
Lysosomal storage diseases
Abstract
Lysosomes are cytoplasmic organelles that contain a variety of different hydrolases. A genetic deficiency in the enzymatic activity of one of these hydrolases will lead to the accumulation of the material meant for lysosomal degradation. Examples include glycogen in the case of Pompe disease, glycosaminoglycans in the case of the mucopolysaccharidoses, glycoproteins in the cases of the oligosaccharidoses, and sphingolipids in the cases of Niemann-Pick disease types A and B, Gaucher disease, Tay-Sachs disease, Krabbe disease, and metachromatic leukodystrophy. Sometimes, the lysosomal storage can be caused not by the enzymatic deficiency of one of the hydrolases, but by the deficiency of an activator protein, as occurs in the AB variant of GM2 gangliosidosis. Still other times, the accumulated lysosomal material results from failed egress of a small molecule as a consequence of a deficient transporter, as in cystinosis or Salla disease. In the last couple of decades, enzyme replacement therapy has become available for a number of lysosomal storage diseases. Examples include imiglucerase, taliglucerase and velaglucerase for Gaucher disease, laronidase for Hurler disease, idursulfase for Hunter disease, elosulfase for Morquio disease, galsulfase for Maroteaux-Lamy disease, alglucosidase alfa for Pompe disease, and agalsidase alfa and beta for Fabry disease. In addition, substrate reduction therapy has been approved for certain disorders, such as eliglustat for Gaucher disease. The advent of treatment options for some of these disorders has led to newborn screening pilot studies, and ultimately to the addition of Pompe disease and Hurler disease to the Recommended Uniform Screening Panel (RUSP) in 2015 and 2016, respectively.
1Lysosomal structure and function
Lysosomes are membrane-enclosed cytoplasmic organelles with a diameter of 0.05 to 0.5 μm [1]. They were discovered serendipitously by Christian de Duve in 1955, in his laboratory in Leuven, Belgium. He was studying the effects of insulin in the liver, and he surmised that knowing the subcellular localization of glucose-6-phosphatase might provide a clue to understanding the hepatic action of insulin. As a control, he tracked the activity and subcellular locatization of acid phosphatase. He homogenized rat livers using two different techniques: Albert Claude’s method, a mild procedure by which cellular organelles are kept intact, and a coarser method using a kitchen blender. The activity of acid phosphatase was much higher when measured in homogenates obtained with the kitchen blender, and also after thawing samples that were kept in the refrigerator. This and subsequent lines of evidence led him to propose that acid phosphatase – and other enzymes- were sequestered within membrane-bound organelles. Refinements of fractionation methods by centrifugation led to the discovery of “lytic bodies”, or lysosomes. He never returned to insulin research, but decided to focus on cell structure instead. Ten years later, in 1965, he also discovered peroxisomes. He received the Nobel Prize in Physiology or Medicine in 1974, for his discovery “concerning the structural and functional organization of the cell” [2, 3].
Lysosomes contain a variety of active hydrolytic enzymes (hydrolases) such as glycosidases, sulfatases, phosphatases, lipases, phospholipases, proteases, and nucleases (lysosomal enzymes) in an acid milieu (pH approximately 5). Most lysosomal enzymes enter a lysosome by means of a recognition signal (usually mannose-6-phosphate) and its corresponding receptors [4].
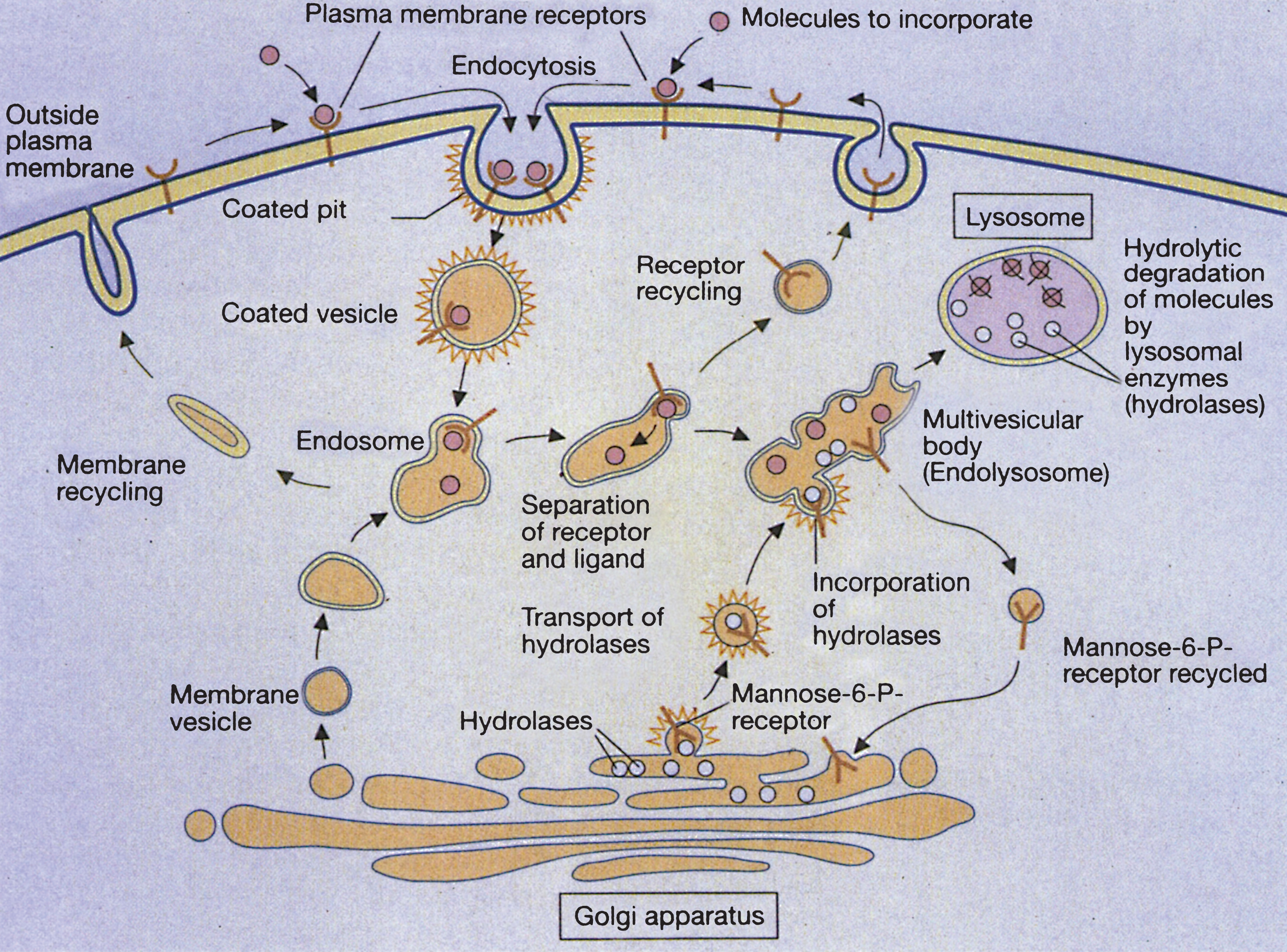
Extracellular molecules to be degraded are taken into the cell by a variety of means, one of which is receptor-mediated endocytosis. First, the molecules are bound to specific cell surface receptors. The loaded receptors are concentrated in an invagination of the plasma membrane (coated pit). This separates from the plasma membrane and forms a membrane-enclosed cytoplasmic compartment (coated vesicle). Hormones, growth factors, energy-delivering proteins, and numerous viruses and toxins also enter cells by receptor-mediated endocytosis. The cytoplasmic lining of the vesicle consists of a network of a trimeric protein called clathrin. The clathrin coat is quickly lost within the cell and an endosome forms; the endosome fuses with membrane vesicles from the Golgi apparatus to form larger endosomal compartments [4]. Once within lysosomes, macromolecules (lipids, carbohydrates, proteins) are degraded by hydrolytic enzymes to form their respective terminal components (fatty acids, monosaccharides, amino acids), which subsequently exit the lysosome.
The deficiency of a single hydrolase will create an inability to degrade the rest of the macromolecule. The end result is a lysosomal storage disease, and lysosomal enzymes undergo a complex multi-step process from gene transcription in the nucleus to functional protein within the lysosome. Messenger RNA, coding for a lysosomal enzyme, is directed to membrane-bound ribosomes of the rough endoplasmic reticulum, at which site the enzyme polypeptide is constructed. Next, an NH2-terminal signal peptide directs the polypeptide into the lumen of the endoplasmic reticulum. Typically, the polypeptide is then targeted to the lysosome by the addition of mannose-6-phosphate to the N-linked oligosaccharide side chains of the polypeptide. Many lysosomal hydrolases undergo further proteolytic cleavage before becoming fully activated. Defects in the targeting of lysosomal enzymes to the lysosome have been documented in mucolipidosis II and III. However, alternative ways of targeting the lysosomes, independent of mannose-6-phosphate, also exist. As an example, glucocerebrosidase enters the lysosome by binding to LIMP-2, while acid sphingomyelinase, prosaposin and the GM2 Activator Protein use sortilin as their receptor [5]. Receptor-mediated endocytosis and lysosome formation is shown in Fig. 1.
Fig.1
Receptor-mediated and lysosome formation. (Courtesy of Dr. Eberhard Passarge and Thieme Medical Publishers.).
For some lysosomal hydrolases to be fully active, activator proteins must be present; mutations affecting these activator proteins mimic deficiency of the hydrolase. The group of activator proteins consists of 4 small nonenzymatic glycoproteins called Sphingolipid Activator Proteins, or SAPs, plus the GM2 Activator Protein [6]. A SAP precursor, or prosaposin, gives rise to the 4 SAP proteins. It contains 524 amino acids and 5 N-glycosylation sites with 4 homologous domains of approximately 80 amino acids each. Most of the precursor is exported to the cell surface and then imported into the lysosomal compartment, where it is processed to the mature glycoproteins sap-A, sap-B, sap-C, and sap-D. The SAP precursor gene resides on chromosome 10 while the ganglioside GM2 activator gene is on chromosome 5 [7]. The primary lysosomal enzymes affected by activator protein deficiencies are listed in Table 1, along with the associated diseases.
Table 1
Sphingolipid activator proteins
| Activator | Activated Enzyme | Disease |
| SAP A | β-Galactosylceramidase | Krabbe |
| SAP B | Arylsulfatase A; α-Galactosidase | Metachromatic Leukodystrophy |
| SAP C | β-Glucosylceramidase | Gaucher |
| SAP D | Sphingomyelinase | Niemann-Pick |
| GM2 Activator Protein | β-Hexosaminidase A | GM2 AB variant* |
*Resembles Tay-Sachs disease.
In addition to lysosomal enzyme and activator defects, there exist five known disorders of lysosomal membrane transport, each of which reflects the inability to carry a small molecule out of the lysosome and into the cytoplasm. In these diseases (i.e., cystinosis, Salla disease, cobalamin F and cobalamin J disease, and mucolipidosis type IV), the intralysosomal material consists of an amino acid, monosaccharide, a cofactor, and cations, respectively, in contrast to the enzyme deficiencies, in which a macromolecule is stored.
Patients with lysosomal storage disorders are generally normal at birth, with symptoms developing in the first year of life. However, pathological findings can appear in the fetus. In a 9-week fetus with Tay-Sachs disease, lamellar and granular inclusions were present in the developing brain neurons. In fetuses from 12 to 22 weeks’ gestation, the lysosomal inclusions are more typical of membranous cytoplasmic bodies and are found in anterior horn cells of the spinal cord, ganglion cells of the retina, enteric plexus, and spinal ganglia [8, 9].
Many different cell types and tissues are affected by lysosomal storage disorders, with involvement at different stages in the disease process. Evaluation of these tissues through histology, enzymatic analysis, or imaging can assist in the diagnosis. Tissues that are useful in the diagnosis of storage diseases are listed in Table 2. Those disorders in which there is recognized peripheral blood, bone marrow, and conjunctival or skin pathology are shown in Table 3 and metabolic disorders with placental and/or fetal pathology are shown in Table 4.
Table 2
Tissues useful in the diagnosis of storage diseases
| Organ | Manifestation | Disease to be Considered | Procedure* | Diagnostic Test* |
| Liver | Hepatomegaly; Elevated liver function tests (occasionally) | Cholesteryl ester storage disease | Liver biopsy | Fibroblast or DBS lipase |
| Mucopolysaccharidoses (MPS) | Urine MPS | Enzyme analysis | ||
| Glycoproteinoses | Urine oligosaccharides | Enzyme analysis | ||
| Mucolipidoses II, III | Blood draw | Serum (increased) or fibroblast (decreased) enzyme analysis | ||
| Glycogen storage disease type II | Liver biopsy | Lymphocyte, DBS or fibroblast α-glucosidase; electron microscopy | ||
| Gaucher disease | Liver, bone marrow biopsy | Leukocyte, DBS or fibroblast β-glucocerebrosidase; electron microscopy | ||
| Niemann-Pick disease | Liver biopsy | Leukocyte, DBS or fibroblast sphingomyelinase | ||
| Wolman disease | Liver biopsy | Electron microscopy; fibroblast or DBS acid lipase | ||
| Spleen | Splenomegaly | Mucopolysaccharidoses | Urine MPS | Enzyme analysis |
| Gaucher disease | Liver, bone marrow biopsy | Leukocyte, DBS or fibroblast β-glucocerebrosidase; electron microscopy | ||
| Niemann-Pick disease | Liver biopsy, blood draw | Leukocyte, DBS or fibroblast sphingomyelinase | ||
| Bone and Joint | Dysostosis multiplex | Mucopolysaccharidoses | Urine MPS | Enzyme analysis |
| Glycoproteinoses | Urine oligosaccharides | Enzyme analysis | ||
| Swollen joints, soft tissue nodules | Farber disease | Tissue biopsy | Fibroblast acid ceramidase | |
| Eye | Macular cherry-red spot | Tay-Sachs disease | Blood draw | Serum, leukocyte or fibroblast hexosaminidase A |
| Sandhoff disease | Blood draw | Serum, leukocyte or fibroblast total hexosaminidase | ||
| Niemann-Pick disease | Liver biopsy | Leukocyte, DBS or fibroblast sphingomyelinase | ||
| GM1 gangliosidosis | Urine oligosaccharides | Leukocyte, DBS or fibroblast β-galactosidase | ||
| Sialidoses | Urine oligosaccharides | Fibroblast sialidase | ||
| Corneal clouding | Mucopolysaccharidoses | Urine MPS | Enzyme assay | |
| Mucolipidoses II, III | Blood draw | Serum (increased) or fibroblast (decreased) enzyme analysis | ||
| Corneal crystals | Cystinosis | Blood draw | Leukocyte cystine | |
| Adrenal gland | Bilateral adrenal calcifications | Wolman disease | Liver biopsy | Fibroblast or DBS acid lipase |
| Muscle-Cardiac Skeletal | Cardiomegaly, heart failure, myopathy | Glycogen storage disease type II | Liver biopsy | Lymphocyte, DBS or fibroblast α-glucosidase; electron microscopy |
| Glycogen storage disease types III, IV | Liver biopsy | Enzyme assay; electron microscopy | ||
| Brain | Mental and motor dysfunction | Krabbe disease | Blood draw; skin biopsy | Leukocyte, DBS or fibroblast β-galactosidase |
| Metachromatic leukodystrophy | Blood draw; skin biopsy | Leukocyte, DBS or fibroblast arylsulfatase A | ||
| Neuronal ceroid lipofuscinosis | Blood draw; skin biopsy | Electron microscopy for ceroid; enzyme assay for the most common forms | ||
| Niemann-Pick disease | Liver biopsy | Leukocyte, DBS or fibroblast sphingomyelinase | ||
| Gaucher disease | Liver, bone marrow biopsy | Leukocyte, DBS or fibroblast β-glucocerebrosidase; electron microscopy | ||
| Mucopolysaccharidoses | Urine MPS | Enzyme assay | ||
| Glycoproteinoses | Urine oligosaccharides | Enzyme assay | ||
| Tay-Sachs disease | Blood draw; skin biopsy | Serum, leukocyte or fibroblast hexosaminidase A | ||
| Sandhoff disease | Blood draw; skin biopsy | Serum, leukocyte or fibroblast total hexosaminidase | ||
| GM1 gangliosidosis | Urine oligosaccharides | Leukocyte, DBS or fibroblast β-galactosidase |
*Most disorders are now diagnosed on molecular grounds, and invasive biopsies are not required. DBS: dried blood spot.
Table 3
Lysosomal storage diseases with peripheral blood, bone marrow, and conjunctival/skin pathology
| Conjunctival/Skin Biopsy | ||||
| Disease | Peripheral Leukocytes | Bone Marrow Foamy Histiocytes | Inclusion Type | Site |
| Niemann-Pick disease | VL | + | Pleomorphic, dense-lucent, lamellar | Ep, En, M, N |
| Gaucher disease | – | + | Non-lamellar clefts and immature lamellar membranes (only in type 2) | Ep |
| Krabbe disease | – | Crystal-like | N | |
| Metachromatic leukodystrophy | – | – | Herringbone | N |
| Farber disease | – | + | Tubular, “banana bodies”, granular, membranous | M, En, N |
| Glycogen storage disease type II | VL | – | Granular | Ep, M, En, N |
| GM1 Gangliosidosis | VL | + | Fibrillogranular, membranous | Ep, En, M, N |
| Tay-Sachs disease | – | – | Membranous, granular | N, En, M |
| Sandhoff disease | – | – | Membranous, granular | N, En, M |
| Fabry disease | – | – | Lamellar | En, M |
| Wolman disease | VL | + | Vacuolar, membranous | En, M |
| Mucopolysaccharidoses I, II, III | NG, VL | + | Fibrillogranular, membranous | Ep, M, N, En |
| Mucolipidosis II | VL | – | Fibrillogranular, lamellar | M, En, N |
| Mucolipidosis III | VL | – | Fibrillogranular, lamellar | M, En, N |
| Mucolipidosis IV | VL | – | Fibrillogranular, lamellar | Ep, En, M, N |
| Fucosidosis | VL | + | Fine granular, sparse | Ep, En, M, N |
| Mannosidosis | VL | + | Fibrillogranular | M, En, N |
| Aspartylglucosaminuria | VL | – | Fibrillogranular | En, M, N |
| Galactosialidosis | VL | + | Fibrillogranular | Ep, M |
| Cystinosis | – | + | Crystals | M |
| Sialic acid storage disease | VL | + | Granular, sparse | En, M |
Ep, epithelial; En, endothelial; M, mesenchymal (histiocytes); N, neural; NG, neutrophil granules; VL, vacuolated lymphocytes.
Table 4
Lysosomal storage disorders with placental and/or fetal pathology
| Disease | Placental Pathology | Fetal Pathology |
| Niemann-Pick disease | + | + |
| Gaucher disease | ± | + |
| Krabbe disease | – | + |
| Metachromatic leukodystrophy | – | + |
| Gangliosidoses | + | + |
| Glycogen storage disease type II | + | + |
| Glycogen storage disease type IV | + | + |
| Wolman disease | + | + |
| Cholesteryl ester storage disease | + | + |
| Mucopolysaccharidoses | + | + |
| Mucolipidoses | + | + |
| Sialidoses | + | + |
| Ceroid lipofuscinosis | + | + |
| Cystinosis | + | + |
| Sialic acid storage diseases | + | + |
2Lysosomal storage diseases due to enzyme defects
Lysosomal enzyme deficiencies can be categorized based upon the macromolecule that fails to be degraded and is consequently stored. Carbohydrates, for example, are stored in glycogen storage disease type II, or Pompe disease. Neutral lipids (triglycerides and cholesteryl esters) are stored in Wolman disease and cholesteryl ester storage disease. Glycolipids accumulate in mucolipidoses. Glycoproteinoses involve deficiencies of enzymes that degrade the glycans that decorate glycoproteins [10]; these are largely N-linked sugar trees. The resulting diseases, named for the sugar that is not hydrolyzed, include fucosidosis, α- and β-mannosidosis, sialidosis, and aspartylglucosaminuria. The former disorders are screened by urine oligosaccharide analysis, performed by thin-layer chromatography,or more recently by mass spectrometry. The amino group from aspartylglucosamine also causes a diagnostic ninhydrin peak on urine amino acid analysis. Lysosomes also contain an abundance of proteases, and the deficiency of one of them, cathepsin K, results in pycnodysostosis, a disorder of increased bone density [11]. Cathepsin K is a cysteine protease.
Most of the remaining lysosomal storage diseases are glycosphingolipidoses. The sphingolipids (Fig. 2) have a long-chain amino alcohol (sphingosine) that is combined with a fatty acid to produce complex lipids (ceramides). When one sugar is added to a ceramide, a cerebroside results. When a sulfate group is added to the sugar, the sphingolipid is a sulfatide. When a polysaccharide is added to the ceramide, the product is a globoside, while the addition of one or more molecules of N-acetylneuraminic acid to a globoside results in a ganglioside. N-acetylneuraminic acid (sialic acid) is always a terminal sugar. The metabolic disorders characterized by sphingolipid storage and their enzyme deficiencies are shown in Fig. 3. In these diseases (Table 5), a variety of lipids can be stored, including ceramides, cerebroside and other ceramide hexosides, sulfatides, sphingomyelins, gangliosides and lipofuscins.
Fig.2
Structure of sphingolipids. Sphingosine (top) plus a fatty acid forms ceramide (middle). Ceramide attached to a single sugar forms a glucocerebroside (bottom). If ceramide is combined with a polysaccharide (complex sugar) with one or more terminal N-acetylneuraminic acids, the result is a ganglioside.
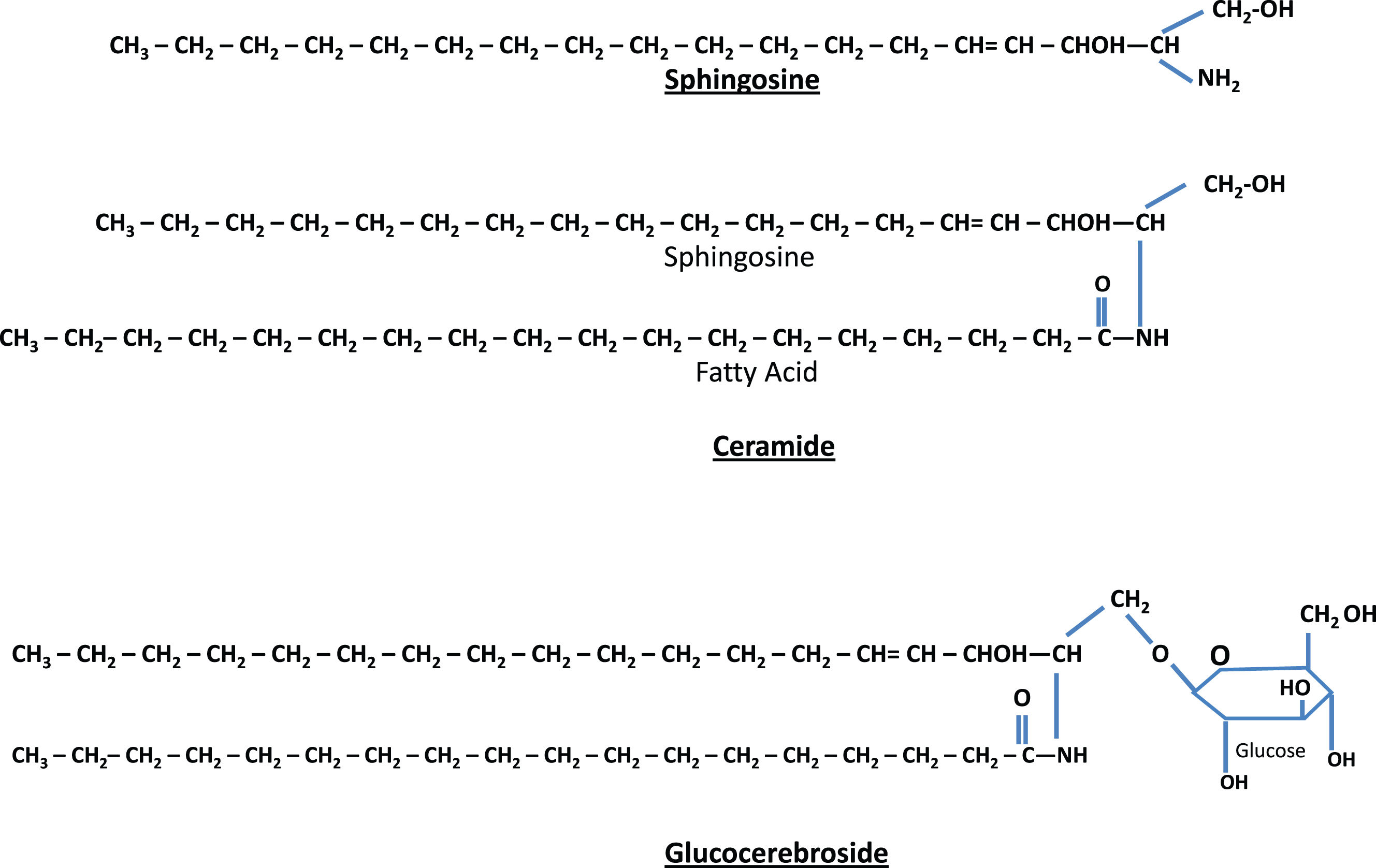
Fig.3
Metabolic disorders characterized by sphingolipid storage and their enzyme deficiencies. Notice accumulation of glucocerebroside in Gaucher disease, sphingomyelin in Niemann-Pick disease, galactocerebroside in Krabbe disease, sulfatide in metachromatic leukodystrophy, globotriaosylceramide in Fabry disease, and GM2 ganglioside in Tay-Sachs disease. In addition to GM2 ganglioside, patients with Sandhoff disease also accumulate globosides. GM1 ganglioside is stored in generalized β-galactosidase deficiency, while the H antigen accumulates in fucosidosis.
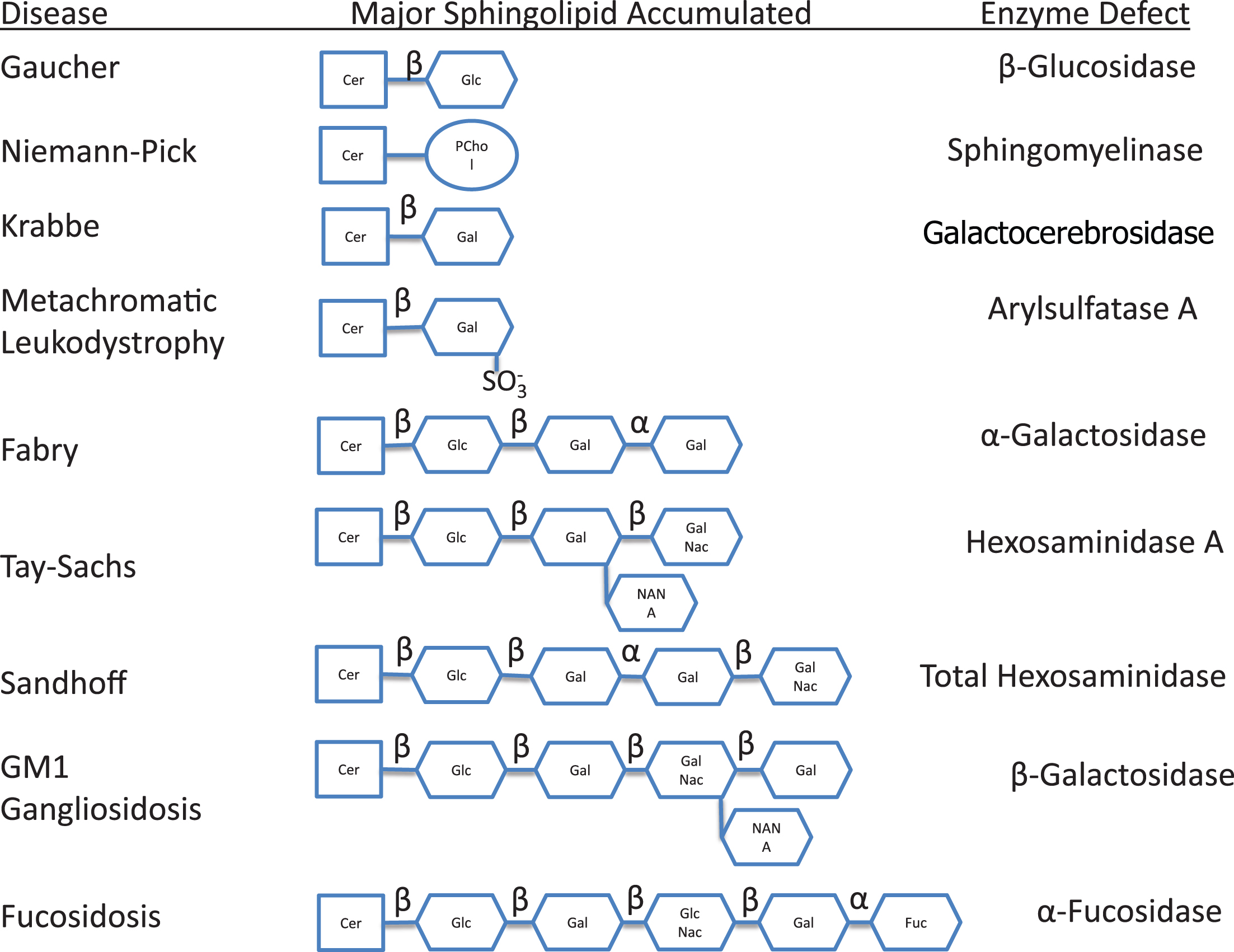
Table 5
Lysosomal lipid storage diseases
| MIM No. | Disease | Inheritance | Deficiency Enzyme | Clinicopathologic Features |
| 257200 | Niemann-Pick A (infantile cerebral type) | AR | Sphingomyelinase | Visceromegaly, cerebral deterioration, rapid course; storage in reticuloendothelial cells (foam cells), hepatocytes, bone marrow, lungs, peripheral and central neurons, Ashkenazi Jews |
| Niemann-Pick B (juvenile noncerebral type) | AR | Sphingomyelinase | Visceromegaly, lung infiltration; storage cells and sea-blue histiocytes; neurons not involved; milder | |
| 257220 | Niemann-Pick C (subacute juvenile or chronic neuronopathic type) | AR | Deficient esterification of exogenous cholesterol | Visceromegaly with storage cells, cerebellar ataxia, seizures, psychotic features; fibroblasts stain for cholesterol |
| 230800 | Gaucher I (adult type) | AD | Glucocerebrosidase (ß-glucosidase) | Hepatosplenomegaly, osteoporosis, fractures; survival to sixth decade; Gaucher cells are macrophages of spleen, liver, nodes, thymus, marrow; Ashkenazi Jews |
| 120900 | Gaucher IIA (infantile cerebral type) | AR | Glucocerebrosidase (ß-glucosidase) | Visceral disease similar to type I, rapidly progressive cerebral damage with brain macrophage cerebroside storage, but only discrete neuronal storage; short survival |
| Gaucher III | AR | Glucerebrosidase (ß-glucosidase) | Both visceral and progressive neurologic dysfunction; survival into second or third decades; storage cells as in Gaucher I and adventitia of vessels of cortex and cerebral white matter | |
| 245200 | Krabbe, infantile type short survival | AR | Galactocerebrosidase | Onset 4–6 mo with rapid, severe neurologic damage; storage “globoid body” cells in central white matter |
| 228000 | Farber Lipogranulomatosis | |||
| A. Early onset | AR | Ceramidase | Subcutaneous nodules, arthritis, laryngeal, cerebral | |
| B. Infantile type | AR | Ceramidase | Periarticular, no lung involvement, can have normal intelligence | |
| C. Late onset | AR | Ceramidase | Survival into second decade, laryngeal, joint but not lung involvement and less cerebral dysfunction | |
| Neonatal type | AR | Ceramidase | Psychomotor delays, hepatomegaly, debility | |
| 301500 | Fabry disease | XLR | α-Galactosidase A | Onset in males in first decade, later with slower course in female heterozygotes; nephrotic syndrome, renal failure, cardiomyopathy (esp. females), abdominal pain, angiokeratomas of skin; storage in glomerular and tubular epithelium, cardiac myocytes, enteric and brainstem/spinal cord neurons, blood vessel walls |
| 230500 | GM1 gangliosidosis type I (infantile) | AR | ß-Galactosidase | Survival 1-2 yr, abnormal facies, skeletal changes (esp. vertebrae), visceromegaly, cerebral deterioration; ganglioside/mucopolysaccharide in reticuloendothelial cells, liver, glomerular and tubular epithelium, pancreatic and mucoserous gland epithelium, central and peripheral neurons |
| 230600 | GM1 gangliosidosis type II (juvenile) | AR | ß-Galactosidase | Later onset; resembles type I |
| 230650 | GM1 gangliosidosis type III (adult) | AR | ß-Galactosidase | Adult onset; mental deterioration, visual loss, myoclonic seizures, angiokeratomas |
| 272800 | GM2 gangliosidosis, type I, infantile Tay-Sachs | AR | Hexosaminidase A (α-unit mutation) | Rapid dementia, blindness, macular cherry-red spot; Ashkenazi Jews |
| 272800 | GM2 gangliosidosis, juvenile Tay-Sachs | AR | Hexosaminidase A (α-unit mutation) | Later onset than infantile form |
| 272750 | GM2 gangliosidosis, Tay-Sachs AB form | AR | Hexosaminidase Activator Protein (Saposin) | Similar to infantile Tay-Sachs disease |
| 268800 | GM2 gangliosidosis type II, infantile Sandhoff | AR | Hexosaminidases A and B | Neuronal storage like that of Tay-Sachs disease with modest visceral storage |
| 230700 | GM2 gangliosidosis, juvenile Sandhoff | AR | Hexosaminidases A and B | Progressive dementia, ataxia |
| 230710 | GM2 gangliosidosis, chronic | AR | Hexosaminidases A and B | Spinocerebellar degeneration resembling Friedreich ataxia, juvenile spinal muscular atrophy resembling Kugelberg-Welander disease, or like amyotrophic lateral sclerosis. Chronic Sandhoff disease can resemble X-linked bulbospinal muscular atrophy |
| 250100 | Metachromatic leukodystrophy, infantile | AR | Arylsulfatase A (cerebroside sulfate sulfatase, or sulfatidase) | Onset ∼2 yr, survival to ∼5 yr; hypotonia, muscle weakness, mental deterioration, peripheral nerve involvement; sulfatide storage in liver, kidney, epithelium, rectal lamina propia, central white matter, nerves |
| Metachromatic leukodystrophy, juvenile | AR | Arylsulfatase A | Onset 4–10 years; slower course | |
| Metachromatic leukodystrophy, adult | AR | Arylsulfatase A | Onset second decade; psychiatric symptoms | |
| Metachromatic leukodystrophy | AR | Saposin B (Sulfatase Activator Protein 1) | Variable | |
| 272200 | Multiple sulfatase deficiency | AR | Deficiency of 7 sulfatases due to inability to convert a cysteine to 2-amino 3-oxopropionic acid | Resembles metachromatic leukodystrophy with ichthyosis and features of mucopolysaccharidosis; variable severity |
2.1Niemann-Pick disease (acid sphingomyelinase deficiency)
The disease was first described by Albert Niemann in 1914 in a child with hepatosplenomegaly, lymphadenopathy and progressive neurological deterioration who died before the age of 2 years [12]. Histopathological studies were later performed by Ludwig Pick, demonstrating the presence of foamy cells, similar but not identical to those found in Gaucher disease [13]. In 1934, Ernst Klenk identified the stored lipid as sphingomyelin [14]. In 1961, Crocker proposed classifying the disease into types A, B, and C [15]. It wasn’t until 1966 that Roscoe Brady found a deficiency of acid sphyngomyelinase in patients affected with Niemann-Pick disease type A and B, but not for type C [16].
The different types of Nieman-Pick disease (NPD) form a continuum of clinical severity [17]. The most common and severe variant is type A, the acute neuronopathic form. Affected individuals, often of Eastern European Jewish ancestry, present early in life. The skin can have a yellow-brown pigmentation, lymph nodes are enlarged, and ocular manifestations (cherry-red macula and corneal opacifications) are evident. Few patients survive beyond 4 years of age. The type B, or non-neuronopathic variant (allelic with type A) resembles type A but spares the CNS. Affected children present at a later age with isolated splenomegaly. In time, a more generalized visceral pattern of involvement is manifest, yet many patients survive several decades. The term NPD type E was used in the past to designate patients with an adult-onset, non-neuronopathic form of the disease with moderate hepatosplenomegaly; however, this designation has fallen out of favor. An NPD type F form was proposed for a relatively benign form characterized by the lack of neurological involvement and the presence of sea-blue histiocytosis [18]; this designation is also no longer in use.
In NPD B, a lipid pro-atherogenic profile has been described, with increased total and LDL cholesterol, increased triglycerides and low HDL cholesterol [19]. Mean splenic size is 12.7 multiples of normal, while mean liver size is 1.91 multiples of normal. Thrombocytopenia is seen in 39%, and leukopenia in 3%. There is stable elevation of liver transaminases and bilirubin, while pulmonary function deteriorates over time [19]. The typical pattern of pulmonary involvement is that of restrictive lung disease [20]. It can present with unexplained diffuse lipid pneumonia, even during adulthood [21]. Joint and/or limb pain is seen in 39% of patients [20]. Short stature, low weight and delayed bone age is also common [22]. There can be macular halo and/or cherry-red spots, but these do not portend neurodegeneration [23].
In both NPD type A and B, storage material is found in reticuloendothelial cells, hepatocytes [24], pulmonary macrophages, and syncytiotrophoblast and villus stromal cells [25]. Greenbaum et al. [26] documented that total extracts of brain glycolipids in NPD type A contain increased levels of glucosylceramide, dihexoside, and trihexoside, as well as GM2. In type A, there is mostly neuronal, less so vascular, sphingomyelin storage, while neutral glycolipid dominates in the vascular wall [27]. Hepatic storage of glycogen has been described in type B [28], and this has led to its misdiagnosis as a glycogen storage disorder [29].
Schoenfeld et al. [25] reported placental ultrasonographic, biochemical, and histochemical studies in human fetuses affected with NPD type A. Focal, opaque, strong echoes in placental tissue with thick and irregular chorionic plates occur as early as 18.5 weeks in placentae of NPD fetuses [25]. Prenatal diagnosis has been achieved by measuring sphingomyelinase activity in cultured amniotic fluid cells [30].
The gene whose mutations are responsible for NPD types A and B is SMPD1. More than 180 different mutations have been reported [31], and sequencing identifies a mutation in 95% of new cases. Three mutations— p.Arg498Leu, p.Leu304Pro, and p.Phe333Serfs*52— account for more than 90% of the mutant alleles in Ashkenazi Jewish patients [17]. Unlike NPD type A, NPD type B does not occur more frequently in the Ashkenazi Jewish population; more than half of patients reported with NPD type B are of Turkish, Arabic, or North African descent [32]. A single deletion, p.Arg610del, accounts for almost 90% of the mutant alleles in type B patients from North Africa [33]. Some degree of genotype-phenotype correlation has been identified [19, 32]. The prevalence of NPD types A and B combined is about 1 in 250,000 [17].
The diagnosis of types A and B NPD can be made readily by enzymatic determination of sphingomyelinase activity in cell and/or tissue extracts. However, heterozygote detection is unreliable by enzyme assay and requires molecular studies. Prenatal diagnosis has been accomplished by enzymatic and/or molecular analyses of cultured amniocytes or chorionic villi.
Although there is no specific therapy for type A or B NPD, bone marrow transplantation has corrected the metabolic defect and reduced the liver and spleen size. However, there has been no effect on the neurological status, and thus it is reserved for patients with NPD type B [34–36]. A recombinant human acid sphingomyelinase (olipudase alpha) is currently in clinical trials in the form of intravenous enzyme replacement therapy [37]. Retrovirus-mediated gene transfer corrects the metabolic defect in cultured NPD fibroblasts, raising the possibility of future treatment of type B NPD by somatic gene therapy [38].
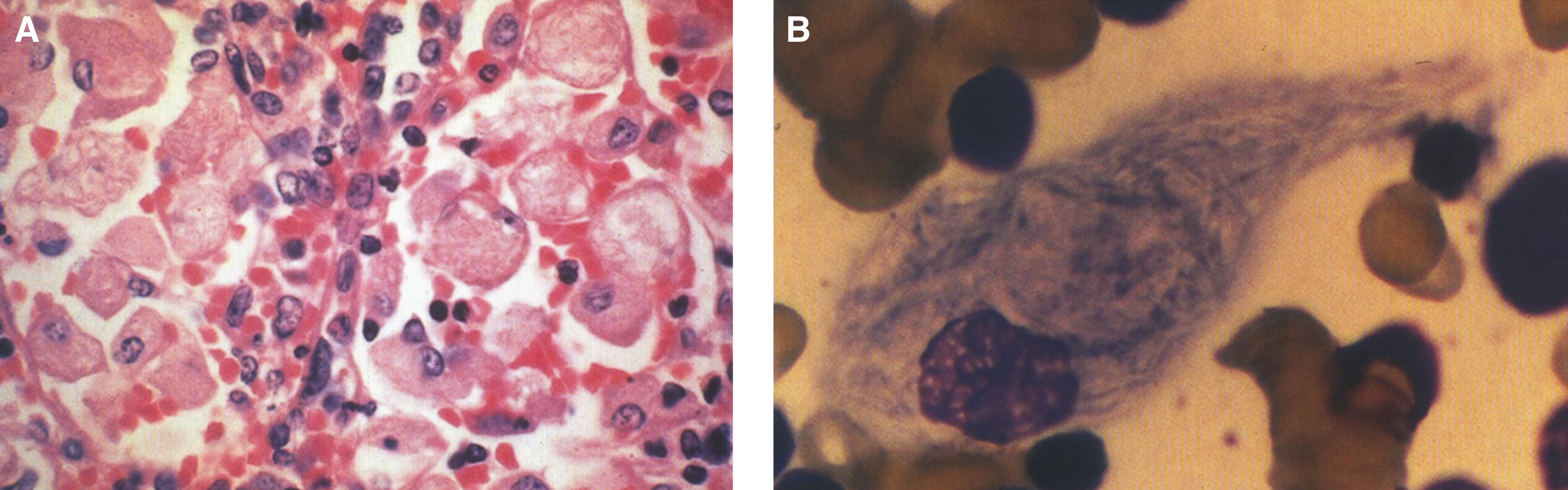
Pathologically, light (LM) and electron microscopic (EM) changes are similar in Niemann-Pick types A and B. The storage cell ranges in diameter from 20 to 90 μm, and the cytoplasm is filled with lipid droplets that impart a “soap bubble” appearance with the nucleus displaced to the periphery (Fig. 4). Cells in the viscera, predominantly liver (Fig. 5), spleen (Fig. 6), lung alveoli (Fig. 7), and ganglion cells of the myenteric plexus (Fig. 8) may contain lipofuscin pigment, sphingomyelin, and lesser quantities of cholesterol and gangliosides [11]. Storage is also present in the macrophages of the lung. The histopathological correlate of the cherry-red spot is that of ballooned, lipid-laden retinal ganglion cells [39]. The stored material is birefringent when examined under a polarized filter, unless lipid solvents have been used. The histochemical staining reactions confirm the presence of a complex phospholipid (Table 6). Differential staining characteristics of Gaucher, NPD, and the gangliosidoses are shown in Table 7. The NPD cells stain positively with oil red O, Sudan black (Fig. 9), Nile blue sulfate, and Luxol fast blue and the OTAN (osmium tetroxide α-naphthyl-amine) reaction after pretreatment with NaOH. The ultrastructural appearance of the lipid inclusions consists of concentrically laminated, myelin-like figures with a periodicity of approximately 50 Å, resembling membranous cytosomes and other pleomorphic lipid profiles. In the viscera, lipid inclusions are frequently membrane-bound and contain stacked membranes, concentrically laminated membranes, and pleomorphic profiles with both electron-dense and electron-lucent cores (Fig. 10) [40–43]. Cultured fibroblasts have similar inclusions and cultured amniotic cells contain storage material (Fig. 11).
Fig.4
Niemann-Pick disease. (A) A sea-blue histiocyte is present in the marrow. These cells are predominantly seen in types C and F. (B) A histiocyte in the bone marrow has a “soap-bubble” appearance and measures 60 to 80 μm in diameter.
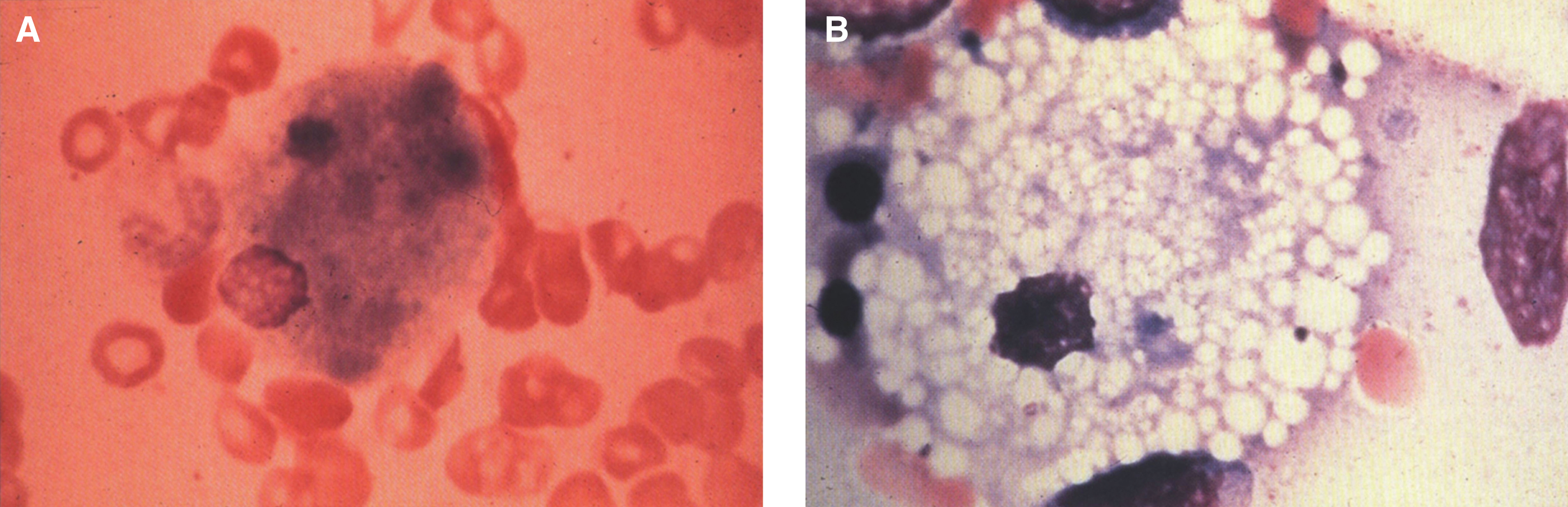
Fig.5
Niemann-Pick disease. Vacuolated storage cells are present in Kupffer cells within the sinusoids of the liver.
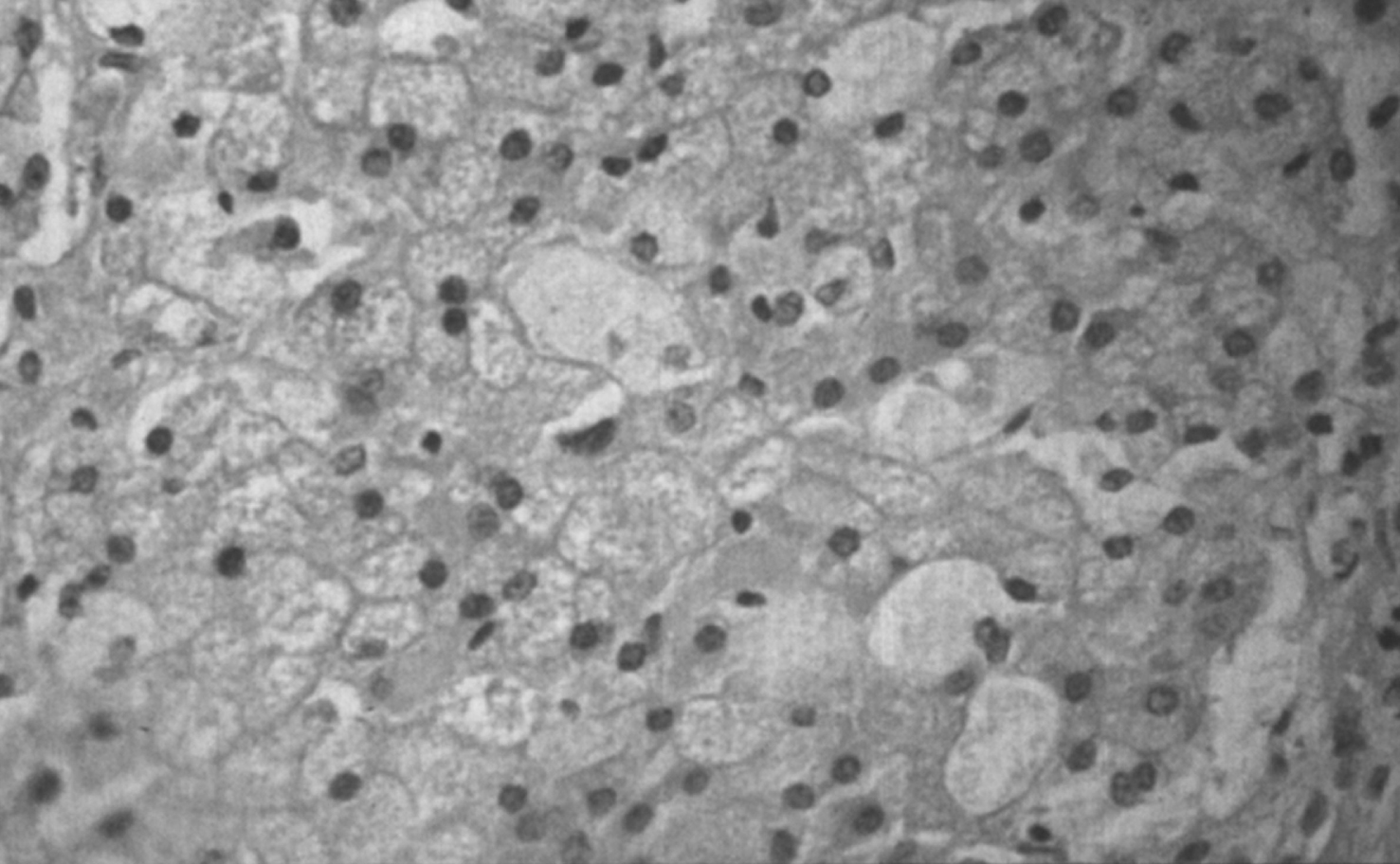
Figs. 6–8
Niemann-Pick disease. The splenic sinusoids are filled with distended vacuolated histiocytes; (7) Niemann-Pick disease. The alveoli of the lungs are filled with storage cells; (8) Niemann-Pick disease. The ganglion cells of the myenteric plexus of the gastrointestinal tract are distended with storage material (Luxol fast blue stain).
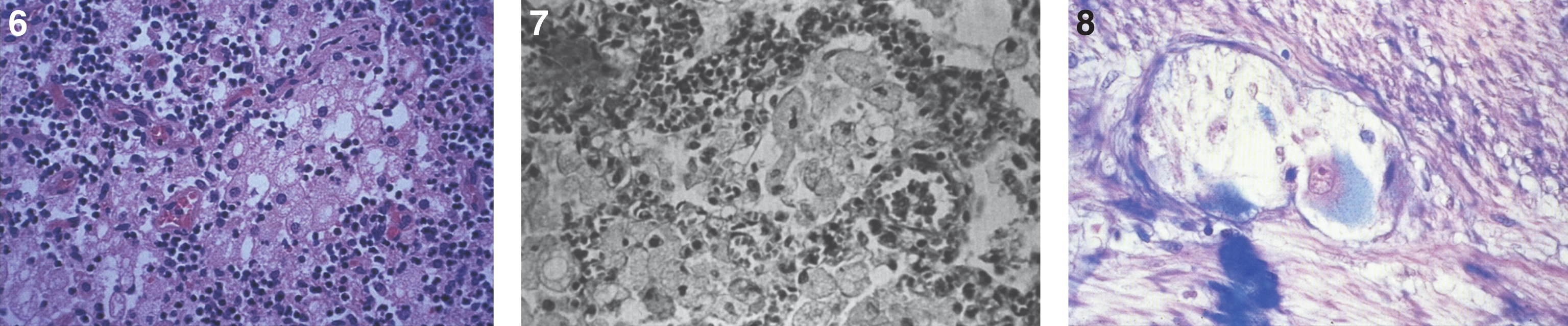
Table 6
Histochemical profile in Niemann-Pick disease
| Neural Tissue | Visceral Organs | Bone Marrow | |
| Luxol fast blue | +++ | ++ | ++ |
| PAS | ++ | +++ | +++ |
| After NaOH | ++ | +++ | +++ |
| After Kbr | ++ | +++ | +++ |
| Sudan black B | ++ | ++ | ++ |
| Oil red O | + | + | + |
| After hot acetone | + | + | + |
| After cold acetone | + | + | + |
| OTAN | +++ | +++ | +++ |
| NaOH-OTAN* | +++ | +++ | +++ |
| Ferric hematoxylin | ++ | ++++ | ++++ |
| After NaOH | ++ | ++++ | ++++ |
| Schultze reaction for cholesterol | + | ++ | ++ |
| AZO dye reaction for acid phosphatase | ++ | ++ | ++ |
| Autofluorescence | +++ | ++ | + |
PAS, periodic acid-Schiff; OTAN, osmium tetroxide α-napthylamine. *The most specific histochemical stain for sphingomyelin.
Table 7
Histochemical staining of three types of lysosomal lipidoses
| Gaucher Disease | Niemann-Pick Disease | Gangliosidoses GM1 and | |
| GM2 (Types 1 and 2) | |||
| PAS | +++ | 0 to + | ++ to +++ |
| PAS-amylase | +++ | 0 to + | +++ |
| Schultz cholesterol technique | 0 | ++ | – |
| Oil red O | + to ++ | +++ | ++ |
| Oil red O after cold acetone | + to ++ | +++ | ++ |
| extraction | |||
| Oil red O after hot acetone | 0 | +++ | + to ++ |
| extraction | |||
| Oil red O after pyrimidine | 0 | 0 | 0 |
| extraction | |||
| Luxol-fast blue | 0 | +++ | ++ |
| OTAN | 0 | +++ | ++ |
| NaOH-OTAN | 0 | +++ | 0 to + |
| Alcian blue | Adult 0 to +/infantile ++ | + | 0 |
| Acid phosphatase | ++ | 0 to + | ++ |
| Cells involved | RE cells (neuron in infantile) | RE cells (neuron in infantile) | Neurons |
| Biopsy tissues of choice | Spleen, marrow | Marrow or spleen | Nerve cells |
PAS, periodic acid-Schiff; OTAN, osmium tetroxide α-napthylamine; RE, reticuloendothelial.
Figs. 9–11
Niemann-Pick disease. Ganglion cells of the myenteric plexus stain strongly for lipid with Sudan black B stain; (10) Niemann-Pick disease. Electron micrograph of a storage cell in the spleen contains pleomorphic lipid profiles; (11) Niemann-Pick disease. Electron micrograph of a cultured fibroblast contains pleomorphic lipid profiles and electron-dense deposits.
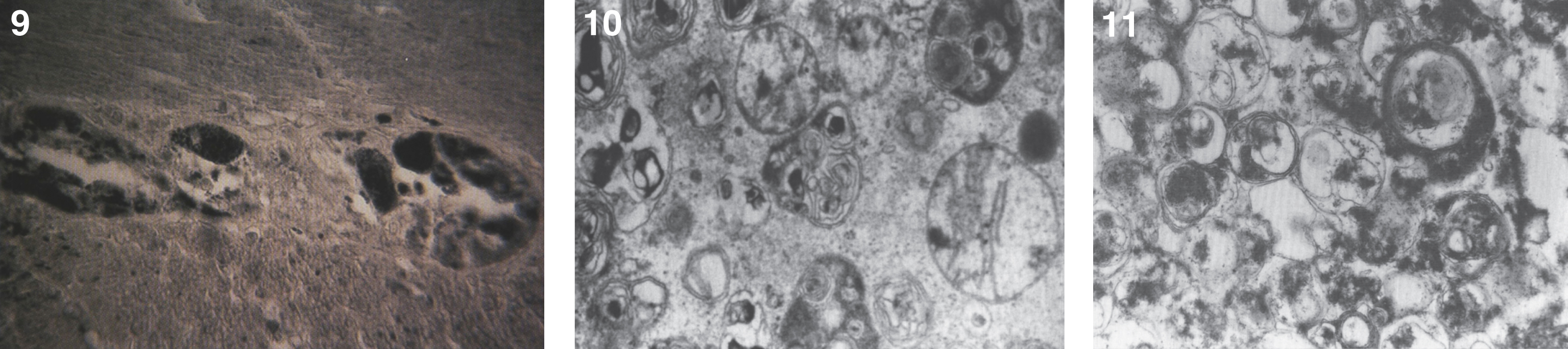
The brain is large (Fig. 12) during the first 6 to 12 months of life because of the accumulation of sphingolipids and gangliosides with membranous concentric bodies (MCBs) (Fig. 13). The neurons are distended (Fig. 14). Later the brain becomes atrophic with enlargement of the ventricles (Fig. 15).
Figs. 12–14
Niemann-Pick disease. The brain is large during the first years of life due to the accumulation of storage material; (13) Niemann-Pick disease. Electron micrograph of a neuron shows membranous concentric bodies of gangliosides; (14) Niemann-Pick disease. Microscopic section of the brain shows large distended neurons.

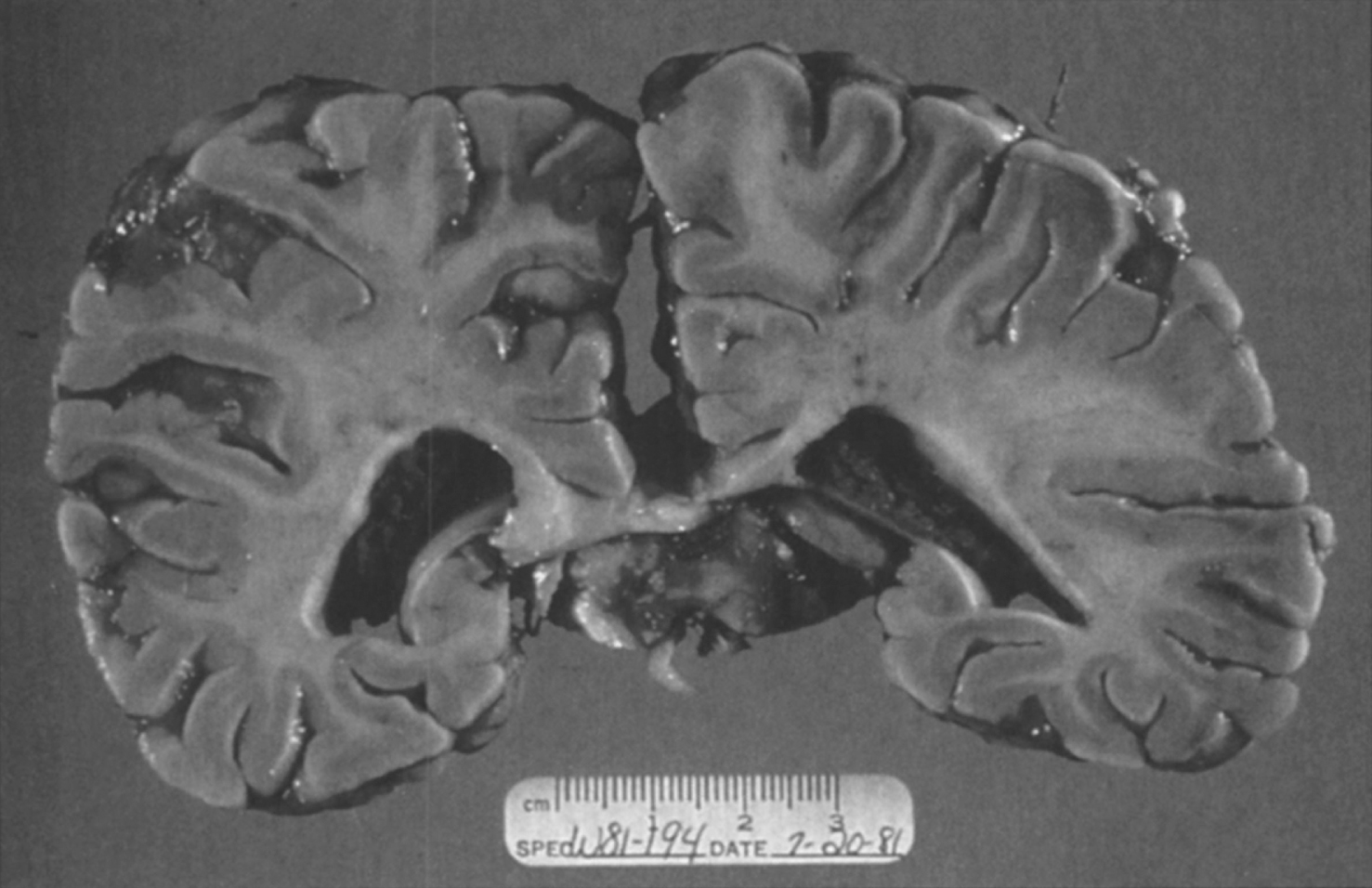
Fig.15
Niemann-Pick disease. A child 2 years of age. The brain is atrophic with enlargement of the ventricles.
The placenta may be enlarged, and there is vacuolation and histochemical evidence of storage in syncytiotrophoblast and villus stromal cells [25]
Many of the lipids that accumulate in NPD are not substrates for sphingomyelinase, including glucocerebroside, GM2, and GM3. The predominant lipid in NPD is phospholipid, but sphingomyelin, cholesterol, bis-(monoacylglycerol) phosphate, and glycosphingolipid also accumulate.
2.2Niemann-Pick disease type C
Niemann-Pick disease type C (NP-C) is not a primary acid sphingomyelinase deficiency, but was given its moniker because sphingomyelinase activity is secondarily reduced. NP-C is an autosomal recessive lipidosis distinguished by a unique error in cellular trafficking of cholesterol that is associated with lysosomal accumulation of unesterified cholesterol. Most patients with NP-C have progressive neurologic disease, and hepatic damage is prominent in certain cases [44]. There is lysosomal accumulation of low-density lipoprotein (LDL)-derived cholesterol, and glycolipid is stored in neurons. At least some cases of lactosylceramidosis, previously thought to be a distinct condition, are now known to be caused by NP-C [45].
There are two known genes in which biallelic mutations result in NP-C. NPC1, responsible for 90% of cases of the disease, contains 25 exons and encodes a 1278 amino acid protein. More than 390 different mutations have been described. The NPC1 protein has sequence similarity to the morphogen receptor PATCHED and the putative sterol-sensing regions of SREBP cleavage-activating protein (SCAP) and ß-hydroxy-ß-methylglutaryl coenzyme A reductase [46]. Niemann-Pick type D, or the Nova-Scotian variant, was found to be caused by a specific mutation in NPC1, and thus this designation is no longer in use [47]. NPC2, with 5 exons encoding a 132-amino acid glycoprotein, accounts for 4% of cases of NP-C. Approximately 20 mutations have been found in the NPC2 gene [48]. Genotype-phenotype correlations have been described for both the NPC1 and NPC2 genes [50].
The clinical manifestations of NP-C are heterogeneous. The classic phenotype is characterized by variable hepatosplenomegaly, vertical supranuclear ophthalmoplegia, progressive ataxia, dystonia, and other neurologic symptoms like cataplexia, ataxia, myoclonus, seizures, and loss of previously acquired speech at about 5 years of age. Organomegaly is less pronounced than in types A and B, and may be lacking altogether. Affected children present in childhood and die in the second decade. Neonates can present with ascites and meconium ileus and have developed biliary atresia or, more commonly, a neonatal hepatitis syndrome [51]. Other phenotypes include early infantile onset with hypotonia and delayed motor development, and adult variants where psychosis and dementia predominate [52]. Patients with NP-C2 typically have marked pulmonary involvement, leading to respiratory failure and early death [50].
The diagnosis of NP-C now relies upon molecular testing; filipin-cholesterol staining in cultured fibroblasts during LDL uptake can also be performed by some laboratories. There is considerable variability in the degree of impairment of cholesterol esterification in NP-C. About 15% of patients show a “variant” biochemical phenotype, with an intermediate-to-normal rate of cholesterol esterification, and a less distinctive filipin staining [53]. Elevated levels of oxysterols in plasma–in particular cholestane-3β,5α,6β-triol and 7-ketocholesterol-as measured by LC-MS/MS can also be used as a diagnostic tool, both in patients with NP-C1 [54] and NP-C2 [48]. Antenatal diagnosis can be achieved through molecular diagnostics. NP-C is estimated to be as frequent as NP-A and NP-B combined, with a prevalence of about 1 in 150,000 [44].
Various drug regimens lower hepatic cholesterol levels in NP-C. It is not known if such treatment influences neurologic progression [55]; one drug currently in clinical trials is 2-hydroxypropyl-β-cyclodextrin [56]. Symptomatic treatment of seizures, dystonia, and cataplexy is effective in many patients with NP-C. Bone marrow transplantation has been performed in patients with NP-C2, leading to resolution of the lung disease, although it can be associated with a severe “graft-versus-substrate” effect requiring intense immunosuppression until resolution [57].
The pathology of NP-C includes the presence of foam cells or sea-blue histiocytes in many tissues. Such cells are not specific for NP-C and may be absent in cases lacking visceromegaly. Neuronal storage with cytoplasmic ballooning and a variety of inclusions is present throughout the nervous system. Meganeurites and axonal spheroids are also seen. Neurofibrillary tangles and neuropil threads composed of tau protein can be detected by Bielschowsky stain or immunohistochemistry [58, 59]. The former vary in shape and size, unlike the torch-like or flame-shaped inclusions seen in patients with Alzheimer’s disease [59]. Characteristic inclusions may be identified in skin and conjunctival biopsies [60–62]. Unesterified cholesterol, sphingomyelin, phospholipids, and glycolipids are stored in excess in the liver and spleen, while only glycolipids are elevated in the brain. Marked storage can be seen as early as in a 20-week fetus [63]. Partial sphingomyelinase deficiency, observed only in cultured cells, represents a variable, secondary consequence of lysosomal cholesterol esterification.
2.3Gaucher disease
In 1882, while just a 27-year-old intern, Phillippe Gaucher presented his MD thesis, in which he described a patient with splenomegaly due to infiltration by abnormal cells. He had speculated that this was a primary splenic epithelioma [64]. Similar cases were subsequently published, but it wasn’t until the first decade of the twentieth century that the eponym Gaucher disease started being used [65]. The underlying enzyme deficiency was identified by Roscoe Brady in 1965 [66].
Gaucher disease, the most common of the lysosomal storage diseases, is an autosomal recessive disorder due to deficiency of glucocerebrosidase (acid β-glucosidase), which converts glucocerebroside to ceramide and glucose [67, 68]. As a consequence of this defect, glucocerebroside (GL1), an intermediate in the catabolism of globoid and gangliosides, accumulates in the reticuloendothlial system (Fig. 16).
The gene encoding acid β-glucosidase, GBA, resides on chromosome 1q21-q22 and has 11 exons; more than 400 mutations have been described [31]. Most of the Gaucher disease alleles are missense mutations that lead to the synthesis of acid β-glucosidases with decreased catalytic function and/or stability. Nonsense and frameshift mutations have also been reported. Four mutations, N370S, 84GG, IVS2+1G>A, and L444P, are responsible for 90% of the mutant alleles in type 1 Gaucher disease in Ashkenazi Jews and over 50% in non-Jewish patients with type 1 Gaucher disease [67]. The clinical manifestations of Gaucher disease are relatively mild in N370S homozygotes, and do not include neurological findings. Homozygotes for L444P, a common mutation also encountered in a geographical isolate in Northern Sweden, generally present with type 3 disease of variable severity.
Fig.16
(A) The pathophysiology of Gaucher disease. It should be noted that this macrophage-centric view of the disease has recently been called into question, since it does not explain certain aspects of the disease such as the predisposition to malignancy, osteoporosis or Parkinson disease (108). (B) The enzymatic defect in Gaucher disease. (From Gaucher Disease Diagnosis Evaluation and Treatment, Genzyme Therapeutics, Parsipanny, NJ; with permission.)
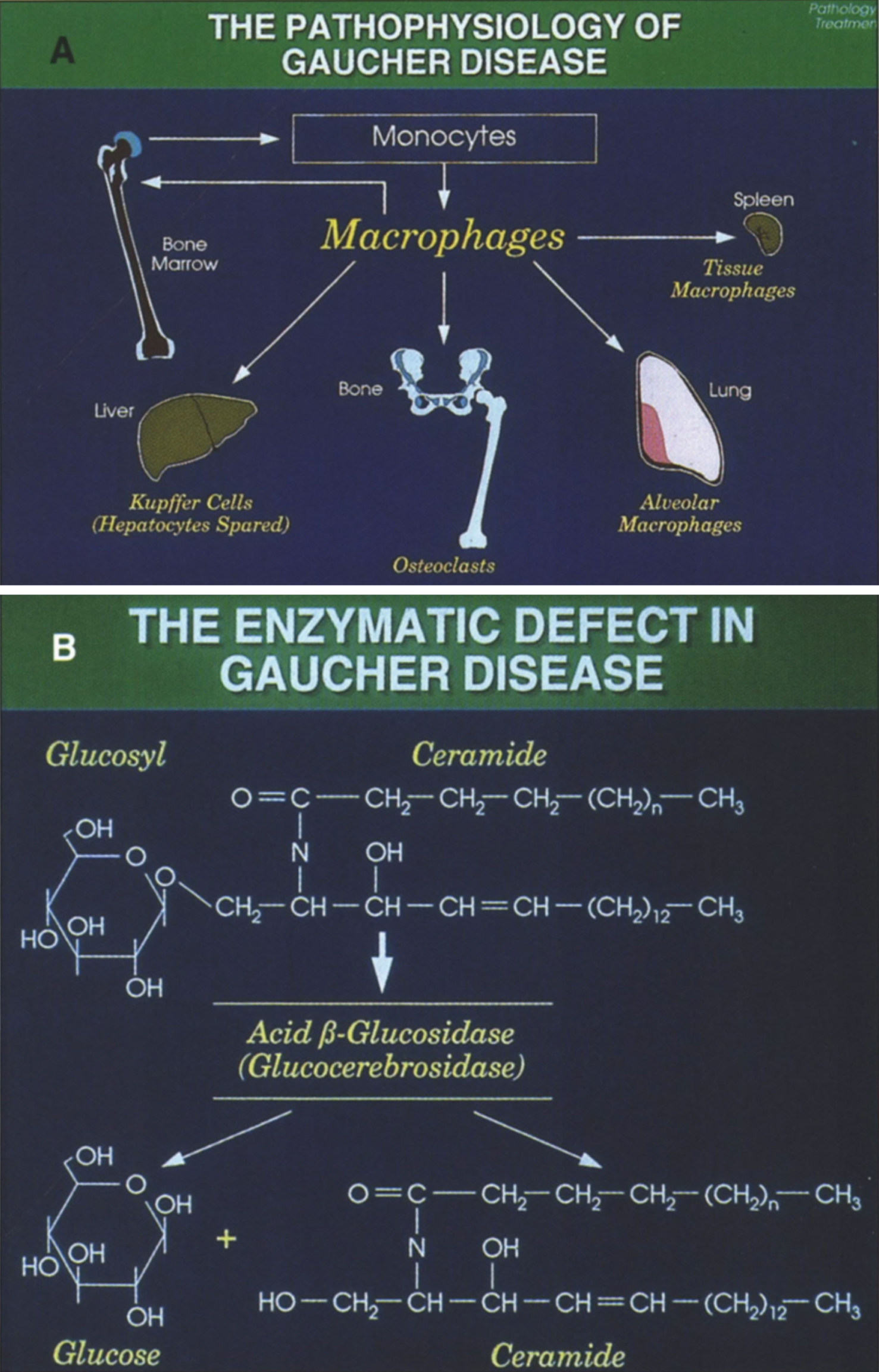
A pseudogene that has maintained a high degree of homology is located approximately 16 kb downstream of the active gene. Several mutations appear to have originated from pseudogene sequences; some appear as fusion genes between the gene and pseudogene [69], or as part of other recombinant alleles that contain varying lengths of additional pseudogene sequences [70].
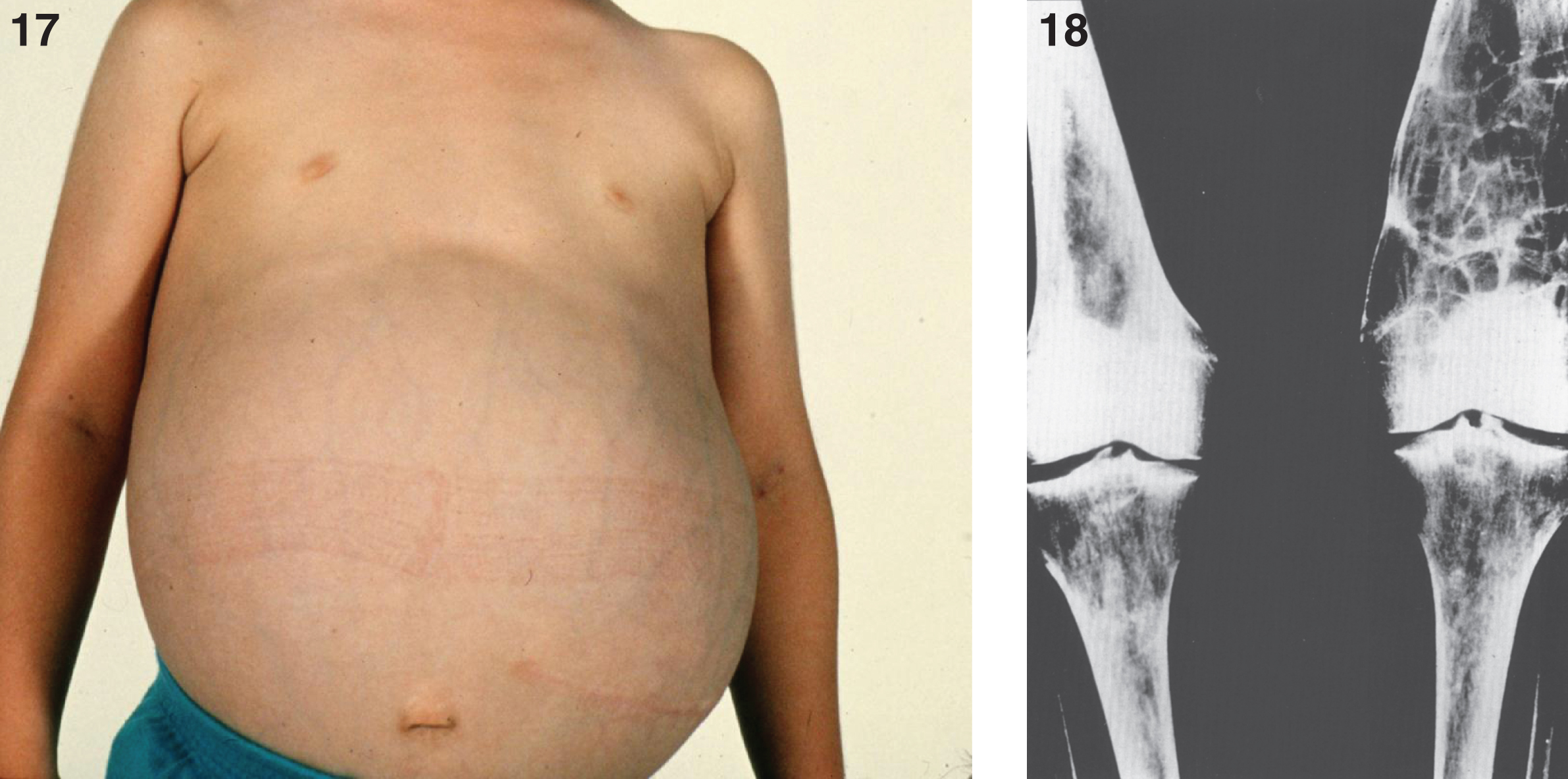
Three types of Gaucher disease (Table 8) reflect the degree of β-glucocerebrosidase deficiency [67, 68], although hepatosplenomegaly, bone lesions, and occasional involvement of lungs and other organs occur in all forms of Gaucher disease. Type 1, by far the most common form, occurs in the Ashkenazi Jewish population with a frequency of approximately 1 in 855 [67], and is distinguished from types 2 and 3 disease by the lack of primary CNS involvement, with the exception of Parkinsonism in later life. The average age at diagnosis for type 1 is 30–40 years, although patients are commonly diagnosed in childhood or adolescence. The chronic manifestations of the disease vary considerably in severity, and Gaucher disease does not always breed true [71]. Milder patients present as adults, and a significant number of individuals with type 1 disease never come to medical attention [72]. The systemic manifestations include hepatosplenomegaly (Fig. 17), thrombocytopenia, anemia, leucopenia, osseous abnormalities, delayed sexual maturation and growth retardation. A radiographic finding of an Erlenmeyer flask deformity of the distal ends of the femur (Fig. 18) is common in Gaucher disease [73]. Renal involvement, pulmonary hypertension, and cardiac abnormalities are less frequent. Patients with type 1 disease can have a normal life expectancy, although an increased risk of malignancy, especially multiple myeloma and hepatocellular carcinoma, has occurred in late adulthood [74].
Table 8
Gaucher disease: Clinical types
| Clinical Features | Type I-Chronic, non- | Type II-Acute, | Type III-Subacute, |
| neuronopathic | neuronopathic | neuronopathic | |
| Age at presentation | Childhood/Adulthood | Infancy | Childhood/Adulthood |
| Splenomegaly | + ⟶ +++ | ++ | + ⟶ +++ |
| Hepatomegaly | + ⟶ +++ | ++ | + ⟶ +++ |
| Skeletal Disease | –⟶+++ | – | ++ ⟶ +++ |
| Primary CNS Disease | Absent | +++ | + ⟶ +++ |
| Lifespan (typically) | 6–80+ years | <2 years | 2–60 years |
| Ethnicity | Panethnic; Ashkenazi Jews | Panethnic | Panethnic; Norbottnian Swedes |
| Frequency | 1/450–1/1000 Ashkenazi Jewish | <1/100000 | <1/50000 |
| 1/40000 – 1/2000000 General Population |
Adapted from Gaucher Disease: Diagnosis Evaluation and Treatment, Genzyme Therapeutics, with permission. (Genzyme Therapeutics, 100 Lackawanna Ave., Parsippany, NJ 07054; Ph: 800/745-4447, ext. 7664; Fax: 617/374-7357; www.genzyme.com/prodserv/welcome.htm.).
Figs. 17,18
Gaucher disease type I. A child 10 years of age showing marked distension of the abdomen due to massive splenomegaly. The spleen was surgically removed and weighed 10 kg; (18) Gaucher disease. Radiograph of lower extremities showing Erlenmeyer flask deformity of the distal ends of the femur.
Type 2 Gaucher disease, the acute neuronopathic form, has an early onset with severe CNS involvement and death usually within the first 2 years of life. It has no ethnic predilection. There are severe neurologic complications and signs of cranial nerve nuclei and extrapyramidal tract involvement can appear at birth or by 6 months of age. Although neuronal cerebroside storage is not a feature, the brain is the site of extensive neuronal cell death, reactive gliosis, and the perivascular accumulation of Gaucher cells. Clinical manifestations include strabismus, oculomotor apraxia, trismus, dysphagia, retroflexion of the head, cortical thumbs and limb rigidity, increased deep tendon reflexes with a positive Babinski sign, pulmonary infections, and failure to thrive. Some patients have seizures. The presence of hepatosplenomegaly in an infant with oculomotor abnormalities and retroflexion of the neck is highly suggestive of type 2 Gaucher disease. Estimates of the incidence of type 2 Gaucher disease range between approximately 1 in 100,000 and 1 in 500,000 births [75].
Therapy of type 2 Gaucher disease has primarily been supportive.
A variant of type 2 Gaucher disease also displays congenital ichthyosis and/or hydrops, sometimes with joint abnormalities [76].
Patients with type 3 (subacute neuropathic) Gaucher disease have neurologic symptoms with a later onset and a more chronic course than that observed in type 2 disease. The median age of onset is one year, with considerable variability. The most common neurologic manifestation is the slowing and looping of the horizontal saccadic eye movements. Some patients may develop ataxia, spasticity, akinetic and myoclonic seizures, and variable degrees of dementia while others only have eye movement abnormalities or learning disabilities. A variant known as type 3c presents with cardiovascular calcifications, including aortic valve, mitral valve, and ascending aorta calcifications. It can also be accompanied by corneal clouding, hydrocephalus and supranuclear ophthalmoplegia. Gaucher disease type 3c is caused by homozygous D409H mutations [77–79].
Recently, Gaucher disease has been associated with Parkinsonism, and even heterozygosity for GBA mutations carries an increased risk for it [80–82]. The penetrance of Parkinsonism in carriers increases with age [83, 84]. In fact, mutations in GBA are now the most common known genetic risk factor for Parkinson disease. However, the majority of adults with Gaucher disease never develop Parkinson disease.
Skeletal manifestations are a major source of disability in Gaucher disease, affecting over 80% of symptomatic patients and resulting in serious complications in many (Table 9). Early assessment and routine monitoring of skeletal involvement, even in asymptomatic patients, are important since changes in bone are progressive and in some cases, may be reversed by enzyme replacement therapy. Several imaging modalities are currently used to evaluate bone disease: plain radiography, magnetic resonance imaging (MRI), MR quantitative chemical-shift imaging (QCSI), computed tomography (CT), and nuclear scans.
Table 9
Bone radiographic findings in Gaucher disease
| •Femoral head and femoral shaft are most frequently involved |
| •The “Erlenmeyer flask” deformity of the distal femur is a common finding |
| •Vertebral bodies, humeral heads, and the ischium may be involved |
| •Vertebral bodies can collapse, creating a gibbus and spinal cord compression |
| •Marrow replacement occurs |
| •Osteopenia can lead to fractures |
| •Bone lesions display focal deposits of Gaucher cells |
| •Lytic lesions may simulate bone tumors and may require biopsy |
The diagnosis of Gaucher disease (Table 10) should be considered in any patient with unexplained splenomegaly. The diagnosis is made by finding deficient glucocerebrosidase activity in leukocytes, dried blood spots, or fibroblasts, i.e., 0–35% of normal depending upon the laboratory [67, 68]. Reliable detection of carriers by enzyme assay is more difficult, as there is considerable overlap between the glucocerebrosidase activity of normal individuals and that of heterozygotes, with up to 20% of obligate heterozygotes possessing enzyme activity in the normal range [33]. Enzyme assay for heterozygote detection is being superseded by molecular techniques that can efficiently detect heterozygotes once the mutations of an affected patient are known. More than 90% of mutations in Ashkenazi Jews can be detected by screening for the most common mutations, and approximately 50% of the mutations in non-Jewish populations can be detected in this manner. However, the exact percentage depends upon the number of common mutations being screened, and upon the population’s ethnicity. In utero diagnosis of Gaucher disease can be made by applying enzyme assay techniques or mutation analysis to cultured amniocytes [24].
Table 10
Diagnosis of Gaucher disease
| •Glucocerebrosidase assay (on leukocytes, DBS, fibroblasts, amniocytes, chorionic villi) is |
| the gold standard. |
| •The assay uses a fluorescent substrate; less than 30% activity makes the diagnosis. |
| •Heterozygotes average 50% of normal β-glucocerebrosidase activity. |
| •20% of carriers demonstrate enzyme activity in the normal range. |
| •DNA screens for selected GBA mutations may detect common mutations but can lead |
| to ambiguous results when variants of unknown significance are found. |
| •DNA analysis is better for heterozygotes if the mutation is known. |
| Associated Findings |
| •Elevated serum acid phosphatase, chitotriosidase and angiotensin-converting enzyme |
| •Elevated plasma ferritin levels are commonly seen |
| •Decreased plasma cholesterol levels in unsplenectomized patients |
The treatment of Gaucher disease can be divided into therapy directed at the basic defect and supportive care. Directed therapy [67] includes enzyme replacement, which has been extremely successful for systemic manifestations, substrate reduction with miglustat or eliglustat, and bone marrow transplantation [85, 86]. Cell and gene therapy have been effective in a mouse model [87, 88]. Recombinant human glucocerebrosidase is modified for enhanced uptake by macrophages. The enzyme is given by periodic intravenous infusions according to various dosing schedules, and is well tolerated albeit extremely expensive with infusions costing several hundred thousand dollars per patient per year. Approximately 15% of patients develop antibodies, depending upon the preparation, but most are not neutralizing. Some individuals can develop urticaria and pruritus related to the infusions, treatable with antihistamines. Enzyme replacement improves quality of life, reduces hepatomegaly, hypersplenism, anemia, thrombocytopenia, and secondary bone marrow failure [67]. It is also reported to reduce the rate of bone loss and bone crises [89] and to stabilize myelofibrosis and cirrhosis. However, no neurological benefit has been proven. Supportive therapy for Gaucher disease involves treating bone crises, the hypersplenism and its consequences, and chronic pain. Table 11 lists some supportive measures.
Table 11
Supportive therapy modalities
| Bone Crises |
| •Intravenous hydration and administration of narcotics |
| •Cultures and antibiotics for osteomyelitis if indicated |
| •Orthopedic procedures (joint replacement) |
| •Analgesics for bone pain |
| •Restriction of activity |
| Hypersplenism |
| •Rarely, splenectomy may be indicated for severe thrombocytopenia and anemia |
| •Transfusions |
| Supplemental treatment |
| •Oral bisphosphonates and vitamin D |
Fig.19
Gaucher disease. (A) Microscopic section of the spleen. The sinusoids are filled with large distended storage cells. (B) The bone marrow contains a large Gaucher cell with cytoplasmic striations with typical “crinkled tissue paper” appearance.
In type 2 Gaucher disease, it appears that the pathology begins in the placenta and fetus. The placenta may be edematous, and storage material has been found within Hofbauer (placental histiocytes) cells and in cells within intravillus vessels [90]. Fetuses at 17 and 20 weeks’ gestation also have storage cells, particularly in the liver and spleen, but do not demonstrate neuropathologic changes. In contrast [91], a 26-week-gestation fetus has shown extensive neuronal loss and gliosis in those areas with the most mature neurons, i.e., spinal cord, brainstem, thalamus, and basal ganglia [91]. The fetal Gaucher cell may not have well-delineated striations as it does in the mature patient. Instead, it may appear foamy, and yet tubular cytosomes are still identifiable by EM. The association of Gaucher disease and hydrops fetalis has been observed in several cases [70, 90–92]. Gaucher cells, present in abundance in hepatic sinusoids and filling the lumens of hepatic veins, may contribute to the fetal ascites.
Table 12
Pathologic findings in Gaucher disease
| SPLEEN-Gross |
| •May be more than 20 times the normal size and have hard texture and surface nodules |
| •Range from deep red (normal) to purple (extramedullary hematopoiesis) to yellow (old infarcts) |
| SPLEEN-Microscopic |
| •Accumulation of Gaucher cells |
| •Fibrosis |
| •Infarcts that account for up to 25% – 50% of a massively enlarged spleen |
| LIVER-Gross |
| •Yellow-brown discoloration |
| •Areas of extramedullary hematopoiesis |
| •Nodules may be present in areas of infarction or Gaucher cell infiltration |
| LIVER-Microscopic |
| •Gaucher cells in the sinusoids and in parenchymal nodules |
| •Fibrosis may be present |
| CENTRAL NERVOUS SYSTEM (CNS) |
| •Spinal cord compression secondary to vertebral collapse |
| •Bleeding due to coagulopathies can cause CNS damage |
| •In type 2 Gaucher disease, Gaucher cells can be seen within the brain parenchyma, especially within occipital lobes |
| including the Virchow Robin spaces of the cortex, deep white matter, gray matter of the thalamus and subependymal |
| tissue of the pons and medulla |
| •Neuronophagia is prominent in the cortex, midbrain nuclei, basal ganglia, brainstem, and dentate nucleus |
| •Neuronal loss is widespread in type 2; the dentate nucleus is severely involved as well as hipocampal layers CA2-4. |
| •PAS-positive inclusions may be seen |
| HEMATOLOGIC FINDINGS |
| •Bleeding secondary to thrombocytopenia, factor XI or factor IX deficiency |
| •Thrombocytopenia due to splenic sequestration; responds to splenectomy |
| •Anemia (normocytic, normochromic); usually mild, with hemoglobin > 8 mg/dL but can be severe |
| •Marrow replacement |
| •Leukopenia |
| •Acquired von Willebrand factor deficiency |
| •Gaucher cells in marrow |
| •Increased iron storage |
| •Increased incidence of multiple myeloma |
| •Necrosis, yellow discolored areas of bone marrow replacement |
| LUNG |
| •Rarely, pulmonary failure may result from infiltration by Gaucher cells, or right to left shunting |
| •Pathology can be interstitial infiltration, alveolar consolidation, or capillary plugging by Gaucher cells |
| •Pulmonary hypertension can develop |
| OTHER PATHOLOGIC FINDINGS |
| •Osteoporosis |
| •Lymph node involved with Gaucher cells |
| •Thymus, Peyer patches, adenoids, and tonsils can be involved |
| •Brown masses of Gaucher cells have been reported in the eye at the corneoscleral limbus |
| •Gaucher cells have been found in a colonic polyp and the maxillary sinus |
| •Type 2 patient autopsies show severe infiltration of the adrenal gland |
| •Rare Gaucher cells have been found in the renal glomerular tufts and renal interstitium |
| •Tubular inclusions have been seen in endothelial cells lining glomerular and interstitial capillaries |
| BIOCHEMICAL ABNORMALITIES |
| •Marked deficiency of lysosomal glucocerebrosidase in leukocytes, fibroblasts, or tissues |
| •Elevated plasma tartrate-resistant acid phosphatase |
| •Decreased or elevated plasma cholesterol |
| •Increased plasma angiotensin converting enzyme |
| •Increased plasma chitotriosidase |
| •Increased plasma glucocerebroside and glucosylsphingosine |
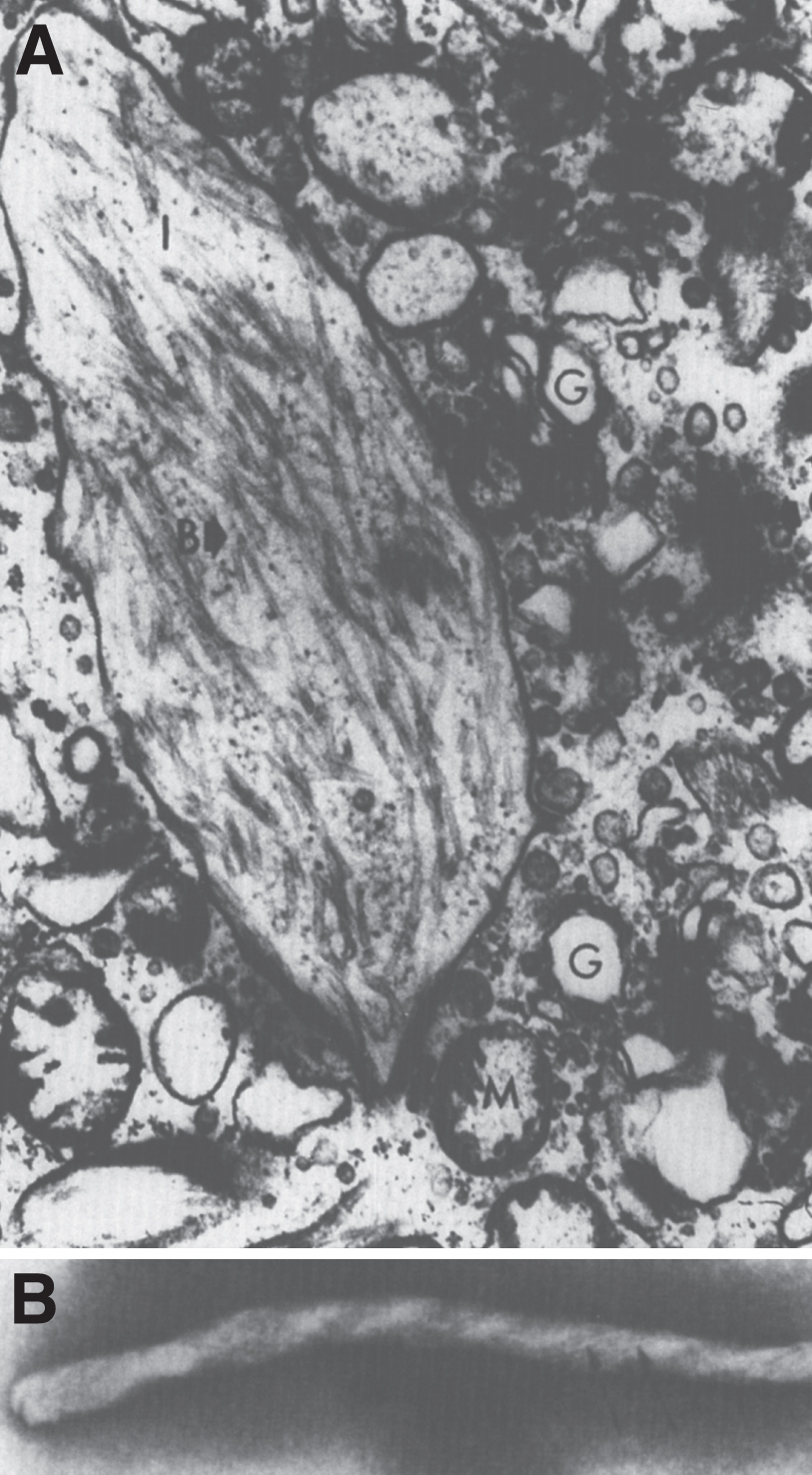
Postnatal pathology in Gaucher disease is characterized by the accumulation of glucocerebroside in histiocytes of the reticuloendothelial system including spleen (Fig. 19A), lymph nodes, bone marrow, and Kupffer cells of the liver (Table 12). Late in the disease, portal fibrosis may progress to cirrhosis. The pathognomonic feature is the Gaucher cell–a large vacuolated histocyte seen in the bone marrow, liver, spleen, and lymph nodes with striated cytoplasm (Fig. 19B) best demonstrated with trichrome or aldehyde fuchsin staining [93]. Gaucher cells are 20 to 100 μm in size and have a small eccentric nucleus with a “wrinkled tissue paper” or “crumpled silk” appearance to their cytoplasm. They stain positively with the periodic acid-Schiff (PAS) reagent. Most give a strongly positive reaction for acid phosphatase. The appearance of the cell is distinctive enough to permit its use as a diagnostic criterion but bone marrow biopsy should not be used as a method of diagnosis. The macrophages of other sphingolipidoses, such as Niemann-Pick disease, possess multiple intracytoplasmic droplets of uniform size, giving them a foamy rather than crumpled silk appearance. Cells resembling Gaucher cells may be found in multiple myeloma, leukemias, thalassemia, and congenital dyserythropoietic anemia. However, Gaucher cells can be distinguished from these so-called pseudo-Gaucher cells by their much stronger expression of HLA-DR on immunohistochemistry [94]. Erythrophagocytosis by Gaucher cells is common, and hemosiderin is frequently found in the cells. Ultrastructurally, the Gaucher cell cytoplasm is packed with rod-shaped inclusions bound by a single membrane. The inclusions contain tubules that run parallel to their long axes and are branched. The tubules have a clockwise spiral (Fig. 20). This membrane-bound cytoplasmic vacuole, containing tubular structures, is known as the “Gaucher body”. The iron dispersed in the cells is seen by EM and stains positively by the Prussian blue stain; individual micelles of ferritin are present [95]. In the bone marrow, large collections of Gaucher cells replace the marrow cells. In the liver [96], the accumulation of the glycolipid occurs within the Kupffer cells and the hepatocytes are spared. In the lungs, alveolar involvement by Gaucher disease can occur, but rarely. Gaucher cells are also frequently observed in both the cortical and medullary portions of the thymus and adrenal gland. Lymph nodes and the lamina propria of the gastrointestinal tract may be involved [97]. Lipid analyses of spleens and livers from patients with Gaucher disease have exhibited 20 to 100-fold increases in glucocerebroside [97, 98]. However, the quantity of lipid stored does not account for the extreme organomegaly seen.
Fig.20
Gaucher disease. Electron micrograph of a Gaucher cell. (A). Branching tubular profiles are present in a lysosome. (B) High magnification of tubule with clockwise spiral in Gaucher disease lysosome.
The systemic pathologic findings in type 2 Gaucher disease resemble those of type 1. Gaucher cells are especially abundant in the red pulp of the spleen, the sinusoids of the liver, the alveolar capillaries, lymph nodes, and bone marrow. The spleen is enlarged with fibrosis, infarcts, and nodules related to vascular malformation of the red pulp or extramedullary hematopoiesis [97]. The gross appearance of the brains of patients with type 2 Gaucher disease is usually not altered. Microscopically, however, Gaucher cells can be observed both perivascularly in the Virchow-Robin spaces and scattered individually or in clumps within the cerebral gray matter [97]. Neuronophagia and neuronal loss in the thalamus, basal ganglia, brainstem, cerebellum, and spinal cord can also be observed [97, 99, 100]. The lesions described are small and focal.
Glucosylsphingosine (lyso-GL1) is also hydrolyzed by the enzyme glucocerebrosidase and is elevated in brains from patients with neuronopathic Gaucher disease [101]. Levels of glucosylsphingosine are also elevated in spleen and liver in patients with all three types of Gaucher disease. It has been postulated that derivatives of glycosphingolipids are potent inhibitors of protein kinase C activity [102] and could disrupt neuronal activity by interfering with signal transduction. The accumulation of substrates such as GL1 and lyso-GL1 is known to cause immune dysregulation [103, 104].
Glucocerebrosidase also plays a role in skin barrier function. Rare patients with type 2 Gaucher disease have been described with congenital ichthyosis. While light microscopic analysis of skin from patients with type 1 and type 3 Gaucher disease is normal, skin samples from patients with type 2 Gaucher disease reveal dense hyperkeratosis, epidermal hyperplasia, and inflammation [105]. The ultrastructure of the epidermis from type 2 Gaucher patients, with or without clinical evidence of ichthyosis, shows abnormal arrays of loosely packed lamellar bodies in the stratum corneum [105–107].
2.4Krabbe disease (globoid cell leukodystrophy)
The disease was first described by the Danish neurologist Knud Haraldsen Krabbe in 1916 [109], with the enzymatic defect identified in 1970 [110]. Krabbe disease is an autosomal recessive disorder caused by the deficiency of galactocerebrosidase, the lysosomal enzyme responsible for the degradation of galactocerebroside to ceramide and galactose. As a consequence, galactosylceramide accumulates in the peripheral and central nervous systems. Galactocerebroside (galactosylceramide) is a sphingoglycolipid consisting of sphingosine, fatty acid, and galactose, and is normally present almost exclusively in the myelin sheath [111]. In Krabbe disease, galactocerebroside does not accumulate in the white matter but is catabolized through an alternative pathway that yields pyschosine (galactosphingosine), which is toxic to oligodendrocytes. Galactocerebroside, however, does accumulate in cerebral macrophages that fuse to form multinucleated, PAS-positive globoid cells. The galactocerebrosidase gene, GALC, has 17 exons encoding 669 amino acids [112]. More than 130 disease-causing mutations have been reported [31]. The prevalence of the disease is estimated at around 1 in 100,000 births [112].
Krabbe disease usually presents between 3 and 6 months of age after a normal neonatal period with a rapidly progressive course involving irritability, hypersensitivity to external stimuli, and severe mental and motor deterioration [111, 112]. Patients rarely survive beyond 2 years. Clinical manifestations are limited to the nervous system with prominent long-tract signs. Hypertonicity with hyperactive reflexes present in the early stages, but patients later become flaccid and hypotonic. Blindness, deafness, and seizures are common. Peripheral neuropathy is almost always detectable. The clinical picture of the classic infantile form is relatively uniform, but atypical or late-onset forms of the disease have more variable, albeit progressive, neurological courses. Pes cavus, optic disc pallor, progressive spastic tetraparesis and moderate CSF protein elevation is common in late-onset forms [113]. Late-onset Krabbe disease has been misdiagnosed as motor neuron disease [114], hereditary spastic paraplegia [115], multiple sclerosis and Charcot-Marie-Tooth disease [116]. Aside from the leukodystrophy seen on MRI, patients with Krabbe disease can have intracranial calcifications, better assessed by CT [117–119].
The diagnosis of Krabbe disease can be made by assay of galactosylceramidase in leukocytes or cultured fibroblasts, using appropriate natural glycolipid substrates. This method can also identify heterozygous carriers. Intrauterine diagnosis of affected fetuses by galactosylceramidase assays on amniotic fluid cells or biopsied chorionic villi has been performed [111]. Umbilical cord blood transplantation in presymptomatic newborns with the infantile form of the disease allowed progressive myelin deposition and developmental gains, while transplantation after the initiation of symptoms provides no benefits [120]. Stem cell transplantation has been performed in individuals with the late-onset form of the disease as late as 16 years after initiation of symptoms, still allowing clinical improvement and halting progression of demyelination on serial neuroimaging [121]. Other modalities under investigation include enzyme replacement therapy, neural stem cell transplantation, chemical chaperone therapy, and substrate reduction [112].
Table 13
Krabbe disease: Pathologic findings
| • Widespread myelin loss throughout the white matter with fibrillary gliosis in these areas |
| • Neurons and axons are not as severely affected, but significant loss may occur |
| • Globoid cells |
| Hallmark of the disease, hence the alternate term globoid cell leukodystrophy |
| Multinucleated giant phagocytic cells up to 50 μm in diameter; up to 20 nuclei |
| PAS-positive due to cerebroside accumulation |
| Concentrated around blood vessels |
| On EM, contain cytoplasmic inclusions with straight or tubular profiles |
| appearing crystalloid in cross section |
| Decrease in number as disease progresses |
The pathology of Krabbe disease (Table 13) is confined to the CNS and is characterized by cerebral atrophy, loss of myelin, gliosis, and globoid cells. The globoid cells are multinucleated microglial macrophages distended with galactocerebroside storage material. There is diffuse demyelination of the white matter (Fig. 21A) and numerous calcifications. An intense gliosis is present in the cortex, basal ganglia, and especially around the perivascular spaces of the white matter. The perivascular spaces contain an accumulation of rounded mononuclear or binucleated PAS-positive globoid cells 15 to 20 μm in diameter (Fig. 21B). The cells are Sudan black-positive and glial fibrillary acidic protein (GFAP)-negative and stain strongly for Ricinus communis agglutinin and less strongly for peanut and wheat germ agglutinin. By EM, tubular structures (Fig. 22A) are seen similar to those observed in Gaucher disease except for the counterclockwise spiral of the tubules (Fig. 22B).
Fig.21
Krabbe disease. (A) Coronal section of the brain stained with oil red O. Only a few myelinated areas stain. The remainder of the white matter does not stain, indicating loss of myelin. (B) Microscopic section of brain with globoid cells in white matter. The brain does not store the substrate, galactosylceramide, but it stimulates infiltration of macrophages which transform to globoid cells. The increased levels of psychosine that occur have cytotoxic effects.
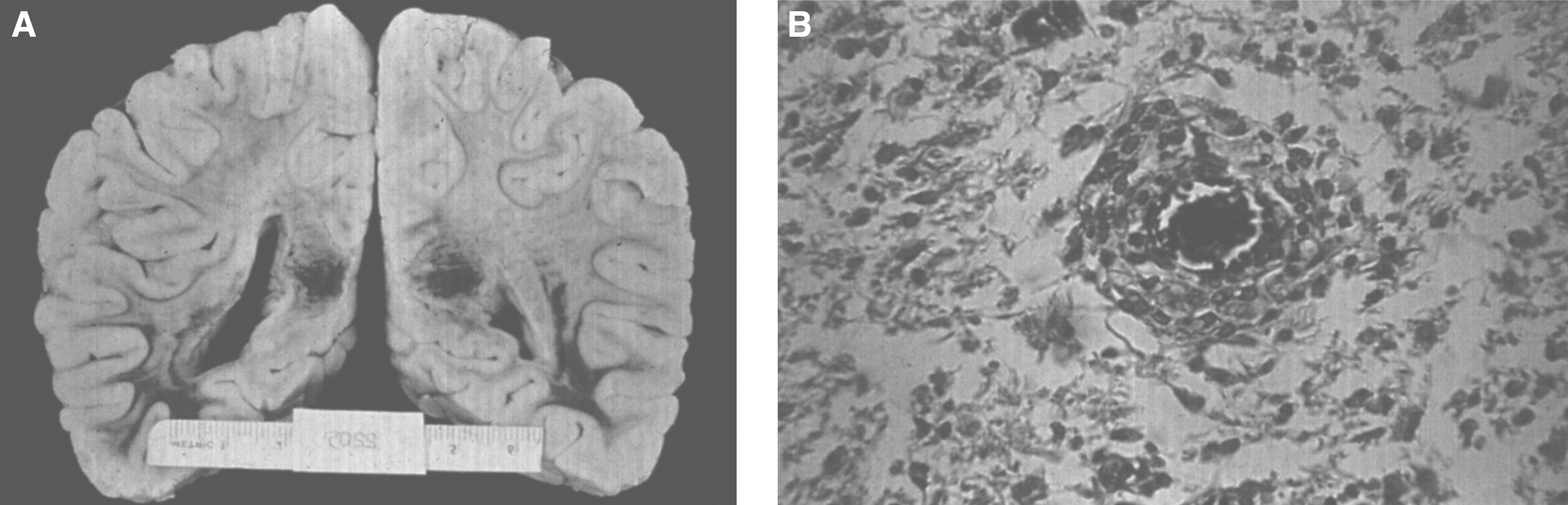
Fig.22
Krabbe disease. Electron micrograph of brain. (A) A globoid cell containing tubules with electron-dense deposits. (B) High magnification of the twisted tubule with a counterclockwise spiral.
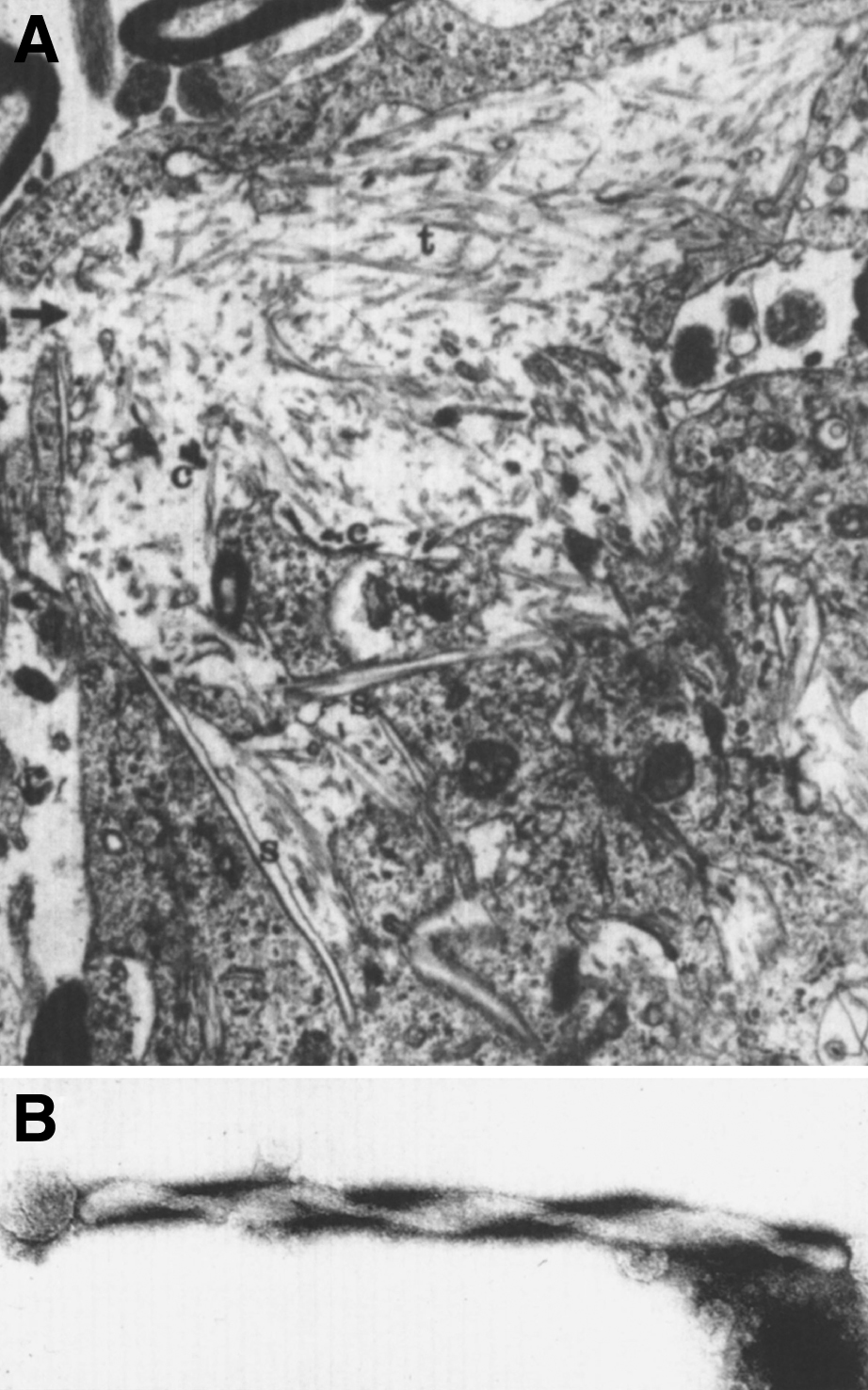
Consistent with the myelin loss, the white matter is evenly depleted of all lipids, particularly glycolipids. The ratio of galactocerebroside to sulfatide is abnormally high. Segmental demyelination, axonal degeneration, fibrosis, and macrophage infiltration are common in the peripheral nervous system [111].
2.5Farber disease (lipogranulomatosis)
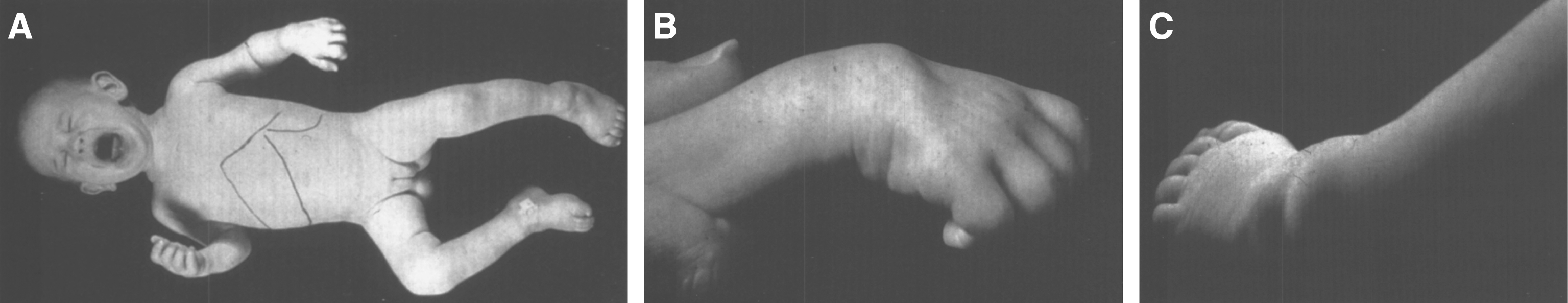
Lipogranulomatosis was initially described at a Mayo Foundation lecture in 1947 by Sidney Farber, a pediatric pathologist working at Boston Children’s Hospital. He first published a short description of the disease in 1952 as a transaction from the 62nd Annual Meeting of the American Pediatric Society [122], and later published an expanded description in 1957 [123]. Dr. Farber proposed that the disease had characteristics intermediate between Niemann-Pick disease (lipid storage) and Hand-Schüller-Christian disease, a histiocytosis (inflammatory infiltrates). The stored lipid was subsequently identified as ceramide in 1969 [124], and the deficient enzyme was noted to be ceramidase in 1972 [125]. Farber disease manifests clinically with the diagnostic triad of hoarseness, painful and swollen joints, and periarticular and subcutaneous nodules (Fig. 23) particularly over pressure points [126]. Cherry-red spots have been occasionally described [127–129]. In most cases, there is no significant visceral involvement. Farber disease often leads to death within the first few years of life due to respiratory infections. The presentation may be in infancy, with hoarseness, respiratory difficulty, vomiting, swollen painful joints, and failure to thrive. There is significant clinical variability among sibs with Farber disease [130], and some patients follow a more prolonged course. These milder cases can be misdiagnosed as juvenile idiopathic arthritis [131].
Fig.23
Farber disease. (A) Child with multiple skin nodules. (B) Large nodules on the wrist and (C) on the ankle. (Courtesy of Dr. Steven Qualman.).
Definitive diagnosis is made by determining lysosomal acid ceramidase activity in leukocytes or cultured skin fibroblasts. Acid ceramidase activity in heterozygotes is usually reduced [126]. Biopsy of periarticular tissues may be diagnostic. Prenatal diagnosis has been accomplished by demonstrating acid ceramidase deficiency in cultured amniotic fluid cells. Farber disease patients have a significant elevation of ceramide in tissue or body fluids [126], but ceramide is difficult to detect histologically because of its lack of specific staining reactions [132]. The nonspecific oil-red-O positivity and periodic acid-Schiff-diastase (PAS-D) positivity may reflect peripheral deposition of ceramide and a ganglioside, respectively. Tissue analysis for stored ceramide may prove especially useful, since the ceramide may be demonstrated in formalin-fixed tissues and has a specific 2-hydroxy fatty acid component. The gene for Farber disease, ASAH1, has been identified. Biallelic mutations in the same gene also cause spinal muscular atrophy with progressive myoclonic epilepsy [133]. There is no specific treatment for Farber disease at present, although a recombinant human acid ceramidase has been produced and is currently in preclinical studies [134].
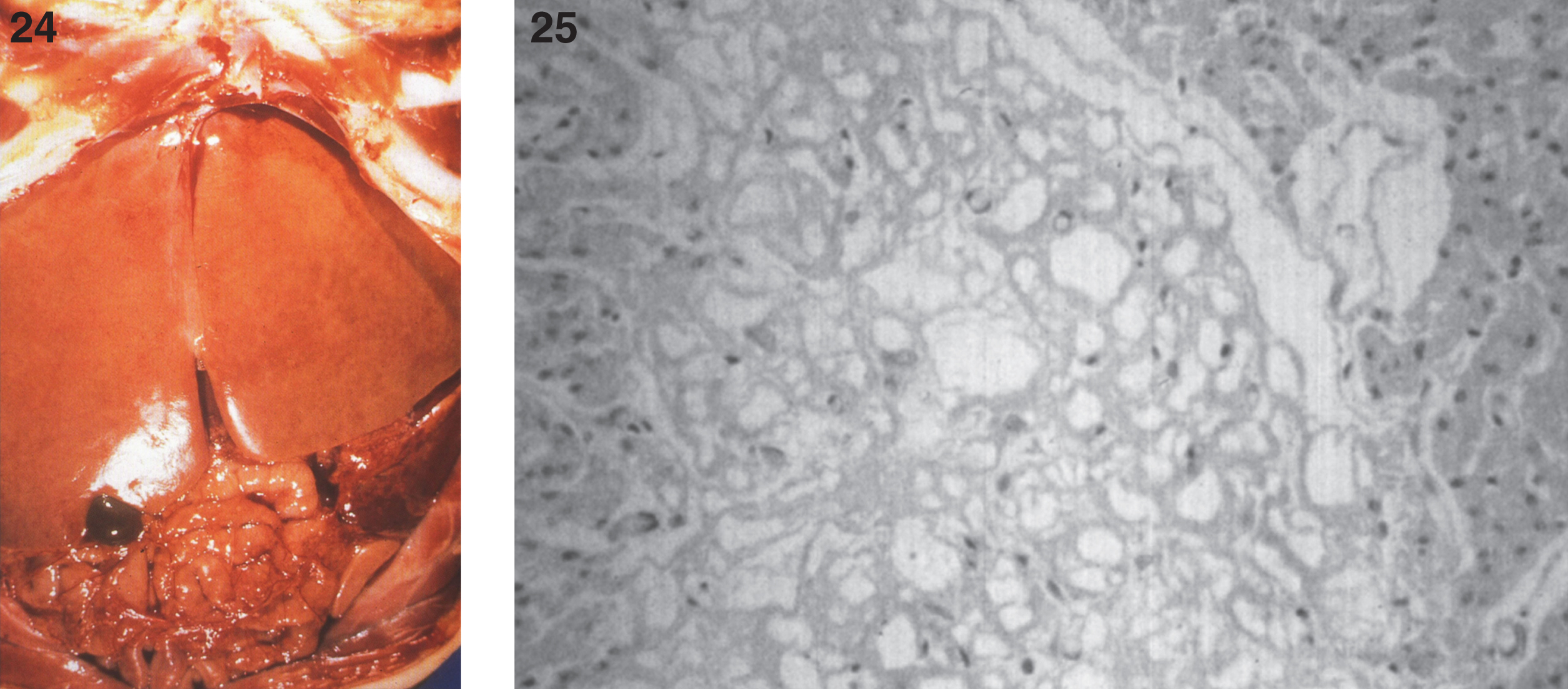
Figs.24, 25
Farber disease. The opened abdominal cavity at autopsy shows a greatly enlarged liver that has a yellow appearance. (Courtesy of Dr. Steven Qualman.); (25) Farber disease. Microscopic section of the liver. The hepatocytes are distended with lipid. (Courtesy of Dr. Steven Qualman.).
Pathologically, foam cells with granulomatous reaction occur in the respiratory system, soft tissues, and joints [135]. The cells are vacuolated and foam cells are present in lymph nodes, bone marrow, spleen, and liver as well as renal proximal tubular epithelial and glomerular cells. Ceramide, mucopolysaccharides, and gangliosides accumulate. By EM, foam cells containing curvilinear tubular structures, membrane-bound reticulogranular material, mucopolysaccharides, and zebra bodies are seen [136]. The characteristic ultrastructural finding is the so-called “Farber body”, a comma-shaped curvilinear, tubular intracytoplasmic inclusion [136] thought to reflect ceramide storage. This inclusion is not present in liver cells but is present in macrophages and Kupffer cells. “Banana bodies” are needle-like, membrane-bound structures found in Schwann cells [137], while zebra bodies are seen in endothelial cells, neurons and secretory cells from sweat glands [138].
In some cases, gross and microscopic autopsy findings can show massive organomegaly. The liver is enlarged and pale and has a yellow appearance (Fig. 24). The hepatocytes are distended with lipid, and histiocytic cells massively infiltrate hepatic sinusoids (Fig. 25) and splenic red pulp with replacement of the white pulp. The lymph nodes are diffusely enlarged by histiocytic infiltrates that stain with PAS (Fig. 26). The histiocytes replace lymphoid parenchyma in the medullary and cortical areas, sparing subcapsular and trabecular sinuses. Patchy histiocytic infiltrates are noted in the bone marrow; the marrow is cellular with all normal hematopoietic elements in adequate numbers. Erythrophagocytosis in lymph nodes, bone marrow, liver, and spleen may be seen. Ultrastructurally, typical curvilinear “Farber” bodies are present (Fig. 27).
Figs. 26–28
Farber disease. Microscopic section of a lymph node showing PAS-positive storage histiocytes. (Courtesy of Dr. Steven Qualman.); (27) Farber disease. Electron micrograph of Farber’s lipogranulomatosis. Typical curvilinear bodies (arrowheads) are shown. (Courtesy of Dr. James Phillips.); (28) Farber disease. Dense white nodules removed from the larynx. (Courtesy of Dr. Steven Qualman.).
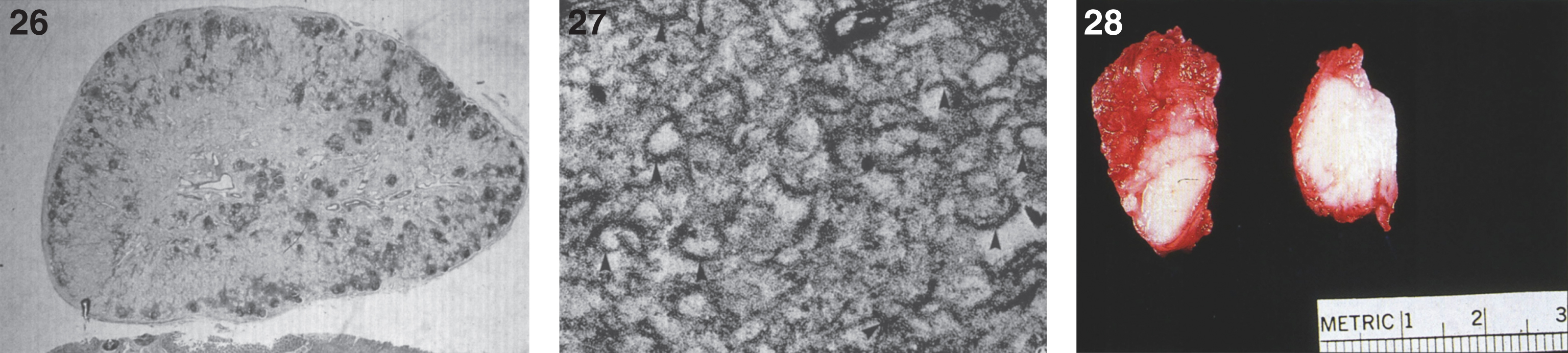
There are nodules (Fig. 28) and significant infiltration of the vocal cords causing marked narrowing of the larynx (Fig. 29) and of the soft tissues and skeletal muscles of the larynx by histiocytes (Fig. 30) with large pools of extracellular storage material causing swelling and obstruction of the laryngeal lumen. Storage material is present in subcutaneous nodules at the wrists, knees, and ankles with a peripheral histiocytic infiltrate and central necrosis [130] and in the joint synovium and lungs (Fig. 31). The heart may show epicardial nodules (Fig. 32) due to histocytic granulomas (Fig. 33), and the lungs may be infiltrated with histiocytic cells (Fig. 34).
Figs. 29–31
Farber disease. Section of larynx greatly narrowed by infiltration of storage histiocytes (right) compared with normal larynx (left). (Courtesy of Dr. Steven Qualman.); (30) Farber disease. Microscopic section of larynx showing large distended vacuolated histiocytes. (Courtesy of Dr. Steven Qualman.); (31) Farber disease. Microscopic section of the joint synovium with infiltration by storage histiocytes.
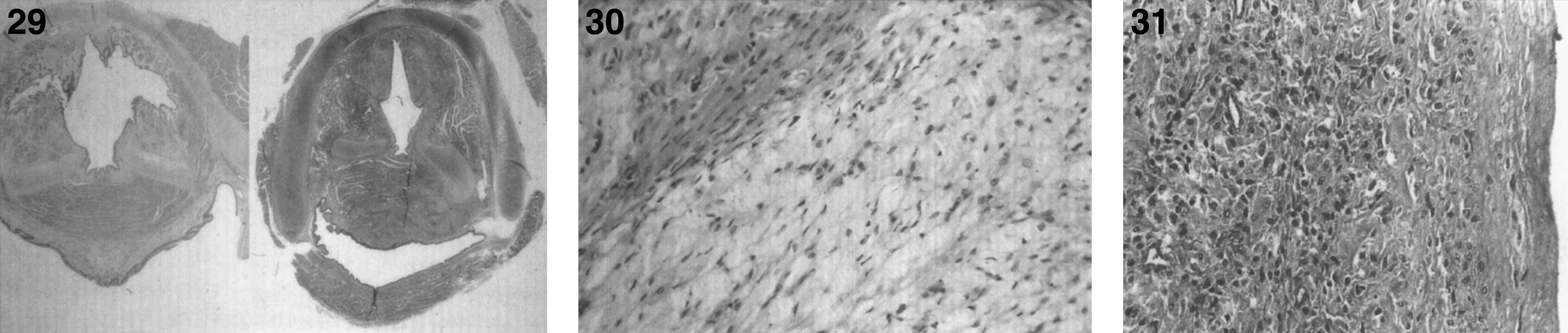
Figs. 32–34
Farber disease. The heart shows epicardial nodules. (Courtesy of Dr. Steven Qualman.); (33) Farber disease. Microscopic section of heart showing epicardial infiltration by storage histiocytes. PAS stain. (Courtesy of Dr. Steven Qualman.); (34) Farber disease. Microscopic section of the lung showing infiltration by storage histiocytes.
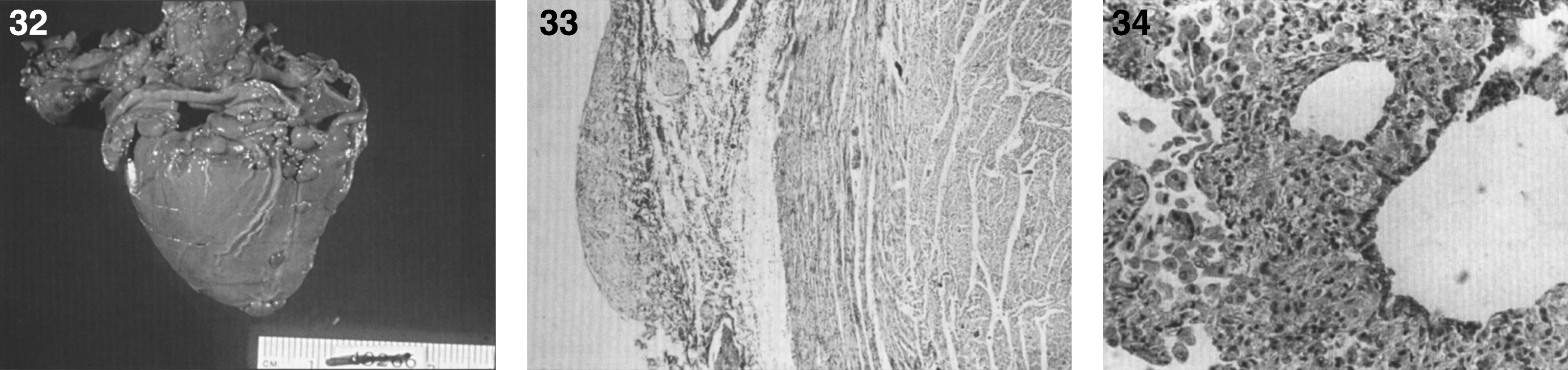
Histiocytic cells in the liver store flocculent, granular material in membrane-bound intracytoplasmic vacuoles. Storage vacuoles are occasionally present within hepatocytes.
Renal storage disease has been documented morphologically and biochemically with elevated ceramide levels in the renal parenchyma or urine in severely affected patients [136, 139] as well as in the fetus. Kidneys show storage material within both glomerular podocytes and proximal tubular epithelial cells with membrane-bound accumulations of flocculent granular material but no curvilinear tubular inclusions. There may be moderate nervous system dysfunction related to the accumulation of ceramide and gangliosides in neurons (Fig. 35) [130] and in the anterior horn cells of the spinal cord (Fig. 36).
Figs. 35,36
Farber disease. Microscopic section of the cerebral cortex of the brain. The neurons are distended with storage material; (36) Farber disease. Microscopic section of spinal cord. The anterior horn cells are distended with storage material. (Courtesy of Dr. Steven Qualman.).
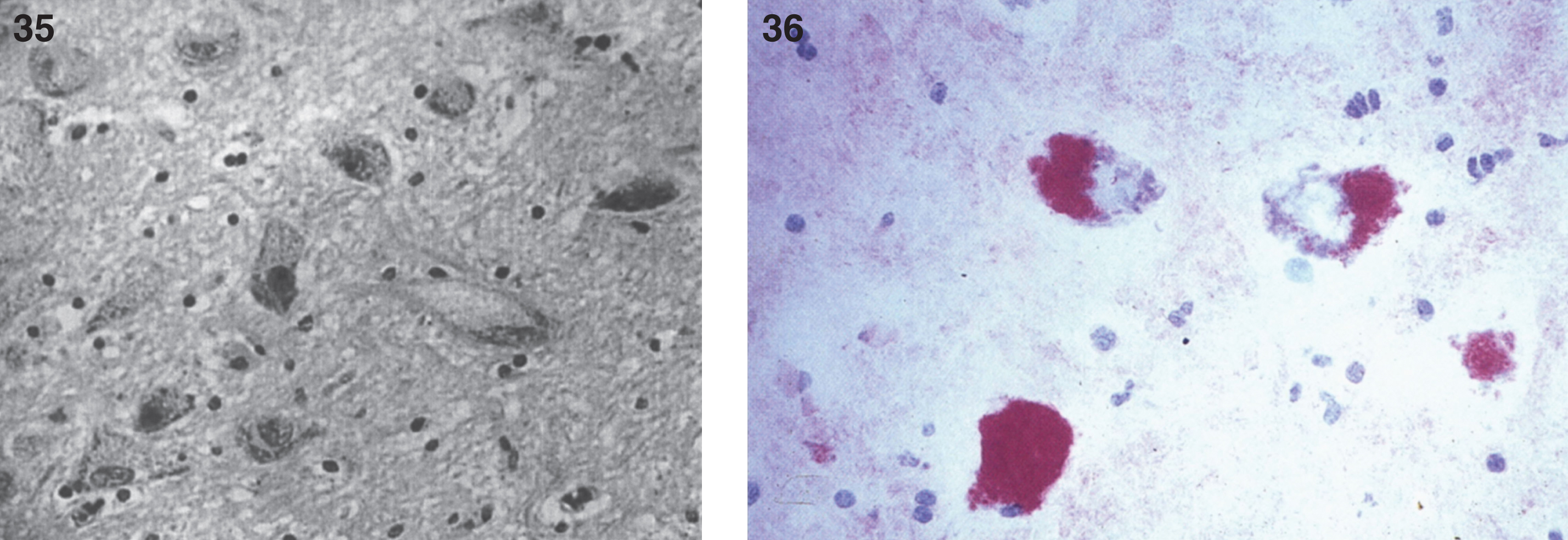
Liver, spleen, and subcutaneous nodules contain a highly water-soluble, PAS-D-resistant cytoplasmic substance within histiocytes. Oil red O-positive lipid, present within histiocytes in formalin-fixed tissue, is lost during paraffin embedding. Distinct PAS-D-positive and oil red O-positive substances are detected in both thickened glomerular capillary loops and vacuolated proximal tubular epithelial cells in the kidneys.
Although subcutaneous nodules of Farber disease have a distinct histopathologic appearance on biopsy, they are not universally diagnostic of Farber disease and may be absent [126]. Lymph nodes may be the most accessible tissue for biopsy and diagnosis, if palpable subcutaneous nodules are absent. Lymph node involvement has been seen more frequently in Farber disease than in other visceral sites of storage. The sparing of lymph node sinuses by the disease is in contradistinction to the histiocytic proliferative disorders, which often involve lymph nodes but primarily involve the sinuses [140].
2.6Fabry disease
The disease was described independently in 1898 by two dermatologists, Johannes Fabry in Germany and William Anderson in England [141, 142]. Fabry disease is an X-linked inborn error of glycosphingolipid catabolism resulting from the deficient activity of the lysosomal hydrolase α-galactosidase A in tissues and fluids [143, 144]. The enzyme defect leads to the systemic deposition of glycosphingolipids with terminal α-galactosyl moieties, predominantly globotriaosylceramide and, to a lesser extent, galactosylceramide and blood group B antigens. Hemizygous males have extensive deposition of these glycosphingolipid substrates in body fluids and in the lysosomes of endothelial, perithelial, and smooth muscle cells of blood vessels. Deposition also occurs in ganglion cells, and in many cell types in the heart, kidneys, eyes, and most other tissues [145].
Fig.37
Fabry Disease. (A) Corneal whorls, representing storage of trihexosylceramide. (B) Angiokeratomata of umbilicus. (C) Tortuous vessels in bulbar conjunctiva.
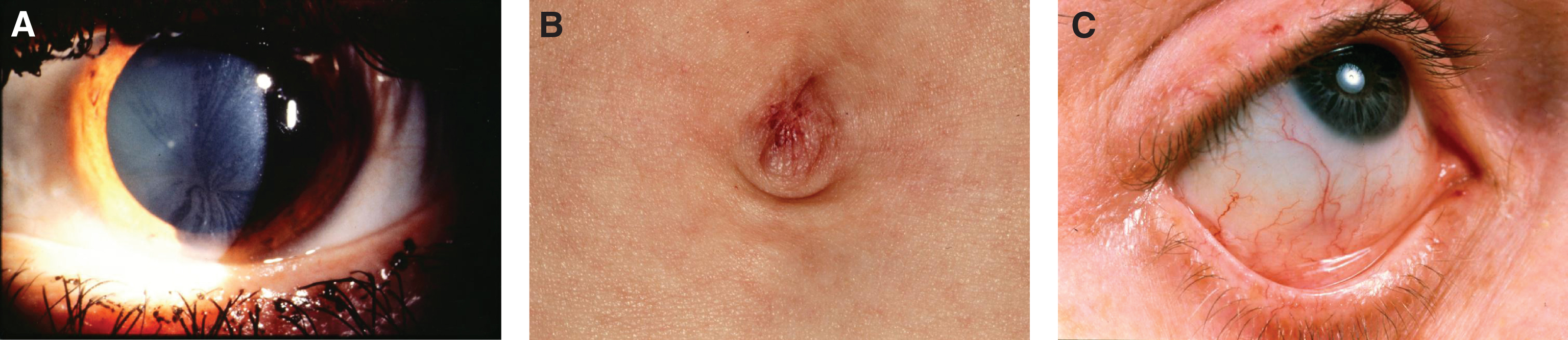
Fabry disease results from mutations in the gene encoding α-galactosidase A, i.e., GLA. This gene, which has been localized to chromosome Xq22, has 7 exons and encodes a 429 amino acid protein with a 31-amino acid signal peptide. More than 750 different mutations in GLA have been identified [143]. Prevalence estimates range from 1 in 50,000 to 1 in 117,000 [143]. A high prevalence of the cardiac variant was found in Taiwan, where it is estimated to affect 1 in 1,600 males [146].
Clinical manifestations in classically affected hemizygotes who have no detectable α-galactosidase A activity include the onset of pain and paresthesias in the distal extremities (acroparesthesias), vessel ectasia (angiokeratoma) in skin and mucous membranes, hypohidrosis during childhood or adolescence, corneal and lenticular opacities, and tortuous conjunctival vessels (Fig. 37). With increasing age, proteinuria, hyposthenuria, and lymphedema appear. Severe renal impairment leads to hypertension and uremia. Death usually occurs from renal failure or from cardiac or cerebrovascular disease. Atypical hemizygotes with residual α-galactosidase A activity may be asymptomatic or have late-onset, mild disease manifestations primarily limited to the heart (the “cardiac variant”) [143].
Heterozygous females may have an attenuated form of Fabry disease. They are usually asymptomatic, although rarely can be as severely affected as hemizygous males. The most frequent clinical finding in females is the characteristic whorl-like corneal epithelial dystrophy observed on slit-lamp examination [143].
Confirmation of the clinical diagnosis in hemizygotes and heterozygotes requires the demonstration of deficient α-galactosidase A activity in plasma, leukocytes, or tears, or increased levels of globotriaosylceramide in urine. Heterozygous females may have intermediate levels of enzyme activity and accumulated substrate. The more accurate diagnosis of heterozygous females can be accomplished by detection of the molecular lesion in GLA. Prenatal diagnosis can be accomplished by demonstration of deficient α-galactosidase A activity and XY karyotype, or by demonstration of the specific α-galactosidase A mutation in chorionic villi or cultured amniotic cells [143, 144].
Enzyme replacement therapy is now the treatment of choice for Fabry disease. In addition, supportive measures include low maintenance dosages of diphenylhydantoin, gabapentin or carbamazepine to relieve the excruciating pain and constant discomfort of acroparesthesias. Oral anticoagulants are recommended for stroke-prone patients. Renal dialysis and transplantation are effective in the treatment of end-stage renal disease.
With respect to the pathology of Fabry disease, glycosphingolipids (principally the trihexosyl ceramide globotriaosylceramide) accumulate in all organs and tissues [147] and may be up to 300-fold higher than normal levels [148]. Immunohistochemistry using antibodies against globotriaosylceramide is possible [149]. The greatest accumulation is observed in kidney, lymph nodes, blood vesels, prostate, and autonomic ganglia (Fig. 38A) [147]. The storage has a predilection for vascular endothelium (Fig. 38B) and smooth muscle cells, which is different from that seen in other forms of glycosphingolipidoses [147]. The major abnormalities of vessels are narrowness, dilation, and motor unresponsiveness. In addition to endothelial proliferation, the swollen endothelial cells may be the precursor of thromboses, ischemia, and infarction, and there may be progressive aneurysmal dilation of the weakened vessel walls. Microaneurysms may form in retinal and conjunctival vessels, and in the skin a transition from telangiectasia to a frank angiokeratoma is one of the hallmarks of the disease [147]. The involvement of peripheral and central autonomic nerve cells may be responsible for paresthesias, pain, gastrointestinal symptoms such as nausea and diarrhea, and other vague neurologic symptoms. In the eye, lipid deposits can be seen in the basal cells of the corneal epithelium and in the epithelial cells of the lens [150]. Lipid storage has also been found in Leydig cells of the testis and basal cells of the epididymal ducts [151, 152]. Pathologic changes occur in the kidney (Fig. 39) with accumulation of glycosphingolipid in the glomerular visceral, glomerular parietal, and tubular epithelial cells. Subendothelial deposits of membrane-like material associated with duplications of the glomerular basement membrane have also been described [153]. Vacuolated epithelial cells known as oval fat bodies can be seen in the urine sediment [154]. A scoring system for the renal pathology has been developed [155].
Fig.38
Fabry disease. (A) Microscopic section of the myenteric plexus showing large distended ganglion cells that are PAS-positive. (B) Microscopic section of a blood vessel showing thickening of the vessel wall by accumulation of glycosphingolipid (PAS stain).
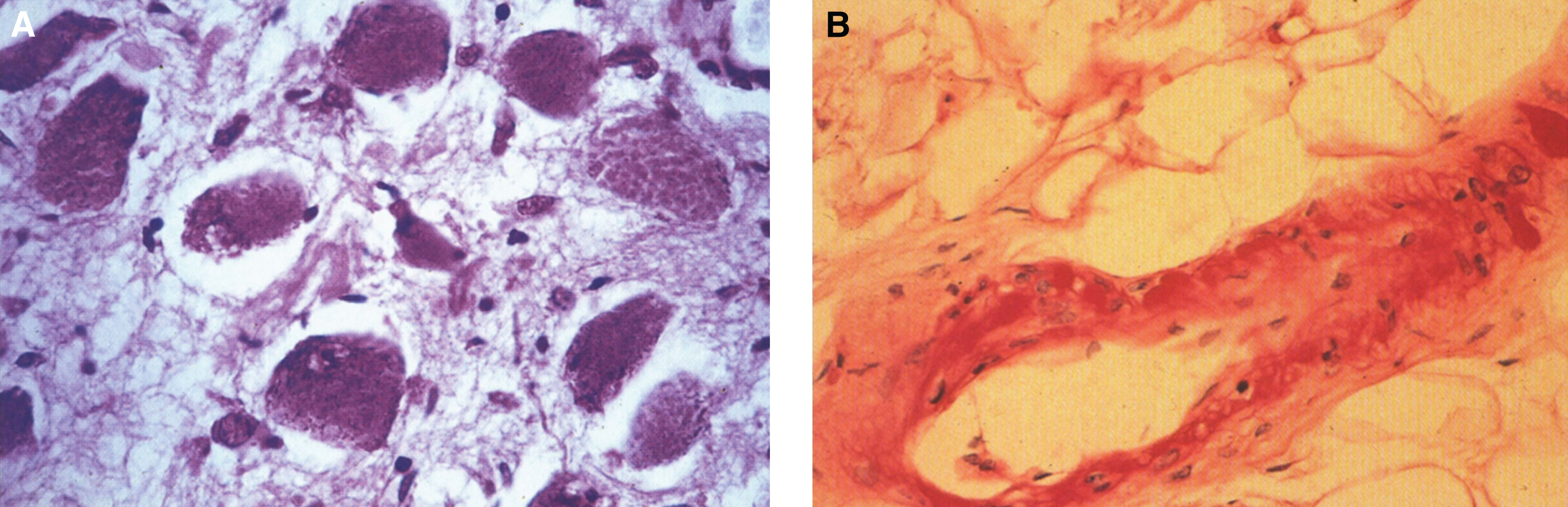
Fig.39
Fabry disease: kidney. (A) Gross appearance of end-stage disease with scarring and cyst formation. External view (left), cut surface (right). (B) Microscopic section of kidney glomerulus. Foamy glomerular epithelial cells are present. (C) Electron micrograph of kidney. Glomerular epithelial cells contain myelin-like figures. (D) Glomerular parietal epithelial cells show pleomorphic lipid inclusions.
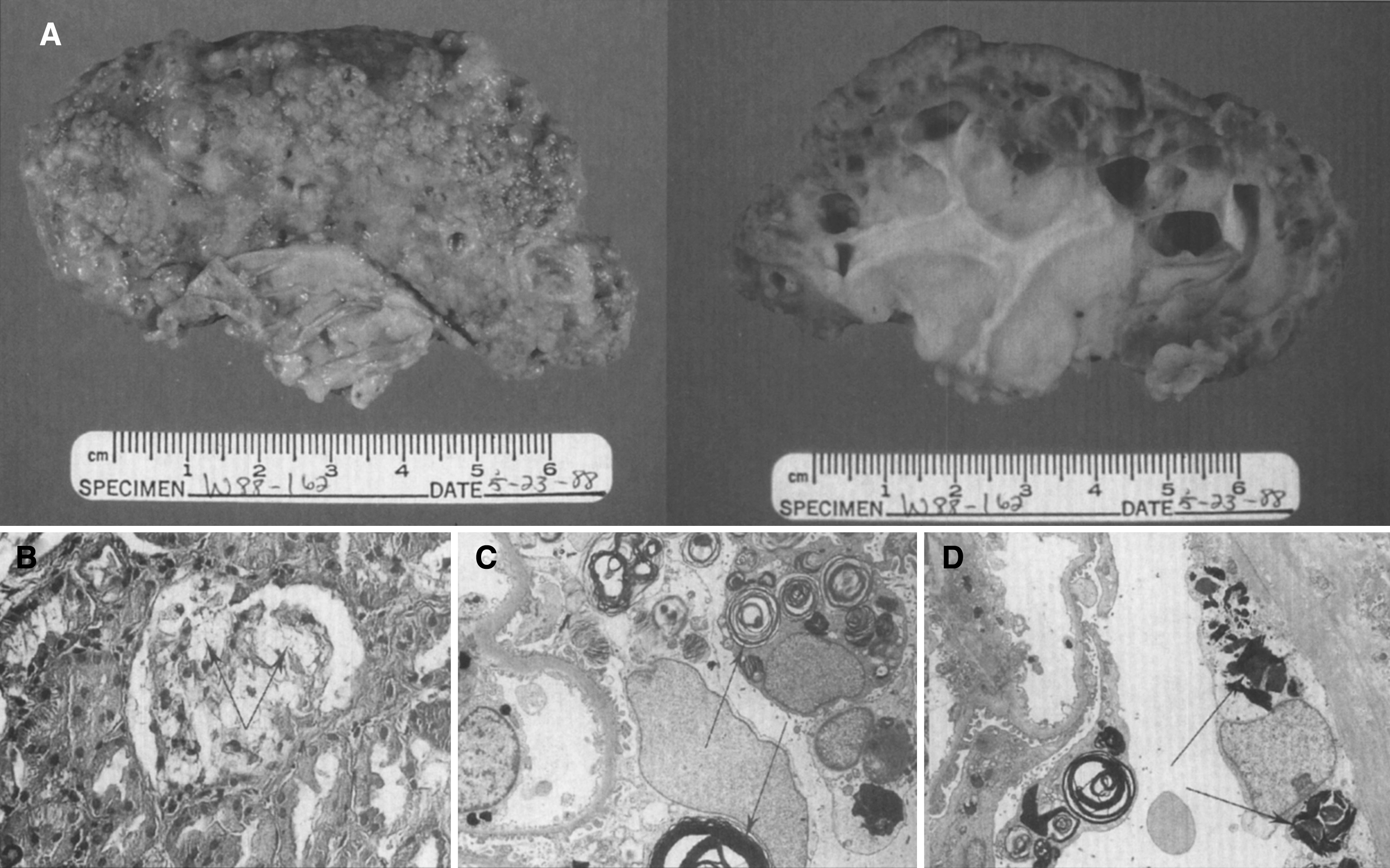
In histologic sections, the deposits of ceramide trihexoside appear as vacuoles, and in frozen sections they are sudanophilic, PAS-positive, and strongly birefringent under polarized microscopy. In cardiac muscle, the deposits occupy the central perinuclear areas of the cytoplasm displacing the contractile elements to the periphery. This results in the histologic appearance of a lacework pattern similar to that seen in type II glycogenosis. Ganglia are distended with storage material (Fig. 40). Ultrastructurally, the storage material is deposited in lysosomes with concentric or parallel lamellae with a periodicity of 4 to 55 nm. These concentric lamellar bodies are known as myelin figures. The lamellae react positively with periodate-thiosemicarbazide silver proteinate and periodate-thiosemicarbazide-osmium tetroxide. These structures can also be demonstrated on freeze-fractured preparations [147].
Fig.40
Fabry disease. (A) Large distended ganglion cells in gasserian ganglion. Hematoxylin and eosin stain (left); Luxol fast blue stain (right). (B) Electron micrograph shows concentric parallel lamellae of the lipid storage substance.
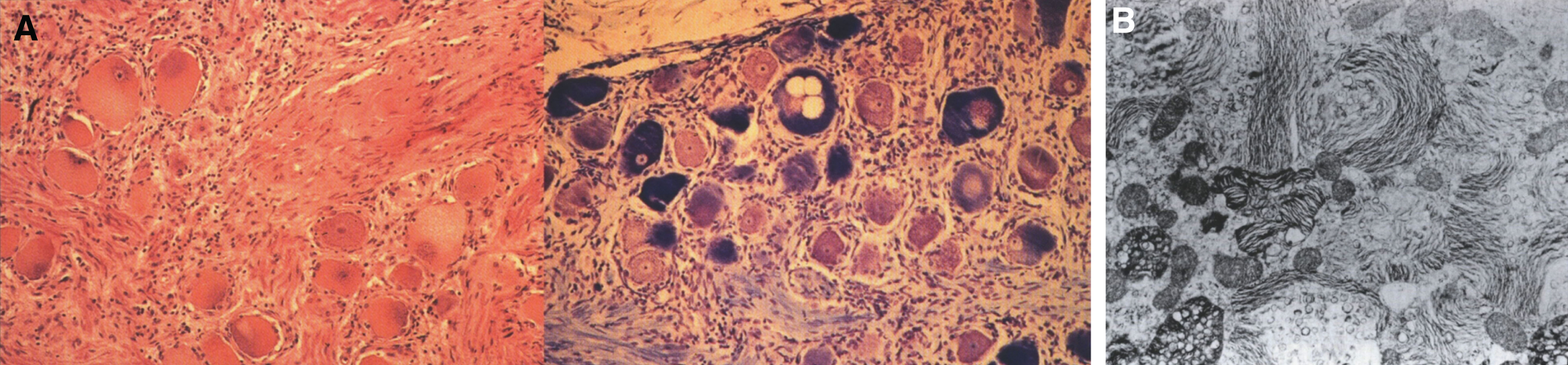
The histopathologic changes frequently involve the heart, especially the myocardial cells, specialized tissues of the atrial ventricular conduction system, and the valves [148, 156] and the coronary arteries, due to deposition of glycosphingolipid. Hypertrophic obstructive cardiomyopathy has been noted [157]. The aorta may show changes suggestive of cystic medionecrosis [147]. In a patient on long-term enzyme replacement therapy, the typical myelin-like figures were lost, being replaced instead by crystalline tubular structures and abnormally-branched glycogen [158].
The severity of this disorder appears to be related to the total amount of the glycosphingolipid stored in the tissues, which in turn depends on time, the presence or absence of residual α-galactosidase A activity, and the individual’s ABO blood type. Hemizygotes and heterozygotes who are type B and AB are more severely affected because of the additional accumulation of B-specific glycosphingolipid, with a terminal α-galactosyl residue (Fig. 41).
The cutaneous lesion (angiokeratoma) is not distinctive for Fabry disease. Angiokeratomas also occur in fucosidosis, adult-onset GM1 gangliosidosis (β-galactosidase deficiency), aspartylglucosaminuria, α-N-acetylgalactosaminidase deficiency (Schindler neuroaxonal dystrophy syndrome and Kanzaki disease), sialidosis type II, galactosialidosis (Goldberg syndrome), and β-mannosidosis.
Fig.41
The terminal residue of blood group antigen B is metabolized by α-galactosidase A–deficient in patients with Fabry disease-while antigen A is cleaved by α-galactosidase B–deficient in patients with Schindler’s disease.
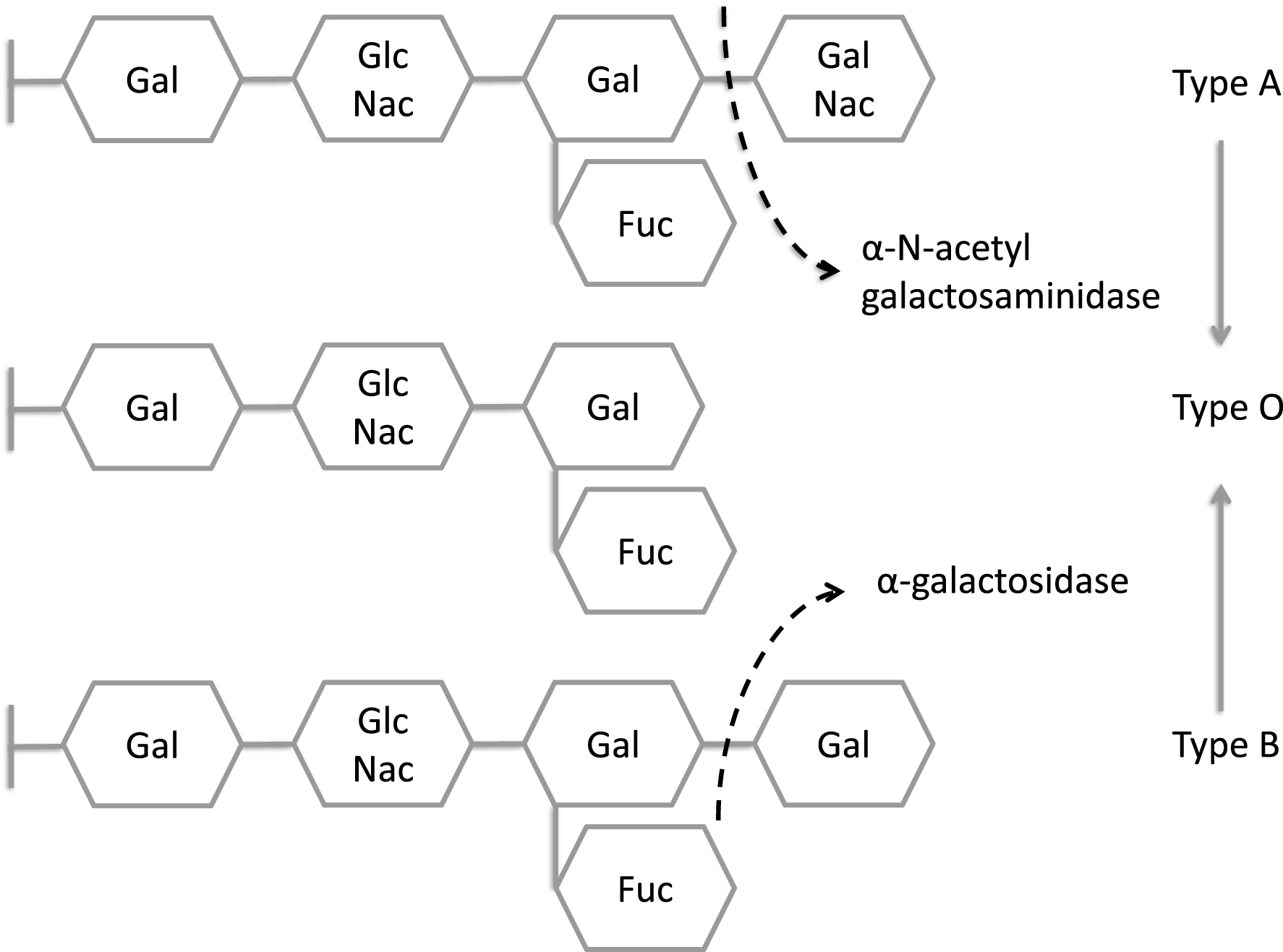
2.7Schindler disease
The disease was first described by Detlev Schindler and colleagues in 1987, who reported two German brothers with profound intellectual disability, seizures, blindness and decorticate posturing caused by α-N-acetylgalactosaminidase (α-NAGAL) deficiency [159]. Thin-layer chromatography of urinary oligosaccharides revealed an abnormal pattern, and the older sibling, whose blood group was type A, was noted to excrete 5 times the normal level of the group A determinant, a trisaccharide with a terminal α-N-acetylgalactosamine. Subsequently, the degradation rate of blood group A glycosphingolipids has been found to be low in fibroblasts from patients with α-NAGAL deficiency [160], supporting the notion that patients with blood group A have worse storage disease (Fig. 37). α-NAGAL is also known as α-galactosidase B, and the NAGA gene encoding for this enzyme shares 46% sequence identity with the GLA gene, encoding for α-galactosidase A (the enzyme deficienct in Fabry disease). In fact, both genes derive from a common ancestor [161].
This rare, autosomal recessive lysosomal storage disease has three main phenotypes: Schindler disease type I, an infantile-onset neuroaxonal dystrophy; type II, an adult-onset disorder with mild intellectual disability, and type III with an intermediate phenotype accompanied by mild to moderate neurologic involvement [162].
Schindler disease type I is characterized by onset around the second year of life with weakness, hypotonia, and areflexia. As the disease progresses, rigidity, spasticity, cerebellar signs, deafness, blindness, and mental deterioration occur. Death occurs typically before the age of 6 years. Clinically and histopathologically, the disease mimics Seitelberger disease, with characteristic spheroids seen in terminal axons.
Type II disease was first reported in 1989 by Kanzaki and colleagues, who described a 46-year-old Japanese woman with angiokeratoma corporis diffusum and glycoprotein storage [163]. Since the glycopeptiduria found in this patient was similar to that found in the original German siblings with Schindler disease, Kanzaki and colleagues measured the activity of α-NAGAL and found it to be deficient [164]. Thus, α-NAGAL deficiency type II is also known as Kanzaki disease. Although the original Japanase proband was initially reported as not having neurologic involvement, more careful phenotyping revealed mild intellectual disability and peripheral neuropathy [165]. The same patient was reported once more in 2004, by which time she had developed sensorineural hearing loss and recurrent vertigo attacks [166]. Other cases of Kanzaki disease have been associated with Ménière’s disease, hearing loss and peripheral neuropathy [167]. Variable features include tortuosity of conjunctival and retinal vessels [165, 168], cardiac involvement in the form of moderate cardiomegaly [168] or hypertrophy of the interventricular septum [167], and lymphedema [168]. Pathologic findings include markedly dilated lysosomes producing vacuolation of endothelial cells, fibroblasts, Schwann cells, and secretory cells of eccrine sweat gland [163, 165, 167, 168]. Small cytoplasmic vacuoles have been found in lymphocytes, granulocytes and monocytes on peripheral blood smears stained with Giemsa [165].
The first case of α-NAGAL deficiency type III was described in 1993 in a girl with generalized seizures starting at 11 months of age, and mild but persistent oligosacchariduria [169]. No vacuolization was seen in lymphocytes, granulocytes or monocytes. Interestingly, her younger brother had a similar degree of α-NAGAL deficiency but remained asymptomatic by the age of 8 year [170]. A similar case of an asymptomatic patient with α-NAGAL deficiency ascertained through an affected sibling with type III disease was subsequently described [170]. Interestingly, these two siblings carried the same homozygous E325K mutation in NAGA as the original German siblings with type I disease, arguing against a genotype-phenotype correlation. In fact, the α-NAGAL activity in fibroblasts from two patients with milder Kanzaki disease was found to be lower than in patients with Schindler disease types I and III [171].
There is no treatment for the disease, although two pharmacological chaperones – 2-acetamido-1,2-dideoxy-D-galactonojirimycin (DGJNAc) and 1-deoxygalactonojirimycin (DGJ) – have been proposed as potential therapeutic agents [172].
2.8GM1 Gangliosidosis type I (Norman-Landing disease)
The gangliosidoses are autosomal recessive conditions and are divided into 2 major groups, GM1 and GM2 gangliosidoses [173]. Until the mid-1960s, GM2 gangliosidosis remained the only ganglioside storage disease known. In 1959, Norman and colleagues described a patient with cherry-red spots, hepatomegaly caused by infiltration of foamy histiocytes, and facial features similar to those seen in Hurler disease [174]. Landing and colleagues reviewed eight similar cases in 1964 [175]. O’Brien and colleagues identified the stored material as a ganglioside, structurally different from the compound that accumulates in Tay-Sachs disease [176]. A deficiency of β-galactosidase was identified as the underlying etiology in 1968 [177]. The same enzymatic deficiency can lead to Morquio disease type B [178]. When glycosaminoglycans fail to have their terminal galactose removed, as in bone and connective tissue, the result is Morquio type B disease. When ganglosides fail to have their terminal galactose removed, as in neurons, the result is GM1 gangliosidosis.
GM1 gangliosidosis is a neurosomatic disease occurring mainly in early infancy (infantile form, type I). Developmental arrest is observed a few months after birth, followed by progressive neurologic deterioration and generalized spasticity with sensorimotor and psychointellectual dysfunctions. Macular cherry-red spots occur in 50% of patients; facial dysmorphism, hepatosplenomegaly, and generalized skeletal dysplasia are usually present in infantile cases. Extrapyramidal signs and dystonia are the major neurologic manifestations in adults with GM1 gangliosidosis [173].
The human β-galactosidase gene, GLB1, has been mapped to chromosome 3 and has 16 exons encoding a 76 kDa protein. More than 180 mutations have been described [31], including the common mutations, R201C, I51T, and W273L, found in Japanese or Caucasian patients with late infantile-juvenile GM1, with adult-chronic GM1 gangliosidosis, and with Morquio B syndrome, respectively. The Romani population also harbors a founder mutation, p.R59H [179, 180]. The estimated prevalence varies from 1 in 100,000 to 1 in 300,000 [178], with a much higher prevalence in southern Brazil [181], the Maltese islands [182], and the Romani population [183].
Glycoconjugates with terminal β-galactose are increased in tissues and urine from patients with GM1 gangliosidosis and Morquio B syndrome. The ganglioside GM1 and its asialo derivative GA1 accumulate in the brain of GM1-gangliosidosis patients. Oligosaccharides derived from keratan sulfate or glycoproteins have been reported in visceral organs and urine from patients with GM1 gangliosidosis. Deficient activity of β-galactosidase results in accumulation of ganglioside in neurons, and in other sites the defect causes accumulation of mucopolysaccharides (glycosaminoglycans) [175].
Definitive diagnosis by β-galactosidase assay is established using leukocytes, dried blood spots, fibroblasts, or amniocytes. A variant of sialidosis (galactosialidosis) has both sialidase and β-galactosidase deficiencies.
The brain pathology involves progressive atrophy with neuronal swelling (Fig. 42) and loss of neurons and gliosis. Neurons contain sudanophilic material (ganglioside) and some PAS-positive material, whereas other viscera, particularly the liver (Fig. 43) and kidney (Fig. 44), accumulate strongly PAS-positive material because of the presence of mucopolysaccharides. By EM [184], MCBs (Fig. 45) as well as pleomorphic membranous vesicular bodies or large compact oval bodies are seen in skin or conjunctival biopsy and reticulogranular material is seen in cultured skin fibroblasts and/or endothelial cells of skin biopsies. Peripheral blood and bone marrow lymphocytes and syncytiotrophoblasts of the placenta are vacuolated. Other pathologic findings are similar to those in GM1 type II (see below).
Figs. 42,43
GM1 gangliosidosis type I: brain. The cortical neurons are distended with ganglioside; (43) GM1 gangliosidosis type I: liver. Some hepatocytes are swollen and contain storage material.
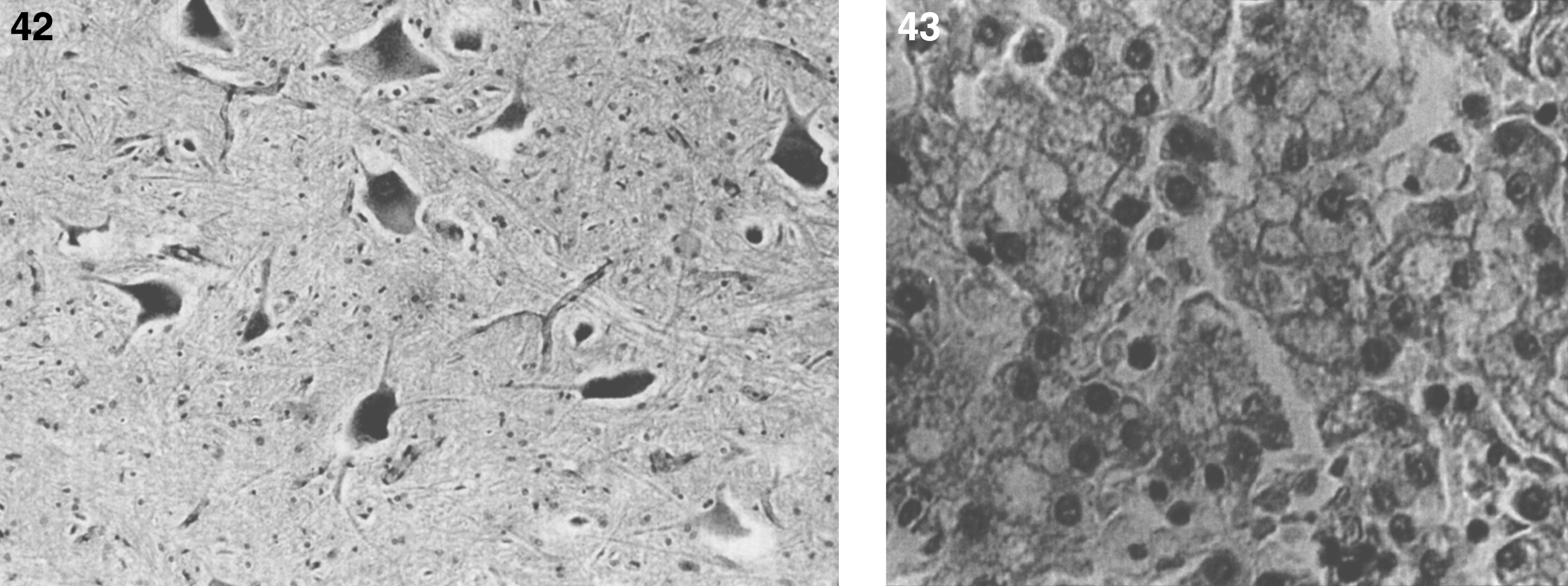
Figs. 44,45
GM1 gangliosidosis type I: kidney. The glomerular epithelial cells are vacuolated; (45) GM1 gangliosidosis type I. Electron micrograph in conjunctival biopsy showing typical membranous concentric bodies (MCBs) of gangliosides. MCBs are the morphologic expression of gangliosides seen in all the gangliosidoses including Tay-Sachs disease.
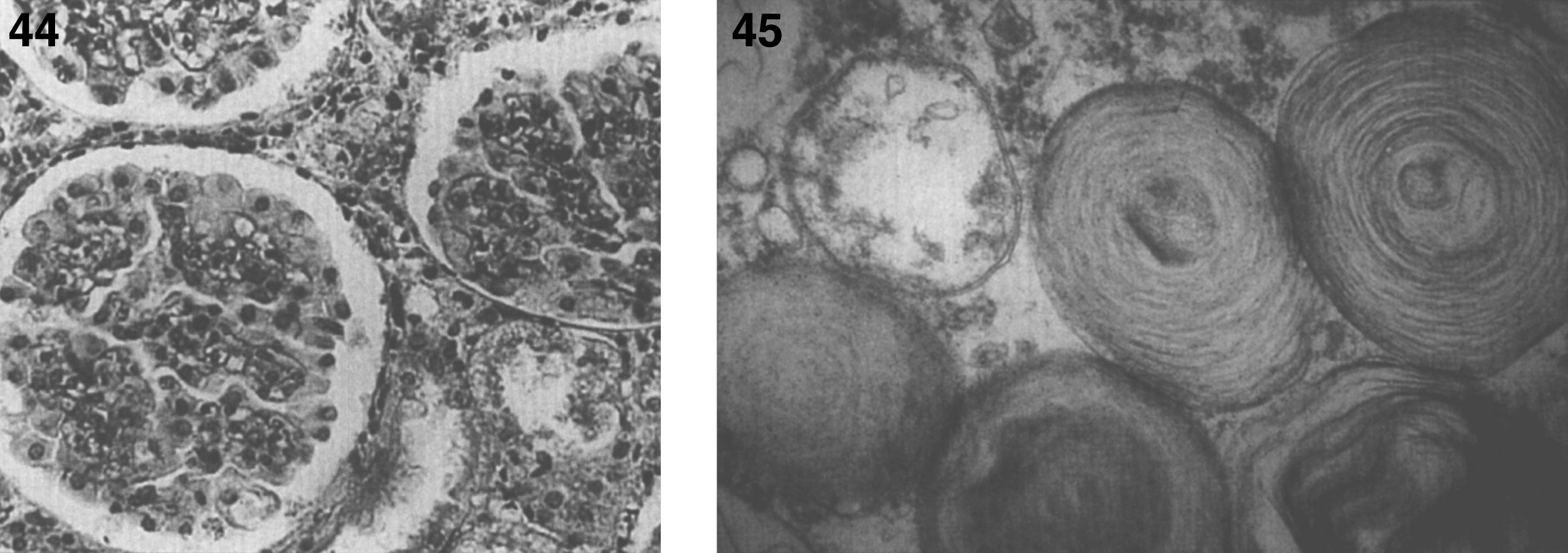
In the fetus at midgestation, nervous system involvement is expressed prominently in the most mature neurons, i.e., in myenteric plexus, spinal cord, cerebellum, and deeper cerebral cortical regions. The lysosomes are mostly empty, with a few fibrils in neurons of the brain, whereas more mature peripheral neurons contain membranous inclusions [185, 186]. Lysosomal inclusions have been described in liver, renal tubules, glomeruli, lymphocytes, and syncytiotrophoblasts of a 17-week-gestation fetus. The lysosomal pathology is most readily detected by EM in these fetal cases. Little may be evident by LM, except in the placenta where the trophoblastic epithelium is vacuolated.
2.9GM1 gangliosidosis type II (Derry’s syndrome)
The late-infantile, or juvenile form of the disease was first described by Derry and colleagues in 1968 [187]. GM1 type II gangliosidosis (juvenile) usually becomes apparent at about 1 year of age and is clinically and pathologically similar to type I gangliosidosis [186]. The facial features are characteristic, with prominent philtrum (Fig. 46), hypertrophic gingivae (Fig. 47), enlarged tongue, and prominent papillae. There are radiologic features of dysostosis multiplex (Fig. 48). Mental and motor deterioration begins after the first year of life. Viscera are usually not significantly enlarged and only subtle bone changes may be present. Deficiency of β-galactosidase results in excessive accumulation of GM1 ganglioside and its asialo derivative in the CNS and of a keratan sulfate-like galactose-containing proteoglycan in viscera.
Figs. 46,47
GM1 gangliosidosis type II. Facial appearance at autopsy. Mild anomalies of the ear and a prominent philtrum are present; (47) GM1 gangliosidosis type II. Appearance of oral cavity at autopsy. Note marked gingival hypertrophy.
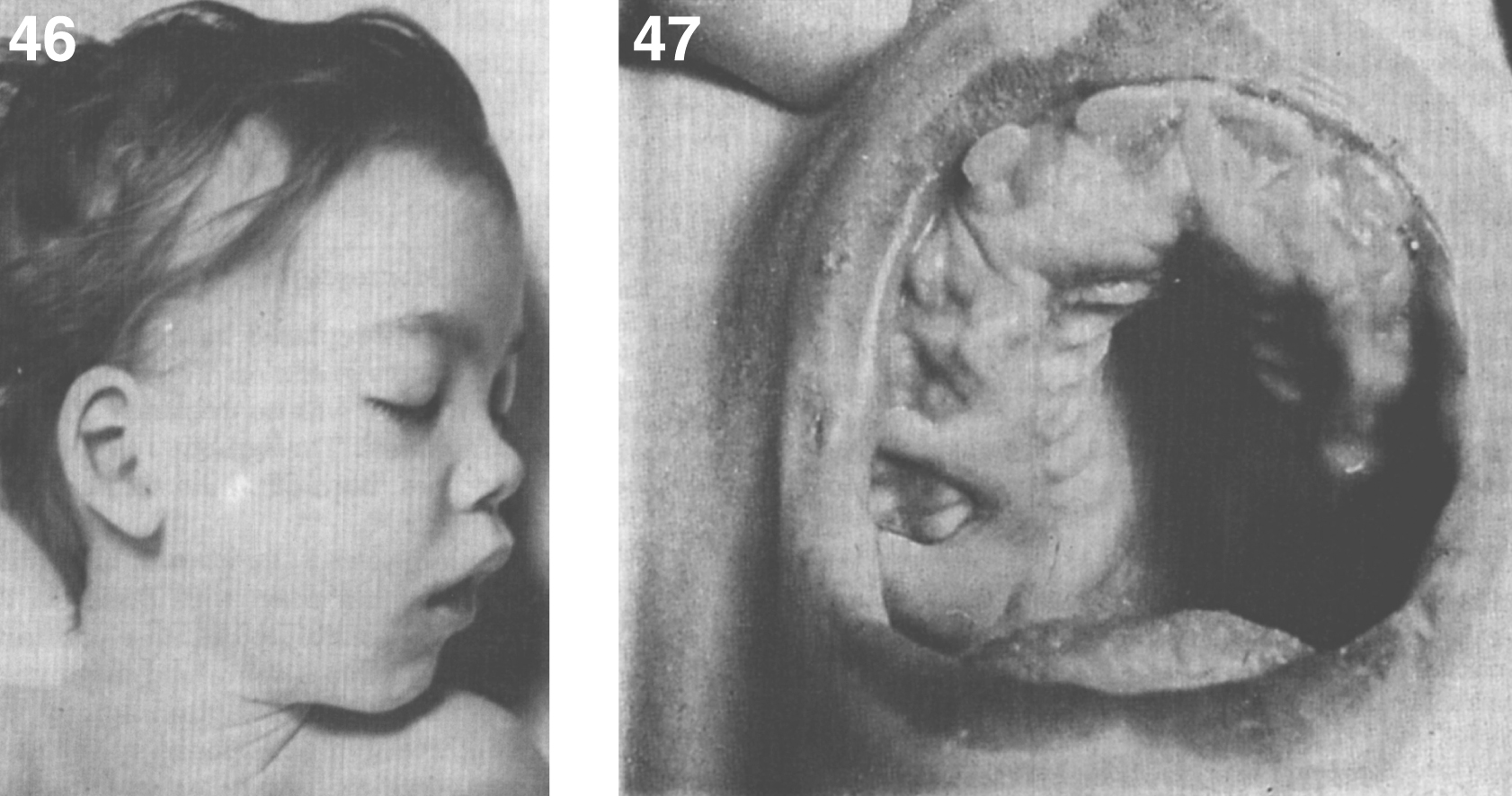
Figs. 48,49
GM1 gangliosidosis type II. Postmortem X-ray. (A) Changes of dysostosis multiplex in skull. (B) Lateral projection of vertebrae that shows mild generalized platyspondyly of cervical vertebrae and tear-drop deformity of vertebrae; (49) GM1 gangliosidosis type II. Gross appearance of heart opened through the left ventricle. The mitral valve leaflets are markedly deformed and thickended.
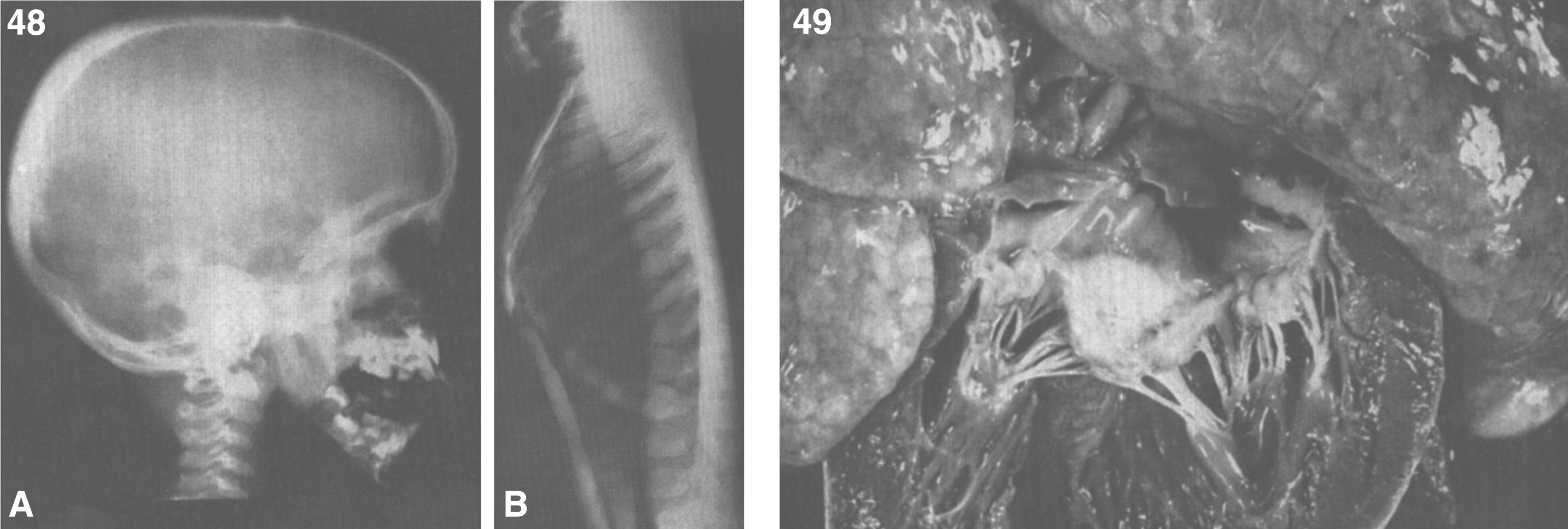
The most consistent pathologic changes of GM1 gangliosidosis (Table 14) include: (i) neuronal lipidosis, swelling of neurons, presence of abnormal neuronal cytoplasmic organelles; (ii) swelling of the renal visceral glomerular epithelium and other renal parenchymal cells; (iii) severe cardiac valvular involvement with excessive deposition of mucopolysaccharide in heart valves; and (iv) the presence of foamy histiocytes in the liver, spleen, lymph nodes, and bone marrow. The cytoplasmic inclusions in visceral histiocytes are PAS-positive, Alcian blue-positive, weakly metachromatic, and weakly sudanophilic. The neuronal lipidosis occurs throughout the cortex, brainstem, and spinal cord and also in the myenteric plexus. The neuronal cytoplasm is ballooned with storage material displacing the nucleus to the periphery. The storage substance appears as finely dispersed particles in LM. These manifestations characterize both forms of the disease, but are more extensive in type I [186].
Table 14
Pathologic manifestations of 3 siblings with GM1 gangliosidosis type II
| Case 1 | Case 2 | Case 3 | |
| General Features | |||
| Contractures | + | + | + |
| Hypertrophy of gums | + | + | + |
| Lumbar kyphosis | + | + | + |
| Exotropia | + | + | + |
| Mildly thickened tongue | + | ||
| Heart | |||
| Cardiomegaly | + | – | + |
| Valves thick, rigid, and retracted | + | + | + |
| Storage histocytes in valve leaflets (Alcian blue, PAS, and toluidine blue positive) | + | + | + |
| Spleen | |||
| Splenomegaly | + | + | + |
| Storage histiocytes | + | + | + |
| Liver | |||
| Hepatomegaly | + | + | + |
| Alcian blue, PAS, and toluidine blue positive material in hepatocytes | – | + | + |
| Alcian blue, PAS, and toluidine blue positive material in Kupffer cells | – | + | + |
| Lipid in hepatocytes | + | + | + |
| Brain | |||
| Weight below normal | + | + | + |
| Leptomeninges thick and fibrotic | + | + | + |
| Marked to severe atrophy of cerebral and cerebellar cortices | + | + | + |
| Loss of cerebral cortical neurons, extensive | + | + | + |
| Loss of neurons, brain stem nuclei | + | + | + |
| Storage material in neurons of cerebral cortex, cerebellar cortex, and brainstem nuclei | + | + | + |
| Spinal cord | |||
| Storage material in anterior horn cells | + | + | + |
| Kidneys | |||
| Vacuolation of glomerular visceral epithelium | + | + | + |
| Vacuolation of tubular epithelium | + | + | + |
| Intestine | |||
| Storage material in myenteric plexus | + | + | + |
| X-Ray Findings in Bones | |||
| Cortical thinning of long bones | + | ||
| Anterior protrusion, lower sternum | + | + | |
| Hypoplasia, vertebral bodies | + | ||
| Osteosclerosis, calvarium and base of anterior fossa | + |
Unlike the myocardial involvement in GM1 type I the myocardial cells of GM1 type II do not appear to contain storage material [186]. Both mitral and aortic valve leaflets are thickened (Fig. 49), retracted, and markedly deformed, and fused with the chordae tendineae. The valve cusps contain vacuolated histiocytes (Fig. 50). The aorta and its major branches show no evidence of intimal thickening or plaques [186]. Electron micrographs of cytoplasm of storage histiocytes from the heart valves show (i) membrane-bound structures filled with reticulo-granular material and (ii) membranous arrays with circular profiles.
Figs. 50–52
GM1 gangliosidosis type II. Microscopic section of mitral valve showing vacuolated histiocytes and intense staining of collagen. Alcian blue stain; (51) GM1 gangliosidosis type II. Gross appearance of brain showing atrophy of cerebral hemisphere, particularly the frontal and occipital lobes; (52) GM1 gangliosidosis type II. Microscopic section of cerebellar cortex showing large distended Purkinje cells. PAS stain.
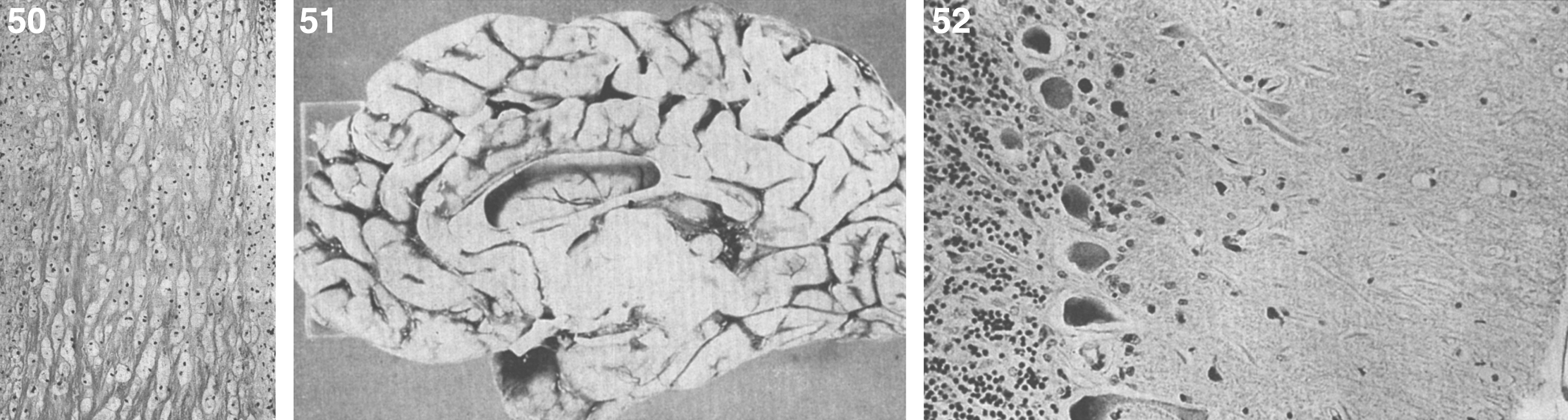
The brain has marked, diffuse atrophy of the cerebral cortex (Fig. 51) and of the cerebellar cortex including the vermis. The consistency of both cerebral and cerebellar tissues is firmer than usual. On section, hydrocephalus ex vacuo may be present. The corpus callosum is extremely thin, measuring only 1 mm in its posterior portion [186]. Most of the nerve cells are distended with granular cytoplasmic inclusions. Similar changes are seen in the cerebellum, where Purkinje cells and their dendritic processes are swollen to varying degrees and contain cytoplasmic granules with the same staining characteristics (Fig. 52). Astrogliosis and microglial mobilization with glitter cell formation are present in all affected parts of the brain [186]. The anterior horn cells of the spinal cord are large, distended, and vacuolated (Fig. 53). Sections from cerebral and cerebellar cortices show inclusions in the perikarya of neurons and glial cells, and also in the processes of Purkinje cells. Three types of inclusions are identified. MCBs (Fig. 54) are present in most neuronal perikarya and their processes and in glial cells. Their size varies, the largest being 3 μm in diameter. They consist of alternating electron-lucent and electron-opaque bands with a periodicity of 60 Å with no limiting membrane [186].
Figs. 53–55
GM1 gangliosidosis type II. Microscopic section of spinal cord showing large vacuolated anterior motor neurons. PAS stain; (54) GM1 gangliosidosis type II. Electron micrograph of cerebral cortical neuron. Membranous cytoplasmic bodies (MCB) in neuronal perikaryon; (55) GM1 gangliosidosis type II. Electron micrograph of cerebral cortical neuron. The cytoplasm is filled with pleomorphic lipid bodies (PLB).
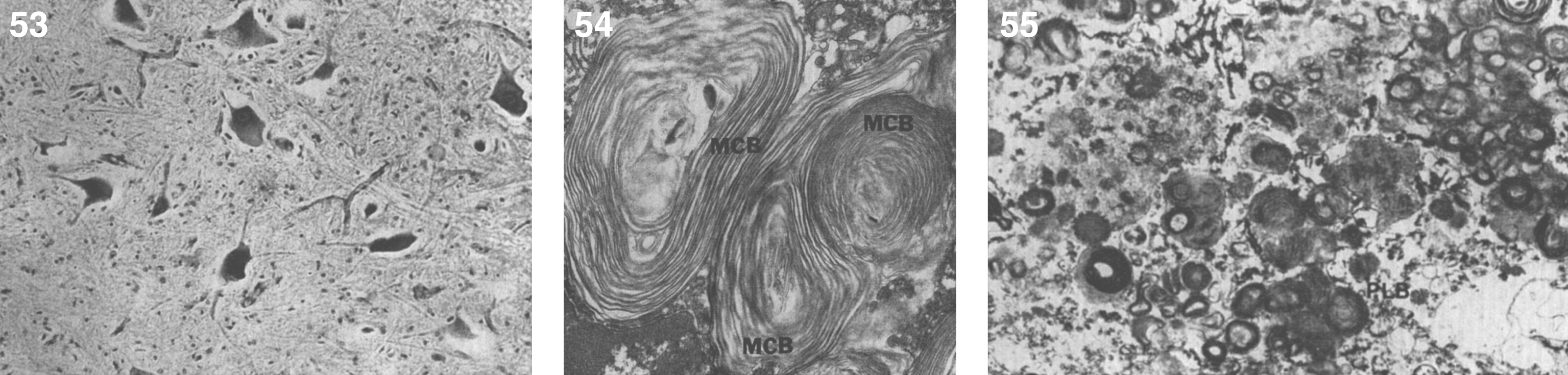
Pleomorphic lipid bodies (PLBs) are also observed in nerve and glial cells (Fig. 55), as are stacked or circularly arranged lamellae (Fig. 56). Occasionally the lamellae are tightly packed with a periodicity of 30 Å. More often, however, the periodicity is 60 Å, as in the MCBs. In some areas the granular material appears to represent fragments of the lamellae. The PLBs are often surrounded by a limiting membrane [186].
Figs. 56,57
GM1 gangliosidosis type II. Electron micrograph of glial cell containing cytoplasmic inclusion displaying unusual lamellar pattern; (57) GM1 gangliosidosis type II. Electron micrographs of tubular inclusions (T) in cytoplasm of splenic macrophage. Mosaic-like pattern of membrane bounded compartments filled with tubules.
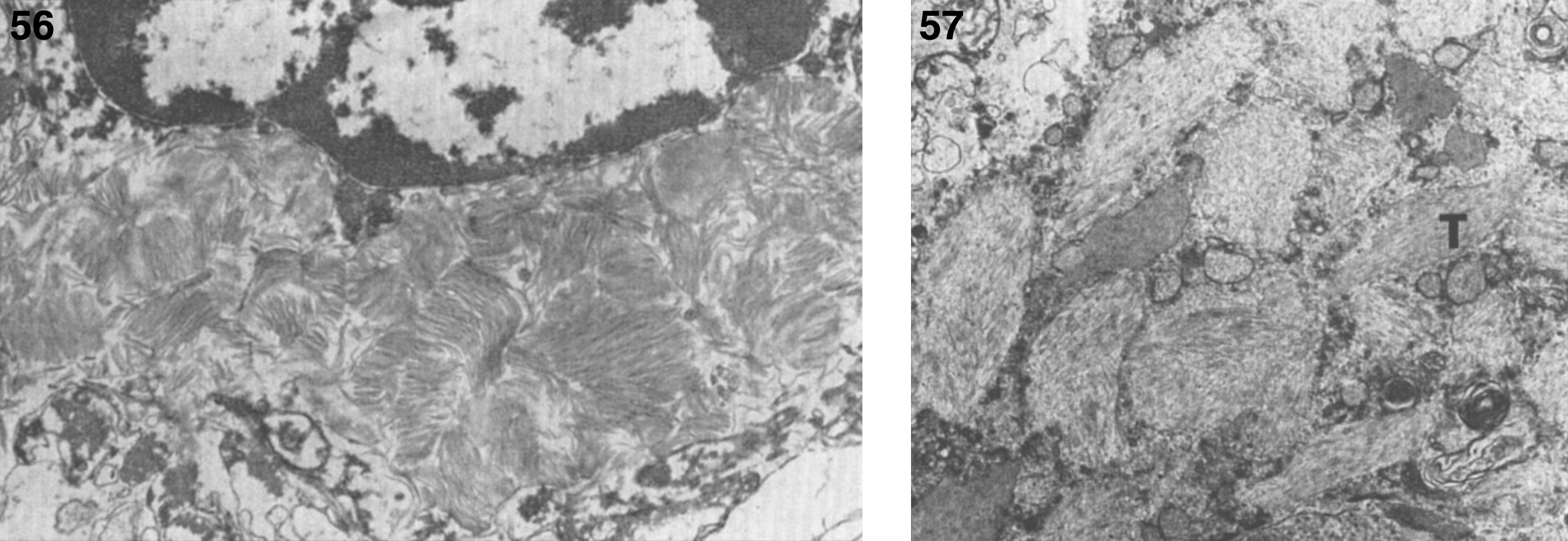
The involvement by PAS-positive deposits in renal glomerular epithelial cells is a characteristic of GM1 gangliosidosis [175] not seen in other types of lipid storage diseases except Fabry disease [188], I-cell disease [189] and Farber disease. The glomerular epithelial cells store large quantities of cytoplasmic PAS-positive material that is excreted in the urine in both the infantile and juvenile types of GM1 gangliosidosis [190]. Hence toluidine blue and Alcian blue tests for urine mucopolysaccharides may be positive.
The visceral histiocytes in GM1 gangliosidosis appear to be enlarged chiefly due to storage of mucopolysaccharides rather than of gangliosides. The Kupffer cells, some hepatocytes, and splenic histiocytes show lipid inclusions with electron-dense or, less frequently, with electron-lucent centers and deposits of tubular structures (Figs. 57 and 58). Suzuki et al. [191] demonstrated that in juvenile GM1 gangliosidosis the liver and spleen contain approximately 10 times the normal amount of mucopolysaccharide, which is similar to keratan sulfate. The concentration of this keratan sulfate-like mucopolysaccharide in the liver is greater in generalized gangliosidosis than in the juvenile form [186].
Lysosomal membrano-granular material, reflecting mucopolysaccharide, accumulates in the visceral histiocytes of GM1 gangliosidosis. The tubules contained within the cytoplasm of Kupffer cells and of splenic macrophages are strikingly similar to those described by Suzuki et al. [192] in macrophages of the liver and spleen and by Lowden et al. [193] in macrophages of liver, spleen, and bone marrow of the same disease. The former investigators give the width of the tubules as approximately 200 Å. Membrane-bound cytoplasmic deposits of tubules have also been reported for Krabbe disease [194] and Gaucher disease [195]. However, the tubules seen in the globoid cells of the brain appeared distinctly twisted in transmission electron micrographs and ranged from 225 to 350 Å in width. Tubules found in Gaucher cells of the liver and spleen also appeared twisted and measured 400 to 600 Å in width. Chemical analysis of the tubules of any of the aforementioned conditions has not been performed. Lee [196] compared by transmission electron microscopy the tubule of purified cerebroside with the tubules in a fractionated preparation of Gaucher cells and concluded that the latter contained a protein component, seen as fine fibrils, that was not present in the cerebroside. The histiocytes of the bone marrow have a “crinkled paper” appearance resembling that of the Gaucher cell [193]. The bone marrow contains large histiocytes (Fig. 59), and the liver (Fig. 60), kidney (Fig. 61), and myenteric plexus (Fig. 62) show storage cells.
Figs. 58–60
GM1 gangliosidosis type II. Electron micrograph of tubular inclusions in cytoplasm of splenic macrophage with curved tubules at higher magnification and cut obliquely; (59) GM1 gangliosidosis type II. Microscopic section of marrow with large histiocytes containing PAS positive material. PAS stain; (60) GM1 gangliosidosis type II. Microscopic section of liver showing Kupffer cells distended with storage material.
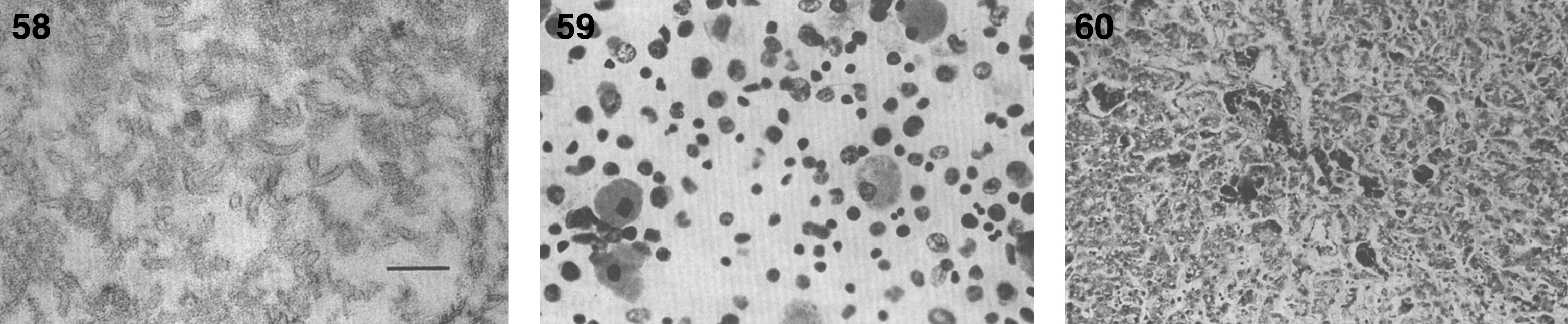
Figs. 61,62
GM1 gangliosidosis type II. Microscopic section of kidney showing enlarged visceral glomerular epithelial cells containing granules; (62) GM1 gangliosidosis type II. Microscopic section of gastrointestinal tract showing ganglion cells in the myenteric plexus. One ganglion cell contains granular deposits stained deeply with Luxol fast blue.
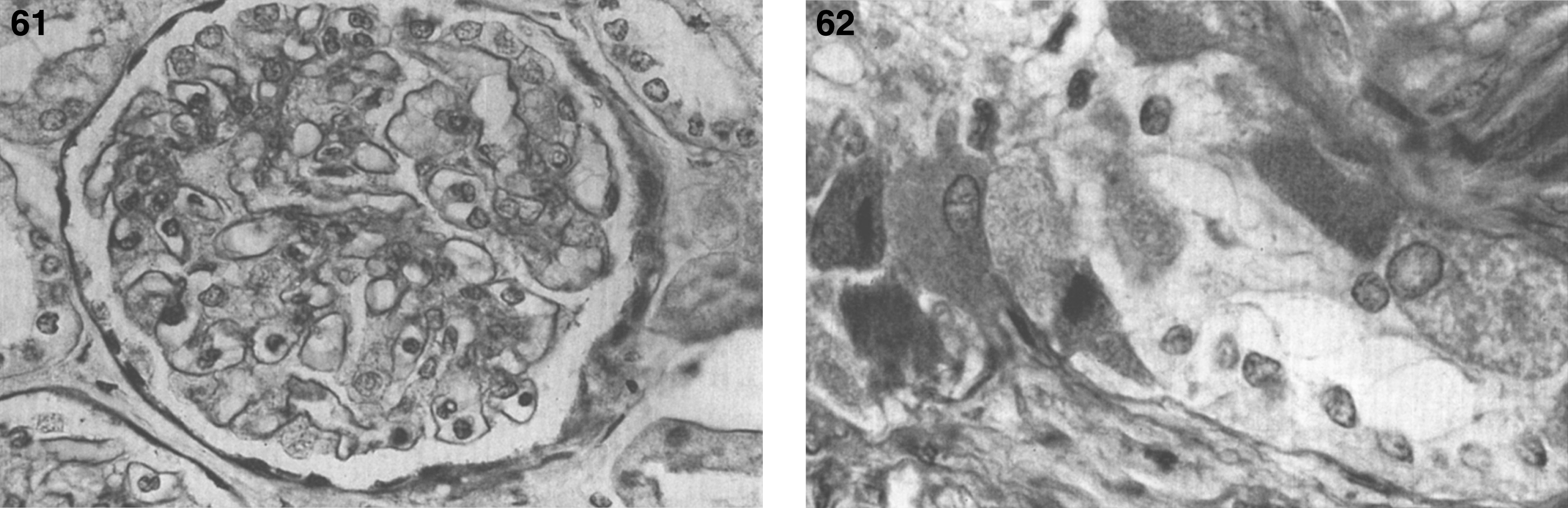
No morphologic differences have been demonstrated between the MCBs seen in GM1 gangliosidosis and those of Tay-Sachs disease; acid phosphatase activity has also been demonstrated in MCBs of Tay-Sachs disease [197].
Ultrastructural studies of the retina in both types of GM1 gangliosidosis demonstrated MCBs in ganglion cells [198]. MCBs have also been observed in the processes of Miller cells [199]. Clinical and LM evidence of retinal involvement has not been documented in the juvenile form of this disease. Neurons of fetal brain in GM1 and GM2 gangliosidosis have been found to contain MCBs [200]. However, in fetal brain, they were described as incompletely developed and as lacking the compactness of mature MCBs [186].
2.10GM1 gangliosidosis type III (adult or chronic)
This disorder is a milder version of types I and II, with onset in the teens and lack of visceral involvement. GM1 gangliosides accumulate in the central nervous system, especially the basal ganglia, and keratan sulfate degradation products accumulate in somatic cells. More than half of all patients reported in the literature are from Japan [201]. The most common presentation is that of progressive generalized dystonia, and almost half of all patients have associated akinetic-rigid parkinsonism [201]. Facial dystonia, seen in approximately 90% of patients, is an important diagnostic clue [202, 203]. Gait and speech disturbances–the latter in the form of dysarthria or anarthria–are almost universal during the course of the disease [201]. Bilateral putaminal hyperintensities on T2-weighted magnetic resonance imaging can be seen in about half of all cases, especially in Japanese patients [201]. In rectal biopsies, histiocytes with PAS-positive granules can be seen in the mucosa, while ultrastructurally the ganglion cells of Meissner’s plexus have osmiophilic lamellar inclusions [204–206].
2.11GM2 gangliosidoses
Tay-Sachs disease was first described by a British ophthalmologist, Waren Tay, in 1881 [207]. The original patient died soon afterwards, but Dr. Tay reported a more complete clinical description of the disease in a second patient from the same family in 1884. An American neurologist, Bernard Sachs, first published the brain cortical pathology of the disease in 1887 [208]. Ernst Klenk, in Cologne, first detected a new glycosphingolid in brains from patients with Tay-Sachs disease and Niemann-Pick disease [209], and later coined the term ganglioside since the same lipid was found in abundance in normal ganglion cells [210]. The deficiency of hexosaminidase enzymatic activity was not discovered until 1969 [211], although a year earlier Sandhoff, Andreae and Jatzkewitz had described a more generalized form of hexosaminidase deficiency, with the additional finding of globoside storage in visceral organs [212].
GM2 gangliosidoses are autosomal recessive disorders caused by deficiency of β-hexosaminidase and, consequently, excessive intralysosomal accumulation of ganglioside GM2 and related glycolipids, particularly in neuronal cells [213]. The enzymatic hydrolysis of ganglioside GM2 by β-hexosaminidase requires that GM2 be complexed with a substrate-specific cofactor, the GM2 activator. There are 2 isoenzymes of β-hexosaminidase: Hex A, consisting of 2 subunits, α and β; and Hex B, a homodimer of β subunits. Only Hex A can act on the ganglioside GM2-GM2 activator complex. Defects in any of 3 genes may lead to GM2 gangliosidosis: HEXA (on chromosome 15), which encodes the α subunit of Hex A; HEXB (on chromosome 5), which encodes the β subunits of Hex A and Hex B; or GM2A (on chromosome 5), which encodes the GM2 activator. There are 3 forms of GM2 gangliosidoses: (i) Tay-Sachs disease and variants, resulting from mutations of the HEXA gene and associated with deficient activity of Hex A but normal Hex B (variant B); (ii) Sandhoff disease and variants, resulting from mutations of the HEXB gene and associated with deficient activity of both Hex A and Hex B (variant O); and (iii) GM2 activator deficiency, due to mutation of the GM2A gene and characterized by normal Hex A and Hex B but the inability to form a functional ganglioside GM2-GM2 activator complex (variant AB). There are also pseudodeficient or clinically benign mutations characterized by a Hex A enzyme that exhibits biochemical defects but has functional activity toward ganglioside GM2. The gross pathologic changes are similar in Tay-Sachs disease, Sandhoff disease, and GM2 activator deficiency, except that visceral organ involvement is evident in Sandhoff disease. The most pronounced cellular change is the presence throughout the nervous system of swollen neurons with massive accumulation of storage material in lysosomes. These form characteristic inclusions, the so-called membranous cytoplasmic bodies [213].
The hexosaminidases and GM2 activator are glycoproteins that are synthesized in the lumen of the endoplasmic reticulum and processed through the Golgi. They are transported via the mannose 6-phosphate receptor to the lysosome, where they are processed further to their final mature forms. Hex A and Hex B hydrolyze a broad spectrum of substrates with terminal N-acetylglucosamine (GlcNAc) residues in β linkage. While only hexosaminidase A hydrolyzes GM2, both isoenzymes hydrolyze glycoproteins, glycosaminoglycans, and glycolipids. Both also hydrolyze synthetic substrates, of which the most sensitive and commonly used is a fluorogenic substrate, the β-GlcNAc derivative of 4-methylumbelliferone (4MUG). 4MUG is recognized by both Hex A and Hex B and does not require the GM2 activator. Hex A and Hex B can be distinguished by different thermal, pH, or electrophoretic characteristics of the isoenzymes. In particular, hexosaminidase A is heat-labile at 50°C, while hexosaminidase B is heat-stable at that temperature. In addition to 4MUG, a related compound cleaved by Hex A, but not Hex B, is 4-methylumbelliferyl-GlcNAc-6-sulfate (4MUGS). Substrates such as 4MUG or 4MUGS are used for routine diagnostic testing and screening for carriers of HEXA or HEXB mutations [213]. The total serum hexosaminidase activity is measured using 4MUG as a substrate. A second serum aliquot is exposed to heat, thus inactivating hexaosaminidase A and reflecting only the activity of hexosaminidase B. The hexosaminidase A activity is then calculated by substracting the hexosaminidase B activity from the total hexosaminidase activity. One limitation of this technique is that certain HEXA mutations make hexosaminidase A thermostable, causing the B1 variant of Tay-Sachs disease, in which hexosaminidase A activity is normal when measured by the standard heat-inactivation technique, but is reduced when using 4MUGS as a substrate. The B1 variant (also known as pseudo-AB, or AMB variant) is more common in patients of Portuguese ancestry [214]. The converse can also be true; a mutation in HEXB can render hexosaminidase B thermolabile, thus mimicking the biochemical phenotype of Sandhoff disease in a normal patient [215], or masking a carrier status for Tay-Sachs disease, since it artificially raises the activity of hexosaminidase A [216]. Another limitation of the heat-inactivation method is that pregnant women or those taking oral contraceptives produce a circulating heat-stable hexosaminidase P, which can falsely decrease the calculated serum activity of hexosaminidase A relative to that of total hexosaminidase. This is why carrier status in pregnant women or those taking OCPs should be assessed in leukocytes, as opposed toserum.
Clinical phenotypes in the GM2 gangliosidoses vary widely. Infantile-onset, rapidly progressive neurodegenerative disease culminates in death before 4 years of age (classic Tay-Sachs disease and Sandhoff disease, as well as GM2 activator deficiency). Later-onset, subacute, or chronic forms exhibit slower neurological progression, compatible with survival into childhood or adolescence (subacute form) or into adulthood (chronic or adult-onset forms) [104]. The most common presenting symptoms in late-onset cases are balance problems and difficulty climbing stairs [217]. A very characteristic pattern of predominant triceps, iliopsoas and quadriceps weakness is seen with late-onset disease [217]. Chronic forms include several different clinical phenotypes, each dominated by symptoms referable to one or another part of the CNS, including progressive dystonia, spinocerebellar degeneration, motor neuron disease [218–221], spinal muscular atrophy [222–225], and psychosis [213, 226]. Patients with late-onset Sandhoff disease can have autonomic neuropathy [227, 228], which can lead to acroparesthesias similar to the ones experienced by patients with Fabry disease [229].
There are more than 175 known mutations scattered over the 14 exons of HEXA [31], including founder mutations among the French Canadians, Cajuns, Irish, and Brazilians [213, 226]. The most common among Ashkenazi Jews, accounting for over 90% of the Tay-Sachs disease in that community, are p.Tyr427Ilefs*5 and a donor splice-junction mutation in intron 12, c.1421+1G>C; these are associated with the severe, infantile-onset disease [226]. The c.1073+1G>A mutation is associated with severe disease among non-Jewish invididuals, and the p.Gly269Ser mutation results in adult-onset disease [226]. Mutations causing subacute or chronic forms allow for low levels of residual activity toward ganglioside GM2. The level of activity correlates inversely with the severity of the disease. Hex A pseudodeficiency is caused by point mutations (p.Arg247Trp and p.Arg249Trp) that leave the Hex A isozyme with reduced but variable activity toward synthetic substrates but with functional ganglioside GM2-hydrolyzing activity [213, 226].
Heterozygotes for any of the defects are completely asymptomatic. In the non-Jewish population, heterozygote frequencies are estimated at 0.006 for HEXA mutations and 0.0036 for HEXB mutations. Among the Ashkenazi Jewish populations of North America and Israel, a heterozygote frequency of 0.033 was found for HEXA mutations. Genetic counseling and monitoring of at-risk pregnancies have reduced the incidence of Tay-Sachs disease in the Ashkenazi Jewish population by 90%.
2.12GM2 gangliosidosis type I (Tay-Sachs disease)
GM2 gangliosidosis type I takes three forms, i.e., variants B and B1 with mutations of the α subunit of β-hexosaminidase, and the AB variant due to mutations in the hexosaminidase activator protein. The prototype, type B, has a frequency in Ashkenazi Jews of 1 in 4,000. Since the α but not β subunit is mutated, the total amount of β-hexosaminidase is normal, but the hexosaminidase A activity relative to the total hexosaminidase activity is reduced. Affected infants are normal at birth, but in the first year of life develop rapidly progressive psychomotor deterioration, seizures, hypotonia, blindness, dementia and death by 3 to 5 years. The typical cherry-red spot (Fig. 63) is a red area that represents a normal segment of the retina rendered vivid by contiguous white areas, which contain the storedmaterial.
The pathologic changes are similar to GM1 gangliosidoses except that in GM2 type I (Tay-Sachs) the changes are confined to the CNS. Ganglioside accumulation results in massive gliosis and intralysosomal accumulation of lipophilic membranous bodies and MCBs by EM (Fig. 64). The brain is enlarged early due to the accumulation of storage material in the neurons but becomes atrophic later (Fig. 65). The Jacob sheep serves as an animal model of Tay-Sachs disease [230]. Carrier screening and prenatal diagnosis have been established [231].
Fig.63
GM2 gangliosidosis type I (Tay-Sachs disease). The retina shows a cherry-red spot in the macula.
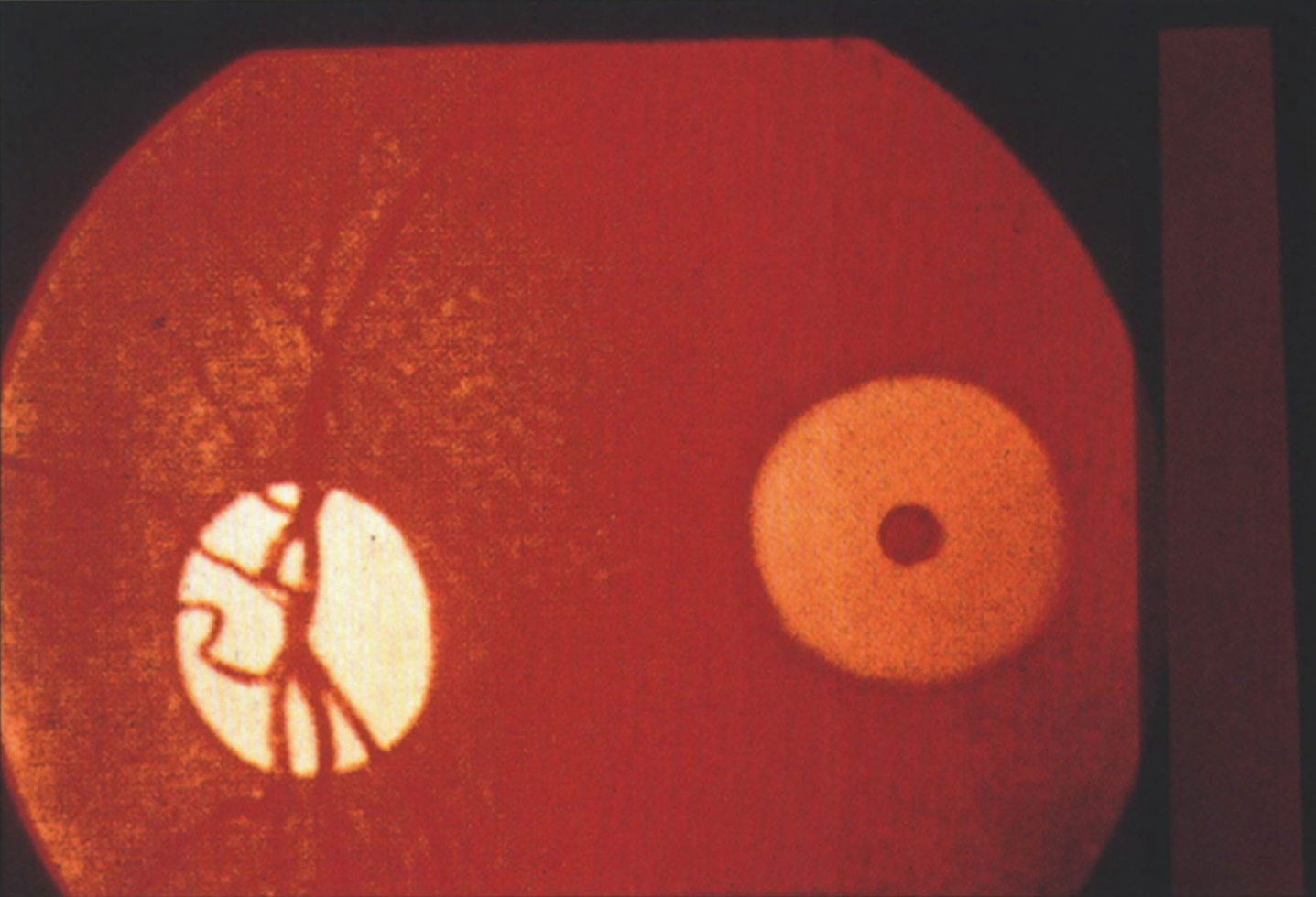
Fig.64
GM2 gangliosidosis type I (Tay-Sachs disease). (A) Microscopic section of the brain shows large swollen neurons due to accumulation of ganglioside. (B) Electron micrograph showing characteristic membranous concentric bodies.
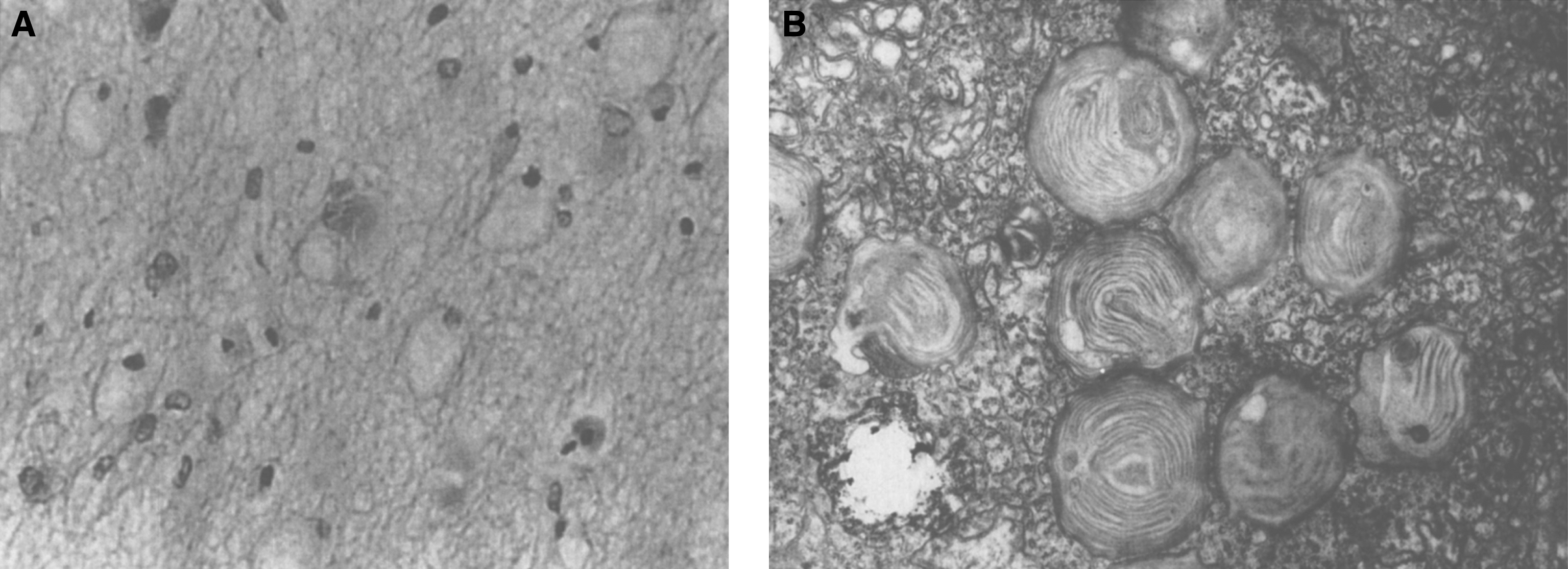
Fig.65
GM2 gangliosidosis type II (Sandhoff disease). (A) The brain shows atrophy of gyri. (B) On cross section, both gray and white matter are atrophic.
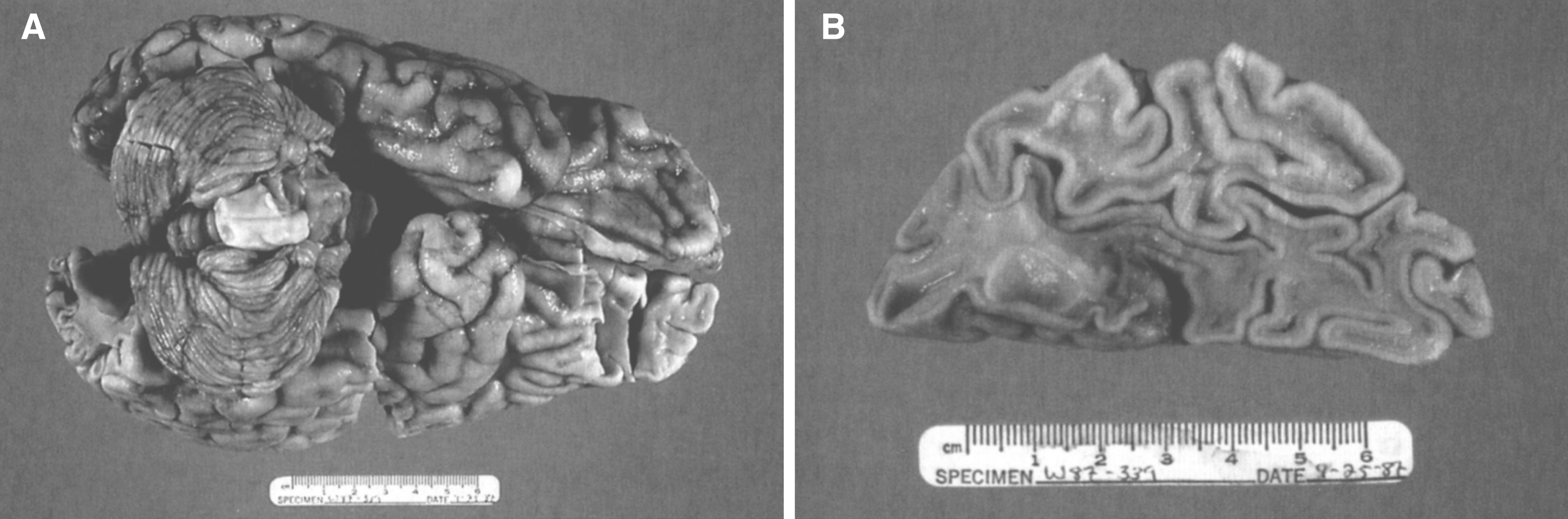
2.13GM2 gangliosidosis type II (Sandhoff-Jatzkewitz disease)
Various phenotypes occur within this group, which features a total deficiency of β-hexosaminidase leading to the extensive neuronal and visceral storage of GM2-gangliosides, glycolipids, glycoproteins, and oligosaccharides [226]. The pattern of inheritance is autosomal recessive, with a predicted incidence of about 1 in 1,000,000 and no Jewish predominance.
Sandhoff disease usually presents clinically at 3 to 6 months. Clinical signs include mental and motor delays, doll-like facial appearance, cherry-red spot of the retina, startle response to sound, macrocephaly, seizures, early blindness, and hypotonia. Death usually occurs by 2 to 5 years [231]. There is also a late-onset variant, with prolonged survival.
Sandhoff disease is caused by a deficiency of the enzymes Hex A and Hex B. Hexosaminidase A cleaves the substrates GM2, GA2, globoside, and hexosamine oligosaccharides. All of these substrates accumulate in Sandhoff disease. Cerebral levels of ganglioside GM2 are increased 100 to 300 times in Tay-Sachs and Sandhoff disease. A derivative of the ganglioside GM2 and GA2 accumulates to levels about 20 times normal. Much higher amounts of GA2 accumulate in the brain and viscera in Sandhoff disease than in Tay-Sachs disease. Globoside accumulates in large quantities in the viscera, especially in the kidney and spleen in Sandhoff disease, and oligosaccharides accumulate in the tissues and are excreted. Oligosaccharides appear to be derived from degradation of glycoproteins, which accumulate because of absence of both Hex A and Hex B [232, 233].
The diagnosis of GM2 gangliosidosis relies upon the assay of hexosaminidase in serum or cultured fibroblasts. Heterozygotes can be detected using serum. Low total hexosaminidase with a low percentage of HexB makes the diagnosis of GM2 or a GM2 carrier [226]. All the gangliosidoses and their variants can be diagnosed prenatally from cultured amniotic fluid cells or chorionic villus biopsies [226].
Several attempts have been made in recent years at enzyme replacement for patients with GM2 gangliosidoses [234], including by an intracerebroventricular route [235]. Other therapies that have been attempted include pyrimethamine [236–238] and substrate reduction with miglustat [239], but neither has shown clinical benefits. Investigational therapies under consideration include gene therapy and chaperone therapy. Supportive treatments include anti-epileptics for seizure control, anti-psychotics, maintenance of nutrition, and management of infectious diseases.
The pathologic changes in GM2 gangliosidosis are similar to those of the GM1 gangliosidoses. The cerebral hemispheres appear symmetrical with atrophic gyri (Fig. 66). The pons, medulla, cerebellum, and spinal cord are atrophic (Fig. 67) and rubbery in consistency. The aortic and mitral valve leaflets are thickened and distorted, and their histiocytes are filled with storage material. The liver, spleen, and lymph nodes are normal grossly, and microscopically show minimal storage material. The bone marrow contains an increased number of foamy, lipid-laden macrophages of storage material on microscopic examination. In the gastrointestinal tract, the ganglia are remarkably swollen and contain storage material [233]. EM examination of the brain, spinal cord, retina, liver and spleen document many intracellular and extracellular concentric laminations within neurons, microglia, ganglion cells and macrophages (Fig. 68) [101].
Figs. 66,67
GM2 gangliosidosis type II (Sandhoff disease). A coronal section of the brain, normal brain (top) compared with severe cortical atrophy (bottom); (67) GM2 gangliosidosis type II (Sandhoff disease). A normal spinal cord (top) is compared with atrophy in Sandhoff disease (bottom).
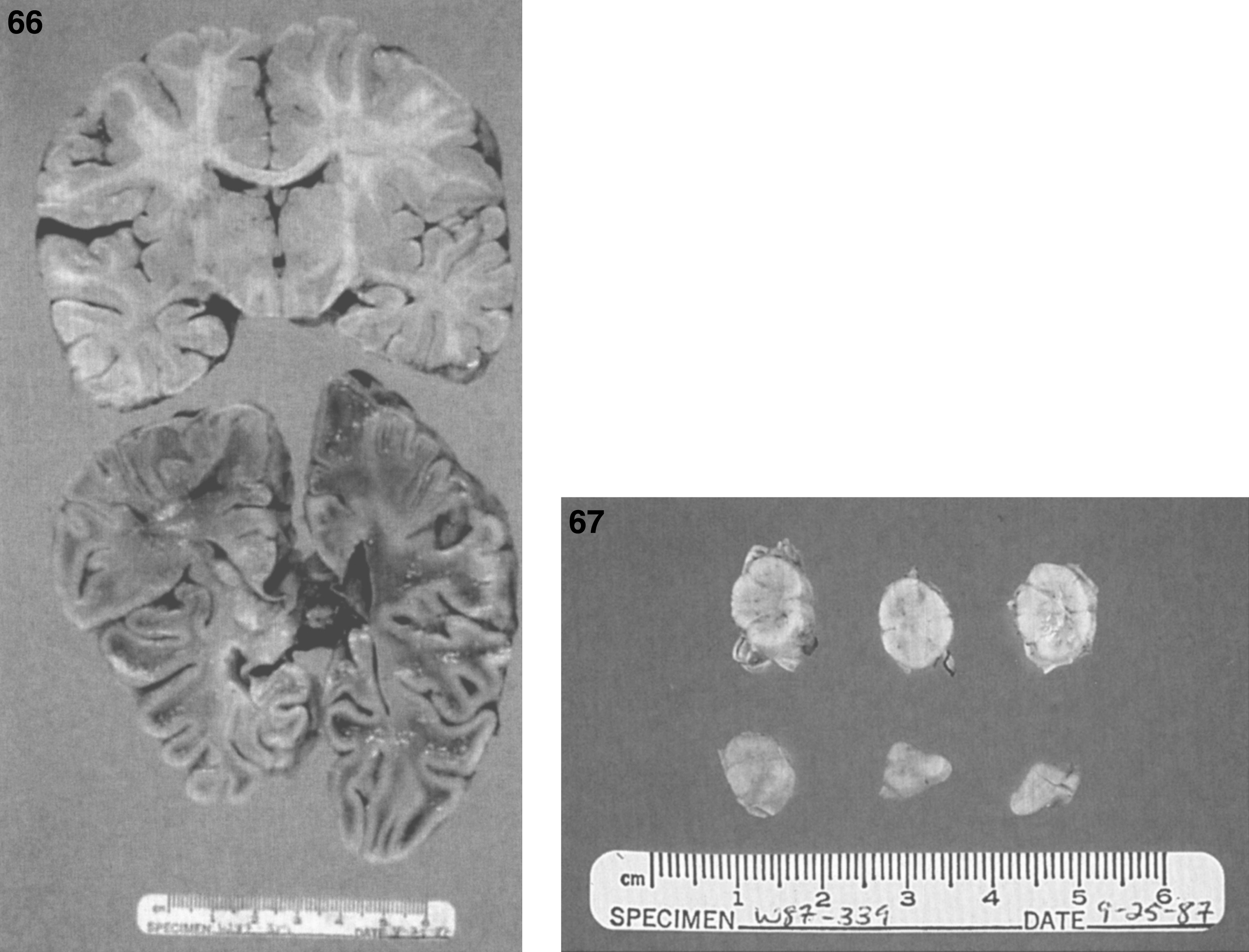
Fig.68
GM2 gangliosidosis type II (Sandhoff disease). Electron micrograph of (A) cerebral cortex showing inclusions of electron-dense profiles with concentric laminations, and (B) liver containing intracellular concentric laminations within hepatocytes.
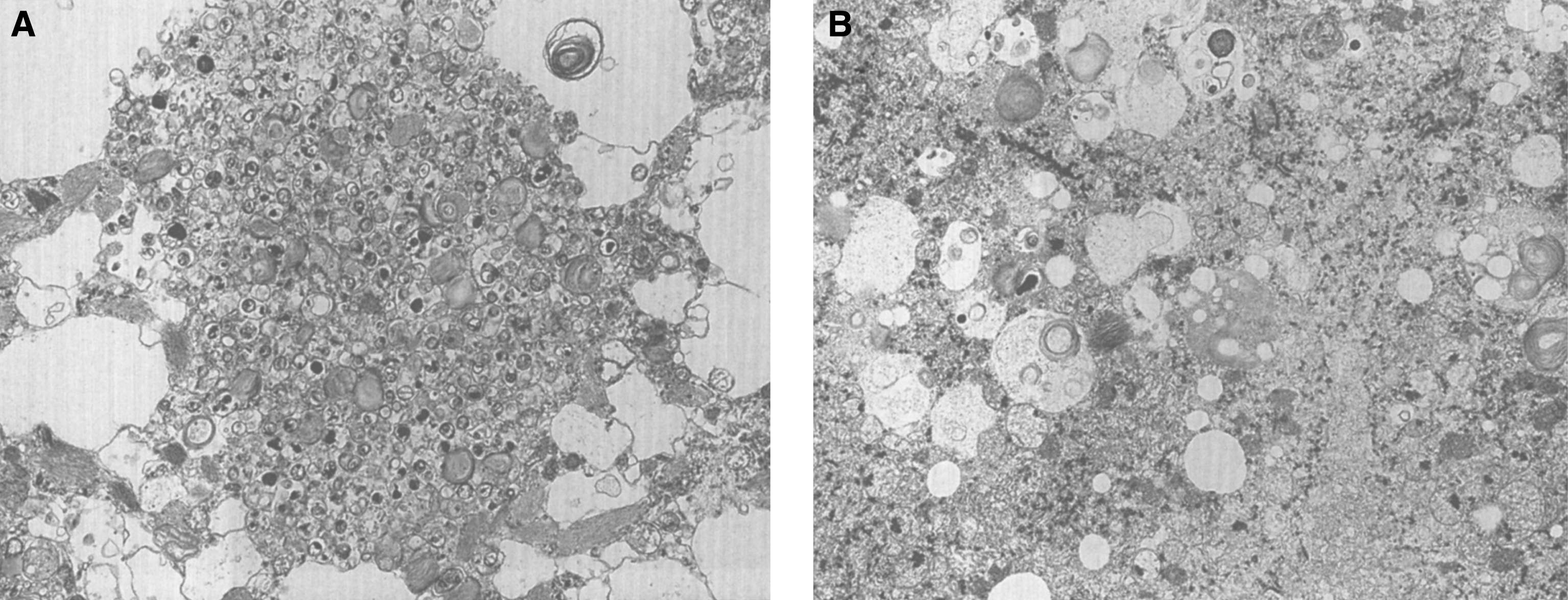
2.14Metachromatic leukodystrophy
The disease was first described by Scholz in 1925 [240], although it wasn’t until decades later that Scholz’s brain samples were shown to stain metachromatically [241]. Similar cases were reported by Bielschowsky and Henneberg in 1928 [242], and by Greenfield in 1933 [243], accounting for the old nomenclature of Scholz-Bielschowsky-Henneberg syndrome, or Greenfield disease. The enzymatic defect was not identified until 1963 [244]. Metachromatic leukodystrophy (MLD) is an autosomal recessive lysosomal storage disease with a frequency of 1 in 40,000 caused by deficiency of arylsulfatase A (EC 3.1.6.8) [245, 246]. This enzyme degrades sulfated glycolipids; one of its major substrates is cerebroside 3-sulfate, a lipid found mainly in myelin membranes, where it accounts for 3% to 4% of total membrane lipids. Arylsulfatase A removes the sulfate from a galactose residue of cerebroside 3-sulfate with the assistance of saposin B [247]. Deficiencies of this activator protein cause a very rare disease that is indistinguishable from metachromatic leukodystrophy [247]. In metachromatic leukodystrophy, galactosyl sulfatide (cerebroside sulfate) accumulates in the white matter of the CNS and in the peripheral nerves. Galactosyl sulfatide, and to a lesser extent lactosyl sulfatide, also accumulate within the kidney, gallbladder, and certain other visceral organs and are excreted in excessive amounts in the urine. In histologic preparations, they form spherical granular masses that stain metachromatically.
The human ARSA gene, on chromosome 22q, contains 8 exons encoding 507 amino acids [246]. Over 180 mutations have been identified [31]. MLD-causing mutations are found in 90-95% of patients [246].
Clinically, metachromatic leukodystrophy is heterogeneous. Three different forms can be distinguished: (i) a severe late-infantile form starting between the ages of 1 and 3 years; (ii) a juvenile form with an age of onset at 3 to 16 years; and (iii) adult forms that may not become apparent before the third decade of life. The progression is slower in the late-onset forms and patients may survive for as much as 20 years after the disease has started. Although the sulfatide storage occurs in all MLD tissues, it mainly affects the nervous system, leading to progressive demyelination. This demyelination is not just seen in the central nervous systems, but it also leads to peripheral neuropathy [248], which can be the presenting sign in adult-onset forms of the disease [249]. Psychiatric symptoms may prevail, particularly in adult patients, before the neurologic symptoms develop [245]. Proximal renal tubular acidosis has been described [250], with subclinical metabolic acidosis at baseline, worsening to a clinically significant acidosis in the acute setting [251]. Sulfatides are known to irritate the gallbladder mucosa, potentially leading to gallbladder papillomatosis [252, 253] and hemobilia [254–256]. A major determinant of the clinical phenotype is the residual enzyme activity that is associated with a particular genotype. In the typical case the disease starts at the age of about 18 months. Children lose acquired capabilities, develop a spastic tetraparesis, dysarthrias, ataxias, dementias, and finally die in a decerebrate state. In each of the variants, gait disturbance, mental regression, and urinary incontinence are among the earliest signs. In the childhood variants, other common signs are blindness, loss of speech, quadriparesis, peripheral neuropathy, and seizures. In the adult, behavioral disturbances and dementia are the major presenting signs, and the disease may progress slowly over several decades [245, 246]. It has been hypothesized that the first description of Alzheimer’s disease, presented by Alois Alzheimer in 1907, was in fact a case of metachromatic leukodystrophy [257].
Arylsulfatase A deficiency can also be observed in individuals who are clinically healthy [258]. This phenomenon has been termed pseudodeficiency. These individuals have only about 10- 20% of normal enzyme activity. The frequency of this arylsulfatase A pseudodeficiency allele is estimated to be between 7% and 15%, which predicts that 0.5% to 2% of the population are homozygous and thus pseudodeficient [259, 260]. Whereas pseudodeficiency is harmless for the carriers, it causes problems in the diagnosis and genetic counseling of MLD because it is difficult to differentiate pseudodeficiency from MLD on enzymatic grounds [259].
The diagnosis of MLD relies upon enzymatic assay of arylsulfatase A in leukocytes, dried blood spots, or fibroblasts, although some patients may have normal activity but deficiency of the enzyme activator protein or a Km mutant. In addition, some normal individuals may have pseudodeficiency. A nerve biopsy or detection of urinary sulfatide excretion based on the finding of brown metachromasia on a filter paper urine spot with cresyl violet may be used for diagnosis (Fig. 69). Although carrier detection can be problematic, prenatal diagnosis based on arylsulfatase A activity in cultured amniotic fluid or chorionic villus cells appears to be reliable.
There are few treatment options for MLD. For selected individuals, hematopoietic stem cell transplantation or bone marrow transplantation are options that may slow the progression of symptoms. In most late-infantile patients, symptoms are progressing rapidly by the time of diagnosis, and the risks of the procedure may outweigh the possible benefits. In instances in which the diagnosis can be made presymptomatically and a well-matched donor is available, bone marrow transplantation may be reasonable. Cell culture modeling suggests that protease inhibitor treatments, enzyme replacement, and gene replacement therapies could be effective [261]. Lentiviral vectors have been used to transfer a functional copy of the gene into the patient’s own hematopoietic stems cells, which are subsequentely reinfused. This approach halted disease progression in three individuals reported to date [262].
Fig.69
Metachromatic leukodystrophy. Urinary spot test showing metachromasia due to the presence of sulfatide (right) compared to normal (left).
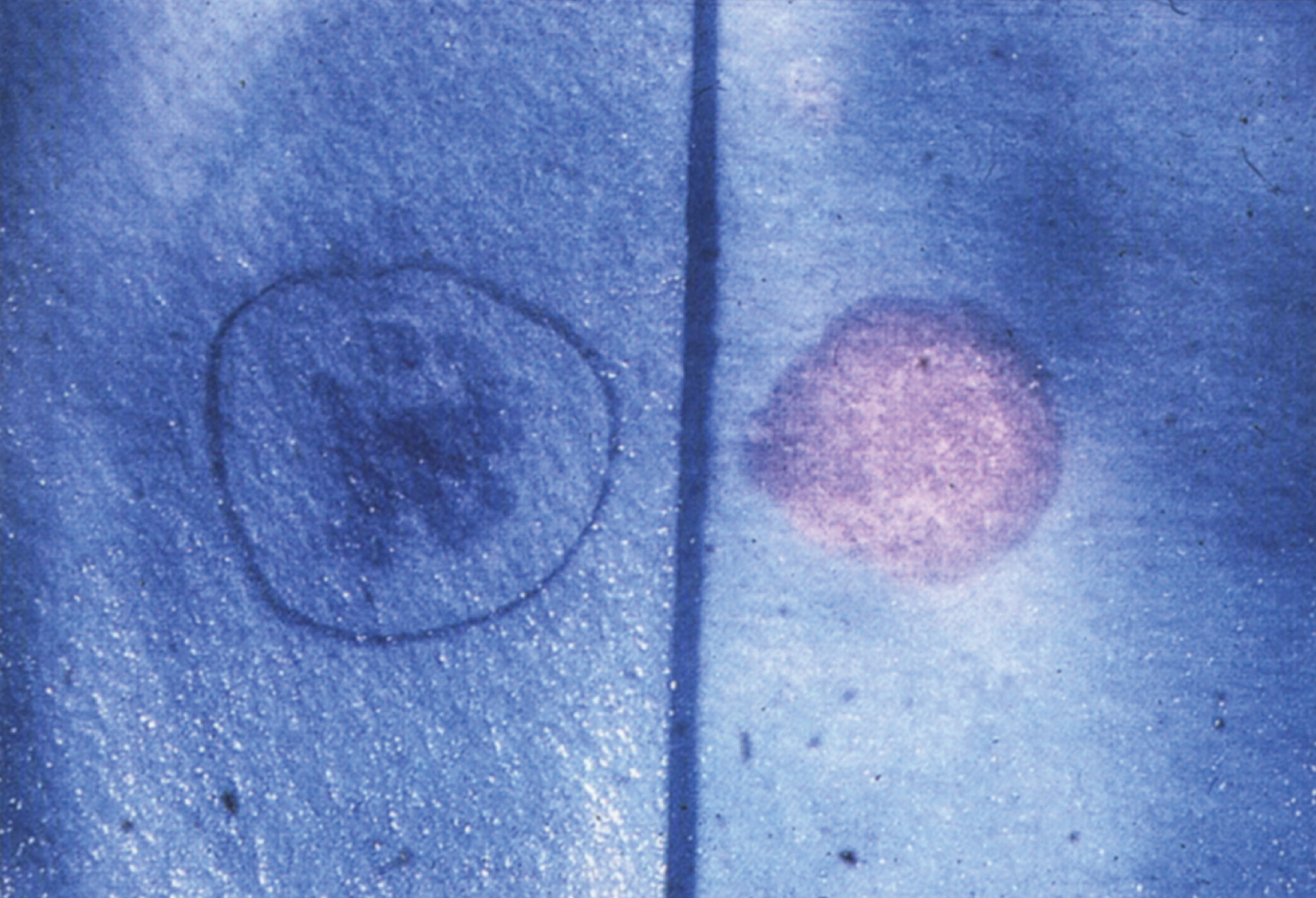
Table 15
Metachromatic leukodystrophy: Pathologic findings
| •Destruction of myelin in the cerebral and cerebellar white matter |
| •Gliosis |
| •Ballooned macrophages (vacuolated) |
| •Metachromatic staining of sulfatides in macrophages and white matter |
| –Basic dyes (e.g., acidified cresyl violet and toluidine blue) stain different colors when |
| they react with the tissue |
| –Frozen section tissue must be used |
| •Subcortical arcuate fibers may be spared |
| •EM: Prismatic inclusions, representing accumulated sulfatides, can be seen in all tissues |
The pathologic findings of MLD, summarized in Table 15, involve the cortex, brainstem, cerebellum (Fig. 70), and spinal cord. There is loss of retinal ganglion cells [263], which accumulate residual bodies surrounded by a trilaminar membrance [264, 265]. MCB-like inclusions can be seen in anterior horn cells, neurons in the globus pallidus and large pyramidal neurons of the motor cortex [266]. Cortical atrophy of the white matter is severe and there is accumulation of sulfatide in the neurons (Fig. 71) and other sites, notably in the lamina propria of the gastrointestinal tract and the gallbladder that may become polypoid (Fig. 72). The storage material is autofluorescent (Fig. 73), and can be seen even in utero [267]. Bile ducts and Kupffer cells in the liver and renal tubular epithelial cells in the kidney contain storage material (Fig. 74). A sural nerve biopsy also shows metachromatic substance (Fig. 75). Brown metachromasia is also seen in the pancreatic acinar cells (Fig. 76). Ultrastructurally cytoplasmic inclusions are seen in oligodendrocytes and astrocytes that may have a herringbone pattern (Fig. 77), amorphous material, and lamellar structures.
Fig.70
Metachromatic leukodystrophy. (A) The white matter in the cerebellum shows brown metachromasia after staining with cresyl violet. (B) Microscopic section of cerebellum showing brown metachromasia. (C) Dense metachromasia in the spinal cord.
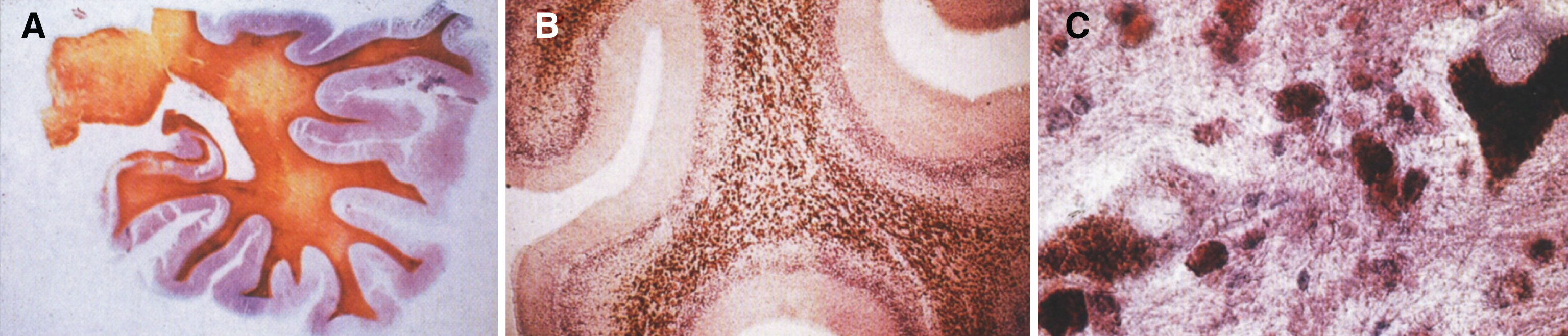
Figs. 71,72
Metachromatic leukodystrophy. (A) The neurons of the cerebral cortex show metachromatic staining with cresyl violet. (B) Anterior horn cell of spinal cord shows metachromasia with cresyl violet; (72) Metachromatic leukodystrophy. Vacuolated storage histiocyte cells are present in the lamina propria of the gallbladder.
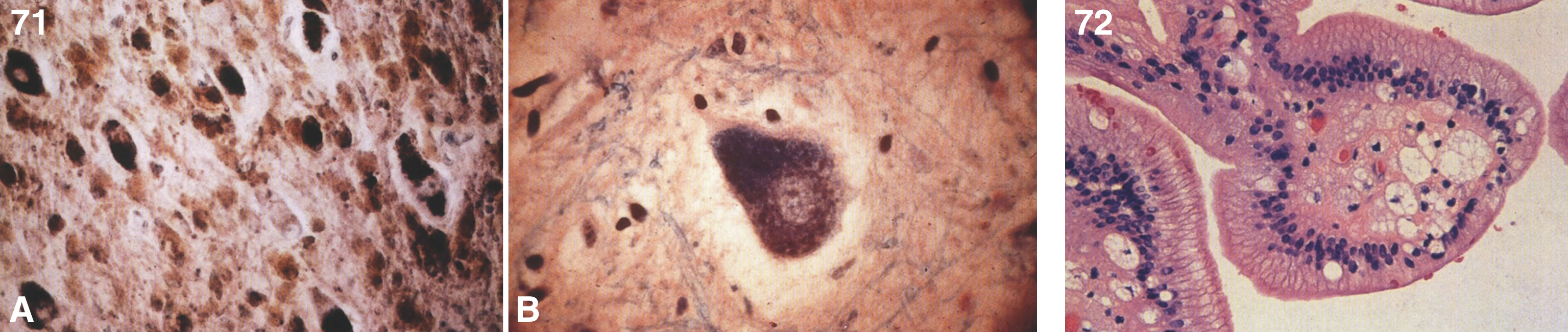
Fig.73
Metachromatic leukodystrophy. Autofluorescence of storage cells.
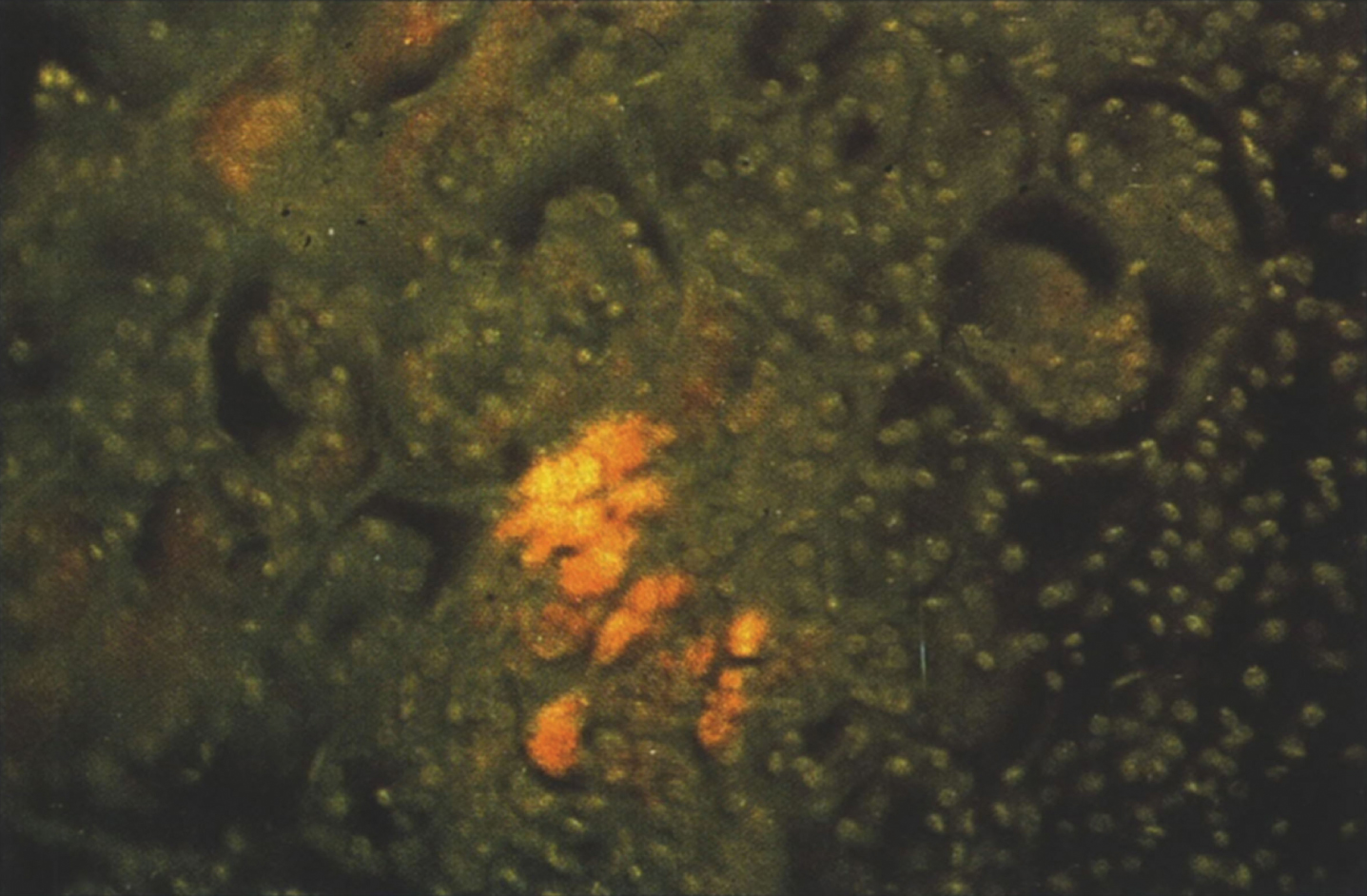
Figs. 74,75
Metachromatic leukodystrophy. Microscopic section of the kidney shows brown metochromasia of the renal tubular epithelial cells; (75) Metachromatic leukodystrophy. A sural nerve biopsy show metachromatic substance within the nerve fibers.
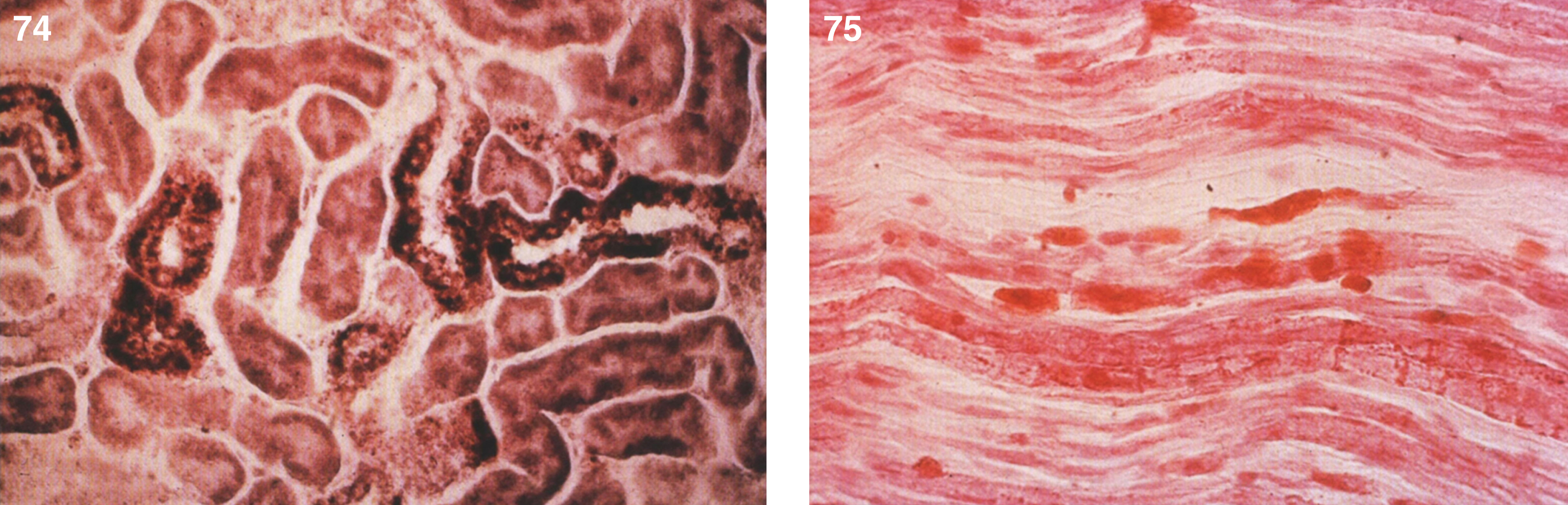
Figs. 76,77
Metachromatic leukodystrophy. Section of the pancreas shows brown metachromasia in the acinar cells; (77) Metachromatic leukodystrophy. Electron micrograph of a neuron of the white matter shows pleomorphic and parallel crystalline lipid profiles. Inset, crystalline arrays at high magnification.
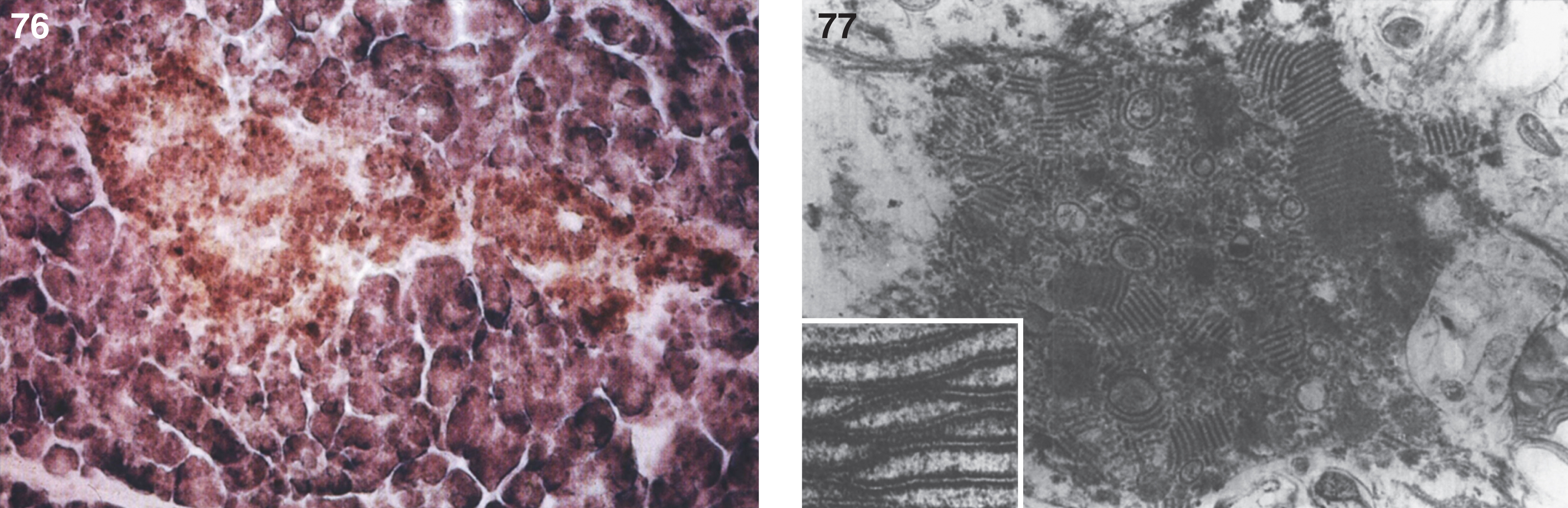
2.15Multiple sulfatase deficiency (Austin disease; Metachromatic leukodystrophy variant O)
This disorder is caused by an inability to convert a cysteine, present in at least 7 different sulfatase enzymes, to 2-amino-3-oxopropionic acid [268]. As a consequence, all of these sulfatases, including arylsulfatase A, B, and C, are deficient; patients express features of MPS and MLD [269], specifically those of late infantile MLD. Increased amounts of MPS and sulfatide are found in the affected tissues and in the urine. Hurler-type features are mild and there is rapid psychomotor deterioration. Peripheral nerve biopsies show brown metachromasia. The disorder may have its onset in the neonatal period. Congenital hydrocephalus, mild chondrodysplasia calcificans, and heart abnormalities have been described; patients may later develop ichthyosis and features of MPS. Most patients die in the first decade.
3Lysosomal storage diseases due to activator defects
In 1971, Sandhoff and colleagues studied 12 autopsy cases of Tay-Sachs disease [270]. One of the cases did not have decreased hexosaminidase activity, and in fact the activities of both hexosaminidases A and B in brain were three to four times higher than in controls. This occurred despite the fact that the GM2 ganglioside concentration in brain tissue was even higher than that seen in the other patients with Tay-Sachs disease. In 1978, Conzelmann and Sandhoff demonstrated that this so-called AB variant of GM2 gangliosidosis was not caused by a hydrolase defect, but by a defective factor needed for the interaction of the sphingolipid substrate with the water-soluble degrading enzyme [271], the first time such a mechanism was described. All proven cases of this GM2 activator protein deficiency have a clinical phenotype indistinguishable from that of classic infantile Tay-Sachs disease [213]. The histopathology is also similar, with widespread storage in the central nervous system, but lack of visceral involvement [272].
There are five known sphingolipid activator proteins, also known as SAPs or saposins. Four of them are derived from the same precursor protein, prosaposin, encoded by the PSAP gene on chromosome 10q22.1. These four saposins are highly homologous small proteins of about 80 amino acids each [273]. The fifth activator protein, GM2 activator protein (also known as SAP-3), is much larger and is encoded by a separate gene on chromosome 5q33.1.
Prosaposin exists in two different forms: a 65 kDa form that is targeted to the lysosomes, and a 70 kDa form that is secreted into extracellular fluids. The 65 kDa form enters the lysosome by a mannose-6-phosphate independent mechanism. Instead, it has a 17 amino acid sequence in the C-terminal region that binds sortilin for lysosomal entry [274]. Prosaposin is transported in tandem with procathepsin D, and once inside the lysosome, it is cleaved into the four saposins A-D by cathepsin D [275]. In addition to being the precursor for the SAPs, prosaposin itself is known to have neurotrophic effects [276–279].
Sap-A is an activator for galactocerebrosidase and glucocerebrosidase [280]. Little is known about its mechanism of action, although it is thought to be similar to that of sap-C (see below) [280]. Only one case of sap-A deficiency has been reported so far, in a patient who had a clinical picture typical of classic infantile Krabbe disease [281]. The activity of galactocerebrosidase in leukocytes was decreased, but it was normal in fibroblasts, which led the authors to suspect an activator protein deficiency. Sequencing of the PSAP gene revealed a mutation in the sap-A coding region.
Sap-B is also known as SAP-1, cerebroside sulfatide activator, or GM1 activator. It activates arylsulfatase A, α-galactosidase, β-galactosidase and sphingomyelinase [280]. The mechanism of action is similar to that of the GM2 activator protein, since it binds 1:1 with glycolipids, exerting a detergent function that solubilizes the lipid so it can interact with its water-soluble hydrolase [273]. The first case of metachromatic leukodystrophy (MLD) with normal arylsulfatase A activity was reported in 1979 [282]. In 1981, it was first proposed that this phenomenon occurred due to an activator protein deficiency [247]. This form of MLD is also known as the AB variant, while the arylsulfatase A deficiency is known as the B variant of MLD, in analogy to the nomenclature for the GM2 gangliosidoses [283]. Interestingly, urine from one patient with MLD due to sap-B deficiency not only revealed increased sulfatide excretion, but also increased levels of globotriaosylceramide, consistent with activation of α-galactosidase [284]. This latter patient subsequently received a bone marrow transplant, with no major clinical improvement noted [285].
Sap-C is also known as SAP-2, and it activates glucocerebrosidase and galactocerebrosidase. Instead of lipid-protein interaction (as in the case of sap-B and GM2AP), its mechanism of action involves protein-protein interaction, since it does not bind to glucosylceramide, but to glucocerebrosidase itself [273]. Its deficiency leads to an atypical form of Gaucher disease with normal glucocerebrosidase enzyme activity. Of the five patients reported so far, three presented with a type 3 phenotype, while two siblings had a type 1 presentation [286].
Sap-D is also known as component C. It activates ceramidase and sphingomyelinase. There is no human case of isolated sap-D deficiency. The mouse with sap-D deficiency exhibits polyuria due to renal tubular degeneration, and ataxia due to progressive and selective loss of Purkinje cells in the cerebellum [287].
The deficiency of prosaposin–also known as combined saposin deficiency, or Farber disease type VII-causes a unique type of lysosomal storage disorder. There are seven known cases, revealing a uniform picture. The first case was reported in 1989, in a patient who presented immediately after birth with hyperkinetic movements, myoclonus, respiratory insufficiency and hepatomegaly. He was initially diagnosed with Gaucher disease since glucocerebrosidase activity was low in leukocytes and fibroblasts, while the activities of arylsulfatase A, α-galactosidase, β-galactosidase and hexosaminidases A and B were normal. The patient died at 16 weeks of age, and post-mortem examination revealed additional deficiency of galactocerebrosidase in leukocytes and fibroblasts and ceramidase in fibroblasts. An amniocentesis was performed during the mother’s next pregnancy, and deficiency of acid ceramidase was found in cultured amniocytes, leading to pregnancy termination. The fetus had contracted joints. Liver samples from both patients showed increased levels of glucosylceramide, lactosylceramide and free ceramide. Immunoblot analysis of fibroblast extracts using antibodies against sap-C revealed absence of staining [288]. Subsequently, a deficiency in sap-B was also detected [289], and a homozygous mutation in the initiation codon of the PSAP gene was found [290]. The third patient described had generalized seizures within minutes after birth, as well as hepatosplenomegaly. He died at 3.5 months. A generalized storage process was found in numerous cells, and ultrastructurally the stored material was membranous with oligolamellar profiles. Neuropathology revealed massive depopulation of cortical neurons with pronounced fibrillary astrocytosis, as well as a paucity of myelin. There were increased monohexosylceramides (mainly glucosylceramide), dihexosylceramide (mainly lactosylceramide), globotriaosylceramide, sulfatides, ceramide and globotetraosylceramide, but with normal cholesterol and sphingomyelin. Immunohistochemistry revealed absence of saposins A-D [291]. The authors showed evidence that a patient who died at 4 weeks, previously reported as a case of lactosylceramidosis due to NPC [292], in fact had prosaposin deficiency. These two unrelated patients were from eastern Slovakia, arguing for the existence of a genetic isolate. A homozygous frameshift mutation leading to nonsense-mediated decay (NMD) was found [291]. The fifth patient had multifocal myoclonus and cyanotic hypoxia immediately after birth, followed by protein-losing enteropathy, seizures, and death at 17 weeks. Brain MRI revealed gray matter heterotopias. The thymus and mesenteric lymph nodes were devoid of lymphocytes. The pathology was otherwise similar to other cases. He was found to have a homozygous initiation codon mutation [293]. The sixth patient had myoclonic bursts, abnormal eye movements, hepatosplenomegaly and seizures, and died at 15 weeks. Brain MRI also revealed gyral pattern abnormalities. He had a nonsense mutation leading to NMD [294]. The last reported case had clonic seizures, myoclonus, hepatosplenomegaly, pulmonary infiltrates, optic disc atrophy and gray matter heterotopias on brain MRI. The patient died at 55 days of life. Analysis of urinary lipids by tandem mass spectrometry revealed elevated excretion of sulfatides, globotriaosylceramide, lactosylceramide, glucosylceramide, and ceramide. He was found to have a homozygous splice mutation leading to NMD [295]. Although this condition is rare, it likely remains underdiagnosed, as cases can be confused with Gaucher disease type II. However, several different tests point towards the right diagnosis, including: 1) lysosomal enzyme assays revealing deficiency not only of glucocerebrosidase but also galactocerebrosidase and ceramidase; 2) sphingolipid analysis in tissues or urine, the latter available on a clinical basis, showing elevated excretion of multiple sphingolipids but with normal cholesterol and sphingomyelin; 3) immunohistochemistry for saposins, revealing lack of staining; and 4) typical histopathology changes, with marked foamy appearance of storage cells in visceral organs, and neuronal non-lipid eosinophilic aggregates inside distended lysosomes that show massive ubiquitination [296]. The combination of generalized lysosomal storage with extreme depletion of cortical neurons is typical for the condition [293]. The unique neuropathology is thought to be related to the loss of trophic effects of prosaposin [293, 296]. The adrenal cortex seems uniformly affected, a feature shared with NPD type A and Wolman disease [293].
4Lysosomal storage diseases due to transport defects
In contrast to lysosomal enzyme deficiencies, which result in the storage of undegraded macromolecules, lysosomal transport deficiencies cause small molecules to accumulate within lysosomes. There are many lysosomal transporters, but only five have been associated with human disease, perhaps because redundancy of small molecule carriers prevents pathological accumulation of material. The five transport defects are cystinosis, Salla disease (free sialic acid storage disease), cobalamin F disease and cobalamin J disease, and mucolipidosis type IV.
4.1Cystinosis
In 1903, Abderhalden reported the first case of the disease, that of an infant with failure to thrive, found on autopsy to have cystine crystals in the liver and spleen. The association with rickets was first noticed by Lignac in 1924 [297]. The first renal transplant was reported in 1970 [298], and the defective lysosomal cystine efflux was discovered in 1982 [299].
Cystinosis is an autosomal recessive lysosomal storage disorder leading to renal tubular Fanconi syndrome in the first year of life, renal glomerular failure by approximately 10 years of age, and growth retardation, photophobia and hypothyroidism in early childhood. Late complications include myopathy, swallowing difficulty, pancreatic insufficiency, pulmonary dysfunction, retinal blindness, male hypogonadism, and neurological deterioration in the second to fifth decades, if untreated [300–302]. The disease occurs worldwide, with an estimated incidence of one in 100-200,000. More than 120 mutations have been identified in the causative gene, CTNS, which encodes the lysosomal membrane transporter cystinosin [303]. One mutation, a 57,257 bp deletion, accounts for half the cystinosis cases in Northern Europe and North America [300–302]. CTNS contains 12 exons within 23 kb of genomic DNA [303].
Cystinosin transports cystine, the disulfide of the free thiol amino acid cysteine, from the lysosome into the cytoplasm [299]; failure to achieve this transport results in the accumulation of free (nonprotein) cystine in the lysosomes of most cells of the body. The crystals are birefringent under cross-polarizing light (Fig. 78) and form in the kidney, spleen, liver, intestine, cornea, conjunctiva, bone marrow, pancreas, testis, muscle, and other tissues due to cystine’s low solubility in aequeous fluids. The classic disease causes renal failure early, but variants exist with milder clinical courses [300–302]. Older patients can present with a distal vacuolar myopathy; the vacuoles stain positively with acid phosphatase, are not birefringent under a polarized filter-indicating they do not contain cystine crystals-and are filled with an amorphous, unidentified material when evaluated by electron microscopy [304].
Figs. 78,79
Light micrograph under birefringent light showing cystine crystals of various shapes. Aequeous fixative has smoothed the hexagonal and rectangular edges; (79) Slit lamp examination of cornea of a child with cystinosis showing a multitude of crystals.
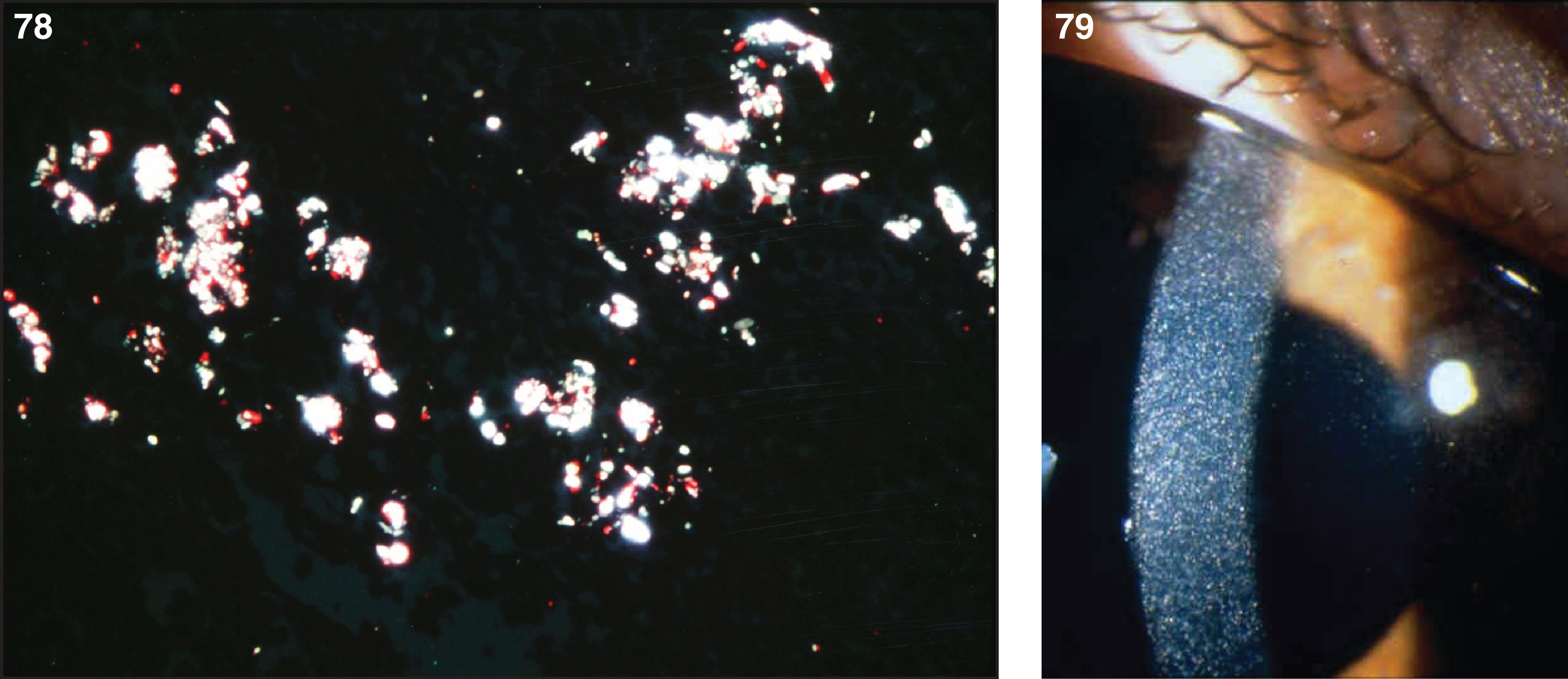
The diagnosis of cystinosis is based upon cystine measurements in circulating leukocytes or by finding corneal crystals on slit lamp examination of the eyes (Fig. 79). Molecular confirmation is also possible. Prenatal diagnosis is available by performing cystine measurements or molecular diagnostics on cultured amniocytes or samples of chorionic villi [300–302].
Treatment is both supportive and directed toward the basic defect. Symptomatic therapy involves replacement of renal losses with supplements such as citrate, phosphate, potassium, calcium, sodium, and carnitine [300–302]. Vitamin D is often helpful in promoting gastrointestinal phosphate absorption. Free access to water is essential. Some patients require L-thyroxine replacement for hypothyroidism and dark glasses for photophobia. Young patients respond to growth hormone injections; some older patients require insulin; occasionally, older males benefit from testosterone supplements. Treatment aimed at the primary defect involves cysteamine, which depletes cystinosis cells of their lysosomal cystine content. This is possible because it severs the disulfide bond of cystine to form cysteine-cysteamine mixed disulfide, structurally similar to lysine, that can then be exported from the lysosome by a cationic amino acid transporter encoded by PQLC2 [305]. Oral cysteamine (Cystagon®) prevents or retards renal glomerular deterioration [306], enhances growth, and prevents most or all of the late complications of cystinosis [307]. A delayed-release cysteamine (Procysbi®) formulation is also available [308]. Cysteamine eyedrops dissolve corneal cystine crystals [309]. If renal function is lost, dialysis serves as a temporizing measure until a renal allograft can be provided. Kidney transplants perform well in cystinosis patients.
4.2Free sialic acid storage disease
Salla disease carries the name of the municipality in Finnish Lapland where the disease was first described in 1979 [310]. Infantile free Sialic acid Storage Disease (ISSD) was first described in 1982 [311]. Salla disease and ISSD are rare allelic disorders of free sialic acid characterized by accumulation of the charged monosaccharide sialic acid (N-acetylneuraminic acid) in lysosomes [312]. The basic defect in these disorders involves failure to transport sialic acid out of lysosomes [313, 314].
Typically, Salla disease patients are normal at birth but develop muscular hypotonia and ataxia between 6 and 12 months of age. Nystagmus and seizures can also occur. Motor development is always delayed, with an average age at walking of 4 years [312]. Other developmental milestones are lost over time. Growth is delayed, head circumference is normal and there is no hepatosplenomegaly. Coarse facial features generally develop later in adulthood. Scoliosis is present in less than half of the patients. Cognitive ability is severely impaired. The mean age at death in Salla disease is 34.6 years [312].
The clinical course of ISSD is more severe than that of Salla disease and usually leads to death in early infancy, with a mean age at death of 13.1 months [315]. Hydrops fetalis and/or ascites can occur [315]. All ISSD patients demonstrate failure to thrive, hypotonia, hepatosplenomegaly, growth retardation and markedly delayed psychomotor development in early infancy [312]. Hypopigmentation of the skin and hair, mild skeletal dysplasias, and coarse facial features are also seen. Nephrotic syndrome is not uncommon [315, 316]. Electron microscopy of renal biopsies can reveal swollen epithelial, endothelial and mesangial cells filled with membrane-bound, electron-lucent vacuoles [315, 316]. Storage material accumulates in CNS neurons and astrocytes, associated with severe demyelination, gliosis and marked hypoplasia of the corpus callosum [315, 317, 318]. Staining of neurons, endothelial cells and Kupffer cells using the sialic acid-specific lectin wheat germ agglutinin, identifies the storage material as sialic acid [317].
The diagnosis of Salla disease or ISSD relies upon the finding of increased urinary excretion of free sialic acid. In Salla disease, the levels range from 5 to 20 times normal (280–2100 nmol/mg creatinine), while in ISSD urine sialic acid excretion is increased up to two-hundred-fold (1071–14230 nmol/mg creatinine) [312]. Sialic acid is also elevated in cultured fibroblasts from Salla disease and ISSD patients. Electron microscopic studies of skin, kidney and liver from sialic acid storage disease patients reveal numerous electron-lucent membrane-bound vacuoles in various cell types [310, 311, 315, 317, 319]. Vacuoles are also present in cultured fibroblasts from patients and can sometimes be observed in peripheral blood lymphocytes [310].
Mutations in SLC17A5, coding for the transport protein sialin, were found in both Salla disease and ISSD patients. A homozygous missense mutation (c.115C>T; p.Arg39Cys) in SLC17A5 is present in all Finnish Salla disease patients [320]. There is a founder mutation causing ISSD in the Inuit population [321]. There is no known treatment for either Salla disease or ISSD.
4.3Cobalamin F and cobalamin J disease
David Rosenblatt first described a defect in lysosomal vitamin B12 egress, leading to accumulation of non-protein bound cobalamin intralysosomally [322]. The underlying molecular defect was not known until 2009, when mutations in the LMBRD1 gene were described [323]. Mutations in ABCD4 were shown to lead to a similar defect in lysosomal cobalamin efflux [324]. It has been postulated that the proteins encoded by LMBRD1 and ABCD4 form a membrane-bound complex that delivers cobalamin from the lysosome directly to MMACHC in the cytoplasm, thus preventing cytoplasmic dilution or inactivation of cobalamin [325].
There are only a handful of known patients in the world with either of these lysosomal cobalamin disorders, and they have a combination of the clinical manifestations of homocystinuria and methylmalonic acidemia, since cobalamin is needed for both of the pathways involved. One feature that differentiates the lysosomal cobalamin disorders from the other inborn errors of cobalamin metabolism is that patients tend to have low serum levels of vitamin B12. In addition, patients with cblF disease commonly have recurrent or persistent stomatitis, while two out of four patients with cblJ disease had skin hyperpigmentation that improved after treatment [326, 327], features not commonly seen in other cobalamin defects.
4.4Mucolipidosis IV (Berman syndrome)
The disease was first described by Berman and colleagues in 1974 [328]. It is characterized by intellectual disabilities, corneal clouding, retinal degeneration, and achloridia leading to hypergastrinemia [329]. Histopathologically, there are lamellar membranous cytoplasmic bodies, as well as single membrane-bound vacuoles filled with fibrillogranular material in various cell types [328]. Mutations in MCOLN1 (also known as TRPML1) were first identified in 2000 [330]. The gene encodes mucolipin-1, a cationic channel located in late endosomes and lysosomes that transports calcium, sodium, potassium [330] and iron [331]. Calcium release from the lumen of late endosomes and lysosomes into the cytosol is needed for fusion of these organelles, a critical step in membrane trafficking. Thus, inactivation of mucolipin-1 leads to abnormal storage of lipids and other compounds within late endosomes, since their content cannot be degraded in the lysosomes due to lack of fusion with these organelles [332]. In addition, lysosomal calcium efflux through mucolipin-1 activates calcineurin, thus promoting the nuclear translocation of TFEB, a master transcriptional regulator of lysosomal biogenesis and autophagy [333].
5Acquired lysosomal storage disorders
Phospholipidosis consists of the exaggerated storage of phospholipids inside lysosomes, which acquire a multilamellar appearance [334]. There are more than fifty drugs that can cause this, including antiarrhythmics, antimalarials, antipsychotics, antibacterials, and others [334]. All these compounds are cationic amphiphilic drugs, with a hydrophobic ring and a hydrophilic side chain charged with a cationic amine group [334]. Drug-induced phospholipidosis is considered an acquired form of lysosomal storage disorder, since the offending drugs are thought to inhibit lysosomal phospholipases [335]. Histopathologically, the cells acquire a foamy appearance, and ultrastructurally there is storage of multi-lamellar whorled membranes, or “myeloid bodies” [335]. As an example, amiodarone can induce the appearance of these inclusion bodies not just in intra-alveolar foamy macrophages, but also in hepatocytes, tissue macrophages, lymphocytes, granulocytes and plasma cells [336].In fact, the cutaneous hyperpigmentation induced by amiodarone is the result of lysosomal storage of this lipid-like material [337]. In the case of chloroquine-induced phospholipidosis, the cell types most commonly involved are renal cells, endothelial cells, cardiomyocytes and skeletal muscle cells [338]. In addition to the whorled inclusions seen in other types of phospholipidoses, curvilinear bodies can be seen in skeletal and cardiac muscle cells, and to some extent in renal cells, in cases of chloroquine toxicity [338]. Heart involvement can lead to hypertrophic [339] or restrictive cardiomyopathy, even necessitating heart transplant in severe cases [340]. Similar myeloid bodies and curvilinear bodies can be seen in cases of hydroxychloroquine-induced cardiomyopathy [341]. The renal phospholipidosis induced by chloroquine mimics the histopathology of Fabry disease [342], and has led to its misdiagnosis [338]. Bis(monoacylglycerol)phosphate (BMP) is an anionic phospholipid found exclusively in lysosomes and late endosomes [335], and di-22:6-BMP is considered a reliable biomarker of drug-induced phospholipidosis that can be monitored in serum and urine [343].
6Treatment and screening of lysosomal storage diseases
There is safe and efficacious treatment for only a fraction of the known lysosomal disorders. Others are being investigated (e.g., Niemann-Pick disease, Sly syndrome, Farber disease). Even for those disorders for which therapy is available, early detection, before the onset of irreversible changes, is critical. This applies to bone marrow transplantation in some of the enzyme deficiencies as well as cysteamine treatment for cystinosis. Hence, newborn screening would be a huge benefit for the treatment of lysosomal storage disorders. Various techniques have been proposed in order to screen for lysosomal diseases, such as 1) direct measurement of enzyme activity, either by tandem mass spectrometry or by digital microfluidics fluorometric assays; 2) measuring the abundance of lysosomal enzymes through immunological assays; or 3) assaying biomarkers. The most studied techniques have involved directly measuring enzyme activity, and mass spectrometry appears to better differentiate affected from non-affected individuals when compared to fluorometric assays [344].
Several states have published their experience with newborn screening for lysosomal storage disorders. After screening 110,000 newborns from Washington state, a prevalence of 1/7,800 was estimated for Fabry disease males, 1/27,800 for Pompe disease, and 1/35,500 for Hurler disease, 2–4 times greater than the prevalence estimated on clinical grounds [345]. Missouri reported their experience with screening 43,701 specimens during the first 6 months of their pilot study, and found a detection rate of 1/2,913 for Fabry disease, 1/5,463 for Pompe disease, 1/14,567 for Hurler disease and 1/43,701 for Gaucher disease [346].
The Advisory Committee on Heritable Disorders in Newborns and Children (ACHDNC) was convened by the United States Secretary of Health and Human Services with the purpose of reviewing the evidence and impact on public health of disorders that might be added to the newborn screening panel. After reviewing the evidence, this committee advises the Secretary on whether or not the condition in question should be added to a Recommended Uniform Screening Panel (RUSP). Although the ACHDNC makes recommendations regarding which conditions to screen for on a national level, each state decides which disorders will be included in their newborn screening panel. Hence, Pompe disease was added to the RUSP in February 2015, and Hurler disease was added to the RUSP in March 2016, but as of mid-2016 only four states were providing universal screening for Pompe disease, and only three states were providing universal screening for Hurler disease.
Newborn screening for LSDs is not unique to the United States, and in fact one of the first large-based pilot studies came from Austria, where the incidence of Fabry disease was 1/3,859 newborns, Pompe disease 1/8,684 births, and Gaucher disease 1/17,368 births, after screening 34,736 newborns [347]. Newborn screening in Taiwan led to the identification of the cardiac variant of Fabry disease in 1/1,600-2,450 male newborns [146, 348]. A study from Italy screened 37,104 newborn males for Fabry disease, and found an incidence of 1/3,100–4,600, with a ratio of later-onset:classic-onset of 7–11:1 [349]. This increased incidence of disease when using newborn screening appears related to an increased diagnosis of later-onset forms of the disease, regardless of the lysosomal storage disorder being screened [345, 347]. This fact raises ethical concerns about performing newborn screening for LSDs, since the majority of cases diagnosed in the newborn period will not manifest any symptoms until adulthood. This is not the only issue that plagues the implementation of newborn screening for LSDs. Since the most common technique used for newborn screening involves the measurement of lysosomal enzyme activity, populations that have a high prevalence of pseudodeficiency alleles will also have a high percentage of false positive results. As an example, homozygosity for the α–glucosidase pseudodeficiency allele is found in 3.3–3.9% of Asians [350], although different algorithms have been created in order to increase the positive predictive value of the test in this population [351]. Perhaps more importantly, the early identification of a disorder does not always lead to a better clinical outcome. As an example, New York state published their experience after screening almost 2 million infants for Krabbe disease, and found five individuals with early infantile Krabbe disease. Of those, four received hematopoietic stem cell transplants, with two dying from complications of the transplant, and the other two still having moderate to severe developmental delay after the transplant [352]. Thus, although the technology exists to permit us to screen for multiple LSDs, early diagnosis will not always lead to clinical benefit, at least until better treatment options become available.
References
[1] | Sabatini D.D. and Adesnik M.B. , The Biogenesis of Membranes and Organelles. In: Valle DL, Beaudet AL, Vogelstein B, Kinzler KW, Antonarakis SE, Ballabio A, et al., editors. The Online Metabolic and Molecular Bases of Inherited Disease [Internet]. New York, NY: The McGraw-Hill Companies, Inc.; (2014) [cited 2015 Jul 27]. Available from: http://mhmedical.com/content.aspx?aid=1102899807 |
[2] | Bowers W.E. , Christian de Duve and the discovery of lysosomes and peroxisomes, Trends Cell Biol 8: (8) ((1998) ), 330–333. |
[3] | Sabatini D.D. and Adesnik M. , Christian de Duve: Explorer of the cell who discovered new organelles by using a centrifuge, Proc Natl Acad Sci U S A 110: (33) ((2013) ), 13234–13235. |
[4] | Passarge E. , Color Atlas of Genetics. Thieme Publishing Group; (1995) . |
[5] | Coutinho M.F. , Prata M.J. and Alves S. , A shortcut to the lysosome: The mannose-6-phosphate-independent pathway, Mol Genet Metab 107: (3) ((2012) ), 257–266. |
[6] | Kolter T. and Sandhoff K. , Principles of lysosomal membrane digestion: Stimulation of sphingolipid degradation by sphingolipid activator proteins and anionic lysosomal lipids, Annu Rev Cell Dev Biol 21: ((2005) ), 81–103. |
[7] | O’Brien J.S. , Kretz K.A. , Dewji N. , Wenger D.A. , Esch F. and Fluharty A.L. , Coding of two sphingolipid activator proteins (SAP-1 and SAP-2) by same genetic locus, Science 241: ((1988) ), 1098–1101. |
[8] | Adachi M. , Schneck L. and Volk B.W. , Ultrastructural studies of eight cases of fetal Tay-Sachs disease, Lab Investig J Tech Methods Pathol 30: (1) ((1974) ), 102–112. |
[9] | Cutz E. , Lowden J.A. and Conen P.E. , Ultrastructural demonstration of neuronal storage in fetal Tay-Sachs disease, J Neurol Sci 21: (2) ((1974) ), 197–202. |
[10] | Thomas G.H. , Disorders of Glycoprotein Degradation: α-Mannosidosis, β-Mannosidosis, Fucosidosis, and Sialidosis. In: Valle DL, Beaudet AL, Vogelstein B, Kinzler KW, Antonarakis SE, Ballabio A, et al., editors. The Online Metabolic and Molecular Bases of Inherited Disease [Internet]. New York, NY: The McGraw-Hill Companies, Inc.; (2014) [cited 2015 Jul 27]. Available from: http://mhmedical.com/content.aspx?aid=1102894718 |
[11] | Gelb B.D. , Shi G.P. , Chapman H.A. and Desnick R.J. , Pycnodysostosis, a lysosomal disease caused by cathepsin K deficiency, Science 273: (5279) ((1996) ), 1236–1238. |
[12] | Niemann A. , Ein unbekanntes krankheitsbild, Jahrb Für Kinderheilkd 79: (1) ((1914) ), 1–10. |
[13] | Pick L. , Der Morbus Gaucher und die ihm ähnlichen Krankheiten (die lipoidzellige Splenohepatomegalie Typus Niemann und die diabetische Lipoidzellenhypoplasie der Milz), Ergeb Inn Med Kinderheilkd 29: ((1926) ), 519–627. |
[14] | Klenk E. , Über die Natur der Phosphatide und anderer Lipoide des Gehirns und der Leber bei der Niemann-Pickschen Krankheit. [12. Mitteilung über Phosphatide.], Hoppe-Seyler's Z Für Physiol Chem 235: (1-2) ((1935) ), 24–36. |
[15] | Crocker A.C. , The cerebral defect in Tay-Sachs disease and Niemann-Pick disease, J Neurochem 7: ((1961) ), 69–80. |
[16] | Brady R.O. , Kanfer J.N. , Mock M.B. and Fredrickson D.S. , The metabolism of sphingomyelin. II. Evidence of an enzymatic deficiency in Niemann-Pick diseae, Proc Natl Acad Sci U S A 55: (2) ((1966) ), 366–369. |
[17] | Wasserstein M.P. and Schuchman E.H. , Acid Sphingomyelinase Deficiency. In: Pagon RA, Adam MP, Ardinger HH, Wallace SE, Amemiya A, Bean LJ, et al., editors. GeneReviews(®) [Internet]. Seattle (WA) University of Washington, Seattle; (1993) [cited 2015 Jul 27]. Available from: http://www.ncbi.nlm.nih.gov/books/NBK1370/ |
[18] | Schneider E.L. , Pentchev P.G. , Hibbert S.R. , Sawitsky A. and Brady R.O. , A new form of Niemann-Pick disease characterised by temperature-labile sphingomyelinase, J Med Genet 15: (5) ((1978) ), 370–374. |
[19] | Wasserstein M.P. , Desnick R.J. , Schuchman E.H. , Hossain S. , Wallenstein S. and Lamm C. , et al., The natural history of type B Niemann-Pick disease: Results from a 10-year longitudinal study, Pediatrics 114: (6) ((2004) ), e672–e677. |
[20] | McGovern M.M. , Wasserstein M.P. , Giugliani R. , Bembi B. , Vanier M.T. and Mengel E. , et al., A prospective, cross-sectional survey study of the natural history of Niemann-Pick disease type B, Pediatrics 122: (2) ((2008) ), e341–e349. |
[21] | Nicholson A.G. , Florio R. , Hansell D.M. , Bois R.M. , Wells A.U. and Hughes P. , et al., Pulmonary involvement by Niemann-Pick disease, A report of six cases, Histopathology 48: (5) ((2006) ), 596–603. |
[22] | Wasserstein M.P. , Larkin A.E. , Glass R.B. , Schuchman E.H. , Desnick R.J. and McGovern M.M. , Growth restriction in children with type B Niemann-Pick disease, J Pediatr 142: (4) ((2003) ), 424–428. |
[23] | McGovern M.M. , Wasserstein M.P. , Aron A. , Desnick R.J. , Schuchman E.H. and Brodie S.E. , Ocular manifestations of Niemann-Pick disease type B, Ophthalmology 111: (7) ((2004) ), 1424–1427. |
[24] | Barness L.A. , Wiederhold S. , Chandra S. , Odell G.B. , Shahidi N.T. and Gilbert E.F. , et al., Clinicopathological conference: One-year-old infant with hepatosplenomegaly and developmental delay, Am J Med Genet 28: (2) ((1987) ), 411–431. |
[25] | Schoenfeld A. , Abramovici A. , Klibanski C. and Ovadia J. , Placental ultrasonographic biochemical and histochemical studies in human fetuses affected with Niemann-Pick disease type A, Placenta 6: (1) ((1985) ), 33–43. |
[26] | Greenbaum M. , Hoffman L.M. and Schneck L. , Ceramide hexosides in Niemann-Pick disease brain, J Neurol 213: (3) ((1976) ), 251–255. |
[27] | Elleder M. and Jirásek A. , Neuropathology of various types of Niemann-Pick disease, Acta Neuropathol Suppl 7: ((1981) ), 201–203. |
[28] | Narita T. , Nakazawa H. , Hizawa Y. and Kudo H. , Glycogen storage disease associated with Niemann-Pick disease: Histochemical, enzymatic, and lipid analyses, Mod Pathol Off J U S Can Acad Pathol Inc 7: (3) ((1994) ), 416–421. |
[29] | Smith W.E. , Kahler S.G. , Frush D.P. , Milov D.E. , Gottfried M.R. and Chen Y.T. , Hepatic storage of glycogen in Niemann-Pick disease type B, J Pediatr 138: (6) ((2001) ), 946–948. |
[30] | Higami S. , Omura K. , Nishizawa K. , Yamashita T. and Tada K. , Prenatal diagnosis and fetal pathology of Niemann-Pick disease, Tohoku J Exp Med 125: (1) ((1978) ), 11–17. |
[31] | Stenson P.D. , Mort M. , Ball E.V. , Shaw K. , Phillips A. and Cooper D.N. , The Human Gene Mutation Database: Building a comprehensive mutation repository for clinical and molecular genetics, diagnostic testing and personalized genomic medicine, Hum Genet 133: (1) ((2014) ), 1–9. |
[32] | Simonaro C.M. , Desnick R.J. , McGovern M.M. , Wasserstein M.P. and Schuchman E.H. , The demographics and distribution of type B Niemann-Pick disease: Novel mutations lead to new genotype/phenotype correlations, Am J Hum Genet 71: (6) ((2002) ), 1413–1419. |
[33] | Vanier M.T. , Ferlinz K. , Rousson R. , Duthel S. , Louisot P. and Sandhoff K. , et al., Deletion of arginine (608) in acid sphingomyelinase is the prevalent mutation among Niemann-Pick disease type B patients from northern Africa, Hum Genet 92: (4) ((1993) ), 325–330. |
[34] | Vellodi A. , Hobbs J.R. , O’Donnell N.M. , Coulter B.S. and Hugh-Jones K. , Treatment of Niemann-Pick disease type B by allogeneic bone marrow transplantation, Br Med J Clin Res Ed 295: (6610) ((1987) ), 1375–1376. |
[35] | Shah A.J. , Kapoor N. , Crooks G.M. , Parkman R. , Weinberg K.I. and Wilson K. , et al., Successful hematopoietic stem cell transplantation for Niemann-Pick disease type B, Pediatrics 116: (4) ((2005) ), 1022–1025. |
[36] | Schneiderman J. , Thormann K. , Charrow J. and Kletzel M. , Correction of enzyme levels with allogeneic hematopoeitic progenitor cell transplantation in Niemann-Pick type B, Pediatr Blood Cancer 49: (7) ((2007) ), 987–989. |
[37] | Wasserstein M.P. , Jones S.A. , Soran H. , Diaz G.A. , Lippa N. and Thurberg B.L. , et al., Successful within-patient dose escalation of olipudase alfa in acid sphingomyelinase deficiency, Mol Genet Metab ((2015) ). |
[38] | Suchi M. , Dinur T. , Desnick R.J. , Gatt S. , Pereira L. and Gilboa E. , et al., Retroviral-mediated transfer of the human acid sphingomyelinase cDNA: Correction of the metabolic defect in cultured Niemann-Pick disease cells, Proc Natl Acad Sci U S A 89: (8) ((1992) ), 3227–3231. |
[39] | Libert J. , Toussaint D. and Guiselings R. , Ocular findings in Niemann-Pick disease, Am J Ophthalmol 80: (6) ((1975) ), 991–1002. |
[40] | Gal A.E. , Brady R.O. , Hibbert S.R. and Pentchev P.G. , A practical chromogenic procedure for the detection of homozygotes and heterozygous carriers of Niemann-Pick disease, N Engl J Med 293: (13) ((1975) ), 632–636. |
[41] | Luse S. , The fine structure of the brain and other organs in Niemann–Pick disease. In: Volk BW, Aronson SM, editors. Inborn Disorders of Shingolipid Metabolism [Internet]. Pergamon; (1967) [cited 2015 Jul 27]. p. 93–105. Available from: http://www.sciencedirect.com/science/article/pii/B9781483198552500101 |
[42] | Lynn R. and Terry R.D. , Lipid histochemistry and electron microscopy in adult Niemann-Pick disease, Am J Med 37: ((1964) ), 987–994. |
[43] | Gumbinas M. , Larsen M. and Mei Liu H. , Peripheral neuropathy in classic Niemann-Pick disease: Ultrastructure of nerves and skeletal muscles, Neurology 25: (2) ((1975) ), 107–113. |
[44] | Patterson M. , Niemann-Pick Disease Type C. In: Pagon RA, Adam MP, Ardinger HH, Wallace SE, Amemiya A, Bean LJ, et al., editors. GeneReviews(®) [Internet]. Seattle (WA) University of Washington, Seattle; (1993) [cited 2015 Jul 27]. Available from: http://www.ncbi.nlm.nih.gov/books/NBK1296/ |
[45] | Vanier M.T. , Wenger D.A. , Comly M.E. , Rousson R. , Brady R.O. and Pentchev P.G. , Niemann-Pick disease group C: Clinical variability and diagnosis based on defective cholesterol esterification. A collaborative study on 70 patients, Clin Genet 33: (5) ((1988) ), 331–348. |
[46] | Carstea E.D. , Morris J.A. , Coleman K.G. , Loftus S.K. , Zhang D. and Cummings C. , et al., Niemann-Pick C1 disease gene: Homology to mediators of cholesterol homeostasis, Science 277: (5323) ((1997) ), 228–231. |
[47] | Greer W.L. , Riddell D.C. , Gillan T.L. , Girouard G.S. , Sparrow S.M. and Byers D.M. , et al., The Nova Scotia (type D) form of Niemann-Pick disease is caused by a G–>T transversion in NPC1, Am J Hum Genet 63: (1) ((1998) ), 52–54. |
[48] | Reunert J. , Lotz-Havla A.S. , Polo G. , Kannenberg F. , Fobker M. and Griese M. , et al., Niemann-Pick Type C-2 Disease: Identification by Analysis of Plasma Cholestane-3β,5α,6β-Triol and Further Insight into the Clinical Phenotype, JIMD Rep 23: ((2015) ), 17–26. |
[49] | Millat G. , Marçais C. , Tomasetto C., Chikh K., Fensom A.H., Harzer K., et al., Niemann-Pick C1 disease: Correlations between NPC1 mutations, levels of NPC1 protein, and phenotypes emphasize the functional significance of the putative sterol-sensing domain and of the cysteine-rich luminal loop, Am J Hum Genet 68: (6) ((2001) ), 1373–1385. |
[50] | Millat G. , Chikh K. , Naureckiene S. , Sleat D.E. , Fensom A.H. and Higaki K. , et al., Niemann-Pick disease type C: Spectrum of HE1 mutations and genotype/phenotype correlations in the NPC2 group, Am J Hum Genet 69: (5) ((2001) ), 1013–1021. |
[51] | Adam G. , Brereton R.J. , Agrawal M. and Lake B.D. , Biliary atresia and meconium ileus associated with Niemann-Pick disease, J Pediatr Gastroenterol Nutr 7: (1) ((1988) ), 128–131. |
[52] | Hulette C.M. , Earl N.L. , Anthony D.C. and Crain B.J. , Adult onset Niemann-Pick disease type C presenting with dementia and absent organomegaly, Clin Neuropathol 11: (6) ((1992) ), 293–297. |
[53] | Sun X. , Marks D.L. , Park W.D. , Wheatley C.L. , Puri V. and O’Brien J.F. , et al., Niemann-Pick C variant detection by altered sphingolipid trafficking and correlation with mutations within a specific domain of NPC1, Am J Hum Genet 68: (6) ((2001) ), 1361–1372. |
[54] | Porter F.D. , Scherrer D.E. , Lanier M.H. , Langmade S.J. , Molugu V. and Gale S.E. , et al., Cholesterol oxidation products are sensitive and specific blood-based biomarkers for Niemann-Pick C1 disease, Sci Transl Med 2: (56) ((2010) ), 56ra81. |
[55] | Patterson M.C. , Vanier M.T. , Suzuki K. , Morris J.A. , Carstea E. and Neufeld E.B. , et al., Niemann-Pick Disease Type C: A Lipid Trafficking Disorder. In: Beaudet AL, Vogelstein B, Kinzler KW, Antonarakis SE, Ballabio A, Gibson KM, et al., editors. The Online Metabolic and Molecular Bases of Inherited Disease [Internet]. New York, NY: The McGraw-Hill Companies, Inc.; (2014) [cited 2015 Jul 27]. Available from: http://mhmedical.com/content.aspx?aid=1102895490 |
[56] | Ottinger E.A. , Kao M.L. , Carrillo-Carrasco N. , Yanjanin N. , Shankar R.K. and Janssen M. , et al., Collaborative development of 2-hydroxypropyl-β-cyclodextrin for the treatment of Niemann-Pick type C1 disease, Curr Top Med Chem 14: (3) ((2014) ), 330–339. |
[57] | Bonney D.K., O’Meara A., Shabani A., Imrie J., Bigger B.W. and Jones S., et al., Successful allogeneic bone marrow transplant for Niemann-Pick disease type C2 is likely to be associated with a severe “graft versus substrate” effect, J Inherit Metab Dis 33: (Suppl 3) ((2010) ), S171–S173. |
[58] | Suzuki K., Parker C.C., Pentchev P.G., Katz D., Ghetti B., D’Agostino A.N., et al., Neurofibrillary tangles in Niemann-Pick disease type C, Acta Neuropathol (Berl) 89: (3) ((1995) ), 227–238. |
[59] | Distl R. , Treiber-Held S. , Albert F. , Meske V. , Harzer K. and Ohm T.G. , Cholesterol storage and tau pathology in Niemann-Pick type C disease in the brain, J Pathol 200: (1) ((2003) ), 104–111. |
[60] | Libert J. and Danis P. , Differential diagnosis of type A, B and C Niemann-Pick disease by conjunctival biopsy, J Submicrosc Cytol 11: ((1979) )143–157. |
[61] | Arsénio-Nunes M.L. and Goutières F. , Morphological diagnosis of Niemann-Pick disease type C by skin and conjunctival biopsies, Acta Neuropathol Suppl 7: ((1981) ), 204–207. |
[62] | Boustany R.N. , Kaye E. and Alroy J. , Ultrastructural findings in skin from patients with Niemann-Pick disease, type C, Pediatr Neurol 6: (3) ((1990) ), 177–183. |
[63] | Dumontel C. , Girod C. , Dijoud F. , Dumez Y. and Vanier M.T. , Fetal Niemann-Pick disease type C: Ultrastructural and lipid findings in liver and spleen, Virchows Arch A Pathol Anat Histopathol 422: (3) ((1993) ), 253–259. |
[64] | Gaucher P.C.E. , De l’epithélioma primitif de la rate: Hypertrophie idiopathique de la rate sans leucémie. Octave Doin (1882) . |
[65] | Beighton P. and Beighton G. , The man behind the syndrome. Springer Science & Business Media; (1986) . |
[66] | Brady R.O. , Kanfer J.N. and Shapiro D. , Metabolism of glucocerebrosides. II. Evidence of an enzymatic deficiency in Gaucher’s disease, Biochem Biophys Res Commun 18: ((1965) ), 221–225. |
[67] | Pastores G.M. and Hughes D.A. , Gaucher Disease. In: Pagon RA, Adam MP, Ardinger HH, Wallace SE, Amemiya A, Bean LJ, et al., editors. GeneReviews(®) [Internet]. Seattle (WA) University of Washington, Seattle; (1993) [cited 2015 Jul 27]. Available from: http://www.ncbi.nlm.nih.gov/books/NBK1269/ |
[68] | Grabowski G.A. , Petsko G.A. and Kolodny E.H. , Gaucher Disease. In: Valle DL, Beaudet AL, Vogelstein B, Kinzler KW, Antonarakis SE, Ballabio A, et al., editors. The Online Metabolic and Molecular Bases of Inherited Disease [Internet]. New York, NY: The McGraw-Hill Companies, Inc.; (2014) [cited 2015 Jul 27]. Available from: http://mhmedical.com/content.aspx?aid=1102895727 |
[69] | Zimran A. , Sorge J. , Gross E. , Kubitz M. , West C. and Beutler E. , A glucocerebrosidase fusion gene in Gaucher disease. Implications for the molecular anatomy, pathogenesis, and diagnosis of this disorder, J Clin Invest 85: (1) ((1990) ), 219–222. |
[70] | Sidransky E. , Tayebi N. , Stubblefield B.K. , Eliason W. , Klineburgess A. and Pizzolato G.P. , et al., The clinical, molecular, and pathological characterisation of a family with two cases of lethal perinatal type 2 Gaucher disease, J Med Genet 33: (2) ((1996) ), 132–136. |
[71] | Biegstraaten M. , van Schaik I.N. , Aerts J.M.F.G., Langeveld M., Mannens M.M.A.M., Bour L.J., et al., A monozygotic twin pair with highly discordant Gaucher phenotypes, Blood Cells Mol Dis 46: (1) ((2011) ), 39–41. |
[72] | Azuri J. , Elstein D. , Lahad A. , Abrahamov A. , Hadas-Halpern I. and Zimran A. , Asymptomatic Gaucher disease implications for large-scale screening, Genet Test 2: (4) ((1998) ), 297–299. |
[73] | Patterson M.C. , Vanier M.T. , Suzuki K. , Morris J.A. , Carstea E. , Neufeld E.B. , et al., Niemann-Pick Disease Type C: A Lipid Trafficking Disorder. In: Valle DL Beaudet AL Vogelstein B Kinzler KW Antonarakis SE Ballabio A, et al., editors. The Online Metabolic and Molecular Bases of Inherited Disease [Internet]. New York, NY: The McGraw-Hill Companies, Inc.; (2014) [cited 2015 Jul 27]. Available from: http://mhmedical.com/content.aspx?aid=1102895490 |
[74] | de Fost M. , Vom Dahl S., Weverling G.J., Brill N., Brett S., Häussinger D., et al., Increased incidence of cancer in adult Gaucher disease in Western Europe, Blood Cells Mol Dis 36: (1) ((2006) ), 53–58. |
[75] | Gupta N. , Oppenheim I. , Kauvar E. , Tayebi N. and Sidransky E. , Type 2 Gaucher disease: Phenotypic variation and genotypic heterogeneity, Blood Cells Mol Dis 46: (1) ((2011) ), 75–84. |
[76] | Sherer D.M. , Metlay L.A. , Sinkin R.A. , Mongeon C. , Lee R.E. and Woods J.R. , Congenital ichthyosis with restrictive dermopathy and Gaucher disease: A new syndrome with associated prenatal diagnostic and pathology findings, Obstet Gynecol 81: (5 (Pt 2)) ((1993) ), 842–844. |
[77] | Uyama E. , Takahashi K. , Owada M. , Okamura R. , Naito M. and Tsuji S. , et al., Hydrocephalus, corneal opacities, deafness, valvular heart disease, deformed toes and leptomeningeal fibrous thickening in adult siblings: A new syndrome associated with beta-glucocerebrosidase deficiency and a mosaic population of storage cells, Acta Neurol Scand 86: (4) ((1992) ), 407–420. |
[78] | Chabás A. , Cormand B. , Grinberg D. , Burguera J.M. , Balcells S. and Merino J.L. , et al., Unusual expression of Gaucher’s disease: Cardiovascular calcifications in three sibs homozygous for the D409H mutation, J Med Genet 32: (9) ((1995) ), 740–742. |
[79] | Abrahamov A. , Elstein D. , Gross-Tsur V. , Farber B. , Glaser Y. and Hadas-Halpern I. , et al., Gaucher’s disease variant characterised by progressive calcification of heart valves and unique genotype, Lancet Lond Engl 346: (8981) ((1995) ), 1000–1003. |
[80] | Sidransky E. , Nalls M.A. , Aasly J.O. , Aharon-Peretz J. , Annesi G. and Barbosa E.R. , et al., Multicenter analysis of glucocerebrosidase mutations in Parkinson’s disease, N Engl J Med 361: (17) ((2009) ), 1651–1661. |
[81] | Goker-Alpan O. , Stubblefield B.K. , Giasson B.I. and Sidransky E. , Glucocerebrosidase is present in α-synuclein inclusions in Lewy body disorders, Acta Neuropathol (Berl) 120: (5) ((2010) ), 641–649. |
[82] | Halperin A. , Elstein D. and Zimran A. , Increased incidence of Parkinson disease among relatives of patients with Gaucher disease, Blood Cells Mol Dis 36: (3) ((2006) ), 426–428. |
[83] | Anheim M. , Elbaz A. , Lesage S. , Durr A. , Condroyer C. and Viallet F. , et al., Penetrance of Parkinson disease in glucocerebrosidase gene mutation carriers, Neurology 78: (6) ((2012) ), 417–420. |
[84] | Rana H.Q. , Balwani M. , Bier L. and Alcalay R.N. , Age-specific Parkinson disease risk in GBA mutation carriers: Information for genetic counseling, Genet Med Off J Am Coll Med Genet 15: (2) ((2013) ), 146–149. |
[85] | Beutler E. , Enzyme Replacement in Gaucher Disease, PLoS Med 1: (2) ((2004) ), e21. |
[86] | Shemesh E. , Deroma L. , Bembi B. , Deegan P. , Hollak C. and Weinreb N.J. , et al., Enzyme replacement and substrate reduction therapy for Gaucher disease, Cochrane Database Syst Rev 3: ((2015) ), CD010324. |
[87] | Enquist I.B. , Nilsson E. , Ooka A. , Månsson J-E. , Olsson K., Ehinger M., et al., Effective cell and gene therapy in a murine model of Gaucher disease, Proc Natl Acad Sci U S A 103: (37) ((2006) ), 13819–13824. |
[88] | McEachern K.A. , Nietupski J.B. , Chuang W.-L. , Armentano D. , Johnson J. and Hutto E. , et al., AAV8-mediated expression of glucocerebrosidase ameliorates the storage pathology in the visceral organs of a mouse model of Gaucher disease, J Gene Med 8: (6) ((2006) ), 719–729. |
[89] | Charrow J. , Dulisse B. , Grabowski G.A. and Weinreb N.J. , The effect of enzyme replacement therapy on bone crisis and bone pain in patients with type 1 Gaucher disease, Clin Genet 71: (3) ((2007) ), 205–211. |
[90] | Sun C.C. , Panny S. , Combs J. and Gutberlett R. , Hydrops fetalis associated with Gaucher disease, Pathol Res Pract 179: (1) ((1984) ), 101–104. |
[91] | Clarke L.E. , Dimmick J.E. and Applegarth D.A. , Pathology of inherited metabolic diseases. In: Dimmick JE, Kalousek DK, editors. Develomental pathology of the embryo and fetus [Internet]. Lippincott; (1992) . Available from: https://books.google.com/books?id=VcFsAAAAMAAJ |
[92] | Reissner K. , Tayebi N. , Stubblefield B.K. , Koprivica V. , Blitzer M. and Holleran W. , et al., Type 2 Gaucher disease with hydrops fetalis in an Ashkenazi Jewish family resulting from a novel recombinant allele and a rare splice junction mutation in the glucocerebrosidase locus, Mol Genet Metab 63: (4) ((1998) ), 281–288. |
[93] | Ishak K.G. , Pathology of inherited metabolic disorders. In: Balistreri WF, Stocker, editors. Pediatric Hepatology. 1 edition. New York: CRC Press; (1990) . |
[94] | Florena A.M. , Franco V. and Campesi G. , Immunophenotypical comparison of Gaucher’s and pseudo-Gaucher cells, Pathol Int 46: (2) ((1996) ), 155–160. |
[95] | Lorber M. and Nemes J.L. , Identification of ferritin within Gaucher cells. An electron microscopic and immunofluorescent study, Acta Haematol 37: (4) ((1967) ), 189–197. |
[96] | James S.P. , Stromeyer F.W. , Chang C. and Barranger J.A. , LIver abnormalities in patients with Gaucher’s disease. Gastroenterology 80: (1) ((1981) ), 126–133. |
[97] | Lee R.E. , The pathology of Gaucher disease, Prog Clin Biol Res 95: ((1982) ), 177–217. |
[98] | Nilsson O. , Månsson J.E. , Håkansson G. and Svennerholm L., The occurrence of psychosine and other glycolipids in spleen and liver from the three major types of Gaucher’s disease, Biochim Biophys Acta 712: (3) ((1982) ), 453–463. |
[99] | Banker B.Q. , Miller J.Q. and Crocker A.C. , The Cerebral Pathology of Infantile Gaucher’s Disease1. In: Aronson SM, Volk BW, editors. Cerebral Shingolipidoses [Internet]. Academic Press; (1962) . p. 73–99. Available from: http://www.sciencedirect.com/science/article/pii/B9781483196480500110 |
[100] | Cervos-Navarro J. and Zimmer C. , Light microscopic and ultrastructural study on CNS lesions in infantile Gaucher’s disease, Clin Neuropathol 9: (6) ((1990) ), 310–313. |
[101] | Nilsson O. and Svennerholm L. , Accumulation of glucosylceramide and glucosylsphingosine (psychosine) in cerebrum and cerebellum in infantile and juvenile Gaucher disease, J Neurochem 39: (3) ((1982) ), 709–718. |
[102] | Hannun Y.A. and Bell R.M. , Lysosphingolipids inhibit protein kinase C: Implications for the sphingolipidoses, Science 235: (4789) ((1987) ), 670–674. |
[103] | Liu J. , Halene S. , Yang M. , Iqbal J. , Yang R. and Mehal W.Z. , et al., Gaucher disease gene GBA functions in immune regulation, Proc Natl Acad Sci U S A 109: (25) ((2012) ), 10018–10023. |
[104] | Pandey M.K. and Grabowski G.A. , Immunological cells and functions in Gaucher disease, Crit Rev Oncog 18: (3) ((2013) ), 197–220. |
[105] | Sidransky E. , Fartasch M. , Lee R.E. , Metlay L.A. , Abella S. and Zimran A. , et al., Epidermal abnormalities may distinguish type 2 from type 1 and type 3 of Gaucher disease, Pediatr Res 39: (1) ((1996) ), 134–141. |
[106] | Holleran W.M. , Ginns E.I. , Menon G.K. , Grundmann J.U. , Fartasch M. and McKinney C.E. , et al., Consequences of beta-glucocerebrosidase deficiency in epidermis. Ultrastructure and permeability barrier alterations in Gaucher disease, J Clin Invest 93: (4) ((1994) ), 1756–1764. |
[107] | Chan A. , Holleran W.M. , Ferguson T. , Crumrine D. , Goker-Alpan O. and Schiffmann R. , et al., Skin ultrastructural findings in type 2 Gaucher disease: Diagnostic implications, Mol Genet Metab 104: (4) ((2011) ), 631–636. |
[108] | Mistry P.K. , Liu J. , Yang M. , Nottoli T. , McGrath J. and Jain D. , et al., Glucocerebrosidase gene-deficient mouse recapitulates Gaucher disease displaying cellular and molecular dysregulation beyond the macrophage, Proc Natl Acad Sci U S A 107: (45) ((2010) ), 19473–19478. |
[109] | Krabbe K.H. , A new familial, infantile form of diffuse brain-sclerosis, Brain 39: (1-2) ((1916) ), 74–114. |
[110] | Suzuki K. and Suzuki Y. , Globoid Cell Leucodystrophy (Krabbe’s Disease) Deficiency of Galactocerebroside β-Galactosidase, Proc Natl Acad Sci 66: (2) ((1970) ), 302–309. |
[111] | Wenger D.A. , Escolar M.L. , Luzi P. and Rafi M.A. , Krabbe disease (Globoid Cell Leukodystrophy). In: Valle DL, Beaudet AL, Vogelstein B, Kinzler KW, Antonarakis SE, Ballabio A, et al., editors. The Online Metabolic and Molecular Bases of Inherited Disease [Internet]. New York, NY: The McGraw-Hill Companies, Inc.; (2014) [cited 2015 Jul 27]. Available from http://mhmedical.com/content.aspx?aid=1102896057 |
[112] | Wenger D.A. , Krabbe Disease. In: Pagon RA, Adam MP, Ardinger HH, Wallace SE, Amemiya A, Bean LJ, et al., editors. GeneReviews(®) [Internet]. Seattle (WA) University of Washington, Seattle; ((1993) ) [cited 2015 Jul 28]. Available from: http://www.ncbi.nlm.nih.gov/books/NBK1238/ |
[113] | Kolodny E.H. , Raghavan S. and Krivit W. , Late-onset Krabbe disease (globoid cell leukodystrophy) Clinical and biochemical features of 15 cases, Dev Neurosci 13: (4-5) ((1991) ), 232–239. |
[114] | Henderson R.D. , MacMillan J.C. and Bradfield J.M. , Adult onset Krabbe disease may mimic motor neurone disease, J Clin Neurosci Off J Neurosurg Soc Australas 10: (5) ((2003) ), 638–639. |
[115] | Bajaj N.P.S. , Waldman A. , Orrell R. , Wood N.W. and Bhatia K.P. , Familial adult onset of Krabbe’s disease resembling hereditary spastic paraplegia with normal neuroimaging. J Neurol Neurosurg Psychiatry 72: (5) ((2002) ), 635–638. |
[116] | Duffner P.K. , Barczykowski A. , Kay D.M. , Jalal K. , Yan L. and Abdelhalim A. , et al., Later onset phenotypes of Krabbe disease: Results of the world-wide registry, Pediatr Neurol 46: (5) ((2012) ), 298–306. |
[117] | Feanny S.J. , Chuang S.H. , Becker L.E. and Clarke. J.T. , Intracerebral paraventricular hyperdensities: A new CT sign in Krabbe globoid cell leukodystrophy, J Inherit Metab Dis 10: (1) ((1987) ), 24–27. |
[118] | Lehman A.M. , Schultz K.R. , Poskitt K. , Bjornson B. , Keyes R. and Waters P.J. , et al., Intracranial calcification after cord blood neonatal transplantation for krabbe disease, Neuropediatrics 40: (4) ((2009) ), 189–191. |
[119] | Livingston J.H. , Graziano C. , Pysden K. , Crow Y.J. , Mordekar S.R. and Moroni I. , et al., Intracranial calcification in early infantile Krabbe disease: Nothing new under the sun, Dev Med Child Neurol 54: (4) ((2012) ), 376–379. |
[120] | Escolar M.L. , Poe M.D. , Provenzale J.M. , Richards K.C. , Allison J. and Wood S. , et al., Transplantation of umbilical-cord blood in babies with infantile Krabbe’s disease, N Engl J Med 352: (20) ((2005) ), 2069–2081. |
[121] | Sharp M.E. , Laule C. , Nantel S. , Mädler B. , Aul R.B. and Yip S. , et al., Stem cell transplantation for adult-onset krabbe disease: Report of a case, JIMD Rep 10: ((2013) ), 57–59. |
[122] | Farber S. , A lipid metabolic disorder: Disseminated lipogranulomatosis; a syndrome with similarity to, and important difference from, Niemann-Pick and Hand-Schüller-Christian disease, AMA Am J Dis Child 84: (4) ((1952) ), 499–500. |
[123] | Farber S. , Cohen J. and Uzman L.L. , Lipogranulomatosis; a new lipo-glycoprotein storage disease, J Mt Sinai Hosp N Y 24: (6) ((1957) ), 816–837. |
[124] | Prensky A.L. , Ferreira G. , Carr S. and Moser H.W. , Ceramide and Ganglioside Accumulation in Farber’s Lipogranulomatosis, Exp Biol Med 126: (3) ((1967) ), 725–728. |
[125] | Sugita M. , Dulaney J.T. and Moser H.W. , Ceramidase deficiency in Farber’s disease (lipogranulomatosis), Science 178: (4065) ((1972) ), 1100–1102. |
[126] | Levade T. , Sandhoff K. , Schulze H. and Medin J.A. , Acid Ceramidase Deficiency: Farber Lipogranulomatosis. In: Valle D.L., Beaudet A.L., Vogelstein B., Kinzler K.W., Antonarakis S.E., Ballabio A., et al., editors. The Online Metabolic and Molecular Bases of Inherited Disease [Internet]. New York, NY: The McGraw-Hill Companies, Inc.; (2014) [cited 2015 Jul 27]. Available from: http://mhmedical.com/content.aspx?aid=1102895115 |
[127] | Nowaczyk M.J. , Feigenbaum A. , Silver M.M. , Callahan J. , Levin A. and Jay V. , Bone marrow involvement and obstructive jaundice in Farber lipogranulomatosis: Clinical and autopsy report of a new case, J Inherit Metab Dis 19: (5) ((1996) ), 655–660. |
[128] | Pellissier J.F. , Berard-Badier M. and Pinsard N. , Farber’s disease in two siblings, sural nerve and subcutaneous biopsies by light and electron microscopy, Acta Neuropathol (Berl) 72: (2) ((1986) ), 178–188. |
[129] | Guseva M.R. and Pavliuk E.I. , The cherry-red spot is an early sign of Farber’s lipogranulomatosis, Vestn Oftalmol 124: (3) ((2008) ), 51–53. |
[130] | Antonarakis S.E. , Valle D. , Moser H.W. , Moser A. , Qualman S.J. and Zinkham W.H. , Phenotypic variability in siblings with Farber disease, J Pediatr 104: (3) ((1984) ), 406–409. |
[131] | Sólyom A. , Karabul N. , Hügle B. , Simonaro C. and Schuchman E. , Polyarticular arthritis as presenting feature of farber disease: A lysosomal storage disease involving inflammation,P, Pediatr Rheumatol Online J 12: (Suppl 1) ((2014) ), 285. |
[132] | Becker H. , Auböck L. , Haidvogl M. and Bernheimer H. , Disinated lipogranulomatosis (Farber). Case report of the 16th case of a ceramidose (author’s transl), Verhandlungen Dtsch Ges Für Pathol ((1976) )254–sem. |
[133] | Zhou J. , Tawk M. , Tiziano F.D. , Veillet J. , Bayes M. and Nolent F. , et al., Spinal muscular atrophy associated with progressive myoclonic epilepsy is caused by mutations in ASAH1, Am J Hum Genet 91: (1) ((2012) ), 5–14. |
[134] | Schuchman E.H. and Galanin I. , Development of enzyme replacement therapy for Farber disease and other disorders with ceramide storage, Mol Genet Metab 111: (2), S94. |
[135] | Tanaka T. , Takahashi K. , Hakozaki H. , Kimoto H. and Suzuki Y. , Farber’s disease. (disseminated lipogranulomatosis)–a pathological, histochemical and ultrastructural study–, Acta Pathol Jpn 29: (1) ((1979) ), 135–155. |
[136] | Qualman S.J. , Moser H.W. , Valle D. , Moser A.E. , Antonarakis S.E. and Boitnott J.K. , et al., Farber disease: Pathologic diagnosis in sibs with phenotypic variability, Am J Med Genet Suppl 3: ((1987) ), 233–241. |
[137] | Rauch H.J. and Auböck L. , “Banana bodies” in disseminated lipogranulomatosis (Farber’s disease), Am J Dermatopathol 5: (3) ((1983) ), 263–266. |
[138] | Zappatini-Tommasi L. , Dumontel C. , Guibaud P. and Girod C. , Farber disease: An ultrastructural study. Report of a case and review of the literature, Virchows Arch A Pathol Anat Histopathol 420: (3) ((1992) ), 281–290. |
[139] | Iwamori M. and Moser H.W. , Above-normal urinary excretion of urinary ceramides in Farber’s disease, and characterization of their components by high-performance liquid chromatography, Clin Chem 21: (6) ((1975) ), 725–729. |
[140] | Williams J.W. and Dorfman R.F. , Lymphadenopathy as the initial manifestation of histiocytosis X, Am J Surg Pathol 3: (5) ((1979) ), 405–421. |
[141] | Fabry J. , Ein Beitrag zur Kenntniss der Purpura haemorrhagica nodularis (Purpura papulosa haemorrhagica Hebrae), Arch Dermatol Res 43: (1) ((1898) ), 187–200. |
[142] | Anderson W. , A case of “angiokeratoma”, Br J Dermatol 10: ((1898) ), 113–117. |
[143] | Mehta A. and Hughes D.A. , In: Pagon R.A., Adam M.P., Ardinger H.H., Wallace S.E., Amemiya A., Bean L.J., et al., editors. GeneReviews(®) [Internet]. Seattle (WA) University of Washington, Seattle; (1993) [cited 2015 Jul 28]. Available from: http://www.ncbi.nlm.nih.gov/books/NBK1292/ |
[144] | Desnick R.J. , Ioannou Y.A. and Eng C.M. , α-Galactosidase A Deficiency: Fabry Disease. In: Valle D.L., Beaudet A.L., Vogelstein B., Kinzler K.W., Antonarakis S.E., Ballabio A., et al., editors. The Online Metabolic and Molecular Bases of Inherited Disease [Internet]. New York, NY: The McGraw-Hill Companies, Inc.; (2014) [cited 2015 Jul 27]. Available from: http://mhmedical.com/content.aspx?aid=1102896680 |
[145] | Brady R.O. , Gal A.E. , Bradley R.M. , Martensson E. , Warshaw A.L. and Laster L. , Enzymatic defect in Fabry’s disease: Ceramidetrihexosidase deficiency, N Engl J Med 276: (21) ((1967) ), 1163–1167. |
[146] | Lin H.-Y. , Chong K.-W. , Hsu J.-H. , Yu H.-C. , Shih C.-C. and Huang C.-H. , et al., High incidence of the cardiac variant of Fabry disease revealed by newborn screening in the Taiwan Chinese population, Circ Cardiovasc Genet 2: (5) ((2009) ), 450–456. |
[147] | Abreo K. , Oberley T.D. , Gilbert E.F. , Opitz J.M. and Updike S.J. , Clinicopathological conference: A 29-yr-old man with recurrent episodes of fever, abdominal pain, and vomiting, Am J Med Genet 18: (2) ((1984) ), 249–264. |
[148] | Schibanoff J.M. , Kamoshita S. and O’Brien J.S. , Tissue distribution of glycosphingolipids in a case of Fabry’s disease, J Lipid Res 10: (5) ((1969) ), 515–520. |
[149] | Ogawa K. , Sugamata K. , Funamoto N. , Abe T. , Sato T. and Nagashima K. , et al., Restricted accumulation of globotriaosylceramide in the hearts of atypical cases of Fabry’s disease, Hum Pathol 21: (10) ((1990) ), 1067–1073. |
[150] | Font R.L. and Fine B.S. , Ocular pathology in Fabry’s disease: Histochemical and electron microscopic observations, Am J Ophthalmol 73: (3) ((1972) ), 419–430. |
[151] | Nistal M. , Paniagua R. and Picazo M.L. , Testicular and epididymal involvement in Fabry’s disease, J Pathol 141: (2) ((1983) ), 113–124. |
[152] | Elleder M. , Sequelae of storage in Fabry disease–pathology and comparison with other lysosomal storage diseases, Acta Paediatr Oslo Nor 1992 Suppl 92: (443) ((2003) ), 46–53 discussion 45. |
[153] | Fischer E.G. , Moore M.J. and Lager D.J. , Fabry disease: A morphologic study of 11 cases, Mod Pathol 19: (10) ((2006) ), 1295–1301. |
[154] | Alroy J. , Sabnis S. and Kopp J.B. , Renal pathology in Fabry disease, J Am Soc Nephrol 13: (suppl 2) ((2002) ), S134–S138. |
[155] | Fogo A.B. , Bostad L. , Svarstad E. , Cook W.J. , Moll S. and Barbey F. , et al., Scoring system for renal pathology in Fabry disease: Report of the International Study Group of Fabry Nephropathy (ISGFN), Nephrol Dial Transplant Off Publ Eur Dial Transpl Assoc - Eur Ren Assoc 25: (7) ((2010) ), 2168–2177. |
[156] | Becker A.E. , Schoorl R. , Balk A.G. and van der Heide R.M. , Cardiac manifestations of Fabry’s disease. Report of a case with mitral insufficiency and electrocardiographic evidence of myocardial infarction, Am J Cardiol 36: (6) ((1975) ), 829–835. |
[157] | Gilbert-Barness E. , Metabolic Cardiomyopathy of Childhood. In: Pomerance H.H., Bercu B.B., editors. Topics in Pediatrics [Internet]. Springer New York; (1990) [cited 2015 Jul 28]. p. 122–53. Available from: http://link.springer.com/chapter/10.1007/978-1-4612-3230-8_12 |
[158] | Owens C.L. , Russell S.D. and Halushka M.K. , Histologic and electron microscopy findings in myocardium of treated Fabry disease, Hum Pathol 37: (6) ((2006) ), 764–768. |
[159] | van Diggelen O.P. , Schindler D., Kleijer W.J., Huijmans J.M., Galjaard H., Linden H.U., et al., Lysosomal alpha-N-acetylgalactosaminidase deficiency: A new inherited metabolic disease, Lancet Lond Engl 2: (8562) ((1987) ), 804. |
[160] | Asfaw B. , Ledvinová J. , Dobrovolńy R. , Bakker H.D. , Desnick R.J. and van Diggelen O.P. , et al., Defects in degradation of blood group A and B glycosphingolipids in Schindler and Fabry diseases, J Lipid Res 43: (7) ((2002) ), 1096–1104. |
[161] | Tomasic I.B. , Metcalf M.C. , Guce A.I. , Clark N.E. and Garman S.C. , Interconversion of the specificities of human lysosomal enzymes associated with Fabry and Schindler diseases, J Biol Chem 285: (28) ((2010) ), 21560–21566. |
[162] | Desnick R.J. and Schindler D. , α-N-Acetylgalactosaminidase Deficiency: Schindler Disease. In: Valle D.L., Beaudet A.L., Vogelstein B., Kinzler K.W., Antonarakis S.E., Ballabio A., et al., editors. The Online Metabolic and Molecular Bases of Inherited Disease [Internet]. New York, NY: McGraw-Hill, The Comanies, Inc.; (2014) [cited 2015 Jul 27]. Available from: http://mhmedical.com/content.aspx?aid=1102894437 |
[163] | Kanzaki T. , Yokota M. , Mizuno N. , Matsumoto Y. and Hirabayashi Y. , Novel lysosomal glycoaminoacid storage disease with angiokeratoma corporis diffusum, Lancet Lond Engl 1: (8643) ((1989) ), 875–877. |
[164] | Kanzaki T. , Wang A.M. and Desnick R.J. , Lysosomal alpha-N-acetylgalactosaminidase deficiency, the enzymatic defect in angiokeratoma corporis diffusum with glycopeptiduria, J Clin Invest 88: (2) ((1991) ), 707–711. |
[165] | Kanzaki T. , Yokota M. , Irie F. , Hirabayashi Y. , Wang A.M. and Desnick R.J. , Angiokeratoma corporis diffusum with glycopeptiduria due to deficient lysosomal alpha-N-acetylgalactosaminidase activity. Clinical, morphologic, and biochemical studies, Arch Dermatol 129: (4) ((1993) ), 460–465. |
[166] | Umehara F. , Matsumuro K. , Kurono Y. , Arimura K. , Osame M. and Kanzaki T. , Neurologic manifestations of Kanzaki disease, Neurology 62: (9) ((2004) ), 1604–1606. |
[167] | Kodama K. , Kobayashi H. , Abe R. , Ohkawara A. , Yoshii N. and Yotsumoto S. , et al., A new case of α-N-acetylgalactosaminidase deficiency with angiokeratoma corporis diffusum, with Ménière’s syndrome and without mental retardation, Br J Dermatol 144: (2) ((2001) ), 363–368. |
[168] | Chabás A. , Coll M.J. and Aparicio M. , Rodriguez Diaz E. Mild phenotypic expression of alpha-N-acetylgalactosaminidase deficiency in two adult siblings, J Inherit Metab Dis 17: (6) ((1994) ), 724–731. |
[169] | de Jong J. , van den Berg C., Wijburg H., Willemsen R., van Diggelen O., Schindler D., et al., alpha-N-acetylgalactosaminidase deficiency with mild clinical manifestations and difficult biochemical diagnosis, J Pediatr 125: (3) ((1994) ), 385–391. |
[170] | Bakker H.D. , de Sonnaville M.L. , Vreken P., Abeling N.G., Groener J.E. and Keulemans J.L., et al., Human alpha-N-acetylgalactosaminidase (alpha-NAGA) deficiency: No association with neuroaxonal dystrophy? Eur J Hum Genet EJHG 9: (2) ((2001) ), 91–96. |
[171] | Keulemans J.L. , Reuser A.J. , Kroos M.A. , Willemsen R. , Hermans M.M. and van den Ouweland A.M. , et al., Human alpha-N-acetylgalactosaminidase (alpha-NAGA) deficiency: New mutations and the paradox between genotype and phenotype, J Med Genet 33: (6) ((1996) ), 458–464. |
[172] | Clark N.E. , Metcalf M.C. , Best D. , Fleet G.W.J. and Garman S.C. , Pharmacological chaperones for human α-N-acetylgalactosaminidase, Proc Natl Acad Sci U S A 109: (43) ((2012) ), 17400–17405. |
[173] | Suzuki Y. , Nanba E. , Matsuda J. , Higaki K. , Oshima A. , β-Galactosidase Deficiency (β-Galactosidosis) GM1 Gangliosidosis and Morquio B Disease. In: Valle D.L., Beaudet AL., Vogelstein B., Kinzler K.W., Antonarakis S.E., Ballabio A., et al., editors. The Online Metabolic and Molecular Bases of Inherited Disease [Internet]. New York, NY: The McGraw-Hill Companies, Inc.; (2014) [cited 2015 Jul 27]. Available from: http://mhmedical.com/content.aspx?aid=1102896957 |
[174] | Norman R.M. , Urich H. , Tingey A.H. and Goodbody R.A. , Tay-Sachs’ disease with visceral involvement and its relationship to Niemann-Pick’s disease, J Pathol Bacteriol 78: ((1959) ), 409–421. |
[175] | Landing B.H. , Silverman F.N. , Craig J.M. , Jacoby M.D. , Lahey M.E. and Chadwick D.L. , Familial neurovisceral lipidosis. An analysis of eight cases of a syndrome previously reported as “Hurler-variant,” “pseudo-Hurler,” and “Tay-Sachs disease with visceral involvement.”, Am J Dis Child 1960 108: ((1964) ), 503–522. |
[176] | O’Brien J.S. , Stern M.B. , Landing B.H. , O’brien J.K. and Donnell G.N. , Generalized Gangliosidosis: Another Inborn Error of Ganglioside Metabolism? Am J Dis Child 1960 109: ((1965) ), 338–346. |
[177] | Okada S. and O’Brien J.S. , Generalized gangliosidosis: Beta-galactosidase deficiency, Science 160: (3831) ((1968) ), 1002–1004. |
[178] | Regier D.S. and Tifft C.J. , GLB1-Related Disorders. In: Pagon R.A., Adam M.P., Ardinger H.H., Wallace S.E., Amemiya A., Bean L.J., et al., editors. GeneReviews(®) [Internet]. Seattle (WA) University of Washington, Seattle; (1993) [cited 2015 Aug 20]. Available from: http://www.ncbi.nlm.nih.gov/books/NBK164500/ |
[179] | Santamaria R. , Chabás A. , Coll M.J. , Miranda C.S. , Vilageliu L. and Grinberg D. , Twenty-one novel mutations in the GLB1 gene identified in a large group of GM1-gangliosidosis and Morquio B patients: Possible common origin for the prevalent p. R59H mutation among gypsies, Hum Mutat 27: (10) ((2006) ), 1060. |
[180] | Santamaria R. , Blanco M. , Chabás A. , Grinberg D. and Vilageliu L. , Identification of 14 novel GLB1 mutations, including five deletions, in 19 patients with GM1 gangliosidosis from South America, Clin Genet 71: (3) ((2007) ), 273–279. |
[181] | Severini M.H. , Silva C.D. , Sopelsa A. , Coelho J.C. and Giugliani R. , High frequency of type 1 GM1 gangliosidosis in southern Brazil, Clin Genet 56: (2) ((1999) ), 168–169. |
[182] | Lenicker H.M. and Vassallo Agius P. , Young E.P. and Attard Montalto S.P., Infantile generalized GM1 gangliosidosis: High incidence in the Maltese Islands, J Inherit Metab Dis Sep 20: (5) ((1997) ), 723–724. |
[183] | Sinigerska I. , Chandler D. , Vaghjiani V. , Hassanova I. , Gooding R. and Morrone A. , et al., Founder mutation causing infantile GM1-gangliosidosis in the Gypsy population, Mol Genet Metab 88: (1) ((2006) ), 93–95. |
[184] | Severi F. , Magrini U. , Tettamanti G. , Bianchi E. and Lanzi G. , Infantile GM1 gangliosidosis. Histochemical, ultrastructural and biochemical studies, Helv Paediatr Acta 26: (2) ((1971) ), 192–209. |
[185] | Kudoh T. , Kikuchi K. , Nakamura F. , Yokoyama S. , Karube K. and Tsugawa S. , et al., Prenatal diagnosis of GM1-gangliosidosis: Biochemical manifestations in fetal tissues, Hum Genet 44: (3) ((1978) ), 287–293. |
[186] | Gilbert E.F. , Varakis J. , Opitz J.M. , ZuRhein G.M. , Ware R. and Viseskul C. , et al., Generalized gangliosidosis type II (juvenile GM1 gangliosidosis). A pathological, histochemical and ultrastructural study, Z Für Kinderheilkd 120: (3) ((1975) ), 151–180. |
[187] | Derry D.M. , Fawcett J.S. , Andermann F. and Wolfe L.S. , Late infantile systemic lipidosis. Major monosialogangliosidosis. Delineation of two types, Neurology 18: (4) ((1968) ), 340–348. |
[188] | Pabico R.C. , Atancio B.C. , McKenna B.A. , Pamukcoglu T. and Yodaiken R. , Renal pathologic lesions and functional alterations in a man with Fabry’s disease, Am J Med 55: (3) ((1973) ), 415–425. |
[189] | Gilbert E.F. , Dawson G. , Rhein G.M. and Opitz J.M. , Spranger null. I-cell disease, mucolipidosis II. Pathological, histochemical, ultrastructural and biochemical observations in four cases, Z Für Kinderheilkd 114: (4) ((1973) ), 259–292. |
[190] | O’Brien J.S. , Ganglioside Storage Diseases. In: Harris H., Hirschhorn K., editors. Advances in Human Genetics [Internet]. Springer US; (1972) [cited 2015 Jul 30]. p. 39–98. Available from: http://link.springer.com/chapter/10.1007%2F978-1-4757-4429-3_2 |
[191] | Suzuki Y. , Crocker A.C. and Suzuki K. , GM1-gangliosidosis. Correlationof clinical and biochemical data, Arch Neurol 24: (1) ((1971) ), 58–64. |
[192] | Suzuki K. and Chen G.C. , GM1-gangliosidosis (gneralized gangliosidosis). Morphology and chemical pathology, Pathol Eur 3: (2) ((1968) ), 389–408. |
[193] | Lowden J.A. , Callahan J.W. , Norman M.G. , Thain M. and Prichard J.S. , Juvenile GM1 gangliosidosis. Occurrence with absence of two beta-galactosidase components, Arch Neurol 31: (3) ((1974) ), 200–203. |
[194] | Yunis E.J. and Lee R.E. , The ultrastructure of globoid (Krabbe) leukodystrophy, Lab Investig J Tech Methods Pathol 21: (5) ((1969) ), 415–419. |
[195] | Pennelli N. , Scaravilli F. and Zacchello F. , The morphogenesis of Gaucher cells investigated by electron microscopy, Blood 34: (3) ((1969) ), 331–347. |
[196] | Lee R.E. , The fine structure of the cerebroside occurring in Gaucher’s disease, Proc Natl Acad Sci U S A 61: (2) ((1968) ), 484–489. |
[197] | Wallace B.J. , Volk B.W. and Lazarus S.S. , Fine structural localization of acid phosphatase activity in neurons of Tay-Sachs disease, J Neuropathol Exp Neurol [Internet] ((1964) ) 23: (4). Available from: http://journals.lww.com/jneuropath/Fulltext/1964/10000/FINE_STRUCTURAL_LOCALIZATION_OF_ACID_PHOSPHATASE.7.aspx |
[198] | Emery J.M. , Green W.R. , Wyllie R.G. and Howell R.R. , GM1-gangliosidosis. Ocular and pathological manifestations, Arch Ophthalmol Chic Ill 1960 85: (2) ((1971) ), 177–187. |
[199] | Goebel H.H. , Fix J.D. and Zeman W. , Retinal Pathology in GM1 Gangliosidosis, Type II, Am J Ophthalmol 75: (3) ((1973) ), 434–441. |
[200] | Percy A.K. , McCormick U.M. , Kaback M.M. and Herndon R.M. , Ultrastructure manifestations of GM1 and GM2 gangliosidosis in fetal tissues, Arch Neurol 28: (6) ((1973) ), 417–419. |
[201] | Roze E. , Paschke E. , Lopez N. , Eck T. , Yoshida K. and Maurel-Ollivier A. , et al., Dystonia and parkinsonism in GM1 type 3 gangliosidosis, Mov Disord Off J Mov Disord Soc 20: (10) ((2005) ), 1366–1369. |
[202] | Muthane U. , Chickabasaviah Y. , Kaneski C. , Shankar S.K. , Narayanappa G. and Christopher R. , et al., Clinical features of adult GM1 gangliosidosis: Report of three Indian patients and review of 40 cases, Mov Disord Off J Mov Disord Soc 19: (11) ((2004) ), 1334–1341. |
[203] | Maciel R.O.H. , Pedroso J.L. and Barsottini O.G.P. , Facial grimacing as a clue for the diagnosis of GM1 type 3 gangliosidosis, Arq Neuropsiquiatr 69: ((2011) ), 406–407. |
[204] | Nakano T. , Ikeda S. , Kondo K. , Yanagisawa N. and Tsuji S. , Adult GM1-gangliosidosis: Clinical patterns and rectal biopsy, Neurology 35: (6) ((1985) ), 875–880. |
[205] | Ushiyama M. , Ikeda S. , Nakayama J. , Yanagisawa N. , Hanyu N. and Katsuyama T. , Type III (chronic) GM1-gangliosidosis. Histochemical and ultrastructural studies of rectal biopsy, J Neurol Sci 71: (2-3) ((1985) ), 209–223. |
[206] | Ikeda S. , Ushiyama M. , Nakano T. , Kikkawa T. , Kondo K. and Yanagisawa N. , Ultrastructural findings of rectal and skin biopsies in adult GM1-gangliosidosis, Acta Pathol Jpn 36: (12) ((1986) ), 1823–1831. |
[207] | Tay W. , Symmetrical changes in the region of the yellow spot in each eye of an infant, Trans Ophthalmol Soc 1: ((1881) ), 55–57. |
[208] | Sachs B. , On Arrested Cerebral Development, With Special Reference To Its Cortical Pathology. 1, J Nerv Ment Dis 14: (9-10) ((1887) ), 541–553. |
[209] | Klenk E. , Die. Fettstoffe des Gehirns bei Amaurotischer Idiotie und Niemann-Pick’scher Krankheit, Ber Ges Physiol 96: ((1937) ), 659–660. |
[210] | Klenk E. , Über die Ganglioside, eine neue Gruppe von zuckerhaltigen Gehirnlipoiden. Hoppe-Seyler', Z Für Physiol Chem 273: (1-2) ((1942) ), 76–86. |
[211] | Okada S. and O’Brien J.S. , Tay-Sachs disease: Generalized absence of a beta-D-N-acetylhexosaminidase component, Science 165: (3894) ((1969) ), 698–700. |
[212] | Sandhoff K. , Andreae U. and Jatzkewitz H. , Deficient hexosaminidase activity in an exceptional case of Tay-Sachs disease with additional storage of kidney globoside in visceral organs, Life Sci 7: (6) ((1968) ), 283–288. |
[213] | Gravel R.A. , Kaback M.M. , Proia R.L. , Sandhoff K. , Suzuki K. and Suzuki K. , The GM2 Gangliosidoses. In: Valle D.L., Beaudet A.L., Vogelstein B., Kinzler K.W., Antonarakis S.E., Ballabio A., et al., editors. The Online Metabolic and Molecular Bases of Inherited Disease [Internet]. New York, NY: The McGraw-Hill Companies, Inc.; (2014) [cited 2015 Jul 27]. Available from: http://mhmedical.com/content.aspx?aid=1102897473 |
[214] | Peleg L. , Meltzer F. , Karpati M. and Goldman. B. , GM2 gangliosidosis B1 variant: Biochemical and molecular characterization of hexosaminidase A, Biochem Mol Med 54: (2) ((1995) ), 126–132. |
[215] | Navon R. , Kopel R. , Nutman J. , Frisch A. , Conzelmann E. and Sandhoff K. , et al., Hereditary heat-labile hexosaminidase B: A variant whose homozygotes synthesize a functional HEX A, Am J Hum Genet 37: (1) ((1995) ), 138–146. |
[216] | Park N.J. , Morgan C. , Sharma R. , Li Y. , Lobo R.M. and Redman J.B. , et al., Improving accuracy of Tay Sachs carrier screening of the non-Jewish population: Analysis of 34 carriers and six late-onset patients with HEXA enzyme and DNA sequence analysis, Pediatr Res 67: (2) ((2010) ), 217–220. |
[217] | Neudorfer O. , Pastores G.M. , Zeng B.J. , Gianutsos J. , Zaroff C.M. and Kolodny E.H. , Late-onset Tay-Sachs disease: Phenotypic characterization and genotypic correlations in 21 affected patients, Genet Med Off J Am Coll Med Genet 7: (2) ((2005) ), 119–123. |
[218] | Mitsumoto H. , Sliman R.J. , Schafer I.A. , Sternick C.S. , Kaufman B. and Wilbourn A. , et al., Motor neuron disease and adult hexosaminidase A deficiency in two families: Evidence for multisystem degeneration, Ann Neurol 17: (4) ((1985) ), 378–385. |
[219] | Mondelli M. , Rossi A. , Palmeri S. , Rizzuto N. and Federico A. , Neurophysiological study in chronic GM2 gangliosidosis (hexosaminidase A and B deficiency), with motor neuron disease phenotype, Ital J Neurol Sci 10: (4) ((1989) ), 433–439. |
[220] | Drory V.E. , Birnbaum M. , Peleg L. , Goldman B. and Korczyn A.D. , Hexosaminidase A deficiency is an uncommon cause of a syndrome mimicking amyotrophic lateral sclerosis, Muscle Nerve 28: (1) ((2003) ), 109–112. |
[221] | Godeiro-Junior C. , Felicio A.C. , Benites V. , Chieia M.A. and Oliveira A.S.B. , Late-onset hexosaminidase A deficiency mimicking primary lateral sclerosis, Arq Neuropsiquiatr 67: (1) ((2009) ), 105–106. |
[222] | Johnson W.G. , Wigger H.J. , Karp H.R. , Glaubiger L.M. and Rowland L.P. , Juvenile spinal muscular atrophy: A new hexosaminidase deficiency phenotype, Ann Neurol 11: (1) ((1982) ), 11–16. |
[223] | Parnes S. , Karpati G. , Carpenter S. , Kin N.M. , Wolfe L.S. and Suranyi L. , Hexosaminidase-A deficiency presenting as atypical juvenile-onset spinal muscular atrophy, Arch Neurol 42: (12) ((1985) ), 1176–1180. |
[224] | Navon R. , Khosravi R. , Melki J. , Drucker L. , Fontaine B. and Turpin J.C. , et al., Juvenile-onset spinal muscular atrophy caused by compound heterozygosity for mutations in the HEXA gene, Ann Neurol 41: (5) ((1997) ), 631–638. |
[225] | Jamrozik Z. , Lugowska A. , Gołębiowski M. , Królicki L., Mączewska J. and Kuźma-Kozakiewicz M., Late onset GM2 gangliosidosis mimicking spinal muscular atrophy, Gene 527: (2) ((2013) ), 679–682. |
[226] | Kaback M.M. and Desnick R.J. , Hexosaminidase A Deficiency. In: Pagon R.A., Adam M.P., Ardinger H.H., Wallace S.E., Amemiya A., Bean L.J., et al., editors. GeneReviews(®) [Internet]. Seattle (WA) University of Washington, Seattle; (1993) [cited 2015 Aug 3]. Available from: http://www.ncbi.nlm.nih.gov/books/NBK1218/ |
[227] | Salman M.S. , Clarke J.T. , Midroni G. and Waxman M.B. , Peripheral and autonomic nervous system involvement in chronic GM2-gangliosidosis, J Inherit Metab Dis 24: (1) ((2001) ), 65–71. |
[228] | Pellegrini M. , Zicari E. , Dotti M.T. and Federico A. , Dysautonomic achalasia in two siblings with Sandhoff disease, J Neurol Sci 241: (1-2) ((2006) ), 107–109. |
[229] | Chow G.C. , Clarke J.T. and Banwell B.L. , Late-onset GM2 gangliosidosis presenting as burning dysesthesias, Pediatr Neurol 25: (1) ((2001) ), 59–61. |
[230] | Torres P.A. , Zeng B.J. , Porter B.F. , Alroy J. , Horak F. and Horak J. , et al., Tay-Sachs disease in Jacob sheep, Mol Genet Metab 101: (4) ((2010) ), 357–363. |
[231] | Kaback M. , Lim-Steele J. , Dabholkar D. , Brown D. , Levy N. and Zeiger K. , Tay-Sachs disease–carrier screening, prenatal diagnosis, and the molecular era. An international perspective, to The International TSD Data Collection Network, JAMA 270: (19) ((1993) ), 2307–2315. |
[232] | Rosengren B. , Månsson J.E. and Svennerholm L., Composition of gangliosides and neutral glycosphingolipids of brain in classical Tay-Sachs and Sandhoff disease: More lyso-GM2 in Sandhoff disease? J Neurochem 49: (3) ((1987) ), 834–840. |
[233] | Bolhuis P.A. , Oonk J.G. , Kamp P.E. , Ris A.J. , Michalski J.C. and Overdijk B. , et al., Ganglioside storage, hexosaminidase lability, and urinary oligosaccharides in adult Sandhoff’s disease, Neurology 37: (1) ((1987) ), 75–81. |
[234] | Tsuji D. , Akeboshi H. , Matsuoka K. , Yasuoka H. , Miyasaki E. and Kasahara Y. , et al., Highly phosphomannosylated enzyme replacement therapy for GM2 gangliosidosis, Ann Neurol 69: (4) ((2011) ), 691–701. |
[235] | Matsuoka K. , Tamura T. , Tsuji D. , Dohzono Y. , Kitakaze K. and Ohno K. , et al., Therapeutic potential of intracerebroventricular replacement of modified human β-hexosaminidase B for GM2 gangliosidosis, Mol Ther J Am Soc Gene Ther 19: (6) ((2011) ), 1017–1024. |
[236] | Clarke J.T.R. , Mahuran D.J. , Sathe S. , Kolodny E.H. , Rigat B.A. and Raiman J.A. , et al., An open-label Phase I/II clinical trial of pyrimethamine for the treatment of patients affected with chronic GM2 gangliosidosis (Tay-Sachs or Sandhoff variants), Mol Genet Metab 102: (1) ((2011) ), 6–12. |
[237] | Osher E. , Fattal-Valevski A. , Sagie L. , Urshanski N. , Amir-Levi Y. and Katzburg S. , et al., Pyrimethamine increases β-hexosaminidase A activity in patients with Late Onset Tay Sachs, Mol Genet Metab 102: (3) ((2011) ), 356–363. |
[238] | Osher E. , Fattal-Valevski A. , Sagie L. , Urshanski N. , Sagiv N. and Peleg L. , et al., Effect of cyclic, low dose pyrimethamine treatment in patients with Late Onset Tay Sachs: An open label, extended pilot study, Orphanet J Rare Dis 10: ((2015) ), 45. |
[239] | Shapiro B.E. , Pastores G.M. , Gianutsos J. , Luzy C. and Kolodny E.H. , Miglustat in late-onset Tay-Sachs disease: A 12-month, randomized, controlled clinical study with 24 months of extended treatment, Genet Med Off J Am Coll Med Genet 11: (6) ((2009) ), 425–433. |
[240] | Scholz W. , Klinische, Pathologisch-Anatomische und Erbbiologische untersuchungen bei Familiärer, Diffuser hirnsklerose im Kindesalter, Z Für Gesamte Neurol Psychiatr 99: (1) ((1925) ), 651–717. |
[241] | Pfeiffer J. , Über die metachromatischen Leukodystrophien (Typ Scholz), Arch Für Psychiatr Nervenkrankh 199: (4) ((1959) ), 386–416. |
[242] | Bielschowsky M. and Henneberg R. , Über familiäre diffuse Sklerose (Leukodystrophia cerebri progressiva hereditaria), J Psychol NeurolLpz 36: ((1928) ), 131–181. |
[243] | Greenfield J.G. , A Form of Progressive Cerebral Sclerosis in Infants associated with Primary Degeneration of the Interfascicular Glia, Proc R Soc Med 26: (6) ((1933) ), 690–697. |
[244] | Austin J.H. , Balasubramanian A.S. , Pattabiraman T.N. , Saraswathi S. , Basu D.K. and Bachhawat B.K. , A controlled study of enzymatic activies in three human disorder of glycolipid metabolism, J Neurochem 10: (11) ((1963) ), 805–816. |
[245] | Gieselmann V. and Ingeborg K.-M. , Metachromatic Leukodystrophy. In: Valle D.L., Beaudet A.L., Vogelstein B., Kinzler K.W., Antonarakis S.E., Ballabio A., et al., editors. The Online Metabolic and Molecular Bases of Inherited Disease [Internet]. New York, NY: The McGraw-Hill Companies, Inc.; (2014) [cited 2015 Jul 27]. Available from: http://mhmedical.com/content.aspx?aid=1102896204 |
[246] | Fluharty A.L. , Arylsulfatase A Deficiency. In: Pagon R.A., Adam M.P., Ardinger H.H., Wallace S.E., Amemiya A., Bean L.J., et al., editors. GeneReviews(®) [Internet]. Seattle (WA) University of Washington, Seattle; (1993) [cited 2015 Aug 3]. Available from: http://www.ncbi.nlm.nih.gov/books/NBK1130/ |
[247] | Stevens R.L. , Fluharty A.L. , Kihara H. , Kaback M.M. , Shapiro L.J. and Marsh B. , et al., Cerebroside sulfatase activator deficiency induced metachromatic leukodystrophy, Am J Hum Genet 33: (6) ((1981) ), 900–906. |
[248] | Bindu P.S. , Mahadevan A. , Taly A.B. , Christopher R. , Gayathri N. and Shankar S.K. , Peripheral neuropathy in metachromatic leucodystrophy. A study of 40 cases from south India, J Neurol Neurosurg Psychiatry 76: (12) ((2005) ), 1698–701. |
[249] | Sabatelli M. , Quaranta L. , Madia F. , Lippi G. , Conte A. and Monaco M.L.o. , et al., Peripheral neuropathy with hypomyelinating features in adult-onset Krabbe’s disease, Neuromuscul Disord NMD 12: (4) ((2002) ), 386–391. |
[250] | Rodriguez-Soriano J. , Rivera J.M. , Vallo A. , Prats-Viñas J.M. and Castillo G. , Proximal renal tubular acidosis in metachromatic leukodystrophy, Helv Paediatr Acta 33: (1) ((1978) ), 45–52. |
[251] | Lorioli L. , Cicalese M.P. , Silvani P. , Assanelli A. , Salvo I. and Mandelli A. , et al., Abnormalities of acid-base balance and predisposition to metabolic acidosis in Metachromatic Leukodystrophy patients, Mol Genet Metab 115: (1) ((2015) ), 48–52. |
[252] | Fock J.M. , Begeer J.H. and Prins T.R. , Metachromatic leukodystrophy and coincidental finding of papillomatosis of the gallbladder. A case report, Neuropediatrics 26: (1) ((1995) ), 55–56. |
[253] | Oak S. , Rao S. , Karmarkar S. , Kulkarni B. , Kalgutkar A. and Malde A. , et al., Papillomatosis of the gallbladder in metachromatic leukodystrophy, Pediatr Surg Int 12: (5-6) ((1997) ), 424–425. |
[254] | Siegel E.G. , Lücke H. , Schauer W. and Creutzfeldt W. , Repeated upper gastrointestinal hemorrhage caused by metachromatic leukodystrophy of the gall bladder, Digestion 51: (2) ((1992) ), 121–124. |
[255] | Vettoretto N. , Giovanetti M. , Regina P. , Baronchelli C. and Giulini S.M. , Hemorrhagic cholecystitis as a likely cause of nontraumatic hemobilia in metachromatic leukodystrophy: Report of a case, Ann Ital Chir 72: (6) ((2001) ), 725–728. |
[256] | Garavelli L. , Rosato S. , Mele A. , Wischmeijer A. , Rivieri F. and Gelmini C. , et al., Massive hemobilia and papillomatosis of the gallbladder in metachromatic leukodystrophy: A life-threatening condition, Neuropediatrics 40: (6) ((2009) ), 284–286. |
[257] | Amaducci L. , Sorbi S. , Piacentini S. and Bick K.L. , The first Alzheimer disease case: A metachromatic leukodystrophy? Dev Neurosci 13: (4-5) ((1991) ), 186–187. |
[258] | Dubois G. , Harzer K. and Baumann N. , Very low arylsulfatase A and cerebroside sulfatase activities in leukocytes of healthy members of metachromatic leukodystrophy family, Am J Hum Genet 29: (2) ((1977) ), 191–194. |
[259] | Herz B. and Bach G. , Arylsulfatase. A in pseudodeficiency. Hum Genet, 66: (2-3) ((1984) ), 147–150. |
[260] | Hohenschutz C. , Eich P. , Friedl W. , Waheed A. , Conzelmann E. and Propping P. , Pseudodeficiency of arylsulfatase A: A common genetic polymorphism with possible disease implications, Hum Genet 82: (1) ((1989) ), 45–48. |
[261] | Gieselmann V. , Zlotogora J. , Harris A. , Wenger D.A. and Morris C.P. , Molecular genetics of metachromatic leukodystrophy. Hum Mutat 4: (4) ((1994) ), 233–242. |
[262] | Biffi A. , Montini E. , Lorioli L. , Cesani M. , Fumagalli F. and Plati T. , et al., Lentiviral hematopoietic stem cell gene therapy benefits metachromatic leukodystrophy, Science 341: (6148) ((2013) ), 1233158. |
[263] | Quigley H.A. and Green W.R. , Clinical and ultrastructural ocular histopathologic studies of adult-onset metachromatic leukodystrophy, Am J Ophthalmol 82: (3) ((1976) ), 472–479. |
[264] | Goebel H.H. , Shimokawa K. , Argyrakis A. and Pilz H. , The ultrastructure of the retina in adult metachromatic leukodystrophy, Am J Ophthalmol 85: (6) ((1978) ), 841–849. |
[265] | Goebel H.H. , Busch-Hettwer H. and Bohl J. , Ultrastructural study of the retina in late infantile metachromatic leukodystrophy, Ophthalmic Res 24: (2) ((1992) ), 103–109. |
[266] | Peng L. and Suzuki K. , Ultrastructural study of neurons in metachromatic leukodystrophy, Clin Neuropathol 6: (5) ((1986) ), 224–230. |
[267] | Wiesmann U.N. , Meier C. , Spycher M.A. , Schmid W. , Bischoff A. and Gautier E. , et al., Prenatal metachromatic leukodystrophy, Helv Paediatr Acta 30: (1) ((1975) ), 31–42. |
[268] | Schmidt B. , Selmer T. , Ingendoh A. and von K. , Figura, A novel amino acid modification in sulfatases that is defective in multiple sulfatase deficiency, Cell 82: (2) ((1995) ), 271–278. |
[269] | Hopwood J.J. and Ballabio A. , Multiple Sulfatase Deficiency and the Nature of the Sulfatase Family. In: Valle D.L., Beaudet A.L., Vogelstein B., Kinzler K.W., Antonarakis S.E., Ballabio A., et al., editors. The Online Metabolic and Molecular Bases of Inherited Disease [Internet]. New York, NY: The McGraw-Hill Companies, Inc.; (2014) [cited 2015 Jul 27]. Available from: http://mhmedical.com/content.aspx?aid=1102896477 |
[270] | Sandhoff K. , Harzer K. , Wässle W. and Jatzkewitz H. , Enzyme alterations and lipid storage in three variants of Tay-Sachs disease, J Neurochem 18: (12) ((1971) ), 2469–2489. |
[271] | Conzelmann E. and Sandhoff K. , AB variant of infantile GM2 gangliosidosis: Deficiency of a factor necessary for stimulation of hexosaminidase A-catalyzed degradation of ganglioside GM2 and glycolipid GA2, Proc Natl Acad Sci U S A 75: (8) ((1978) ), 3979–3983. |
[272] | Goldman J.E. , Yamanaka T. , Rapin I. , Adachi M. , Suzuki K. and Suzuki K. , The AB-variant of GM2-gangliosidosis. Clinical, biochemical, and pathological studies of two patients, Acta Neuropathol (Berl) 52: (3) ((1980) ), 189–202. |
[273] | Schuette C.G. , Pierstorff B. , Huettler S. and Sandhoff K. , Sphingolipid activator proteins: Proteins with complex functions in lipid degradation and skin biogenesis, Glycobiology 11: (6) ((2001) ), 81R–90R. |
[274] | Yuan L. and Morales C.R. , A stretch of 17 amino acids in the prosaposin C terminus is critical for its binding to sortilin and targeting to lysosomes, J Histochem Cytochem Off J Histochem Soc 58: (3) ((2010) ), 287–300. |
[275] | Gopalakrishnan M.M. , Grosch H.-W. , Locatelli-Hoops S. , Werth N. , Smolenová E. and Nettersheim M. , et al., Purified recombinant human prosaposin forms oligomers that bind procathepsin D and affect its autoactivation, Biochem J 383: (Pt. 3) ((2004) ), 507–515. |
[276] | O’Brien J.S. , Carson G.S. , Seo H.C. , Hiraiwa M. and Kishimoto Y. , Identification of prosaposin as a neurotrophic factor, Proc Natl Acad Sci U S A 91: (20) ((1994) ), 9593–9596. |
[277] | O’Brien J.S. , Carson G.S. , Seo H.C. , Hiraiwa M. , Weiler S. and Tomich J.M. , et al., Identification of the neurotrophic factor sequence of prosaposin, FASEB J Off Publ Fed Am Soc Exp Biol 9: (8) ((1995) ), 681–685. |
[278] | Kotani Y. , Matsuda S. , Wen T.C. , Sakanaka M. , Tanaka J. and Maeda N. , et al., A hydrophilic peptide comprising 18 amino acid residues of the prosaposin sequence has neurotrophic activity in vitro and in vivo, J Neurochem 66: (5) ((1996) ), 2197–2200. |
[279] | Misasi R. , Hozumi I. , Inuzuka T. , Capozzi A. , Mattei V. and Kuramoto Y. , et al., Biochemistry and neurobiology of prosaposin: A potential therapeutic neuro-effector, Cent Nerv Syst Agents Med Chem 9: (2) ((2009) ), 119–131. |
[280] | Fürst W. and Sandhoff K. , Activator proteins and topology of lysosomal sphingolipid catabolism, Biochim Biophys Acta 1126: (1) ((1992) ), 1–16. |
[281] | Spiegel R. , Bach G. , Sury V. , Mengistu G. , Meidan B. and Shalev S. , et al., A mutation in the saposin A coding region of the prosaposin gene in an infant presenting as Krabbe disease: First report of saposin A deficiency in humans, Mol Genet Metab 84: (2) ((2005) ), 160–166. |
[282] | Shapiro L.J. , Aleck K.A. , Kaback M.M. , Itabashi H. , Desnick R.J. and Brand N. , et al., Metachromatic leukodystrophy without arylsulfatase A deficiency, Pediatr Res 13: (10) ((1979) ), 1179–1181. |
[283] | Hahn A.F. , Gordon B.A. , Gilbert J.J. and Hinton G.G. , The AB-variant of metachromatic leukodystrophy (postulated activator protein deficiency). Light and electron microscopic findings in a sural nerve biopsy, Acta Neuropathol (Berl) 55: (4) ((1981) ), 281–287. |
[284] | Henseler M. , Klein A. , Reber M. , Vanier M.T. , Landrieu P. and Sandhoff K. , Analysis of a splice-site mutation in the sap-precursor gene of a patient with metachromatic leukodystrophy, Am J Hum Genet 58: (1) ((1996) ), 65–74. |
[285] | Landrieu P. , Blanche S. , Vanier M.T. , Metral S. , Husson B. and Sandhoff K. , et al., Bone marrow transplantation in metachromatic leukodystrophy caused by saposin-B deficiency: A case report with a 3-year follow-up period, J Pediatr 133: (1) ((1998) ), 129–132. |
[286] | Tylki-Szymańska A. , Czartoryska B. , Vanier M.-T. , Poorthuis B.J.M.H. , Groener J.A.E. and Ługowska A. , et al., Non-neuronopathic Gaucher disease due to saposin C deficiency, Clin Genet 72: (6) ((2007) ), 538–542. |
[287] | Matsuda J. , Kido M. , Tadano-Aritomi K. , Ishizuka I. , Tominaga K. and Toida K. , et al., Mutation in saposin D domain of sphingolipid activator protein gene causes urinary system defects and cerebellar Purkinje cell degeneration with accumulation of hydroxy fatty acid-containing ceramide in mouse, Hum Mol Genet 13: (21) ((2004) ), 2709–2723. |
[288] | Harzer K. , Paton B.C. , Poulos A. , Kustermann-Kuhn B. , Roggendorf W. and Grisar T. , et al., Sphingolipid activator protein deficiency in a 16-week-old atypical Gaucher disease patient and his fetal sibling: Biochemical signs of combined sphingolipidoses, Eur J Pediatr 149: (1) ((1989) ), 31–39. |
[289] | Paton B.C. , Schmid B. , Kustermann-Kuhn B. , Poulos A. and Harzer K. , Additional biochemical findings in a patient and fetal sibling with a genetic defect in the sphingolipid activator protein (SAP) precursor, prosaposin. Evidence for a deficiency in SAP-1 and for a normal lysosomal neuraminidase, Biochem J 285: (Pt 2) ((1992) ), 481–488. |
[290] | Schnabel D. , Schröder M. , Fürst W. , Klein A. , Hurwitz R. and Zenk T. , et al., Simultaneous deficiency of sphingolipid activator proteins 1 and 2 is caused by a mutation in the initiation codon of their common gene, J Biol Chem 267: (5) ((1992) ), 3312–3315. |
[291] | Hulková H. , Cervenková M. , Ledvinová J. , Tochácková M. , Hrebícek M. and Poupetová H. , et al., A novel mutation in the coding region of the prosaposin gene leads to a complete deficiency of prosaposin and saposins, and is associated with a complex sphingolipidosis dominated by lactosylceramide accumulation, Hum Mol Genet 10: (9) ((2001) ), 927–940. |
[292] | Elleder M. , Jirásek A. , Smíd F. , Ledvinová J. , Besley G.T. and Stopeková M. , Niemann-Pick disease type C with enhanced glycolipid storage. Report on further case of so-called lactosylceramidosis, Virchows Arch A Pathol Anat Histopathol 402: (3) ((1984) ), 307–317. |
[293] | Elleder M. , Jerábková M. , Befekadu A. , Hrebícek M. , Berná L. and Ledvinová J. , et al., Prosaposin deficiency – a rarely diagnosed, rapidly progressing, neonatal neurovisceral lipid storage disease. Report of a further patient, Neuropediatrics 36: (3) ((2005) ), 171–180. |
[294] | Millat G. , Verot L. , Rodríguez-Lafrasse C. , DiMarco J. , Rimet Y. and Poujol A. , et al., Fourth reported family with prosaposin deficiency. In: Elleder M., Ledvinová J., Hřebíček M., Poupětová H. and Kožich V., editors. Book of Abstracts 14th ESGLD Workshop (September 18th-21st, 2003). Pod?brady/Prague: Guarant Ltd; (2003) . p. 81. |
[295] | Kuchar L. , Ledvinová J. , Hrebícek M. , Mysková H. , Dvoráková L. and Berná L. , et al., Prosaposin deficiency and saposin B deficiency (activator-deficient metachromatic leukodystrophy) Report on two patients detected by analysis of urinary sphingolipids and carrying novel PSAP gene mutations, Am J Med Genet A 149A: (4) ((2009) ), 613–621. |
[296] | Sikora J. , Harzer K. and Elleder M. , Neurolysosomal pathology in human prosaposin deficiency suggests essential neurotrophic function of prosaposin, Acta Neuropathol (Berl) 113: (2) ((2007) ), 163–175. |
[297] | Lignac G. , Über störung des Cystinstoffwechsels bei Kindern, Deut Arch Klin Med 145: ((1924) ), 139–150. |
[298] | Mahoney C.P. , Striker G.E. , Hickman R.O. , Manning G.B. and Marchioro T.L. , Renal transplantation for childhood cystinosis, N Engl J Med 283: (8) ((1970) ), 397–402. |
[299] | Gahl W.A. , Bashan N. , Tietze F. , Bernardini I. and Schulman J.D. , Cystine transport is defective in isolated leukocyte lysosomes from patients with cystinosis, Science 217: (4566) ((1982) ), 1263–1265. |
[300] | Gahl W.A. , Thoene J.G. and Schneider J.A. , Cystinosis, N Engl J Med 347: (2) ((2002) ), 111–121. |
[301] | Gahl W.A. and Thoene J.G. , Cystinosis: A Disorder of Lysosomal Membrane Transport. In: Valle D.L., Beaudet A.L., Vogelstein B., Kinzler K.W., Antonarakis S.E., Ballabio A., et al., editors. The Online Metabolic and Molecular Bases of Inherited Disease [Internet]. New York, NY: The McGraw-Hill Companies, Inc.; (2014) [cited 2015 Jul 27]. Available from: http://mhmedical.com/content.aspx?aid=1102907102 |
[302] | Nesterova G. and Gahl W.A. . Cystinosis. In: Pagon R.A. , Adam M.P. , Ardinger H.H. , Wallace S.E. , Amemiya A. and Bean L.J. , et al., editors. GeneReviews(®) [Internet]. Seattle (WA) University of Washington, Seattle; (1993) [cited 2015 Aug 6]. Available from: http://www.ncbi.nlm.nih.gov/books/NBK1400/ |
[303] | Town M. , Jean G. , Cherqui S. , Attard M. , Forestier L. and Whitmore S.A. , et al., A novel gene encoding an integral membrane protein is mutated in nephropathic cystinosis, Nat Genet 18: (4) ((1998) ), 319–324. |
[304] | Charnas L.R. , Luciano C.A. , Dalakas M. , Gilliatt R.W. , Bernardini I. and Ishak K. , et al., Distal vacuolar myopathy in nephropathic cystinosis, Ann Neurol 35: (2) ((1994) ), 181–188. |
[305] | Jézégou A. , Llinares E. , Anne C. , Kieffer-Jaquinod S. , O’Regan S. and Aupetit J. , et al., Heptahelical protein PQLC2 is a lysosomal cationic amino acid exporter underlying the action of cysteamine in cystinosis therapy, Proc Natl Acad Sci U S A 109: (50) ((2012) ), E3434–E3443. |
[306] | Markello T.C. , Bernardini I.M. and Gahl W.A. , Improved renal function in children with cystinosis treated with cysteamine, N Engl J Med 328: (16) ((1993) ), 1157–1162. |
[307] | Gahl W.A. , Balog J.Z. and Kleta R. , Nephropathic cystinosis in adults: Natural history and effects of oral cysteamine therapy, Ann Intern Med 147: (4) ((2007) ), 242–250. |
[308] | Langman C.B. , Greenbaum L.A. , Sarwal M. , Grimm P. , Niaudet P. and Deschênes G. , et al., A randomized controlled crossover trial with delayed-release cysteamine bitartrate in nephropathic cystinosis: Effectiveness on white blood cell cystine levels and comparison of safety, Clin J Am Soc Nephrol CJASN 7: (7) ((2012) ), 1112–1120. |
[309] | Kaiser-Kupfer M.I. , Fujikawa L. , Kuwabara T. , Jain S. and Gahl W.A. , Removal of corneal crystals by topical cysteamine in nephropathic cystinosis, N Engl J Med 316: (13) ((1987) ), 775–779. |
[310] | Aula P. , Autio S. , Raivio K.O. , Rapola J. , Thodén C.J. and Koskela S.L. , et al., “Salla disease”: A new lysosomal storage disorder, Arch Neurol 36: (2) ((1979) ), 88–94. |
[311] | Tondeur M. , Libert J. , Vamos E. , Van Hoof F. , Thomas G.H. and Strecker G. , Infantile form of sialic acid storage disorder: Clinical, ultrastructural, and biochemical studies in two siblings, Eur J Pediatr 139: (2) ((1982) ), 142–147. |
[312] | Aula P. , Gahl W.A. , Disorders of Free Sialic Acid Storage. In: Valle D.L. , Beaudet A.L. , Vogelstein B. , Kinzler K.W. , Antonarakis S.E. , Ballabio A. , et al., editors. The Online Metabolic and Molecular Bases of Inherited Disease [Internet]. New York NY: The McGraw-Hill Comanies, Inc.; (2014) [cited 2015 Jul 27]. Available from: http://mhmedical.com/content.aspx?aid=1102907714 |
[313] | Renlund M. , Tietze F. and Gahl W.A. , Defective sialic acid egress from isolated fibroblast lysosomes of patients with Salla disease, Science 232: (4751) ((1986) ), 759–762. |
[314] | Tietze F. , Seppala R. , Renlund M. , Hopwood J.J. , Harper G.S. and Thomas G.H. , et al., Defective lysosomal egress of free sialic acid (N-acetylneuraminic acid) in fibroblasts of patients with infantile free sialic acid storage disease, J Biol Chem 264: (26) ((1989) ), 15316–15322. |
[315] | Lemyre E. , Russo P. , Melançon S.B. , Gagné R. , Potier M. and Lambert M. , Clinical spectrum of infantile free sialic acid storage disease, Am J Med Genet 82: (5) ((1999) ), 385–391. |
[316] | Sperl W. , Gruber W. , Quatacker J. , Monnens L. , Thoenes W. and Fink F.M. , et al., Nephrosis in two siblings with infantile sialic acid storage disease, Eur J Pediatr 149: (7) ((1990) ), 477–482. |
[317] | Pueschel S.M. , O’Shea P.A. , Alroy J. , Ambler M.W. , Dangond F. and Daniel P.F. , et al., Infantile sialic acid storage disease associated with renal disease, Pediatr Neurol 4: (4) ((1988) ), 207–212. |
[318] | Stevenson R.E. , Lubinsky M. , Taylor H.A. , Wenger D.A. , Schroer R.J. and Olmstead P.M. , Sialic acid storage disease with sialuria: Clinical and biochemical features in the severe infantile type, Pediatrics 72: (4) ((1983) ), 441–449. |
[319] | Kleta R. , Morse R.P. , Orvisky E. , Krasnewich D. , Alroy J. and Ucci A.A. , et al., Clinical, biochemical, and molecular diagnosis of a free sialic acid storage disease patient of moderate severity, Mol Genet Metab 82: (2) ((2004) ), 137–143. |
[320] | Verheijen F.W. , Verbeek E. , Aula N. , Beerens C.E. , Havelaar A.C. and Joosse M. , et al., A new gene, encoding an anion transporter, is mutated in sialic acid storage diseases, Nat Genet 23: (4) ((1999) ), 462–465. |
[321] | Lines M.A. , Rupar C.A. , Rip J.W. , Baskin B. , Ray P.N. and Hegele R.A. , et al., Infantile Sialic Acid Storage Disease: Two Unrelated Inuit Cases Homozygous for a Common Novel SLC17A5 Mutation, JIMD Rep 12: ((2014) ), 79–84. |
[322] | Rosenblatt D.S. , Hosack A. , Matiaszuk N.V. , Cooper B.A. and Laframboise R. , Defect in vitamin B12 release from lysosomes: Newly described inborn error of vitamin B12 metabolism, Science 228: (4705) ((1985) ), 1319–1321. |
[323] | Rutsch F. , Gailus S. , Miousse I.R. , Suormala T. , Sagné C. and Toliat M.R. , et al., Identification of a putative lysosomal cobalamin exporter altered in the cblF defect of vitamin B12 metabolism, Nat Genet 41: (2) ((2009) ), 234–239. |
[324] | Coelho D. , Kim J.C. , Miousse I.R. , Fung S. , du Moulin M. and Buers I. , et al., Mutations in ABCD4 cause a new inborn error of vitamin B12 metabolism, Nat Genet 44: (10) ((2012) ), 1152–1155. |
[325] | Deme J.C. , Hancock M.A. , Xia X. , Shintre C.A. , Plesa M. and Kim J.C. , et al., Purification and interaction analyses of two human lysosomal vitamin B12 transporters: LMBD1 and ABCD4, Mol Membr Biol 31: (7-8) ((2014) ), 250–61. |
[326] | Kim J.C. and Lee N.-C. , Hwu PW.-L. , Chien Y.-H. , Fahiminiya S. and Majewski J. , et al., Late onset of symptoms in an atypical patient with the cblJ inborn error of vitamin B12 metabolism: Diagnosis and novel mutation revealed by exome sequencing, Mol Genet Metab 107: (4) ((2012) ), 664–668. |
[327] | Takeichi T. , Hsu C.-K. , Yang H.-S. , Chen H.-Y. , Wong T.-W. and Tsai W.-L. , et al., Progressive hyperpigmentation in a Taiwanese child due to an inborn error of vitamin B12 metabolism (cblJ), Br J Dermatol 172: (4) ((2015) ), 1111–1115. |
[328] | Berman E.R. , Livni N. , Shapira E. , Merin S. and Levij I.S. , Congenital corneal clouding with abnormal systemic storage bodies: A new variant of mucolipidosis, J Pediatr 84: (4) ((1974) ), 519–526. |
[329] | Schiffmann R. , Grishchuk Y. , Goldin E. , Mucolipidosis IV. In: Pagon R.A. , Adam M.P. , Ardinger H.H. , Wallace S.E. , Amemiya A. , Bean L.J. , et al., editors. GeneReviews(®) [Internet]. Seattle (WA) University of Washington, Seattle; (1993) [cited 2015 Aug 23]. Available from: http://www.ncbi.nlm.nih.gov/books/NBK1214/ |
[330] | Sun M. , Goldin E. , Stahl S. , Falardeau J.L. , Kennedy J.C. and Acierno J.S. , et al., Mucolipidosis type IV is caused by mutations in a gene encoding a novel transient receptor potential channel, Hum Mol Genet 9: (17) ((2000) ), 2471–2478. |
[331] | Dong X.-P. , Cheng X. , Mills E. , Delling M. , Wang F. and Kurz T. , et al., The type IV mucolipidosis-associated protein TRPML1 is an endolysosomal iron release channel, Nature 455: (7215) ((2008) ), 992–996. |
[332] | LaPlante J.M. , Ye C.P. , Quinn S.J. , Goldin E. , Brown E.M. and Slaugenhaupt S.A. , et al., Functional links between mucolipin-1 and Ca2+-dependent membrane trafficking in mucolipidosis IV, Biochem Biophys Res Commun 322: (4) ((2004) ), 1384–1391. |
[333] | Medina D.L. , Di Paola S. , Peluso I. , Armani A. , De Stefani D. and Venditti R. , et al., Lysosomal calcium signalling regulates autophagy through calcineurin and TFEB, Nat Cell Biol Mar 17: (3) ((2015) ), 288–299. |
[334] | Halliwell W.H. , Cationic amphiphilic drug-induced phospholipidosis, Toxicol Pathol 25: (1) ((1997) ), 53–60. |
[335] | Shayman J.A. and Abe A. , Drug induced phospholipidosis: An acquired lysosomal storage disorder, Biochim Biophys Acta 1831: (3) ((2013) ), 602–611. |
[336] | Dake M.D. , Madison J.M. , Montgomery C.K. , Shellito J.E. , Hinchcliffe W.A. and Winkler M.L. , et al., Electron microscopic demonstration of lysosomal inclusion bodies in lung, liver, lymph nodes, and blood leukocytes of patients with amiodarone pulmonary toxicity, Am J Med 78: (3) ((1985) ), 506–512. |
[337] | Rappersberger K. , Konrad K. , Wieser E. , Weber H. and Wolff K. , Morphological changes in peripheral blood cells and skin in amiodarone-treated patients, Br J Dermatol 114: (2) ((1986) ), 189–196. |
[338] | Albay D. , Adler S.G. , Philipose J. , Calescibetta C.C. , Romansky S.G. and Cohen A.H. , Chloroquine-induced lipidosis mimicking Fabry disease, Mod Pathol Off J U S Can Acad Pathol Inc 18: (5) ((2005) ), 733–738. |
[339] | Baguet J.P. , Tremel F. and Fabre M. , Chloroquine cardiomyopathy with conduction disorders, Heart Br Card Soc 81: (2) ((1999) ), 221–223. |
[340] | Freihage J.H. , Patel N.C. , Jacobs W.R. , Picken M. , Fresco R. and Malinowska K. , et al., Heart transplantation in a patient with chloroquine-induced cardiomyopathy, J Heart Lung Transplant Off Publ Int Soc Heart Transplant 23: (2) ((2004) ), 252–255. |
[341] | Soong T.R. , Barouch L.A. , Champion H.C. , Wigley F.M. and Halushka M.K. , New clinical and ultrastructural findings in hydroxychloroquine-induced cardiomyopathy–a report of 2 cases, Hum Pathol 38: (12) ((2007) ), 1858–1863. |
[342] | Müller-Höcker J. , Schmid H. , Weiss M. , Dendorfer U. and Braun G.S. , Chloroquine-induced phospholipidosis of the kidney mimicking Fabry’s disease: Case report and review of the literature, Hum Pathol 34: (3) ((2003) ), 285–289. |
[343] | Liu N. , Tengstrand E.A. , Chourb L. and Hsieh F.Y. , Di-22:6-bis(monoacylglycerol)phosphate: A clinical biomarker of drug-induced phospholipidosis for drug development and safety assessment, Toxicol Appl Pharmacol 279: (3) ((2014) ), 467–476. |
[344] | Gelb M.H. , Scott C.R. and Turecek F. , Newborn screening for lysosomal storage diseases, Clin Chem 61: (2) ((2015) ), 335–346. |
[345] | Scott C.R. , Elliott S. , Buroker N. , Thomas L.I. , Keutzer J. and Glass M. , et al., Identification of infants at risk for developing Fabry, Pompe, or mucopolysaccharidosis-I from newborn blood spots by tandem mass spectrometry, J Pediatr 163: (2) ((2013) ), 498–503. |
[346] | Hopkins P.V. , Campbell C. , Klug T. , Rogers S. , Raburn-Miller J. and Kiesling J. , Lysosomal storage disorder screening implementation: Findings from the first six months of full population pilot testing in Missouri, J Pediatr 166: (1) ((2015) ), 172–177. |
[347] | Mechtler T.P. , Stary S. , Metz T.F. , De Jesús V.R. , Greber-Platzer S. and Pollak A. , et al., Neonatal screening for lysosomal storage disorders: Feasibility and incidence from a nationwide study in Austria, Lancet Lond Engl 379: (9813) ((2012) ), 335–341. |
[348] | Hwu W.-L. , Chien Y.-H. , Lee N.-C. , Chiang S.-C. , Dobrovolny R. and Huang A.-C. , et al., Newborn screening for Fabry disease in Taiwan reveals a high incidence of the later-onset GLA mutation c.936+919G>A (IVS4+919G>A), Hum Mutat 30: (10) ((2009) ), 1397–1405. |
[349] | Spada M. , Pagliardini S. , Yasuda M. , Tukel T. , Thiagarajan G. and Sakuraba H. , et al., High incidence of later-onset fabry disease revealed by newborn screening, Am J Hum Genet 79: (1) ((2006) ), 31–40. |
[350] | Shigeto S. , Katafuchi T. , Okada Y. , Nakamura K. , Endo F. and Okuyama T. , et al., Improved assay for differential diagnosis between Pompe disease and acid α-glucosidase pseudodeficiency on dried blood spots, Mol Genet Metab 103: (1) ((2011) ), 12–17. |
[351] | Chiang S.-C. , Hwu W.-L. , Lee N.-C. , Hsu L.-W. and Chien Y.-H. , Algorithm for Pompe disease newborn screening: Results from the Taiwan screening program, Mol Genet Metab 106: (3) ((2012) ), 281–286. |
[352] | Wasserstein M.P. , Andriola M. , Arnold G. , Aron A. , Duffner P. and Erbe R.W. , et al., Clinical outcomes of children with abnormal newborn screening results for Krabbe disease in New York State, Genet Med 12: ((2016) ). [Epub ahead of print]. |


