
Gene therapy for metabolic diseases
Abstract
Gene therapy has recently shown great promise as an effective treatment for a number of metabolic diseases caused by genetic defects in both animal models and human clinical trials. Most of the current success has been achieved using a viral mediated gene addition approach, but gene-editing technology has progressed rapidly and gene modification is being actively pursued in clinical trials. This review focuses on viral mediated gene addition approaches, because most of the current clinical trials utilize this approach to treat metabolic diseases.
1Introduction
In the early 1970s, the concept of gene therapy became a reality when an exogenous gene was delivered to a mammalian cell resulting in protein expression [1]. In theory, gene addition, the delivery of a normal gene to a diseased cell or organ, could achieve some clinical benefit by allowing the expression of the normal protein. The sequencing of the human genome accompanied by advances in DNA sequence technology has resulted in the identification of thousands of genes responsible for different genetic diseases [2]. Unfortunately, the identification of disease causing genes has outpaced the development of new treatments, creating a void between definition and therapy. Gene therapy is a direct approach to the treatment of genetic diseases, for which the gene causing the underlying condition has been identified, regardless of how well the underlying pathophysiology is understood. Hence, this approach can be viewed as a universal therapeutic alternative.
The first federally sanctioned clinical gene therapy trial was performed at the National Institutes of Health in 1990 to treat severe combined immunodeficiency caused by adenosine deaminase deficiency (ADA-SCID) [3]. However, until recently poor efficacy of viral gene delivery and genotoxicity observed after the administration of integrating vectors has hampered the use of gene therapy as a treatment for genetic diseases. In 1999, a gene therapy clinical trial for ornithine transcarbamylase deficiency resulted in the death of patient, which was caused by a severe immune response to the therapeutic adenoviral vector [4]. Then in 2003, it was reported that patients in a gene therapy trial for X-linked severe combined immunodeficiency (SCID-X1) developed a T-cell leukemia caused by insertional mutagenesis of the vector [5, 6]. These severe adverse events retarded the progress of and dampened the enthusiasm for gene therapy as treatment for genetic disease in both the public and private sector.
More recently, clinical trials for gene therapy have yielded encouraging short-term safety and efficacy in a broad spectrum of genetic diseases that include: ADA-SCID, Leber’s congenital amaurosis, X-linked adrenoleukodystrophy, Parkinson’s disease, hemophilia B, and aromatic aminoacid decarboxylase deficiency [7–12]. In 2003, The Chinese State Food and Drug Administration was the first to endorse gene therapy for clinical use when it approved Gendicine, an adenoviral vector designed to express p53, for the treatment of head and neck cancer [13]. In 2012 Glybera®, an adeno-associated viral gene therapy vector, was approved to treat lipoprotein lipase deficiency by the European Commission [14]. To date in the United States, there have been no approved gene therapytreatments.
With the many scientific advances since the inception of gene therapy, the potential uses of gene therapy have been extended beyond the realm of gene addition for monogenic disorders. Gene therapy now includes the delivery of DNA coding for antigens as vaccines, DNA editing enzymes to repair genetic defects, and small interfering RNA (siRNA) to treat cancer by modulating protein expression. It would be impossible to review all the potential applications of gene therapy in a single chapter. Therefore, this chapter will focus on gene addition for genetic and inherited metabolic diseases with an emphasis on methodologies that have shown clinical promise.
Gene therapy by gene addition requires that a genetic coding sequence for a protein be delivered to the cells of a patient. The ideal gene therapy vector would have the following characteristics: efficient and specific transduction of the target cell regardless of cell cycle, a therapeutic level and proper duration of gene expression, no associated genotoxicity, absent pre-existing immunity against the vector and transgene, and a non-invasive delivery route. A number of methods have been established to accomplish gene delivery. Each of these methods has its distinct advantages and disadvantages. The characteristics of a genetic disease such as the size of therapeutic gene to be transferred and the tissue affected help determine the factors in which type of gene delivery method is best suited to treat a disease.
2Therapeutic Nucleotide Sequence (TNS)
Gene addition is accomplished by the delivery of a ssDNA, dsDNA, or RNA sequence that codes for the normal protein that is defective in the patient. In addition to the nucleotide coding sequence (transgene), the therapeutic sequence typically contains regulator elements that include a promoter, an enhancer, an intron, and a polyadenylation signal (Fig. 1). The promoter drives the expression of the transgene and can be ubiquitous (expressed in all tissues) or tissue specific. The enhancer and intron are elements used to increase the expression of the transgene. The polyadenylation signal is necessary for the proper translation of the transcript. In the case of viral vectors, the transgene and the sequence for transcriptional elements are packaged into viral vectors along with certain viral elements such as those necessary for proper packaging (this will be discussed in more detail later).
3Delivery methods
Gene delivery can be accomplished using an ex vivo or in vivo delivery approach. Ex vivo gene delivery is performed by transferring a gene to a stem, progenitor or differentiated cell line outside the patient’s body, typically in tissue culture. After gene transfer, the cells can then be selected, expanded and transplanted back into the patient. This approach requires the use of gene and cell therapy, and is appropriate for tissue specific diseases for which cell expansion and transplantation have been established as a treatment. For these reasons, ex vivo gene therapy has largely focused on genetic diseases that involve hematopoietic derived cells such as ADA-SCID, X-linked SCID, and leukodystrophies [15, 16]. However, a recent study in mice has demonstrated the correction of hemophilia B afterin vivo correction of hepatocytes, and selection to expand the corrected hepatocytes [17]. This type of approach may be ideal for metabolic diseases for which liver transplantation is a treatment.
The delivery of a gene directly to a patient is referred to as in vivo gene delivery. In vivo gene delivery encompasses directed tissue injection and/or systemic delivery of a gene. This approach has been used to target diseases such as Leber’s congenital amaurosis, a retinal disease, where the gene therapy vector was delivered by subretinal injection [8]. Alternatively, a recent hemophilia B clinical trial used a less invasive systemic approach where the gene therapy vector was delivered by intravenous injection [11]. The success of the hemophilia B trial relied upon the tropism (ability of the vector to target and transduce a specific cell or tissue type) of the AAV serotype 8 capsid toward hepatocytes, which produce the clotting factor defective in hemophilia.
4Non-viral gene delivery
Non-viral gene transfer has several advantages over the use of viral vectors. Non-viral vectors tend to be less immunogenic, to have a larger packing capacity and are easier to produce at scale than viral vectors. Unfortunately, the uptake of nucleic acid from non-viral vectors by cells is not very efficient. Some physical delivery methods such as hydrodynamic delivery, electroporation, ultrasound and magnetofection have shown encouraging results in vivo. The use polymeric systems, such as cationic lipids or polycations to condense the DNA and facilitate uptake of nucleic acids, have also been shown to significantly increase transfection efficacy. Polymeric compounds can also be modified with molecules that result in tissue tropism. Although these non-viral delivery methods have shown promise, their gene transfer efficacy is less than that of viral mediated gene delivery systems and is not high enough to be therapeutic for most genetic diseases. For this reason, gene therapy using viral mediated gene transfer is predominately pursued as a treatment for genetic disease. However, efforts to further improve the efficacy of gene delivery by non-viral approaches may some day result in a shift from the use of viral delivery to the use of non-viral delivery as the preferred treatment for genetic diseases.
5Viral gene delivery
Viral gene delivery takes advantage of the ability of a gene therapy vector to infect a cell, which eventually leads to the expression of the therapeutic transgene (Fig. 2). The viral life cycle requires proteins from both the virus and host. Most of viral vectors used in gene therapy are, in fact, derived from pathogenic viruses. However, the viruses used in gene therapy are recombinant viruses that have significant amounts of the viral genome removed and replaced with a therapeutic transgene: the removal of viral sequence creates space for the transgene and increases safety by eliminating the ability of viral vector to replicate. Replication competent viruses are sometimes used in the treatment of cancer. The viral sequences that remain in a gene therapy vector code for viral elements, which are required for the packaging of the transgene into the viral capsid. Vector production is then accomplished by transfecting multiple plasmids that encode the transgene or TNS, the viral capsid and proteins required for the assembly of the recombinant virus into a packaging cell line. After production and packaging, the recombinant virus is purified, concentrated, and titrated prior to use. A schematic of this process in the production of an adeno-associated virus (AAV) is presented in Fig. 3.
Numerous viruses have been engineered as recombinant gene therapy vectors. The natural properties of these viruses such as their life cycles, genome structure and composition, viral coat and non-structural viral proteins can differ greatly. Such properties account for the unique characteristics that individual viruses exhibit as gene therapy vectors (Table 1). It is important to remember that selecting a gene therapy vector for the treatment of a disease is a balance between the characteristics of the genetic disease to be treated and the properties of the vectors available for treatment. There are numerous viral vectors being developed or currently in use for gene delivery such as baculoviruses, poxviruses, herpes viruses and foamy viruses. The subsequent sections will focus on a select group of the most commonly used viral vectors: simple retroviruses, lentiviruses, adenoviruses, and adeno-associated viruses.
6Simple retrovirus
Retroviruses are classified as part of the family Retroviridae and are named for their ability reverse-transcribe their genome from RNA to DNA. This family of viruses is responsible for a variety of human diseases including cancer, AIDS, autoimmunity and diseases of central nervous system, bone and joints. A number of agricultural and animal diseases are also caused by retroviruses. The Retroviridae family can be further divided into ‘simple’ and ‘complex’ retrovirus according to genome organization [18]. Simple retroviruses contain only gag, pol, env, and pro genes, while complex retroviruses, such as lentiviruses, contain extra non-structural genes having regulatory or auxiliary functions. First, simple retroviruses will be explored and then the complex retroviruses, specifically lentiviruses, will be discussed in the following section.
The retroviral genome consists of single stranded RNA 7–12 kilobases. All retroviral genomes contain a common set of genes: the gag gene that encodes the viral core proteins which form the matrix, the capsid, and the nucleoprotein structures; the pol gene that encodes reverse transcriptase and integrase; the env gene that encodes surface and transmembrane proteins of the viral envelope; and pro gene, which encodes the virion protease, reviewed in [19]. These genes are flanked by long inverted terminal repeats (LTR) that serve as an enhancer/promoter for viral gene expression and contain the packaging (ψ) signals required for viral assembly. The viral genome, integrase and reverse transcriptase are covered by a capsid protein shell that is 80–100 nm in diameter. A lipid bilayer envelope, which originates from the host cell’s membrane during viral budding, surrounds the capsid. The lipid bilayer envelope contains virus-encoded surface glycoproteins and transmembrane glycoproteins.
The life cycle of a retrovirus begins when the glycoproteins of the viral envelope bind to specific receptors on the host cell; the specific host receptors varies between retroviruses and the presence or absence of the receptor dictates cell tropism. Next, the retroviral envelope fuses with the membrane of the host cell and the viral core is released into the cell cytoplasm. Once inside the cytoplasm, reverse transcriptase synthesizes a dsDNA genome from the single-stranded RNA genome. Entry of the nascent dsDNA into the nucleus requires the breakdown of the nuclear membrane, which occurs only during cell division. Therefore, retroviruses can only infect dividing cells. After gaining access to the nucleus, viral integrase facilitates the integration of the newly formed dsDNA into the host cell genome. The integrated viral genome is called a provirus and it is replicated along with the host genome during the S-phase of the cell cycle. Retroviral replication uses the host’s RNA polymerase II to transcribe the provirus to mRNA, which codes for viral genome and proteins, which are assembled into virons in the cytoplasm. Finally, virons escape the cell by budding out from the cellular membrane (Fig. 4).
As a gene therapy vector, retroviruses are capable of yielding long-term transgene expression because of their ability to integrate and replicate with the host cell’s genome. Other viral vectors, which do not integrate and remain episomal, will not replicate with the host genome and therefore do not result in long-term transgene expression. Sustained expression of the transgene is a desirable characteristic of a gene therapy vector for the treatment of genetic diseases because these disorders are commonly chronic in nature. The limitations of the simple retroviruses as gene therapy vectors include a limited packaging capacity, an inability to transduce non-dividing cells, and a potential for insertional mutagenesis. The term insertional mutagenesis refers to the integration of a provirus into the host genome in a location that alters the expression of a host gene(s). Altered expression of the host gene expression can have pathological consequences; the integration of the provirus near or in a micro-RNA genes involved in cell cycling can cause carcinogenesis.
Retroviruses are commonly used as gene therapy vectors because they can transduce hematopoietic stem cells (HSC) and deliver the corrective gene for diseases such as SCID-X1 and ADA-SCID [20]. Although both forms of SCID can be treated by HSC transplantation, the long-term success of the HSC engraftment is highly dependent on the availability of allogeneic (HLA-matched) donors [21]. Correcting the patient’s own HSC by gene transfer allows autologous corrected cells to be transplanted back into the patient, thereby increasing the chance for successful engraftment. Typically, HSC are extracted, expanded, transduced ex vivo, and then transplanted back into the patient. A retroviral vector allows the therapeutic transgene to be inherited by progeny cells and yields lasting and, in theory, permanent correction because of integration.
There are several examples of genetic disorders that have been treated by retroviral gene therapy. SCID-X1 is caused by a mutation in the γc cytokine receptor subunit that disrupts lymphocyte differentiation leading to a lethal immunodeficiency [22–24]. After successful pre-clinical studies for X-linked SCID, two clinical trials were initiated beginning in the late 1990s [25, 26]. In these clinical trials, a simple retrovirus-derived vector was used for ex vivo gene deliver of γc cytokine receptor cDNA to CD34+ cells. Gene expression was driven using the viral LTR enhancer/promoter. The corrected autologous cells were then transplanted back into the patient. Because γc cytokine receptor subunit is involved in survival and proliferation signaling in lymphocytes, the corrected stem cells were posited to have a selective advantage, which would allow them to repopulate the bone marrow. After treatment, 18 of the 20 trial participants showed significant and sustained γc cytokine receptor expression, sustained T-cell reconstitution, improved immunologic response and were able to leave protective immunological isolation. The observed clonal expansion of the corrected CD34+ cells in patients suggested a possible survival advantage for the treated cells following engraftment. Of the two subjects who did not show significant improvement following treatment, one subject was unable to undergo the procedure because of pre-existing infection and the second subject had a low CD34+ transduction efficacy, which hindered engraftment.
Between 2–5 years post gene therapy, an acute T-cell leukemia developed in 5 of the subjects and the clinical trials were halted [6, 27]. Chemotherapy was successful in treating 4 of the 5 patients that developed leukemia; the fifth patient failed to respond to the chemotherapy and did not survive the leukemia. Surprisingly, non-malignant T-cells expressing the γc cytokine receptor reappeared in all 4 patients following chemotherapy and the remissions of leukemia. The integration of the transgene in or near a known proto-oncogene, LMO2, was shown to have up-regulated the expression of LMO2 and triggered leukemia in four of the patients [27, 28]. In the fifth patient, the up-regulation of the proto-oncogenes CCND2 and BMI-1 was found to be responsible for the leukemia. The location of these integration events is explained by retroviruses’ preferential integration into the promoter or coding region of active gene loci and allowed for the transactivation of local proto-oncogenes by the strong viral LTR enhancer. To reduce the potential for transactivation by the viral enhancer, self-inactivating (SIN) vectors have been developed, in which the viral enhancer is missing [29]. The increased safety of SIN vectors has yet to be proven in humans.
Gene therapy trials were also conducted for ADA-SCID using a similar simple retroviral vector, ex vivo CD34+ cell transduction and transplantation of corrected cells. In the ADA-SCID gene therapy trial, the retroviral vector delivered the adenosine deaminase cDNA. Correction of immunodeficiency and sustained T-cell populations were observed in 20 of the 30 gene therapy treated patients at 2–10 years post-treatment [7, 30, 31]. Interesting, no severe adverse events (SAEs), such as acute T-cell leukemia, have been observed in any of these patients despite the detection of retroviral integration sites located near or within proto-oncogenes like LMO-2 [7, 31]. These results indicate that the disease being treated as well as the vector being used play a yet to be understood role in the occurrence of SAEs. Despite the SAEs, the overall outcome for the treatment of ADA-SCID and SCID-X1 can be viewed as positive when taking into consideration the severity of these diseases and the poor outcomes of patients for whom HLA-matched donors are not available.
7Lentivirus
The lentiviruses are complex retroviruses that have a life cycle, envelope capsid, and a single stranded RNA genome of 7–12 kb similar to the simple retroviruses previously described (Fig. 4). However, there are some important differences in the life cycle, genome, and protein components of the lentiviruses. In addition to encoding the gag, pol and env proteins like a simple retrovirus, the lentiviral genome encodes accessory proteins (tat, rev, vif, vpr, nef, and vpu) that are important for viral replication, binding, infection, and release, reviewed in [19]. Another difference is that lentiviral gene expression occurs in two separate phases, known as the early and late phases, which are separated by the binding of the rev protein. Unlike simple retroviruses that tend to integrate in or near promoters, lentiviruses tend to integrate into introns, which may lessen the possibility of transactivation of a proto-oncogene. Lastly, the lentivirus, unlike a simple retrovirus, can infect dividing and non-dividing cells, a function that is dependent on the expression of proteins from the vpr gene.
Simian, equine, and feline and lentiviral vectors have been developed for gene delivery. Of these vectors, the human immunodeficiency virus type 1 (HIV-1) derived vectors have been the most extensively studied and utilized as lentiviral gene therapy vectors. The accessory genes are nonessential for lentiviral vector production and transduction, and they are deleted from the recombinant gene therapy vector. Because lentiviruses integrate into the host genome, insertional mutagenesis like that observed with simple retrovirus, is a potential problem. To reduce the risk of insertional mutagenesis through transactivation of proto-oncogenes, HIV-1 vectors have been designed as self-inactivating vectors by eliminating U3 enhancer-promoter contained in the LTRs. HIV-1, a human pathogen that attacks cells of the immune system, is an efficient gene delivery vector for transducing HSC and lymphocytes cells, but not other tissue types. In order to expand the tropism of HIV-1, its viral envelope, which contains viral specific glycoproteins, has been pseudo-typed (substituted) with viral envelope proteins from vesicular stomatitis virus glycoprotein (VSV-G) as well as glycoprotein from other viruses such as hepatitis C [32, 33]. Pseudotyping changes the tropism of the vector and can expand the range of cell types that are infectable by any viral vector.
Phase I and Phase II lentivirus clinical trials have demonstrated efficacy and safety in the treatment of numerous genetic diseases including acquired immune deficiency syndrome (AIDS) [34], β-thalassemia [35], Parkinson disease [10] and X-linked adrenoleukodystrophy (X-ALD) [9]. For example, X-ALD is a fatal demyelinating disease of the central nervous system (CNS) [36]. It is caused by mutations of the ABCD1 gene encoding the ALD protein, an adenosine triphosphate–binding cassette transporter localized in the peroxisomal membrane. Defects in ALD inhibit peroxisomal degradation of very-long-chain fatty acids in oligodendrocytes and microglia, and deficiency of ALD causes active multifocal brain demyelination in affected boys. Early allogeneic hematopoietic cell transplantation is the only proven effective therapy for X-ALD and this effect is mediated by the replacement of brain microglial cells derived from the transplanted bone marrow cells [37, 38]. Unfortunately, allogeneic hematopoietic cells are not often available, and mismatched-HLA hematopoietic cell transplantation is less effective and runs a greater risk of graft versus host diseases. The X-ALD gene therapy trial was designed to create ALD corrected HSC by using ex vivo gene delivery of the ABCD1 cDNA and then to transplant the autologous cells back into the patient. Because no selective advantage was observed in pre-clinical studies, both trial participants under went a full myeloablative procedure with cyclophosphamide and busulfan to increase transplant engraftment. Both participants show significant clinical benefits 14 to 16 months after infusion of the ALD corrected HSCs [9] including cessation of progressive cerebral demyelination, a clinical outcome comparable to that achieved by allogeneic HCT.
8Adenovirus
Adenovirus, a group I virus (double-stranded linear DNA genome) in the Adenovirdae family, is a common human pathogen responsible for upper respiratory infections in children as well adults. Its capsid is a non-enveloped, icosahedral protein shell (70–100 nm in diameter) that surrounds the inner DNA-containing core. There have been 57 serotypes described in humans as well as numerous additional serotypes that have been isolated from other animal species [39]. The wild-type adenoviral genome is approximately 26–40 kilobases in length and it is flanked by inverted terminal repeats that are essential for packaging. Based on their temporal expression patterns, the genome can be subdivided into early (E1a, E1b, E2a, E2b, E3 and E4) and late expression genes (L1-5), reviewed in [39]. Early viral genes code for viral DNA polymerase, as well as proteins involved in modulation of the host’s response to viral infection, viral gene expression, replication, and interaction with host’s cellular components. The late genes encode structural proteins used for DNA packaging, viral capsids and cell lysis.
As a gene therapy vector, adenoviral vectors have several advantages over other vectors including the ability to efficiently transduce varied cell types, transduce both dividing and quiescent cells, and package a large coding sequence of approximately 37 kilobases. This allows for the delivery of large therapeutic genes, multiple transgenes and large cis-acting elements, which can enhance, prolong and regulate transgene expression (Fig. 2). Because this virus is non-integrating and the adenoviral genome remains episomal following transduction, the risk of genotoxicity from insertional mutagenesis is greatly reduced.
The first-generation of adenoviruses (FGAd) used for gene delivery retained many of the viral coding sequences and therefore expressed not only the transgene protein but also the viral proteins. The expression of the viral proteins caused an immune responses against transduced cells displaying viral proteins, which resulted in the elimination of the transduced cells and transgene expression [40]. To overcome this problem, additional viral coding sequences were eliminated from subsequent generations of adenoviral vectors, and these new vectors were found to be less immunogenic [41, 42].
Unfortunately, in 1999 a phase I adenoviral gene therapy trial for ornithine transcarbamylase (OTC) deficiency, a urea cycle disorder, resulted in an acute immune response and the first death of a gene therapy subject [4]. In this trial, a total of 18 subjects received a systemic dose of 2×109–6×1011 viral particles per kilogram of a second-generation adenovirus (E1a-, E1b- and E4- deleted). This tragic event temporarily halted all clinical gene therapy trials and dampened enthusiasm for gene therapy as a viable treatment for genetic disorders.
More recently, helper dependent adenoviruses (HDAd) that do not encode for any viral proteins have been developed [43]. Compared with previous generation adenoviruses, these newer vectors are associated with prolonged transgene expression, significantly reduced acute and chronic hepatotoxicity, and reduced inflammatory responses [44]. Although the HDAd do not express viral proteins, they have the same viral coat as the FGAd, which can cause an acute toxic immune response [45, 46]. The immune response to the viral coat upon systemic delivery is dose dependent and the toxic viral dose for systemic delivery is≥1×1013 viral particles per kilogram (vp/kg). A single patient was treated with a low dose (1×1010 vp/kg) of HDAd in a gene therapy clinical trial for hemophilia A, a bleeding disorder. This patient developed transient fever, chills, achiness, back pain, and headache and experienced thrombocytopenia and elevations in liver enzymes values with values peaking 7 days after intravenous infusion [47]. An immune response to the virus was concluded to be the cause of the toxicity. Given the low dose of HDAd that caused this episode and the toxicity previously observed with this vector in the clinical trial for OTC, this trial was terminated.
Although early gene therapy clinical trials using adenoviral vectors to treat metabolic disorders have not been fruitful, new strategies are being explored to overcome the toxicity that has been observed. Due to the immunogenicity and toxicity that can result from systemic delivery of HDAd at levels that would be necessary to see a therapeutic effect, more recent efforts to use HDAd as treatment for genetic disorders have focused on liver directed delivery of this vector. The liver is an ideal target for numerous metabolic disorders and new methods that use balloon occlusion of the liver to target the vector have demonstrated high transduction efficiency and less toxicity [48]. Results in non-human primates have shown that this method can achieve greater transgene expression at lower and presumably less toxic doses. Moreover, the treatment with immunosuppressants prior to infusion of adenoviral vectors may also reduce or negate the toxicity this vector causes following systemic delivery; however, this approach still needs to be explored [49].
Interestingly, adenoviral vectors have been used as a gene delivery vectors in more clinical trials than any other viral vector. They have been used safely and shown efficacy in the treatment of cancer and as a genetic vaccine. In these clinical trials the vector is typically delivered directly to a tissue and the toxicity observed with systemic delivery is not usually an issue.
9Adeno-associated virus
Use of adeno-associated viruses (AAV) is one of the most promising avenues for viral mediated in vivo gene therapy; these vectors have been successful in correcting a number of murine models of metabolic disease [50]. To date, hundreds of different natural occurring AAVs have been isolated from numerous species as well as genetically engineered. A number of AAVs have been shown to exhibit tissue specific tropisms for hepatocytes, myocytes and neuronal cells. This recombinant virus has several advantages over other commonly used viruses for gene delivery, such as retrovirus, lentivirus and adenovirus. These advantages include highly efficient in vivo cell transduction, sustained transgene expression, tropisms for disease specific tissues, low immunogenicity, and a history of clinical safety.
AAV, which belongs to the genus Parvoviridae and the Dependovirus family, is a helper-dependent virus, classified as a Group II virus (single-stranded linear DNA). The adeno-associated species name arises not from any relationship to the adenovirus, which is a Group 1 virus (double-stranded DNA), but rather because it was first discovered as a contaminant in an adenoviral isolate [51]. The virus is referred to as helper-dependent virus because it requires proteins from either an adenovirus or herpes simplex virus in order to replicate [52, 53].
The AAV genome consists of two open reading frames coding for the Rep and the Cap viral proteins, which are flanked by inverted terminal repeats (ITR), reviewed in [54–56]. The viral genome is 4.7 kilobases in size and its relatively small size restricts the packaging capacity of the AAV to less than 5 kilobases, one of the major disadvantages of this vector. The ITRs are DNA elements that are approximately 145 base pairs in length and they are integral to the AAV life cycle, including genome replication, hairpin formation, self-priming, site-specific viral integration and proper DNA encapsidation. The ITRs are the only portion of the viral genome that is packaged into recombinant AAVs used in gene therapy, thereby greatly reducing the chance of an immune response to the expression of a viral protein following transduction.
The Rep proteins, which consist of Rep40, Rep52, Rep68 and Rep78, are non-structural proteins necessary for AAV regulation and replication, reviewed in [54–56]. These non-structural proteins have been shown to exhibit sequence specific DNA binding activity, endonuclease activity, ATP-dependent helicase activity, and the ability to increase DNA synthesis. The three viral proteins made by AAV- VP1, VP2 and VP3- form the non-enveloped icosahedral capsid with a size of approximately 20 nanometers, which is relatively small in comparison to other virus such as adenovirus with a size of 90–100 nanometers [57]. Although variations in VPs are responsible for tissue tropisms between different AAV serotypes, the variation between VPs of different AAVs serotypes is relatively small at the amino acid sequence level. This raises a theoretical concern that pre-existing antibodies against one AAV serotype might cross-react with another serotype and prevent cell transduction; there is a high prevalence of pre-existing antibodies against AAV2 in humans. However, studies in both mice and non-human primates have demonstrated that cross-reactivity between certain serotypes does not occur.
The life cycle of AAV of a natural infection includes both a latent and lytic phase. During the latent stage the provirus preferentially integrates into a specific location on human chromosome 19 [58]. The location where AAV integrates is not thought to cause insertional mutagenesis; AAV infections are non-pathogenic and are not usually associated with cancer. In fact, the non-pathogenic nature of this virus and its ability to integrate into a safe harbor in chromosome 19 while giving long-term transgene expression make AAV an ideal candidate for gene therapy. However, when the virus is genetically engineered to carry a transgene, all the viral genome except for the ITRs is removed, and the virus loses its ability on integrate into the host genome. Therefore, the AAV vectors used in gene therapy exist predominantly in the host cell’s nucleus as non-replicating episomes where they are lost over time because of an inability to replicate with the host cell’s DNA. A low number of random integrations have been reported to occur following gene delivery using AAV vectors.
Unlike other adenoviral and retroviral vectors, AAV has been used in numerous clinical trials with no vector related adverse events. A gene therapy pilot safety study using an adenovirus to treat a patient with ornithine transcarbamylase deficiency resulted in a fatality thought to be the result of a systemic inflammatory response to the adenoviral capsid [4]. An ex vivo gene transfer clinical trial that utilized a retrovirus to treat X-linked severe combined immunodeficiency resulted in cancer caused by insertional mutagenesis [28]. Because an AAV vector exists in a episomal state and integrates at very low frequency following transduction, the chances of insertional mutagenesis are greatly reduced in comparison to a retrovirus and lentivirus, which integrate as part of the life cycle. However, two independent studies have observed an increased incidence of hepatic carcinoma after a neonatal injection of a high dose of AAV in mice with Sly disease (MPS VII) [59] and methylmalonic acidemia [60] and data suggests that insertional mutagenesis may have been the cause. While AAV has been considered to be non-pathogenic for decades, a recent study suggests that wild-type AAV2 insertions can cause hepatic cancer in humans [61]. The contribution of recombinant AAV exposure to the development of hepatic cancer by insertional mutagenesis is debated but the many studies conducted in both mice and larger animals indicates the risk is likely very low.
One important consideration when using AAV in gene therapy applications is the wide range of serotypes, each with the ability to confer unique tropisms upon a given vector, available for use. For example, an AAV serotype 8 vector has greater than a 100-fold increase in the transduction of hepatocytes compared to AAV vectors of other serotypes, such as AAV serotype 2. Several AAV vectors have demonstrated great preclinical efficacy and one, Glybera – an AAV serotype 1 vector delivered by intra-muscular injection to express a lipoprotein lipase transgene - has recently been approved by the European Commission to treat lipoprotein lipase deficiency. Currently, this is the only approved gene therapy treatment for a metabolic disease [14]. However, many metabolic disorders affect enzymes that are expressed predominately or exclusively in the liver. An AAV with a liver tropism – such as a serotype 8 vector – thus would be ideal for treating many inborn errors of metabolism. In addition, liver transplantation has been successful in treating a number of metabolic diseases and reported to improve the condition of patients, suggesting that liver-directed gene therapy could be a beneficial treatment approach for a variety of inborn errors of metabolism. Because the liver often exhibits disease related metabolic abnormalities and pathology, replacing some of the lost enzymatic activity using gene delivery might lessen or reverse pathology in this tissue.
Non-human primate studies have documented that AAV serotype 8 (AAV8) vectors can efficiently transduce liver, muscle, and cardiac tissues, and express a transgene for a period of over five years following gene delivery [62, 63]. Preliminary results from an AAV8 liver restricted clinical gene therapy trial for hemophilia B have also demonstrated stable and persistent transgene expression in some treated patients [11, 64]. An earlier hemophilia B clinical trial utilized an AAV2 vector, but the transgene expression was transient because of a suspected immune response to the AAV2 capsid, something that might have been predicted given the fact that AAV2 is a common non-pathogenic virus found in humans [65, 66]. The clinical protocol for the successful AAV8 trial screened the patient cohort for the existence of pre-existing anti-AAV8 antibodies. Antibodies against AAV2 are detected at a higher frequency in human serum samples than antibodies against AAV8 and pre-existing immunity to AAV2 may explain why the AAV8 clinical trial has been much more successful in achieving long–term transgene expression and therapeutic levels of factor IX, the clotting factor which is deficient in hemophilia B. The successful long-term transgene expression achieved after systemic delivery of AAV8 targeting the liver in the hemophilia B trial establishes an approach for liver – directed gene therapy in humans with metabolic disorders.
10Future of gene therapy
Though numerous other viral vectors exist, this abridged review of gene therapy only discussed the most commonly used viral vectors and selected applications. It is unlikely that a single vector will dominate the field of gene therapy and new vectors are constantly under development. Vectors capable of targeted integration and DNA editing exist and have shown some promise in vitro and in pre-clinical studies. Targeted integration could decrease the chance of insertional mutagenesis and DNA editing should afford the genomic repair of the disease causing genetic lesion in situ. The development of immunosuppression regiments for gene therapy might also improve the safety and transduction efficacy of viral vectors, which are often pathogens that can elicit immune responses.
The early clinical trials for gene therapy were initiated with the hope that a new era for the treatment of inherited diseases would begin. Unfortunately, early gene therapy clinical trials showed poor vector efficacy and severe adverse events, casting doubt on the potential of gene therapy to emerge as a viable treatment for genetic disorders. Despite the fact that thousands of participants have received gene therapy without complications, most people might still associate gene therapy with the severe adverse events seen in a few patients. More recently, gene therapy clinical trials have demonstrated safety and efficacy in treating genetic disorders. Currently, two gene therapy treatments are approved for clinical use: one to treat head and neck cancers in China and another to treat lipoprotein lipase deficiency in Europe. Based on the successes being reported from a number of clinical trials without the occurrence of severe adverse events, more gene therapy treatments will likely be approved for clinical use in the coming years. When one considers that there are no or inadequate treatments for a majority of patients with genetic diseases, gene therapy is certain to represent a new and exciting therapeutic option for those afflicted by inborn errors of metabolism.
References
[1] | Merril C.R. , Geier M.R. and Petricciani J.C. , Bacterial virus gene expression in human cells, Nature 233: ((1971) ), 398–400. |
[2] | Lander E.S. , Linton L.M. , Birren B. , Nusbaum C. , Zody M.C. , Baldwin J. , Devon K. , Dewar K. , Doyle M. , FitzHugh W. , Funke R. , Gage D. , Harris K. , Heaford A. , Howland J. , Kann L. , Lehoczky J. , LeVine R. , McEwan P. , McKernan K. , Meldrim J. , Mesirov J.P. , Miranda C. , Morris W. , Naylor J. , Raymond C. , Rosetti M. , Santos R. , Sheridan A. , Sougnez C. , Stange-Thomann N. , Stojanovic N. , Subramanian A. , Wyman D. , Rogers J. , Sulston J. , Ainscough R. , Beck S. , Bentley D. , Burton J. , Clee C. , Carter N. , Coulson A. , Deadman R. , Deloukas P. , Dunham A. , Dunham I. , Durbin R. , French L. , Grafham D. , Gregory S. , Hubbard T. , Humphray S. , Hunt A. , Jones M. , Lloyd C. , McMurray A. , Matthews L. , Mercer S. , Milne S. , Mullikin J.C. , Mungall A. , Plumb R. , Ross M. , Shownkeen R. , Sims S. , Waterston R.H. , Wilson R.K. , Hillier L.W. , McPherson J.D. , Marra M.A. , Mardis E.R. , Fulton L.A. , Chinwalla A.T. , Pepin K.H. , Gish W.R. , Chissoe S.L. , Wendl M.C. , Delehaunty K.D. , Miner T.L. , Delehaunty A. , Kramer J.B. , Cook L.L. , Fulton R.S. , Johnson D.L. , Minx P.J. , Clifton S.W. , Hawkins T. , Branscomb E. , Predki P. , Richardson P. , Wenning S. , Slezak T. , Doggett N. , Cheng J.F. , Olsen A. , Lucas S. , Elkin C. , Uberbacher E. , Frazier M. , Gibbs R.A. , Muzny D.M. , Scherer S.E. , Bouck J.B. , Sodergren E.J. , Worley K.C. , Rives C.M. , Gorrell J.H. , Metzker M.L. , Naylor S.L. , Kucherlapati R.S. , Nelson D.L. , Weinstock G.M. , Sakaki Y. , Fujiyama A. , Hattori M. , Yada T. , Toyoda A. , Itoh T. , Kawagoe C. , Watanabe H. , Totoki Y. , Taylor T. , Weissenbach J. , Heilig R. , Saurin W. , Artiguenave F. , Brottier P. , Bruls T. , Pelletier E. , Robert C. , Wincker P. , Smith D.R. , Doucette-Stamm L. , Rubenfield M. , Weinstock K. , Lee H.M. , Dubois J. , Rosenthal A. , Platzer M. , Nyakatura G. , Taudien S. , Rump A. , Yang H. , Yu J. , Wang J. , Huang G. , Gu J. , Hood L. , Rowen L. , Madan A. , Qin S. , Davis R.W. , Federspiel N.A. , Abola A.P. , Proctor M.J. , Myers R.M. , Schmutz J. , Dickson M. , Grimwood J. , Cox D.R. , Olson M.V. , Kaul R. , Shimizu N. , Kawasaki K. , Minoshima S. , Evans G.A. , Athanasiou M. , Schultz R. , Roe B.A. , Chen F. , Pan H. , Ramser J. , Lehrach H. , Reinhardt R. , McCombie W.R. , de la Bastide M. , Dedhia, N., Blocker, H., Hornischer, K., Nordsiek, G., Agarwala, R., Aravind, L., Bailey, J.A., Bateman, A., Batzoglou, S., Birney, E., Bork, P., Brown, D.G., Burge, C.B., Cerutti, L., Chen, H.C., Church, D., Clamp, M., Copley, R.R., Doerks, T., Eddy, S.R., Eichler, E.E., Furey, T.S., Galagan, J., Gilbert, J.G., Harmon, C., Hayashizaki, Y., Haussler, D., Hermjakob, H., Hokamp, K., Jang, W., Johnson, L.S., Jones, T.A., Kasif, S., Kaspryzk, A., Kennedy, S., Kent, W.J., Kitts, P., Koonin, E.V., Korf, I., Kulp, D., Lancet, D., Lowe, T.M., McLysaght, A., Mikkelsen, T., Moran, J.V., Mulder, N., Pollara, V.J., Ponting, C.P., Schuler, G., Schultz, J., Slater, G., Smit, A.F., Stupka, E., Szustakowski, J., Thierry-Mieg, D., Thierry-Mieg, J., Wagner, L., Wallis, J., Wheeler, R., Williams, A., Wolf, Y.I., Wolfe, K.H., Yang, S.P., Yeh, R.F., Collins, F., Guyer, M.S., Peterson, J., Felsenfeld, A., Wetterstrand, K.A., Patrinos, A., Morgan, M.J., de Jong, P., Catanese, J.J., Osoegawa, K., Shizuya, H., Choi, S. and Chen, Y.J. , Initial Sequencing and Analysis of the Human Genome, Nature 409: ((2001) ), 860–921. |
[3] | Blaese R.M. , Culver K.W. , Miller A.D. , Carter C.S. , Fleisher T. , Clerici M. , Shearer G. , Chang L. , Chiang Y. , Tolstoshev P. , Greenblatt J.J. , Rosenberg S.A. , Klein H. , Berger M. , Mullen C.A. , Ramsey W.J. , Muul L. , Morgan R.A. and Anderson W.F. , T lymphocyte-directed gene therapy for ADA- SCID: Initial trial results after 4 years, Science 270: ((1995) ), 475–480. |
[4] | Raper S.E. , Chirmule N. , Lee F.S. , Wivel N.A. , Bagg A. , Gao G.P. , Wilson J.M. and Batshaw M.L. , Fatal systemic inflammatory response syndrome in a ornithine transcarbamylase deficient patient following adenoviral gene transfer, Mol Genet Metab 80: ((2003) ), 148–158. |
[5] | Hacein-Bey-Abina S. , von Kalle C. , Schmidt, M., Le, Deist F., Wulffraat, N., McIntyre, E., Radford, I., Villeval, J.L., Fraser, C.C., Cavazzana-Calvo, M. and Fischer, A., A serious adverse event after successful gene therapy for X-linked severe combined immunodeficiency, N Engl J Med 348: ((2003) ), 255–256. |
[6] | Hacein-Bey-Abina S. , Hauer J. , Lim A. , Picard C. , Wang G.P. , Berry C.C. , Martinache C. , Rieux-Laucat F. , Latour S. , Belohradsky B.H. , Leiva L. , Sorensen R. , Debre M. , Casanova J.L. , Blanche S. , Durandy A. , Bushman F.D. , Fischer A. and Cavazzana-Calvo M. , Efficacy of gene therapy for X-linked severe combined immunodeficiency, N Engl J Med 363: ((2010) ), 355–364. |
[7] | Aiuti A. , Cattaneo F. , Galimberti S. , Benninghoff U. , Cassani B. , Callegaro L. , Scaramuzza S. , Andolfi G. , Mirolo M. , Brigida I. , Tabucchi A. , Carlucci F. , Eibl M. , Aker M. , Slavin S. , Al-Mousa H. , Al Ghonaium A. , Ferster A., Duppenthaler A., Notarangelo L., Wintergerst U., Buckley R.H., Bregni M., Marktel S., Valsecchi M.G., Rossi P., Ciceri F., Miniero R., Bordignon C. and Roncarolo M.G., Gene therapy for immunodeficiency due to adenosine deaminase deficiency, N Engl J Med 360: ((2009) ), 447–458. |
[8] | Maguire A.M. , High K.A. , Auricchio A. , Wright J.F. , Pierce E.A. , Testa F. , Mingozzi F. , Bennicelli J.L. , Ying G.S. , Rossi S. , Fulton A. , Marshall K.A. , Banfi S. , Chung D.C. , Morgan J.I. , Hauck B. , Zelenaia O. , Zhu X. , Raffini L. , Coppieters F. , De Baere E. , Shindler K.S., Volpe N.J., Surace E.M., Acerra C., Lyubarsky A., Redmond T.M., Stone E., Sun J., McDonnell J.W., Leroy B.P., Simonelli F. and Bennett J., Age-dependent effects of RPE65 gene therapy for Leber’s congenital amaurosis: A phase 1 dose-escalation trial, Lancet 374: ((2009) ), 1597–1605. |
[9] | Cartier N. , Hacein-Bey-Abina S. , Bartholomae C.C. , Veres G. , Schmidt M. , Kutschera I. , Vidaud M. , Abel U. , Dal-Cortivo L. , Caccavelli L. , Mahlaoui N. , Kiermer V. , Mittelstaedt D. , Bellesme C. , Lahlou N. , Lefrere F. , Blanche S. , Audit M. , Payen E. , Leboulch P. , l’Homme B. , Bougneres P. , Von Kalle C. , Fischer A., Cavazzana-Calvo M. and Aubourg P., Hematopoietic stem cell gene therapy with a lentiviral vector in X-linked adrenoleukodystrophy, Science 326: ((2009) ), 818–823. |
[10] | Interim Update On ProSavin® Phase I/II Study In Parkinson’s Disease, http://www.oxfordbiomedica.co.uk/page.asp?pageid=59&newsid=643, Scientist Microscope, OxfordBioMedica, (2011) . |
[11] | Nathwani A.C. , Tuddenham E.G. , Rangarajan S. , Rosales C. , McIntosh J. , Linch D.C. , Chowdary P. , Riddell A. , Pie A.J. , Harrington C. , O’Beirne J. , Smith K. , Pasi J. , Glader B. , Rustagi P. , Ng C.Y. , Kay M.A. , Zhou J. , Spence Y. , Morton C.L. , Allay J. , Coleman J. , Sleep S. , Cunningham J.M. , Srivastava D. , Basner-Tschakarjan E. , Mingozzi F. , High K.A. , Gray J.T. , Reiss U.M. , Nienhuis A.W. and Davidoff A.M. , Adenovirus-associated virus vector-mediated gene transfer in hemophilia B, N Engl J Med 365: ((2011) ), 2357–2365. |
[12] | Hwu W.L. , Muramatsu S. , Tseng S.H. , Tzen K.Y. , Lee N.C. , Chien Y.H. , Snyder R.O. , Byrne B.J. , Tai C.H. and Wu R.M. , Gene therapy for aromatic L-amino acid decarboxylase deficiency, Sci Transl Med 4: ((2012) ), 134ra161. |
[13] | Peng Z. , Current status of gendicine in China: Recombinant human Ad-p53 agent for treatment of cancers, Hum Gene Ther 16: ((2005) ), 1016–1027. |
[14] | Gallagher J. , Gene therapy: Glybera approved by European Commission, http://www.bbc.co.uk/news/health-20179561, News Health, BBC News, (2012) . |
[15] | Biffi A. , Aubourg P. and Cartier N. , Gene therapy for leukodystrophies, Hum Mol Genet 20: ((2011) ), R42–53. |
[16] | Cavazzana-Calvo M. and Fischer A. , Gene therapy for severe combined immunodeficiency: Are we there yet? J Clin Invest 117: ((2007) ), 1456–1465. |
[17] | Nygaard S. , Barzel A. , Haft A. , Major A. , Finegold M. , Kay M.A. and Grompe M. , A universal system to select gene-modified hepatocytes in vivo, Sci Transl Med 8: ((2016) ), 342ra379. |
[18] | Coffin J.M. , Structure and classification of retroviruses, in: Levy J.A. (Ed.), The retroviridae, Plenum Press, New York, (1992) , pp. 19–49. |
[19] | Coffin J.M. , Hughes S.H. and Varmus H.E. , Retroviruses, Cold Spring Harbor Laboratory Press, Cold Spring Harbor (NY), (1997) . |
[20] | Fischer A. , Hacein-Bey-Abina S. and Cavazzana-Calvo M. , Strategies for Retrovirus-Based Correction of Severe, Combined Immunodeficiency (SCID), Elsevier Inc., San Diego, (2012) . |
[21] | Neven B. , Leroy S. , Decaluwe H. , Le Deist F. , Picard C., Moshous D., Mahlaoui N., Debre M., Casanova J.L., Dal Cortivo L., Madec Y., Hacein-Bey-Abina S., de Saint Basile G., de Villartay J.P., Blanche S., Cavazzana-Calvo M. and Fischer A., Long-term outcome after hematopoietic stem cell transplantation of a single-center cohort of 90 patients with severe combined immunodeficiency, Blood 113: ((2009) ), 4114–4124. |
[22] | Sugamura K. , Asao H. , Kondo M. , Tanaka N. , Ishii N. , Ohbo K. , Nakamura M. and Takeshita T. , The interleukin-2 receptor gamma chain: Its role in the multile cytokine receptor complexes and T cell development in XSCID, Annu Rev Immunol 14: ((1996) ), 179–205. |
[23] | Leonard W.J. , The molecular basis of X-linked severe combined immunodeficiency: Defective cytokine receptor signaling, Annu Rev Med 47: ((1996) ), 229–239. |
[24] | Noguchi M. , Yi H. , Rosenblatt H.M. , Filipovich A.H. , Adelstein S. , Modi W.S. , McBride O.W. and Leonard W.J. , Interleukin-2 receptor gamma chain mutation results in X-linked severe combined immunodeficiency in humans, Cell 73: ((1993) ), 147–157. |
[25] | Cavazzana-Calvo M. , Hacein-Bey S. , de Saint Basile G. , Gross F., Yvon E., Nusbaum P., Selz F., Hue C., Certain S., Casanova J.L., Bousso P., Deist F.L. and Fischer A., Gene therapy of human severe combined immunodeficiency (SCID)-X1 disease, Science 288: ((2000) ), 669–672. |
[26] | Gaspar H.B. , Parsley K.L. , Howe S. , King D. , Gilmour K.C. , Sinclair J. , Brouns G. , Schmidt M. , Von Kalle C. , Barington T., Jakobsen M.A., Christensen H.O., Al Ghonaium A., White H.N., Smith J.L., Levinsky R.J., Ali R.R., Kinnon C. and Thrasher A.J., Gene therapy of X-linked severe combined immunodeficiency by use of a pseudotyped gammaretroviral vector, Lancet 364: ((2004) ), 2181–2187. |
[27] | Howe S.J. , Mansour M.R. , Schwarzwaelder K. , Bartholomae C. , Hubank M. , Kempski H. , Brugman M.H. , Pike-Overzet K. , Chatters S.J. , de Ridder D. , Gilmour K.C., Adams S., Thornhill S.I., Parsley K.L., Staal F.J., Gale R.E., Linch D.C., Bayford J., Brown L., Quaye M., Kinnon C., Ancliff P., Webb D.K., Schmidt M., von Kalle C., Gaspar H.B. and Thrasher A.J., Insertional mutagenesis combined with acquired somatic mutations causes leukemogenesis following gene therapy of SCID-X1 patients, J Clin Invest 118: ((2008) ), 3143–3150. |
[28] | Hacein-Bey-Abina S. , Von Kalle C. , Schmidt M., McCormack M.P., Wulffraat N., Leboulch P., Lim A., Osborne C.S., Pawliuk R., Morillon E., Sorensen R., Forster A., Fraser P., Cohen J.I., de Saint Basile G., Alexander I., Wintergerst U., Frebourg T., Aurias A., Stoppa-Lyonnet D., Romana S., Radford-Weiss I., Gross F., Valensi F., Delabesse E., Macintyre E., Sigaux F., Soulier J., Leiva L.E., Wissler M., Prinz C., Rabbitts T.H., Le Deist F., Fischer A. and Cavazzana-Calvo M. , LMO2-associated clonal T cell proliferation in two patients after gene therapy for SCID-X1, Science 302: ((2003) ), 415–419. |
[29] | Thornhill S.I. , Schambach A. , Howe S.J. , Ulaganathan M. , Grassman E. , Williams D. , Schiedlmeier B. , Sebire N.J. , Gaspar H.B. , Kinnon C. , Baum C. and Thrasher A.J. , Self-inactivating gammaretroviral vectors for gene therapy of X-linked severe combined immunodeficiency, Mol Ther 16: ((2008) ), 590–598. |
[30] | Ferrua F. , Brigida I. and Aiuti A. , Update on gene therapy for adenosine deaminase-deficient severe combined immunodeficiency, Curr Opin Allergy Clin Immunol 10: ((2010) ), 551–556. |
[31] | Gaspar H.B. , Cooray S. , Gilmour K.C. , Parsley K.L. , Zhang F. , Adams S. , Bjorkegren E. , Bayford J. , Brown L. , Davies E.G. , Veys P. , Fairbanks L. , Bordon V. , Petropolou T. , Kinnon C. and Thrasher A.J. , Hematopoietic stem cell gene therapy for adenosine deaminase-deficient severe combined immunodeficiency leads to long-term immunological recovery and metabolic correction, Sci Transl Med 3: ((2011) ), 97ra80. |
[32] | Bartosch B. , Dubuisson J. and Cosset F.L. , Infectious hepatitis C virus pseudo-particles containing functional E1-E2 envelope protein complexes, J Exp Med 197: ((2003) ), 633–642. |
[33] | Burns J.C. , Friedmann T. , Driever W. , Burrascano M. and Yee J.K. , Vesicular stomatitis virus G glycoprotein pseudotyped retroviral vectors: Concentration to very high titer and efficient gene transfer into mammalian and nonmammalian cells, Proc Natl Acad Sci U S A 90: ((1993) ), 8033–8037. |
[34] | Levine B.L. , Humeau L.M. , Boyer J. , MacGregor R.R. , Rebello T. , Lu X. , Binder G.K. , Slepushkin V. , Lemiale F. , Mascola J.R. , Bushman F.D. , Dropulic B. and June C.H. , Gene transfer in humans using a conditionally replicating lentiviral vector, Proc Natl Acad Sci U S A 103: ((2006) ), 17372–17377. |
[35] | Cavazzana-Calvo M. , Payen E. , Negre O. , Wang G. , Hehir K. , Fusil F. , Down J. , Denaro M. , Brady T. , Westerman K. , Cavallesco R. , Gillet-Legrand B. , Caccavelli L. , Sgarra R. , Maouche-Chretien L. , Bernaudin F. , Girot R. , Dorazio R. , Mulder G.J. , Polack A. , Bank A. , Soulier J. , Larghero J. , Kabbara N. , Dalle B. , Gourmel B. , Socie G. , Chretien S. , Cartier N. , Aubourg P. , Fischer A. , Cornetta K. , Galacteros F. , Beuzard Y. , Gluckman E. , Bushman F. , Hacein-Bey-Abina S. and Leboulch P. , Transfusion independence and HMGA2 activation after gene therapy of human beta-thalassaemia, Nature 467: ((2010) ), 318–322. |
[36] | Moser H.W. , Mahmood A. and Raymond G.V. , X-linked adrenoleukodystrophy, Nat Clin Pract Neurol 3: ((2007) ), 140–151. |
[37] | Aubourg P. , Blanche S. , Jambaque I. , Rocchiccioli F. , Kalifa G. , Naud-Saudreau C. , Rolland M.O. , Debre M. , Chaussain J.L. , Griscelli C. , et al., Reversal of early neurologic and neuroradiologic manifestations of X-linked adrenoleukodystrophy by bone marrow transplantation, N Engl J Med 322: ((1990) ), 1860–1866. |
[38] | Shapiro E. , Krivit W. , Lockman L. , Jambaque I. , Peters C. , Cowan M. , Harris R. , Blanche S. , Bordigoni P. , Loes D. , Ziegler R. , Crittenden M. , Ris D. , Berg B. , Cox C. , Moser H. , Fischer A. and Aubourg P. , Long-term effect of bone-marrow transplantation for childhood-onset cerebral X-linked adrenoleukodystrophy, Lancet 356: ((2000) ), 713–718. |
[39] | Shenk T. , Adenoviridae: The viruses and their replication, in Fields Virology, Lippincott-Raven Publishers, Philadelphia, (1996) . |
[40] | McConnell M.J. and Imperiale M.J. , Biology of adenovirus and its use as a vector for gene therapy, Hum Gene Ther 15: ((2004) ), 1022–1033. |
[41] | Jones N. and Shenk T. , Isolation of adenovirus type 5 host range deletion mutants defective for transformation of rat embryo cells, Cell 17: ((1979) ), 683–689. |
[42] | Berkner K.L. , Development of adenovirus vectors for the expression of heterologous genes, Biotechniques 6: ((1988) ), 616–629. |
[43] | Amalfitano A. , Next-generation adenoviral vectors: New and improved, Gene Ther 6: ((1999) ), 1643–1645. |
[44] | Palmer D.J. and Ng P. , Helper-dependent adenoviral vectors for gene therapy, Hum Gene Ther 16: ((2005) ), 1–16. |
[45] | Brunetti-Pierri N. , Palmer D.J. , Beaudet A.L. , Carey K.D. , Finegold M. and Ng P. , Acute toxicity after high-dose systemic injection of helper-dependent adenoviral vectors into nonhuman primates, Hum Gene Ther 15: ((2004) ), 35–46. |
[46] | Muruve D.A. , Cotter M.J. , Zaiss A.K. , White L.R. , Liu Q. , Chan T. , Clark S.A. , Ross P.J. , Meulenbroek R.A. , Maelandsmo G.M. and Parks R.J. , Helper-dependent adenovirus vectors elicit intact innate but attenuated adaptive host immune responses in vivo, J Virol 78: ((2004) ), 5966–5972. |
[47] | Brunetti-Pierri N. and Ng P. , Helper-dependent adenoviral vectors for liver-directed gene therapy, Hum Mol Genet 20: ((2011) ), R7–13. |
[48] | Brunetti-Pierri N. , Stapleton G.E. , Law M. , Breinholt J. , Palmer D.J. , Zuo Y. , Grove N.C. , Finegold M.J. , Rice K. , Beaudet A.L. , Mullins C.E. and Ng P. , Efficient, long-term hepatic gene transfer using clinically relevant HDAd doses by balloon occlusion catheter delivery in nonhuman primates, Mol Ther 17: ((2009) ), 327–333. |
[49] | Seregin S.S. , Appledorn D.M. , McBride A.J. , Schuldt N.J. , Aldhamen Y.A. , Voss T. , Wei J. , Bujold M. , Nance W. , Godbehere S. and Amalfitano A. , Transient pretreatment with glucocorticoid ablates innate toxicity of systemically delivered adenoviral vectors without reducing efficacy, Mol Ther 17: ((2009) ), 685–696. |
[50] | Alexander I.E. , Cunningham S.C. , Logan G.J. and Christodoulou J. , Potential of AAV vectors in the treatment of metabolic disease, Gene Ther 15: ((2008) ), 831–839. |
[51] | Atchison R.W. , Casto B.C. and Hammon W.M. , Adenovirus-Associated Defective Virus Particles, Science 149: ((1965) ), 754–756. |
[52] | Richardson W.D. and Westphal H. , A cascade of adenovirus early functions is required for expression of adeno-associated virus, Cell 27: ((1981) ), 133–141. |
[53] | Mishra L. and Rose J.A. , Adeno-associated virus DNA replication is induced by genes that are essential for HSV-1 DNA synthesis, Virology 179: ((1990) ), 632–639. |
[54] | Wietzman M.D. and Linden R.M. , Adeno-Assoitaed Virus Biology in Adeno-Associated Viruses: Methods & Protocols Springer Science, New York, (2011) . |
[55] | Berns K.I. and Giraud C. , Biology of adeno-associated virus, Curr Top Microbiol Immunol 218: ((1996) ), 1–23. |
[56] | Kotin R.M. , Prospects for the use of adeno-associated virus as a vector for human gene therapy, Hum Gene Ther 5: ((1994) ), 793–801. |
[57] | Rose J.A. , Maizel J.V. Jr. , Inman J.K. and Shatkin A.J., Structural proteins of adenovirus-associated viruses, J Virol 8: ((1971) ), 766–770. |
[58] | Kotin R.M. , Linden R.M. and Berns K.I. , Characterization of a preferred site on human chromosome 19q for integration of adeno-associated virus DNA by non-homologous recombination, Embo J 11: ((1992) ), 5071–5078. |
[59] | Donsante A. , Miller D.G. , Li Y. , Vogler C. , Brunt E.M. , Russell D.W. and Sands M.S. , AAV vector integration sites in mouse hepatocellular carcinoma, Science 317: ((2007) ), 477. |
[60] | Chandler R.J. , LaFave M.C. , Varshney G.K. , Trivedi N.S. , Carrillo-Carrasco N. , Senac J.S. , Wu W. , Hoffmann V. , Elkahloun A.G. , Burgess S.M. and Venditti C.P. , Vector design influences hepatic genotoxicity after adeno-associated virus gene therapy, J Clin Invest 125: ((2015) ), 870–880. |
[61] | Nault J.C. , Datta S. , Imbeaud S. , Franconi A. , Mallet M. , Couchy G. , Letouze E. , Pilati C. , Verret B. , Blanc J.F. , Balabaud C. , Calderaro J. , Laurent A. , Letexier M. , Bioulac-Sage P. , Calvo F. and Zucman-Rossi J. , Recurrent AAV2-related insertional mutagenesis in human hepatocellular carcinomas, Nat Genet 47: (10) ((2015) ), 1187–1193. |
[62] | Gao G.P. , Alvira M.R. , Wang L. , Calcedo R. , Johnston J. and Wilson J.M. , Novel adeno-associated viruses from rhesus monkeys as vectors for human gene therapy, Proc Natl Acad Sci U S A 99: ((2002) ), 11854–11859. |
[63] | Nathwani A.C. , Rosales C. , McIntosh J. , Rastegarlari G. , Nathwani D. , Raj D. , Nawathe S. , Waddington S.N. , Bronson R. , Jackson S. , Donahue R.E. , High K.A. , Mingozzi F. , Ng C.Y. , Zhou J. , Spence Y. , McCarville M.B. , Valentine M. , Allay J. , Coleman J. , Sleep S. , Gray J.T. , Nienhuis A.W. and Davidoff A.M. , Long-term safety and efficacy following systemic administration of a self-complementary AAV vector encoding human FIX pseudotyped with serotype 5 and 8 capsid proteins, Mol Ther 19: ((2011) ), 876–885. |
[64] | Nathwani A.C. , Reiss U.M. , Tuddenham E.G. , Rosales C. , Chowdary P. , McIntosh J. , Della Peruta M. , Lheriteau E., Patel N., Raj D., Riddell A., Pie J., Rangarajan S., Bevan D., Recht M., Shen Y.M., Halka K.G., Basner-Tschakarjan E., Mingozzi F., High K.A., Allay J., Kay M.A., Ng C.Y., Zhou J., Cancio M., Morton C.L., Gray J.T., Srivastava D., Nienhuis A.W. and Davidoff A.M., Long-term safety and efficacy of factor IX gene therapy in hemophilia B, N Engl J Med 371: ((2014) ), 1994–2004. |
[65] | Manno C.S. , Pierce G.F. , Arruda V.R. , Glader B. , Ragni M. , Rasko J.J. , Ozelo M.C. , Hoots K. , Blatt P. , Konkle B. , Dake M. , Kaye R. , Razavi M. , Zajko A. , Zehnder J. , Rustagi P.K. , Nakai H. , Chew A. , Leonard D. , Wright J.F. , Lessard R.R. , Sommer J.M. , Tigges M. , Sabatino D. , Luk A. , Jiang H. , Mingozzi F. , Couto L. , Ertl H.C. , High K.A. and Kay M.A. , Successful transduction of liver in hemophilia by AAV-Factor IX and limitations imposed by the host immune response, Nat Med 12: ((2006) ), 342–347. |
[66] | Mingozzi F. , Maus M.V. , Hui D.J. , Sabatino D.E. , Murphy S.L. , Rasko J.E. , Ragni M.V. , Manno C.S. , Sommer J. , Jiang H. , Pierce G.F. , Ertl H.C. and High K.A. , CD8(+) T-cell responses to adeno-associated virus capsid in humans, Nat Med 13: ((2007) ), 419–422. |
Figures and Tables
Fig.1
Diagram of therapeutic nucleotide sequences (TNS). A. Non-viral TNS have an enhancer (optional) for increased transgene expression, a promoter (necessary) to drive transgene expression, an intron (optional) for increase gene expression, a transgene to correct the genetic defect and a polyadenylation (poly-A) signal (necessary) for proper translation. B. Adeno-associated virus TNS contain all the elements of non-viral TNS as well as inverted terminal repeats (ITR), which are necessary for the packaging of the TNS into the virus. C. Retroviral TNS contain the elements necessary for proper transgene expression, and retroviral elements and coding sequences. The Ψ element is a retroviral packaging signal. The retroviral long terminal repeats (LTR) mediated integration of retroviral DNA and have promoter activity, which is sometimes inactivated to prevent insertional mutagenesis. The rev-responsive element (RRE) is necessary for post-transcriptional transport of viral RNA during retroviral packaging.
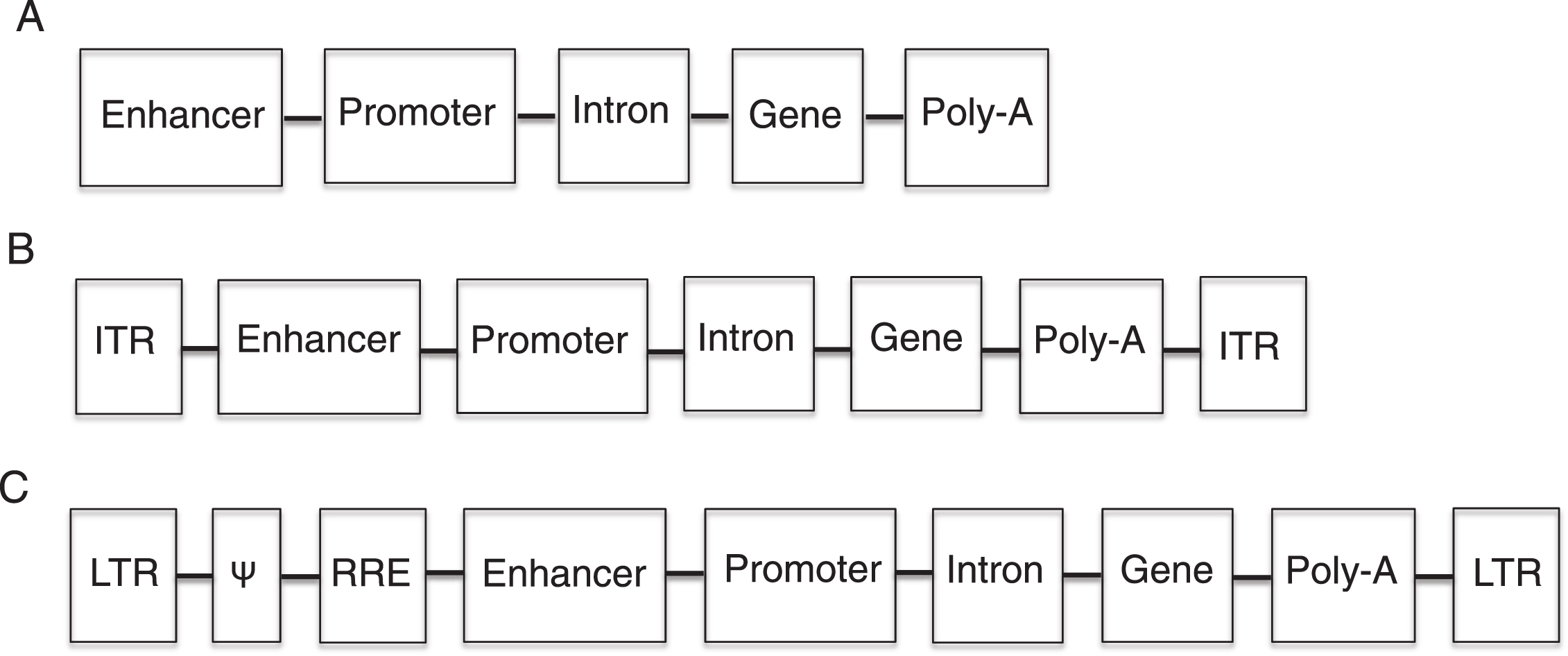
Fig.2
Gene delivery by a viral gene therapy vector. The diagram depicts gene delivery to a cell by an adenoviral vector. The adenovirus binds to the cell membrane and then enters the cells cytoplasm. After enter the cell, the virus traffics to the nucleus where it delivers its transgene. Once inside the nucleus, the transgene remains episomal and is transcribe to RNA, which is transported to the cytoplasm where it is translated into protein. The newly form protein can then become active in the cytosol or can be transported to cellular organs or excreted into the extracellular space depending on the function of the protein.
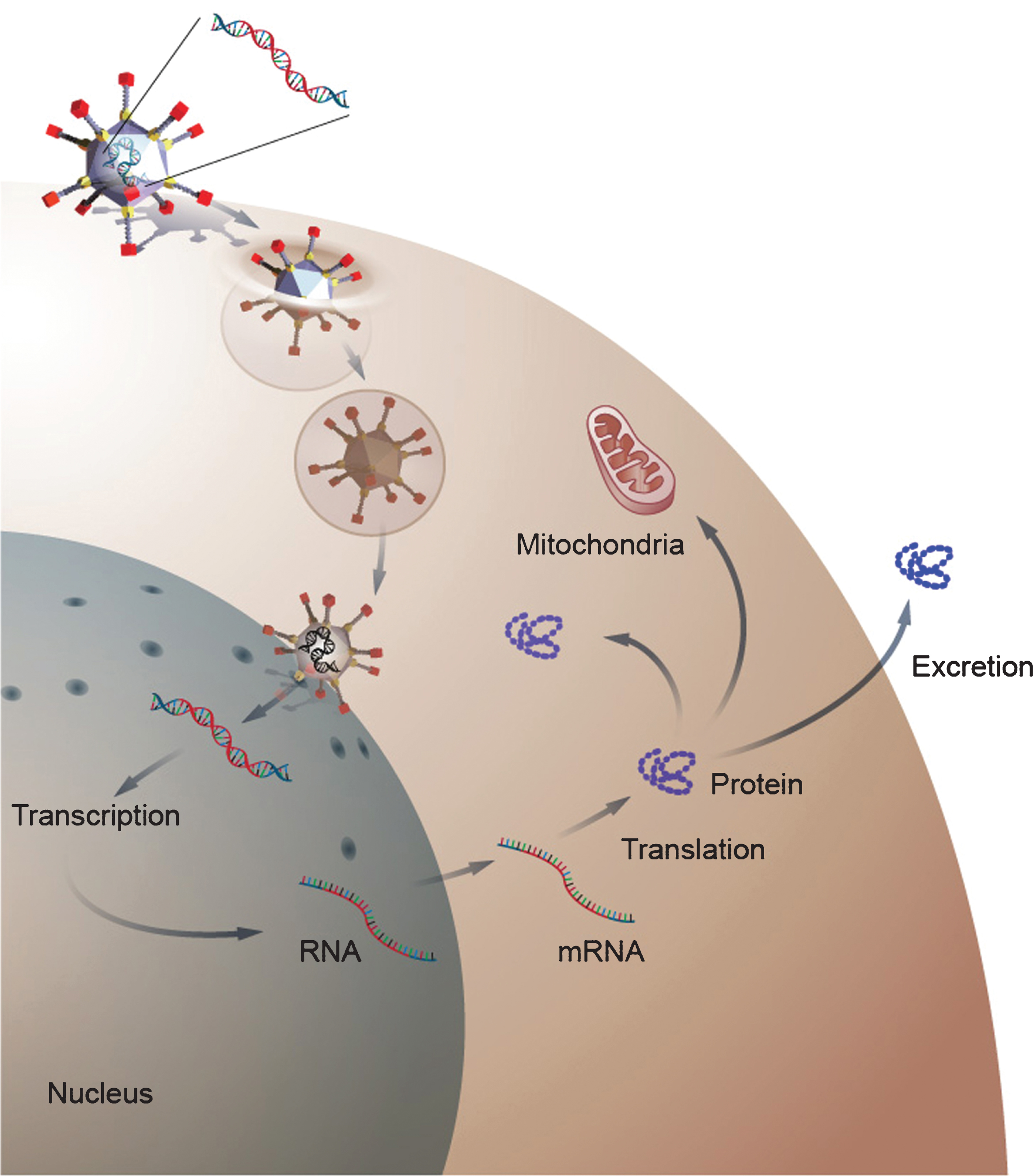
Fig.3
Production of recombinant adeno-associated virus (AAV). Three separate plasmids that carry the rep and cap sequences, the adenovirus (AD) helper genes and the TNS depicted in Fig. 1B are transfected into cell lines. Viral genome replication, protein translation and the assembly of the virons occur in the cell line. The AAV virons are then collected, purified and tittered.
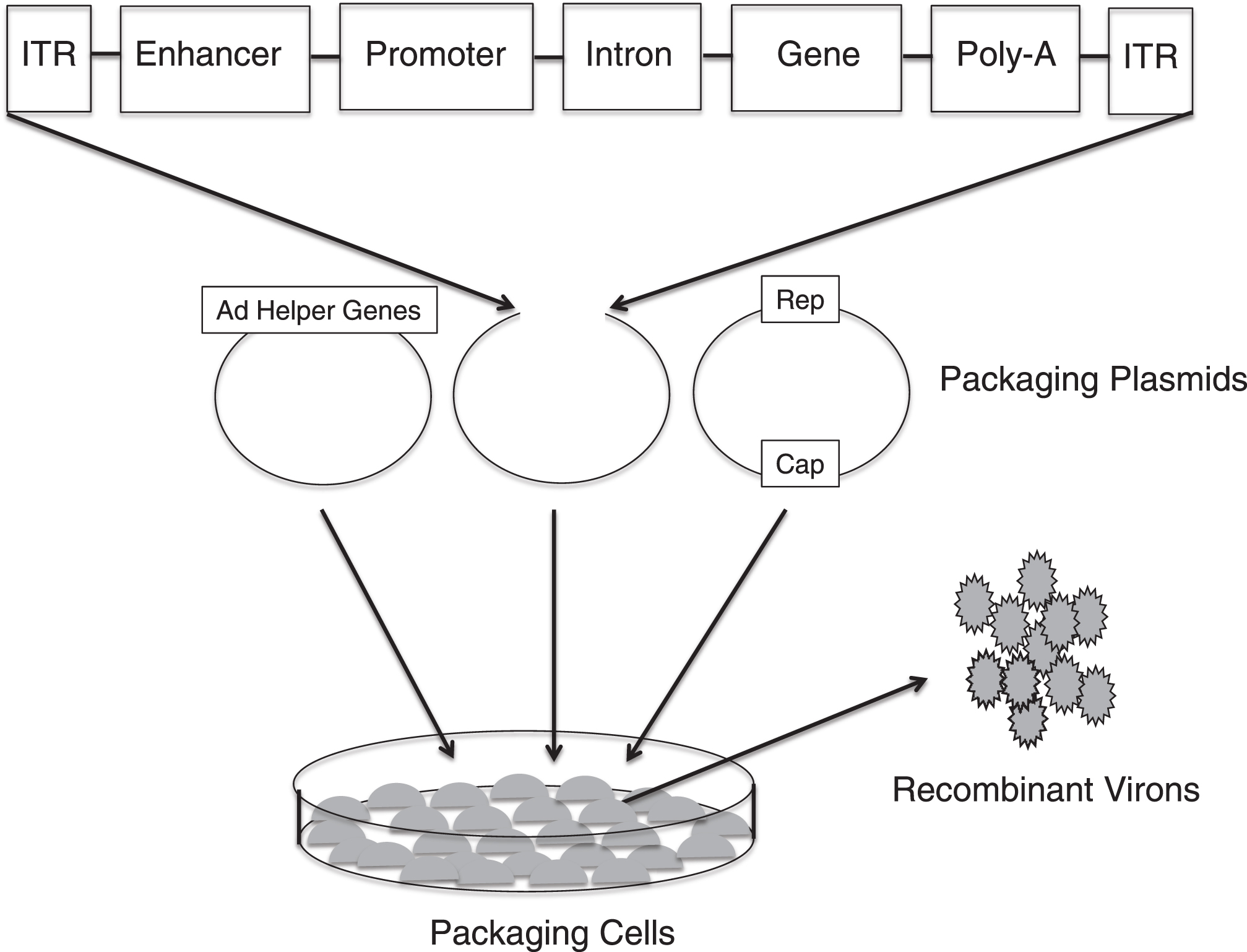
Fig.4
A depiction of the life cycle of the Human immunodeficiency Virus (HIV). HIV is a complex retrovirus. The HIV life cycle includes viral binding, entry, reverse transcription, integration, transcription, translation, protein modification, assembly and budding. This figure is courtesy of Wikimedia Commons and is re-printed under the GNU free documentationlicense.
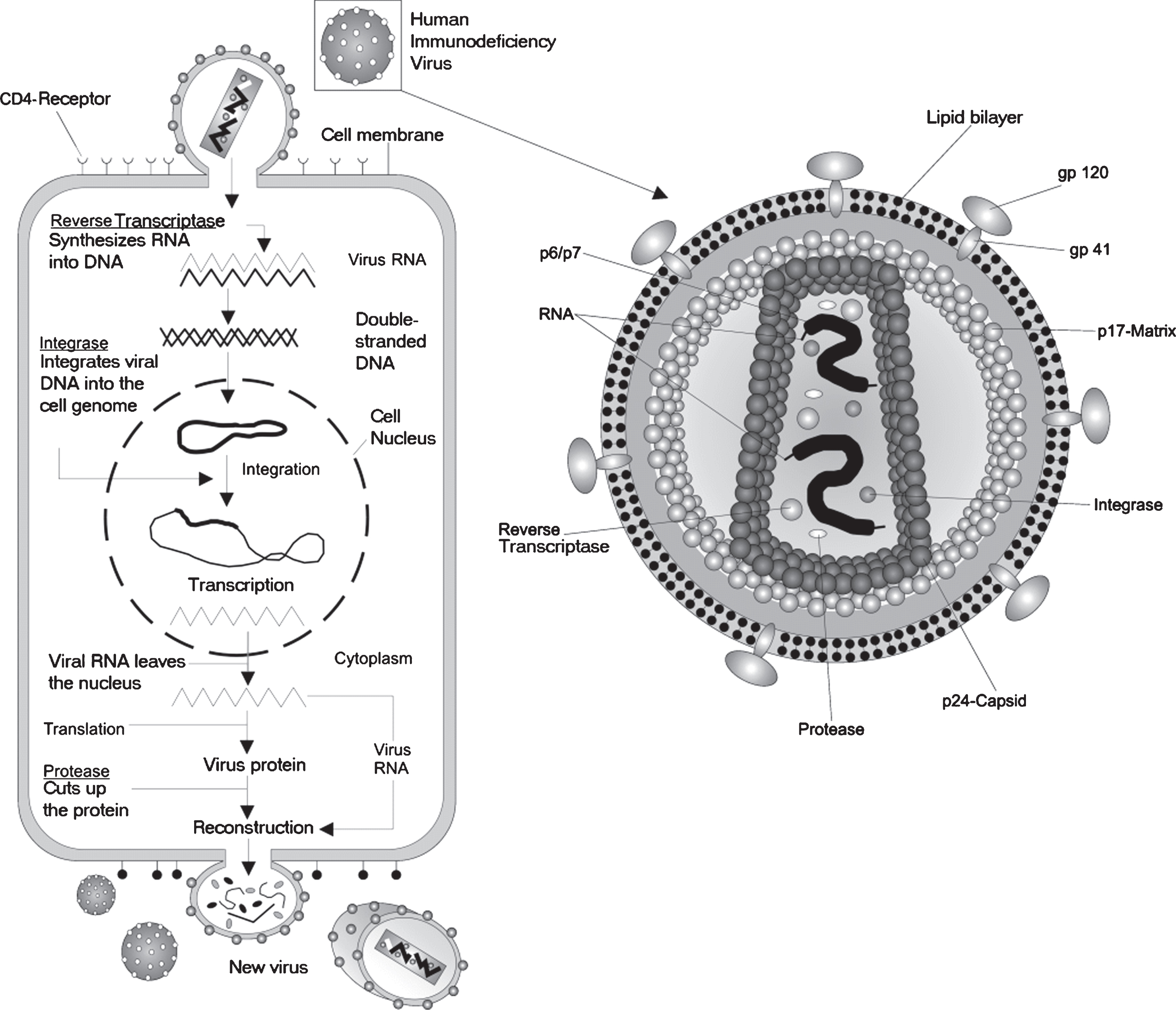
Table 1
Characteristics of viral gene therapy vectors
| Virus | Adenovirus | Adeno-associated virus | Retrovirus | Lentivirus |
| Family | Adenoviridae | Parvoviridae | Retroviridae | Retroviridae |
| Genome | dsDNA | ssDNA | ssRNA | ssRNA |
| Genome Size | 26–40 kb | 5 kb | 7–12 kb | 7–12kb |
| Coat | naked | naked | enveloped | enveloped |
| Viron Diameter | 70–100 nm | 18–26 nm | 80–100 nm | 80–100 nm |
| Pathogenic | yes | No | yes | yes |
| Infects | dividing and non-dividing | dividing and non-dividing | dividing | dividing and non-dividing |
| Interaction with | non-integrating | non-integrating | integrating | integrating |
| Host Genome | ||||
| Transgene Expression | transient | sustained | sustained | sustained |
| Packaging Capacity | 7.5 kb | 4.5 kb | 8 kb | 7 kb |

