
Defects of the urea cycle
Abstract
The elimination of waste nitrogen as urea is a final and central step of amino acid catabolism. It is accomplished by one of the most essential pathways of terrestrial animals, the urea cycle. This article describes the basic function of the 5 catalytic enzymes, the two transporters, and the cofactor synthesizing enzyme that comprise the urea cycle as well as the consequences of their deficiencies and ways to treat them. The article then elaborates on related disorders of metabolism that may either cause hyperammonemia or elevation of an amino acid that is key to urea cycle function.
1Urea cycle disorders
The urea cycle consists of one cofactor synthesizing enzyme, five catalytic enzymes, and two transporters responsible for the elimination of nitrogen as urea, these are: N-acetylglutamate synthase (NAGS), carbamoyl phosphate synthetase I (CPS1), ornithine transcarbamylase (OTC), argininosuccinate synthetase (ASS1), argininosuccinate lyase (ASL), arginase 1 (ARG1), the mitochondrial aspartate/glutamate carrier SLC25A13 also called Citrin, and the mitochondrial ornithine transporter SLC25A15, also called ORNT1 (Fig. 1, Table 1). When the urea cycle does not function normally nitrogen builds up as ammonia. NAGS, CPS1 and OTC are localized in the mitochondrial matrix, whereas the other 3 catalytic enzymes are in the cytosol, the two transporters are located in the inner mitochondrial membrane. The complete urea cycle is found only in the liver where all 5 catalytic enzymes are induced in the perinatal period in a coordinated manner. Induction of the urea cycle enzymes is stimulated by dietary protein and hormones such as glucagon and glucocorticoids. In extrahepatic tissues, CPS1 and OTC are expressed moderately in the small intestine, and ASS1 and ASL are expressed strongly in the intestines, the kidney and many other tissues.
Waste nitrogen is derived from amino acid breakdown. Muscle catabolism is another major source of nitrogen. The urea cycle is the only pathway for excreting waste nitrogen. Glycine and benzoate to form hippurate, or phenylbutyrate via phenylacetate, and glutamine to form phenylacetylglutamine, increase waste nitrogen excretion when the urea cycle is impaired.
Ammonia is probably derived principally from glutamine and is converted to carbamoylphosphate by carbamoyl phosphate synthetase, formed by the condensation of bicarbonate, ATP, and glutamine or ammonia (Fig. 1). Carbamoyl phosphate condenses with ornithine to form citrulline in a reaction catalyzed by OTC. Citrulline condenses with aspartate to produce argininosuccinate catalyzed by ASS1, and argininosuccinate catalyzed by ASL is then split into arginine and fumarate. Arginine is cleaved by ARG1, releasing urea, and ornithine is formed.
Hyperammonemic crises due to urea cycle disorders are characterized by the triad of encephalopathy, respiratory alkalosis, and hyperammonemia. A urea cycle disorder should be considered a diagnostic possibility in any patient of any age with acute encephalopathy [1–3].
Hyperammonemia in the preterm infant may be due to “transient hyperammonemia of the newborn”. In the newborn and older person, hyperammonemia occurs with many inborn errors of metabolism, including organic acidemias, fatty acid oxidation disorders, neonatal herpes simplex, liver diseases, and urea cycle defects (Table 2). The latter is distinguished by absence of acidosis and ketosis and a very low serum urea level [4]. At a few days of age an affected neonate develops lethargy, coma, convulsions after vomiting, and refusal to eat— signs frequently mistaken for infection or sepsis.
Complete deficiency of CPS1 and OTC result in the most severe forms of urea cycle disorders while ASS1 and ASL deficiencies maintain the ability to excrete nitrogen in the form of citrulline, arginine, or argininosuccinic acid and hyperammonemic crises in these two conditions are a little bit easier to manage. However, all four of these enzyme deficiencies when severe present in the neonatal period with severe hyperammonemia and coma. ARG1 deficiency very rarely presents in the neonatal period, Citrin deficiency (Citrullinemia type II) may present as severe neonatal cholestasis, and ORNT1 deficiency (hyperornithinemia, hyperammonemia, homocitrillinuria (HHH) syndrome) does not usually cause symptoms in neonates. Patients with partial deficiency of the catalytic enzymes present at any age from early infancy to late adulthood depending on the degree of enzyme deficiency and the magnitude of the stress their bodies are exposed to. This includes males hemizygous and females heterozygous for partial OTC deficiency.
The estimated incidence of each of the urea cycle disorders is shown in Table 3. The overall incidence of UCD is 1 per 35,000 life birth, at an annual birth rate of 4 million there are about 114 children with UCD born each year, approximately 26% will have neonatal onset disease and 69% late onset disease while about 5% will remain asymptomatic.
The severity of the first hyperammonemic crisis plays an important role in the neuropsychological outcome. Among survivors of the first hyperammonemic crisis, not only patients with neonatal-onset disease but also some patients with postneonatal-onset disease manifest intellectual disability the more so when they experience repeated hyperammonemic crises [5].
The pathophysiology of hyperammonemia in the newborn is shown in Fig. 2.
Presentation in the neonatal period is as a catastrophic illness or later as intermittent hyperammonemic episodes and neuropsychiatric signs including attention deficit, hyperactivity, migraine, and behavioral disorders (Table 4).
To prevent brain damage secondary to cerebral edema and increased intracranial pressure, a urea cycle disorder should be considered a diagnostic possibility in any patient of any age with acute encephalopathy [1]. The hyperammonemic encephalopathy characteristic of urea cycle disorders is manifested by progressive lethargy, vomiting and irritability, and in older children and adults, ataxia and combative and bizarre behavior.
Respiratory alkalosis early in the course is almost pathognomonic for a urea cycle disorder, although in the later stages of hyperammonemia when brainstem compression and hemodynamic instability ensue, metabolic acidosis will occur. Quantitative analysis of plasma amino acids and urine orotate and qualitative analysis of urine for organic acids by gas chromotography/mass spectrometry (GC/MS) should be done (Table 5). Low serum urea increases the likelihood of a urea cycle disorder.
2Diagnosis of urea cycle defects
Any neonate that was born full-term without complications and suddenly developed a severe, life threatening illness should have the following diagnostic studies: plasma ammonium and amino acid levels, qualitative tests for urine ketones, and GS/MS analysis of urine for organic acids. DNA analysis or specific enzyme activity measurement may provide the definitive diagnosis.
3General pathologic features of urea cycle disorders
Pathology of urea cycle disorders is nonspecific and includes mild steatosis and focal hepatocellular necrosis in the liver (Table 6). In the brain, after a severe hyperammonemic crisis, there may be spongiosis, Alzheimer type II astrocytic reaction, and superimposed lesions of anoxic-ischemic injury [6]. Heterotopias; hypomyelination, and multiple cystic lesions have been described [7, 8].
Postmortem examination reveals cerebral edema and herniation of the cerebellar tonsils as well as generalized neuronal cell loss. The extent of neuropathologic findings depends on the duration of the coma and the interval between coma and death. Histologically, swollen astrocytes were one of the first observations made in an animal model for hepatic encephalopathy, leading to the hypothesis that swollen astrocytes may be the cellular correlate of the brain edema in acute hyperammonemia. Cerebral edema is regularly found in clinical hyperammonemic states, e.g., hepatic encephalopathy Reye syndrome, and urea cycle disorders. The brain lacks an effective urea cycle and relies exclusively on glutamine synthesis in astrocytes for ammonia removal. Cerebral edema in hyperammonemic states may thus be a consequence of astrocyte accumulation of glutamine, which is an osmolyte.
Studies by Brusilow et al. [2] support a role for glutamine in producing cerebral edema. Plasma glutamine concentration increases before the onset of hyperammonemia [9] and hyperammonemia is uncommon when glutamine levels are normal or nearly so [10]. Brain glutamine accumulation can be prevented in hyperammonemic rats by pretreatment with methionine sulfoxime (MSO) [11]. Astrocyte swelling was the key observation reported in hyperammonemic rats that served as a model for hepatic encephalopathy [12].
Brain levels of myo-inositol, an intracellular regulatory osmolyte, are decreased in hepatic encephalopathy patients [13]. Indeed, there is a negative correlation between the glutamine level and the myo-inositol level in brain [14]. During hyperammonemia, myo-inositol levels may be down-regulated to oppose the osmotic effects of glutamine accumulation.
Hyperammonemia in the adult results in encephalopathy and edema whereas the exposure of developing brain to millimolar ammonia concentrations results in seizures, edema, and intellectual disability.
As stated above, neuropathologic evaluation of the brains of patients with UCD focused on astrocytes. Several pathophysiologic mechanisms (Fig. 2) have been proposed to explain the deleterious effects of ammonia on central nervous system (CNS) function. Such mechanisms include: (i) direct effects on the generation of excitatory and inhibitory post-synaptic potentials; (ii) a direct effect on expression of the astrocytic glutamate transporter GLT-l, a transport that is necessary for glutamate removal from the extracellular space; (iii) a direct effect of ammonia on “peripheral-type” benzodiazepine receptor expression. Peripheral-type benzodiazepine receptors mediate the synthesis of neurosteroids, some of which are γ-amino butyric acid or glutamate receptor agonists; (iv) a direct effect on cerebral energy metabolism via the inhibition of α-ketoglutarate dehydrogenase and stimulation of phosphofructokinase resulting in increased formation of lactate; and (v) an indirect role (via glutamine formation in the brain) in the pathophysiology of brain edema.
However, more recently altered water transport between blood and brain and brain and cerebrospinal fluid as well as abnormal flux of potassium ions in brain was implicated [15] and demonstrated [16] in an animal model for UCD. Indicating that hyperammonemia disturbs potassium and water homeostasis in brain. Finally hyperammonemia also causes oxidative and nitrosative stress [17].
4Prenatal diagnosis of urea cycle disorders
Citrullinemia, ASS1 deficiency, and argininosuccinic aciduria (ASL deficiency) can be diagnosed by enzyme assay in the first trimester of pregnancy using chorionic villi (CV) or later by using cultured amniocytes when the enzyme assay is clinically available. Alternatively, metabolite concentrations may be measured in amniotic fluid at 14 to 16 weeks or earlier. Mutation analysis is now the commonly used method for prenatal diagnosis but requires knowledge of the disease causing mutations in the index patient, which may be unique in families.
In contrast to ASS1 and ASL, enzyme analysis is complicated in OTC and the rare deficiencies of CPS1, N-acetylglutamate synthetase (NAGS), and arginase, because these enzymes are not expressed in CV or cultured amniotic cells. Fetal liver (OTC, CPS1, and NAGS) and fetal blood (arginase) have been used for prenatal enzyme analysis, but this involves a greater risk and delayed diagnosis. DNA analysis is the method of choice whenever possible. For the X-linked OTC deficiency, DNA analysis also allows the detection or exclusion of heterozygosity in females. When mutation analysis for OTC deficiency is inconclusive carrier testing can also be done by an allopurinol loading test and measuring increase in urinary orotic acid and orotidine [18].
Citrullinemia type 2 and HHH syndrome can also be diagnosed by mutation analysis.
5Newborn screening for urea cycle disorders
With the advent of expanded newborn screening (NBS) ASS1 deficiency (Citrullinemia), ASL deficiency (argininosuccinic aciduria (ASA)), Arg1 deficiency (Argininemia), Citrin deficiency (Citrullinemia type 2), and HHH syndrome can be detected by newborn screen. In Maine, Massachusetts, New Hampshire, Rhode Island, and Vermont OTC deficiency is also screened for. There is some concern about the sensitivity and specificity of the screening marker used; however, the false positive rate may be similar to that for other screening markers [19] and might be improved by using several amino acid markers as well as amino acid ratios [20]. The most severe catalytic enzyme deficiencies typically manifest in the first days of life before NBS results are available, but NBS results still make a contribution to the care of these neonates by aiding with speedy diagnosis confirmation in these cases.
6Long term outcomes
In the 1980s, neonatal hyperammonemic coma due to UCD had a mortality of 46% and 79% of the survivors had developmental disability. There was a negative correlation between duration of stage III and IV neonatal hyperammonemic coma and result of the Bayley Scales of Infant Development (BSID) at 12 months of age but not between peak ammonia level and the BSID [21]. The introduction of nitrogen scavenger therapy for the chronic treatment of UCDs in 1987 has improved the long-term outcome of patients with UCDs [22] however no systematic analysis of the relationship between the quality of the control of nitrogen metabolism and outcome of UCDs was conducted until 2006.
Since 2006, a NIH funded Rare Diseases Clinical Research Network, the Urea Cycle Disorders Consortium (UCDC), is performing a longitudinal study of urea cycle disorders (UCDs) which allows for the first time an assessment of the natural history of UCD as well as of currently used treatment regiments. By 2014 six hundred patients had been enrolled.
The first analyses of the neuropsychological data collected by the UCDC showed that 66% of children with neonatal onset disease 4 years of age and older [23] and 25% of children with late onset disease [24] have intellectual disability, and problems with emotional/behavioral regulation, attention, and executive function underscoring that the neuropsychological outcome is the preeminent problem.
Subsequently, analyses of the longitudinal study data of the first 8 years showed that hyperammonemic episodes after the neonatal period are most often caused by intercurrent infections, that both plasma ammonia and glutamine elevations may be biomarkers for neuropsychological outcome, that low protein diet causes reduced height in UCD patients, that high dose arginine has a negative impact on liver function in ASA patients, that phenylbutyrate causes low levels of branched chain amino acids, and that patients with OTC deficiency tend to have hepatic dysfunction even liver failure. In addition, at study sites the mortality of neonatal onset disease was 24% and that of post-neonatal onset disease 11% [25]. The effect of the disruption of nitric oxide synthesis in urea cycle disorders is a new research focus and explains high blood pressure and possibly also part of the intellectual disability in patients with ASL [26].
7General considerations for treatment of urea cycle defects
Prevention of hyperammonemia and treatment of UCDs rely heavily on ammonia-lowering strategies with administration of nitrogen scavengers such as, benzoate, and phenylacetate or phenylbutyrate as well as low protein diet.
Aggressive therapy to control ammonia is necessary to affect neurodevelopmental outcome. Long-term treatment consists chiefly of a low protein diet and the use of oral nitrogen scavengers, while monitoring carefully that sufficient protein and calories are supplied to provide adequate nutrients for growth and prevention of catabolism.
In a hyperammonemic crisis glucose infusion, protein restriction, intravenous sodium phenylacetate and sodium benzoate, or hemodialysis or continuous veno-venous hemofiltration (CVVH) should be started immediately. Intravenous sodium benzoate and phenylacetate is effective in lowering ammonia levels of up to 300μmol/L. The use of high-carbohydrate fluids alone is not sufficient to treat a hyperammonemic crisis in UCD; if intravenous sodium benzoate and phenylacetate are not available, or at ammonia levels of 500μmol/L or higher hemodialysis or CVVH should be started immediately. Peritoneal dialysis, and exchange transfusion are not efficient enough in removing ammonia rapidly and should therefore not be attempted.
The outlook for patients with urea cycle disorders has changed from certain death in the first weeks of life to one of prolonged survival and acquisition of cognitive function since alternative pathway therapy with nitrogen scavengers was developed [27]. The oral nitrogen scavenger sodium phenylbutyrate was approved by the Food and Drug administration in 1996 and glycerol phenylbutyrate in 2013. Administration of phenylbutyrate and either arginine or citrulline, depending on the specific disorder in question, has allowed these children to attend school and make developmental progress. However, intercurrent episodes of hyperammonemia, frequently attendant on an intercurrent viral infection, remain a constant threat [28]. Arginine or citrulline are important medications for these children, however, since they are amino acids they are considered dietary supplements and not drugs which causes insurance companies to deny coverage and makes them less tightly controlled by the FDA. Although there is less FDA supervision for dietary supplements, the suppliers are still bound by ‘Current Good Manufacturing Practice’ (CGMP) and the label on the product dispensed has to be consistent with the content of the vial or can and its concentration; otherwise dispensing the product is illegal [29].
Treatment can provide intervals free of hyperammonemic episodes, during which the child grows and makes developmental progress. Early orthotopic liver transplantation prevents further hyperammonemic episodes and cures CPSI and OTC deficiency. Gene therapy is also under investigation [30].
8CPS deficiency
CPS1 (EC 6.3.4.16) is located within the inner mitochondrial matrix and in a monomeric form catalyzes the initial rate-limiting step of the urea cycle the formation of carbamoyl phosphate from ammonia and bicarbonate. Unless treated, complete deficiency of CPS1, a rare autosomal recessive disease, causes death in newborn infants. Survivors are often intellectually disabled and suffer frequent hyperammonemic crises during intercurrent illness or other catabolic stresses.
Besides by markedly elevated ammonia levels, the disease is characterized by reduced plasma citrulline, arginine, and urea. Urine contains no orotate and no citrulline. Liver function tests are normal [27]. In the severe neonatal onset form of the disease symptoms usually appear within 24 hours after birth and include vomiting, lethargy, hypothermia, hypotonia, irritability, and opisthotonus. Symptoms in patients with the late-onset form are similar but milder than in the neonatal-onset form.
CPS1 is only detectable in liver and in the intestinal mucosa where it is located in the mitochondrial matrix. The definitive diagnosis is made by determination of enzyme activity in liver parenchyma obtained by biopsy or mutation analysis. More than 220 mutations have been described in the human CPS1 gene [31].
8.1Pathology
No consistent pathologic abnormalities have been described at autopsy.
The liver may show mild steatosis and focal cellular necrosis that progresses to mild portal fibrosis but changes in the liver are variable. There have been reports of increased glycogen deposition and swollen mitochondria with dramatic loss of cristae in hepatocytes, as well as reports of reduced glycogen content of hepatocytes with fatty change, and an increase of fibrous tissue. However, the lack of any visible liver change in patients with CPS1 deficiency has also been reported. In the brain Alzheimer type II astrocytic changes and spongiosis are the usual findings in hepatic encephalopathy (Figs. 3 and 4) [32].
Light microscopy (LM) of the kidney shows no pathologic change, but electron microscopy (EM) of glomeruli reveals foot process effacement [6]. EM of the proximal tubule shows increased glycogen deposition, numerous protein resorption droplets, and swollen mitochondria with disorganized cristae.
8.2Prenatal diagnosis
Mutation analysis of the CPS1 gene.
8.3Treatment
Therapy consists of hemodialysis, restriction of dietary protein intake, and activation of alternative pathways of waste nitrogen excretion, as in OTC deficiency. Despite therapy, the mortality rate is high, and morbidity, primarily due to brain damage, is significant. Early liver transplantation preserves quality of life if the initial crisis did not devastate the patient.
9OTC deficiency
OTC deficiency is the most common of the urea cycle disorders. It is inherited as an X-linked trait (the others are inherited as autosomal recessive traits) and is also expressed in some heterozygous females. Males with severe deficiency typically present within 24–72 hours after birth with severe hyperammonemia and encephalopathy.
Males and females with partial OTC deficiency may present at any age and may have normal development. There is some degree of phenotype/genotype correlation with regard to the severity of the disease [33], however, the same mutation can be associated with different severity and age of onset especially since both are influenced by the constellation of the precipitating events of the initial crisis.
9.1Diagnosis
OTC deficiency occurs as a neonatal form with very severe manifestations in 25–30% of patients, and a late-onset form that accounts for approximately 70% of patients, 66% of whom are heterozygous females. The gene locus has been mapped to Xp.11.4. Although the different mutations at the OTC locus may contribute to the spectrum of disease seen in females too, their variability is more likely a consequence of the proportion of hepatocytes in which the active X chromosome carries the normal or mutant OTC gene, i.e. of the X-inactivation pattern in their livers.
Hemizygous males with severe deficiency have a fulminant neonatal course with hypotonia, lethargy, coma, and seizures. Heterozygous females and males with partial deficiency are asymptomatic or have variable expression and a less severe form of the disease. In infants, hyperammonemia is marked and serum urea is low. There is no acidosis or ketosis initially but acidosis may occur as the patient’s status is deteriorating. Plasma citrulline is low or absent, and orotic acid is markedly elevated in urine. The diagnosis may be made by enzyme assay of liver tissue or mutation analysis. The clinical spectrum for time of onset and severity is continuous, ranging from neonatal hyperammonemic coma to asymptomatic adults. The OTC enzyme has been crystallized and analyzed by X-ray diffraction. It is a trimer with 1 active site per monomer.
Large deletions of one exon or more are seen in approximately 8% of patients, small deletions or insertions in about 10%, and the remaining mutations are single base substitutions. Approximately 18% of all mutations affect splice sites. Some mutations have been expressed in bacteria or mammalian cells. Examples of expressed mutations include R277W and R277Q, which are associated with late-onset disease. R141Q (neonatal disease) disrupts the active site while the purified R40H mutant enzyme has a normal catalytic function, and this mutation is likely therefore to affect posttranslational processing such a mitochondrial targeting. Uncommonly, mild mutations are transmitted by asymptomatic males to their daughters, resulting in clinical disease in some of the male offspring in future generations [34].
9.2Pathology
The pathology in neonates is nonspecific. In males, the liver is enlarged with focal cellular necrosis and predominantly microvesicular steatosis (Fig. 5). In older heterozygous females, there may be focal piecemeal necrosis, inflammation, steatosis, fibrosis, and focal nodular hyperplasia of the liver [35]. Peroxisomal swelling and matrix diffraction are the usual ultrastructural changes observed. Elongated, tortuous, or branching mitochondria may display a paracrystalline arrangement of cristae [36]. The CNS changes [32] are due to the hyperammonemic effects and include Alzheimer type II astrocytes particularly in the segmental area of the pons and midbrain with spongiosis, and hypomyelination. Changes with cystic lesions are frequent.
9.3Treatment
Recommended crisis treatment for ammonia levels up to 300μmol/L is intravenous sodium benzoate plus sodium phenylacetate each 250 mg/kg as a bolus followed by the same dose given over 24 hours for patients up to 25 kg body weight and 5500 mg/m2 given as a bolus followed by the same dose over 24 hours for patients with more than 25 kg body weight, supplemented with 10% arginine hydrochloride providing 200 mg/kg of arginine as a bolus followed by the same dose given over 24 hours for patients up to 25 kg body weight and 4000 mg/m2 given as a bolus followed by the same dose over 24 hours for patients greater than 25 kg body weight. When the ammonia level is normalized, the patient can be transitioned from 24 h dosing of continuous IV nitrogen scavengers to oral phenylbutyrate at 450 mg - 600 mg/kg/day for patients up to 20 kg body weight and 9900 – 13 000 mg/m2/day for patients greater than 20 kg body weight supplemented with L-Citrulline (instead of Arginine) at 170 mg/kg/day for patients up to 20 kg body weight and 3800 mg/m2/day for patients greater than 20 kg body weight [37].
10Citrullinemia
Citrullinemia (ASS1 deficiency) is an autosomal recessive trait and is characterized by marked hyperammonemia with low arginine and high plasma citrulline levels due to deficiency of ASS1. The disease usually manifests in the first days of life with life-threatening hyperammonemia and blood citrulline levels 50 to 100 times normal. Enzyme assays may be performed on fibroblast or amniotic cell cultures. Pathologic changes include fatty metamorphosis, focal necrosis, and patchy cholestasis in the liver [38]. Neuropathologic changes are similar to those of the other urea cycle disorders.
10.1Treatment
Currently treatment entails restriction of protein intake and the administration of oral phenylbutyrate as in OTC deficiency and oral arginine base, the current recommendation is for 400–700 mg/kg/day of arginine base in patients up to 20 kg body weight and 8800 – 15 400 mg/m2/day for patients over 20 kg body weight [37]. Although this therapy is effective, repeated episodes of hyperammonemia result in a less-than-satisfactory final outcome and the use of less Arginine might be beneficial.
11Citrin deficiency/citrullinemia type 2
Citrullinemia type II or Citrin deficiency is caused by mutations in SLC25A13, the gene for the mitochondrial aspartate/glutamate carrier at the inner mitochondrial membrane also called citirn. In this autosomal recessive condition the lack of aspartate in the cytosol to conjugate with citrulline causes decreased urea cycle function. Citrulline levels can be elevated up to 400μmol/L, plasma ammonia levels are less of a concern.
Citrin deficiency manifests in adults with cyclical erratic behavior, dysarthria, seizures, weakness, and coma. Treatment consists of nitrogen-scavengers (arginine and phenylbutyrate) and liver transplant. A neonatal-onset form exists that manifests with intrahepatic cholestasis.
The children may also have aminoacidemia, galactosemia, hypoproteinemia, and hypoglycemia. Treatment consists of high-protein/low-carbohydrate diet, and the symptoms often disappear after the first year of life. Rarely liver transplant is needed in such a child. Hyperammonemia is not a major component of this neonatal presentation.
12Argininosuccinicaciduria
Argininosuccinicaciduria (ASA; ASL deficiency) is an autosomal recessive disorder due to deficiency of ASL [39]. In the newborn, hyperammonemia develops with increased argininosuccinic acid in plasma and urine and elevation of plasma citrulline levels. Hepatomegaly and increased levels of liver enzymes develop. Microvesicular steatosis may be confused with Reye syndrome; however, the mitochondrial changes seen in Reye syndrome are not present. A nodular liver with severe septal fibrosis can be the long-term outcome [40] Ultrastructural changes appear to be nonspecific [41]. In the brain, hypomyelination is present. Trichorrhexis nodosa is another symptom of the disease.
The usual neonatal presentation of ASA is that of a full-term infant who develops feeding problems, vomiting, and lethargy, sliding into coma. As in all other enzyme deficiencies patients with partial deficiency have a milder phenotype [42]. ASA is now detected by newborn screen and children with a milder degree of ASA that remain asymptomatic have been identified. Current treatment recommendations are for monotherapy with high dose arginine free base enterally (400–700 mg/kg/day of arginine base in patients up to 20 kg body weight and 8800 – 15 400 mg/m2/day for patients over 20 kg body weight). However, a recent clinical trial showed benefit of low dose arginine (100 mg/kg/d) combined with phenylbutyrate versus high dose arginine monotherapy with regard to hepatic function tests in ASA patients [43].
13Arginase deficiency (ARGININEMIA)
Arginase is the final enzyme of the urea cycle (Fig. 6). Its deficiency in the liver results in hyperargininemia and progressive mental and neurologic deterioration. Orotic aciduria may occur. It is the most rare one of the enzyme deficiencies of the urea cycle, but is easily overlooked as ‘developmental delay/intellectual disability with spasticity of unknown cause’ [44].The typical patient is thought to be normal at birth and in infancy, but between the age of 2 and 3 years, growth and development slow and spasticity, especially of the lower extremities, is seen. The disorder is progressive, and may be punctuated by episodes of hyperammonemia. Most affected patients have survived, some into their late thirties. The infrequency of apparent hyperammonemia, and the unique and stereotypic clinical findings have led to the idea that arginine or one of its products is primarily responsible for the symptoms [44]. Nitric oxide (NO) is synthesized from arginine by NO synthase. Arginine is converted to NO and citrulline. The reaction is a minor source of citrulline but NO is an important neurotransmitter and regulator of immune function and blood vessel dilation.
13.1Pathology
In the liver the hepatocytes are swollen, and steatosis and moderate periportal fibrosis may be present [45]. Ultrastructural changes are nonspecific and the mitochondria are large in liver cells [36].
Biochemical studies in patients and animals led to the hypothesis that at least one other form of arginase existed in mammals and that its activity might be responsible for the persistent ureagenesis in affected patients. This was proven when liver arginase AI-deficient patients were found to have enhanced levels of an immunologically distinct arginase in kidney [44, 46].
AI, cloned in the mid 1980 s, was found to have 8 exons and to be about 12 kb in length. The cDNA specified a protein of 322 amino acids, which shares substantial homology in highly conserved regions to the arginases of a number of lower species. High-level expression in liver may be mediated by an enhancer in intron 7. It is not known to be regulated, in liver, independently of other urea cycle enzymes [44, 47].
AII, sometimes referred to as kidney arginase, has been localized to the mitochondrion and is thought to be involved in ornithine biosynthesis. It was cloned, shares all the major conserved regions with the other arginases, and has a mitochondrial leader sequence cleaved off during internalization into this organelle. Its posttranslational amino acid length is 354. It is expressed most abundantly in kidney and prostate and is greatly enhanced when nitric oxide synthetase (NOS) is induced in macrophage cells. It may be regulated by arginine levels. It appears to be highly expressed in a number of cancers [44].
No human phenotype has been associated with AII deficiency. The spectrum of functions related to AII remains to be defined, as does its role in macrophage function [44, 48].
13.2Treatment
Patients respond to nitrogen-scavenger therapy and a protein restricted diet (0.5–1.0 g/kg/d) supplemented with essential amino acids (0.5 g/kg) which can halt the progression of the spasticity when it succeeds in lowering the arginine level, and botulinum toxin (Botox) and surgical tendon release help improve function. Hyperammonemia is managed as previously indicated.
14Hyperornithinemia Hyperammonemia Homocitrullinuria (HHH) syndrome
HHH (hyperornithinemia, hyperammonemia, homocitrullinuria) syndrome is an autosomal recessive disorder characterized by post-prandial or intermittent hyperammonemia with increased plasma ornithine concentration [49]. Homocitrulline, not ordinarily found in urine, and orotic acid are present in urine [50]. Symptoms may begin in the newborn period or at any time into adulthood. Hypotonia, ataxia, vomiting, coma, seizures, or developmental delay may occur as well as hypertonia/spasticity in especially the lower extremities.
Ornithine transport into the mitochondria is decreased. Ornithine accumulates in the cytoplasm. Reduced intramitochondrial ornithine causes impaired urea formation and orotic aciduria.
14.1Pathology
Liver biopsy on LM shows no abnormalities, but on EM liver mitochondria are elongated with bizarre shapes and with crystalloid inclusions (Fig. 7). Abnormal mitochondria may also be found in skeletal muscle, leukocytes, and cultured fibroblasts [49].
Patients improve on a protein restricted diet, with citrulline supplementation, and ammonia scavenger therapy Disturbance in fetal development with intellectual disability may occur with maternal HHH syndrome [51, 52]. Neonatal onset of HHH syndrome has been reported [53].
15Hyperinsulinism and hyperammonemia
Patients with a syndrome of hyperinsulinism and hyperammonemia (HA/HI) have been reported [54, 55] that have fasting and protein sensitive hypoglycemia in combination with elevated ammonia levels. The condition was shown to be caused by gain of function mutations in the glutamate dehydrogenase (GDH) gene [55] and called HA/HI syndrome. It is an example of an inborn error of metabolism that is due to a gain of function mutation in an enzyme.
16Lysinuric protein intolerance
Lysinuric protein intolerance (LPI) is due to a defect in the transport of dibasic amino acids with urinary excretion of ornithine, arginine, and large amounts of lysine [56]. It is caused by mutations in the SLC7A7 gene which codes for the light chain of system y+L. There is an increased incidence in Finland and a second geographical concentration in Italy.
Patients present with hypotonia, recurrent vomiting and diarrhea, lethargy and somnolence even coma after a protein rich meal, hepatosplenomegaly, aversion to high-protein food, failure to thrive, and intermittent elevation of serum ammonia. Acute pancreatitis may also occur. Long-term the patient may develop interstitial lung disease with alveolar proteinosis, kidney disease with glomerular and proximal tubular dysfunction, and macrophage activation syndrome. Improvement occurs with diets low in protein, and most infants do well on breast milk. Besides the effects of ammonia on the CNS, the pathologic effects are predominantly in the bones, lungs, and kidneys. Fractures due to osteoporosis, renal rickets, and alveolar proteinosis have been described. Autoimmune disorders including systemic lupus erythematosus and a T-cell abnormality occur. The dibasic (cationic) amino acids are poorly absorbed from the intestine and lost in excess in the urine, and their plasma concentrations are usually decreased. Plasma citrulline concentration is moderately increased (1.5 to 3 times the upper reference limit). Excess orotic acid is excreted in urine after protein ingestion or prolonged fasting, and during acute febrile illnesses [57].
The transport of cationic amino acids at the basolateral membrane of enterocytes and renal tubular cells is affected. The impairment of urea cycle function is probably caused by reduced intracellular availability of ornithine in hepatocytes. System y+L activity is reduced in monocytes and alveolar macrophages possibly explaining the effect of the disease on the lung and kidney [58]. Sebastio and Nunes [58] also discuss the possibility that the transport defect may lead to an intracellular entrapment of arginine and the consequences this would have for nitric oxide metabolism [58].
16.1Clinical findings
Patients with LPI suffer from symptoms of periodic hyperammonemia after protein ingestion. The children are usually symptom-free after birth if breast-fed, but introduction of high-protein formula or other high-protein foods can cause severe hyperammonemia. The children typically develop marked protein aversion at about 1 year of age, fail to thrive, and show mild growth retardation. The child may present with alopecia, diminished subcutaneous fat and loose skin folds, anemia, and leukopenia. Osteoporosis may lead to fractures, and some patients develop chronic renal failure. Skin lesions may be reminiscent of acrodermatitis enteropathica. The liver and spleen are often enlarged. Serum lactate dehydrogenase and ferritin concentrations are almost always markedly elevated. If episodes of hyperammonemia are avoided, mental development is normal. Truncal obesity is common, but the extremities are thin and muscle strength is poor. Most patients develop pulmonary changes that first resemble mild interstitial pneumonia but may suddenly progress to fatal or life-threatening pulmonary failure, characterized histologically by alveolar proteinosis(Fig. 8).
16.2Laboratory findings
Laboratory studies may show increased levels of lysine, arginine, and ornithine in the urine. Twenty-four-hour urine collection should reveal lysinuria. Plasma cationic amino acids (lysine, arginine, and ornithine) are usually decreased but may be normal and glutamine, alanine, glycine, and citrulline are increased. There is an increase in post-prandial serum ammonia and orotic aciduria and an increase in serum lactate dehydrogenase and ferritin.
16.3Treatment
Some patients benefit from repeated bronchoalveolar lavages. Hyperammonemia in LPI is preventable with dietary protein limitation and citrulline supplementation; patients may also receive nitrogen scavengers [57] and L-lysine.
17Gyrate atrophy of the choroid and retina
Gyrate atrophy (GYA) of the choroid and retina is not a urea cycle disorder and does not cause hyperammonemia but like HHH syndrome it is marked by hyperornithinemia [49]. It presents in the first decade with myopia, night blindness, and loss of peripheral vision. Blindness occurs in the third to fourth decade. Plasma ammonia is not elevated.
On examination, the normally red fundus appears gray. Atrophic areas appear gray or white; subcapsular cataracts and constricted visual fields develop [59].
Ornithine is markedly elevated in plasma, cerebrospinal fluid, eye structures, and urine. The condition is caused by deficiency of ornithine aminotransferase (OAT). OAT catalyzes the transamination of ornithine to glutamate semialdehyde. Pyridoxine is a cofactor.
Deficiency of OAT is detected in fibroblasts, lymphocytes, muscle cultures, and liver biopsy.
17.1Treatment
While no treatment is curative, some patients have a pyridoxine responsive form of the disease and improve with large doses of pyridoxine and limited dietary intake of protein [59].
References
[1] | Masataka M. , Regulation of the urea cycle genes in urea and nitric oxide synthesis [abstract]. In Advances in Inherited Urea Cycle Disorders, Vienna. Austria, ((1997) ), 20–21. |
[2] | Brusilow S.W. and Maestri N.E. , Urea cycle disorders: Diagnosis, pathophysiology, and therapy. In Barness LA, editor: Advances in Pediatrics, St. Louis, Mosby-Year Book, 43: , ((1996) ) 127. |
[3] | Brusilow S.W. , Urea cycle disorders: Clinical paradigm of hyperammonemic encephalopathy. In Boyer JL, Ockner RK, editors: Progress in Liver Disease, Philadelphia, WB, Saunders 5: ((1995) ). |
[4] | Morrow G. , Barness L.A. and Efron M.L. , Citrullinemia with defective urea production, Pediatrics 40: ((1967) ), 565. |
[5] | Uchino T. , Endo F. and Ichiro M. , Neurodevelopmental outcome of long-term therapy in Japan [abstract]. In Advances in Inherited Urea Cycle Disorders, Vienna, Austria, ((1997) ), 20–21. |
[6] | Dimmick J.E. and Applegarth D.A. , Pathology of inherited metabolic disease. In Stocker J, Dehner L, editors: Pediatric Pathology, Philadelphia, JB, Lippincott ((1992) ). |
[7] | Filloux F. , Townsend J.J. and Leonard C. , Ornithine transcarbamylase deficiency: Neuropathologic changes acquired in utero, J Pediatr 108: ((1986) ), 942. |
[8] | Harding B.N. , Leonard J.V. and Erdohazi M. , Ornithine transcarbamylase deficiency: Neuropathological study, Eur J Pediatr 141: ((1984) ), 215. |
[9] | Batshaw M.L. , Walser M. and Brusilow S.W. , Plasma alpha-ketoglutarate in urea cycle enzyrnopathies and its role as a harbinger of hyperammonemic coma, Pediatr Res 14: ((1992) ), 1316. |
[10] | Maestri N.E. , McGowan D.K. and Brusllow S.W. , Plasma glutamine concentration: A guide in the management of urea cycle disorders, J Pediatr 121: ((1992) ), 259. |
[11] | Takahashi H. , Koehler R.C. , Brusilow S.W. and Traystman R.J. , Inhibition of brain glutamine accumulation prevents cerebral edema in hyperammonemic rats, Am J PhysioI 261: ((1991) ), H825–9. |
[12] | Norenberg M.D. , A light and electron microscopic study of experimental portal-systemic (ammonia) encephalopathy, Progression and reversal of the disorder, Lab Invest 36: (6) ((1977) ), 618–627. |
[13] | Kries R. , Ross B.D. , Farrow N.A. and Ackerman Z. , Metabolic disorders of the brain in chronic hepatic encephalopathy 7 detected with H-l MR spectroscopy, Radiology 182: ((1992) ), 19. |
[14] | Zwingmann C. , Chatauret N. , Rose C. , Leibfritz D. and Butterworth R.F. , Selective alterations of brain osmolytes in acute liver failure: Protective effect of mild hypothermia, Brain Res 999: (1) ((2004) ), 118–123. |
[15] | Lichter-Konecki U. , Mangin J.M. , Gordish-Dressman H. , Hoffman E.P. and Gallo V. , Gene expression profiling of astrocytes from hyperammonemic mice reveals altered pathways for water and potassium homeostasis in vivo, Glia 56: (4) ((2008) ), 365–377. |
[16] | Rangroo Thrane V. , Thrane, A.S., Wang, F., Cotrina, M.L., Smith, N.A., Chen, M., Xu, Q., Kang, N., Fujita, T., Nagelhus, E.A. and Nedergaard, M., Ammonia triggers neuronal disinhibition and seizures by impairing astrocyte potassium buffering, Nat Med 19: (12) ((2013) ), 1643–1648. |
[17] | Rao V.L. , Audet R.M. and Butterworth R.F. , Increased neuronal nitric oxide synthase expression in brain following portacaval anastomosis, Brain Res 765: (1) ((1997) ), 169–172. |
[18] | Hauser E.R. , Finkelstein J.E. , Valle D. and Brusllow S.W. , Allo-purinol-induced orotinuria, A test for mutations at the ornithine carbamoyltransferase locus in women, N Eng/ J Med 322: ((1990) ), 1641. |
[19] | Hall P.L. , Marquardt G. , McHugh D.M. , Currier R.J. , Tang H. , Stoway S.D. and Rinaldo P. , Postanalytical tools improve performance of newborn screening by tandem mass spectrometry, Genet Med 16: (12) ((2014) ), 889–895. |
[20] | The Collaborative Laboratory Integrated Reports (CLIR), an interactive web tool [mycharhttps://clir.mayo.edu/Home/About]. |
[21] | Msall M. , Batshaw M.L. , Suss R. , Brusilow S.W. and Mellits E.D. , Neurologic outcome in children with inborn errors of urea synthesis, Outcome of urea-cycle enzymopathies, N Engl J Med 310: (23) ((1984) ), 1500–1505. |
[22] | Maestri N.E. , Hauser E.R. , Bartholomew D. , et al., Prospective therapy of urea cycle disorders, J Pediatr 119: ((1991) ), 923. |
[23] | Ah Mew N. , Krivitzky, L., McCarter, R., Batshaw, M. and Tuchman, M., Urea Cycle Disorders Consortium of the Rare Diseases Clinical Research Network. Clinical outcomes of neonatal onset proximal versus distal urea cycle disorders do not differ, J Pediatr 162: (2) ((2013) ), 324–329.e1. PMID:2290174124. |
[24] | Krivitzky L. , Babikian T. , Lee H.S. , Thomas N.H. , Burk-Paull K.L. and Batshaw M.L. , Intellectual, adaptive, and behavioral functioning in children with urea cycle disorders, Pediatr Res 66: (1) ((2009) ), 96–101. PMID:19287347. |
[25] | Batshaw M.L. , Tuchman M. , Summar M. and Seminara J. , Members of the Urea Cycle Disorders Consortium: A longitudinal study of urea cycle disorders, Mol Genet Metab 113: (1-2) ((2014) ), 127–130. PMID: 251356. |
[26] | Nagamani S.C. , Campeau P.M. , Shchelochkov O.A. , Premkumar M.H. , Guse K. , Brunetti-Pierri N. , Chen Y. , Sun Q. , Tang Y. , Palmer D. , Reddy A.K. , Li L. , Slesnick T.C. , Feig D.I. , Caudle S. , Harrison D. , Salviati L. , Marini J.C. , Bryan N.S. and Erez A. , Lee B: Nitric-oxide supplementation for treatment of long-term complications in argininosuccinic aciduria, Am J Hum Genet 90: (5) ((2012) ), 836–846. |
[27] | Brusilow S.W. and Horwich A.L. , Urea cycle enzymes. In Scriver C, Beaudet A, Sly W, Valle D, editors: Metabolic and Molecular Bases of Inherited Disease, 7th ed, New York, McGraw-Hill, ((1995) ), 1187. |
[28] | Thoene J.G. , Treatment of urea cycle disorders, J Pedlatr 134: ((1999) ), 255. |
[29] | D’Aco K. , Mooney R. , Cusmano-Ozog K. , Hofherr S. and Lichter-Konecki U. , What is in the can? The dilemma with dietary supplements, Mol Genet Metab 113: (4) ((2014) ), 239–240. |
[30] | Yang Y. , Wang L. , Bell P. , McMenamin D. , He Z. , White J. , Yu H. , Xu C. , Morizono H. , Musunuru K. , Batshaw M.L. and Wilson J.M. , A dual AAV system enables the Cas9-mediated correction of a metabolic liver disease in newborn mice, Nat Biotechnol 34: (3) ((2016) ), 334–338. |
[31] | Häberle J. , Shchelochkov O.A. , Wang J. , Katsonis P. , Hall L. , Reiss S. , Eeds A. , Willis A. , Yadav M. , Summar S. , Lichtarge O. , Rubio V. , Wong L.J. and Summar M. , Molecular defects in human carbamoy phosphate synthetase I: Mutational spectrum, diagnostic and protein structure considerations, Hum Mutat 32: (6) ((2011) ), 579–589. |
[32] | Zimmerman A. , Bachmann C. , Colombo J.P. , Ultrastructural pathology in congenital defects of the urea cycle: Ornithine transcarbamylase and carbamyl phosphate synthetase deficiency, Virchows Arch Pathol Anat 393: ((1981) ), 321. |
[33] | Garcia M.J. , Merinero B. and Perez-Cerdá C. , et al., Mild phenotype in two male patients with OTC deficiency [abstract], In Advances in Inherited Urea Cycle Disorders, Vienna, Austria, ((1997) ), 20–21. |
[34] | Matsuda I. and Tanase S. , The ornithine transcarbamylase (OTC) gene: Mutations in 50 Japanese families with OTC deficiency, Am J Med Genet 71: ((1997) ), 378. |
[35] | Ishak K.G. , Pathology of inherited metabolic disorders in hepatic disease in children. In Balistreri and Stocker JT, editors: Pediatric Hepatology, New York, Hemisphere Publishing ((1990) ). |
[36] | Phillips M.J. , Powell S. , Paterson J. and Valencia P. , The Liver, An Atlas and Text of Ultrastructural Pathology New York, Raven Press, ((1987) ). |
[37] | Batshaw M.L. , MacArthur R.B. and Tuchman M. , Alternative pathway therapy for urea cycle disorders: Twenty years later, J Pediatr 138: (1 Suppl) ((2001) ), S46–S54; discussion S54–5. |
[38] | Sharp H.L. , Inherited disorders with metabolic hepatic dysfunction. In Berk JE, editor: Bochus Gastroenterlogy, 4th ed, Philadelphia, WB, Saunders, ((1985) ), 3236. |
[39] | Simard L. , O’Brien W.E. and McInnes R.R. , Argiininosuccinate lyase deficiency, Proc Natl Acad Sci USA 81: ((1984) ), 44. |
[40] | Zimmerman A. , Cachmann C. and Baumgartner R. , Severe liver fibrosis in argininosuccinic acidura, Arch Pathol Lab Med 128: ((1986) ), 136. |
[41] | Travers H. , Reed J.S. and Kennedy J.A. , Ultrastructural study of the liver in argininosuccinate deficiency, Pediatr Pathol 5: ((1986) ), 307. |
[42] | Schuller E. , Lazarowa D. , Item C. , et al., Mutation analysis in patients with partial argininosuccinate lyase deficiency identified by newborn screening [abstract], In Advances in Inherited Urea Cycle Disorders, Vienna, Austria, ((1997) ), 20–21. |
[43] | Nagamani S.C. , Shchelochkov O.A. , Mullins M.A. , Carter S. , Lanpher B.C. , Sun Q. , Kleppe S. , Erez A. , O’Brian Smith E. and Marini J.C., Members of the Urea Cycle Disorders Consortium, Lee B, A randomized controlled trial to evaluate the effects of high-dose versus low-dose of arginine therapy on hepatic function tests in argininosuccinic aciduria, Mol Genet Metab 107: (3) ((2012) ), 315–321. |
[44] | Cederbaum S. , The human arginases and arginase deficiency. In Advances in Inherited Urea Cycle Disorders, Vienna, Austria, ((1997) ), 20–21. |
[45] | Jorda A. , Portoles M. , Rubio V. , Capdevila A. , Vilas J. and García-Piño J. , Liver fibrosis in arginase deficiency, Arch Pathol Lab Med 111: ((1987) ), 691. |
[46] | Jenkinson C.P. , Grody W.W. and Cederbaum S.D. , Comparative properties of arginases, Comp Biochem Physiol B Biochem Mol Biol 114: ((1996) ), 107. |
[47] | Gotoh T. , Molecular cloning of cDNA for nonhepatic mitochondrial arginase (arginase II) and comparison of its induction with nitric oxide synthase in a murine macrophage-like cell line, FEBS Lett 395: ((1996) ), 119. |
[48] | Vockley J.G. , Jenkinson C.P. , Shukla H. , Kern R.M. , Grody W.W. and Cederbaum S.D. , Cloning and characterization of the human type II arginase gene, Genomics 38: ((1996) ), 118. |
[49] | Valle D. and Simell O. , The hyperonithinemias. In Scriver CR, Beaudet AL, Sly WS, Valle S, editors: The Metabolic and Molecular Bases of Inherited Disease, 7th ed, New York, McGraw-Hill, ((1995) ), 1147. |
[50] | Haust D.M. and Gordon B.A. , Possible pathogenetic mechanism in hyperornithinemia, hyperammonemia and homocitrullinuria syndrome, Birth Defects Orig Artic Ser 23: ((1987) ), 17. |
[51] | Wong P. , Lessick M. , Kang S. , et al., Maternal hyperornithinemia-homocitrullinemia-hyperammonemia (HHH syndrome), Am J Hum Genet 45: ((1989) ), A14. |
[52] | Clarke L.A. , Dimmick J.E. and Applegarth D.A. , Pathology of inherited metabolic disease. In Dimmick JE, Kalousek DK, editors: Developmental Pathology of the Embryo and Fetus, Philadelphia, Lippincott ((1993) ). |
[53] | Zammarchi E. , Ciani F. , Pasquini E. , Buonocore G. , Shih V.E. and Donati M.A. , Neonatal onset of hyperornithinemia-hyperammonemia-homocitrullinuria syndrome with favorable outcome, Clin Lab Outcomes 131: ((1996) ), 440. |
[54] | Weinzimer S.A. , Stanley C.A. , Berry G.T. , Yudkoff M. , Tuchman M. and Thornton P.S. , A syndrome of congenital hyperinsulinism and hyperammonemia, J Pediatr 130: (4) ((1997) ), 661. |
[55] | Stanley C.A. , Lieu Y.K. , Hsu B.Y.L. , Burlina A.B. , Greenberg C.R. , Hopwood N.J. , Perlman K. , Rich B.H. , Zammarchi E. and Poncz M. , Hyperinsulinism and hyperammonemia in infants with regulatory mutations of the glutamate dehydrogenase gene, New Eng J Med 338: ((1998) ), 1352–1357. |
[56] | Cox R.P. and Dancis J. , Errors of lysine metabolism. In Scriver CR Beaudet AL Sly WS Valle D, editors: The Metabolic and Molecular Bases of Inherited Disease, 7th ed, New York, McGraw-Hill, ((1995) ). |
[57] | Simell O. , Lysinuric protein intolerance and metabolite transport. In Advances in Inherited Urea Cycle Disorders, Vienna, Austria, ((1997) ), 20–21. |
[58] | Sebastio G. , Nunes V. , Lysinuric Protein Intolerance. Editors In: Pagon RA, Adam MP, Ardinger HH, Wallace SE, Amemiya A, Bean LJH, Bird TD, Fong CT, Mefford HC, Smith RJH, Stephens K, editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2016. 2006 Dec 21 [updated 2011 Oct 13]. |
[59] | Kaiser-Kupfer M. , De Monasteri F. , Valle D., Walser M. and Brusilow S., Visual results of a long-term trial of a low arginine diet in gyrate atrophy of the choroid and retina, Ophthalmology 88: ((1981) ), 307. |
Figures and Tables
Fig.1
Defects in catalytic urea cycle enzymes.
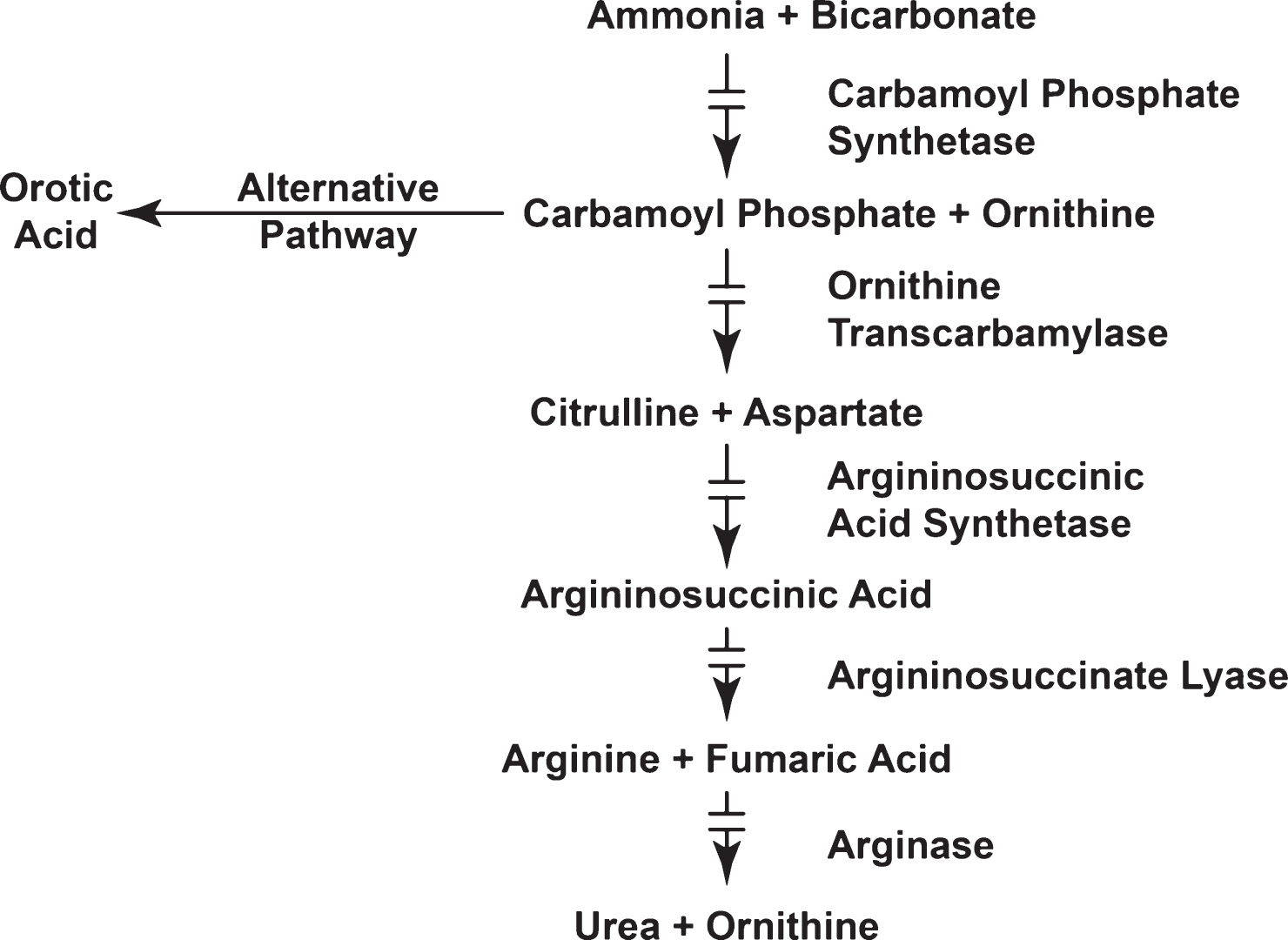
Fig.2
Pathophysiologic mechanisms responsible for hyperammonemic encephalopathy.
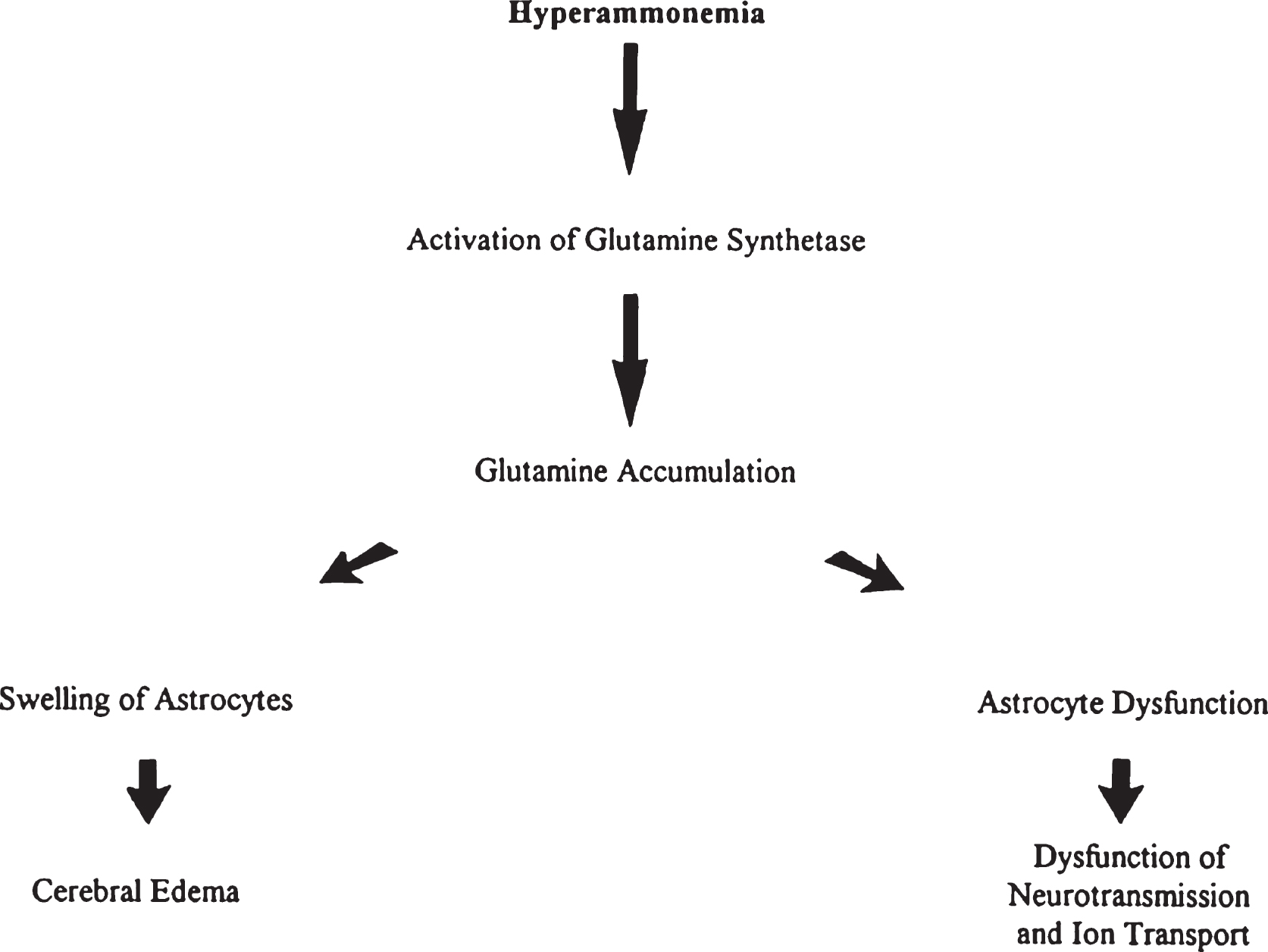
Fig.3
CPS deficiency. Microscopic section of the cortex of the brain showing Alzheimer type II astrocytes in the white matter. Nuclei are swollen and appear as naked nuclei.
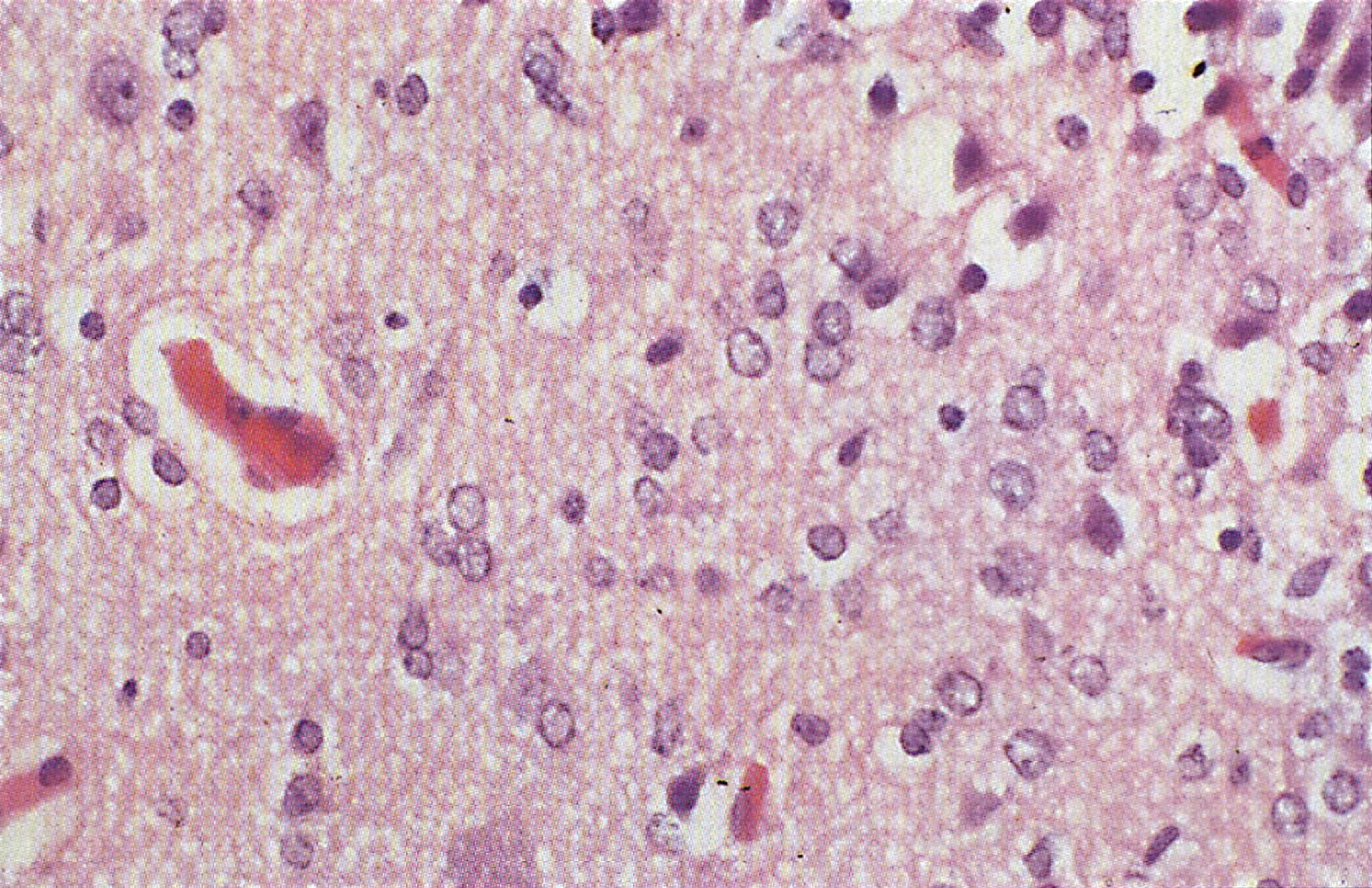
Fig.4
CPS deficiency. Microscopic section of cortex showing clusters of glial cells around neurons— “eggs in a basket” pattern.
Fig.5
Microscopic section of liver in OTC deficiency. Microsteatosis of the hepatocytes in present.
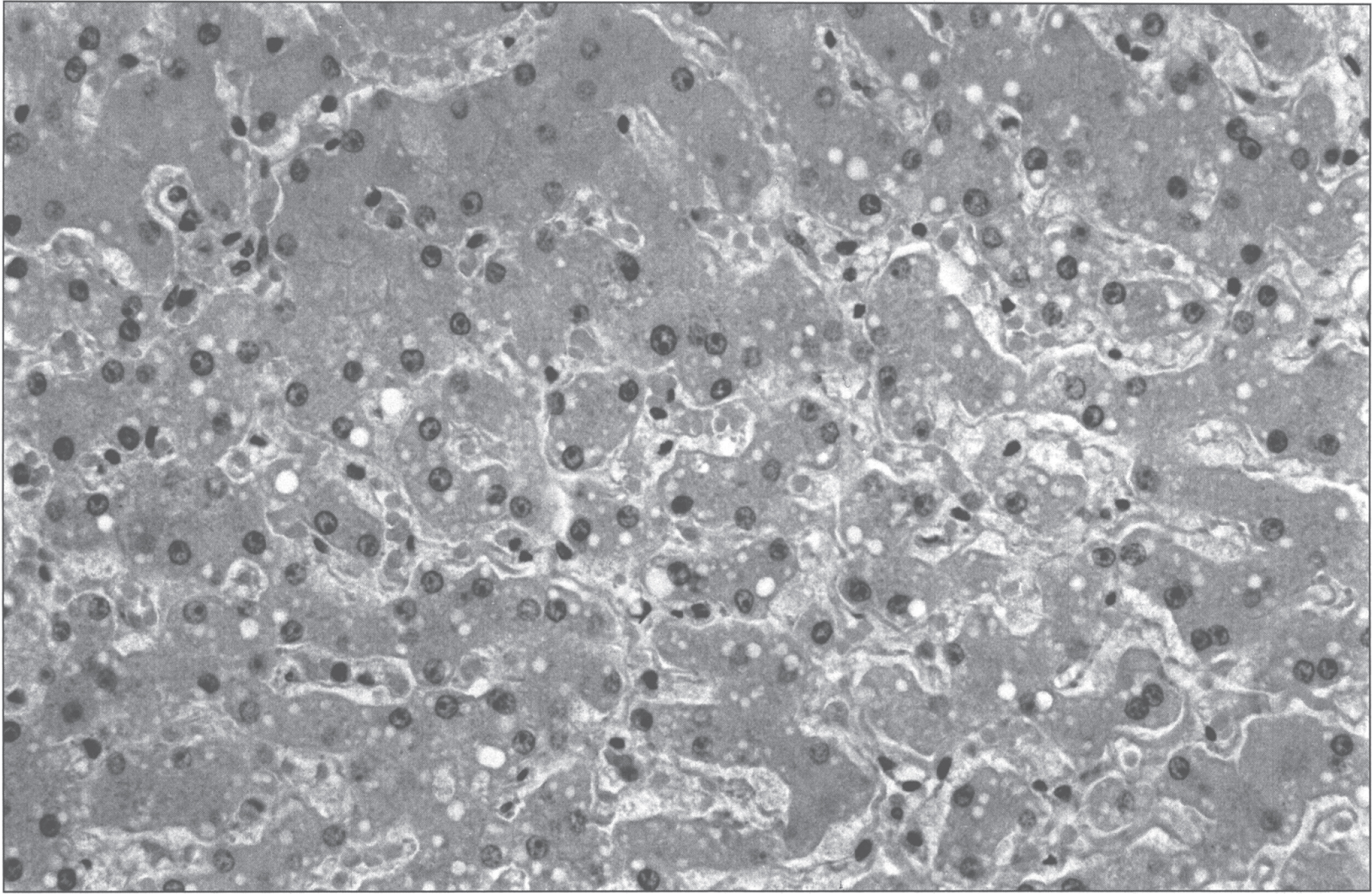
Fig.6
Conversion of arginine to ornithine and urea by the enzymatic activity of arginase.
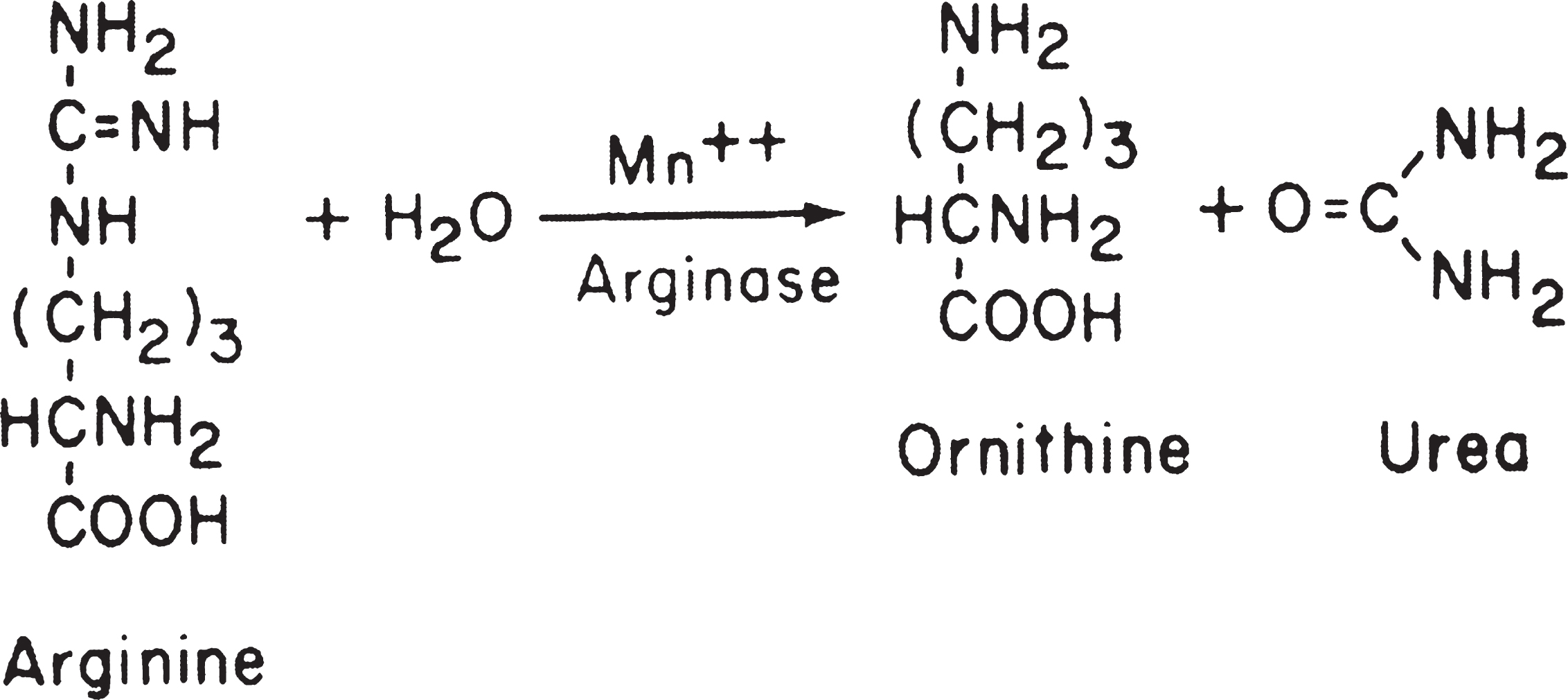
Fig.7
HHH syndrome. Increased number of mitochondria in the perinuclear region of a hepatocyte display altered forms and length. The long forms show tubules running parallel to the longitudinal axis, a thick structure applied to the inner mitochondria memberane (arrow) with sieve-like appearance on a slightly tangential cut (double arrow), and several small bulges protruding from the surface (arrowheads). Note the peripheral arrangement of the tubules on cross-sections. (Courtesy of Dr. M. Daria Haust.)
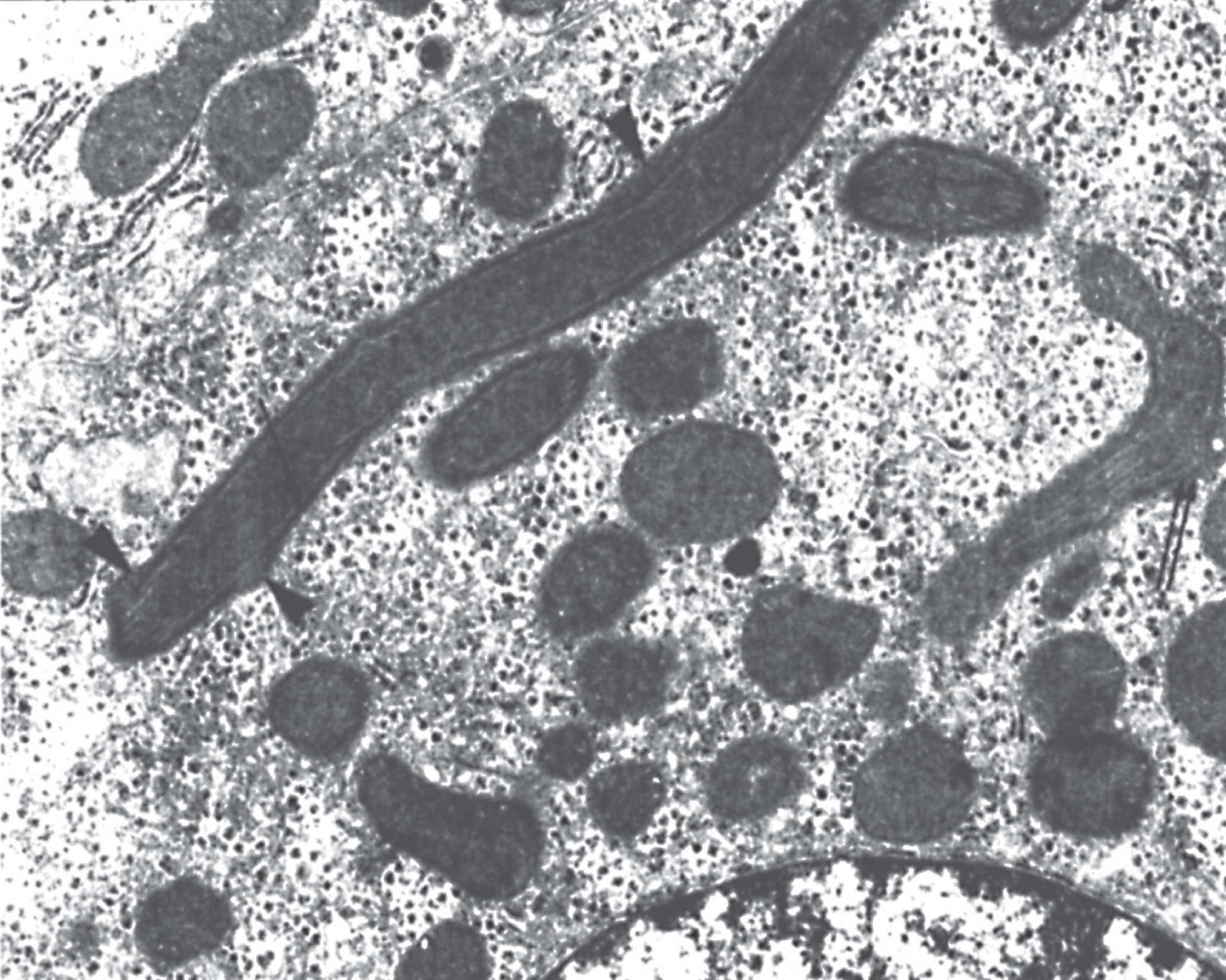
Fig.8
Microscopic section of lung showing alveolar proteinosis as may be seen in lysinuric protein intolerance. The alveoli are filled with proteinaceous material that is periodic acid-Schiff (PAS) positive.
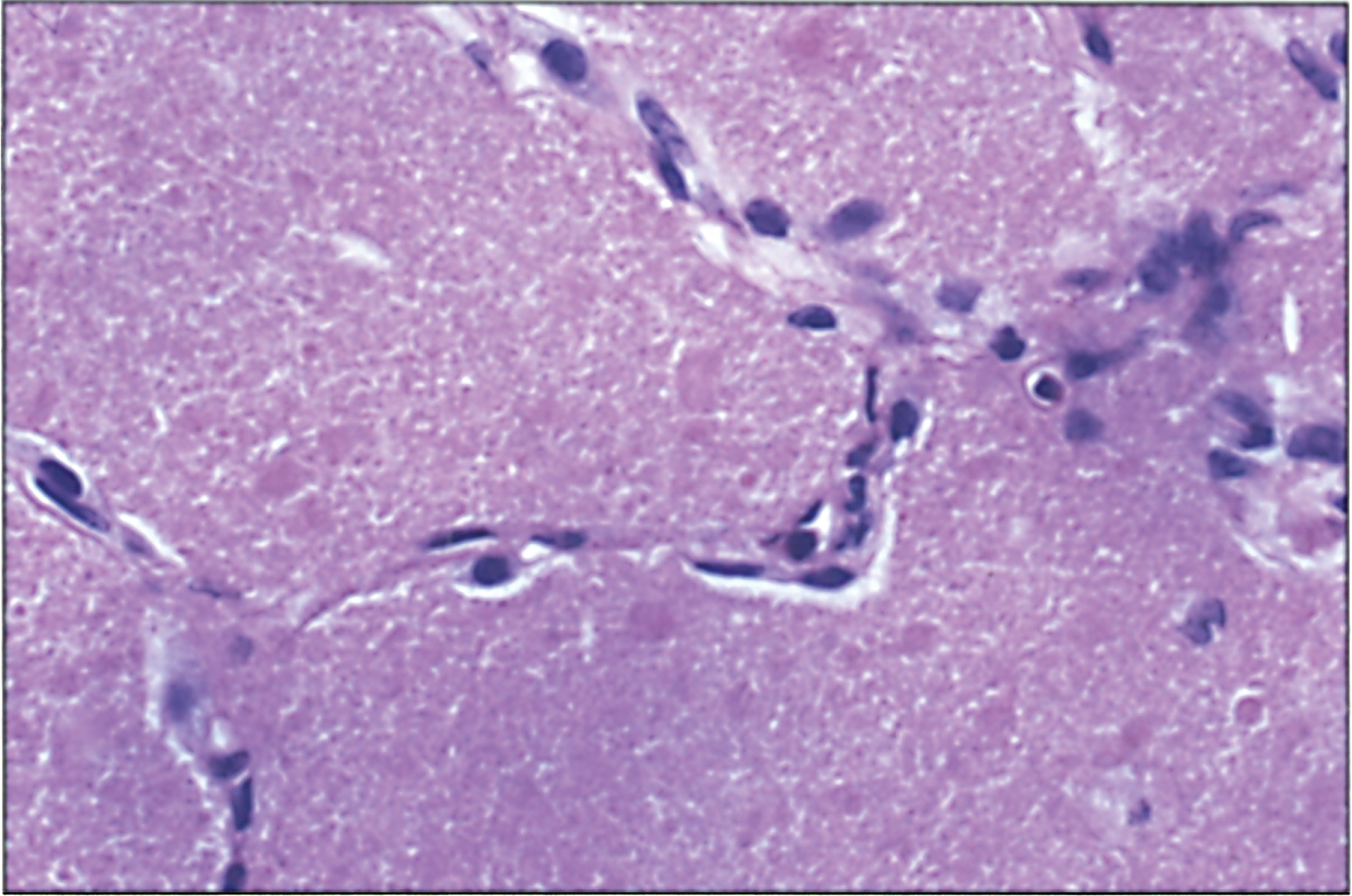
Table 1
Urea cycle
| Catalytic Enzyme Defect | Reaction Catalyzed |
| Carbamoyl phosphate synthetase | Ammonia + bicarbonate + 2ATP ⟶ carbamoyl phosphate + 2ADP + Pi |
| Ornithine transcarbamylase | Carbamoyl phosphate + ornithine ⟶ citrulline + Pi |
| Argininosuccinate synthetase | Citrulline + aspartate + ATP ⟶ argininosuccinic acid + AMP + pyrophosphate |
| Argininosuccinate lyase | Argininosuccinic acid ⟶ arginine + fumaric acid |
| Arginase | Arginine + water ⟶ urea + ornithine |
Table 2
Differential diagnosis of hyperammonemia
| Urea Cycles Enzyme Defects |
| Carbamoyl phosphate synthetase (CPS1) deficiency |
| Ornithine transcarbamylase (OTC) deficiency |
| Argininosuccinate synthetase (AS) deficiency (Citrullinemia) |
| Argininosuccinate lyase (AL) deficiency (argininosuccinic acidura (ASA)) |
| Arginase deficiency (Argininemia) |
| N-acetylgutamate synthase (NAGS) deficiency |
| Transport Defects |
| Citrin deficiency (Citrullinemia II) |
| Hyperammonemia-Hyperornithinemia-Homocitrullinuria (HHH) Syndrome |
| Lysinuric protein intolerance (LPI) |
| Fatty Acid Oxidation Defects |
| Medium-chain acyl-CoA dehydrogenase (MCAD) deficiency |
| other Acyl-CoA dehydrogenase deficiencies |
| Carnitine Palmitoyltransferase 2 (CPT2) deficiency |
| Organic Acidemias |
| Propionic acidemia |
| Methylmalonic acidemia |
| Isovaleric acidemia |
| Glutaric acidemia II |
| Others |
| Hyperinsulinism/Hyperammonemia |
| Congenital lactic acidosis/Energy Metabolism Defects |
| ... ... ... ... ... ... ... ... ... ... ... ... |
| Acquired |
| Transient hyperammonemia of newborn |
| Reye syndrome/Reye-like Syndrome |
| Liver failure |
| Herpes simplex in newborn |
| Short-gut Syndrome |
| Arginine Deficiency |
| Toxins/Poisenings |
Table 3
Estimation of the incidence of each urea cycle disorder (per Live Births)
| Enzyme Deficiency | Incidence |
| Carbamoyl phosphate synthase and N-acetylglutamate synthase | 1 per 975 000 |
| Ornithine transcarbamylase | 1 per 63 000 |
| Argininosuccinate synthetase | 1 per 250 000 |
| Argininosuccinate lyase | 1 per 220 000 |
| Arginase | <1 per 1 000 000 |
| All urea cycle disorders | 1 per 35 000 |
Table 4
Diagnostic clues for urea cycle defects
| Carbamoyl Phosphate Synthetase Deficiency |
| 1. Coma with extreme hyperammonemia after first day of life |
| 2. Partial deficiency later depending on environmental stressors |
| 3. Acidosis or ketosis absent at least initially |
| 4. Low plasma citrulline level |
| 5. Low urinary orotic acid level |
| 6. No significant evidence of liver disease |
| N-Acetylglutamate Synthetase Deficiency |
| 1. Hyperammonemic Episodes / Coma typically after neonatal period |
| 2. Fatty liver |
| 3. Hyperammonemia, no acidosis or ketosis |
| Ornithine Transcarbamylase Deficiency |
| 1. Coma with extreme hyperammonemia after first day of life in males |
| 2. Altered mental status, irritability, lethargy, vomiting, hepatomegaly and transaminase elevation |
| At any time during life depending on environmental stressors in partial deficiency |
| 3. Acidosis or ketosis absent at least initially |
| 4. High plasma glycine and alanine level |
| 5. Low plasma citrulline level |
| 6. High urinary orotic acid level |
| 7. High urinary orotidine level after oral allopurinol in heterozygous females |
| Argininosuccinate Synthetase Deficiency |
| 1. Coma with extreme hyperammonemia after first day of life in newborn or |
| 2. Episodic symptoms in childhood due to hyperammonemia |
| 3. Acidosis or ketosis absent at least initially |
| 4. Plasma citrulline level 50–100 x normal |
| 5. Decreased arginine level in plasma and possibly increased orotic acid level in urine |
| 6. No significant evidence of liver disease |
| Arginosuccinate Lyase Deficiency |
| 1. Coma with extreme hyperammonemia after first day of life |
| 2. Psychomotor retardation / intellectual disability even if no history of hyperammonemia |
| 3. Possible hepatomegaly, abnormal liver function tests, coagulopathy with fibrosis on liver biopsy |
| 4. Dry and short brittle hair (in 50% of patients) |
| 5. Acidosis or ketosis absent at least initially |
| 6. Elevated plasma citrulline levels typically in the 100–300μmol/L range |
| 7. Plasma and urine argininosuccinate and anhydrides and low plasma arginine level |
| Argininemia |
| 1. Irritability and developmental delay during infancy with subsequent progressive neurologic damage |
| 2. Spasticity especially in lower extremities |
| 3. Elevated arginine level |
Table 5
Plasma amino acids and urinary orotic acid and tissue for enzyme analyses in the urea cycle disorders
| Enzyme Deficient | Plasma Amino Acids | Urine Orotic Acid | Tissue for Enzyme Analysis |
| Carbamoyl phosphate synthetase (CPS) | ↑glutamine | Normal | Liver |
| ↑alanine | |||
| ↓citrulline | |||
| ↓arginine | |||
| Ornithine transcarbamylase (OTC) | ↑glutamine | Increase | Liver |
| ↑alanine | |||
| ↓citrulline | |||
| ↓arginine | |||
| Argininosuccinate synthetase (ASS, citrullinemia) | ↑citrulline | Increase | Liver |
| ↓arginine | |||
| Argininosuccinate lyase (ASL) | ↑citrulline | Increase | Red Blood Cells |
| (Argininosuccinic aciduria -ASA) | ↑argininosuccinate | Liver | |
| ↓arginine | |||
| Arginase | ↑arginine | Increase | Red Blood Cells |
| (Argininemia) | Liver | ||
| N-acetylglutamine | ↑glutamine | Normal | Liver |
| synthase (NAGS) | ↑alanine | ||
| Hyperornithinemia, Hyperammonemia, Homocitrullinuria (HHH) | ↑ornithine | ||
| ↑homocitrulline | |||
| Citrin deficiency (Citrullinemia 2) | ↑citrulline | ||
| ↑galactose | |||
| Aminoacidemia | |||
| Hypoglycemia | |||
| Neonatal Cholestasis |
Table 6
Urea cycle and related disorders
| MIM# | Disorder | Gene Locus, | Enzyme | Diagnosis | Symptoms | Pathology |
| Inheritance, | Defect | |||||
| Incidence | ||||||
| 237310 | N-AcetylglutamateSynthetaseDeficiency | 17q21.31AR | NAGS | hyperammonemia low citrulline, high glutamine (blood); mutation analysis | Irritability altered mental status coma | |
| 237300 | Carbamoyl phosphate synthase deficiency | 2q34, AR | CPS | hyperammonemia, low citrulline, arginine, high glutamine (blood); enzyme (liver) mutation analysis | hypotonia, irritability lethargy coma | Alzheimer type II; astrocytosis and spongiosis brain |
| 311250 | Ornithine transcarbamylase deficiency | Xp11.4, XL | OTC | hyperammonemia, orotic acid (urine), low citrulline (plasma); enzyme (liver) mutation analysis | hypotonia, seizures, coma altered mental status irritability | liver: focal necrosis, steatosis; brain; Alzheimer type II, spongiosis |
| 215700 | Citrullinemia | 9q34.11, AR | arginino-succinate synthetase (ASS) | hyperammonemia; ↑citrulline 50–100 fold mutation analysis | same as OTC | same as OTC |
| 207900 | Argininosuccinic aciduria | 7q11.21, AR | arginino-succinate lyase (ASL) | ↑citrulline, argininosuccinate (plasma, urine); enzyme (red blood cells) mutation analysis | same as OTC intellectual disability also without hyperammonemia | same as OTC |
| 207800 | Argininemia | 6q23.2, AR | arginase | ↑arginine, enzyme (liver) mutation analysis | Developmental delay, spasticity | |
| 238970 | Hyperornithinemia hyperammonemia, homocitrullineuria | 13q14.11, AR | SLC25A15, ORNT1 transport defect | hyperammonemia, ornithinemia, homocitrullinuria mutation analysis | hypotonia, developmental delay | |
| 603471 | Citrullinemia 2 Citrin deficiency | 7q21.3, AR | SLC25A13 Citrin transport defect | mutation analysis | neonatal cholestasis irritability, mental status change adults | |
| 222700 | Lysinuric protein intolerance | 14q11.2, AR | SLC7A7 transport defect | transport dibasic amino acids, low serum lysine, high urine arginine, omithine, mutation analysis | hypotonia, osteoporosis, lens opacity Interstitial lung disease |
AR, autosomal recessive; XL, X-linked; ID, intellectual disability; CPS, carbamoyl phosphate synthase; OTC, ornithine transcarbamylase.


