
Glycogen storage diseases: Diagnosis, treatment and outcome
Abstract
The glycogen storage diseases (GSDs) are a group of inherited metabolic disorders that result from a defect in any one of several enzymes required for either glycogen synthesis or glycogen degradation. The GSDs can be divided into those with hepatic involvement, which present as hypoglycemia, and those which are associated with neuromuscular disease and weakness. The severity of the GSDs range from those that are fatal in infancy if untreated to mild disorders with a normal lifespan. The diagnosis, treatment, and prognosis for the common types of GSDs are reviewed.
1Glycogen storage diseases
The glycogen storage diseases (GSDs) are a group of inherited metabolic disorders that result from a defect in any one of several enzymes required for either glycogen synthesis or glycogen degradation. Broadly speaking, the GSDs can be divided into those with hepatic involvement, which present as hypoglycemia, and those which are associated with neuromuscular disease and weakness (Table 1) [1]. The severity of the GSDs range from those that are fatal in infancy if untreated to mild disorders with a normal lifespan. While some forms of GSD affect a single tissue type (for example, skeletal muscle in McArdle disease), others affect multiple systems.
The GSDs have traditionally been diagnosed using a combination of clinical symptoms, biochemical results, and pathology findings. Standard studies performed by the pathologist include muscle or liver histology findings in combination with electron microscopy and enzyme studies. Depending on the specific GSD, enzyme deficiency may be detected in liver, muscle, skin fibroblasts, and, rarely, blood cells. Within the last decade, DNA mutation analysis has become the primary method for diagnosing glycogen storage disease. While such testing was initially performed to complement enzymatic activity studies and clarify ambiguous results, such testing is now becoming the gold standard to confirm a suspected diagnosis. The benefits of such testing are numerous and include the following:
• Molecular analysis obviates the need for invasive biopsy.
• Unlike enzyme studies which may require a significant amount of fresh frozen tissue, DNA-based testing does not usually require extremely careful handling of sensitive specimens.
• DNA mutation analysis can be used for prenatal diagnosis.
• Molecular analysis may help differentiate affected patients with higher levels of residual enzyme activity from heterozygous carriers not predicted to develop disease.
This review will begin with an overview of glucose metabolism followed by a description of the various GSDs. A particular emphasis will be placed on those glycogenoses with hepatic presentation (see Table 2).
1.1Overview of glycogen metabolism and glucose homeostasis
Glucose is the preferred energy source of the brain. Thus, a constant source of blood glucose is essential for human life. Blood glucose can be obtained from three primary sources: diet, degradation of glycogen, and gluconeogenesis. Because dietary intake of glucose (and glucose precursors) is sporadic and gluconeogenesis cannot occur in rapid response to falling blood glucose levels, glycogen serves as a means for storing glucose in a form that can be readily mobilized.
Glycogen is a branched-chain homopolysaccharide synthesized from α-D-glucose molecules [2]. In the absence of dietary glucose, liver glycogen is rapidly broken down to glucose and released into the blood; similarly, skeletal muscle glycogen is degraded and used to generate ATP for muscle contraction. When glycogen stores are depleted, gluconeogenesis can occur in specific tissues, allowing synthesis of glucose de novo using amino acids from protein along with lactate from both the kidney and muscles.
A review of glycogen metabolism can be found in [2]. Glycogen is synthesized in the cytosol of liver parenchymal cells in an ATP-dependent process. Glucose freely enters the liver and is rapidly phosphorylated to glucose-6-phosphate by glucokinase. Glucose-6-phosphate is then converted to glucose-1-phosphate via the enzyme phosphoglucomutase. Glucose-1-phosphate serves as the starting point for glycogen synthesis. In the presence of uridine triphosphate (UTP), UDP-glucose pyrophosphorylase converts glucose-1-phosphate to UDP-glucose. The glucose portion of UDP-glucose can then be added to existing glycogen, or can be added to the protein glycogenin to create a new glycogen molecule. Glycogen synthase catalyzes the formation of α-1,4-linkages necessary for elongating glucose chains. After every eight to ten glucose residues, a second enzyme, amylo-(α-1,4⟶α-1,6)-transglycosylase (also known as glucosyl 4 : 6 transferase), creates a branch point by joining glucose molecules via α-1,6-linkage. With the formation of many long chains and branch points, a tree-like glycogen molecule is created; the numerous branches allow for the addition or removal of multiple glucose molecules at once as needed by the body.
In the early stages of fasting, the liver provides a steady source of glucose from glycogen breakdown. Glycogen phosphorylase is activated via phosphorylation by phosphorylase b kinase. Glycogen phosphorylase cleaves the α-1,4-glycosidic bonds, releasing glucose 1-phosphate. A second enzyme, debrancher enzyme, is required for removal of branch point glucose residues attached via α-1,6-linkage. Glucose-1-phosphate is subsequently converted by phosphoglucomutase to glucose-6-phosphate, and glucose 6-phosphatase catalyzes the last step of glycogenolysis; it hydrolyzes the phosphate group from glucose-6-phosphate to create free glucose that can be released from the liver into the systemic circulation. Of note, glucose-6-phosphatase is not present in the muscles so the muscle only forms of GSD are not associated with hypoglycemia. Normally, only with prolonged fasting is glucose generated in the liver from noncarbohydrate precursors through gluconeogenesis, but this can be an important source of endogenous glucose production in the ketotic forms of GSD.
2Glycogen storage disease type I
Glycogen storage disease type I, also known as von Gierke disease, is an inborn error of metabolism due to deficiency of the glucose-6-phosphatase complex. This multi-component complex, referred to at the G6Pase system, or G6Pase-α, was hypothesized by Arion et al. to consist of four separate proteins, including the G6Pase-α catalytic subunit (G6PC), the glucose-6-phosphate transporter (G6PT), an inorganic phosphate transporter, and a glucose transporter [3]. There are at least two known forms of GSD type I: GSD Types Ia and Ib; these are due to defects in the G6PC and G6PT, respectively. The existence of a third and fourth type, GSD Types Ic and Id, have been largely debated since they do not differ from GSD Type Ib clinically, enzymatically, or genetically [4–6]. Approximately 80% of GSD Type I cases are of the Type Ia variety and result from mutations in the G6PC gene which encodes the glucose-6-phosphatase-α catalytic subunit (G6PC = OMIM 613742). Almost all other remaining cases (GSD Type Ib) are due to mutations in the SLC37A4 gene encoding the glucose-6-phosphate transporter (G6PT = OMIM 602671).
2.1Glycogen storage disease type Ia
GSD Ia (OMIM 232200) was the first inborn error of metabolism proven to be caused by an enzyme deficiency. In 1952, Gerty and Carl Cori demonstrated deficiency of glucose-6-phosphatase activity in liver homogenate from five patients with a clinical diagnosis of von Gierke disease [7, 8]. In two of these cases, which were fatal, there was virtual absence of enzyme activity.
The glucose-6-phosphatase-α catalytic subunit is expressed in the liver, kidneys, and intestinal mucosa. It is the key enzyme in homeostatic regulation of blood glucose levels, and GSD type Ia has the distinction of being the only glycogen storage disease to be both a disorder of glycogenolysis and gluconeogenesis.
2.1.1Clinical presentation
Affected individuals usually present in the first year of life with severe fasting hypoglycemia, hepatomegaly, failure to thrive, growth retardation, and developmental delay. Other common findings related to hypoglycemia include sweating, irritability, muscle weakness, drowsiness, and seizures. Symptoms usually become apparent as infants are weaned from frequent feeds. In addition to severe fasting hypoglycemia, biochemical studies reveal hyperlactatemia, hyperuricemia, and hypertriglyceridemia. Children often experience bruising and epistaxis due to impaired platelet function, and normochromic anemia may be present.
Children with GSD type Ia develop a markedly protuberant abdomen due to massive stores of liver glycogen. The spleen, however, remains normal in size and cirrhosis does not develop. Other physical findings include truncal obesity, doll-like facies, short stature, and hypotrophic muscles. With optimal metabolic control, the hepatomegaly improves and growth normalizes. Complications including hepatic adenomas, osteoporosis, focal segmental glomerulosclerosis, and a small fiber neuropathy used to be common in the 2nd and 3rd decades of life, but the frequency of these complications has markedly decreased with improvements in therapy and good metabolic control [9, 10].
Management of hepatic adenomas when they occur remains a source of debate. Although these tumors are mostly benign, malignant transformation to carcinoma may occur in 5–10% of patients. Most adenomas appear during puberty, and they stabilize following adolescence if metabolic control is optimized. Recently, regression of hepatic adenomas has been reported with improvement in patients whose metabolic control improved [11]. Since hepatocellular carcinoma in GSD Ia arises from adenomas, frequent imaging of adenomas with MRI and ultrasounds is commonly used. If changes are appreciated in adulthood (especially in lesions over 5 cm), surgical resection of the liver lesion or transplantation is usually recommended.
Since glucose-6-phosphatase is also in the kidneys, renal complications can also occur. “Silent” glomerular hyperfiltration almost universally develops in the teen years and may be followed by microalbuminuria and later overt proteinuria. Decreased glomerular filtration rate is due to focal segmental glomerulosclerosis and interstitial fibrosis. Other renal abnormalities include dysfunction of the proximal and/or distal tubules. Dysfunction of the proximal tubules leads to Type II renal tubular acidosis, and distal tubular dysfunction is associated with hypercalciuria. Furthermore, metabolically compensated patients show hypocitraturia that worsens with age [12]. Treatment with ACE inhibitors can slow the progression of kidney damage, and improved metabolic control may slow or even reverse renal disease. Unlike other complications in GSD Ia, kidney stone formation is not primarily related to metabolic control. Hypocitraturia develops in most people with GSD Ia during adolescence, and citrate supplementation has been successful at preventing renal calcification.
Patients with large hepatic adenomas may have severe, iron refractory anemia. This anemia has been observed to resolve spontaneously after adenoma resection or liver transplantation. Based upon these findings, it was determined that large adenomas may express inappropriately high levels of hepcidin mRNA [13]. Hepcidin is a peptide hormone that has been implicated as the key regulator of iron by controlling iron absorption across the enterocyte and macrophage recycling of iron. The increased hepcidin expression in the GSD adenomas is thought to interrupt iron availability and cause iron restricted anemia.
2.1.2Genetics
GSD Type Ia has a disease incidence of approximately 1 in 100,000 births and a carrier rate of approximately 1 in 150. The disorder is found in ethnic groups from all over the world, and the disease is more common in people of Ashkenazi Jewish, Mormon, Mexican, and Chinese heritage [14–16]. The disorder is associated with mutations in the G6PC gene on chromosome 17q21 which encodes the glucose-6-phosphatase-α catalytic subunit. GSD Ia has classic autosomal recessiveinheritance.
G6PC spans 12.6 kilobases and is composed of five exons which encode for a hydrophobic, endoplasmic reticulum (ER)-associated glycoprotein [17]. Despite the gene’s compact size, almost 100 different disease-causing mutations have been reported [18, 19]. Sequencing analysis of all coding exons plus flanking intronic sequence is predicted to detect approximately 94% of cases [20, 21].
2.1.3Pathology
While liver biopsies are no longer required for diagnosing this condition, glycogen filled hepatocytes with prominent steatosis are seen in GSD type Ia. Unlike other forms of GSD, however, fibrosis and cirrhosis do not occur. Hepatocellular carcinoma appears to arise from inflammatory adenomas, and chromosomal alterations have been described in the cancerous lesions with proto-oncogene activation leading to dysregulation of insulin-glucagon-growth hormone signaling [22]. In patients with von Gierke disease, the inability to convert glucose-6-phosphate to glucose results in shunting of G6P to the pentose phosphate shunt and the glycolytic pathway. This, in turn, results in increased synthesis of uric acid, fatty acids and triglycerides. The block in gluconeogenesis also leads to a buildup of lactate and pyruvate which can enter the Krebs cycle and lead to alterations in ATP/ADP and NADH/NAD+ ratios that may favor tumorigenesis [23, 24].
2.1.4Treatment
Dietary treatment has immensely improved prognosis. The aim of treatment is to prevent hypoglycemia and counter-regulation thereby minimizing the secondary metabolic derangements. Therapy may consist of continuous gastric tube feeds or uncooked cornstarch may be used depending on the age of the child and patient/family preferences.
Cornstarch therapy can be introduced as early as six months of age, as soon as the baby’s pancreatic amylase activity has matured. Due to intestinal immaturity, diarrhea can limit cornstarch use, and children less than 2 years of age usually require feeds every 3 to 3-1/2 hours. Cornstarch feeds can be spaced usually to every 4-5 hours in older children and adults. The dose of cornstarch can be estimated by calculating the basal glucose production rate using the following formula: y = 0.0014x3 – 0.214x2+10.411 – 9.084 where y = mg/kg/min of glucose and x = weight in kg. Doses of cornstarch are typically individualized based upon home and clinic based glucose and lactate monitoring in order to maintain glucose concentrations above 75 mg/dL and lactate less than 2.2 mmol/L. Cornstarch is mixed with water or a sugar-free liquid in a starch:water ratio of 1 : 2. Adding glucose is not recommended since it stimulates insulin production and offsets the advantage of the starch. Of note, a new extended release formulation (Glycosade) was recently introduced for night feeds, and it has allowed older children and adults to have a 7–10 hour period of coverage without sacrificing metabolic control [25].
Intake of galactose, sucrose, and fructose is restricted since these sugars will worsen the hepatomegaly and metabolic derangements. The GSD diet is very prohibitive, and it can be difficult for individuals to get all required nutrients without multivitamin supplementation. Other medications are also commonly used to prevent complications. Allopurinol is prescribed when serum urate concentrations are elevated, and fish oil supplementation or a prescription fibrate may be used to lower triglycerides and reduce the risk of pancreatitis. Treatment with an angiotensin-converting enzyme (ACE) inhibitor is used in patients with proteinuria to reduce intraglomerular capillary pressure and provide renoprotection. Preventive calcium and vitamin D3 supplementation is also recommended to prevent osteoporosis.
Most patients with GSD Ia are clinically doing well into adulthood, and complications are becoming uncommon as metabolic control has improved. Many successful pregnancies have occurred [26]. At times, intravenous glucose support may be required. Patients with even minor infections or intercurrent illnesses who are vomiting and/or unable to eat should be taken to the hospital. Surgery should be undertaken with caution due to a bleeding tendency and risk of intraoperative lactic acidosis. Orthotopic liver transplantation has been performed for some individuals with unresectable adenomas or hepatocellular carcinoma. Liver transplantation, however, is deemed a treatment of last resort since renal failure has been a common complication due to the impact of immunosuppression on abnormal kidneys [27].
2.2Glycogen storage disease type Ib
2.2.1Clinical presentation
Early in life, patients with GSD Ib may be clinically and metabolically identical to those with GSD Ia. With aging, however, most patients develop neutropenia and inflammatory bowel disease. The neutropenia is the hallmark feature of GSD Ib, but the age of onset and clinical course are variable. It may be present at birth or not appear until late in childhood as cyclic or permanent neutropenia.
Inflammatory bowel disease resembling Crohn’s disease is a major complication. This nearly universal complication usually appears between 5–12 years of age, but cases as young as 13 months have been reported. Unlike inflammatory bowel disease in the general population, GSD enterocolitis is most commonly located in the small intestine [28]. Diarrhea and abdominal pain may be late manifestations of the co-morbidity, and it often presents as growth failure, severe anemia, or perioral infections. A normal colonoscopy does not rule out the condition, and a capsule endoscopy sometimes is required to establish its presence.
2.2.2Genetics
Type Ib accounts for only 10–15% of type I GSD. While rare in the general population (1 in 1,000,000 individuals), high risk populations include people of Native American, Iranian Jewish, and Italian heritage. The SLC37A4 gene is located on 11q23.3 and the gene has nine exons [29–31]. Mutations in exon 8 are found in 50% of the documented cases, but full gene sequencing may be required to establish the diagnosis since over 90 mutations have been reported [19, 32].
2.2.3Pathology
The histologic appearance of a GSD Ib liver is identical to that of GSD Ia. Establishing the diagnosis of GSD Ib is therefore a challenge since enzymatic testing cannot be relied upon. While almost all glycogenolytic enzymes are found in the cytoplasm, glucose-6-phosphatase is localized to the inner luminal wall of the endoplasmic reticulum. This means that glucose-6-phosphate must cross the membrane of the endoplasmic reticulum in order to act as substrate for glucose-6-phosphatase. This transport protein for glucose-6-phosphate is defective in GSD Ib. Measurement of glucose-6-phosphate translocase activity is difficult to measure, however, and requires fresh unfrozen liver tissue. While liver sample with intact hepatocytes and microsomes will show deficient glucose-6-phosphatase activity because the translocase cannot deliver the G6P substrate to the ER lumen, microsomes disrupted by solubilization or damage from freezing will show normal glucose-6-phosphatase enzyme activity because the substrate is now readily accessible. Due to the difficulty of the biochemical assay, most clinical diagnostic laboratories do not offer such testing and diagnosis by molecular genetic testing is recommended [21].
2.2.4Treatment
Treatment guidelines for patients with GSD Ib are similar to those for GSD Ia with the addition of therapy for the neutropenia and GSD enterocolitis. Recombinant human granulocyte-colony-stimulating factor (GCSF), a cytokine that induces proliferation and differentiation of bone marrow precursor cells into mature neutrophils, should be used to treat neutropenia if infections, severe mouth ulcers, or chronic diarrhea are occurring. The GSD Ib population has been prone to untoward effects (massive splenomegaly, splenic sequestration, splenic rupture, and portal hypertension) with GCSF therapy. Therefore, a starting dose of 2.5 micrograms/kg/day is recommended, and the lowest dose that controls infections should be used [33]. Supplementation with high dose vitamin E appears to boost the neutrophil count and improve function in GSD Ib, and supplementation may allow lower GCSF doses to be used [34]. Non-absorbable salicylates (Pentasa, Asacol, and Lialda) are the first line therapies for GSD enterocolitis. Steroids and immunomodulators must be used with caution due to the metabolic consequences and associated immune dysfunction [34].
3Glycogen storage disease type II – Pompe disease
Glycogen storage disease type II (acid maltase deficiency, or Pompe disease) (OMIM 232300) is caused by a deficiency of α-1,4 glucosidase, an enzyme required for the degradation of lysosomal glycogen [35]. The disorder was initially described by Johannes Pompe in 1932 [36]. It is the only form of GSD to be classified as a lysosomal storage disorder. Pompe disease is purely a neuromuscular form of GSD which does not present with metabolic abnormalities because the lysosomal enzyme defect lies outside of intermediary metabolism. Instead, storage of glycogen occurs mainly in skeletal muscle and leads to loss of muscle function.
3.1Clinical presentation
Pompe disease has a broad clinical spectrum with variable age of onset, severity of symptoms, and rate of disease progression. The disorder encompasses a continuum of phenotypes ranging from a rapidly progressive infantile form to a slowly progressive late-onset form. In general, however, Pompe disease is classified into three different subtypes, including infantile, juvenile, and adult forms. There is clinical correlation with the amount of α-1,4-glucosidase expression: residual enzyme activity is found in the adult form, while enzyme activity is completely absent in the severe infantile form. It is important to note that mental development and blood glucose concentrations are normal in all forms of Pompe disease.
The classic infantile form is the most severe. Affected infants present shortly after birth with profound hypotonia, muscle weakness, and hyporeflexia. An enlarged tongue and hypertrophic cardiomyopathy are characteristic. Diagnosis may be based on typical EKG findings which include large QRS complexes and shortened PR intervals [37]. The liver is normal in size. Sensorineural hearing loss is also prevalent and a less recognized feature [38, 39]. Without therapy, the disease is rapidly fatal with children usually dying of cardiopulmonary failure or aspiration pneumonia by two years of age.
In the juvenile form of the disease, affected children have hypotonia and weakness of limb girdle and truncal muscles. Motor milestones are delayed, and the myopathy is more gradual in nature. There is no overt cardiac disease, and the patient usually dies from respiratory failure before adulthood without therapy.
The vast majority of patients with Pompe disease are adults. Adult-onset Pompe disease has a long latency and affected individuals may live to old age. Decreased muscle strength and weakness develop in the third or fourth decade, but cardiac involvement, if any, is minimal. Glycogen accumulates in vascular smooth muscle cells and there are rare reports of death from ruptured aneurysms [40, 41]. Slow, progressive weakness of the pelvic girdle, paraspinal muscles, and diaphragm leads to loss of mobility and respiratory function. Most patients eventually become confined to wheelchairs and/or ventilator-dependent. Respiratory muscle weakness is the leading cause of death.
3.2Genetics
The incidence of Pompe disease is estimated to be approximately 1 in 40,000 to 1 in 50,000. The disorder can be found in ethnically diverse populations, including European Caucasians, Hispanics, and Asians, and several mutations are more common in some populations due to founder effects. For more information, the reader is referred to the Pompe Disease Mutation Database at www.pompecenter.nl/ [42]. α-1,4-glucosidase is encoded by the GAA gene located on the long arm of chromosome 17 at 17q25.3 [43–45]. The gene is composed of 20 exons and over 350 different mutations have been reported [19]. Of note, while most mutations will be picked up by gene sequencing, at least 11 different gross deletions and one gross insertion have been reported which would not be detectable using this method [19]. Prenatal diagnosis is possible via enzyme assay or DNA analysis of chorionic villi obtained between 10–12 weeks gestation.
There appears to be genotype-phenotype correlation, with specific mutations associated with infantile, juvenile, and adult-onset disease [46–48]. Severe mutations which lead to complete loss of enzyme activity are associated with severe, infantile Pompe disease, while mutations which allow partial enzyme expression are associated with adult onset disease. One very common mutation in intron 1 of the GAA gene, defined as c. -32-13 T>G, has been found in almost two-thirds of patients with adult onset disease [49]. This particular mutation affects pre-mRNA splicing but allows for correct splicing approximately 10% of the time. Affected individuals who are compound heterozygotes for this particular mutation plus a null allele show approximately 5% of normal enzyme activity [50].
3.3Pathology
The site of glycogen accumulation is different for all three forms of Pompe disease. Furthermore, the amount varies greatly in different organs and even in different muscles [51]. Histological examination of muscle will reveal large glycogen-filled vacuoles as well as freely dispersed glycogen outside the lysosomes. As lysosomes accumulate with glycogen, cell function becomes impaired.
Mutation analysis is now the preferred method of diagnosis. Enzymatic studies can be performed, however, on muscle tissue or fibroblasts. It is imperative that α-1,4-glucosidase, also known as acid maltase due to its optimum pH lying between 4.0 and 4.5, be differentiated from neutral maltase found in the cytoplasm.
Acid maltase is initially an inactive enzyme that is transported to the prelysosomal and lysosomal compartment via the mannose-6-phosphate receptor [52–54]. The enzyme is eventually processed into a fully active form that normally degrades glycogen that enters lysosomes via autophagy. Deficiency of enzyme causes glycogen to overload the lysosomal system and leads to progressive and irreversible cellular damage.
3.4Treatment
Before the advent of enzyme replacement therapy, treatment was generally supportive in nature and respiratory insufficiency was treated with assisted ventilation. For patients with juvenile Pompe disease, dysarthria and dysphagia caused by severe weakness of the facial muscles might necessitate feeding by G-tube. A high-protein diet, particularly a high-protein diet fortified with branched-chain amino acids, is recommended to help diminish catabolism of muscle protein.
In 2006, enzyme replacement therapy (ERT) became a commercially available option [55]. Myozyme® (alglucosidase alfa) is indicated for use in patients with infantile-onset Pompe disease and has been shown to improve ventilator-free survival. In contrast, for patients who are eight years and older and do not have an enlarged heart, Lumizyme® (alglucosidase alfa) is available and may help to preserve respiratory function and walking ability. Nevertheless, ERT is very expensive, and approximately 1% of patients undergoing ERT experienced severe anaphylactic shock and/or cardiac arrest associated with infusion treatment [56–58]. ERT has proven to be less effective in the infantile Pompe patients than in the other populations. Since most people with the infantile form have no enzyme activity, the enzyme is recognized as foreign by the body, and a robust immune response develops against the ERT. Immunosuppression may help blunt this response and increase efficacy. Gene therapy using AAV-8 injected into the diaphragm is also being attempted in humans with the disease [59].
3.5Glycogen storage disease type IIb – Danon disease
Glycogen storage disease type IIb (Danon Disease) (OMIM 300257) is a multisystem disorder characterized by hypertrophic cardiomyopathy, heart arrhythmias, skeletal myopathy, retinal abnormalities, and variable degree of mental retardation [60–63]. Disease onset typically occurs in adolescence, with rapid progression toward end-stage heart failure in early adulthood [62]. Although the disease was initially classified as a glycogen storage disorder, glycogen is not always elevated in patients [64].
3.5.1Pathology
The biochemical hallmark of the disease is the accumulation of pathologic vacuoles containing glycogen or intermediary metabolites, mainly in skeletal and myocardial muscle with no evidence of enzyme deficiency.
3.5.2Genetics
Danon disease is quite rare and good estimates of the incidence are not available. The disorder is X-linked dominant in nature and is due to LAMP-2 (lysosome-associated membrane protein-2) deficiency. Although biochemical analysis is possible in male patients, diagnosis in females requires DNA mutation analysis [65]. Over fifty different mutations in the LAMP-2 gene have been identified [19, 66].
4Glycogenoses types III and IV – disorders of abnormally structured glycogen
Glycogenoses types III and IV are clinically heterogeneous disorders caused by buildup of abnormally structured glycogen in the liver and muscle.
4.1GSD type III – Cori disease
Glycogen storage disease type III (Cori disease or Forbes disease) (OMIM 232400) was initially discovered in 1952 when a patient being followed by Dr. Gilbert Forbes was found to have excessive amounts of abnormally structured glycogen in liver and muscle tissue [67, 68]. Type III GSD varies widely in clinical presentation and can be divided into two types: type IIIa, with both hepatic and muscle involvement, and type IIIb, which primarily presents with liver disease [69]. Both GSD IIIa and GSD IIIb result from an enzyme deficiency in the glycogen debranching enzyme (GDE). This enzyme is encoded by the AGL gene located on chromosome 1p21.
4.1.1Clinical presentation
GSD type III is a phenotypically heterogeneous disorder with a wide clinical spectrum. While patients with GSD type IIIb mainly present with hepatic findings, affected individuals with type IIIa have both liver and muscle involvement. For both IIIa and IIIb, liver disease predominates in infancy and early childhood including hepatomegaly, hypoglycemia, hyperlipidemia, and growth retardation. For those with type IIIa GSD, there is additional variable skeletal and/or cardiac myopathy which often, though not always, may be detected via elevated serum CK concentrations once children are ambulating. Mild hypotonia and delayed motor development are usually the only manifestation during early childhood. By late childhood and adolescence, decreased stamina and pain with exertion can be noted. Muscle wasting is slowly progressive in adulthood and may be severe by the 3rd or 4th decade of life [70]. Although ventricular hypertrophy is a frequent finding, symptomatic cardiomyopathy leading to death is relatively rare. Unlike muscle disease which is a progressive process, the hypertrophic cardiomyopathy is reversible and appears to be due to excessive storage of glycogen. With a diet restricting intake of simple sugars, the hypertrophic cardiomyopathy can resolve and cardiac function normalize [71, 72].
Childhood hepatic symptoms tend to become milder with age. Complications aside from the myopathy are rare. A minority of patients (4–10%) develop hepatic adenomas [73]. Cirrhosis can also develop in patients with GSD III, and rare cases of hepatocellular carcinoma have been reported [73, 74]. Unlike in GSD Ia, hepatocellular carcinoma can develop anywhere in the liver, and it is not the result of malignant transformation of a hepatic adenoma [23]. Although all individuals with GSD type III show liver involvement, in rare instances the hepatic symptoms are mild and the diagnosis is not made until adulthood when individuals show signs of neuromuscular disease. Other clinical findings include abnormal nerve conduction studies and osteoporosis. Successful pregnancies have been reported.
4.1.2Genetics
GSD Types IIIa and IIIb are autosomal recessive allelic disorders caused by mutations in the AGL gene on the short arm of chromosome 1 [75]. The incidence of GSD III is estimated to be 1 in 100,000 live births, but high risk populations have been identified. Unusually high rates of GSD IIIa occur in the First Nation of Canada and on the Faroe Islands where there are carrier frequencies of 1 : 18 and 1 : 22, respectively [76, 77]. GSD IIIa is also more common on the Indian subcontinent (India, Pakistan, Afghanistan). Type IIIb accounts for 15% of all GSD III, and it is unusually frequent in Jews of Northern Africa (carrier frequency of 1 : 35) [66, 78]. To date, at least 140 different pathogenic AGL mutations have been reported [19].
4.1.3Pathology
The encoded enzyme, glycogen debranching enzyme (GDE), together with glycogen phosphorylase, is responsible for the complete degradation of glycogen. GDE has a presumed glycogen binding site at the carboxy terminal end, as well as two separate sites responsible for independent catalytic activities. These activities include 4-α-glucanotransferase activity (1,4-α-D-glucan:1,4-α-D-glucan 4-α-D glycosyltransferase activity) responsible for the transfer of three glucose units to the outer end of an adjacent chain, and an amylo-1,6-glucosidase activity responsible for hydrolysis of branch point glucose residues.
The variable phenotype seen in GSD type III is partly explained by differences in tissue-specific expression. When the enzyme is deficient in both liver and muscle, GSD type IIIa results; in contrast, when AGL is deficient only in the liver and enzyme activity is retained in muscle, then GSD type IIIb results. Rare cases have also been reported where only one of two GDE catalytic activities is lost [79–81]. When there is loss of only glucosidase activity, the patient is classified as having GSD Type IIIc, and when there is only loss of transferase activity, the patient is classified as having GSD type IIId.
4.1.4Treatment
While glycogenolysis is impaired in GSD III, gluconeogenesis is intact allowing lactate, amino acids, and glycerol (from fatty acid oxidation) to be used to maintain blood glucose concentrations. Protein is used as the primary source of energy in GSD type III since it also can be used directly by the muscles and has been associated with improvement in the myopathy. A diet with 3-4 grams/kg of protein is recommended through dietary protein and supplements [69]. This is supplemented with cornstarch therapy which is dosed to maintain blood glucose concentrations above 70 mg/dL and beta-OH-butyrate concentrations less than 0.3 mmol/L. The frequency of cornstarch doses varies with age. In infancy, frequent cornstarch administration may be required with therapy similar to that used in GSD type I. With older children and adults, cornstarch frequently is only required 2-3 times per day, and sometimes it is only administered prior to bedtime. For patients with moderate to severe hypertrophic cardiomyopathy, a high-protein nocturnal enteral therapy may be beneficial. Intake of simple sugars is limited to 5 grams per meal to minimize postprandial hyperinsulinemia and avoid over-storage of glycogen.
4.2GSD type IV (Andersen disease) and adult polyglucosan body disease
Glycogen storage disease type IV (Andersen disease) (OMIM 232500) and Adult Polyglucosan Body Disease (APBD) (OMIM 263570) are allelic disorders caused by a deficiency of the glycogen branching enzyme encoded by the GBE1 gene. While GSD type IV is a clinically heterogeneous disorder that severely affects liver and/or muscle, APBD is a late-onset slowly progressive disorder affecting the central and peripheral nervous systems. GSD type IV is quite rare, representing 0.3% of all glycogenoses.
4.2.1Clinical presentation
GSD type IV shows significant variability in terms of age of onset and extent of organ and tissue involvement [82–85]. In its common classic form, patients have failure to thrive and hepatosplenomegaly. Portal hypertension and ascites develop, and progressive cirrhosis often occurs in early childhood. Without a liver transplant, death usually occurs by five years of age. Unlike the other liver forms of GSD, hypoglycemia is a late manifestation of GSD IV.
Neuromuscular forms of GSD type IV are quite variable and may be classified into several different phenotypes; interestingly, they represent the most severe and the most mild forms of GSD type IV. The most severe and relatively rare form of GSD type IV presents perinatally as fetal akinesia deformation sequence with arthrogryposis, hydrops, polyhydramnios, and pulmonary hypoplasia. In this form of the disease, death occurs at an early age due to cardiac or pulmonary insufficiency. Other severe forms of neuromuscular GSD type IV present congenitally or in early infancy with hypotonia and skeletal muscle atrophy. Prognosis varies for these forms of the disease, usually depending on the extent of cardiac and hepatic involvement. Finally, in its milder forms, GSD type IV may present in late childhood, adolescence, or even adulthood as myopathy or adult polyglucosan body disease (APBD) with central and peripheral nervous system dysfunction [85]. APBD is an allelic variant of GSD Type IV characterized by adult-onset progressive neurogenic bladder, gait difficulties due to spasticity and weakness, distal lower extremity sensory loss, and mild cognitive difficulties (OMIM 263570) [86].
4.2.2Genetics
GSD type IV is the result of a deficiency of glycogen branching enzyme which is encoded by the GBE1 gene located on chromosome 3p12.3. This gene is the only gene known to be associated with GSD type IV. Deficiency or absence of the encoded enzyme leads to excessive deposition of abnormally-structured, amylopectin-like glycogen in affected tissues. Because the accumulated glycogen lacks multiple branch points, it has poor solubility and causes irreversible tissue and organ damage.
Residual enzyme activity may confound the results of enzyme analysis; therefore, mutation analysis is often recommended to confirm the diagnosis. Thus far, at least thirty-nine different mutations have been reported across the entire length of the gene, including nonsense, missense, splice site changes, micro insertions and deletions, and several gross deletions spanning multiple exons [19]. At least ten different mutations have been reported to involve exon 12, and therefore this exon is considered a mutation “hotspot” [85, 87].
At present, there does not appear to be a strong genotype-phenotype correlation, and patients with the same mutation may show a wide range of clinical severity. In general, patients with two missense mutations have a milder form of disease than individuals with two null mutations. There is one GBE1 exon 7 missense variant that is predicted to result in the amino acid substitution p.Tyr329Ser. This founder mutation, which decreases branching enzyme activity to approximately 50% of normal, has been documented to cause Adult Polyglucosan Body Disease and is commonly found in the homozygous state in individuals with Ashkenazi Jewish ancestry [88, 89]. It should be noted that there is at least one case report of an individual with APBD who was a manifesting heterozygote with 48% branching enzyme activity [90].
4.2.3Pathology
While the hepatic scarring is the most severe of the glycogenoses, hepatic transaminase elevation is variable. Hepatic dysfunction occurs as the disease progresses. Creatine kinase levels range from normal to very elevated, and electromyography may show diffuse fibrillations.
GSD type IV is characterized by amylopectinosis. Histologic examination of liver tissue reveals periodic acid-Schiff (PAS)-positive, diastase-resistant intracytoplasmic inclusions consistent with abnormal glycogen. Characteristic findings in hematoxylin and eosin stained liver tissue include distorted hepatic architecture with diffuse interstitial fibrosis and wide fibrous septa surrounding micronodular areas of parenchyma. Hepatocytes are generally two to three times normal size with basophilic cytoplasmic inclusions. Electron microscopy of affected tissue reveals normal glycogen particles plus abnormal fibrillary aggregates typical of amylopectin (polyglucosan bodies). Muscle fibers from affected patients demonstrate severe depletion of myofibrils, and there may be amyloplasia with total fatty replacement of skeletal muscle. Polyglucosan bodies are invariably seen which are resistant to diastase digestion. In contrast to classical GSD type IV, the pathologic hallmark of adult polyglucosan body disease is the widespread accumulation of round, intracellular polyglucosan bodies throughout the nervous system, which are confined to neuronal and astrocytic processes [89].
4.2.4Treatment
Treatment of GSD IV is typically supportive. A high protein diet may have some benefit, but it has not prevented progression of the liver disease. Cornstarch is beneficial if hypoglycemia is occurring, but it similarly does not change the natural history of the disease. Due to the poor prognosis, liver transplantation remains the primary treatment for the child with early-onset, classic hepatic presentation. Individuals with adult-onset APBD may require antispasmodic bladder medications or bladder catheterization. Gait assist devices may also help to minimize the risk of falls [86].
5Glycogen storage diseases types V, VI and IX due to deficiency of glycogen phosphorylase or its activating enzyme phosphorylase b kinase (Phk)
Glycogen storage diseases types V (McArdle Disease) and VI (Hers Disease) are the result of a deficiency of glycogen phosphorylase, while glycogen storage disease Type IX is due to deficiency of phosphorylase b kinase, the activating enzyme of glycogen phosphorylase. Glycogen phosphorylase enzyme catalyzes the rate-limiting step in glycogenolysis and shows tissue-specific expression, with different forms of the enzyme being expressed in liver and muscle.
5.1Glycogen storage disease type V – McArdle disease
Glycogen storage disease type V (OMIM 232600) is a pure myopathic form of GSD affecting skeletal muscle. This disease was the first metabolic myopathy to be recognized and was described by Dr. Brian McArdle in 1951 after studying a young man with exercise intolerance and muscle cramps [91]. McArdle concluded that his patient’s disorder was the result of a failure to break down glycogen into lactic acid.
5.1.1Clinical presentation
The clinical severity of McArdle disease is highly variable. Virtually all people with GSD V describe lifelong exercise intolerance, but the diagnosis is not usually made until the second to third decade of life when cramping becomes more prominent. Patients present with exercise-induced fatigue, painful muscle cramps, myalgia, and myoglobinuria. Approximately 90% of patients have elevated serum creatine kinase levels, even at rest. Diagnosis can be made by demonstration of failure of venous lactate to rise with an ischemic forearm test or following exercise. Electromyography does not demonstrate specific abnormalities.
In 2003, Vissing and Haller published a diagnostic test for McArdle disease based on moderate cycle exercise [92]. The authors noted that in contrast to patients with other metabolic myopathies, McArdle disease patients show decreased heart rate 7 to 15 minutes into moderate, constant-workload aerobic activity. This “second wind” phenomenon is a defining feature of McArdle disease and is thought to be due to the body’s ability to utilize alternative energy sources, including fatty acids, hepatic glycogen, and protein.
5.1.2Genetics
McArdle disease results from a deficiency of muscle-expressed glycogen phosphorylase, or myophosphorylase [93, 94]. Myophosphorylase is encoded by the PYGM gene on the long arm of chromosome 11 [95, 96]. The gene is comprised of 20 exons, and over 140 mutations have been described [19, 97–99]. Two mutations, Arg50Stop (R50X) in exon 1 (which also has been commonly reported in the research literature as R49X) and Gly205Ser in exon 5 are common mutations in patients with European heritage [91, 100]. Although no strict genotype-phenotype correlations have been made, there have been reports of more severe phenotypes in patients homozygous for both R50X mutations in PYGM and Q12X mutations in the AMPD1 gene encoding muscle adenylate deaminase [101, 102]. Cases of muscle symptoms in heterozygous carriers have been reported [103].
5.1.3Pathology
Enzyme studies on muscle biopsy will reveal absence of myophosphorylase in muscle fibers. Microscopy may also reveal acid-Schiff stained glycogen.
5.1.4Treatment
Because the metabolic block in McArdle disease impairs glycogen breakdown but glucose utilization remains intact, patients with GSD type V benefit from glucose or sucrose loading before exercise [104, 105]. Intense exercise should be avoided as it can lead to rhabdomyolysis with concomitant myoglobinuria and renal failure. Statin usage is contraindicated, and it should be noted that even heterozygous carriers may show adverse side effects to these medications [106]. Oral vitamin B6 has been reported to impart greater resistance to fatigue, and a high protein diet may also help [107].
5.2GSD type VI – Hers disease
Glycogen storage disease type VI (Hers disease) (OMIM 232700) was reported by Henry-Gery Hers in 1959 [108]. This disorder is the result of a deficiency of liver glycogen phosphorylase, which is encoded by the PYGL gene located on chromosome 14q22 [109].
5.2.1Clinical presentation
Patients present in infancy or early childhood with varying degrees of growth retardation and prominent hepatomegaly secondary to excessive liver glycogen. Ketotic hypoglycemia or just hyperketosis occur with prolonged fasting or strenuous exercise [110]. Because gluconeogenesis is preserved, hypoglycemia tends to be mild. Hypotonia may lead to delayed motor development even though there is no intrinsic muscle involvement. Mild hyperlipidemia is common, and liver function tests may reveal elevated serum transaminases. Unlike other types of GSD, lactic acid and uric acid concentrations are normal. While patients with GSD VI have a milder course with few complications, treatment improves growth, stamina, and normalizes the biochemical abnormalities. Rarely, liver fibrosis develops in GSD VI, and a cardiomyopathy can occur from over storage of carbohydrate [111]. Most adults are asymptomatic, but adult females may experience hypoglycemia during pregnancy or with alcohol consumption.
5.2.2Genetics
Liver glycogen phosphorylase catalyzes the phosphorylysis of an α-1,4-glycosidic bond in glycogen to yield glucose-1-phosphate that helps meet the body’s energy needs. The enzyme exists as a homodimer of the PYGL protein and requires pyridoxal phosphate (PLP) as a cofactor. The enzyme switches between an active conformation (GPa) and an inactive conformation (GPb), with activation dependent upon phosphorylation of a serine located at amino acid position 14. Such phosphorylation occurs in response to the hormones glucagon and epinephrine.
GSD type VI is inherited in an autosomal recessive fashion. The PYGL gene on chromosome 14 spans over 39,000 base pairs, consists of 20 coding exons, and encodes a protein that is 846 amino acids in length [112]. Thus far, thirty disease-causing mutations have been reported [19, 113–115]. The vast majority of pathogenic variants are missense mutations [116]. No affected individuals have been described with two null alleles, suggesting that complete absence of liver glycogen phosphorylase activity may be incompatible with life. The estimated disease incidence ranges from 1 in 65,000 to 1 in 85,000 births, but many people with this condition are undiagnosed. In the Mennonite community, however, there is a founder mutation (c.1620+1G>A, at the intron 13 splice donor site), and the prevalence is approximately 0.1% (1 in 1,000 individuals) [112].
5.2.3Pathology
Although hepatic glycogen phosphorylase enzyme is expressed in several cell types and its activity can be assayed using erythrocytes, leukocytes, or hepatocytes, such testing is neither highly sensitive nor specific. False negative results are common because enzyme activity is significantly reduced in Hers disease but is never completely absent. False positive results also are not rare, because reduced liver phosphorylase activity may be due to mutations in the PYGL gene or mutations in several other genes (including PHKA2, PHKB, and PHKG2) that encode phosphorylase b kinase, the activating enzyme for hepatic glycogen phosphorylase. As a result, mutation detection is now the preferred method to differentiate liver phosphorylase deficiency from the much more common deficiency of the phsophorylase b kinase activating enzyme. Liver biopsies demonstrate glycogen filled hepatocytes with or without fibrosis, but DNA analysis or enzymatic testing is needed to differentiate GSD VI from the other forms of GSD.
5.2.4Treatment
Affected individuals should avoid prolonged fasting, and eat frequent small meals. Uncooked cornstarch (1–4 times per day) and protein supplementation may help stabilize blood glucose levels and prevent complications such as short stature, delayed puberty, and osteoporosis. Protein supplementation typically is lower than in GSD III (2–2.5 g/kg/day) with a goal of normalizing prealbumin concentrations. Rarely, cirrhosis and hepatocellular carcinoma can occur in Hers disease [111, 117]. Patients should avoid excessive amounts of simple sugars. In addition, growth hormone therapy should not be used to treat short stature since it will lead to increased ketone production. To assess metabolic control, blood glucose levels and blood ketones should be routinely monitored. Height and weight measurements should also be assessed regularly since growth is normal when treatment is optimized.
5.3Glycogen storage disease type IX
Glycogen storage disease type IX is due to a deficiency of the enzyme phosphorylase b kinase, and it constitutes approximately 25% of all GSD cases. Because phosphorylase b kinase is required to activate the enzyme glycogen phosphorylase, GSD Types VI and IX show significant clinical overlap. Nevertheless, these two glycogenoses are very different disorders from a genetic standpoint, and this may have important implications for accurate genetic counseling and recurrence risk.
5.3.1Clinical presentation
GSD type IX has the most heterogeneous clinical picture of all of the glycogen storage diseases. Most patients are diagnosed after hepatomegaly is incidentally found, and it is the most common identifiable cause of ketotic hypoglycemia in males [118]. While most patients are relatively mild, a severe variant exists that mimics type I GSD in infancy with severe fasting hypoglycemia. Patients with GSD type IX may present with hepatic and/or muscle disease, and many patients suffer from a combination of the two. There is at least one form of GSD type IX which is strictly muscle-specific, and affected patients may present with muscle pain and weakness, exercise intolerance, and myoglobinuria. Another form of GSD type IX strictly presents as hepatic disease that typically begins in the first few months of life, and affected individuals may have ketotic hypoglycemia, hepatomegaly due to elevated glycogen content, liver disease, growth retardation, hypotonia, abnormal lipid profile, and increased lactate and uric acid. In its mildest hepatic form, patients may have a phenotype similar to GSD Type VI and symptoms may gradually subside with age. In patients with hepatic transaminase elevation, liver complications can develop including fibrosis, cirrhosis, adenomas, and hepatocellular carcinoma [111].
5.3.2Genetics
Glycogen storage disease type IX is a genetically heterogeneous disorder. The phosphorylase kinase (Phk) enzyme is a hexadecameric structure comprised of four copies each of four different polypeptides, including alpha (α), beta (β), gamma (γ), and delta (δ) subunits [119]. To add to the molecular complexity, various tissue-specific isoforms exist for each subunit; these isoforms may be due to expression from separate genes or from alternative splicing of a single gene. α-associated GSD Type IX may result from mutations in one of two X-linked genes: PHKA1 or PHKA2. PHKA1 is located on the long arm of chromosome X at Xq13 while PHKA2 is located on the short arm of the X chromosome at Xp22.13 [120]. PHKA1 expression is confined to muscle, and therefore PHKA1 mutations are associated with exercise intolerance, muscle pain, weakness, and myoglobinuria [121–123]. To date, there are only seven reported mutations in the PHKA1 gene [19]. In contrast, PHKA2 gene expression is confined to liver and blood cells, and patients with PHKA2 mutations strictly have a hepatic presentation with ketotic hypoglycemia, hepatomegaly, chronic liver disease, retarded growth and motor development, and elevated lipids [124–128].
In contrast to the α-subunit, there is only one gene known to encode the β-subunit of the Phk enzyme. This gene, PHKB, is located on the long arm of chromosome 16 at 16q12.1 [120]. Alternative splicing of several exons gives rise to tissue-specific transcripts, and PHKB mutations have been associated with phosphorylase kinase enzyme deficiency in both liver and muscle [129–133]. Most mutations identified in PHKB have been severe null mutations expected to lead to premature protein truncation or mRNA decay; nevertheless, patients generally have mild symptoms including hypoglycemia after prolonged fasting, hepatomegaly, and mild hypotonia [19]. Cirrhosis and other major complications have not been reported in patients with PHKB mutations to date. It is important to note that PHKB mutations have not been found in patients with only muscle disease [134].
The PHKG2 gene on chromosome 16 encodes the liver- and testis-specific form of the γ-subunit [135]. This subunit contains the phosphorylase kinase enzyme’s active site responsible for the phosphorylation and activation of glycogen phosphorylase [136]. PHKG2-linked disease is associated with a more severe phenotype, which may include fasting hypoglycemia, impaired glucagon response, muscle weakness and fatigue, hepatomegaly, liver fibrosis and cirrhosis [133, 137–139].
5.3.3Pathology
Since hepatomegaly is often the presenting symptom, the diagnosis of GSD IX is still commonly made by liver biopsy. As with the other forms of GSD, glycogen filled hepatocytes with prominent steatosis is seen, but fibrosis is usually present in GSD IX. Clinical diagnostic laboratories in the United States require samples from affected tissues (i.e., liver and/or muscle) for enzyme studies. However, non-invasive analysis of phosphorylase kinase enzyme has been reported using blood cells [133]. Although biochemical testing may be used to provide a GSD type IX diagnosis, enzyme analysis cannot determine which gene is causing disease. Mutation analysis is therefore recommended in individuals suspected to have GSD type IX. Mutations in PHKA2 cause GSD IX in over 80% of males with the disease. Sequencing of PHKB should be considered first in females.
5.3.4Treatment
Although GSD type IX was once considered a benign condition, it is now clear that patients may experience more long-term complications. Patients who have elevated hepatic transaminases and post-prandial hyperlactatemia are particularly at risk for development of cirrhosis, and aggressive management is imperative [140]. In all patients with GSD IX, treatment improves growth, stamina, and normalizes biochemical tests.
Affected individuals should restrict intake of simple sugars and eat frequent small meals. A high protein diet is used (2-3 g/kg), and uncooked cornstarch supplementation is used to maintain glucose concentrations and avoid ketosis. While most patients with GSD type IX can make it through the night with cornstarch and protein, overnight feeds are sometimes needed in patients with mutations in PHKA2 and PHKG2. For these patients, the extended release cornstarch preparation (Glycosade) can be considered [141]. Growth hormone therapy should not be used to treat short stature since it will lead to increased ketone production. Liver ultrasound examinations are recommended beginning in childhood since patients are at risk for developing hepatic adenomata and cirrhosis.
6Glycogen storage disease type VII – Tarui disease
Glycogen storage disease type VII, otherwise known as Tarui disease, was first described in three Japanese adult siblings in 1965 [142]. The disorder is the result of a deficiency of muscle-specific phosphofructokinase. This disease is one of the rarest forms of GSD, and symptoms are usually similar to those seen in GSD type V (McArdle disease).
6.1Clinical presentation
There are at least three different subtypes of GSD type VII, including classic, infantile onset, and late onset [143, 144]. In the infantile form, babies have myopathy, joint contractures, seizures, psychomotor retardation, and blindness due to cataracts; death occurs during childhood. In contrast, late onset disease may manifest in adulthood with progressive muscle weakness.
In classic disease, which typically presents in childhood, exercise intolerance is the key feature. CPK levels are elevated and affected children may experience undue fatigue, muscle pain, cramps, and nausea. Intense exercise may lead to myoglobinuria and acute renal failure. Because a defect in muscle phosphofructokinase (known as PFK-M) results in a partial defect in PFK activity in erythrocytes, patients may present with hemolytic anemia. The anemia is usually compensated because the metabolic block causes a decrease in 2,3 diphosphoglycerate and enhanced oxygen affinity of hemoglobin which leads to an increase in erythrocyte formation. The enzyme deficiency also results in elevated levels of glucose-6-phosphate. This may result in enhanced nucleotide formation and increased levels of uric acid. Increased reticulocytes, hyperbilirubinemia, jaundice, gallstones, and gout may help provide diagnostic clues.
6.2Genetics
Phosphofructokinase is a glycolytic enzyme that catalyzes the irreversible conversion of fructose-6-phosphate to fructose-1,6-bisphosphate. Because glycolysis follows glycogenolysis in muscle, muscle tissue in patients with Tarui disease cannot utilize glycogen-derived glucose.
Human phosphofructokinase functions as a homotetrameric or heterotetrameric enzyme. The various subunits are tissue-specific and include PFK-M (muscle), PFK-L (liver and kidneys), and PFK-P (platelets); these are respectively encoded by three different genes, PFKM, PFKL, and PFKP. Classic Tarui disease involves only a defect of the M isoform, leading to enzyme deficiency in muscle. Because erythrocytes express both M and L isoforms and are still capable of producing tetramers of L isozyme, the phosphofructokinase activity in erythrocytes is approximately 50% of normal and patients may have hemolysis.
GSD type VII is an autosomal recessive genetic disorder. The gene which encodes muscle phosphofructokinase, PFKM, consists of 22 exons and lies on the long arm of chromosome 12 [145, 146]. Two founder mutations account for approximately 95% of known mutations in patients of Ashkenazi Jewish descent. These include a G>A transition which disrupts the consensus GT splice donor site at the exon 5/intron 5 border and results in a deletion of exon 5, as well as a frameshift mutation caused by deletion of a single nucleotide in exon 22 [147, 148].
6.3Pathology
Muscle biopsy reveals glycogen accumulation in the subsarcolemmal space plus variation in myofibril size [107]. In addition, there may be pockets of abnormal polysaccharide consistent with polyglucosan that is PAS-positive but only partially digested by diastase [104]. Electron microscopy may reveal finely granular and fibrillar material similar to the amylopectin-like storage material found in GSD Type IV. It has been hypothesized that the metabolic block leads to high glucose-6-phophate (G6P), and that elevated G6P abnormally activates glycogen synthase and alters the ratio of glycogen synthase to branching enzyme [104]. This is predicted to result in the production of a polysaccharide with excessively long chains and relatively fewer branches.
6.4Treatment
In contrast to patients with McArdle disease, individuals with Tarui disease do not benefit from carbohydrate-rich meals [104]. In fact, consuming carbohydrates exacerbates exercise intolerance because glucose decreases the blood concentration of alternative fuels such as free fatty acids and ketones by increasing insulin concentrations. Strenuous exercise is contraindicated. Nutritional therapy, including a high protein diet and vitamin B6 supplementation, may help rebuild damaged muscle [107].
7GSD type XI – Fanconi-Bickel syndrome
Glycogen storage disease type XI (Fanconi-Bickel syndrome) (OMIM 227810) results from defects in a transport protein, the GLUT2 glucose transporter [149–153]. This syndrome was first described in 1949 [154].
7.1Clinical presentation
Abnormal glucose absorption in the intestines is associated with diarrhea and failure to thrive. Defective glucose transport into the pancreas leads to hypoinsulinemia and postprandial hyperglycemia, and children with GSD XI often are confused with type 1 diabetes. Impairment in GLUT2-mediated efflux of glucose leads to glycogen accumulation in renal tubules and failure to reabsorb multiple filtered solutes. Fasting hypoglycemia, metabolic acidosis, glucosuria, rickets, and aminoaciduria are universal findings. While glucose uptake into the liver is abnormal, glycogen can be synthesized from other sugars and from glucose-6-phosphate generated by gluconeogenesis. Impaired release of the glycogen leads to the massive hepatomegaly and nephromegaly.
7.2Genetics
The GLUT2 transporter is expressed in hepatocytes, pancreatic β-cells, renal epithelial cells, and the basolateral membrane of the intestines [149, 155]. The GLUT2 gene which encodes this transporter lies on the long arm of chromosome 3 at 3q26.2 [156, 157]. At least 46 different mutations have been reported in the GLUT2 gene [19, 158, 159].
7.3Pathology
The constellation of liver enlargement, hyperglycemia, and failure to thrive can be confused as Mauriac syndrome, but fasting hypoglycemia and hyperlactatemia can be used to distinguish it from diabetes. Due to the multiorgan involvement, liver biopsies are rarely needed to make this diagnosis. When a biopsy is performed, glycogen filled hepatocytes are found with hepatic steatosis. There is no fibrosis, and it can be difficult to distinguish from other forms of GSD. Enzymatic studies will be normal. Sequencing of the GLUT2 gene is therefore the preferred diagnostic method.
7.4Treatment
Patients are treated with a high protein diet supplemented with cornstarch dosed to prevent hypoglycemia. Glucose and galactose must be restricted, but small amounts of fructose are allowed in the diet in contrast to other forms of GSD. Supplementation with vitamin D is critical, and replacement of renal losses of bicarbonate is needed.
8Glycogen storage disease type 0
Glycogen storage disease type 0 (OMIM 240600) is caused by a deficiency of the enzyme hepatic glycogen synthase [160–164]. This enzyme is required for glycogen synthesis, and is encoded by the GYS2 gene on chromosome 12. Unlike other forms of glycogen storage disease, GSD type 0 does not involve the storage of excessive or abnormal glycogen; instead, it is characterized by moderately decreased stores of normal-structured glycogen in the liver. The inability of patients to store glucose as glycogen in the liver leads to postprandial hyperglycemia and hyperlactatemia, as well as fasting ketotic hypoglycemia.
8.1Clinical presentation
Glycogen storage disease type 0 is usually diagnosed during late infancy and childhood. Affected infants may be diagnosed after having trouble being weaned from nighttime feeds, or children may be diagnosed after experiencing ketotic hypoglycemia during an acute gastrointestinal illness. Since post-prandial hyperglycemia occurs, children are often misdiagnosed as having diabetes although the duration of hyperglycemia is usually not long enough to cause polyuria or polydipsia [165].
Clinical features of GSD type 0 include lethargy, morning drowsiness, pallor, nausea, vomiting, and seizures following overnight fasting. Abdominal examination may be normal or may reveal paradoxically mild hepatomegaly from a fatty liver. Growth failure is common with both height and weight percentiles below average. In addition, 22% of reported patients have been described with developmental delay [166].
8.2Genetics
Glycogen storage disease type 0 is rare, representing less than 1% of all GSD cases. However, there is some indication that this disease may be underdiagnosed, since asymptomatic siblings have been identified in several GSD type 0 families [163].
Patients with GSD type 0 have deficient hepatic enzyme activity, but normal activity of muscle glycogen synthase (which is encoded by the GYS1 gene) [165, 167]. The gene which encodes hepatic glycogen synthase, GYS2, lies on the short arm of chromosome 12 at 12p12.2 [168]. GYS2 is composed of 16 exons and encodes an enzyme consisting of 703 amino acids.
8.3Pathology
All patients with GSD 0 have elevated ketones in the blood after an overnight fast. Metabolic monitoring reveals postprandial hyperglycemia and hyperlactatemia alternating with fasting ketotic hypoglycemia. Although disease confirmation previously required a liver biopsy to provide evidence of decreased hepatic glycogen content and decreased glycogen synthase activity, genetic testing for GSD type 0 is now the preferred method for diagnosing this condition. GYS2 mutations have been found in affected individuals throughout the world, including Austria, Argentina, England, Germany, and the United States. To date, at least 17 different GYS2 mutations have been identified [166].
8.4Treatment
The goal of treatment for GSD type 0 is to prevent hypoglycemia and acidosis by avoidance of fasting. A high protein diet is recommended (3 g/kg/day), and sugar intake is restricted since it will cause hyperglycemia, hyperinsulinemia, and lactate elevation. The prognosis for this condition is outstanding, and long-term complications have not been described to date.
9Summary
With a better understanding of the biochemical defects underlying the glycogen storage diseases, therapy has improved and patients are living longer and with a better quality of life. Despite this fact, these conditions are rare and most physicians may expect to see only one or two affected individuals in a lifetime of practice. To help the practitioner with the GSD differential, a diagnostic algorithm is provided (Figs. 1 and 2). Because ethnicity may provide an important clue to diagnosis, Table 3 lists populations in which the hepatic glycogenoses are common. With tremendous advances in molecular biology, gene therapy may become possible in the near future for this challenging group of metabolic disorders.
References
[1] | Online Mendelian Inheritance in Man, OMIM®. McKusick-Nathans Institute of Genetic Medicine, Johns Hopkins University. Baltimore, MD, (2012) . World Wide Web URL: http://omim.org/ |
[2] | Champe P.C. , Harvey R.A. , Glycogen Metabolism, In: Biochemistry, 2nd ed, Philadelphia, (1994) , JB Company Lippincott, p. 135. |
[3] | Arion W.J. and Canfield W.K. , Glucose-6-phosphatase and type 1 glycogen storage disease: Some critical considerations,S, Eur J Pediatr 152: (Suppl 1) ((1993) ), 7. |
[4] | Veiga-da-Cunha M. , Gerin I. and Van Schaftingen E. , How many forms of glycogen storage disease type I? Eur J Pediatr 159: ((2000) ), 314. |
[5] | Veiga-da-Cunha M. , Gerin I. , Chen Y.T. , et al., The putative glucose 6-phosphate translocase gene is mutated in essentially all cases of glycogen storage disease type I non-a, Eur J Hum Genet 7: ((1999) ), 717. |
[6] | Veiga-da-Cunha M. , Gerin I. , Chen Y.T. , et al., A gene on chromosome 11q23 coding for a putative glucose- 6-phosphate translocase is mutated in glycogen-storage disease types Ib and Ic, Am J Hum Genet 63: ((1998) ), 976. |
[7] | Cori G.T. and Cori C.F. , Glucose-6-phosphatase of the liver in glycogen storage disease, J Biol Chem 199: ((1952) ), 661. |
[8] | Harper P.S. , Glycogen Storage Disease, In: Harer PS, editor: Landmarks in Medical Genetics. New York, (2004) , Oxford University Press, p. 187. |
[9] | Wang D.Q. , Carreras C.T. , Fiske L.M. , et al., Characterization and pathogenesis of anemia in glycogen storage disease type Ia and Ib, Genet Med 14: ((2012) ), 795. |
[10] | Minarich L.A. , Kirpich A. , Fiske L.M. , Weinstein D.A. , Bone mineral density in glycogen storage disease type Ia and Ib. Genet Med Epub 04/07/2012; PMID: 22481133. |
[11] | Beegle R.D. , Brown L.M. and Weinstein D.A. , Regression of hepatocellular adenomas with strict dietary therapy in patients with glycogen storage disease type I, JIMD Rep 18: ((2015) ), 23. |
[12] | Weinstein D.A. , Somers M.J. , Wolfsdorf J.I. , Decreased urinary citrate excretion in type 1a glycogen storage disease, J Pediatr 138: ((2001) ), 378. |
[13] | Weinstein D.A. , Roy C.N. , Fleming M.D. , et al., Inappropriate expression of hepcidin is associated with iron refractory anemia: Implications for the anemia of chronic disease, Blood 100: ((2002) ), 3776. |
[14] | Lei K.J. , Chen Y.T. , Chen H. , et al., Genetic basis of glycogen storage disease type 1a: Prevalent mutations at the glucose-6-phosphatase locus, Am J Hum Genet 57: ((1995) ), 766. |
[15] | Wong L.J. , Hwu W.L. , Dai P. and Chen T.J. , Molecular genetics of glycogen-storage disease type 1a in Chinese patients of Taiwan, Mol Genet Metab 72: ((2001) ), 175. |
[16] | Ekstein J. , Rubin B.Y. , Anderson S.L. , et al., Mutation frequencies for glycogen storage disease Ia in the Ashkenazi Jewish population, Am J Med Genet A 129A: ((2004) ), 162. |
[17] | Lei K.J. , Shelly L.L. , Pan C.J. , et al., Mutations in the glucose-6-phosphatase gene that cause glycogen storage disease type 1a, Science 262: ((1993) ), 580. |
[18] | Chou J.Y. and Mansfield B.C. , Mutations in the glucose-6-phosphatase-alpha (G6PC) gene that cause type Ia glycogen storage disease, Hum Mutat 29: ((2008) ), 921. |
[19] | Cooper D.N. , Ball E.V. , Stenson P.D. , et al., Human Gene Mutation Database (HGMD): Institute of Medical Genetics. Cardiff, Wales, (2012) . World Wide Web URL: http://www.hgmd.org/ |
[20] | Tamhankar P.M. , Boggula V. , Girisha K.M. and Phadke S.R. , Profile of patients with Von Gierke disease from India, Indian Pediatr 49: ((2012) ), 228. |
[21] | Bali D.S. , Chen Y.T. and Goldstein J.L. , Glycogen Storage Disease Type I, In: Pagon RA, Bird TD, Dolan CR, Stephens K, Adam MP, editors: GeneReviews. Seattle, (1993) . |
[22] | Kishnani P.S. , Chuang T.P. , Bali D. , et al., Chromosomal and genetic alterations in human hepatocellular adenomas associated with type Ia glycogen storage disease, Hum Mol Genet 18: ((2009) ), 4781. |
[23] | Erez A. , Shchelochkov O.A. , Plon S.E. , et al., Insights into the pathogenesis and treatment of cancer from inborn errors of metabolism, Am J Hum Genet 88: ((2011) ), 402. |
[24] | Di Rocco M. , Calevo M.G., Taro M., et al., Hepatocellular adenoma and metabolic balance in patients with type Ia glycogen storage disease, Mol Genet Metab 93: ((2008) ), 398. |
[25] | Ross K.M. , Brown L.M. , Corrado M.M. , et al., Safety and efficacy of chronic extended release cornstarch therapy for glycogen storage disease type Ia, JIMD Rep 26: ((2016) ), 85. |
[26] | Ferrecchia I.A. , Guenette G. , Potocik E.A. and Weinstein D.A. , Pregnancy in women with glycogen storage disease Ia and Ib, J Perinat Neonatal Nurs 28: ((2014) ), 26. |
[27] | David M.K. and Weinstein D.A. , Liver transplantation in children with glycogen storage disease: Controversies and evaluation of the risk/benefit of this procedure, Pediatr Transplant 12: ((2008) ), 137. |
[28] | Davis M.K. , Valentine J.F. , Weinstein D.A. and Polyak S. , Antibodies to CBir1 are associated with glycogen storage disease type Ib, J Pediatr Gastroenterol Nutr 51: ((2010) ), 14. |
[29] | Gerin I. , Veiga-da-Cunha M. , Achouri Y. , et al., Sequence of a putative glucose 6-phosphate translocase, mutated in glycogen storage disease type Ib, FEBS Lett 419: ((1997) ), 235. |
[30] | Marcolongo P. , Barone V. , Priori G. , et al., Structure and mutation analysis of the glycogen storage disease type 1b gene, FEBS Lett 436: ((1998) ), 247. |
[31] | Lam C.W. , Chan B.Y. and Tong S.F. , Definition of the correct sequence in the donor splice site of intron 2 in the human glucose 6-phosphate translocase gene, FEBS Lett 445: ((1999) ), 449. |
[32] | Santer R. , Rischewski J. , Block G. , et al., Molecular analysis in glycogen storage disease 1 non-A: DHPLC detection of the highly prevalent exon 8 mutations of the G6PT1 gene in German patients, Hum Mutat 16: ((2000) ), 177. |
[33] | Kishani P.S. , Austin S.L. , Abdenur J.E. , et al., Diagnosis and management of glycogen storage disease type I: A practive guidline of the American College of Medical Genetics and Genomics,e, Genet Med 16: ((2014) ), 1. |
[34] | Melis D. , Minopoli G. , Balivo F. , Marcolongo P. , Parini R. , Paci S. , et al., Vitamin E clinical outcome of patients affected by glycogen storage disease type Ib, JIMD Rep Epub 07/01/2015, PMID: 26122627. |
[35] | Hers H.G. , alpha-Glucosidase deficiency in generalized glycogen storage disease (Pompe’s disease), Biochem J 86: ((1963) ), 11. |
[36] | Pompe J. , Over idiopatische hypertrophie van het hart, Ned Tijdshr Geneeskd 76: ((1932) ), 304. |
[37] | Kishnani P.S. , Steiner R.D. , Bali D. , et al., Pompe disease diagnosis and management guideline, Genet Med 8: ((2006) ), 267. |
[38] | van Capelle C.I. , Goedegebure A., Homans N.C., et al., Hearing loss in Pompe disease revisited: Results from a study of 24 children, J Inherit Metab Dis 33: ((2010) ), 597. |
[39] | Kamphoven J.H. , de Ruiter M.M. , Winkel L.P., et al., Hearing loss in infantile Pompe’s disease and determination of underlying pathology in the knockout mouse, Neurobiol Dis 16: ((2004) ), 14. |
[40] | Makos M.M. , McComb R.D. , Hart M.N. and Bennett D.R. , Alpha-glucosidase deficiency and basilar artery aneurysm: Report of a sibship, Ann Neurol 22: ((1987) ), 629. |
[41] | Matsuoka Y. , Senda Y. , Hirayama M. , et al., Late-onset acid maltase deficiency associated with intracranial aneurysm, J Neurol 235: ((1988) ), 371. |
[42] | Kroos M. , Hoogeveen-Westerveld M. , Michelakakis H. , et al., Update of the Pompe disease mutation database with 60 novel GAA sequence variants and additional studies on the functional effect of 34 previously reported variants, Hum Mutat Epub May 31: ((2012) ). |
[43] | D’Ancona G.G. , Wurm J. and Croce C.M. , Genetics of type II glycogenosis: Assignment of the human gene for acid alpha-glucosidase to chromosome 17, Proc Natl Acad Sci U S A 76: ((1979) ), 4526. |
[44] | Solomon E. , Swallow D. , Burgess S. and Evans L. , Assignment of the human acid alpha-glucosidase gene (alphaGLU) to chromosome 17 using somatic cell hybrids, Ann Hum Genet 42: ((1979) ), 273. |
[45] | Kuo W.L. , Hirschhorn R. , Huie M.L. and Hirschhorn K. , Localization and ordering of acid alpha-glucosidase (GAA) and thymidine kinase (TK1) by fluorescence in situ hybridization, Hum Genet 97: ((1996) ), 404. |
[46] | Kroos M. , Hoogeveen-Westerveld M. , van der Ploeg A. and Reuser A.J., The genotype-phenotype correlation in Pompe disease, Am J Med Genet C Semin Med Genet 160: ((2012) ), 59. |
[47] | Wan L. , Lee C.C. , Hsu C.M. , et al., Identification of eight novel mutations of the acid alpha-glucosidase gene causing the infantile or juvenile form of glycogen storage disease type II, J Neurol 255: ((2008) ), 831. |
[48] | Hermans M.M. , van Leenen D. , Kroos M.A., et al., Twenty-two novel mutations in the lysosomal alpha-glucosidase gene (GAA) underscore the genotype-phenotype correlation in glycogen storage disease type II, Hum Mutat 23: ((2004) ), 47. |
[49] | Huie M.L. , Chen A.S. , Tsujino S. , et al., Aberrant splicing in adult onset glycogen storage disease type II (GSDII), molecular identification of an IVS1 (–13T–>G) mutation in a majority of patients and a novel IVS10 (+1GT–>CT) mutation, Hum Mol Genet 3: ((1994) ), 2231. |
[50] | Kroos M.A. , Pomponio R.J. , Hagemans M.L. , et al., Broad spectrum of Pompe disease in patients with the same c.-32-13T->G haplotype, Neurology 68: ((2007) ), 110. |
[51] | Fernandes J. , The Glycogen Storage Diseases. In: Fernandes J, Saudubray J-M, van den Berghe G, editors: Inborn Metabolic Diseases: Diagnosis and Treatment, 3rd ed, Heidelberg, Germany, (2000) , Springer-Verlag, 86. |
[52] | Reuser A.J. , Kroos M.A. , Ponne N.J. , et al., Uptake and stability of human and bovine acid alpha-glucosidase in cultured fibroblasts and skeletal muscle cells from glycogenosis type II patients, Exp Cell Res 155: ((1984) ), 178. |
[53] | Hoefsloot L.H. , Willemsen R. , Kroos M.A. , et al., Expression and routeing of human lysosomal alpha-glucosidase in transiently transfected mammalian cells, Biochem J 272: ((1990) ), 485. |
[54] | Wisselaar H.A. , Kroos M.A. , Hermans M.M. , et al., Structural and functional changes of lysosomal acid alpha-glucosidase during intracellular transport and maturation, J Biol Chem 268: ((1993) ), 2223. |
[55] | van der Ploeg A.T. and Reuser A.J., Pompe’s disease, Lancet 372: ((2008) ), 1342. |
[56] | But W.M. , Lee S.H. , Chan A.O. and Lau G.T. , Enzyme replacement therapy for infantile Pompe disease during the critical period and identification of a novel mutation, Hong Kong Med J 15: ((2009) ), 474. |
[57] | Tinkle B.T. , Leslie N. , Glycogen Storage Disease Type II (Pompe Disease). In: Pagon RA, Bird TD, Dolan CR, Stephens K, Adam MP, editors: GeneReviews. Seattle, (1993) . |
[58] | McDowell R. , Li J.S. , Benjamin D.K. Jr , Morgan C., et al., Arrhythmias in patients receiving enzyme replacement therapy for infantile Pompe disease, Genet Med 10: ((2008) ), 758. |
[59] | Byrne B.J. , Falk D.J. , Pacak C.A. , et al., Pompe disease gene therapy, Hum Mol Genet 20: ((2011) ), R61. |
[60] | Danon M.J. , Oh S.J. , DiMauro S. , et al., Lysosomal glycogen storage disease with normal acid maltase, Neurology 31: ((1981) ), 51. |
[61] | Prall F.R. , Drack A. , Taylor M. , et al., Ophthalmic manifestations of Danon disease, Ophthalmology 113: ((2006) ), 1010. |
[62] | Arad M. , Maron B.J. , Gorham J.M. , et al., Glycogen storage diseases presenting as hypertrophic cardiomyopathy, N Engl J Med 352: ((2005) ), 362. |
[63] | Sugie K. , Yamamoto A. , Murayama K. , et al., Clinicopathological features of genetically confirmed Danon disease, Neurology 58: ((2002) ), 1773. |
[64] | Nishino I. , Fu J. , Tanji K. , et al., Primary LAMP-2 deficiency causes X-linked vacuolar cardiomyopathy and myopathy (Danon disease), Nature 406: ((2000) ), 906. |
[65] | Fanin M. , Nascimbeni A.C. , Fulizio L. , et al., Generalized lysosome-associated membrane protein-2 defect explains multisystem clinical involvement and allows leukocyte diagnostic screening in Danon disease, Am J Pathol 168: ((2006) ), 1309. |
[66] | Dougu N. , Joho S. , Shan L. , et al., Novel LAMP-2 mutation in a family with Danon disease presenting with hypertrophic cardiomyopathy, Circ J 73: ((2009) ), 376. |
[67] | Illingworth B. and Cori G.T. , Structure of glycogens and amylopectins. III. Normal and abnormal human glycogen, J Biol Chem 199: ((1952) ), 653. |
[68] | Forbes G.B. , Glycogen storage disease; report of a case with abnormal glycogen structure in liver and skeletal muscle, J Pediatr 42: ((1953) ), 645. |
[69] | Kishnani P.S. , Austin S.L. , Arn P. , et al., Glycogen storage disease type III diagnosis and management guidelines, Genet Med 12: ((2010) ), 446. |
[70] | Sentner C.P. , Hoogeveen I.J. , Weinstein D.A. , et al., Glycogen storage disease type III: Diagnosis, genotype, management, clinical course, and outcome, J Inherit Metab Dis Epub Apr 22 ((2016) ), PMID 27106217. |
[71] | Dagli A.I. , Zori R.T. , McCune H. , et al., Reversal of glycogen storage disease type IIIa-related cardiomyopathy with modification of diet, J Inherit Metab Dis 32: (Suppl 1) ((2009) ), S103. |
[72] | Sentner C.P. , Caliskan K. , Vletter W.B. and Smit G.P. , Heart failure due to severe hypertrophic cardiomyopathy reversed by low calorie, high protein dietary adjustments in a glycogen storage disease type IIIa patient, JIMD Rep 5: ((2012) ), 13. |
[73] | Demo E. , Frush D. , Gottfried M. , et al., Glycogen storage disease type III-hepatocellular carcinoma a long-term complication? J Hepatol 46: ((2007) ), 492. |
[74] | Siciliano M. , De Candia E. , Ballarin S., et al., Hepatocellular carcinoma complicating liver cirrhosis in type IIIa glycogen storage disease, J Clin Gastroenterol 31: ((2000) ), 80. |
[75] | Yang-Feng T.L. , Zheng K. , Yu J. , et al., Assignment of the human glycogen debrancher gene to chromosome 1p21, Genomics 13: ((1992) ), 931. |
[76] | Rousseau-Nepton I. , Okubo M. , Grabs R. , et al., A founder AGL mutation causing glycogen storage disease type IIIa in Inuit identified through whole-exome sequencing: A case series, CMAJ 187: ((2015) ), E68. |
[77] | Santer R. , Kinner M. , Steuerwald U. , et al., Molecular genetic basis and prevalence of glycogen storage disease type IIIA in the Faroe Islands, Eur J Hum Genet 9: ((2001) ), 388. |
[78] | Parvari R. , Moses S. , Shen J. , et al., A single-base deletion in the 3’-coding region of glycogen-debranching enzyme is prevalent in glycogen storage disease type IIIA in a population of North African Jewish patients, Eur J Hum Genet 5: ((1997) ), 266. |
[79] | Van Hoof F. and Hers H.G., The subgroups of type 3 glycogenosis, Eur J Biochem 2: ((1967) ), 265. |
[80] | Ding J.H. , de Barsy T. , Brown B.I. et al., Immunoblot analyses of glycogen debranching enzyme in different subtypes of glycogen storage disease type III, J Pediatr 116: ((1990) ), 95. |
[81] | Sugie H. , Fukuda T. , Ito M. , et al., Novel exon 11 skipping mutation in a patient with glycogen storage disease type IIId, J Inherit Metab Dis 24: ((2001) ), 535. |
[82] | Bruno C. , van Diggelen O.P. , Cassandrini D., et al., Clinical and genetic heterogeneity of branching enzyme deficiency (glycogenosis type IV), Neurology 63: ((2004) ), 1053. |
[83] | Nambu M. , Kawabe K. , Fukuda T. , et al., A neonatal form of glycogen storage disease type IV, Neurology 61: ((2003) ), 392. |
[84] | Tay S.K. , Akman H.O. , Chung W.K. , et al., Fatal infantile neuromuscular presentation of glycogen storage disease type IV, Neuromuscul Disord 14: ((2004) ), 253. |
[85] | Li S.C. , Chen C.M. , Goldstein J.L. , et al., Glycogen storage disease type IV: Novel mutations and molecular characterization of a heterogeneous disorder, J Inherit Metab Dis Epub January 09 ((2010) ). |
[86] | Klein C.J. , Adult Polyglucosan Body Disease. In Pagon RA, Bird TD, Dolan CR, Stephens K, Adam MP, editors: GeneReviews. Seattle, (1993) . |
[87] | Moses S.W. and Parvari R. , The variable presentations of glycogen storage disease type IV: A review of clinical, enzymatic and molecular studies, Curr Mol Med 2: ((2002) ), 177. |
[88] | Bao Y. , Kishnani P. , Wu J.Y. and Chen Y.T. , Hepatic and neuromuscular forms of glycogen storage disease type IV caused by mutations in the same glycogen-branching enzyme gene, J Clin Invest 97: ((1996) ), 941. |
[89] | Lossos A. , Meiner Z. , Barash V. , et al., Adult polyglucosan body disease in Ashkenazi Jewish patients carrying the Tyr329Ser mutation in the glycogen-branching enzyme gene, Ann Neurol 44: ((1998) ), 867. |
[90] | Ubogu E.E. , Hong S.T. , Akman H.O. , et al., Adult polyglucosan body disease: A case report of a manifesting heterozygote, Muscle Nerve 32: ((2005) ), 675. |
[91] | McArdle B. , Myopathy due to a defect in muscle glycogen breakdown, Clin Sci 10: ((1951) ), 13. |
[92] | Vissing J. and Haller R.G. , A diagnostic cycle test for McArdle’s disease, Ann Neurol 54: ((2003) ), 539. |
[93] | Mommaerts W.F. , Illingworth B. , Pearson C.M. , et al., A functional disorder of muscle associated with the absence of phosphorylase, Proc Natl Acad Sci U S A 45: ((1959) ), 791. |
[94] | Schmid R. and Mahler R. , Chronic progressive myopathy with myoglobinuria: Demonstration of a glycogenolytic defect in the muscle, J Clin Invest 38: ((1959) ), 2044. |
[95] | Tsujino S. , Shanske S. and DiMauro S. , Molecular genetic heterogeneity of myophosphorylase deficiency (McArdle’s disease), N Engl J Med 329: ((1993) ), 241. |
[96] | Kubisch C. , Wicklein E.M. and Jentsch T.J. , Molecular diagnosis of McArdle disease: Revised genomic structure of the myophosphorylase gene and identification of a novel mutation, Hum Mutat 12: ((1998) ), 27. |
[97] | Martin M.A. , Rubio J.C. , Buchbinder J. , et al., Molecular heterogeneity of myophosphorylase deficiency (McArdle’s disease): A genotype-phenotype correlation study, Ann Neurol 50: ((2001) ), 574. |
[98] | Bruno C. , Cassandrini D. , Martinuzzi A. , et al., McArdle disease: The mutation spectrum of PYGM in a large Italian cohort, Hum Mutat 27: ((2006) ), 718. |
[99] | Andreu A.L. , Nogales-Gadea G. , Cassandrini D. , et al., McArdle disease: Molecular genetic update, Acta Myol 26: ((2007) ), 53. |
[100] | Arenas J. , Martin M.A. , Andreu A.L. , Glycogen Storage Disease Type V. In: Pagon RA, Bird TD, Dolan CR, Stephens K, Adam MP, editors: GeneReviews. Seattle, (1993) . |
[101] | Tsujino S. , Shanske S. , Carroll J.E. , et al., Double trouble: Combined myophosphorylase and AMP deaminase deficiency in a child homozygous for nonsense mutations at both loci, Neuromuscul Disord 5: ((1995) ), 263. |
[102] | Martinuzzi A. , Sartori E. , Fanin M. , et al., Phenotype modulators in myophosphorylase deficiency, Ann Neurol 53: ((2003) ), 497. |
[103] | Manfredi G. , Silvestri G. , Servidei S. , et al., Manifesting heterozygotes in McArdle’s disease: Clinical, morphological and biochemical studies in a family, J Neurol Sci 115: ((1993) ), 91. |
[104] | Di S. , Mauro, Muscle glycogenoses: An overview, Acta Myol 26: ((2007) ), 35. |
[105] | Vissing J. and Haller R.G. , The effect of oral sucrose on exercise tolerance in patients with McArdle’s disease, N Engl J Med 349: ((2003) ), 2503. |
[106] | Vladutiu G.D. , Simmons Z. , Isackson P.J. , et al., Genetic risk factors associated with lipid-lowering drug-induced myopathies, Muscle Nerve 34: ((2006) ), 153. |
[107] | Hicks J. , Wartchow E. and Mierau G. , Glycogen storage diseases: A brief review and update on clinical features, genetic abnormalities, pathologic features, and treatment, Ultrastruct Pathol 35: ((2011) ), 183. |
[108] | Hers H.G. , Enzymatic studies of hepatic fragments; application to the classification of glycogenoses, Rev Int Hepatol 9: ((1959) ), 35. |
[109] | Newgard C.B. , Fletterick R.J. , Anderson L.A. and Lebo R.V. , The polymorphic locus for glycogen storage disease VI (liver glycogen phosphorylase) maps to chromosome 14, Am J Hum Genet 40: ((1987) ), 351. |
[110] | Hoogeveen I.J. , Van der Ende R.M. , van SpronsenF.J., et al., Normoglycemic ketonemia as biochemical presentation in ketotic glycogen storage disease, JIMD Rep Epub Nov 3 ((2015) ), PMID 26526422. |
[111] | Roscher A. , Patel J. , Hewson S. , et al., The natural history of glycogen storage disease types VI and IX: Long-term outcome from the largest metabolic center in Canada, Mol Genet Metab 113: ((2014) ), 171. |
[112] | Chang S. , Rosenberg M.J. , Morton H. , et al., Identification of a mutation in liver glycogen phosphorylase in glycogen storage disease type VI, Hum Mol Genet 7: ((1998) ), 865. |
[113] | Burwinkel B. , Bakker H.D. , Herschkovitz E. , et al., Mutations in the liver glycogen phosphorylase gene (PYGL) underlying glycogenosis type VI, Am J Hum Genet 62: ((1998) ), 785. |
[114] | Tang N.L. , Hui J. , Young E. , et al., A novel mutation (G233D) in the glycogen phosphorylase gene in a patient with hepatic glycogen storage disease and residual enzyme activity, Mol Genet Metab 79: ((2003) ), 142. |
[115] | Dagli A.I. , Weinstein D.A. , Glycogen Storage Disease Type VI. In: Pagon RA, Bird TD, Dolan CR, Stephens K, Adam MP, editors: GeneReviews. Seattle, (1993) . |
[116] | Beauchamp N.J. , Taybert J. , Champion M.P. , et al., High frequency of missense mutations in glycogen storage disease type VI, J Inherit Metab Dis 30: ((2007) ), 722. |
[117] | Manzia T.M. , Angelico R. , Toti L. , et al., Glycogen storage disease type Ia and VI associated with hepatocellular carinoma: Two case reports, Tranplant Proc 43: ((2011) ), 1181. |
[118] | Brown L.M. , Corrado M.M. , van der Ende R.M. , et al., Evaluation of glycogen storage disease as a cause of ketotic hypoglycemia in children, J Inherit Metab Dis 38: ((2015) ), 489. |
[119] | Beauchamp N.J. , Dalton A. , Ramaswami U. , et al., Glycogen storage disease type IX: High variability in clinical phenotype, Mol Genet Metab 92: ((2007) ), 88. |
[120] | Francke U. , Darras B.T. , Zander N.F. and Kilimann M.W. , Assignment of human genes for phosphorylase kinase subunits alpha (PHKA) to Xq12-q13 and beta (PHKB) to 16q12-q13, Am J Hum Genet 45: ((1989) ), 276. |
[121] | Schneider A. , Davidson J.J. , Wullrich A. and Kilimann M.W. , Phosphorylase kinase deficiency in I-strain mice is associated with a frameshift mutation in the alpha subunit muscle isoform, Nat Genet 5: ((1993) ), 381. |
[122] | Wehner M. , Clemens P.R. , Engel A.G. and Kilimann M.W. , Human muscle glycogenosis due to phosphorylase kinase deficiency associated with a nonsense mutation in the muscle isoform of the alpha subunit, Hum Mol Genet 3: ((1994) ), 1983. |
[123] | Wuyts W. , Reyniers E. , Ceuterick C. , et al., Myopathy and phosphorylase kinase deficiency caused by a mutation in the PHKA1 gene, Am J Med Genet A 133A: ((2005) ), 82. |
[124] | Hendrickx J. , Coucke P. , Dams E. , et al., Mutations in the phosphorylase kinase gene PHKA2 are responsible for X-linked liver glycogen storage disease, Hum Mol Genet 4: ((1995) ), 77. |
[125] | van den Berg I.E. , van Beurden E.A., Malingre H.E., et al., X-linked liver phosphorylase kinase deficiency is associated with mutations in the human liver phosphorylase kinase alpha subunit, Am J Hum Genet 56: ((1995) ), 381. |
[126] | Hendrickx J. , Lee P. , Keating J.P. , et al., Complete genomic structure and mutational spectrum of PHKA2 in patients with x-linked liver glycogenosis type I and II, Am J Hum Genet 64: ((1999) ), 1541. |
[127] | Burwinkel B. , Shin Y.S. , Bakker H.D. , et al., Mutation hotspots in the PHKA2 gene in X-linked liver glycogenosis due to phosphorylase kinase deficiency with atypical activity in blood cells (XLG2), Hum Mol Genet 5: ((1996) ), 653. |
[128] | Burwinkel B. , Amat L. , Gray R.G. , et al., Variability of biochemical and clinical phenotype in X-linked liver glycogenosis with mutations in the phosphorylase kinase PHKA2 gene, Hum Genet 102: ((1998) ), 423. |
[129] | Wullrich-Schmoll A. and Kilimann M.W. , Structure of the human gene encoding the phosphorylase kinase beta subunit (PHKB), Eur J Biochem 238: ((1996) ), 374. |
[130] | van den Berg I.E. , van Beurden E.A., de Klerk J.B., et al., Autosomal recessive phosphorylase kinase deficiency in liver, caused by mutations in the gene encoding the beta subunit (PHKB), Am J Hum Genet 61: ((1997) ), 539. |
[131] | Burwinkel B. , Maichele A.J. , Aagenaes O. , et al., Autosomal glycogenosis of liver and muscle due to phosphorylase kinase deficiency is caused by mutations in the phosphorylase kinase beta subunit (PHKB), Hum Mol Genet 6: ((1997) ), 1109. |
[132] | Burwinkel B. , Moses S.W. and Kilimann M.W. , Phosphorylase-kinase-deficient liver glycogenosis with an unusual biochemical phenotype in blood cells associated with a missense mutation in the beta subunit gene (PHKB), Hum Genet 101: ((1997) ), 170. |
[133] | Davit-Spraul A. , Piraud M. , Dobbelaere D. , et al., Liver glycogen storage diseases due to phosphorylase system deficiencies: Diagnosis thanks to non invasive blood enzymatic and molecular studies, Mol Genet Metab 104: ((2011) ), 137. |
[134] | Burwinkel B. , Hu B. , Schroers A. , et al., Muscle glycogenosis with low phosphorylase kinase activity: Mutations in PHKA1, PHKG1 or six other candidate genes explain only a minority of cases, Eur J Hum Genet 11: ((2003) ), 516. |
[135] | Maichele A.J. , Burwinkel B. , Maire I. , et al., Mutations in the testis/liver isoform of the phosphorylase kinase gamma subunit (PHKG2) cause autosomal liver glycogenosis in the gsd rat and in humans, Nat Genet 14: ((1996) ), 337. |
[136] | Owen D.J. , apageorgiou P.A.C. , Garman E.F. , et al., Expression, purification and crystallisation of phosphorylase kinase catalytic domain, J Mol Biol 246: ((1995) ), 374. |
[137] | Burwinkel B. , Shiomi S. , Al Zaben A. and Kilimann M.W., Liver glycogenosis due to phosphorylase kinase deficiency: PHKG2 gene structure and mutations associated with cirrhosis, Hum Mol Genet 7: ((1998) ), 149. |
[138] | Burwinkel B. , Tanner M.S. and Kilimann M.W. , Phosphorylase kinase deficient liver glycogenosis: Progression to cirrhosis in infancy associated with PHKG2 mutations (H144Y and L225R), J Med Genet 37: ((2000) ), 376. |
[139] | Burwinkel B. , Rootwelt T. , Kvittingen E.A. , et al., Severe phenotype of phosphorylase kinase-deficient liver glycogenosis with mutations in the PHKG2 gene, Pediatr Res 54: ((2003) ), 834. |
[140] | Tsilianidis L.A. , Fiske L.M. , Siegel S. , et al., Aggressive therapy improves cirrhosis in glycogen storage disease type IX, Mol Genet Metab 109: ((2013) ), 179. |
[141] | Ross K. , Brown L.M. , Corrado M.M. and Weinstein D.A. , Safety and efficacy of long-term use of extended release cornstarch therapy for glycogen storage disease types 0, III, VI, and IX, J Nutr Therap 4: ((2015) ), 137. |
[142] | Tarui S. , Okuno G. , Ikura Y. , et al., Phosphofructokinase deficiency in skeletal muscle. A new type of glycogenosis, Biochem Biophys Res Commun 19: ((1965) ), 517. |
[143] | Nakajima H. , Raben N. , Hamaguchi T. and Yamasaki T. , Phosphofructokinase deficiency; past, present and future, Curr Mol Med 2: ((2002) ), 197. |
[144] | Toscano A. and Musumeci O. , Tarui disease and distal glycogenoses: Clinical and genetic update, Acta Myol 26: ((2007) ), 105. |
[145] | Nakajima H. , Noguchi T. , Yamasaki T. , et al., Cloning of human muscle phosphofructokinase cDNA, FEBS Lett 223: ((1987) ), 113. |
[146] | Sharma P.M. , Reddy G.R. , Babior B.M. and McLachlan A. , Alternative splicing of the transcript encoding the human muscle isoenzyme of phosphofructokinase, J Biol Chem 265: ((1990) ), 9006. |
[147] | Raben N. , Sherman J. , Miller F. , et al., A 5’ splice junction mutation leading to exon deletion in an Ashkenazic Jewish family with phosphofructokinase deficiency (Tarui disease), J Biol Chem 268: ((1993) ), 4963. |
[148] | Sherman J.B. , Raben N. , Nicastri C. , et al., Common mutations in the phosphofructokinase-M gene in Ashkenazi Jewish patients with glycogenesis VII–and their population frequency, Am J Hum Genet 55: ((1994) ), 305. |
[149] | Santer R. , Schneppenheim R. , Dombrowski A. , et al., Mutations in GLUT2, the gene for the liver-type glucose transporter, in patients with Fanconi-Bickel syndrome, Nat Genet 17: ((1997) ), 324. |
[150] | Santer R. , Schneppenheim R. , Suter D. , et al., Fanconi-Bickel syndrome–the original patient and his natural history, historical steps leading to the primary defect, and a review of the literature, Eur J Pediatr 157: ((1998) ), 783. |
[151] | Burwinkel B. , Sanjad S.A. , Al-Sabban E. , et al., A mutation in GLUT2, not in phosphorylase kinase subunits, in hepato-renal glycogenosis with Fanconi syndrome and low phosphorylase kinase activity, Hum Genet 105: ((1999) ), 240. |
[152] | Santer R. , Steinmann B. and Schaub J. , Fanconi-Bickel syndrome–a congenital defect of facilitative glucose transport, Curr Mol Med 2: ((2002) ), 213. |
[153] | Elpeleg O.N. , The molecular background of glycogen metabolism disorders, J Pediatr Endocrinol Metab 12: ((1999) ), 363. |
[154] | Fanconi G. and Bickel H. , Chronic aminoaciduria (amino acid diabetes or nephrotic-glucosuric dwarfism) in glycogen storage and cystine disease, Helv Paediatr Acta 4: ((1949) ), 359. |
[155] | Mueckler M. , Facilitative glucose transporters, Eur J Biochem 219: ((1994) ), 713. |
[156] | Fukumoto H. , Seino S. , Imura H. , et al., Sequence, tissue distribution, and chromosomal localization of mRNA encoding a human glucose transporter-like protein, Proc Natl Acad Sci U S A 85: ((1988) ), 5434. |
[157] | Permutt M.A. , Koranyi L. , Keller K. , et al., Cloning and functional expression of a human pancreatic islet glucose-transporter cDNA, Proc Natl Acad Sci U S A 86: ((1989) ), 8688. |
[158] | Santer R. , Groth S. , Kinner M. , et al., The mutation spectrum of the facilitative glucose transporter gene SLC2A2 (GLUT2) in patients with Fanconi-Bickel syndrome, Hum Genet 110: ((2002) ), 21. |
[159] | Grunert S.C. , Schwab K.O. , Pohl M. , et al., Fanconi-Bickel syndrome: GLUT2 mutations associated with a mild phenotype, Mol Genet Metab 105: ((2012) ), 433. |
[160] | Orho M. , Bosshard N.U. , Buist N.R. , et al., Mutations in the liver glycogen synthase gene in children with hypoglycemia due to glycogen storage disease type 0, J Clin Invest 102: ((1998) ), 507. |
[161] | Lewis G.M. , Spencer-Peet J. and Stewart K.M. , Infantile Hypoglycaemia due to inherited deficiency of glycogen synthetase in liver, Arch Dis Child 38: ((1963) ), 40. |
[162] | Aynsley-Green A. , Williamson D.H. and Gitzelmann R. , Hepatic glycogen synthetase deficienc. Definition of syndrome from metabolic and enzyme studies on a 9-year-old girl, Arch Dis Child 52: ((1977) ), 573. |
[163] | Gitzelmann R. , Spycher M.A. , Feil G. , et al., Liver glycogen synthase deficiency: A rarely diagnosed entity, Eur J Pediatr 155: ((1996) ), 561. |
[164] | Rutledge S.L. , Atchison J. , Bosshard N.U. and Steinmann B. , Case report: Liver glycogen synthase deficiency–a cause of ketotic hypoglycemia, Pediatrics 108: ((2001) ), 495. |
[165] | Bachrach B.E. , Weinstein D.A. , Orho-Melander M. , et al., Glycogen synthase deficiency (glycogen storage disease type 0) presenting with hyperglycemia and glucosuria: Report of three new mutations, J Pediatr 140: ((2002) ), 781. |
[166] | Weinstein D.A. , Correia C.E. , Saunders A.C. and Wolfsdorf J.I. , Hepatic glycogen synthase deficiency: An infrequently recognized cause of ketotic hypoglycemia, Mol Genet Metab 87: ((2006) ), 284. |
[167] | Orho M. , Nikula-Ijas P. , Schalin-Jantti C. , et al., Isolation and characterization of the human muscle glycogen synthase gene, Diabetes 44: ((1995) ), 1099. |
[168] | Nuttall F.Q. , Gannon M.C. , Kubic V.L. and Hoyt K.J. , The human liver glycogen synthase isozyme gene is located on the short arm of chromosome 12, Genomics 19: ((1994) ), 404. |
Figures and Tables
Fig.1
Suspected glycogen storage disease.
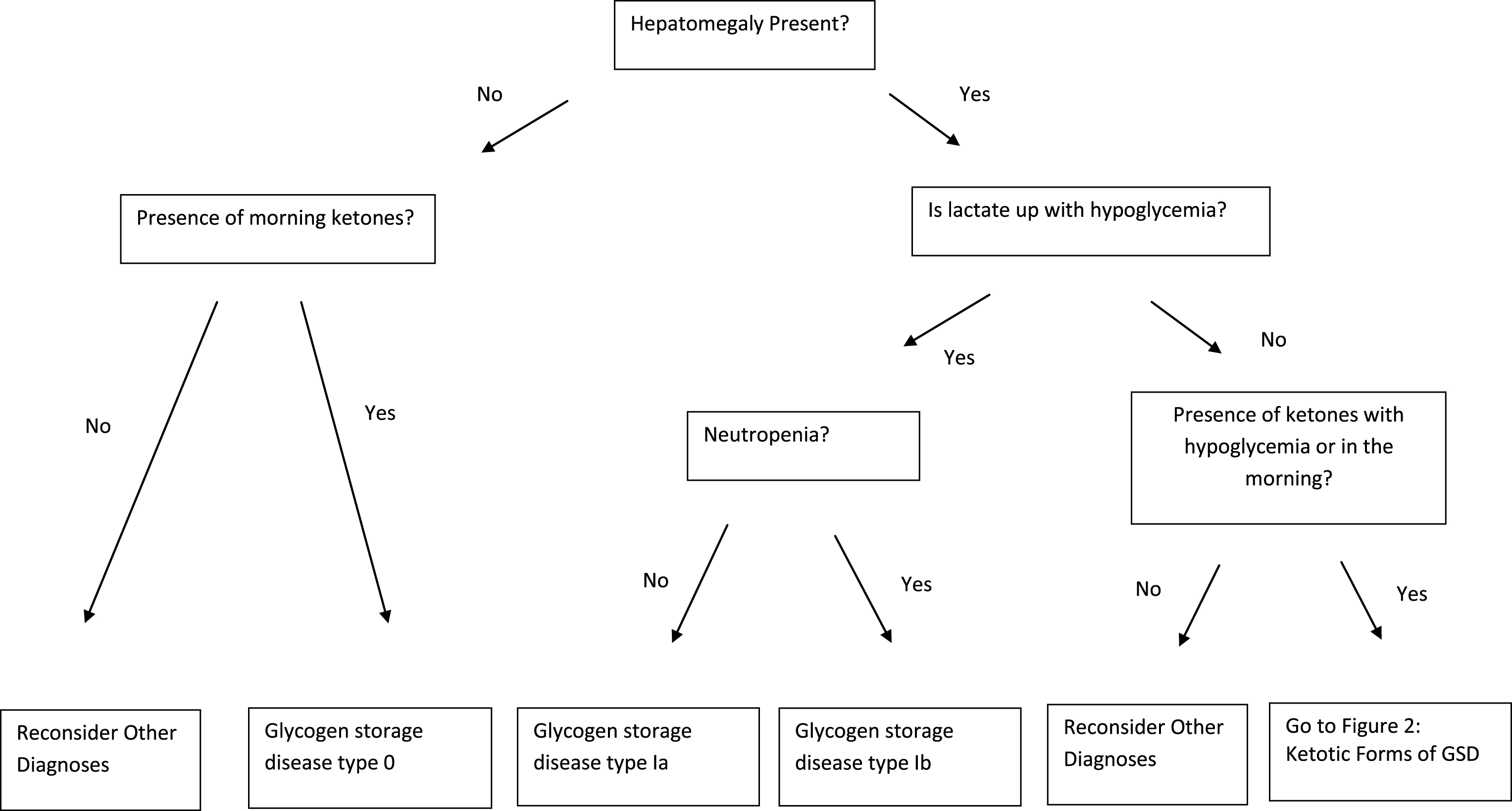
Fig.2
Ketotic forms of GSD.
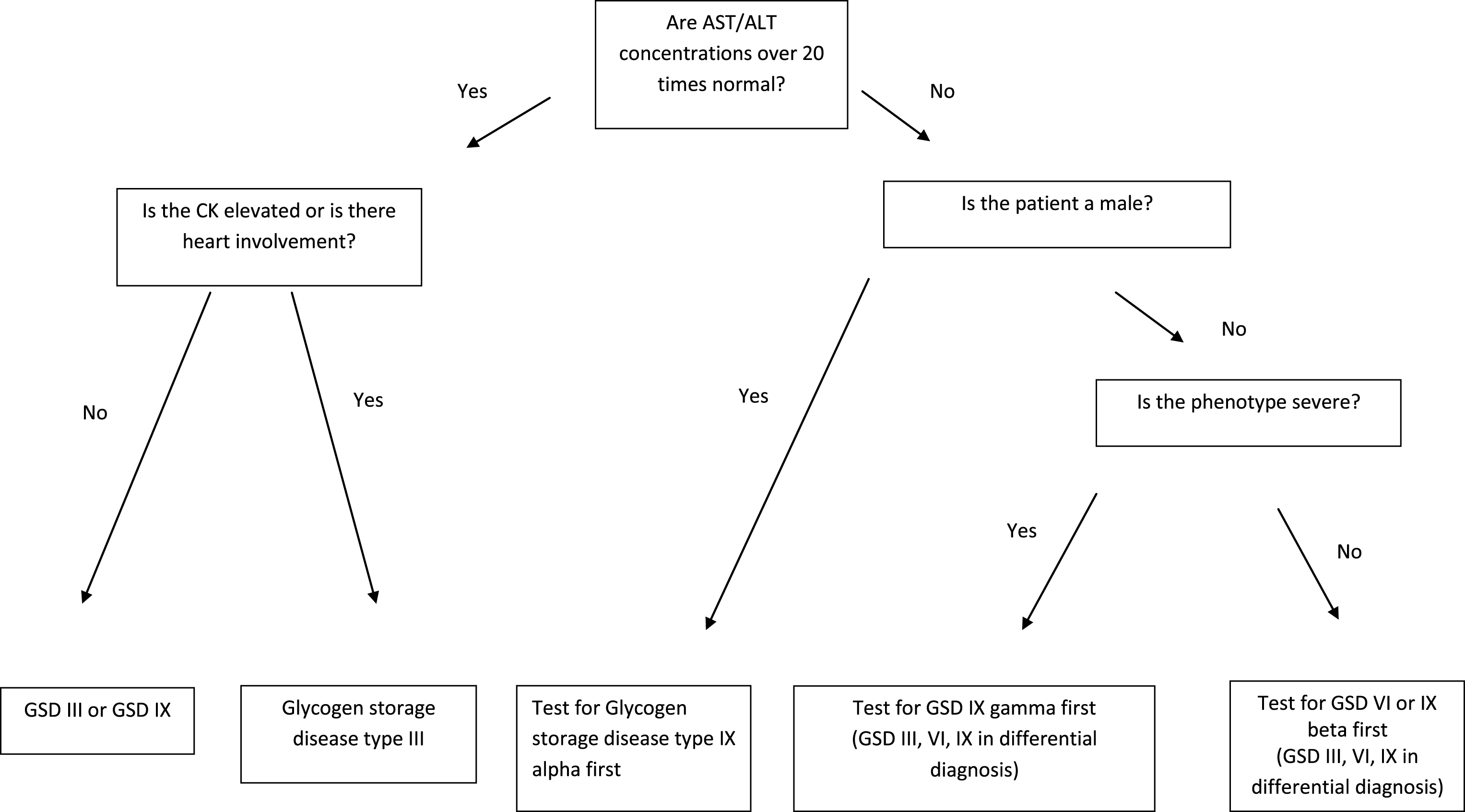
Table 1
Summary of the glycogenoses
| GSD Type | OMIM# (Online | Enzyme/Protein | Gene | Chromosome | Mode of | Main |
| Mendelian | Deficiency | Location | Inheritance | Presentation: | ||
| Inheritance | Hepatic vs. | |||||
| in Man)1 | Neuro-muscular? | |||||
| GSD 0 | 240600 | Glycogen synthase | GYS2 | 12p12.2 | Autosomal recessive | Hepatic |
| GSD Ia | 232200 | Glucose-6-phosphatase-α catalytic subunit | G6PC | 17q21.31 | Autosomal recessive | Hepatic |
| GSD Ib | 232220 | Glucose-6-phosphate transporter | SLC37A4 | 11q23.3 | Autosomal recessive | Hepatic |
| GSD II | 232300 | α-1,4 glucosidase | GAA | 17q25.3 | Autosomal recessive | Neuro-muscular |
| GSD IIb | 300257 | LAMP-2 protein | LAMP2 | Xq24 | X-linked dominant | Neuro-muscular |
| GSD III | 232400 | Glycogen debranching enzyme (includes 4-alpha-glucanotransferase and amylo-1,6-glucosidase activities) | AGL | 1p21.2 | Autosomal recessive | Both |
| GSD IV | 232500 | Glycogen branching enzyme | GBE | 3p12.3 | Autosomal recessive | Both |
| GSD V | 232600 | Glycogen phosphorylase (muscle) | PYGM | 11q13.1 | Autosomal recessive | Neuro-muscular |
| (a.k.a. Myophosphorylase) | ||||||
| GSD VI | 232700 | Glycogen phosphorylase (liver) | PYGL | 14q22.1 | Autosomal recessive | Hepatic |
| GSD VII | 232800 | Phosphofructokinase | PFKM | 12q13.11 | Autosomal recessive | Neuro-muscular |
| (muscle) | ||||||
| GSD IXa | 306000 | Phosphorylase kinase, α subunit (liver) | PHKA2 | Xp22.13 | X-linked recessive | Hepatic |
| GSD IXb | 261750 | Phosphorylase kinase, β subunit | PHKB | 16q12.1 | Autosomal recessive | Hepatic |
| GSD IXc | 613027 | Phosphorylase kinase, γ subunit | PHKG2 | 16p11.2 | Autosomal recessive | Hepatic |
| GSD IXd | 300559 | Phosphorylase kinase, α subunit (muscle) | PHKA1 | Xq13 | X-linked recessive | Neuro-muscular |
| GSD XI | 227810 | GLUT2 transporter | SLC2A2 | 3q26.2 | Autosomal recessive | Hepatic |
1Online Mendelian Inheritance in Man, OMIM®. McKusick-Nathans Institute of Genetic Medicine, Johns Hopkins University. Baltimore, MD, 2012. World Wide Web URL: http://omim.org/
Table 2
Presentation of hepatic glycogen storage disease
| Type | Enzyme | Presenting Symptoms |
| 0 | Glycogen synthase | Ketotic hypoglycemia |
| Diabetes | ||
| I | Glucose-6-Phosphatase | Hypoglycemia |
| FTT /developmental delay | ||
| Hepatomegaly | ||
| III | Debranching | Hepatomegaly |
| Hypoglycemia | ||
| Hepatitis | ||
| VI | Phosphorylase | Hepatomegaly |
| Ketotic Hypoglycemia | ||
| IX | Phosphorylase kinase | Hepatomegaly |
| Ketotic Hypoglycemia |
Table 3
Epidemiology of the hepatic glycogen storage diseases
| Type | Defect | High Risk Ethnic Groups |
| 0 | Hepatic Glycogen Synthase | Italian |
| Ia | Glucose-6-Phosphatase | Ashkenazi Jewish |
| Mormon | ||
| Mexican | ||
| Ib | Glucose-6-Phosphate Transporter | Native American |
| Italian | ||
| III | Debranching Enzyme | Faroe Islands / Scandinavian |
| Indian Subcontinent | ||
| Sephardic Jewish (North African) | ||
| VI | Glycogen Phosphorylase | Mennonite |
| IX | Phosphorylase Kinase | Mayan |


