATD.
• The study must balance the level of risk versus benefit.
• I am very willing to be part of the cure.
• I would like to help improve medications.
From a patient perspective and as a clinical trial participant, I listed some expectations regarding informed consent and participation in a clinical trial. Those expectations follow:
• Expect scientific validity with goals and endpoints.
• Trust that the institutional review board (IRB) has confirmed that informed consent procedures match the study protocol.
• Ensure that implementation/commercialization is feasible (ask about return on investment and if patient population is adequate to power the study).
• Patients/patients advocates were involved in the trial design and IRB review.
A number of questions that patients considering participation in a clinical trial might ask where subsequently listed:
• How did you find me, and why was I asked?
• What are the direct/indirect benefits of the trial for individual participants and the AATD community?
• What are you going to do to me? Will it hurt, make me sick, or damage my health?
• How will this study impact my current treatment, and what if I want to withdraw?
• What happens if there is a complication? Is there insurance, reimbursement, or compensation?
• How will I be kept informed of the progress, timeline, and final results?
• What if an unrelated health issue or genetic mutation is found? How will I be informed? And do I have a choice?
• How will you manage privacy? How secure is “secure”?
• How does my consent impact future use of my data/samples? Are my data de-identified?
Potential reasons for opting out of or withdrawing from a trial were also presented from my perspective as a research participant and through the analysis of the survey responses:
• Misleading statements regarding unexpected out-of-pocket costs, for example, “It will not cost you anything to participate in the study.”
• Lack of relationship between patient and researcher.
• Research outcomes that don’t ensure therapeutic benefit.
• Inadequate coverage for adverse events.
• Lack of attention to various target audiences.
• Participant transition and informed consent—between childhood and becoming an adult.
• Insufficient consideration of privacy issues and de-identification of patient data/samples.
• Risk of being enrolled in the placebo arm of the trial.
• Excessive invasive procedures.
From a patient’s perspective and in summary, suggestions for trial designers included the following:
• Consider the burden placed on patients and include input from patients as you design the study.
• Realize that burden may vary greatly across a disease community.
• Include the patient’s perspective on the practical impact of study design.
• Be conscious of costs to patient (time, financial cost, anxiety costs, effort to complete paperwork, etc.) and consider ways to reduce those costs.
Overall, this survey, administered as part of the FDA’s Patient-Focus Drug Development Initiative, indicates that in the AATD community parents of liver-affected children and adults with lung and/or liver disease have a positive attitude towards clinical trials for new therapeutic solutions. The survey identified several concerns relative to trial design. The survey also showed that patients want to participate in the development of clinical studies and in the IRB review process. It can be concluded that the AATD patient community and patient advocacy groups are motivated to make clinical research in AATD a success. In their view, this requires a close collaboration between them and the researchers in the design and implementation of the trials.
The day following the workshop, members of the ELSI Working Group met for an in-depth discussion of several of the topics that speakers addressed at the workshop. Several themes emerged during their discussion, as well as potential action items related to them. The following research questions were identified as the most critical:
• How are the unique ethical issues encountered in “small ‘n”’ research best recognized and addressed in trial design, review, and conduct?
• How can investigators best anticipate and accommodate the changing risk benefit calculations that families will make over time as they participate in multiple rare disease clinical trials over the course of several years?
• Since many rare disease communities enjoy active and long-term relationships with private industry, can and should rare disease advocacy organizations attempt to alter the current “one size fits all” approach to the management of financial conflicts of interest in research?
• Given that there can be limited capacity in many rare disease communities to support clinical trials, how should these communities deal with the challenges that arise when there are multiple trials seeking enrollment at the same time?
• What are the best mechanisms for including individuals affected by rare diseases on IRBs so that rare disease communities have more effective representation of their interests and concerns in the design, conduct, and review of research on rare diseases?
The ELSI Working Group hopes to encourage work in these areas in the future, both among its own membership and through the annual research funding mechanisms sponsored by the Alpha-1 Foundation.
Reference
26 | Sharp RR, Yarborough M, Walsh JW. Responsible patient advocacy: perspectives from the Alpha-1 Foundation. Am J Med Genet A (2008) ;146A: : 2845–50. |
Walsh carried the flag for millions of COPD patients and helped change the course of history for a little known disease – Alpha–1 Antitrypsin Deficiency (Alpha-1), a condition from which he and his twin brother, Fred, suffered and to which he previously lost his mother.
He co-founded the Alpha–1 Foundation in 1995 and later that year launched AlphaNet, a non-profit organization that would provide innovative health management programs for Alpha–1 patients and fund more than $65 million to the Alpha-1 Foundation for support of scientists and research into the rare disease.
A true visionary, Walsh not only helped raise the profile of COPD and Alpha–1, he changed the paradigm from which respiratory diseases, rare diseases and the impacted patients were viewed and more importantly, how they were helped. The Ethical, Legal and Social Issues Working group (ELSI) was fundamental in Walsh’s efforts to ensure progress in pulmonary medicine and rare disease research was centrally focused on the patient.
From his leadership position, Walsh quietly raised double digit millions while successfully launching innovative tools and programs to not only educate but invigorate the scientific space. The programs he created and influenced would become a model for patient engagement and Walsh, a model for what non-profit leaders could and should be in order to change the world for their disease.


