
Peroxisome biogenesis disorders
1Introduction
Peroxisome biogenesis disorders (PBD) are a group of conditions caused by a partial or generalized defect in peroxisome biogenesis. They encompass two phenotypic groups: 1. the Zellweger spectrum disorders (ZSD) including severe, intermediate and milder forms [previously known respectively as Zellweger syndrome (ZS), Neonatal Adrenoleukodystrophy (NALD) and Infantile Refsum Disease (IRD)] and 2. Rhizomelic Chondrodysplasia Punctata type 1 (RCDP1), as well as the variant phenotypes now being described for both groups. PBD represent the paradigm for metabolic malformation syndromes. In a little over 50 years, an intense, concerted multidisciplinary effort has led to elucidating the fundamental biochemical, pathophysiological and molecular bases for these disorders effectively paving the way for therapeutic interventions and trials.
Peroxisomes are membrane bound organelles derived from the endoplasmic reticulum, numbering up to several hundred per mammalian cell, mostly spherical in shape, up to 1μm in diameter. They are indispensable for normal life and highly conserved throughout evolution amongst most eukaryotes. Peroxisomes were first identified by biochemist Christian DeDuve in the 1960 s, as cytoplasmic particles containing hydrogen peroxide-generating oxidases [1]. Later, the neurologist Hans Zellweger reported a group of children with similar craniofacial dysmorphism and malformations in the brain, eye, liver, and kidney [2]. These cases, initially described under the eponym of “cerebro-hepato-renal-syndrome”, were subsequently termed Zellweger syndrome (ZS) [3]. ZS is considered today the prototype for ZSD. The reported absence of morphologically identifiable peroxisomes in hepatocytes and renal tubular cells of affected individuals using the peroxisomal matrix enzyme marker, catalase, [4] demonstrated a connection between ZS and peroxisome dysfunction. However, the etiological connection was not recognized until the identification of multiple peroxisomal enzymes and their deficiencies in affected patients. Similar findings were later demonstrated in the intermediate and milder ZSD phenotypes and RCDP1, which were also being reported at that time [5–7].
2Peroxisome biochemistry
There are several hundred peroxisomes in all mammalian cells, each containing more than 50 matrix enzymes required for multiple vital metabolic pathways, including the catabolism and synthesis of important cellular lipids [reviewed in [8]]. The best characterized peroxisomal pathways include β-oxidation of very long chain fatty acids (VLCFA), α-oxidation of methyl-branched phytanic acid and the biosynthesis of ether phospholipid (plasmalogen) (see Table 1). The metabolites of these pathways are routinely screened when a peroxisome biogenesis defect is suspected.
Peroxisomal β-oxidation utilizes a variety of different fatty acid substrates in catabolic and synthetic processes: very long (≥ C22) straight chain saturated and unsaturated fatty acids (VLCFA), (2 S) methyl-branched chain pristanic acid and (25 S) di- and tri-hydroxycholestanoic acids (D/THCA). Docosahexanoic acid (DHA), a critical polyunsaturated omega-3 fatty acid, is synthesized in the peroxisome after β-oxidation of its precursor [9–11]. D/THCA are C27 intermediary bile acids that undergo one step of β-oxidation to form the primary C24 bile acids, cholic and chenodeoxycholic acid. Peroxisomal β-oxidation also participates in the regulation of other complex lipids, including pro-inflammatory molecules such as eicosanoids, leukotrienes, and prostaglandins [12–14] [reviewed in [8]]. Furthermore, peroxisomes are the site of β-oxidation of dicarboxylic acids [15]. The initial oxidation step is performed in peroxisomes by acyl- CoA oxidase 1 (ACOX1 for VLCFA, or ACOX2 for pristanic and D/THCA), generating hydrogen peroxide (hence the name, peroxisome) as a byproduct that is detoxified by peroxisomal catalase [16–19] [reviewed in [20]]. D-bifunctional protein (DBP or HSD17B4) catalyzes the second (hydration) and third (dehydration) step of peroxisomal β-oxidation [21–23]. The final step is catalyzed by thiolase (either 3-oxoacyl-CoA thiolase (ACAA1) or SCPX in humans) [reviewed in [8]]. The peroxisomal fatty acid oxidation system is generally not able to degrade fatty acids to completion, so C16 or shorter fatty acids are transported as an acylcarnitine derivative to mitochondria for complete oxidation [11, 24–27].
3-methyl branched fatty acids (predominantly phytanic acid), because of the position of the methyl group, require a preliminary α-oxidation step in order to undergo β-oxidation [28, 29]. This requires the peroxisomal enzymes, phytanyl-CoA hydroxylase (PHYH), phytanyl-CoA lyase, and pristanal dehydrogenase, to generate pristanic acid [30–33] [reviewed in [8]]. Both pristanic acid and D/THCA are generated as (R) and (S) enantiomers, which require conversion to the (S) form by peroxisomal alpha-methyl-CoA racemase (AMACR) before β-oxidation [34]. Phytanic acid is exclusively dietary in origin and obtained from meats and dairy products of ruminant animals and certain fatty fishes [35, 36]. It accumulates in patients with ZSD and RCDP1, as well as in patients with single enzyme defects in PHYH. For a detailed review, see [37].
The committing steps for ether phospholipid synthesis occur in the peroxisome to generate the alkylglycerol backbone and require the peroxisomal enzymes fatty acyl-CoA reductase (FAR1), dihydroxyacetonephosphate acyltransferase (GNPAT), and alkyl-dihydroxy-acetonephosphate synthase (AGPS). Further metabolism in the ER generates the mature ether phospholipids, the majority of which are plasmalogens and contain a fatty alcohol linked by a vinyl ether bond to the sn-1 position of the glycerol backbone and are enriched in DHA and arachidonic acid (AA) at the sn-2 position [reviewed in [38, 39]]. Plasmalogens are a unique class of membrane glycerophospholipids by virtue of their vinyl ether bond, which provides specialized and critical functions to cellular membranes [40–42]. These properties include unique physical membrane attributes [43], signal transduction through released DHA and AA, as well as the remaining lysophospholipid [44], and the ability to provide oxidant protection to other membrane lipids [reviewed in [38, 39]]. Plasmalogens are enriched in heart and skeletal muscles, kidney, brain, and erythrocytes, whereas liver has very low concentration of plasmalogens [45]. Plasmalogen deficiency causes the RCDP phenotype [46–48]. In ZSD, the phenotype is more complex due to multiple enzyme deficiencies. In addition, peroxisomes contain enzymes involved in the catabolism of D-amino acids (D- amino acid oxidase) and L-lysine (pipecolic acid oxidase), polyamine oxidation, detoxification of glyoxylate (alanine-glyoxylate aminotransferase, AGXT), reactive oxygen species, and xenobiotics. [For a full catalog of peroxisomal enzymes see [8]]. Cholesterol levels tend to be low in PBD patients [49–51], although the underlying mechanism remains to be resolved [52]. Recent studies suggest that peroxisome dysfunction leads to chronic ER stress and consequent dysregulation of the sterol response pathway [53].
The catalog of peroxisomal enzymes continues to increase through the use of bioinformatic searches for proteins containing the conserved targeting signals [54–57], as well as proteomics and mass spectrometry techniques [58, 59]. Most peroxisomal enzymes are unique to peroxisomes, but some are shared with other cellular compartments, including mitochondria and cytosol. These multiple subcellular localizations may be due to the presence of multiple targeting signals within the same protein, as shown for 3-hydroxymethylglutaryl-CoA lyase [60], alpha methyl-CoA racemase [61], malonyl-CoA decarboxylase [62] and others, or relate to properties of the oligomeric protein complex as in epoxide hydrolase [63], copper-zinc superoxide dismutase [64], and alanine glyoxylate aminotransferase [65].
Shared enzymes and the movement of substrates and products through various cellular compartments position peroxisomes to be versatile organelles that dynamically adapt their number and metabolic functions in response to cellular needs. This is observed in yeast when grown on fatty acid media, by concomitant transcriptional up-regulation of peroxisomal β-oxidation enzymes and peroxisome proliferation. Rodent peroxisomes also proliferate secondary to increased β-oxidation activity and this is controlled by PPARα [66]. PPARα belongs to a transcription factor superfamily that binds to, and is activated by, a broad range of fatty acids and is a key regulator of peroxisomal and mitochondrial β-oxidation of fatty acids. Nevertheless, PPARα agonists have thus far not been associated with peroxisome proliferation in humans [67]. However, a metabolic control of peroxisome abundance in humans is expected considering the reduced number and enlargement of peroxisomes observed in fibroblast cultures from both ZSD patients and patients with single enzyme defects in β-oxidation pathways (ACOX1 and DBP) [68]. Human peroxisome numbers can also be increased through a PPARα independent mechanism that may rely on PEX11 activation [reviewed in [69]]. Significantly, the PPARγ co-activator (PGC)-1α, a regulator of cellular energy metabolism and mitochondrial biogenesis, was shown also to stimulate peroxisome biogenesis by upregulating PEX11, as well as other PEXproteins [70].
3Peroxisome biogenesis
Peroxisome biogenesis encompasses the different processes required to assemble and maintain functional peroxisomes including matrix protein import, synthesis of new organelles, and fission of existing organelles. The coordinated activity of 16 PEX proteins, or peroxins, encoded by their corresponding PEX genes, is required for this process in mammals. There is evidence that peroxisome movement and assembly utilizes the cytoskeleton [71][reviewed in [72]]. Current models of eukaryotic peroxisome biogenesis have been recently reviewed [69, 73–75] and briefly are summarized below with emphasis on mammalian systems (see Fig. 1 for diagram of matrix protein import). Peroxisomal enzymes are synthesized on cytosolic ribosomes and contain one of two peroxisomal targeting signals (PTS) required for their import into the peroxisome [reviewed in [76]]. PTS1, the most common, is a C-terminal tripeptide, -Ser-Lys-Leu or consensus variant thereof [77–79]. PTS2 is a N-terminal nonapeptide found in at least 3 mammalian enzymes: 3-oxoacyl-CoA thiolase (ACAA1), PhyH, and AGPS [54, 80–84]. PTS1 proteins are recognized by the cytosolic receptor, PEX5, which also acts as a chaperone for PTS1 proteins [85–87]. The crystal structure of PEX5 demonstrating the PTS1 binding site has been solved [88]. There are two isomers of PEX5: PEX5S and PEX5L, resulting from differential splicing of the primary transcript of the PEX5 gene [89]. The PTS2 sequence forms a structural motif that is recognized by the cytosolic receptor PEX7 [90]. Comparative homology modeling has identified the PEX7-PTS2- binding groove [54]. In mammals, PEX7 needs to interact with PEX5L for targeting to the peroxisomal membrane [89, 91]. PEX5L and PEX7 receptors form cytosolic complexes with their ligands and efficiently ferry them to the peroxisome membrane [91]. The receptor/cargo complex binds at the peroxisome membrane to the PEX13 and PEX14 docking complex [92–96]. The cargo proteins are subsequently translocated into the matrix, possibly through a transient pore, which assembles at the peroxisome membrane to enable matrix protein translocation, before it is disassembled afterwards and its components recycled for further rounds of protein import [97, 98]. Direct evidence for the existence of a transient import pore was shown by electrophysiological studies, which demonstrated that a Pex5p-Pex14p sub-complex of S. cerevisiae harbors pore forming activity [99]. Through this transient pore, which can widen to 9 nm, peroxisomes can import folded, oligomeric and cofactor bound proteins [99–101]. An alternate emerging model postulates that the peroxisomal import machinery displays a preference for monomeric proteins. In this model: (i) newly synthesized peroxisomal proteins are folded by cytosolic chaperones and released as soluble monomers; (ii) PEX5 binding to these monomers (PTS1) blocks their oligomerization; and finally, (iii) these monomeric cargoes are translocated by a single PEX5 molecule into the matrix of the organelle where oligomerization occurs [102].
After translocation, the cargo dissociates from the PTS-receptors and is released into the peroxisomal lumen [103]. The PTS receptors are released from the peroxisomal membrane for another import cycle or directed to the proteasome for their degradation [104–106]. Translocation of the ligands into the peroxisome matrix is also associated with the integral membrane complex: PEX2, PEX10, and PEX12. PEX2/10/12 are ubiquitin ligase (E3)-like proteins containing RING (really interesting new gene) domains and are involved in facilitating the mono- or polyubuiquitination based PTS receptor recycling and degradation [107]. In humans, PEX2 mono-ubiquitination of PEX5 occurs on a conserved cysteine residue in the N-terminal region, and is mediated by redundant ubiquitin-conjugating (E2)-enzymes (UbcH5a, UbcH5b and UbcH5c) [108, 109]. This mono-ubiquitination could provide a handle for PEX5 recycling back to the cytosol [110, 111]. If the monoubiquitination dependent recycling pathway is impaired, the PTS-receptors enter a polyubiquitination-dependent alternative pathway where PEX10/12 poly-ubiquitinate PEX5 on lysine residues, targeting it for degradation by the 26 S proteasome [111, 112]. In wild type cells there is very little receptor turnover by degradation [112, 113]. For a complete review of PEX5 ubiquitination and dislocation, see [114, 115].
The ‘exportomer’ complex of PEX1, PEX6, and PEX26 is involved in the recycling of the PTS1-receptor, PEX5, and presumably PEX7, back to the cytosol for additional rounds of import [116]. PEX1 and PEX6 are members of the AAA protein family (ATPases Associated with various cellular Activities) that contain one or two AAA cassettes responsible for ATP-binding and ATP hydrolysis. PEX1 and PEX6 each contain two ATPase domains (D1 and D2), and are therefore members of the double-ring ATPase family [reviewed in [117]]. These proteins typically form hexameric rings that change conformation during the ATPase cycle, providing the motion and force required for the variety of cellular functions they perform [118–120]. ATP binding is required for the interaction of PEX1 and PEX6 [121], and association with the peroxisome membrane occurs by interaction of the N-terminal region of PEX6 with PEX26 [122]. PEX5 export, but not import, is an ATP driven process, with the energy of ATP hydrolysis providing the conformational change required in the PEX1/6 complex to pull monoubiquitinated PEX5 out of the peroxisome membrane [123]. Recently, cryo-electron microscopy studies have shown that yeast Pex1/Pex6 ATPases form a unique double-ring structure in which the two proteins alternate around the ring [124–126]. These data shed light on the mechanism and function of this ATPase complex and suggest a role in peroxisomal protein import analogous to that of p97 in ER-associated protein degradation, in which the ATPase extracts poly-ubiquitinated proteins from the ER membrane [discussed in [117, 127]].
Peroxisomes can form de novo or by growth and division of pre-existing peroxisomes. PEX3, PEX16, and PEX19 are required for these processes and for correct localization of peroxisome membrane proteins (PMPs) [128–136]. In the de novo pathway, PEX16 is first incorporated into the ER, followed by PEX3 and other PMPs [137]. This leads to the formation of specialized “peroxisome-like” domains, or pre-peroxisomal vesicles, which can detach from the ER to form peroxisomes [137, 138] [reviewed in [139]]. During peroxisome growth, PEX19, a cytosolic protein, recognizes newly synthesized PMPs via their internal targeting signal motifs and binds them [140]. The complex then docks to PEX3 and PEX16, located in the peroxisomal membrane [141, 142]. In this pathway, PEX19-PEX3-PEX16 mediates import for most PMPs, including metabolite transporters and PEX proteins [143][reviewed in [144]]. Peroxisome fission requires the integral membrane protein PEX11β and shares several components of its division machinery with mitochondria. These include dynamin-like proteins (DLPs), Fis1, Mff and ganglioside-induced differentiation-associated protein 1 (GDAP1) [145–148]. PEX11β is required for membrane elongation, and binds to Fis1, which recruits DLPs [reviewed in [69]]. PEX11β is constitutively expressed in all tissues, whereas its isoform PEX11α is inducible [149, 150], and PEX11γ is constitutively expressed in liver [151]. PEX11γ is involved in peroxisome membrane protrusion [152] and may function in the de novo pathway to mediate the import of peroxisomal matrix proteins into the pre-peroxisomal vesicles that are derived from the ER [153]. The relative contribution of the ER-derived versus fission pathway to the total peroxisome population in vivo, and the detailed mechanisms of ER entry and exit of PMPs are controversially discussed. For a detailed review, see [154–156].
Peroxisome turnover, termed pexophagy, is by general mechanisms related to autophagy of cellular contents, as well as mechanisms selective for peroxisomes [reviewed in [157, 158]]. Deficiencies in various PEX proteins increase pexophagy, including PEX3 [159] and PEX14 [160]. Studies in yeast show that deficiencies in exportomer components Pex1, Pex6, or Pex15 (the yeast ortholog of PEX26) increase pexophagy more than other peroxin deficiencies [161]. The constitutive turnover of peroxisomes is estimated to be around 30% per day [162].
4Clinical disorders
Biallelic pathogenic mutations in any one of 14 PEX genes cause disorders of peroxisome assembly. These are collectively and classically known as peroxisome biogenesis disorders. No human disease has yet been associated with defects in PEX11α and PEX11γ. Defects in PEX1, PEX2, PEX3, PEX5, PEX6, PEX10, PEX11β, PEX12, PEX13, PEX14, PEX16, PEX19 and PEX26 genes cause Zellweger spectrum disorder (ZSD) including Zellweger syndrome (ZS), Neonatal Adrenoleukodystrophy (NALD), and Infantile Refsum Disease (IRD). These historically distinct phenotypes are now considered different presentations within the same clinical and biochemical spectrum with ZS being the most severe presentation, NALD intermediate and IRD milder [4, 163, 164]. The term ZSD is preferred, given the overlap in individual clinical presentations, and the atypical phenotypes currently being identified by new sequencing technologies. In general, disease severity correlates with patient age at onset of symptoms and the predicted consequence of the mutation on peroxin function, capacity for matrix protein import, residual enzyme functions, and peroxisome numbers. At the cellular level, defects causing ZSD lead to decreased numbers of enlarged peroxisomes that show reduced import of matrix enzymes [165]. These structures are termed peroxisome ‘ghosts’ [165, 166]. Defects in PEX7 cause the distinct phenotype of RCDP1 [167]. Since only PTS2 protein import is disrupted in RCDP1, peroxisome number and morphology are normal and there is deficiency of only a subset of matrix enzymes. A phenotypic spectrum of severity is also present in RCDP1 and correlates to residual plasmalogen levels [46–48]. The overall estimated birth incidence for ZSD is 1/50, 000 [168] and 1/100, 000 for RCDP1 [169]. The prevalence of both ZSD and RCDP1 varies among different populations [170–173]. These disorders are autosomal recessive and panethnic, however founder mutations account for the high frequency of the PEX1-c.[2528G>A] allele (p.[Gly843Asp], or p.G843D), the PEX1-c.[2097_2098insT] allele (p.[Ile700Tyrfs42*]) [174, 175], and the PEX7-c.[875T>A] allele (p.[Leu292*]) [172, 176, 177] in individuals of Northern European heritage. Other founder alleles include PEX10-c.[815-815delCT] in the Japanese population [171], PEX6-c.[802_815del] in French Canadians [170], and PEX12- c.[–26G>A; 102A>T] in individuals of Persian Jewish descent [178] (The PEX12-c.[–26G>A] cis mutation is provided by N. Braverman, personal communication).
4.1ZSD
Severe ZSD (Zellweger syndrome) is a classic malformation syndrome described by its characteristic features as cerebro-hepato-renal syndrome [reviewed in [144, 179, 180]]. Patients present as newborns with severe hypotonia, seizures, and characteristic craniofacial dysmorphisms with high forehead, large anterior fontanel, hypertelorism, epicanthic folds, high arched palate and micrognathia. Brain MRI shows microgyria, pachygyria and heterotopia consequent to neuronal migration defects [reviewed in [181]]. Generalized brain atrophy and subependymal germinolytic cysts [182] have also been described. The pattern of medial pachygyria and lateral (perisylvian) polymicrogyria is a distinguishing feature [183]. Eye disease includes cataracts, congenital glaucoma, retinal dystrophy and optic atrophy. Sensorineural deafness is present. The liver is enlarged and hepatic functions show elevated transaminases, cholestasis and coagulopathy. Cortical renal cysts are observed on ultrasound, but are not usually clinically significant. Chondrodysplasia punctata especially in the knees and hips, can be seen on skeletal x-rays. Cardiovascular malformations and pulmonary hypoplasia have also been reported [184]. Fetuses with severe ZSD, as early as 13 and 21 weeks’ gestation, show brain dysplasia, renal cysts, and calcific stippling of cartilage [185]. Fetal ultrasound examinations have detected verntriculomegaly in the 3rd trimester and follow-up MRI at 35 weeks gestation showed gyral malformations, renal cysts and hepatosplenomegaly [186]. Thus, the most severe degree of peroxisome dysfunction affects prenatal organ and tissue development. In contrast, hepatic fibrosis is a late event that is not present in the fetus, but can be severe postnatally [184]. These infants generally do not survive beyond the first year of life [180].
The intermediate and milder (previously termed NALD and IRD) group of patients present after the newborn period with more varied symptomatology (see Table 2 for a list of ZSD features by age of onset). Hypotonia, failure to thrive, developmental delays, sensory deficits, amelogenesis imperfecta and history of neonatal cholestasis are usually present. The characteristic Zellweger facies is attenuated or absent. Subtle neuronal migration defects have been described in some intermediate patients, but are absent in the milder forms of ZSD [187, 188] [reviewed in [181]]. Overall, this group isclinically distinguished from severe ZSD by the absence of congenital malformations, and is instead a disorder marked by progressive peroxisome dysfunction over time. Most individuals have progressive sensorineural hearing loss and visual loss due to retinal degeneration, leading to deafness and blindness. Other complications include seizures, adrenal insufficiency, leukodystrophy, and osteopenia, the latter leading to pathological fractures [189]. Nephrolithiasis from hyperoxaluria due to deficiency of AGXT has been reported [190]. Liver dysfunction can quiesce, or less frequently, worsen [191]. The prediction of which children will develop these complications and when, as well as the timeline of their progression remains unknown. However, children with the intermediate phenotype generally develop more complications at earlier times, almost all develop adrenal insufficiency, and some will develop leukodystrophy. Lifespan is usually shortened, and many of these individuals do not survive past late childhood. Most individuals with milder ZSD have better cognitive functions, fewer and less severe symptoms of disease and can survive through adulthood [191].
Leukodystrophy was classically described in intermediate ZSD patients in late infancy and early childhood. It manifests with active demyelination in the cerebrum, midbrain and cerebellum and consequent psychomotor regression [192–194]. The boundaries of the demyelinated areas are usually sharply demarcated and there is reactive gliosis; many cases have been associated with perivascular accumulation of lymphocytes as in X-linked adrenoleukodystrophy [187]. In some cases, mild initial symptoms are later followed by severe CNS demyelination and death of the patient [187, 192, 194]. However, leukodystrophy can occur at any age, remain stable, or progress [191, 195]. Limited pathological studies in brain from intermediate patients have shown reduced neuron number and cerebellar atrophy without active demyelination. There can be dysplastic olives and irregular, heterotopic, clumps of Purkinje cells [187, 196]. In the milder forms of ZSD, where patients can survive into adulthood, brain MRI has also revealed white matter abnormalities, although it can be normal. A study including neuroimaging findings in 19 such patients demonstrated high proportion of white matter abnormalities restricted to the cerebellar hilus of the dentate nucleus and/or the peridentate region. Interestingly, at the time of diagnosis, 17 of these patients had a blood metabolite profile typical of a peroxisomal disorder; but at later time points the concentration of many originally accumulating metabolites had declined and, in some patients, even a complete normalization was observed [191]. Histopathological analysis of the brain from an milder patient showed hypoplasia of the cerebellar granular layer and ectopic location of purkinje cells in the molecular layer without active demyelination [197].
Retinal degeneration is a prominent feature of all ZSD patients and can manifest as early as the first year of life in the intermediate and milder spectrum [198]. Cataracts can also present [180, 199]. Clinical case descriptions have included Leopard spot retinal pigmentation [200], congenital amaurosis [201, 202], Usher syndrome [203, 204], as sensorineural hearing loss is typically also present, and retinitis pigmentosa with a predominant macular dystrophic component [201, 202, 205, 206]. ERG recordings typically show severely reduced or extinguished cone and rod responses [198, 206, 207]. The course and progression of the retinal degeneration is unknown, but in general the progression is slower in the milder phenotypes. Histological reports showed degeneration of photoreceptor outer segments and reduced inner segments, and extensive loss of nerve fiber and ganglion cell layers [208]. Areas of photoreceptor degeneration were associated with pigmentary changes. The retinal pigment epithelium can show patchy hypertrophy, nodular hyperplasia, and atrophy. Pigmented cells can be found in the subretinal space and in other layers in the retina. Macrophages may contain pigment and degenerated photoreceptors. Lamellar lipid inclusions were observed in most layers of the retina and within the optic nerve by electron microscopy [208]. These inclusions have also been observed in brain, adrenal glands, and liver [197, 200]. Recently, peroxisomes were shown to cluster at the base of photoreceptor outer segments, suggesting that peroxisomes are intimately involved in maintaining outer segments [203]. Cochlear histology has shown degeneration of the sensory epithelium, with preservation of ganglion cells and nerve fibers [197].
4.2Atypical ZSD presentations
Recently, atypical and milder phenotypes have been reported that may relate to specific mutations in specific PEX genes. These children and adults with normal or near normal intellect, slow disease course and prolonged survival, illustrate that the phenotypic spectrum of these disorders is much broader than initially thought [153, 178, 188, 199, 204, 205, 209–219]. Table 3 summarizes the pertinent findings reported in this novel group of patients. These patients are arbitrarily classified according to major presenting symptomology: those with prominent sensory deficits (visual and hearing loss), and those with cerebellar dysfunction, peripheral neuropathy or relapsing encephalopathy. These individuals inspire fundamental reflections on PBDs and highlight some of the molecular, biochemical and cellular mechanisms by which these variant phenotypes arise.
The underlying molecular mechanisms of these atypical presentations often involve hypomorphic PEX alleles encoding peroxins with residual protein function. This can include bi-allelic hypomorphic alleles, as well as a mono-allelic, hypomorphic mutation in trans with a severe, usually loss of function mutation [220]. Mutations in specific domains (ring finger) of PEX2, PEX10 and PEX12 can lead to specific phenotypes of cerebellar dysfunction and peripheral neuropathy [discussed in [211]]. In addition, an adult with milder disease was identified with two null mutations in PEX11β. It was suggested that functional redundancy of PEX11α and PEX11γ supported the absence of PEX11β [153]. In most of these cases, routine peroxisome metabolite testing in blood (VLCFA, plasmalogen and phytanic acid levels) showed only subtle dysfunction or none at all [153, 188, 209, 210, 220, 221] (Table 3). In this context, if a PBD is strongly suspected, additional peroxisome functions should be investigated. Pipecolic acid, D/THCA and pristanic acid levels, as well as fibroblast enzymatic testing, morphological analysis of cellular peroxisomes and catalase distribution has targeted PBD in difficult cases [222, 223].
Morphological analyses of peroxisomes in fibroblast cultures in milder cases have shown more pero-xisomes with residual function, mosaicism, and temperature sensitivity, phenomena that likely reflect a partially functional peroxin [220]. Mosaicism refers to the observation of a heterogeneous cell population (by both immunohistochemistry and electron microscopy techniques) in fibroblast cultures and in hepatocytes from liver cell biopsies, in which some cells contain functional peroxisomes that import matrix proteins, while adjacent cells contain peroxisomes (ghosts) that show minimal or no import of matrix proteins [224–226]. Temperature sensitivity refers to improvement in peroxisome formation and biochemical function in ZSD fibroblasts cultured at 30°C rather than 37°C [227–229]. It has been suggested that temperature sensitivity is a consequence of an imbalance of folding and unfolding kinetics of mutant proteins, resulting in an increase in the correctly folded protein as temperature decreases [230]. The etiology for peroxisome mosaicism is not known, but could reflect microenvironment or cell cycle differences that enable recovery of a partially functional peroxin, or a partially functional import system. Taken together, these data imply residual peroxisome functions in body tissues that are dependent on a threshold of temperature, microenvironment and other unknown factors which can improve function. In fibroblast cultures from some patients with atypical phenotypes and normal peroxisomal morphology, increasing the culture temperature to 40°C unmasks the underlying peroxisome dysfunction [178]. Finally, peroxisome morphology can provide a clue to the underlying gene defect. For example, fibroblast cultures from patients with defects in DLP1 or Mff exhibit elongated, constricted peroxisomes that are unable to divide (i.e. “pearls on a string” phenotype) [231, 232]. Fibroblasts from a PEX11β patient have enlarged, elongated peroxisomes, indicating a defect in pero-xisome constriction and/or division [153]. Cells from patients with milder variants in PEX16 show a reduced number of enlarged, but import competent peroxisomes [199]. Thus far, peroxisomes in patient cell lines with PEX7 defects (RCDP1) have normal morphology and do not show a temperature response.
4.3Other disorders that affect peroxisome biogenesis
A few patients were reported with mutations in the shared mitochondrial and peroxisome fission proteins, DLP1 [231], Mff [232], and GDAP1 [148]. These individuals displayed clinical evidence of combined mitochondrial and peroxisome dysfunction. Mitochondrial and peroxisomal morphology indicated fission dysfunction. The patient with DLP1 deficiency presented with affected brain development including an abnormal gyral pattern in both frontal lobes associated with dysmyelination, microcephaly, optic atrophy. She died a few weeks after birth. The patients with Mff deficiency showed delayed psychomotor development, microcephaly, pale optic discs, and mild hypertonia. Brain MRI revealed an abnormality in the globus pallidus. The eldest of the 2 patients (brothers) was 4.5 years old at the time of publication. GDAP1 mutations affect peroxisome and mitochondrial dynamics and cause the peripheral neuropathy Charcot-Marie-Tooth disease (CMT) [148, 233]. Interestingly, peroxisome metabolic functions (as measured by biomarkers indicative of classic peroxisomal disorders) were not altered in the tested Mff patient. The DLP1 patient showed only a slight elevation of VLCFA. This minimal or nonexistent biochemical evidence of peroxisomal dysfunction highlights the importance of using alternate methods in patient diagnosis (discussed previously). It is likely that defects in other proteins involved in peroxisome assembly and maintenance that can result in a clinical disease remain to be identified.
The increased use of next-generation sequencing technologies has led to the identification of many of these atypical patients [see Table 3] [153, 178, 188, 199, 205, 209, 211, 212, 216–219] and will continue to do so. However, analysis of peroxisome biochemical and cellular functions remains necessary to prove a peroxisome etiology, especially in cases where there are novel PEX gene variations, novel phenotypes, and defects identified in other pathways involved in peroxisome assembly and maintenance. All this will lead an improved understanding of peroxisome biology in various tissues over time and thus enable more precise genotype and phenotype correlations.
4.4RCDP
The majority of patients with RCDP have defects in PEX7 (RCDP1) [48, 167, 177]. The 3 well-characterized mammalian PTS2 enzymes transported by PEX7 are peroxisomal thiolase (ACAA1), phytanoyl CoA hydroxylase (PHYH), and AGPS [81–84]. ACAA1 is a redundant enzyme, and its functions are assumed by SCPX, which has thiolase activity and is transported by PEX5 [235]. Thus β-oxidation defects have not been detected and VLCFA levels are normal in RCDP1 plasma and fibroblasts. Defects in PHYH result in accumulation of phytanic acid over time, causing the distinct phenotype of Adult Refsum Disease (ARD) [83]. In RCDP1 patients, who typically do not survive to adulthood, the phenotypic features of ARD are absent. In contrast, certain defects in PEX7 result in normal plasmalogen synthesis but retain reduced phytanic acid α-oxidation, resulting in an ARD phenotype [48, 234].
RCDP can also result from defects in the peroxisomal enzymes required for plasmalogen synthesis: GNPAT (RCDP2), and AGPS (RCDP3) [236–243]. RCDP1, 2 and 3 share identical phenotypes and phenotype severity depends on the degree of residual plasmalogen levels [46–48, 236]. Plasmalogen deficiency is thus considered the primary pathophysiological etiology. Newly discovered causes of RCDP phenotypes are PEX5L mutations in the PEX7 binding domain region (RCDP5), which causes a loss of PEX7 transport to the peroxisome, but retains all other PEX5 functions [244]. As expected, the clinical and biochemical phenotype in the two patients reported was identical to classical RCDP1. In addition, mutations in the FAR1 gene (RCDP4), which supplies the fatty alcohol substrate to AGPS, also result in plasmalogen deficiency but the RCDP phenotype was attenuated, perhaps due to overlapping functions of the FAR2 enzyme [245].
RCDP is clinically characterized by proximal shortening of the humerus and to a lesser degree the femur (rhizomelia), punctate calcifications in epiphyseal cartilage (chondrodysplasia punctata), with progressive metaphyseal abnormalities, coronal clefts of the vertebral bodies, and congenital cataracts [246–248]. Typically, there is severe postnatal growth and intellectual deficiency [249]. Up to 52% of patients have congenital heart malformations [250, 251]. Cleft palate, renal malformations, and ichthyosis have also been reported [249]. Life span is broad although most affected children do not survive the first decade of life. A proportion die in the neonatal period and this might be related to primary cardiac abnormalities or pulmonary hypoplasia [252]. Neuroimaging studies show various abnormalities including ventriculomegaly without increase in intracranial pressure, multilevel cervical canal stenosis [253, 254], hypomyelination [46, 255], other supratentorial (involving the parieto-occipital regions) white matter signal anomalies [46, 256, 257], and progressive cerebellar atrophy. Head growth deceleration is seen in all patients with severe RCDP, possibly reflecting myelin or neuronal loss [46]. The striking neuronal migration defects seen in ZSD are not typically observed in RCDP. Pathological studies of RCDP brain show a general decrease in neuron number and white matter, dyplastic olives and progressive cerebellar degeneration [247, 258–260]. Although most reported RCDP patients have a severe, classical phenotype, milder phenotypes are now being recognized in which affected individuals have congenital cataracts and epiphyseal dysplasia, but lack rhizomelia and have less severe intellectual and growth deficiency [48]. In these patients, MRI can be normal or show slowly progressive cerebellar atrophy. The severity of the cerebellar atrophy, as with all RCDP features, correlates directly to the degree of plasmalogen deficiency and not to the phytanic acid elevation[46, 48].
5Diagnosis
Clinical suspicion of PBD requires confirmation by determining peroxisome functions (Table 4). The fundamental problem in ZSD is the inability to generate and maintain functional peroxisomes [4, 5].Consequently, the typical biochemical signature of ZSD is concomitant dysfunction of multiple peroxisomal pathways. This is detected in blood, urine and cells by accumulation of VLCFA, phytanic, pristanic and pipecolic acids, D/THCA and deficiency of plasmlaogens, docosahexanoic and primary bile acids (cholic and chenodeoxycholic acids) [diagnosis reviewed in [261]]. In RCDP1, the PEX7 defect impairs only PTS2 protein import, causing reduced plasmalogens and increased phytanic acid levels. Around 10–15% of patients with ZSD phenotypes have single enzyme defects in fatty acid β-oxidation (ACOX1, DBP, SCPX, AMACR), and a similar number of patients with RCDP phenotypes have single enzyme defects in plasmalogen synthesis (GNPAT, AGPS, FAR1). Thus, it is critical to evaluate several peroxisome functions to tease out true PBD from phenocopies due to single enzyme defects. The fact that patients with single enzyme defects in the β-oxidation pathway have phenotypes identical to ZSD [213, 218, 262, 263] implies that β-oxidation defects contribute in large part to the pathology of ZSD. Similarly, defects in plasmalogen synthesis are directly responsible for the RCDP phenotype [46–48, 236]. Metabolite testing is usually followed by enzymatic confirmation of PBD in fibroblast cultures and/or molecular confirmation of the PEX gene defect.
PEX gene sequencing was previously limited due to the challenge of multiple genes involved[174, 264]. However, with the advent of next generation sequencing, molecular diagnosis has become cost efficient, comprehensive, and has enabled additional genotype-phenotype correlations in atypical patients. Over the past decade, studies on more than 800 patients show that defects in PEX1 represent around 60% of all ZSD alleles [174, 264, 265]. In these reports, 12–30% of ZSD alleles are PEX1-p.G843D, and 5–20% are PEX1-p.[Ile700Tyrfs42*]. About 50% of all RCDP alleles are PEX7-p.[Leu292*]. The high frequency of these alleles is due to a founder effect in persons of Northern European extraction [175, 176]. PEX1-p.G843D is a missense protein with residual functions; the presence of at least one allele predicts an intermediate phenotype, and homozygosity generally results in milder phenotypes [227, 266–268]. The large variations in phenotype severity observed amongst PEX1-p.G843D homozygotes (unrelated, as well as intra-familial) suggest the influence of modifier genes [191, 205].
PEX1-p.[Ile700Tyrfs42*] and PEX7-p.[Leu292*], are null alleles and predict a severe phenotype when homozygous [48, 177, 269]. Overall, bi-allelic mutations in PEX1, 6, 26, 10, 12, and 2 contribute to the majority of ZSD alleles [174]. When previously unknown or uncharacterized variants are identified, functional biochemical studies remain a necessary tool to determine causality and pathogenicity [reviewed in [144, 223]].
In addition to diagnoses of affected individuals, early diagnoses of relative asymptomatic infants will be possible with the advent of newborn screening. The combination of liquid chromatography and tandem mass spectrometry (LC–MS/MS) to detect elevated levels of VLCFAs in newborn blood spots was validated as a diagnostic approach for X-linked adrenoleukodystrophy (X-ALD) [270, 271], a single enzyme/protein defect of peroxisome function in which there is only accumulation of VLCFA [reviewed in [272]]. X-ALD screening was recently included in the US Recommended Uniform Screening Panel [273]. The implications of X-ALD newborn screening include the ability to perform clinical surveillance for early detection and treatment of leukodystrophy in affected males, and counseling for carrier females [274]. Newborn screening for X-ALD should also detect the majority of PBD-ZSD cases that feature elevated blood VLCFA levels, thereby permitting early diagnosis and thus more timely institution of supportive management. It will also enable family counseling, determination of accurate incidence figures and continued expansion of the clinical phenotype as variant patients are identified.
6Management
There are no curative therapies for PBD. Management is multidisciplinary, symptomatic, and based on surveillance of the multiple systems involved. This includes regular developmental, neurological, auditory, visual, nutritional, and orthopedic assessment, and monitoring of liver and adrenal functions [261, 275]. Vitamin K and other fat soluble vitamin supplements are usually prescribed. Care is often palliative for those with severe disease. Genetic counselling and family support organizations provide crucial assistance to parents in their decision making process. The geneticist, neurologist and nutritionist should be involved early in the management of patients with PBD.
Dietary interventions have included restriction of metabolites that accumulate and replacement of those that are deficient. Reports of these approaches in patients are mostly anecdotal and have not been systematically studied. Furthermore, the extensive inter- and intrafamilial variations in the intermediate and milder ZSD patients requires better understanding of the natural history of these disorders and the establishment of reliable clinical endpoints for future interventional trials.
In a small case series in ZSD, recovery of erythrocyte plasmalogens was achieved by administration of the oral plasmalogen precursor batyl alcohol [276], but this did not improve clinical outcome. Oral bile acid supplementation (chenodeoxycholic and ursodeoxycholic acids) in two infants with severe ZSD reduced C27 bile acid intermediates (D/THCA) and improved hepatobiliary function, but did not change the clinical course [278]. In a third infant with severe ZSD, cholic acid supplementation also improved histology with a decrease in bile duct proliferation and inflammation [277]. An infant with AMACR deficiency presenting with cholestasis has done well on long term cholic acid supplementation with complete suppression of C27 bile acids and improvement in liver histology [279]. However, the few other AMACR patients reported were adults with neurological symptoms attributed to pristanic acid accumulation and improved on a phytanic acid restricted diet [213, 280]. Thus the clinical significance of early intervention with bile acid therapy remains unknown.
Patients have also received DHA supplementation, with controversial clinical benefit. In a small case series, improvements in electroretinograms (ERG) and growth were shown [281]. In a double-blind randomized placebo controlled trial that included 48 patients over one year of study, there were no overall improvements in ERG and growth in the treated group compared to the placebo group [207]. Due to the benefit of dietary phytanic acid restriction in adult Refsum disease [282], and the fact that it is not a difficult diet regimen, this is often prescribed for surviving PBD patients but formal studies have not been done to determine if this improves the disease course.
In two siblings with an intermediate-milder ZSD phenotype, the older child received an hepatocyte transplantation [283] and the younger received a liver transplant [284]. Peroxisome biochemical functions improved, but clinical outcome of the younger sibling was relatively better. Another patient received a liver transplant, and biochemical functions improved but it is not yet clear that the clinical outcome has changed [285].
7Research tools: mouse models
In order to gain further insight into the pathogenesis of ZSD, mouse models with complete deficiency of Pex2, Pex5, Pex13, and Pex11β have been engineered [286]. These mice recapitulate many aspects of severe ZSD, including the metabolic consequences of peroxisome elimination and abnormalities in brain formation, hypotonia, and early demise. Detailed investigations of these models revealed neuronal migration defects commencing early in the cytogenetic epoch and demonstrated by birth dating experiments using 5-BrdU. Other abnormalities include delayed neuron differentiation and extensive neuronal apoptosis (Pex5, Pex11β). In the cerebellum, there was delayed neuronal layering, increased apoptosis in granular neurons and abnormal morphology of Purkinje cells (Pex2) [287]. In Pex11β null mice, there were reduced numbers of importing peroxisomes and only mild deficiencies of β-oxidation and plasmalogen biosynthesis, challenging the idea that PBD clinical features are directly related to the severity of the metabolic abnormalities [288, 289]. Overall, the null models survive gestation, but die soon thereafter, indicating that severe peroxisome deficiency is compatible with embryonic, but not postnatal life in both mice and humans. To gain further information, conditional Pex5 null mice were engineered to evaluate peroxisome dysfunction in key organs. Targeted elimination of peroxisomes in neural precursor cells results in moderate cortical migration defects that correct postnatally and might reflect migration delay [290]. Peroxisome elimination in hepatocytes results in postnatal arrest of neuron migration, particularly in the cerebellum, which still develops in the postnatal period [290]. Thus, peroxisome metabolism in the liver influences the development of the murine brain. It was suggested that elevated C27 bile acids, which are more hydrophobic than the terminal conjugated forms, may enter the brain and impair cerebellar development [290]. Survival of the mice with selective inactivation of Pex5 in neural cells, although neurodevelopmental defects were mild, resulted in a progressive motor and cognitive impairment with death before 6 months of age [291]. Myelinated axons and glial cells were severely affected in the brain and spinal cord, showing decreased compaction of myelin, loss of myelin and axonal damage. Mice with Pex5 inactivation in oligodendrocytes also developed similar axonal damage and demyelination [292]. Thus, peroxisomes may not be required for myelination, but for the preservation of axonal integrity and maintainence of myelin, and this role may be specific to oligodendrocyte peroxisomes [292].
Considering the interrelationship in mitochondrial and peroxisome functions, and the mitochondrial abnormalities noted in tissues from severe ZSD patients [4], mitochondrial abnormalities were investigated in the liver selective Pex5 null mouse model [293]. In this model, morphologically abnormal mitochondria with decreased respiratory chain complex activity were observed in liver. However, oxidative damage was not observed, possibly due to a compensatory increase in glycolysis and mitochondrial proliferation [293]. The decreased gluconeogenesis and glycogen synthesis resulted in the requirement for additional carbohydrates, increased food intake and lower body weights [294]. AMACR deficient mice show a metabolic adaptation that allows them to enhance excretion of C27 bile acids[295].
A PEX1-p.G843D equivalent knock-in mouse model (Pex1-p.G844D) was recently engineered to mimic the common human mutation, and to generate a model for the intermediate and milder phenoytpes that feature progressive peroxisome dysfunction over time. Pex1-p.G844D homozygous mice exhibit many of the typical ZSD manifestations, including growth retardation, progressive retinopathy, bile acid defects resulting in fatty liver with cholestasis, elevated VLCFA, and decreased plasmalogens [296]. Further characterization of the course and mechanisms of disease pathophysiology are underway.
Mice engineered to study RCDP include Pex7 and Gnpat null mice, and Pex7 and Agps hypomorphic models [297–300]. The similarity of phenotypes amongst these models supports plasmalogen deficiency as the primary etiology for RCDP. Thus, they have been used to explore the normal functions of plasmalogens, which are largely unknown and likely to include tissue specific functions [299, 301]. Studies of these mice show requirements for plasmalogens in cerebellar, lens, and skeletal development, as well as spermatogenesis.
The PBD mouse models have also been used to test potential therapeutic interventions. In the Pex5 null models, supplementation with DHA, beginning in utero, normalizes DHA levels in brain but does not improve the neuronal migration disorder [302]. Bile acid supplementation in Pex2 null mice improves cerebellar abnormalities, implicating the accumulation of C27 bile acid intermediates in the cerebellar toxicity observed postnatally in these mice [303]. Supplementation with alkylglycerol, a plasmalogen precursor, can recover plasmalogen levels in PBD patient fibroblast lines [276]. Early alkylglycerol supplementation in the Pex7 null model improved growth and minimized cataract formation [45]. Although plasmalogen levels recovered and tissue pathology improved in somatic tissues, this was not the case in brain, where only around 1% of control plasmalogens were present after 2 months of treatment, and around 2% at 4 months of treatment.
8Pharmacological therapies under development
8.1Plasmalogen precursor supplementation
As discussed, oral alkylglycerol supplementation was shown to have some clinical efficacy in the Pex7 null murine model [45]. Additional plasmalogen precursor molecules are now being designed with properties that may improve clinical benefit, especially to recover plasmalogen levels in brain [304].
8.2Induction of peroxisome numbers
Proliferation of peroxisomes in ZSD fibroblasts has been achieved using 4-phenylbutyrate and related compounds that act in a PPARα independent fashion through PEX11 stimulation [305, 306]. In cell lines from patients with intermediate and milder phenotypes, peroxisome proliferation results in improvement in peroxisome enzyme functions [305]. Presumably, an increased number of peroxisomes with partial function can improve overall cell functions. High throughput cell-based phenotype assays to identify additional compounds that can induce human peroxisome proliferation have been reported [67].
8.3Nonsense suppression
The nonsense suppressor aminoglycoside drug, G418 (geneticin), was shown to promote peroxisome recovery in fibroblast lines from patients with PEX2 and PEX12 mutations that produce stable nonsense transcripts [307]. Thus, the development of safe and effective nonsense suppressor therapies might benefit a significant number of PBD patients.
8.4Pharmacological enhancement of activity of a defective PEX protein
Studies on PEX1-p.G843D fibroblasts suggest that this is a misfolded protein amendable to improved folding and function when cells are cultured at 30°C or treated with chaperones drugs [268, 308]. The chemical chaperone arginine and betaine improved peroxisome biogenesis and functions in patient cell lines with missense mutations in PEX1, PEX6, and PEX12. Chemical chaperones are small-molecules that non-selectively stabilize mutant proteins by altering the microenvironment around the protein, while pharmacologic chaperones are small molecules designed to selectively bind target proteins. The latter include enzyme substrates or inhibitors that bind to and facilitate folding of non-native protein intermediates to their native state. To determine if pharmacologic chaperones could be identified for PEX1-p.G843D, a high throughput cell-based phenotype assay was designed using a patient fibroblast cell line with this allele and expressing a GFP-tagged PTS1 reporter that was cytosolic at baseline [308]. After drug treatment the cells were evaluated for recovery of GFP-PTS1 peroxisome importation. Hit compounds, a flavonoid and a protein kinase C inhibitor, were previously shown to bind the ATP binding site of ABC transporter proteins, suggesting they might act as pharmacological chaperones by binding to the ATP binding sites in the PEX1 protein [310, 311]. The advantages of using a cell-based assay include demonstrating that the molecules are able to cross at least one cell membrane and are functional in the cellular milieu. By measuring the downstream effect of peroxisome protein import recovery, these assays should enable the identification of any drug recovery mechanism. Hit compounds identified can be further developed into lead compounds if efficacious in animal models, and this task is currently being undertaken.
9Conclusion
Enormous progress has been made in understanding mammalian peroxisome biology, biochemistry, and the molecular defects that underlie PBD. The PBD mouse models have already shown enormous utility in determining the disease pathophysiology, and are poised to contribute additional information in the future. Finally, pharmacological interventions for PBD that improve clinical outcomes may be possible. Additional therapeutic interventions can now be explored. The natural history of PBDs needs to be better defined and accurate clinical endpoints established in order to move forward in future systematic clinical trials.
Acknowledgments
We acknowledge the support of the Fonds de recherche du Québec – Santé (FRQS 32105) to CA, and the Canadian Institutes of Health Research (CIHR 126108) to NB. We also acknowledge the family organization: the Global Foundation for Peroxisomal Disorders, for their constant collaboration with us to focus research endeavours for best patient care.
References
[1] | Bowers W.E. , Christian de Duve and the discovery of lysosomes and peroxisomes, Trends Cell Biol 8: (8) ((1998) ), 330–333. |
[2] | Zellweger H. , Maertens P. , Superneau D. and Wertelecki W. , History of the cerebrohepatorenal syndrome of Zellweger and other peroxisomal disorders, South Med J 81: (3) ((1988) ), 357–364. |
[3] | Opitz J.M. , ZuRhein G.M. , Vitale L. , Shahidi N.T. , Howe J.J. , Chou S.M. and Shanklin D.R. et al., The Zellweger syndrome (cerebro-hepato-renal syndrome), Birth Defects Orig Art Ser 2: (2) ((1969) ), 144–160. |
[4] | Goldfischer S. , Moore C.L. , Johnson A.B. , Spiro A.J. , Valsamis M.P. and Wisniewski H.K. et al., Peroxisomal and mitochondrial defects in the cerebro-hepato-renal syndrome, Science 182: (4107) ((1973) ), 62–64. |
[5] | Heymans H.S. , Schutgens R.B. , Tan R. , van den Bosch H. and Borst P. , Severe plasmalogen deficiency in tissues of infants without peroxisomes (Zellweger syndrome), Nature 306: (5938) ((1983) ), 69–70. |
[6] | Heymans H.S. , Oorthuys J.W. , Nelck G. , Wanders R.J. and Schutgens R.B. , Rhizomelic chondrodysplasia punctata: Another peroxisomal disorder, N Engl J Med 313: (3) ((1985) ), 187–188. |
[7] | Moser A.E. , Singh I. , Brown F.R. , Solish G.I. , Kelley R.I. and Benke P.J. et al., The cerebrohepatorenal (Zellweger) syndrome, Increased levels and impaired degradation of very-long-chain fatty acids and their use in prenatal diagnosis, N Engl J Med 310: (18) ((1984) ), 1141–1146. |
[8] | Wanders R.J. and Waterham H.R. , Biochemistry of mammalian peroxisomes revisited, Annu Rev Biochem 75: ((2006) ), 295–332. |
[9] | Ferdinandusse S. , Denis S. , Dacremont G. and Wanders R.J. , Studies on the metabolic fate of n-3 polyunsaturated fatty acids, J Lipid Res 44: (10) ((2003) ), 1992–1997. |
[10] | Moore S.A. , Hurt E. , Yoder E. , Sprecher H. and Spector A.A. , Docosahexaenoic acid synthesis in human skin fibroblasts involves peroxisomal retroconversion of tetracosahexaenoic acid, J Lipid Res 36: (11) ((1995) ), 2433–2443. |
[11] | Wanders R.J. , van Roermund C.W. , van Wijland M.J. , Heikoop J. , Schutgens R.B. and Schram A.W. , et al., Peroxisomal very long-chain fatty acid beta-oxidation in human skin fibroblasts: Activity in Zellweger syndrome and other peroxisomal disorders, Clin Chim Acta 166: (2-3) ((1987) ), 255–263. |
[12] | Pahan K. , Smith B.T. and Singh I. , Epoxide hydrolase in human and rat peroxisomes: Implication for disorders of peroxisomal biogenesis, J Lipid Res 37(1): ((1996) ), 159–167. |
[13] | Arand M. , Knehr M. , Thomas H. , Zeller H.D. and Oesch F. , An impaired peroxisomal targeting sequence leading to an unusual bicompartmental distribution of cytosolic epoxide hydrolase, FEBS Lett 294: (1-2) ((1991) ), 19–22. |
[14] | Waechter F. , Bentley P. , Bieri F. , Stäubli W. , Völkl A. and Fahimi H.D. , Epoxide hydrolase activity in isolated peroxisomes of mouse liver, FEBS Lett 158: (2) ((1983) ), 225–228. |
[15] | Nguyen S.D. , Baes M. and Van Veldhoven P.P. , Degradation of very long chain dicarboxylic polyunsaturated fatty acids in mouse hepatocytes, a peroxisomal process, Biochim Biophys Acta 1781: (8) ((2008) ), 400–405. |
[16] | Singh H. and Poulos A. , Distinct long chain and very long chain fatty acyl CoA synthetases in rat liver peroxisomes and microsomes, Arch Biochem Biophys 266: (2) ((1988) ), 486–495. |
[17] | Street J.M. , Johnson D.W. , Singh H. and Poulos A. , Metabolism of saturated and polyunsaturated fatty acids by normal and Zellweger syndrome skin fibroblasts, Biochem J 260: (3) ((1989) ), 647–655. |
[18] | Uchiyama A. , Aoyama T. , Kamijo K. , Uchida Y. , Kondo N. and Orii T. et al., Molecular cloning of cDNA encoding rat very long-chain acyl-CoA synthetase, J Biol Chem 271: (48) ((1996) ), 30360–30365. |
[19] | Schepers L. , Van Veldhoven P.P. , Casteels M. , Eyssen H.J. and Mannaerts G.P. , Presence of three acyl-CoA oxidases in rat liver peroxisomes, An inducible fatty acyl-CoA oxidase, a noninducible fatty acyl-CoA oxidase, and a noninducible trihydroxycoprostanoyl-CoA oxidase, J Biol Chem 265: (9) ((1990) ), 5242–5246. |
[20] | Ferdinandusse S. , Denis S. , Faust P.L. and Wanders R.J. , Bile acids: The role of peroxisomes, J Lipid Res 50: (11) ((2009) ), 2139–2147. |
[21] | Baes M. , Huyghe S. , Carmeliet P. , Declercq P.E. , Collen D. and Mannaerts G.P. et al., Inactivation of the peroxisomal multifunctional protein-2 in mice impedes the degradation of not only 2-methyl-branched fatty acids and bile acid intermediates but also of very long chain fatty acids, J Biol Chem 275: (21) ((2000) ), 16329–16336. |
[22] | Ferdinandusse S. , Denis S. , Van Roermund C.W. , Wanders R.J. and Dacremont G. , Identification of the peroxisomal beta-oxidation enzymes involved in the degradation of long-chain dicarboxylic acids, J Lipid Res 45: (6) ((2004) ), 1104–1111. |
[23] | Ferdinandusse S. , Meissner T. , Wanders R.J. and Mayatepek E. , Identification of the peroxisomal beta-oxidation enzymes involved in the degradation of leukotrienes, Biochem Biophys Res Commun 293: (1) ((2002) ), 269–273. |
[24] | Wanders R.J. , van Roermund C.W. , van Wijland M.J. , Schutgens R.B. , Heikoop J. and van den Bosch H. , et al., Peroxisomal fatty acid beta-oxidation in relation to the accumulation of very long chain fatty acids in cultured skin fibroblasts from patients with Zellweger syndrome and other peroxisomal disorders, J Clin Invest 80: (6) ((1987) ), 1778–1783. |
[25] | Wanders R.J. , van Roermund C.W. , van Wijland M.J. , Schutgens R.B. , van den Bosch H. and Schram A.W. , et al., Direct demonstration that the deficient oxidation of very long chain fatty acids in X-linked adrenoleukodystrophy is due to an impaired ability of peroxisomes to activate very long chain fatty acids, Biochem Biophys Res Commun 153: (2) ((1988) ), 618–624. |
[26] | Su H.M. , Moser A.B. , Moser H.W. and Watkins P.A. , Peroxisomal straight-chain Acyl-CoA oxidase and D-bifunctional protein are essential for the retroconversion step in docosahexaenoic acid synthesis, J Biol Chem 276: (41) ((2001) ), 38115–38120. |
[27] | Jakobs B.S. and Wanders R.J. , Conclusive evidence that very-long-chain fatty acids are oxidized exclusively in peroxisomes in human skin fibroblasts, Biochem Biophys Res Commun 178: (3) ((1991) ), 842–847. |
[28] | Mize C.E. , Steinberg D. , Avigan J. and Fales H.M. , A pathway for oxidative degradation of phytanic acid in mammals, Biochem Biophys Res Commun 25: (3) ((1966) ), 359–365. |
[29] | Eldjarn L. , Heredopathia atactica polyneuritiformis (Refsum’s disease)–a defect in the omega-oxidation mechanism of fatty acids, Scand J Clin Lab Invest 17: ((1965) ), 178–181. |
[30] | van den Brink D.M. , van Miert J.N. , Dacremont G. , Rontani J.F. and Wanders R.J. , Characterization of the final step in the conversion of phytol into phytanic acid, J Biol Chem 280: (29) ((2005) ), 26838–26844. |
[31] | Jansen G.A. , van den Brink D.M. , Ofman R. , Draghici O. , Dacremont G. and Wanders R.J. , Identification of pristanal dehydrogenase activity in peroxisomes: Conclusive evidence that the complete phytanic acid alpha-oxidation pathway is localized in peroxisomes, Biochem Biophys Res Commun 283: (3) ((2001) ), 674–679. |
[32] | Mukherji M. , Chien W. , Kershaw N.J. , Clifton I.J. , Schofield C.J. and Wierzbicki A.S. et al., Structure-function analysis of phytanoyl-CoA 2-hydroxylase mutations causing Refsum’s disease, Hum Mol Genet 10: (18) ((2001) ), 1971–1982. |
[33] | Foulon V. , Antonenkov V.D. , Croes K. , Waelkens E. , Mannaerts G.P. and Van Veldhoven P.P. , et al., Purification, molecular cloning, and expression of 2-hydroxyphytanoyl-CoA lyase, a peroxisomal thiamine pyrophosphate-dependent enzyme that catalyzes the carbon-carbon bond cleavage during alpha-oxidation of 3-methyl-branched fatty acids, Proc Natl Acad Sci U S A 96: (18) ((1999) ), 10039–10044. |
[34] | Schmitz W. , Fingerhut R. and Conzelmann E. , Purification and properties of an alpha-methylacyl-CoA racemase from rat liver, Eur J Biochem 222: (2) ((1994) ), 313–323. |
[35] | Steinberg D. , Avigan J. , Mize C. , Eldjarn L. , Try K. and Refsum S. , Conversion of U-C14-phytol to phytanic acid and its oxidation in heredopathia atactica polyneuritiformis, Biochem Biophys Res Commun 19: (6) ((1965) ), 783–789. |
[36] | Brown J. , Mei G. , Gibberd F.B. , Burston D. , Mayne P.D. , McClinchy J. and et al., Diet and Refsum’s disease. The determination of phytanic acid and phytol in certain foods and the application of this knowledge to the choice of suitable convenience foods for patients with Refsum’s disease, Journal of Human Nutrition and Dietetics 6: ((1993) ), 295–305. |
[37] | Wanders R.J. , Komen J. and Ferdinandusse S. , Phytanic acid metabolism in health and disease, Biochim Biophys Acta 1811: (9) ((2011) ), 498–507. |
[38] | Braverman N.E. and Moser A.B. , Functions of plasmalogen lipids in health and disease, Biochim Biophys Acta 1822: (9) ((2012) ), 1442–1452. |
[39] | da Silva T.F. , Sousa V.F. , Malheiro A.R. and Brites P. , The importance of ether-phospholipids: A view from the perspective of mouse models, Biochim Biophys Acta 1822: (9) ((2012) ), 1501–1508. |
[40] | Marinetti G.V. and Erbland J. , The structure of pig heart plasmalogen, Biochim Biophys Acta 26: (2) ((1957) ), 429–430. |
[41] | Han X.L. and Gross R.W. , Plasmenylcholine and phosphatidylcholine membrane bilayers possess distinct conformational motifs, Biochemistry 29: (20) ((1990) ), 4992–4996. |
[42] | Rog T. and Koivuniemi A. , The biophysical properties of ethanolamine plasmalogens revealed by atomistic molecular dynamics simulations, Biochim Biophys Acta 1858: (1) ((2016) ), 97–103. |
[43] | Hermetter A. , Rainer B. , Ivessa E. , Kalb E. , Loidl J. and Roscher A. et al., Influence of plasmalogen deficiency on membrane fluidity of human skin fibroblasts: A fluorescence anisotropy study, Biochim Biophys Acta 978: (1) ((1989) ), 151–157. |
[44] | Farooqui A.A. and Horrocks L.A. , Plasmalogens, phospholipase A2, and docosahexaenoic acid turnover in brain tissue, J Mol Neurosci 16: (2-3) ((2001) ), 263–272; discussion 79–84. |
[45] | Brites P. , Ferreira A.S. , da Silva T.F. , Sousa V.F. , Malheiro A.R. and Duran M. , et al., Alkyl-glycerol rescues plasmalogen levels and pathology of ether-phospholipid deficient mice, PLoS One 6: (12) ((2011) ), e28539 . |
[46] | Bams-Mengerink A.M. , Majoie C.B. , Duran M. , Wanders R.J. , Van Hove J. and Scheurer C.D. , et al., MRI of the brain and cervical spinal cord in rhizomelic chondrodysplasia punctata, Neurology 66: (6) ((2006) ), 798–803; discussion 789. |
[47] | Bams-Mengerink A.M. , Koelman J.H. , Waterham H. , Barth P.G. and Poll-The B.T. , The neurology of rhizomelic chondrodysplasia punctata, Orphanet J Rare Dis 8: ((2013) ), 174. |
[48] | Braverman N. , Chen L. , Lin P. , Obie C. , Steel G. and Douglas P. et al., Mutation analysis of PEX7 in 60 probands with rhizomelic chondrodysplasia punctata and functional correlations of genotype with phenotype, Hum Mutat 20: (4) ((2002) ), 284–297. |
[49] | Leroy J.G. , Espeel M. , Gadisseux J.F. , Mandel H. , Martinez M. and Poll-The B.T. et al., Diagnostic work-up of a peroxisomal patient, J Inherit Metab Dis 18: (Suppl 1) ((1995) ), 214–222. |
[50] | Mandel H. , Berant M. , Meiron D. , Aizin A. , Oiknine J. and Brook J.G. et al., Plasma lipoproteins and monocyte-macrophages in a peroxisome-deficient system: Study of a patient with infantile refsum disease, J Inherit Metab Dis 15: (5) ((1992) ), 774–784. |
[51] | Poll-The B.T. , Saudubray J.M. , Ogier H.A. , Odièvre M. , Scotto J.M. and Monnens L. et al., Infantile Refsum disease: An inherited peroxisomal disorder, Comparison with Zellweger syndrome and neonatal adrenoleukodystrophy, Eur J Pediatr 146: (5) ((1987) ), 477–483. |
[52] | Kovacs W.J. , Shackelford J.E. , Tape K.N. , Richards M.J. , Faust P.L. and Fliesler S.J. et al., Disturbed cholesterol homeostasis in a peroxisome-deficient PEX2 knockout mouse model, Mol Cell Biol 24: (1) ((2004) ), 1–13. |
[53] | Faust P.L. and Kovacs W.J. , Cholesterol biosynthesis and ER stress in peroxisome deficiency, Biochimie 98: ((2014) ), 75–85. |
[54] | Kunze M. , Neuberger G. , Maurer-Stroh S. , Ma J. , Eck T. and Braverman N. et al., Structural requirements for interaction of peroxisomal targeting signal 2 and its receptor PEX7, J Biol Chem 286: (52) ((2011) ), 45048–45062. |
[55] | Chowdhary G. , Kataya A.R. , Lingner T. and Reumann S. , Non-canonical peroxisome targeting signals: Identification of novel PTS1 tripeptides and characterization of enhancer elements by computational permutation analysis, BMC Plant Biol 12: ((2012) ), 142. |
[56] | Lingner T. , Kataya A.R. and Reumann S. , Experimental and statistical post-validation of positive example EST sequences carrying peroxisome targeting signals type 1 (PTS1), Plant Signal Behav 7: (2) ((2012) ), 263–268. |
[57] | Mizuno Y. , Kurochkin I.V. , Herberth M. , Okazaki Y. and Schönbach C. , Predicted mouse peroxisome-targeted proteins and their actual subcellular locations, BMC Bioinformatics 9: (Suppl 12) ((2008) ), S16. |
[58] | Wiese S. , Gronemeyer T. , Ofman R. , Kunze M. , Grou C.P. and Almeida J.A. et al., Proteomics characterization of mouse kidney peroxisomes by tandem mass spectrometry and protein correlation profiling, Mol Cell Proteomics 6: (12) ((2007) ), 2045–2057. |
[59] | Wiese S. , Gronemeyer T. , Brites P. , Ofman R. , Bunse C. and Renz C. et al., Comparative profiling of the peroxisomal proteome of wildtype and Pex7 knockout mice by quantitative mass spectrometry, International Journal of Mass Spectrometry 312: ((2012) ), 30–40. |
[60] | Ashmarina L.I. , Pshezhetsky A.V. , Branda S.S. , Isaya G. and Mitchell G.A. , 3-Hydroxy-3-methylglutaryl coenzyme A lyase: Targeting and processing in peroxisomes and mitochondria, J Lipid Res 40: (1) ((1999) ), 70–75. |
[61] | Ferdinandusse S. , Denis S. , IJlst L. , Dacremont G. , Waterham H.R. and Wanders R.J. , Subcellular localization and physiological role of alpha-methylacyl-CoA racemase, J Lipid Res 41: (11) ((2000) ), 1890–1896. |
[62] | Wightman P.J. , Santer R. , Ribes A. , Dougherty F. , McGill N. and Thorburn D.R. et al., MLYCD mutation analysis: Evidence for protein mistargeting as a cause of MLYCD deficiency, Hum Mutat 22: (4) ((2003) ), 288–300. |
[63] | Luo B. , Norris C. , Bolstad E.S. , Knecht D.A. and Grant D.F. , Protein quaternary structure and expression levels contribute to peroxisomal-targeting-sequence-1-mediated peroxisomal import of human soluble epoxide hydrolase, J Mol Biol 380: (1) ((2008) ), 31–41. |
[64] | Islinger M. , Li K.W. , Seitz J. , Völkl A. and Lüers G.H. , Hitchhiking of Cu/Zn superoxide dismutase to peroxisomes–evidence for a natural piggyback import mechanism in mammals, Traffic 10: (11) ((2009) ), 1711–1721. |
[65] | Leiper J.M. and Danpure C.J. , The role of dimerization of alanine:Glyoxylate aminotransferase 1 in its peroxisomal and mitochondrial import, Ann N Y Acad Sci 804: ((1996) ), 765–767. |
[66] | Gonzalez F.J. and Shah Y.M. , PPARalpha: Mechanism of species differences and hepatocarcinogenesis of peroxisome proliferators, Toxicology 246: (1) ((2008) ), 2–8. |
[67] | Sexton J.Z. , He Q. , Forsberg L.J. and Brenman J.E. , High content screening for non-classical peroxisome proliferators, Int J High Throughput Screen 2010: (1) ((2010) ), 127–140. |
[68] | Chang C.C. , South S. , Warren D. , Jones J. , Moser A.B. and Moser H.W. et al., Metabolic control of peroxisome abundance, J Cell Sci 112: (Pt 10) ((1999) ), 1579–1590. |
[69] | Schrader M. , Costello J.L. , Godinho L.F. , Azadi A.S. and Islinger M. , Proliferation and fission of peroxisomes - An update, Biochim Biophys Acta 1863: (5) ((2016) ), 971–983. |
[70] | Bagattin A. , Hugendubler L. and Mueller E. , Transcriptional coactivator PGC-1alpha promotes peroxisomal remodeling and biogenesis, Proc Natl Acad Sci U S A 107: (47) ((2010) ), 20376–20381. |
[71] | Schrader M. , Thiemann M. and Fahimi H.D. , Peroxisomal motility and interaction with microtubules, Microsc Res Tech 61: (2) ((2003) ), 171–178. |
[72] | Neuhaus A. , Eggeling C. , Erdmann R. and Schliebs W. , Why do peroxisomes associate with the cytoskeleton? Biochim Biophys Acta 1863: (5) ((2016) ), 1019–1026. |
[73] | Agrawal G. and Subramani S. , De novo peroxisome biogenesis: Evolving concepts and conundrums, Biochim Biophys Acta 1863: (5) ((2016) ), 892–901. |
[74] | Farr R.L. , Lismont C. , Terlecky S.R. and Fransen M. , Peroxisome biogenesis in mammalian cells: The impact of genes and environment, Biochim Biophys Acta 1863: (5) ((2016) ), 1049–1060. |
[75] | Dias A.F. , Francisco T. , Rodrigues T.A. , Grou C.P. and Azevedo J.E. , The first minutes in the life of a peroxisomal matrix protein, Biochim Biophys Acta 1863: (5) ((2016) ), 814–820. |
[76] | Emmanouilidis L. , Gopalswamy M. , Passon D.M. , Wilmanns M. and Sattler M. , Structural biology of the import pathways of peroxisomal matrix proteins, Biochim Biophys Acta 1863: (5) ((2016) ), 804–813. |
[77] | Gould S.J. , Keller G.A. , Hosken N. , Wilkinson J. and Subramani S. , A conserved tripeptide sorts proteins to peroxisomes, J Cell Biol 108: (5) ((1989) ), 1657–1664. |
[78] | Gould S.J. , Keller G.A. and Subramani S. , Identification of peroxisomal targeting signals located at the carboxy terminus of four peroxisomal proteins, J Cell Biol 107: (3) ((1988) ), 897–905. |
[79] | Gould S.G. , Keller G.A. and Subramani S. , Identification of a peroxisomal targeting signal at the carboxy terminus of firefly luciferase, J Cell Biol 105: (6 Pt 2) ((1987) ), 2923–2931. |
[80] | Petriv O.I. , Tang L. , Titorenko V.I. and Rachubinski R.A. , A new definition for the consensus sequence of the peroxisome targeting signal type 2, J Mol Biol 341: (1) ((2004) ), 119–134. |
[81] | Swinkels B.W. , Gould S.J. , Bodnar A.G. , Rachubinski R.A. and Subramani S. , A novel, cleavable peroxisomal targeting signal at the amino-terminus of the rat 3-ketoacyl-CoA thiolase, EMBO J 10: (11) ((1991) ), 3255–3262. |
[82] | Osumi T. , Tsukamoto T. , Hata S. , Yokota S. , Miura S. and Fujiki Y. et al., Amino-terminal presequence of the precursor of peroxisomal 3-ketoacyl-CoA thiolase is a cleavable signal peptide for peroxisomal targeting, Biochem Biophys Res Commun 181: (3) ((1991) ), 947–954. |
[83] | Mihalik S.J. , Morrell J.C. , Kim D. , Sacksteder K.A. , Watkins P.A. and Gould S.J. , Identification of PAHX, a Refsum disease gene, Nat Genet 17: (2) ((1997) ), 185–189. |
[84] | de Vet E.C. , van den Broek B.T. and van den Bosch H. , Nucleotide sequence of human alkyl-dihydroxyacetonephosphate synthase cDNA reveals the presence of a peroxisomal targeting signal 2, BiochimBiophys Acta 1346: (1) ((1997) ), 25–29. |
[85] | Freitas M.O. , Francisco T. , Rodrigues T.A. , Alencastre I.S. , Pinto M.P. and Grou C.P. et al., PEX5 protein binds monomeric catalase blocking its tetramerization and releases it upon binding the N-terminal domain of PEX14, J Biol Chem 286: (47) ((2011) ), 40509–40519. |
[86] | Brocard C. , Kragler F. , Simon M.M. , Schuster T. and Hartig A. , The tetratricopeptide repeat-domain of the PAS10 protein of Saccharomyces cerevisiae is essential for binding the peroxisomal targeting signal-SKL, Biochem Biophys Res Commun 204: (3) ((1994) ), 1016–1022. |
[87] | Terlecky S.R. , Nuttley W.M. , McCollum D. , Sock E. and Subramani S. , The Pichia pastoris peroxisomal protein PAS8p is the receptor for the C-terminal tripeptide peroxisomal targeting signal, EMBO J 14: (15) ((1995) ), 3627–3634. |
[88] | Gatto G.J. , Geisbrecht B.V. , Gould S.J. and Berg J.M. , Peroxisomal targeting signal-1 recognition by the TPR domains of human PEX5, Nat Struct Biol 7: (12) ((2000) ), 1091–1095. |
[89] | Braverman N. , Dodt G. , Gould S.J. and Valle D. , An isoform of pex5p, the human PTS1 receptor, is required for the import of PTS2 proteins into peroxisomes, Hum Mol Genet 7: (8) ((1998) ), 1195–1205. |
[90] | Rehling P. , Marzioch M. , Niesen F. , Wittke E. , Veenhuis M. and Kunau W.H. , The import receptor for the peroxisomal targeting signal 2 (PTS2) in Saccharomyces cerevisiae is encoded by the PAS7 gene, EMBO J 15: (12) ((1996) ), 2901–2913. |
[91] | Dodt G. , Warren D. , Becker E. , Rehling P. and Gould S.J. , Domain mapping of human PEX5 reveals functional and structural similarities to Saccharomyces cerevisiae Pex18p and Pex21p, J Biol Chem 276: (45) ((2001) ), 41769–41781. |
[92] | Gould S.J. , Kalish J.E. , Morrell J.C. , Bjorkman J. , Urquhart A.J. and Crane D.I. , Pex13p is an SH3 protein of the peroxisome membrane and a docking factor for the predominantly cytoplasmic PTs1 receptor, J Cell Biol 135: (1) ((1996) ), 85–95. |
[93] | Elgersma Y. , Kwast L. , Klein A. , Voorn-Brouwer T. and van den Berg M. and Metzig B. , et al., The SH3 domain of the Saccharomyces cerevisiae peroxisomal membrane protein Pex13p functions as a docking site for Pex5p, a mobile receptor for the import PTS1-containing proteins, J Cell Biol 135: (1) ((1996) ), 97–109. |
[94] | Erdmann R. and Blobel G. , Identification of Pex13p a peroxisomal membrane receptor for the PTS1 recognition factor, J Cell Biol 135: (1) ((1996) ), 111–121. |
[95] | Albertini M. , Rehling P. , Erdmann R. , Girzalsky W. , Kiel J.A. and Veenhuis M. et al., Pex14p, a peroxisomal membrane protein binding both receptors of the two PTS-dependent import pathways, Cell 89: (1) ((1997) ), 83–92. |
[96] | Otera H. , Harano T. , Honsho M. , Ghaedi K. , Mukai S. and Tanaka A. et al., The mammalian peroxin Pex5pL, the longer isoform of the mobile peroxisome targeting signal (PTS) type 1 transporter, translocates the Pex7p, PTS2 protein complex into peroxisomes via its initial docking site, Pex14p, J Biol Chem 275: (28) ((2000) ), 21703–21714. |
[97] | Rucktäschel R. , Girzalsky W. and Erdmann R. , Protein import machineries of peroxisomes, Biochim Biophys Acta 1808: (3) ((2011) ), 892–900. |
[98] | Erdmann R. and Schliebs W. , Peroxisomal matrix protein import: The transient pore model, Nat Rev Mol Cell Biol 6: (9) ((2005) ), 738–742. |
[99] | Meinecke M. , Cizmowski C. , Schliebs W. , Krüger V. , Beck S. and Wagner R. et al., The peroxisomal importomer constitutes a large and highly dynamic pore, Nat Cell Biol 12: (3) ((2010) ), 273–277. |
[100] | McNew J.A. and Goodman J.M. , An oligomeric protein is imported into peroxisomes in vivo, J Cell Biol 127: (5) ((1994) ), 1245–1257. |
[101] | Glover J.R. , Andrews D.W. and Rachubinski R.A. , Saccharomyces cerevisiae peroxisomal thiolase is imported as a dimer, Proc Natl Acad Sci U S A 91: (22) ((1994) ), 10541–10545. |
[102] | Freitas M.O. , Francisco T. , Rodrigues T.A. , Lismont C. , Domingues P. and Pinto M.P. et al., The peroxisomal protein import machinery displays a preference for monomeric substrates, Open Biol 5: (4) ((2015) ), 140236. |
[103] | Alencastre I.S. , Rodrigues T.A. , Grou C.P. , Fransen M. , Sá-Miranda C. and Azevedo J.E. , Mapping the cargo protein membrane translocation step into the PEX5 cycling pathway, J Biol Chem 284: (40) ((2009) ), 27243–27251. |
[104] | Dodt G and Gould SJ . Multiple PEX genes are required for proper subcellular distribution and stability of Pex5p, the PTS1 receptor: Evidence that PTS1 protein import is mediated by a cycling receptor, J Cell Biol 135: (6 Pt 2) ((1996) ), 1763–1774. |
[105] | Dammai V. and Subramani S. , The human peroxisomal targeting signal receptor, Pex5p, is translocated into the peroxisomal matrix and recycled to the cytosol, Cell 105: (2) ((2001) ), 187–196. |
[106] | Kiel J.A. , Emmrich K. , Meyer H.E. and Kunau W.H. , Ubiquitination of the peroxisomal targeting signal type 1 receptor, Pex5p, suggests the presence of a quality control mechanism during peroxisomal matrix protein import, J Biol Chem 280: (3) ((2005) ), 1921–1930. |
[107] | Prestele J. , Hierl G. , Scherling C. , Hetkamp S. , Schwechheimer C. and Isono E. et al., Different functions of the C3HC4 zinc RING finger peroxins PEX10, PEX2, and PEX12 in peroxisome formation and matrix protein import, Proc Natl Acad Sci U S A 107: (33) ((2010) ), 14915–14920. |
[108] | Williams C. , van den Berg M. , Sprenger R.R. and Distel B. , A conserved cysteine is essential for Pex4p-dependent ubiquitination of the peroxisomal import receptor Pex5p, J Biol Chem 282: (31) ((2007) ), 22534–22543. |
[109] | Costa-Rodrigues J. , Carvalho A.F. , Gouveia A.M. , Fransen M. , Sá-Miranda C. and Azevedo J.E. , The N terminus of the peroxisomal cycling receptor, Pex5p, is required for redirecting the peroxisome-associated peroxin back to the cytosol, J Biol Chem 279: (45) ((2004) ), 46573–46579. |
[110] | Carvalho A.F. , Pinto M.P. , Grou C.P. , Alencastre I.S. , Fransen M. and Sá-Miranda C. et al., Ubiquitination of mammalian Pex5p, the peroxisomal import receptor, J Biol Chem 282: (43) ((2007) ), 31267–31272. |
[111] | Platta H.W. , El Magraoui F. , Bäumer B.E. , Schlee D. , Girzalsky W. and Erdmann R., , Pex2 and pex12 function as protein-ubiquitin ligases in peroxisomal protein import, Mol Cell Biol 29: (20) ((2009) ), 5505–5516. |
[112] | Platta H.W. , Girzalsky W. and Erdmann R. , Ubiquitination of the peroxisomal import receptor Pex5p, Biochem J 384: (Pt 1) ((2004) ), 37–45. |
[113] | Kragt A. , Voorn-Brouwer T. , van den Berg M. and Distel B. , The Saccharomyces cerevisiae peroxisomal import receptor Pex5p is monoubiquitinated in wild type cells, J Biol Chem 280: (9) ((2005) ), 7867–7874. |
[114] | Platta H.W. , Brinkmeier R. , Reidick C. , Galiani S. , Clausen M.P. and Eggeling C. , Regulation of peroxisomal matrix protein import by ubiquitination, Biochim Biophys Acta 1863: (5) ((2016) ), 838–849. |
[115] | Platta H.W. , Hagen S. , Reidick C. and Erdmann R. , The peroxisomal receptor dislocation pathway: To the exportomer and beyond, Biochimie 98: ((2014) ), 16–28. |
[116] | Platta H.W. , Grunau S. , Rosenkranz K. , Girzalsky W. and Erdmann R. , Functional role of the AAA peroxins in dislocation of the cycling PTS1 receptor back to the cytosol, Nat Cell Biol 7: (8) ((2005) ), 817–822. |
[117] | Tan D. , Blok N.B. , Rapoport T.A. and Walz T. , Structures of the double-ring AAA ATPase Pex1-Pex6 involved in peroxisome biogenesis, FEBS J 283: (6) ((2016) ), 986–992. |
[118] | Carroni M. , Kummer E. , Oguchi Y. , Wendler P. , Clare D.K. and Sinning I. et al., Head-to-tail interactions of the coiled-coil domains regulate ClpB activity and cooperation with Hsp70 in protein disaggregation, Elife 3: ((2014) ), e02481. |
[119] | Wendler P. , Shorter J. , Snead D. , Plisson C. , Clare D.K. and Lindquist S. et al., Motor mechanism for protein threading through Hsp104, Mol Cell 34: (1) ((2009) ), 81–92. |
[120] | Martin A. , Baker T.A. and Sauer R.T. , Rebuilt AAA + motors reveal operating principles for ATP-fuelled machines, Nature 437: (7062) ((2005) ), 1115–1120. |
[121] | Birschmann I. , Rosenkranz K. , Erdmann R. and Kunau W.H. , Structural and functional analysis of the interaction of the AAA-peroxins Pex1p and Pex6p, FEBS J 272: (1) ((2005) ), 47–58. |
[122] | Tamura S., Yasutake S., Matsumoto N. and Fujiki Y., Dynamic and functional assembly of the AAA peroxins, Pex1p and Pex6p, and their membrane receptor Pex26p, J Biol Chem 281: (38) ((2006) ), 27693–27704. |
[123] | Miyata N. and Fujiki Y. , Shuttling mechanism of peroxisome targeting signal type 1 receptor Pex5: ATP-independent import and ATP-dependent export, Mol Cell Biol 25: (24) ((2005) ), 10822–10832. |
[124] | Gardner B.M. , Chowdhury S. , Lander G.C. and Martin A. , The Pex1/Pex6 complex is a heterohexameric AAA+ motor with alternating and highly coordinated subunits, J Mol Biol 427: (6 Pt B) ((2015) ), 1375–1388. |
[125] | Blok N.B. , Tan D. , Wang R.Y. , Penczek P.A. , Baker D. and DiMaio F. et al., Unique double-ring structure of the peroxisomal Pex1/Pex6 ATPase complex revealed by cryo-electron microscopy, Proc Natl Acad Sci U S A 112: (30) ((2015) ), E4017–E4025. |
[126] | Ciniawsky S. , Grimm I. , Saffian D. , Girzalsky W. , Erdmann R. and Wendler P. , Molecular snapshots of the Pex1/6 AAA+ complex in action, Nat Commun 6: ((2015) ), 7331. |
[127] | Schliebs W. , Girzalsky W. and Erdmann R. , Peroxisomal protein import and ERAD: Variations on a common theme, Nat Rev Mol Cell Biol 11: (12) ((2010) ), 885–890. |
[128] | Baerends R.J. , Rasmussen S.W. , Hilbrands R.E. , van der Heide M. , Faber K.N. and Reuvekamp P.T. , et al., The Hansenula polymorpha PER9 gene encodes a peroxisomal membrane protein essential for peroxisome assembly and integrity, J Biol Chem 271: (15) ((1996) ), 8887–8894. |
[129] | Honsho M. , Tamura S. , Shimozawa N. , Suzuki Y. , Kondo N. and Fujiki Y. , Mutation in PEX16 is causal in the peroxisome-deficient Zellweger syndrome of complementation group D, Am J Hum Genet 63: (6) ((1998) ), 1622–1630. |
[130] | Götte K. , Girzalsky W. , Linkert M. , Baumgart E. , Kammerer S. and Kunau W.H. et al., Pex19p, a farnesylated protein essential for peroxisome biogenesis, Mol Cell Biol 18: (1) ((1998) ), 616–628. |
[131] | Matsuzono Y. , Kinoshita N. , Tamura S. , Shimozawa N. , Hamasaki M. and Ghaedi K. et al., Human PEX19: cDNA cloning by functional complementation, mutation analysis in a patient with Zellweger syndrome, and potential role in peroxisomal membrane as sembly, Proc Natl Acad Sci U S A 96: (5) ((1999) ), 2116–2121. |
[132] | Ghaedi K. , Honsho M. , Shimozawa N. , Suzuki Y. , Kondo N. and Fujiki Y. , PEX3 is the causal gene responsible for peroxisome membrane assembly-defective Zellweger syndrome of complementation group G, Am J Hum Genet 67: (4) ((2000) ), 976–981. |
[133] | Sacksteder K.A. , Jones J.M. , South S.T. , Li X. , Liu Y. and Gould S.J. , PEX19 binds multiple peroxisomal membrane proteins, is predominantly cytoplasmic, and is required for peroxisome membrane synthesis, J Cell Biol 148: (5) ((2000) ), 931–944. |
[134] | Hettema E.H. , Girzalsky W. , van Den Berg M. , Erdmann R. and Distel B. , Saccharomyces cerevisiae pex3p and pex19p are required for proper localization and stability of peroxisomal membrane proteins, EMBO J 19: (2) ((2000) ), 223–233. |
[135] | South S.T. and Gould S.J. , Peroxisome synthesis in the absence of preexisting peroxisomes, J Cell Biol 144: (2) ((1999) ), 255–266. |
[136] | Otzen M. , Perband U. , Wang D. , Baerends R.J. , Kunau W.H. and Veenhuis M. et al., Hansenula polymorpha Pex19p is essential for the formation of functional peroxisomal membranes, J Biol Chem 279: (18) ((2004) ), 19181–19190. |
[137] | Kim P.K. , Mullen R.T. , Schumann U. and Lippincott-Schwartz J. , The origin and maintenance of mammalian peroxisomes involves a de novo PEX16-dependent pathway from the ER, J Cell Biol 173: (4) ((2006) ), 521–532. |
[138] | Geuze H.J. , Murk J.L. , Stroobants A.K. , Griffith J.M. , Kleijmeer M.J. and Koster A.J. et al., Involvement of the endoplasmic reticulum in peroxisome formation, Mol Biol Cell 14: (7) ((2003) ), 2900–2907. |
[139] | Tabak H.F. , Braakman I. and van der Zand A. , Peroxisome formation and maintenance are dependent on the endoplasmic reticulum, Annu Rev Biochem 82: ((2013) ), 723–744. |
[140] | Jones J.M. , Morrell J.C. and Gould S.J. , PEX19 is a predominantly cytosolic chaperone and import receptor for class 1 peroxisomal membrane proteins, J Cell Biol 164: (1) ((2004) ), 57–67. |
[141] | Fang Y. , Morrell J.C. , Jones J.M. and Gould S.J. , PEX3 functions as a PEX19 docking factor in the import of class I peroxisomal membrane proteins, J Cell Biol 164: (6) ((2004) ), 863–875. |
[142] | Honsho M. , Hiroshige T. and Fujiki Y. , The membrane biogenesis peroxin Pex16p, Topogenesis and functional roles in peroxisomal membrane assembly, J Biol Chem 277: (46) ((2002) ), 44513–44524. |
[143] | Matsuzaki T. and Fujiki Y. , The peroxisomal membrane protein import receptor Pex3p is directly transported to peroxisomes by a novel Pex19p- and Pex16p-dependent pathway, J Cell Biol 183: (7) ((2008) ), 1275–1286. |
[144] | Waterham H.R. , Ferdinandusse S. and Wanders R.J. , Human disorders of peroxisome metabolism and biogenesis, Biochim Biophys Acta 1863: (5) ((2016) ), 922–933. |
[145] | Koch A. , Yoon Y. , Bonekamp N.A. , McNiven M.A. and Schrader M. , A role for Fis1 in both mitochondrial and peroxisomal fission in mammalian cells, Mol Biol Cell 16: (11) ((2005) ), 5077–5086. |
[146] | Li X. and Gould S.J. , The dynamin-like GTPase DLP1 is essential for peroxisome division and is recruited to peroxisomes in part by PEX11, J Biol Chem 278: (19) ((2003) ), 17012–17020. |
[147] | Koch A. , Thiemann M. , Grabenbauer M. , Yoon Y. , McNiven M.A. and Schrader M. , Dynamin-like protein 1 is involved in peroxisomal fission, J Biol Chem 278: (10) ((2003) ), 8597–8605. |
[148] | Huber N. , Guimaraes S. , Schrader M. , Suter U. and Niemann A. , Charcot-Marie-Tooth disease-associated mutants of GDAP1 dissociate its roles in peroxisomal and mitochondrial fission, EMBO Rep 14: (6) ((2013) ), 545–552. |
[149] | Schrader M. , Reuber B.E. , Morrell J.C. , Jimenez-Sanchez G. , Obie C. and Stroh T.A. et al., Expression of PEX11beta mediates peroxisome proliferation in the absence of extracellular stimuli, J Biol Chem 273: (45) ((1998) ), 29607–29614. |
[150] | Abe I. and Fujiki Y. , cDNA cloning and characterization of a constitutively expressed isoform of the human peroxin Pex11p, Biochem Biophys Res Commun 252: (2) ((1998) ), 529–533. |
[151] | Tanaka A. , Okumoto K. and Fujiki Y. , cDNA cloning and characterization of the third isoform of human peroxin Pex11p, Biochem Biophys Res Commun 300: (4) ((2003) ), 819–823. |
[152] | Koch J and Brocard C. , PEX11 proteins attract Mff and human Fis1 to coordinate peroxisomal fission, J Cell Sci 125: (Pt 16) ((2012) ), 3813–3826. |
[153] | Ebberink M.S. , Koster J. , Visser G. , Spronsen F. , Stolte-Dijkstra I. and Smit G.P. et al., A novel defect of peroxisome division due to a homozygous non-sense mutation in the PEX11β gene, J Med Genet 49: (5) ((2012) ), 307–313. |
[154] | Mayerhofer P.U. , Targeting and insertion of peroxisomal membrane proteins: ER trafficking versus direct delivery to peroxisomes, Biochim Biophys Acta 1863: (5) ((2016) ), 870–880. |
[155] | Agrawal G. and Subramani S. , Emerging role of the endoplasmic reticulum in peroxisome biogenesis, Front Physiol 4: ((2013) ), 286. |
[156] | Dimitrov L. , Lam S.K. and Schekman R. , The role of the endoplasmic reticulum in peroxisome biogenesis, Cold Spring Harb Perspect Biol 5: (5) ((2013) ), a013243. |
[157] | Katarzyna Z.R. and Suresh S. , Autophagic degradation of peroxisomes in mammals, Biochem Soc Trans 44: (2) ((2016) ), 431–440. |
[158] | Honsho M. , Yamashita S. and Fujiki Y. , Peroxisome homeostasis: Mechanisms of division and selective degradation of peroxisomes in mammals, Biochim Biophys Acta 1863: (5) ((2016) ), 984–991. |
[159] | van Zutphen T. , Veenhuis M. and van der Klei I.J. , Damaged peroxisomes are subject to rapid autophagic degradation in the yeast Hansenula polymorpha, Autophagy 7: (8) ((2011) ), 863–872. |
[160] | Hara-Kuge S. and Fujiki Y. , The peroxin Pex14p is involved in LC3-dependent degradation of mammalian peroxisomes, Exp Cell Res 314: (19) ((2008) ), 3531–3541. |
[161] | Nuttall J.M. , Motley A.M. and Hettema E.H. , Deficiency of the exportomer components Pex1, Pex6, and Pex15 causes enhanced pexophagy in Saccharomyces cerevisiae, Autophagy 10: (5) ((2014) ), 835–845. |
[162] | Huybrechts S.J. , Van Veldhoven P.P. , Brees C. , Mannaerts G.P. , Los G.V. and Fransen M. , Peroxisome dynamics in cultured mammalian cells, Traffic 10: (11) ((2009) ), 1722–1733. |
[163] | Kelley R.I. , Datta N.S. , Dobyns W.B. , Hajra A.K. , Moser A.B. and Noetzel M.J. et al., Neonatal adrenoleukodystrophy: New cases, biochemical studies, and differentiation from Zellweger and related peroxisomal polydystrophy syndromes, Am J Med Genet 23: (4) ((1986) ), 869–901. |
[164] | Poll-The B.T. , Saudubray J.M. , Ogier H. , Schutgens R.B. , Wanders R.J. and Schrakamp G. et al., Infantile Refsum’s disease: Biochemical findings suggesting multiple peroxisomal dysfunction, J Inherit Metab Dis 9: (2) ((1986) ), 169–174. |
[165] | Santos M.J. , Imanaka T. , Shio H. and Lazarow P.B. , Peroxisomal integral membrane proteins in control and Zellweger fibroblasts, J Biol Chem 263: (21) ((1988) ), 10502–10509. |
[166] | Santos M.J. , Imanaka T. , Shio H. , Small G.M. and Lazarow P.B. , Peroxisomal membrane ghosts in Zellweger syndrome–aberrant organelle assembly, Science 239: (4847) ((1988) ), 1536–1538. |
[167] | Braverman N. , Steel G. , Obie C. , Moser A. , Moser H. and Gould S.J. et al., Human PEX7 encodes the peroxisomal PTS2 receptor and is responsible for rhizomelic chondrodysplasia punctata, Nat Genet 15: (4) ((1997) ), 369–376. |
[168] | Gould S.J. , Raymond G.V. , Valle D. , The peroxisome biogenesis disorders, In: The Metabolic and Molecular Bases of Inherited Disease McGraw Hill International Book Company, New York, NY, ((2001) ), 3181–3217. |
[169] | Stoll C. , Dott B. , Roth M.P. and Alembik Y. , Birth prevalence rates of skeletal dysplasias, Clin Genet 35: (2) ((1989) ), 88–92. |
[170] | Levesque S. , Morin C. , Guay S.P. , Villeneuve J. , Marquis P. and Yik W.Y. et al., A founder mutation in the PEX6 gene is responsible for increased incidence of Zellweger syndrome in a French Canadian population, BMC Med Genet 13: ((2012) ), 72. |
[171] | Shimozawa N. , Nagase T. , Takemoto Y. , Suzuki Y. and Kondo N. , Genetic heterogeneity in Japanese patients with peroxisome biogenesis disorders and evidence for a founder haplotype for the most common mutation in PEX10 gene, Adv Exp Med Biol 544: ((2003) ), 71. |
[172] | Mathijssen I.B. , Henneman L. , van Eeten-Nijman J.M. , Lakeman P. , Ottenheim C.P. and Redeker E.J. , et al., Targeted carrier screening for four recessive disorders: High detection rate within a founder population, Eur J Med Genet 58: (3) ((2015) ), 123–128. |
[173] | Shaheen R. , Al-Dirbashi O.Y. , Al-Hassnan Z.N. , Al-Owain M. , Makhsheed N. and Basheeri F. et al., Clinical, biochemical and molecular characterization of peroxisomal diseases in Arabs, Clin Genet 79: (1) ((2011) ), 60–70. |
[174] | Steinberg S. , Chen L. , Wei L. , Moser A. , Moser H. and Cutting G. et al., The PEX Gene Screen: Molecular diagnosis of peroxisome biogenesis disorders in the Zellweger syndrome spectrum, Mol Genet Metab 83: (3) ((2004) ), 252–263. |
[175] | Collins C.S. and Gould S.J. , Identification of a common PEX1 mutation in Zellweger syndrome, Hum Mutat 14: (1) ((1999) ), 45–53. |
[176] | Braverman N. , Steel G. , Lin P. , Moser A. , Moser H. and Valle D. , PEX7 gene structure, alternative transcripts, and evidence for a founder haplotype for the frequent RCDP allele, L292ter, Genomics 63: (2) ((2000) ), 181–192. |
[177] | Motley A.M. , Brites P. , Gerez L. , Hogenhout E. , Haasjes J. and Benne R. et al., Mutational spectrum in the PEX7 gene and functional analysis of mutant alleles in 78 patients with rhizomelic chondrodysplasia punctata type 1, Am J Hum Genet 70: (3) ((2002) ), 612–624. |
[178] | Zeharia A. , Ebberink M.S. , Wanders R.J. , Waterham H.R. , Gutman A. and Nissenkorn A. et al., A novel PEX12 mutation identified as the cause of a peroxisomal biogenesis disorder with mild clinical phenotype, mild biochemical abnormalities in fibroblasts and a mosaic catalase immunofluorescence pattern, even at 40 degrees C, J Hum Genet 52: (7) ((2007) ), 599–606. |
[179] | Braverman N.E. , D’Agostino M.D. and Maclean G.E. , Peroxisome biogenesis disorders: Biological, clinical and pathophysiological perspectives, Dev Disabil Res Rev 17: (3) ((2013) ), 187–196. |
[180] | Govaerts L. , Monnens L. , Tegelaers W. , Trijbels F. and van A. , Raay-Selten, Cerebro-hepato-renal syndrome of Zellweger: Clinical symptoms and relevant laboratory findings in 16 patients, Eur J Pediatr 139: (2) ((1982) ), 125–128. |
[181] | Berger J. , Dorninger F. , Forss-Petter S. and Kunze M. , Peroxisomes in brain development and function, Biochim Biophys Acta 1863: (5) ((2016) ), 934–955. |
[182] | Unay B. , Kendirli T. and Ataç K. , Gül D. , Akin R. and Gökçay E. , Caudothalamic groove cysts in Zellweger syndrome, Clin Dysmorphol 14: (3) ((2005) ), 165–167. |
[183] | Barkovich A.J. and Peck W.W. , MR of Zellweger syndrome, AJNR Am J Neuroradiol 18: (6) ((1997) ), 1163–1170. |
[184] | Gilchrist K.W. , Gilbert E.F. , Goldfarb S. , Goll U. , Spranger J.W. and Opitz J.M. , Studies of malformation syndromes of man XIB: The cerebro-hepato-renal syndrome of Zellweger: Comparative pathology, Eur J Pediatr 121: (2) ((1976) ), 99–118. |
[185] | Powers J.M. , Moser H.W. , Moser A.B. , Upshur J.K. , Bradford B.F. and Pai S.G. et al., Fetal cerebrohepatorenal (Zellweger) syndrome: Dysmorphic, radiologic, biochemical, and pathologic findings in four affected fetuses, Hum Pathol 16: (6) ((1985) ), 610–620. |
[186] | Mochel F. , Grébille A.G. , Benachi A. , Martinovic J. , Razavi F. and Rabier D. et al., Contribution of fetal MR imaging in the prenatal diagnosis of Zellweger syndrome, AJNR Am J Neuroradiol 27: (2) ((2006) ), 333–336. |
[187] | Aubourg P. , Scotto J. , Rocchiccioli F. , Feldmann-Pautrat D. and Robain O. , Neonatal adrenoleukodystrophy, J Neurol Neurosurg Psychiatry 49: (1) ((1986) ), 77–86. |
[188] | Sevin C. , Ferdinandusse S. , Waterham H.R. , Wanders R.J. and Aubourg P. , Autosomal recessive cerebellar ataxia caused by mutations in the PEX2 gene, Orphanet J Rare Dis 6: ((2011) ), 8. |
[189] | Rush E.T. , Goodwin J.L. , Braverman N.E. and Rizzo W.B. , Low bone mineral density is a common feature of Zellweger spectrum disorders, Mol Genet Metab 117: (1) ((2016) ), 33–37. |
[190] | Poll-The B.T. , Gootjes J. , Duran M. , De Klerk J.B. , Wenniger-Prick L.J. and Admiraal R.J. , et al., Peroxisome biogenesis disorders with prolonged survival: Phenotypic expression in a cohort of 31 patients, Am J Med Genet A 126A: (4) ((2004) ), 333–338. |
[191] | Berendse K. , Engelen M. , Ferdinandusse S. , Majoie C.B. , Waterham H.R. and Vaz F.M. et al., Zellweger spectrum disorders: Clinical manifestations in patients surviving into adulthood, J Inherit Metab Dis 39: (1) ((2016) ), 93–106. |
[192] | Baes M. and Aubourg P. , Peroxisomes, myelination, and axonal integrity in the CNS, Neuroscientist 15: (4) ((2009) ), 367–379. |
[193] | Kulkarni K.S. , Baranano K.W. , Lin D.D. and Raymond G.V. , Contrast enhancement of brainstem tracts in Zellweger spectrum disorder: Evidence of inflammatory demyelination? Neuropediatrics 42: (1) ((2011) ), 32–34. |
[194] | Poll-The B.T. and Engelen M. , Peroxisomal leukoencephalopathy, Semin Neurol 32: (1) ((2012) ), 42–50. |
[195] | Barth P.G. , Majoie C.B. , Gootjes J. , Wanders R.J. , Waterham H.R. and van der Knaap M.S. , et al., Neuroimaging of peroxisome biogenesis disorders (Zellweger spectrum) with prolonged survival, Neurology 62: (3) ((2004) ), 439–444. |
[196] | Powers J.M. and Moser H.W. , Peroxisomal disorders: Genotype, phenotype, major neuropathologic lesions, and pathogenesis, Brain Pathol 8: (1) ((1998) ), 101–120. |
[197] | Torvik A. , Torp S. , Kase B.F. , Ek J. , Skjeldal O. and Stokke O. , Infantile Refsum’s disease: A generalized peroxisomal disorder, Case report with postmortem examination, J Neurol Sci 85: (1) ((1988) ), 39–53. |
[198] | Verma N.P. , Hart Z.H. and Nigro M. , Electrophysiologic studies in neonatal adrenoleukodystrophy, Electroencephalogr Clin Neurophysiol 60: (1) ((1985) ), 7–15. |
[199] | Ebberink M.S. , Csanyi B. , Chong W.K. , Denis S. , Sharp P. and Mooijer P.A. et al., Identification of an unusual variant peroxisome biogenesis disorder caused by mutations in the PEX16 gene, J Med Genet 47: (9) ((2010) ), 608–615. |
[200] | Lyons C.J. , Castano G. , McCormick A.Q. and Applegarth D. , Leopard spot retinal pigmentation in infancy indicating a peroxisomal disorder, Br J Ophthalmol 88: (2) ((2004) ), 191–192. |
[201] | Michelakakis H.M. , Zafeiriou D.I. , Moraitou M.S. , Gootjes J. and Wanders R.J. , PEX1 deficiency presenting as Leber congenital amaurosis, Pediatr Neurol 31: (2) ((2004) ), 146–149. |
[202] | Ek J. , Kase B.F. , Reith A. , Björkhem I. and Pedersen J.I. , Peroxisomal dysfunction in a boy with neurologic symptoms and amaurosis (Leber disease): Clinical and biochemical findings similar to those observed in Zellweger syndrome, J Pediatr 108: (1) ((1986) ), 19–24. |
[203] | Zaki M.S. , Heller R. , Thoenes M. , Nürnberg G. , Stern-Schneider G. and Nürnberg P. et al., PEX6 is Expressed in Photoreceptor Cilia and Mutated in Deafblindness with Enamel Dysplasia and Microcephaly, Hum Mutat 37: (2) ((2016) ), 170–174. |
[204] | Raas-Rothschild A. , Wanders R.J. , Mooijer P.A. , Gootjes J. , Waterham H.R. and Gutman A. et al., A PEX6-defective peroxisomal biogenesis disorder with severe phenotype in an infant, versus mild phenotype resembling Usher syndrome in the affected parents, Am J Hum Genet 70: (4) ((2002) ), 1062–1068. |
[205] | Majewski J. , Wang Z. , Lopez I. , Al v S. , Ren H. and Racine J. , et al., A new ocular phenotype associated with an unexpected but known systemic disorder and mutation: Novel use of genomic diagnostics and exome sequencing, J Med Genet 48: (9) ((2011) ), 593–596. |
[206] | Weleber R.G. , Tongue A.C. , Kennaway N.G. , Budden S.S. and Buist N.R. , Ophthalmic manifestations of infantile phytanic acid storage disease, Arch Ophthalmol 102: (9) ((1984) ), 1317–1321. |
[207] | Paker A.M. , Sunness J.S. , Brereton N.H. , Speedie L.J. , Albanna L. and Dharmaraj S. et al., Docosahexaenoic acid therapy in peroxisomal diseases: Results of a double-blind, randomized trial, Neurology 75: (9) ((2010) ), 826–830. |
[208] | Cohen S.M. , Green W.R. and de la Cruz Z.C. , Brown F.R. , Moser H.W. and Luckenbach M.W. , et al., Ocular histopathologic studies of neonatal and childhood adrenoleukodystrophy, Am J Ophthalmol 95: (1) ((1983) ), 82–96. |
[209] | Mignarri A. , Vinciguerra C. , Giorgio A. , Ferdinandusse S. , Waterham H. and Wanders R. et al., Zellweger Spectrum Disorder with Mild Phenotype Caused by PEX2 Gene Mutations, JIMD Rep 6: ((2012) ), 43–46. |
[210] | Régal L. , Ebberink M.S. , Goemans N. , Wanders R.J. , De Meirleir L. and Jaeken J. , et al., Mutations in PEX10 are a cause of autosomal recessive ataxia, Ann Neurol 68: (2) ((2010) ), 259–263. |
[211] | Steinberg S.J. , Snowden A. , Braverman N.E. , Chen L. , Watkins P.A. and Clayton P.T. et al., A PEX10 defect in a patient with no detectable defect in peroxisome assembly or metabolism in cultured fibroblasts, J Inherit Metab Dis 32: (1) ((2009) ), 109–119. |
[212] | Schabhüttl M. , Wieland T. , Senderek J. , Baets J. , Timmerman V. and De Jonghe P. , et al., Whole-exome sequencing in patients with inherited neuropathies: Outcome and challenges, J Neurol 261: (5) ((2014) ), 970–982. |
[213] | Ferdinandusse S. , Denis S. , Clayton P.T. , Graham A. , Rees J.E. and Allen J.T. et al., Mutations in the gene encoding peroxisomal alpha-methylacyl-CoA racemase cause adult-onset sensory motor neuropathy, Nat Genet 24: (2) ((2000) ), 188–191. |
[214] | Clarke C.E. , Alger S. , Preece M.A. , Burdon M.A. , Chavda S. and Denis S. et al., Tremor and deep white matter changes in alpha-methylacyl-CoA racemase deficiency, Neurology 63: (1) ((2004) ), 188–189. |
[215] | Thompson S.A. , Calvin J. , Hogg S. , Ferdinandusse S. , Wanders R.J. and Barker R.A. , Relapsing encephalopathy in a patient with α-methylacyl-CoA racemase deficiency, BMJ Case Rep 2009: , 2009. |
[216] | Lieber D.S. , Hershman S.G. , Slate N.G. , Calvo S.E. , Sims K.B. and Schmahmann J.D. et al., Next generation sequencing with copy number variant detection expands the phenotypic spectrum of HSD17B4-deficiency, BMC Med Genet 15: ((2014) ), 30. |
[217] | Lines M.A. , Jobling R. , Brady L. , Marshall C.R. , Scherer S.W. and Rodriguez A.R. et al., Peroxisomal D-bifunctional protein deficiency: Three adults diagnosed by whole-exome sequencing, Neurology 82: (11) ((2014) ), 963–968. |
[218] | Pierce S.B. , Walsh T. , Chisholm K.M. , Lee M.K. , Thornton A.M. and Fiumara A. et al., Mutations in the DBP-deficiency protein HSD17B4 cause ovarian dysgenesis, hearing loss, and ataxia of Perrault Syndrome, Am J Hum Genet 87: (2) ((2010) ), 282–288. |
[219] | McMillan H.J. , Worthylake T. , Schwartzentruber J. , Gottlieb C.C. , Lawrence S.E. and Mackenzie A. et al., Specific combination of compound heterozygous mutations in 17β-hydroxysteroid dehydrogenase type 4 (HSD17B4) defines a new subtype of D-bifunctional protein deficiency, Orphanet J Rare Dis 7: ((2012) ), 90. |
[220] | Ratbi I. , Falkenberg K.D. , Sommen M. , Al-Sheqaih N. , Guaoua S. and Vandeweyer G. et al., Heimler Syndrome Is Caused by Hypomorphic Mutations in the Peroxisome-Biogenesis Genes PEX1 and PEX6, Am J Hum Genet 97: (4) ((2015) ), 535–545. |
[221] | Moser A.B. , Rasmussen M. , Naidu S. , Watkins P.A. , McGuinness M. and Hajra A.K. et al., Phenotype of patients with peroxisomal disorders subdivided into sixteen complementation groups, J Pediatr 127: (1) ((1995) ), 13–22. |
[222] | Waterham H.R. and Ebberink M.S. , Genetics and molecular basis of human peroxisome biogenesis disorders, Biochim Biophys Acta 1822: (9) ((2012) ), 1430–1441. |
[223] | Ferdinandusse S. , Ebberink M.S. , Vaz F.M. , Waterham H.R. and Wanders R.J. , The important role of biochemical and functional studies in the diagnostics of peroxisomal disorders, J Inherit Metab Dis 2016. |
[224] | Mandel H. , Espeel M. , Roels F. , Sofer N. , Luder A. and Iancu T.C. et al., A new type of peroxisomal disorder with variable expression in liver and fibroblasts, J Pediatr 125: (4) ((1994) ), 549–555. |
[225] | Roels F. , Saudubray J.M. , Giros M. , Mandel H. , Eyskens F. and Saracibar N. et al., Peroxisome mosaics, Adv Exp Med Biol 544: ((2003) ), 97–106. |
[226] | Steinberg S.J. , Dodt G. , Raymond G.V. , Braverman N.E. , Moser A.B. and Moser H.W. , Peroxisome biogenesis disorders, Biochim Biophys Acta 1763: (12) ((2006) ), 1733–1748. |
[227] | Imamura A. , Tamura S. , Shimozawa N. , Suzuki Y. , Zhang Z. and Tsukamoto T. et al., Temperature-sensitive mutation in PEX1 moderates the phenotypes of peroxisome deficiency disorders, Hum Mol Genet 7: (13) ((1998) ), 2089–2094. |
[228] | Imamura A. , Tsukamoto T. , Shimozawa N. , Suzuki Y. , Zhang Z. and Imanaka T. et al., Temperature-sensitive phenotypes of peroxisome-assembly processes represent the milder forms of human peroxisome-biogenesis disorders, Am J Hum Genet 62: (6) ((1998) ), 1539–1543. |
[229] | Osumi T. , Imamura A. , Tsukamoto T. , Fujiwara C. , Hashiguchi N. and Shimozawa N. et al., Temperature sensitivity in peroxisome assembly processes characterizes milder forms of peroxisome biogenesis disorders., Cell Biochem Biophys 32: ((2000) ), Spring: 165–170. |
[230] | Hashimoto K. , Kato Z. , Nagase T. , Shimozawa N. , Kuwata K. and Omoya K. et al., Molecular mechanism of a temperature-sensitive phenotype in peroxisomal biogenesis disorder, Pediatr Res 58: (2) ((2005) ), 263–269. |
[231] | Waterham H.R. , Koster J. , van Roermund C.W. , Mooyer P.A. , Wanders R.J. and Leonard J.V. , A lethal defect of mitochondrial and peroxisomal fission, N Engl J Med 356: (17) ((2007) ), 1736–1741. |
[232] | Shamseldin H.E. , Alshammari M. , Al-Sheddi T. , Salih M.A. , Alkhalidi H. and Kentab A. et al., Genomic analysis of mitochondrial diseases in a consanguineous population reveals novel candidate disease genes, J Med Genet 49: (4) ((2012) ), 234–241. |
[233] | Niemann A. , Berger P. and Suter U. , Pathomechanisms of mutant proteins in Charcot-Marie-Tooth disease, Neuromolecular Med 8: (1-2) ((2006) ), 217–242. |
[234] | van den Brink D.M. , Brites P. , Haasjes J. , Wierzbicki A.S. , Mitchell J. and Lambert-Hamill M. , et al., Identification of PEX7 as the second gene involved in Refsum disease, Am J Hum Genet 72: (2) ((2003) ), 471–477. |
[235] | Otera H. , Nishimura M. , Setoguchi K. , Mori T. and Fujiki Y. , Biogenesis of nonspecific lipid transfer protein and sterol carrier protein x: Studies using peroxisome assembly-defective pex cell mutants, J Biol Chem 276: (4) ((2001) ), 2858–2864. |
[236] | Itzkovitz B. , Jiralerspong S. , Nimmo G. , Loscalzo M. , Horovitz D.D. and Snowden A. et al., Functional characterization of novel mutations in GNPAT and AGPS, causing rhizomelic chondrodysplasia punctata (RCDP) types 2 and 3, Hum Mutat 33: (1) ((2012) ), 189–197. |
[237] | Nimmo G. , Monsonego S. , Descartes M. , Franklin J. , Steinberg S. and Braverman N. , Rhizomelic chrondrodysplasia punctata type 2 resulting from paternal isodisomy of chromosome 1, Am J Med Genet A 152A: (7) ((2010) ), 1812–1817. |
[238] | Ofman R. , Lajmir S. and Wanders R.J. , Etherphospholipid biosynthesis and dihydroxyactetone-phosphate acyltransferase: Resolution of the genomic organization of the human gnpat gene and its use in the identification of novel mutations, Biochem Biophys Res Commun 281: (3) ((2001) ), 754–760. |
[239] | Thai T.P. , Rodemer C. , Jauch A. , Hunziker A. , Moser A. and Gorgas K. et al., Impaired membrane traffic in defective ether lipid biosynthesis, Hum Mol Genet 10: (2) ((2001) ), 127–136. |
[240] | Ofman R. , Hettema E.H. , Hogenhout E.M. , Caruso U. , Muijsers A.O. and Wanders R.J. , Acyl-CoA:Dihydroxyacetonephosphate acyltransferase: Cloning of the human cDNA and resolution of the molecular basis in rhizomelic chondrodysplasia punctata type 2, Hum Mol Genet 7: (5) ((1998) ), 847–853. |
[241] | de Vet E.C. , Ijlst L. , Oostheim W. , Wanders R.J. and van den Bosch H. , Alkyl-dihydroxyacetonephosphate synthase, Fate in peroxisome biogenesis disorders and identification of the point mutation underlying a single enzyme deficiency, J Biol Chem 273: (17) ((1998) ), 10296–10301. |
[242] | Thai T.P. , Heid H. , Rackwitz H.R. , Hunziker A. , Gorgas K. and Just W.W. , Ether lipid biosynthesis: Isolation and molecular characterization of human dihydroxyacetonephosphate acyltransferase, FEBS Lett 420: (2-3) ((1997) ), 205–211. |
[243] | Elias E.R. , Mobassaleh M. , Hajra A.K. and Moser A.B. , Developmental delay and growth failure caused by a peroxisomal disorder, dihydroxyacetonephosphate acyltransferase (DHAP-AT) deficiency, Am J Med Genet 80: (3) ((1998) ), 223–226. |
[244] | Barøy T. , Koster J. , P. Strømme, Ebberink M.S. , Misceo D. and Ferdinandusse S. , et al., A novel type of rhizomelic chondrodysplasia punctata, RCDP5, is caused by loss of the PEX5 long isoform, Hum Mol Genet 24: (20) ((2015) ), 5845–5854. |
[245] | Buchert R. , Tawamie H. , Smith C. , Uebe S. , Innes A.M. and Al Hallak B. , et al., A peroxisomal disorder of severe intellectual disability, epilepsy, and cataracts due to fatty acyl-CoA reductase 1 deficiency, Am J Hum Genet 95: (5) ((2014) ), 602–610. |
[246] | Spranger J.W. , Opitz J.M. and Bidder U. , Heterogeneity of Chondrodysplasia punctata, Humangenetik 11: (3) ((1971) ), 190–212. |
[247] | Gilbert E.F. , Opitz J.M. , Spranger J.W. , Langer L.O. , Wolfson J.J. and Viseskul C. , Chondrodysplasia punctata–rhizomelic form, Pathologic and radiologic studies of three infants, Eur J Pediatr 123: (2) ((1976) ), 89–109. |
[248] | Briggs J.N. , Emery J.L. and Illingworth R.S. , Congenital stippled epiphyses, Arch Dis Child 28: (139) ((1953) ), 209–212. |
[249] | White A.L. , Modaff P. , Holland-Morris F. and Pauli R.M. , Natural history of rhizomelic chondrodysplasia punctata, Am J Med Genet A 118A: (4) ((2003) ), 332–342. |
[250] | Huffnagel I.C. , Clur S.A. , Bams-Mengerink A.M. , Blom N.A. , Wanders R.J. and Waterham H.R. et al., Rhizomelic chondrodysplasia punctata and cardiac pathology, J Med Genet 50: (7) ((2013) ), 419–424. |
[251] | Duker A.L. , Eldridge G. , Braverman N.E. and Bober M.B. , Congenital heart defects common in rhizomelic chondrodysplasia punctata, Am J Med Genet A 170A: (1) ((2016) ), 270–272. |
[252] | Oswald G. , Lawson C. , Raymond G. , Golden W.C. and Braverman N. , Rhizomelic chondrodysplasia punctata type 1 and fulminant neonatal respiratory failure, a case report and discussion of pathophysiology, Am J Med Genet A 155A: (12) ((2011) ), 3160–3163. |
[253] | Khanna A.J. , Braverman N.E. , Valle D. and Sponseller P.D. , Cervical stenosis secondary to rhizomelic chondrodysplasia punctata, Am J Med Genet 99: (1) ((2001) ), 63–66. |
[254] | Karabayir N. , Keskindemirci G. , Adal E. and Korkmaz O. , A case of rhizomelic chondrodysplasia punctata in newborn, Case Rep Med 2014: ((2014) ), 879679;. |
[255] | Alkan A. , Kutlu R. , Yakinci C. , Sigirci A. , Aslan M. and Sarac K. , Delayed myelination in a rhizomelic chondrodysplasia punctata case: MR spectroscopy findings, Magn Reson Imaging 21: (1) ((2003) ), 77–80. |
[256] | Sztriha L. , Al-Gazali L.I. , Wanders R.J. , Ofman R. , Nork M. and Lestringant G.G. , Abnormal myelin formation in rhizomelic chondrodysplasia punctata type 2 (DHAPAT-deficiency), Dev Med Child Neurol 42: (7) ((2000) ), 492–495. |
[257] | Viola A. , Confort-Gouny S. , Ranjeva J.P. , Chabrol B. , Raybaud C. and Vintila F. et al., MR imaging and MR spectroscopy in rhizomelic chondrodysplasia punctata, AJNR Am J Neuroradiol 23: (3) ((2002) ), 480–483. |
[258] | Powers J.M. , Kenjarski T.P. , Moser A.B. and Moser H.W. , Cerebellar atrophy in chronic rhizomelic chondrodysplasia punctata: A potential role for phytanic acid and calcium in the death of its Purkinje cells, Acta Neuropathol 98: (2) ((1999) ), 129–134. |
[259] | Hebestreit H. , Wanders R.J. , Schutgens R.B. , Espeel M. , Kerckaert I. and Roels F. et al., Isolated dihydroxyacetonephosphate-acyl-transferase deficiency in rhizomelic chondrodysplasia punctata: Clinical presentation, metabolic and histological findings, Eur J Pediatr 155(12): ((1996) ), 1035–1039. |
[260] | Poulos A. , Sheffield L. , Sharp P. , Sherwood G. , Johnson D. and Beckman K. et al., Rhizomelic chondrodysplasia punctata: Clinical, pathologic, and biochemical findings in two patients, J Pediatr 113(4): ((1988) ), 685–690. |
[261] | Braverman N.E. , Raymond G.V. , Rizzo W.B. , Moser A.B. , Wilkinson M.E. and Stone E.M. et al., Peroxisome biogenesis disorders in the Zellweger spectrum: An overview of current diagnosis, clinical manifestations, and treatment guidelines, Mol Genet Metab 117: (3) ((2016) ), 313–321. |
[262] | Ferdinandusse S. , Kostopoulos P. , Denis S. , Rusch H. , Overmars H. and Dillmann U. et al., Mutations in the gene encoding peroxisomal sterol carrier protein X (SCPx) cause leukencephalopathy with dystonia and motor neuropathy, Am J Hum Genet 78: (6) ((2006) ), 1046–1052. |
[263] | Dick D. , Horvath R. and Chinnery P.F. , AMACR mutations cause late-onset autosomal recessive cerebellar ataxia, Neurology 76: (20) ((2011) ), 1768–1770. |
[264] | Ebberink M.S. , Mooijer P.A. , Gootjes J. , Koster J. , Wanders R.J. and Waterham H.R. , Genetic classification and mutational of more than 600 patients with a Zellweger syndrome spectrum disorder, Hum Mutat 32: (1) ((2011) ), 59–69. |
[265] | Yik W.Y. , Steinberg S.J. , Moser A.B. , Moser H.W. and Hacia J.G. , Identification of novel mutations and sequence variation in the Zellweger syndrome spectrum of peroxisome biogenesis disorders, Hum Mutat 30: (3) ((2009) ), E467–E480. |
[266] | Preuss N. , Brosius U. , Biermanns M. , Muntau A.C. , Conzelmann E. and Gartner J. , PEX1 mutations in complementation group 1 of Zellweger spectrum patients correlate with severity of disease, Pediatr Res 51: (6) ((2002) ), 706–714. |
[267] | Gärtner J. , Preuss N. , Brosius U. and Biermanns M. , Mutations in PEX1 in peroxisome biogenesis disorders: G843D and a mild clinical phenotype, J Inherit Metab Dis 22: (3) ((1999) ), 311–313. |
[268] | Walter C. , Gootjes J. , Mooijer P.A. , Portsteffen H. , Klein C. and Waterham H.R. et al., Disorders of peroxisome biogenesis due to mutations in PEX1: Phenotypes and PEX1 protein levels, Am J Hum Genet 69: (1) ((2001) ), 35–48. |
[269] | Maxwell M.A. , Nelson P.V. , Chin S.J. , Paton B.C. , Carey W.F. and Crane D.I. , A common PEX1 frameshift mutation in patients with disorders of peroxisome biogenesis correlates with the severe Zellweger syndrome phenotype, Hum Genet 105: (1-2) ((1999) ), 38–44. |
[270] | Theda C. , Gibbons K. , Defor T.E. , Donohue P.K. , Golden W.C. and Kline A.D. et al., Newborn screening for X-linked adrenoleukodystrophy: Further evidence high throughput screening is feasible, Mol Genet Metab 111: (1) ((2014) ), 55–57. |
[271] | Hubbard W.C. , Moser A.B. , Liu A.C. , Jones R.O. , Steinberg S.J. and Lorey F. et al., Newborn screening for X-linked adrenoleukodystrophy (X-ALD): Validation of a combined liquid chromatography-tandem mass spectrometric (LC-MS/MS) method, Mol Genet Metab 97: (3) ((2009) ), 212–220. |
[272] | Moser H.W. , Mahmood A. and Raymond G.V. , X-linked adrenoleukodystrophy, Nat Clin Pract Neurol 3(3): ((2007) ), 140–151. |
[273] | Children ACoHDiNa. Recommended Uniform Screening Panel http://www.hrsa.gov/advisorycommittees/mchbadvisory/heritabledisorders/recommendedpanel/: U.S. Department of Health and Social Services 2016 [Available from: http://www.hrsa.gov/advisorycommittees/mchbadvisory/heritabledisorders/recommendedpanel/. |
[274] | Raymond G.V. , Jones R.O. and Moser A.B. , Newborn screening for adrenoleukodystrophy: Implications for therapy, Mol Diagn Ther 11: (6) ((2007) ), 381–384. |
[275] | Klouwer F.C. , Berendse K. , Ferdinandusse S. , Wanders R.J. , Engelen M. and Poll-The B.T. , Zellweger spectrum disorders: Clinical overview and management approach, Orphanet J Rare Dis 10: ((2015) ), 151. |
[276] | Das A.K. , Holmes R.D. , Wilson G.N. and Hajra A.K. , Dietary ether lipid incorporation into tissue plasmalogens of humans and rodents, Lipids 27: (6) ((1992) ), 401–405. |
[277] | Setchell K.D. , Bragetti P. , Zimmer-Nechemias L. , Daugherty C. , Pelli M.A. and Vaccaro R. et al., Oral bile acid treatment and the patient with Zellweger syndrome, Hepatology 15: (2) ((1992) ), 198–207. |
[278] | Maeda K. , Kimura A. , Yamato Y. , Nittono H. , Takei H. and Sato T. et al., Oral bile Acid treatment in two Japanese patients with Zellweger syndrome, J Pediatr Gastroenterol Nutr 35: (2) ((2002) ), 227–230. |
[279] | Setchell K.D. , Heubi J.E. , Bove K.E. , O’Connell N.C. , Brewsaugh T. and Steinberg S.J. et al., Liver disease caused by failure to racemize trihydroxycholestanoic acid: Gene mutation and effect of bile acid therapy, Gastroenterology 124: (1) ((2003) ), 217–232. |
[280] | Smith E.H. , Gavrilov D.K. , Oglesbee D. , Freeman W.D. , Vavra M.W. and Matern D. et al., An adult onset case of alpha-methyl-acyl-CoA racemase deficiency, J Inherit Metab Dis 33: (Suppl 3) ((2010) ), S349–S53. |
[281] | Noguer M.T. and Martinez M. , Visual follow-up in peroxisomal-disorder patients treated with docosahexaenoic Acid ethyl ester, Invest Ophthalmol Vis Sci 51: (4) ((2010) ), 2277–2285. |
[282] | Baldwin E.J. , Gibberd F.B. , Harley C. , Sidey M.C. , Feher M.D. and Wierzbicki A.S. , The effectiveness of long-term dietary therapy in the treatment of adult Refsum disease, J Neurol Neurosurg Psychiatry 81: (9) ((2010) ), 954–957. |
[283] | Sokal E.M. , Smets F. , Bourgois A. , Van Maldergem L. , Buts J.P. and Reding R. , et al., Hepatocyte transplantation in a 4-year-old girl with peroxisomal biogenesis disease: Technique, safety, and metabolic follow-up, Transplantation 76: (4) ((2003) ), 735–738. |
[284] | Van Maldergem L. , Moser A.B. , Vincent M.F. , Roland D. , Reding R. and Otte J.B. , et al., Orthotopic liver transplantation from a living-related donor in an infant with a peroxisome biogenesis defect of the infantile Refsum disease type, J Inherit Metab Dis 28: (4) ((2005) ), 593–600. |
[285] | Matsunami M. , Shimozawa N. , Fukuda A. , Kumagai T. , Kubota M. and Chong P.F. et al., Living-Donor Liver Transplantation From a Heterozygous Parent for Infantile Refsum Disease, Pediatrics (2016) . |
[286] | Baes M. and Van Veldhoven P.P. , Generalised and conditional inactivation of Pex genes in mice, Biochim Biophys Acta 1763: (12) ((2006) ), 1785–1793. |
[287] | Faust P.L. , Abnormal cerebellar histogenesis in PEX2 Zellweger mice reflects multiple neuronal defects induced by peroxisome deficiency, J Comp Neurol 461: (3) ((2003) ), 394–413. |
[288] | Li X. , Baumgart E. , Morrell J.C. , Jimenez-Sanchez G. , Valle D. and Gould S.J. , PEX11 beta deficiency is lethal and impairs neuronal migration but does not abrogate peroxisome function, Mol Cell Biol 22: (12) ((2002) ), 4358–4365. |
[289] | Li X. and Gould S.J. , PEX11 promotes peroxisome division independently of peroxisome metabolism, J Cell Biol 156: (4) ((2002) ), 643–651. |
[290] | Krysko O. , Hulshagen L. , Janssen A. , Schütz G. , Klein R. and De Bruycker M. , et al., Neocortical and cerebellar developmental abnormalities in conditions of selective elimination of peroxisomes from brain or from liver, J Neurosci Res 85: (1) ((2007) ), 58–72. |
[291] | Hulshagen L. , Krysko O. , Bottelbergs A. , Huyghe S. , Klein R. and Van Veldhoven P.P. , et al., Absence of functional peroxisomes from mouse CNS causes dysmyelination and axon degeneration, J Neurosci 28: (15) ((2008) ), 4015–4027. |
[292] | Kassmann C.M. , Lappe-Siefke C. , Baes M. , Brügger B. , Mildner A. and Werner H.B. et al., Axonal loss and neuroinflammation caused by peroxisome-deficient oligodendrocytes, Nat Genet 39: (8) ((2007) ), 969–976. |
[293] | Dirkx R. , Vanhorebeek I. , Martens K. , Schad A. , Grabenbauer M. and Fahimi D. et al., Absence of peroxisomes in mouse hepatocytes causes mitochondrial and ER abnormalities, Hepatology 41: (4) ((2005) ), 868–878. |
[294] | Peeters A. , Fraisl P. , van den Berg S. , Ver Loren van Themaat E. , Van Kampen A. and Rider M.H. , et al., Carbohydrate metabolism is perturbed in peroxisome-deficient hepatocytes due to mitochondrial dysfunction, AMP-activated protein kinase (AMPK) activation, and peroxisome proliferator-activated receptor γ coactivator 1α (PGC-1α) suppression, J Biol Chem 286: (49) ((2011) ), 42162–42179. |
[295] | Selkälä E.M. , Kuusisto S.M. , Salonurmi T. , Savolainen M.J. , Jauhiainen M. and Pirilä P.L. et al., Metabolic adaptation allows Amacr-deficient mice to remain symptom-free despite low levels of mature bile acids, Biochim Biophys Acta 1831: (8) ((2013) ), 1335–1343. |
[296] | Hiebler S. , Masuda T. , Hacia J.G. , Moser A.B. , Faust P.L. and Liu A. et al., The Pex1-G844D mouse: A model for mild human Zellweger spectrum disorder, Mol Genet Metab 111: (4) ((2014) ), 522–532. |
[297] | Brites P. , Motley A.M. , Gressens P. , Mooyer P.A. , Ploegaert I. and Everts V. et al., Impaired neuronal migration and endochondral ossification in Pex7 knockout mice: A model for rhizomelic chondrodysplasia punctata, Hum Mol Genet 12: (18) ((2003) ), 2255–2267. |
[298] | Teigler A. , Komljenovic D. , Draguhn A. , Gorgas K. and Just W.W. , Defects in myelination, paranode organization and Purkinje cell innervation in the ether lipid-deficient mouse cerebellum, Hum Mol Genet 18: (11) ((2009) ), 1897–1908. |
[299] | Braverman N. , Zhang R. , Chen L. , Nimmo G. , Scheper S. , Tran T. et al., A Pex7 hypomorphic mouse model for plasmalogen deficiency affecting the lens and skeleton, Mol Genet Metab 99: (4) ((2010) ), 408–416. |
[300] | Liegel R. , Chang B. , Dubielzig R. and Sidjanin D.J. , Blind sterile 2 (bs2), a hypomorphic mutation in Agps, results in cataracts and male sterility in mice, Mol Genet Metab 103: (1) ((2011) ), 51–59. |
[301] | Gorgas K. , Teigler A. , Komljenovic D. and Just W.W. , The ether lipid-deficient mouse: Tracking down plasmalogen functions, Biochim Biophys Acta 1763: (12) ((2006) ), 1511–1526. |
[302] | Janssen A. , Baes M. , Gressens P. , Mannaerts G.P. , Declercq P. and Van Veldhoven P.P. , Docosahexaenoic acid deficit is not a major pathogenic factor in peroxisome-deficient mice, Lab Invest 80: (1) ((2000) ), 31–35. |
[303] | Keane M.H. , Overmars H. , Wikander T.M. , Ferdinandusse S. , Duran M. and Wanders R.J. et al., Bile acid treatment alters hepatic disease and bile acid transport in peroxisome-deficient PEX2 Zellweger mice, Hepatology 45: (4) ((2007) ), 982–997. |
[304] | Wood P.L. , Khan M.A. , Smith T. , Ehrmantraut G. , Jin W. and Cui W. , et al., In vitroand in vivo plasmalogen replacement evaluations in rhizomelic chrondrodysplasia punctata and Pelizaeus-Merzbacher disease using PPI-an ether lipid plasmalogen precursor, Lipids Health Dis 10: ((2011) ), 182. |
[305] | Wei H. , Kemp S. , McGuinness M.C. , Moser A.B. and Smith K.D. , Pharmacological induction of peroxisomes in peroxisome biogenesis disorders, Ann Neurol 47: (3) ((2000) ), 286–296. |
[306] | Li X. , Baumgart E. , Dong G.X. , Morrell J.C. , Jimenez-Sanchez G. and Valle D. et al., PEX11alpha is required for peroxisome proliferation in response to 4-phenylbutyrate but is dispensable for peroxisome proliferator-activated receptor alpha-mediated peroxisome proliferation, Mol Cell Biol 22: (23) ((2002) ), 8226–8240. |
[307] | Dranchak P.K. , Di Pietro E. , Snowden A. , Oesch N. , Braverman N.E. and Steinberg S.J. , et al., Nonsense suppressor therapies rescue peroxisome lipid metabolism and assembly in cells from patients with specific PEX gene mutations, J Cell Biochem 112: (5) ((2011) ), 1250–1258. |
[308] | Zhang R. , Chen L. , Jiralerspong S. , Snowden A. , Steinberg S. and Braverman N. , Recovery of PEX1-Gly843Asp peroxisome dysfunction by small-molecule compounds, Proc Natl Acad Sci U S A 107: (12) ((2010) ), 5569–5574. |
[309] | Berendse K. , Ebberink M.S. , Ijlst L. , Poll-The B.T. , Wanders R.J. and Waterham H.R. , Arginine improves peroxisome functioning in cells from patients with a mild peroxisome biogenesis disorder, Orphanet J Rare Dis 8: ((2013) ), 138. |
[310] | Lu H. , Chang D.J. , Baratte B. , Meijer L. and Schulze-Gahmen U. , Crystal structure of a human cyclin-dependent kinase 6 complex with a flavonol inhibitor, fisetin, J Med Chem 48: (3) ((2005) ), 737–743. |
[311] | Katayama K. , Masuyama K. , Yoshioka S. , Hasegawa H. , Mitsuhashi J. and Sugimoto Y. , Flavonoids inhibit breast cancer resistance protein-mediated drug resistance: Transporter specificity and structure-activity relationship, Cancer Chemother Pharmacol 60: (6) ((2007) ), 789–797. |
Figures and Tables
Fig.1
Peroxisome matrix protein import. (1) PTS receptor binding. PEX5 and PEX7 cytosolic receptors bind their cognate ligands (PTS1 and PTS2 enzymes, respectively) in the cytosol. PEX5 has two isoforms that differ by alternative splicing. The longer isoform, PEX5L, binds both PTS1 enzymes and PEX7 and delivers them to the peroxisome membrane. (2) Docking. The receptor-ligand complex docks at the peroxisome membrane by binding PEX13 and PEX14. (3) Matrix enzyme translocation. PEX5, together with PEX14, forms a dynamic membrane pore through which the ligands are transported into the peroxisome matrix. (4) Receptor recycling. PEX2, PEX10, and PEX12 mono-ubiquitinate PEX5, allowing its removal from the membrane. The PEX1-PEX6 AAA-ATPase heterohexamer (anchored to the peroxisome membrane by PEX26) uses the energy from ATP hydrolysis to remove PEX5-Ub from the peroxisome membrane for another round of import. PEX7 is recycled to the cytosol after PEX5 in an ATP independent manner. Note that defects in PEX7 prevent import of PTS2 enzymes, but do not disrupt the PEX5/PTS1 import pathway.
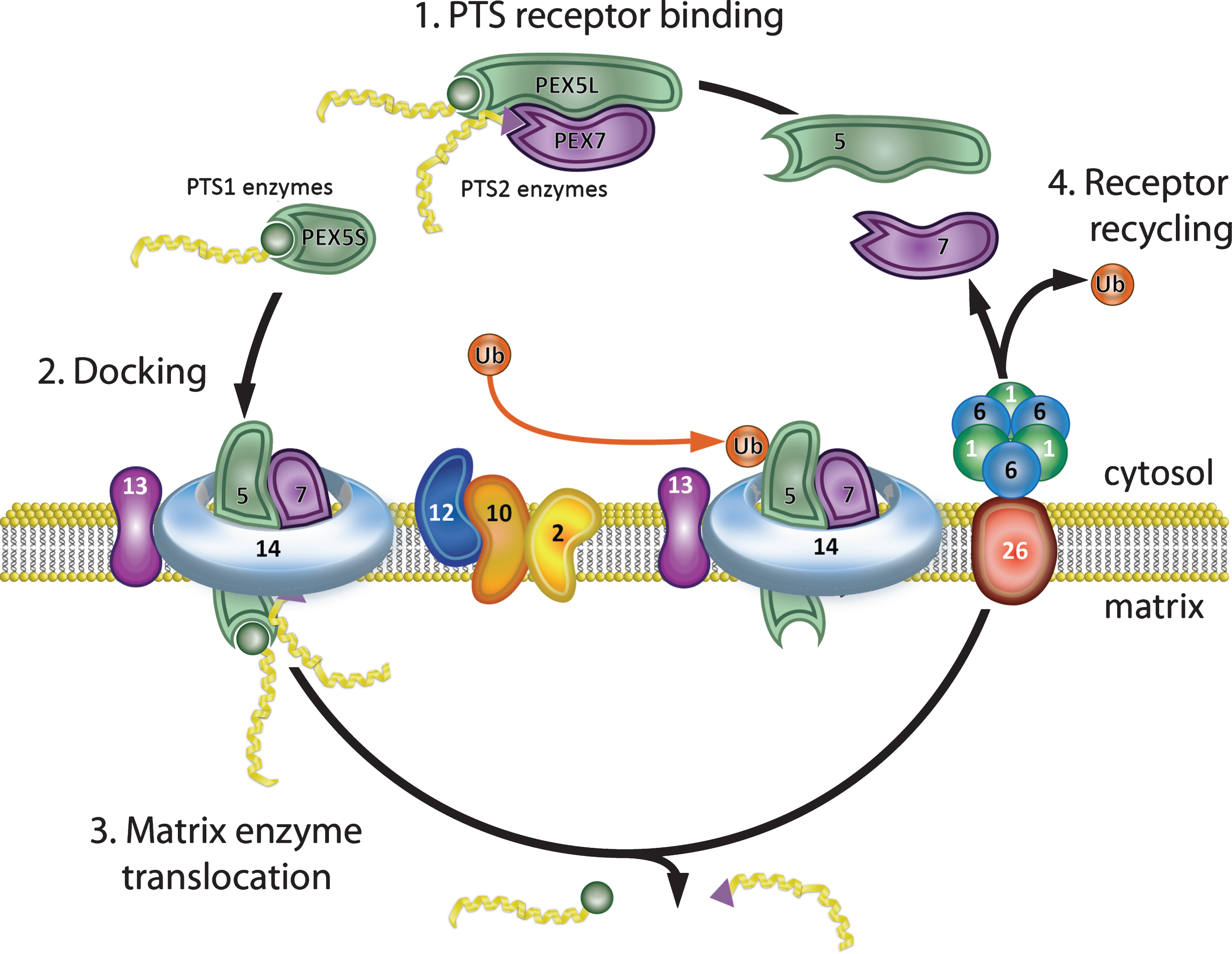
Table 1
Functions of peroxisomal enzymes
| Biochemical pathways | Enzymes1 | Features | Relevance to disease |
| β-oxidation of Straight, very long chain (≥C22) fatty acids | ACOX1, DBP, ACAA1, [SCPX] | Chain shortening of very long chain fatty acids (VLCFA), final step in the synthesis of docosohexanoic acid (DHA) | Tissue accumulation of VLCFA cause brain, nerve and adrenal damage. Deficiency of DHA affects brain function and vision |
| β-oxidation of Methyl-branched chain fatty acid (pristanic) and C27 bile acid precursors, di- and trihydroxycholestanoic acids (D/THCA) | ACOX2, DBP, SCPX | Chain shorteninig of pristanic acid and D/THCA utlizes a different oxidase than that used in straight chain shortening | Accumulation of pristanic acid affects brain and nerve. Increased D/THCA cause liver toxicity |
| β-oxidation of Dicarboxylic fatty acids | ACOX1, LBP, [DBP] ACAA1, [SCPX] | Chain shortening of dicaboxylic acids utilizes L-bifunctional protein (LBP) in contrast to DBP used in straight chain shortening | Unknown |
| α-oxidation of Methyl-branched chain fatty acid (phytanic) | PHYH | Degradation of methyl-branched phytanic acid requires an additional α- oxidation before entering the β-oxidation pathway as pristanic acid | Tissue accumulation of phytanic acid causes retinal degeneration, cerebellar ataxia, peripheral neuropathy |
| Racemization of Pristanic and D/THCA acids from (R) to (S) enantiomers | AMACR | β-oxidation requires the (S) enantiomers of pristanoyl-CoA and C27-bile acyl-CoAs | Causes tissue accumulation of (R) forms of pristanic acid and D/THCA (and secondary elevation of phytanic acid) |
| Biosynthesis of Ether phospholipids (plasmalogen) | GNPAT, AGPS, FAR1, [FAR2] | Initial steps of plasmalogen synthesis occur in the peroxisome | Deficiency causes skeletal dysplasia, cataracts, profound growth and intellectual deficiency |
| Detoxification of Glyoxylate | AGXT | Prevents the conversion of glyoxylate into the toxic metabolite, oxalate | Accumulation of oxalate results in calcium oxalate renal stones |
| Detoxification of Hydrogen Peroxide | CAT | Required for catabolism of hydrogen peroxide, produced as a by-product of oxidase enzymes | Increased oxidant damage |
| Amino acid oxidation L-Pipecolic acid and D-amino acids | PIPOX, DAO | Regulation of lysine degradation through L-pipecolic acid, and D-serine and glycine levels | Pipecolic acid levels are used as a biomarker for peroxisome dysfunction, but the relationship to disease processes is unknown |
1Redundant enzymes are in brackets []. Full enzyme names: ACOX1, acyl-CoA oxidase 1; ACOX2, acyl-CoA oxidase 2;DBP, D-bifunctional protein; LBP, L-bifunctional protein; ACAA1, acetyl-CoA acyltransferase 1 (peroxisomal thiolase); SCPX, sterol carrier protein-peroxisomal thiolase; PHYH, phytanyl-CoA hydroxylase; AMACR, alpha-methylacyl-CoA racemase; GNPAT, glyceronephosphate O-acyltransferase or dihydroxyacetonephosphate acyltransferase); AGPS, alkylglycerone phosphate synthase or alkyl-dihydroxy-acetonephosphate synthase); FAR1, fatty acyl-CoA reductase; FAR2, fatty acyl-CoA reductase 2; AGXT, alanine-glyoxylate aminotransferase; CAT, catalase; PIPOX, L-pipecolic acid oxidase; DAO, D-amino acid oxidase.
Table 2
Clinical features of ZSD by severity and age of onset
| Features | Neonate | 1-6 mos | 6 mos-4 yrs | >4 yrs |
| Neuronal migration defects | S | |||
| Chondrodysplasia punctata | S | |||
| Renal cortical cysts | S | |||
| Respiratory compromise | S | |||
| Craniofacial dysmorphism | S, I, M | I, M | I, M | |
| Jaundice | S, I, M | I | ||
| Hepatomegaly, liver dysfunction, cholestasis | S | I, M | I, M | I, M |
| Hypotonia, failure to thrive | S, I | I, M | I, M | M |
| Sensorineural hearing loss | S | I, M | I, M | M |
| Cataracts | S | I, M | I, M | M |
| Retinal degeneration | S | I, M | I, M | M |
| Psychomotor retardation | S | I, M | I, M | M |
| Seizures | S | I | I | M |
| Leukodystrophy | I | I | M | |
| Adrenal insufficiency | I, M | M | ||
| Osteopenia | I | I, M | ||
| Calcium oxalate renal stones | I, M | I, M | ||
| Peripheral neuropathy | M | M | ||
| Cerebellar ataxia, atrophy | M | M | ||
| Ameliogenesis Imperfecta | I, M | I, M |
Abbreviation for clinical severity classes: S, severe; I, intermediate; M, mild.
Table 3
Clinical features of ZSD by severity and age of onset
| PEX gene | Distinguishing phenotype in group | Peroxisome biochemistry | Molecular Findings | |||||
| # patients (sibs/families) | Plasma/erythrocytes | Fibroblasts | ||||||
| Vision and hearing loss | ||||||||
| PEX1 | Cognition | Normal | VLCFA | N↑ | VLCFA ox. | N | 1. c.[2528G>A];[2528G>A] (p.[G843D];[G843D])205 | |
| 7 (4 sibs/2 families) | Development | Normal | Pristanic a. | N | Pristanic a. ox. | N | ||
| CNS | Leber congenital amaurosis, retinitis pigmentosa | D/THCA | N | Phytanic acid ox. | N↓ | 2. c.[3750G>A];[3750G>A] (p.[W1250*];[W1250*])220 | ||
| PNS | SNHL | Phytanic a. | N | GNPAT activity | N | |||
| Other | Enamel anomalies, Heimler syndrome | Plasmalogens | N | Soluble catalase | ↑ | 3. c.[2114T>G];[2097dup] (p.[L705W]; [I700Yfs*42]220 | ||
| Imaging | Arnold-Chiari malformation, multicystic hydro-syringomyelia | Pipecolic a. | ↑ | Immunofluorescence: Peroxisome mosaicism | 4. c.[1742G>C];[1239+1G>T] (p.[R581P];[0])220 | |||
| 5. c.[2097dup];[1742G>C] (p.[I700Yfs*42];[R581P])220 | ||||||||
| PEX6 | Cognition | Normal/borderline | VLCFA | N↑ | VLCFA ox. | N↓ | 1. c.[1715C>T];[1715C>T] (p.[T572I];[T572I))204 | |
| 6 (4 sibs/2 families) | Development | Normal | Pristanic a. | N↑ | Pristanic a. ox. | N↓ | ||
| CNS | Retinitis pigmentosa | D/THCA | N | GNPAT activity | N↓ | |||
| PNS | SNHL | Phytanic a. | N↑ | Phytanic a. ox. | N↓ | 2. c.[2094+2T>C];[2426C>T; 2534T>C] (p.[A809V;I845T];[0])204 | ||
| Other | Enamel anomalies, Heimler | Plasmalogens | N | Immunofluorescence: Peroxisome mosaicism | 3. c.[821C>T];[1930C>T] (p.[P274L]; [R644W])220 | |||
| syndrome/cholestatic liver disease | Pipecolic a. | N | ||||||
| Imaging | N/A | 4. c.[1802G>A];[c.1841del] (p.[R601Q];[L614Rfs*5])220 | ||||||
| HSD17B4 (DBP) | Cognition | Normal/mild delay/cognitive decline | VLCFA | N | VLCFA ox. | N | 1. c.[715-1207del];[1628G>C] (p.[239_403del];[A196V])216 | |
| 8 (7 sibs/3 families) | Development | Mild delay, learning difficulties | Pristanic | N | Pristanic a. ox. | N↓ | 2. c.[1537C>A];[c.587C>T] (p.[P513T];[R543P])217 | |
| CNS | Retinitis pigmentosa, cerebellar dysfunction, spastic paraparesis | D/THCA | N↑ | Phytanic a. ox. | N↓ | 3. c.[650A>G];[1704T>A] (p.[Y217C];[Y568stop])218 | ||
| PNS | SNHL, axonal/demyelinating sensory-motor neuropathy | Phytanic a. | N↑ | DBP hydratase / dehydrogenase activity | ↓ | 4. c.[101C>T];[1547T>C] (p.[A34V];[I516T])219 | ||
| Other | Perrault syndrome, short stature | Immunofluorescence: Normal morphology/slightly enlarged peroxisomes | ||||||
| Imaging | Progressive cerebellar atrophy, white matter changes | |||||||
| Pure cerebellar dysfunction | ||||||||
| PEX2 | Cognition | Normal/borderline | VLCFA | N | VLCFA ox. | N | 1. c.[865_866insA];[865_866insA] (p.[S289Kfs*36]; [S289Kfs*36])188 | |
| 2 (sibs) | Development | Normal | Pristanic a. | ↑ | Pristanic a. ox. | N | ||
| CNS | Cerebellar dysfunction | D/THCA | ↑ | GNPAT activity | N | |||
| PNS | Normal | Phytanic a. | ↑ | Phytanic a. ox. | ↓ | |||
| Other | Normal review of systems | Pipecolic a. | N | Plasmalogen synth. | N | |||
| Imaging | Cerebellar atrophy | |||||||
| Cerebellar dysfunction “plus” | ||||||||
| PEX2 | Cognition | Normal | VLCFA | ↑ | VLCFAs ox. | N | 1. c.[355C>T];[c.865_866insA] (p.[R119*];[S289Kfs*36])209 | |
| 1 | Development | Normal | Pristanic a. | ↑ | Pristanic a. ox. | N | ||
| CNS | Cerebellar dysfunction | D/THCA | ↑ | GNPAT activity | N | |||
| PNS | Axonal sensory-motor neuropathy | Phytanic a. | ↑ | Phytanic a. ox. | ↓ | |||
| Other | Strabismus, deafness | Plasmalogens | N | Immunofluorescence: Peroxisome mosaicism | ||||
| Imaging | Cerebellar and supratentorial atrophy, decreased NAA/Cr | |||||||
| PEX10 | Cognition | Normal | VLCFA | N↑ | VLCFA ox. | N↓ | 1. c.[992G>A];[764_765insA] (p.[R331E];[L236Afs*102])210 | |
| 3 | Development | Normal | Pristanic a. | ↑ | Pristanic a. ox. | ↓ | ||
| CNS | Cerebellar and posterior column dysfunction | Pristanic a. | ↑ | Pristanic a. ox. | N↓ | 2. c.[2T>C];[790C>T] (p.[0];[R264*])210 | ||
| D/THCA | ↑ | GNPAT activity | N | 3. c.[337delC];[890T>C] (p.[L113fs*39];[L297P)]211 | ||||
| PNS | Axonal motor neuropathy | Phytanic a. | ↑ | Phytanic a. ox. | N↓ | |||
| Other | Neonatal jaundice,↑LFT,↑CK, ↓cholesterol, delayed menarche | Plasmalogens | N↓ | Immunofluorescence: Peroxisome mosaicism | ||||
| Imaging | Cerebellar atrophy, leukodystrophy | Pipecolic a. | ↑ | |||||
| PEX16 | Cognition | Normal | VLCFA | ↑ | VLCFA ox. | ↓ | 1. c[984delG];[984delG] (p.[I330Sfs*27]; [I330Sfs*27])199 | |
| 6 (2 sibs) | Development | Normal, mild delay with catch-up | Pristanic a. | ↑ | Pristanic a. ox. | N | ||
| CNS | Optic atrophy, impaired visual pathways, cerebellar and bulbar dysfunction, spastic paraparesis | D/THCA Phytanic a. | ↑ ↑ | GNPAT activity Phytanic a. ox. | N N | 2. c.[753_755delTGT]; [753_755delTGT] (p.[V252del];[V252del])199 | ||
| Plasmalogens | N | Immunofluorescence: Enlarged peroxisomes, reduced in number | 3. c.[865C>A];[865C>A] (p.[P289T];[P289T])199 | |||||
| PNS | Demyelinating sensory-motor neuropathy | |||||||
| Other | Cataract | 4. c.[992A>G];[992A>G] (p.[Y331C];[Y331C])199 | ||||||
| Imaging | Cerebellar and corpus callosum atrophy, leukodystrophy | 5. c.[952+118_1011+80]; [952+118_1011+80] (p.[R318Sfs*138, R318Ifs*38, E296Dfs*33]199 | ||||||
| Neuropathy | ||||||||
| PEX11β | Cognition | Mild delay | VLCFA | N | VLCFA a. ox. | N | c.[64C>T];[64C>T] (p.[Q22*];[Q22*])153 | |
| 1 | Development | Early normal | ↑C26:0/C22:0 after migraine attack | Pristanic a. ox. | N | |||
| CNS | Nystagmus, migraine-like episodes | GNPAT activity | N | |||||
| PNS | SNHL, demyelinating sensory-motor neuropathy | Pristanic a. | N | Phytanic, a. ox. | N | |||
| Immunofluorescence: Enlarged, elongated peroxisomes, reduced in number | ||||||||
| Other | Cataract, skin scaling, gastro-intestinal problems | D/THCA | N | |||||
| Imaging | Arnold-Chiari malformation | Phytanic a. | N | |||||
| Plasmalogens | N | |||||||
| Pipecolic a. | N | |||||||
| PEX12 | Cognition | Normal/moderate delay | VLCFA | ↑ | VLCFA a. ox. | ↓ | 1. c.[538 C>T];[569C>T] (p.[R180*];[S190L])212 | |
| 4 (2 sibs) | Development | Normal/moderate delay | Pristanic a. | N | Pristanic a. ox. | ↓ | ||
| CNS | Normal | D/THCA | ↑ | GNPAT activity | ↓ | 2. c.[-26G>A;102A>T]; [-26G>A;102A>T] (p.[Met1ext-9fs33*; R34S];[Met1ext-9fs33*; R34S])178 | ||
| PNS | Intermediate sensory-motor neuropathy | Phytanic a. | N | Phytanic, a. ox. | ↓ | |||
| Immunofluorescence: Peroxisome mosaicism | ||||||||
| Other | Tremor, mild proximal lower extremity weakness | |||||||
| Imaging | N/A | |||||||
| Relapsing encephalopathy | ||||||||
| AMACR | Cognition | Normal, cognitive decline | VLCFA | N | VLCFA a. ox. | N | 1. c.[154T>C];[154T>C] (p.[S52P];[S52P])213-215 | |
| 3 | Development | Normal | (2R)-Pristanic | ↑ | Pristanic a. ox. | N | ||
| CNS | Retinitis pigmentosa, optic atrophy, epilepsy, migraine-like episodes, spastic paraparesis | (25R)-D/THCA | ↑ | GNPAT activity | N | |||
| Phytanic | ↑ | Phytanic, a. ox. | N | |||||
| Pipecolic a. | N | AMACR activity | ↓ | |||||
| PNS | Axonal sensory-motor neuropathy | |||||||
| Other | Primary hypogonadism, tremor, depression, mild myopathy | |||||||
| Imaging | Cerebral atrophy, white matter changes | |||||||
Superscript numbers beside mutations correspond to reference list.
Note: Cases diagnosed by whole exome sequencing are those described in references 153, 205, 212, 217, and 220.
Abbreviations: a., acid; AMACR, alpha methylacyl-CoA racemase; CK, creatine kinase; CNS, central nervous system; DBP, D-bifunctional protein; D/THCA, dihydroxycholestanoic acid and trihydroxycholestanoic bile acids; GNPAT, dihydroxyacetonephosphate acyltransferase; DTR, deep tendon refl exes; LCA, Leber congenital amaurosis; LFT, liver function tests; N, normal; NAA, N-acetylaspartate; Cr, creatine; N/A, not reported; ox., oxidation; sibs, siblings; SNHL, sensorineural hearing loss; PNS, peripheral nervous system; VLCFA, very long chain fatty acids.
Table 4
Diagnostic laboratory studies in PBD
| Test | Metabolite/gene | PBD | Note |
| Plasma VLCFA | Increased C26:0, C26:1 and C24/C22, C26/C22 ratio | ZSD | False positives in ketogenic diet, non-fasting or hemolyzed samples, normal in RCDP |
| Plasma phytanic and pristanic acid | Increased | ZSD, RCDP1,5 | Phytanic acid is normal in newborns and accumulates only through dietary intake; pristanic acid is normal in RCDP |
| RBC plasmalogens | Reduced | ZSD, RCDP1,5 | Greatest reduction in RCDP |
| Urine/plasma bile acid intermediates | Increased D/THCA | ZSD | Adjunct to VLCFA and plasmalogen testing |
| Plasma/urine pipecolic acid | Increased | ZSD | Adjunct to VLCFA and plasmalogen testing |
| Cultured fibroblasts | Confirm abnormal metabolites by enzymatic assays, evaluate peroxisome morphology and catalase distribution | ZSD, RCDP1,5 | Allows more complete characterization of peroxisome functions, number and morphology in difficult cases. Validates disease due to novel PEX alleles |
| Molecular genetic testing+, * | PEX 1, 2, 3, 5, 6, 10, 11β, 12, 13, 14, 16, 19, 26 | ZSD | Defects in PEX1, 2, 6, 10, 12, 26 account for ∼80% of ZSD174 |
| PEX 7 | RCDP1 | Four common alleles in exons 7 and 9, account for ∼70% of cases48 |
+Several PEX mutations are more common due to founder effects in persons of Northern European origin: PEX1-p. [Gly843Asp], PEX1-p.[Ile700Tyrfs42*], PEX7-p.[Leu292*]. *Genetic testing allows for carrier detection, preimplantation genetic diagnosis, improved prenatal diagnoses and can also provide prognostic information. It is required for diagnosis when the clinical and biochemical phenotypes do not fit the classic criteria. 174,48Superscript numbers correspond to reference list.


