
Post-market surveillance of medical devices: A review
Abstract
BACKGROUND:
Medical devices (MDs) represent the backbone of the modern healthcare system. Considering their importance in daily medical practice, the process of manufacturing, marketing and usage has to be regulated at all levels. Harmonized evidence-based conformity assessment of MDs during PMS relying on traceability of medical device measurements can contribute to higher reliability of MD performance and consequently to higher reliability of diagnosis and treatments.
OBJECTIVE:
This paper discusses issues within MD post-market surveillance (PMS) mechanisms in order to set a path to harmonization of MD PMS.
METHODS:
Medline (1980–2021), EBSCO (1991–2021), and PubMed (1980–2021) as well as national and international legislation and standard databases along with reference lists of eligible articles and guidelines of relevant regulatory authorities such as the European Commission and the Food and Drug Administration were searched for relevant information. Journal articles that contain information regarding PMS methodologies concerning stand-alone medical devices and relevant national and international legislation, standards and guidelines concerning the topic were included in the review.
RESULTS:
The search strategy resulted in 2282 papers. Out of those only 24 articles satisfied the eligibility criteria and were finally included in the review. Papers were grouped per categories: medical device registry, medical device adverse event reporting, and medical device performance evaluation. In addition to journal articles, national and international legislation, standards, and guidelines were reviewed to assess the state of PMS in different regions of the world.
CONCLUSION:
Although the regulatory framework prescribes PMS of medical devices, the process itself is not harmonized with international standards. Particularly, conformity assessment of MDs, as an important part of PMS, is not measured and managed in a traceable, evidence-based manner. The lack of harmonization within PMS results in an environment of increased adverse events involving MDs and overall mistrust in medical device diagnosis and treatment results.
1.Introduction
Medical devices (MDs) prove to be the backbone of the healthcare system nowadays. Given their high significance in maintaining human health, the processes of manufacturing, marketing and usage have been regulated globally and nationally. According to the definitions and requirements stated in directives and regulations, as well as international standards, activities related to MDs can be divided into pre-market processes and post-market surveillance (PMS).
For instance, in the European Union (EU), pre-market and PMS processes have been defined by Medical Device Directives (MDD) [1, 2, 3] since 1992 and recently updated by Medical Device Regulation (MDR) [4, 5, 6]. The directives and regulations define all aspects from ideation, design and development phase, up to testing, approval and certification before the production process, production itself and PMS of medical devices (Fig. 1). To support the implementation of directives and regulations international standards have been developed. For instance, according to the ISO 13485 standard, which manufacturers are obliged to comply with, during the pre-market phase, the manufacturer who produces MDs needs to demonstrate its ability to provide MDs and related services that consistently meet customer and applicable regulatory requirements [7]. During the development phase, MD functionality, safety and performance needs to be designed to satisfy multiple requirements defined in a variety of international standards such as basic safety standards [8], electro compatibility standards [9], biocompatibility standards [10], environmental protection standards [11], etc. Certification process confirms conformity of the producer and the device itself to these requirements. In the EU CE mark is introduced [12] and serves as an administrative marking that indicates conformity with health, safety, and environmental protection standards for products sold within the European Economic Area (EEA). It is issued by the European Notified Bodies [13], organizations appointed by EU countries to assess the compliance of products before they are placed on the market. Therefore, as it can be seen from Fig. 1, regulators authorize independent notified bodies to do the conformity assessment in order to prove the manufacturer’s and product’s compliance with directives and regulations. Similar methodology is used in other regions and countries worldwide.
![Medical device process – from idea to market (pre-market process) [4, 5, 6].](https://content.iospress.com:443/media/thc/2022/30-6/thc-30-6-thc220284/thc-30-thc220284-g001.jpg)
In the United States of America (USA), pre-market and PMS processes have been defined by the Food and Drug Administration (FDA) [14]. Similarly, to EU legislation, FDA also defines all aspects from ideation to PMS and checks compliance with regulation. In the United States, equivalent to Europe’s CE marking is UL (Underwriters Laboratories) marking [15].
MD regulations for Middle East and North Africa (MENA) countries are heterogeneous as MENA region countries derive their regulations from the EU or USA regulations. Following that practice, in the MENA region, compliance with regulatory requirements for production and marketing of MDs is proven by ISO 13485 certificate. As the most developed countries in this region, Saudi Arabia and the United Arab Emirates (UAE) have very good medical device regulations. Saudi Arabia Food and Drug Authority (SFDA) is responsible for regulating the medical device market and is doing so through Medical Devices Interim Regulations (MDIR) from 2008. The responsible authority for medical devices in the UAE is the Ministry of Health whose legal basis founded upon the International Medical Device Regulators Forum (IMDRF) as well as EU Medical Devices Directives [16].
Generally, in the countries worldwide, MDs undergo a distinctive process from ideation, design and development phase, up to testing, approval and certification before the production, the production itself and PMS of MDs. Administrative marking that indicates conformity with health, safety, and environmental protection standards is introduced differently from country to country. For example, in China, CCC (China Compulsory Certification) [17] marking is used for labeling of approved devices. Japan uses PSE (Product Safety Electrical Appliance & Material) marking [18]. In Australia, there are various types of CE markings, or their equivalents used [19]. Since leaving the EU, the United Kingdom (UK) has introduced the new mark UKCA (United Kingdom Conformity Assessed) which is enforced from January 1st, 2021 [20]. Canada enforces its own CSA (Canadian Standards Association) marking [21]. In all regions, the manufacturer governs the overall process from ideation to market, whereas independent authorized third parties check for compliance, give approval for commercialization and certification before placing the device on the market. So, it is evident that MDs are produced in a strict process harmonized with international standards, and put on the market after authorization and marking.
After market placement a PMS mechanism is initiated. PMS is a collection of processes and activities used to monitor the performance of a medical device once it is placed in healthcare institutions. These activities are designed to generate information regarding the utilization of the device, to expediently identify device design and/or usage problems and accurately characterize the device behavior in practice [22]. Despite the fact that premarket processes are very well defined and standardized and that PMS is conducted by manufacturers or distributors as defined within directives and regulations, it is not unlikely for MDs to cause an error during their usage resulting in patient injury or in the worst-case scenario resulting in patient death [23]. The fact is that there is no difference in medical devices between countries however their performance differs which is noticeable by the number of incidents reported. So, if medical devices are the same the only cause of such state can be found in different approaches to management including preventive service and surveillance. This indicates that current PMS processes possibly have issues.
This paper discusses issues within MD PMS mechanisms recognized through a research review of scientific articles, as well as national and international legislation and standard databases.
2.Methodology
Preferred Reporting Items for Systematic Reviews and Meta-Analyses (PRISMA) methodology was used for the study [24]. Two researchers (LSB and AD), who were blinded to the articles’ author information, conducted the study inclusion, data extraction, and assessment of the risk of bias independently. Other authors (LGP and AB) reviewed the extracted data and were consulted in case of disagreements.
2.1Search strategy
Medline (1980–2021) [25], EBSCO (1991–2021) [26], and PubMed (1980–2021) [27] as well as national and international legislation and standard databases along with reference lists of eligible articles and guidelines of referent regulatory authorities such as European Commission, Food and Drug Administration and national regulatory bodies were screened in the time period from August to October of 2021 with an aim of identifying data sources that have evaluated the importance of MD PMS.
The following search criteria for scientific article databases was used to limit the research:
a) Journal articles that contain keywords: standard, post-market medical device, FDA, EU;
b) Journal articles that concern stand-alone devices (i.e. not implantable devices);
c) Language of publication: English;
d) Publication time: unlimited;
e) Full-text journal publications;
The reference lists of the included studies were checked for other relevant studies and authors were contacted if necessary.
Duplicates from the initial search results were removed using EndNote 20X (Clarivate analytics) and additionally persisting duplicates were manually removed. The sifting of the articles was performed in three stages: by title, by abstract, and finally by full text against the exclusion criteria. Finally, LSB and AD independently screened the remaining studies firstly by title, then keywords and finally abstract. After this step, the full texts of the studies were selected, and the content was compared against the inclusion and exclusion criteria. The main consideration for inclusion of a study was mentioning PMS in the body of the article in a reflective manner.
2.2Data extraction and outcomes of interest
After the search was finalized, two authors have extracted the vital information from the selected studies in the following manner:
• Literature data (first author, publication date);
• Region of the world;
• The problem of PMS evaluated by the study;
• Information regarding the standard or guideline used in the study (if any).
The authors have independently grouped the studies into three groups of interest: (1) medical device registry, (2) adverse event reporting, and (3) performance evaluation. Figure 2 represents the PRISMA guided flow of the review.
Figure 2.
PRISMA flowchart of literature search: Included/excluded titles, abstracts, and full papers.
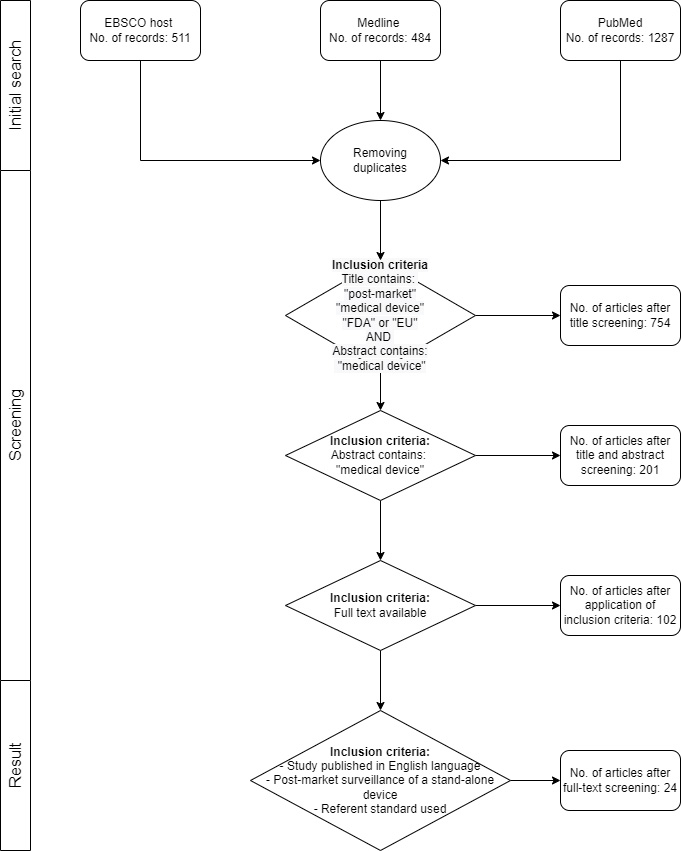
The initial search has returned a total of 2282 articles. After the removal of duplicates and applying the inclusion criteria, a total of 24 papers (Supplementary Table 1.) were included in the meta-analysis. Out of the 24 articles included in the review, 3 of them are dealing with the topic of medical device registry, 3 with adverse event reporting, and 18 with performance evaluation. Recognized limitations of the study can be seen in the selection and exclusion of articles.
3.Results
COVID-19 is the latest in a series of cases to show that raising the global awareness and knowledge about the importance of respecting the essential requirements is a precondition for assuring the appropriate quality, performance, and safety of medical products, especially in case of high demand. The most prominent example of consequences brought upon by the lack of PMS are mechanical ventilators that have been placed on the market without being tested in a clinical setting [28]. All these devices have successfully passed all steps necessary for their placement on the market, however, they have been proven to be faulty in clinical settings. In countries where PMS has not been properly regulated, such devices have been found in healthcare facilities and were actively used on patients. Today, therefore, lawsuits are being filed in many countries against those responsible for the deaths of many patients [29, 30, 31]. As regulators are becoming much more focused on MD PMS and risk management, this review presents the result of the analysis of PMS globally and provides insight into the future work that needs to be done in addressing the found issues within PMS mechanisms. This is important to address because the healthcare sector is one of the most important aspects for the functionality of each country. As such, the healthcare system directly affects the life of each individual and takes a lot of national resources to govern.
The Global Harmonization Task Force (GHTF) [32] discussed PMS and defined conformity assessment principles for MDs with a scope to present and introduce a problem of non-overseen medical devices. They suggested procedures that could be performed by manufacturers to maintain a proper function of their devices on the market. Their system was based on establishing a quality management system (QMS) before the device is even placed on the market, to ensure that the manufacturer will in fact proceed with conformity assessments even after the device is marketed. This proposed system included handling of the complaints by customers, handling the device malfunction reports and providing preventive and corrective actions for the device maintenance. Conclusions were drafted in a report, which defined the specific tasks needed for PMS in the industry and discussed how the requirements for each task could be harmonized across regulatory environments. This report was later updated by the International Medical Device Regulators Forum (IMDRF) [33] to elaborate on reporting guidelines for adverse events. IMDRF has provided a document with a scope to guide their members when to exchange the information about the device, what procedures to follow during this phase and which forms to use to fill a medical device report.
In 2020, the World Health Organization (WHO) published a draft guidance of a proposed PMS plan to be done by the manufacturers [34]. Their proposed plan includes information about how and when to do PMS and what to do based on the data obtained. Their plan consists of following points:
• Manufacturer must state which types of their medical devices are undergoing PMS;
• Manufacturer should state the exact method of data collection through the device’s lifecycle;
• Manufacturer must state the exact method of data analysis on the obtained information;
• Manufacturer must use the obtained results in a way to improve its development phase and decrease possible risks associated with.
The recommendations of these international organizations have been taken into consideration by regulators since PMS is included as part of legislation concerning MDs globally. Supplementary Table 2 presents the list of legislation for MD PMS worldwide. As it can be seen from the Supplementary Table 2, existing regulations on PMS generally oblige stakeholders to monitor the quality, performance, and safety of a device throughout the product lifecycle and to apply corrective or preventive actions when necessary. PMS generally relies on manufacturers, clinicians, and patients to report incidents including medical devices used to assess experience gained from medical devices that have been placed on the market, as well as to determine the need for any action. The PMS strategy generally includes medical device registry, adverse event reporting and medical device performance evaluation. This has been found out in the review of scientific articles in this field. The results of the search in PubMed, EBSCOhost and Medline databases regarding these topics are summarized in Supplementary Table 1. General comments are presented below for each category.
3.1Analysis of the current state in medical device PMS
3.1.1Medical device registry
Generally, countries worldwide have successfully implemented medical device registry through national authority that is usually within the jurisdiction of a body called “Medicine Agency”. As it can be concluded from the review, professionals in the field agree that establishing a national medical device registry is the first step towards creating a PMS [35]. However, the steps towards achieving a functional PMS system go beyond just a creation of a comprehensive database containing all MD manufacturers, distributors, and MDs on the market. For achieving an effective MD PMS establishing a framework of operation is necessary [35, 36, 37]. In their research, Bisdas et al. [36] concluded that patient registries are preferred over medical device registries because they offer more information with respect to risk stratification. Kramer et al. [37], have pointed out that MD registries streamline the procedure of surveillance and diminish the risk of faulty medical devices. Based on this, one can conclude that both patient and medical device registries are equally important to risk stratification in the healthcare sector to ensure safety and quality of care provided to patients.
The analysis of regional/national legislation [38, 39, 40, 41, 42, 43, 44, 45, 46, 47, 48] suggests that authorized regulatory bodies, such as the Medicines Agency, maintain medical devices and medicines registries. According to the legislation, manufacturers/distributors are obliged to make a registration of their products within the registry upon import and market placement. In this process national regulatory bodies actually do the initial registration and take the results of initial product conformity assessment made by independent third-party bodies. This registration is a prerequisite for placement of MD on the national market, and the placement itself is prerequisite for all other PMS strategies which follow MD on the market. In some countries, who adopted different schemes of PMS, as will be shown in part c), different medical device registries exist [49, 50, 51]. Gurbeta et al. [50] explain how an independent inspection body performing PMS developed digital medical device registries for MD safety and performance inspection management. This registry, in particular, contains only information on 11 different groups of medical devices, but the data on safety and performance serves as good input for artificial intelligence tools which is seen as the next step in MD management globally. This opens the potential of enhancing medical device registries in the future.
3.1.2Adverse event reporting
Reporting adverse events, especially in the case of high-risk MDs is a responsibility that medical professionals owe to their patients. This contributes to MD registries as it enriches the pool of information engineers can use to further improve products and PMS mechanisms [52]. Legislation worldwide foresees clinicians in their duty to report any adverse events, or near adverse events. Manufacturers are also required to do a follow up of these reports [53]. Manufacturer and User Facility Device Experience (MAUDE) database regulated by the FDA and European Databank on Medical Devices (EUDAMED) database provide information regarding reported adverse events in healthcare institutions.
Our review reports a study conducted by Kvanagh et al. [54] which showed that 96.6% of adverse events in the US are reported by the manufacturers themselves while the patients and their families have been found to submit adverse event reports five times more often than physicians even though physicians should be the ones governing adverse-event reporting. By comparing results for US and UK the Kvanagh et al. [54] concluded that the regulatory mechanism for adverse event reporting is better in the UK. Based on this, one can conclude that an unambiguous regulatory framework is needed for effective PMS. Also, to prevent discrepancies among countries harmonization within methodology of adverse event reporting is needed. GHTF, IMDRF and WHO set a very good reference for PMS principles including adverse event reporting that now need to be harmonized globally to reach its full effect. On the other hand, this adverse event reporting part of PMS unfortunately means that some unfortunate event involving MD had taken place and that possibly a harm to a patient has been done. To minimize the rate of these events, MD performance should be monitored through their usage in healthcare institutions.
3.1.3Performance evaluation
Collection of information regarding the utilization of the device and the device behavior in practice is the third key component of a PMS mechanism. Kumar et al. [55] highlight the importance of extracting data and signals related to the performance of medical devices on the market. According to the current regional/national legislation this part of the PMS mechanism is a formal obligation of the manufacturer/distributor. The study by Ross et al. [56] reports that out of 47 manufacturers required to approach performance inspection of their MDs, only 22 responded which is less than 50%. This report is a sound for alarm as it shows that manufacturers are not so consistent in MD performance evaluation once the MDs are placed in healthcare institutions. The performance evaluation is done during the preventive maintenance, assuming that the healthcare institution is not doing that ‘in-house’ or during corrective maintenance when some damage potentially has already been done. Also, given that manufacturers have different distributors in different countries the collection of information regarding the utilization of the device and the device behavior in practice is aggravated. This study highlights an important issue in the PMS mechanism, and that is the lack of harmonized evidence-based methodology that will allow collection of MD performance data once they are used in healthcare institutions on a regular basis in order to assess the device behavior in practice and in different environments. This has already been recognized by the professional community. For instance, Tarricone et al. [57] conducted a review of methods used for MD PMS and concluded that well developed strategies for PMS would significantly reduce the risks of medical devices.
This is seen by the results of existing research focusing on MD performance evaluation. For instance, an analysis of results of PMS in the US, conducted in 2012, found that, most commonly, recalls on the US market were due to software failures in medical devices [58]. In the study by Badnjevic et al. [59] it has been reported that 30% of tested mechanical ventilators and infant incubators are not operating properly and should be serviced, recalibrated and/or removed from daily application. Gurbeta et al. [60] reported that 13.84% of tested anesthesia machines and 14.91% of defibrillators device performance is not in accordance with requirements and should either have the device removed from use or scheduled for corrective maintenance. On the other hand, for dialysis machines [61], the results show that 12.6% of inspected devices do not meet electrical safety requirements or have performance outside the specifications. Specifically, 2% of tested devices did not pass the safety inspection in accordance with IEC 60601. For approximately 22.64% of devices that do not meet the performance specification, malfunction of heating systems was detected. Additionally, 11.32% of devices from this group had performance that was not in accordance with device specifications although malfunction was not reported.
These specific studies on performance evaluation of MDs which have been used and managed within healthcare institutions best show how much the devices actually deviate from the reference values and how important it is to develop harmonized evidence-based methodology that will allow collection of MD performance data once they are used in healthcare institutions on a regular basis in order to assess the device behavior in practice and in different environments and prevent adverse events.
The conducted review study resulted in following PMS gaps recognized:
• Limited function of existing medical device registries as currently they have administrative purpose – only facilitating registration and import of medical devices;
• Adverse event reporting mechanism is managed differently globally with varying response of stakeholders;
• MD performance evaluation reveals significant variation in MD performance;
• Utilization of MDs and their performance evaluation currently cannot be assessed with great certainty since data collection and performance evaluation is not done in the same manner through healthcare institutions.
The PMS mechanism is therefore the whitespace for further improvements but with direct impact on quality of care in healthcare institutions and patient safety.
4.Discussion
Comparison of regional/national regulations in the field of PMS worldwide depicts a significant level of heterogeneity and fragmentation when it comes to MD performance evaluation. This review suggests that MD PMS mechanism, unlike pre-market, lacks harmonization. Crucial components of PMS being medical device registry, adverse event reporting and post-market performance evaluation should be harmonized, with the same approach as it has been done for MD pre-market processes, in order to achieve specific goal of PMS and that is increased quality of care to patients.
Figure 3.
White-space for standardization and harmonization of PMS framework.
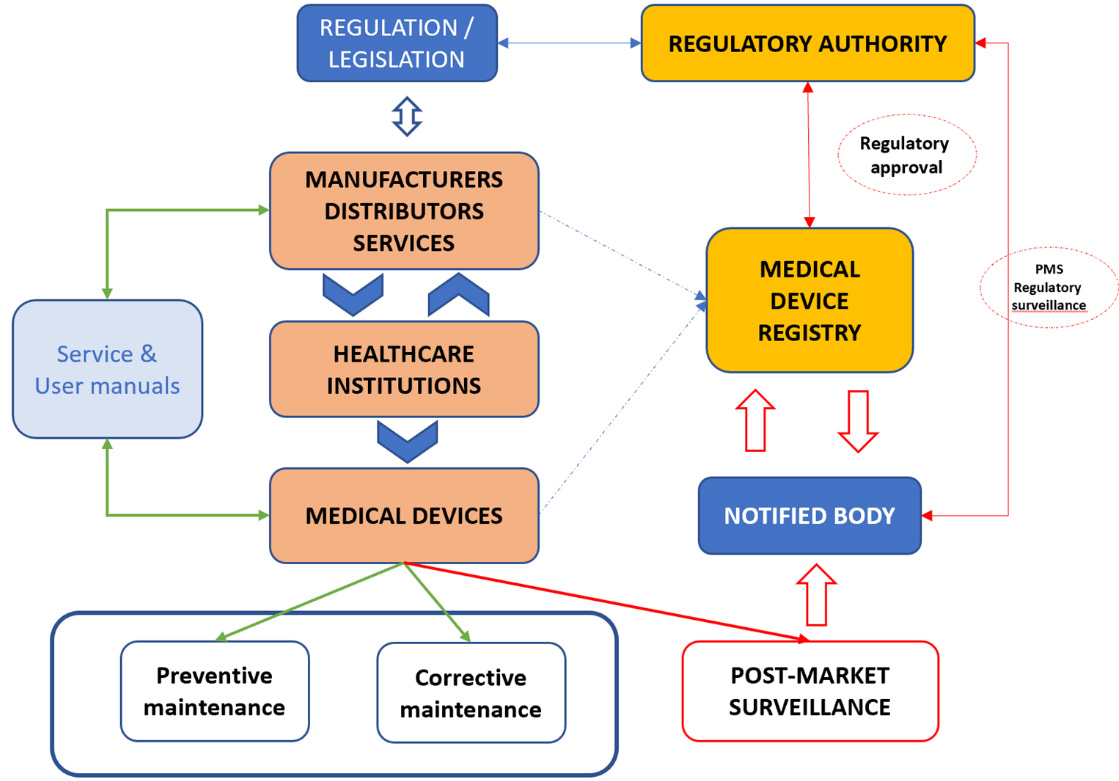
Figure 3 presents the current PMS mechanisms depicting key stakeholders. The most noticeable feature is that under current PMS MD performance evaluation is done by the manufacturer, as mentioned before. The fact is that standard ISO 13485:2016 [7] requires manufacturers to maintain a PMS system regardless of how their medical device is classified. However, this approach has its limitations which can be seen through variability of MD performance as reported in studies mentioned above [59, 60, 61, 62, 63, 64], as well as by the number of reported incidents of the patient injuries and deaths including medical devices [65, 66, 67]. Given these facts, one can conclude that current status of PMS results in duplication of resources and human effort, lack of strategic coordination, mistrust in medical performance and reliability as well as poor responses to global challenges such as the COVID-19 pandemic which only emphasizes the need for harmonization of MD performance evaluation during PMS.
What is interesting to see is that some countries in the world have recognized this issue and have taken steps toward answering the recognized issues. For instance, in China, aside from reports on MDs made by manufacturers, new State Council Order Number 739 [68] enforces more stringent PMS which resulted in more field inspectors in different levels from manufacturing sites to research and development sites, distribution sites and the sites of device usage (healthcare institutions) to see if requirements for every specific device have been met. Additionally, China has developed MD testing institutes on various levels to ensure PMS of MDs [69]. Japan’s regulatory body in charge of development and implementation of safety standards for drugs and medical devices prescribes the PMS which relies on adverse event reporting, information storing and analysis with the aim of ensuring the safety of MDs in healthcare institutions. What is interesting to see is that in Japan novel methodologies such as data mining and sentinel medical institution networks have been considered as a way to enable effective track of MDs, providing prompt response to potential problems and performing preventive activities to ensure safety of all medical devices [70, 71].
Some countries have bridged the identified issues regarding PMS and MD performance evaluation by expanding the regulatory framework and introducing MDs within legal metrology [64]. This approach has been implemented in Spain, Portugal, Saudi Arabia, Republic of Serbia, Bosnia and Herzegovina, Chezch Republic, etc. [72]. According to this legal metrology framework, an independent authorized third body measures MD performance through periodic inspections and the results of inspection are reported in the medical device registry developed for this purpose. Particularly, in Republic of Serbia and Bosnia and Herzegovina independent inspection bodies perform performance evaluation of 12 types of MDs: ECG devices, defibrillators, patient monitors, infant incubators, therapeutic ultrasounds, dialysis machines, anesthesia machines, mechanical ventilators, infusomats, perfusion pumps, high-frequency surgical units and blood pressure devices [59, 60, 61, 62, 63]. The results of these inspections are reported in medical device registry [49, 50, 51] and are used for MD surveillance. Similarly to the intentions of regulators in Japan, the MD performance evaluation data in this registry is considered as a way to enable effective track of MD performance providing prompt response to potential problems as it is shift to evidence-based surveillance of a specific device without generalization to model or manufacturer taking into account the environment in which MD is used. So far, the analysis of these databases [59, 60, 61, 62, 63] for individual medical devices led to a conclusion that MD performance deteriorates over time and that MDs tend to be out of the accuracy limits while still in use which in some cases cannot be recognized by MD users, but it is risky for the patients who are being treated with such devices.
As is known, standards are suggested practices produced by subject matter experts with the intention of describing the best feasible way to achieve a specific end goal, increase efficiency, and eliminate product failures. Therefore, to undoubtedly ensure MD safety through their usage in healthcare institutions harmonization of their performance evaluation during PMS is needed. Harmonized evidence-based conformity assessment of MDs during PMS relying on traceability of medical device measurements will contribute to higher reliability of MD performance and consequently to higher reliability of diagnosis and treatments (Fig. 4).
Figure 4.
Suggested methodology for post-market surveillance of medical devices.
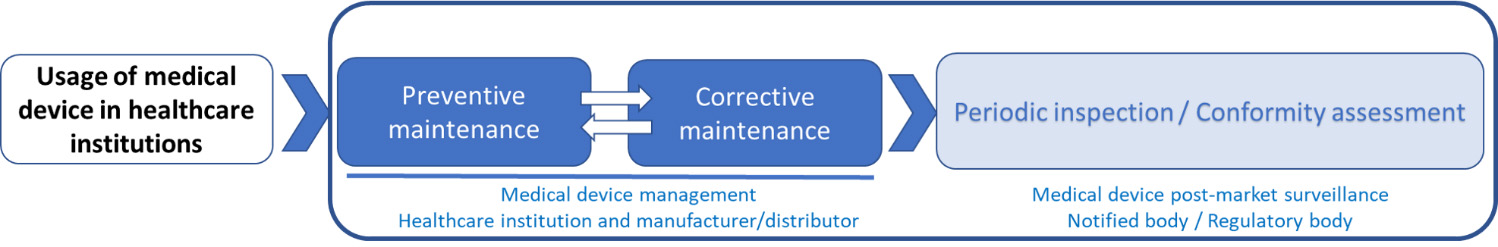
This is not a novel approach since this has been done for a wide range of products in industry. For those devices, for instance, reliability in safety and performance is ensured by periodic inspections or calibration by independent third-party bodies according to the ISO 17020 or ISO 17025 [73, 74].
This method, along with existing preventive service provided by manufacturers would lead to increase in medical device reliability resulting in decrease in adverse events such as injuries and death. It is important to emphasize that mentioned harmonization of PMS would not exclude manufacturers and distributors from doing the PMS. In fact, according to the ISO 17020 an inspection body can be classified in three categories: (1) Type A bodies provide third party services; (2) Type B bodies are separate and identifiable entities, and only provide inspections to its parent company; and (3) Type C bodies are identifiable entities but may not be a separate part of the organization, supply inspection services to parties other than the parent organization. With respect to the EUDAMED or MAUDE database this standardization means that surveillance of MDs can be performed on performance and safety parameters which opens new possibilities toward application of artificial intelligence in this field. As mentioned above, the countries which adopted PMS as part of the legal metrology framework already have national medical device registries with safety and performance measurements [49] which enables data management and introduction of novel tools for data analysis and even performance prediction [75, 76, 77, 78, 79, 80, 81]. However, these medical device registries are not as wide as EUDAMED or MAUDE databases for instance.
5.Conclusion
Based on the conducted review study, it can be seen that MD registry, adverse event reporting and performance evaluation can be improved by strategic work. Harmonization of PMS by adopting evidence-based conformity assessment of MDs during PMS relying on traceability of medical device measurements will not only contribute to higher reliability of MD performance and consequently to higher reliability of diagnosis and treatments but will also open opportunities to improve the function of MD registry by adoption of novel data mining techniques. Some countries have already undertaken steps in this way and adopted PMS regulations that require notified bodies to perform inspection according to international standards for bodies performing testing and inspection.
Given the rising challenges in the healthcare sector MD PMS should be considered more significantly. Strategic collaboration between the professional community and regulatory authorities is needed in order to respond to these challenges. International organizations such as International Federation on Medical and Biological Engineering (IFMBE), European Alliance on Medical and Biological Engineering and Science (EAMBES), American College of Clinical Engineers (ACCE) and others have the expert know-how from the practice that is invaluable for regulators. Such strategic cooperation can result in an environment where MD surveillance is done in a standardized, impartial, evidence-based way. That would ensure that every MDs conformity assessment is derived upon the same parameters regardless of who is performing the evaluation or where, just as is done in pre-market approval of MDs. IEEE has begun to work on harmonization of MD PMS performance evaluation within the Medical Device with Measuring Function (MDMF) group whose first result is the P2727 – Standard for General Vocabulary for Conformity Assessment of Medical Devices with Measuring Function [82, 83].
Conflict of interest
None to report.
Supplementary data
The supplementary files are available to download from http://dx.doi.org/10.3233/THC-220284.
References
[1] | EU Medical Device Directives: Directive 98/79/EC of the European Parliament and of the Council of 27 October 1998 on in vitro diagnostic medical devices. Official Journal of the European Communities, L 331, (December 07, (1998) ). |
[2] | EU Medical Device Directives: Council Directive 93/42/EEC on Medical Devices (MDD). Official Journal of the European Communities, L 169, (July 12, (1993) ). |
[3] | EU Medical Device Directives: Council Directive 90/385/EEC on Active Implantable Medical Devices (AIMDD). Official Journal of the European Communities, L 189, (July 20, (1990) ). |
[4] | EU Medical Device Regulation: Regulation (EU) 2017/745 of the European Parliament and of the Council of 5 April 2017 on medical devices, amending Directive 2001/83/EC. Official Journal of the European Union, L 117, (May 05, (2017) ). |
[5] | EU Medical Device Regulation: Regulation (EC) No 178/2002 of the European Parliament and of the Council of 28 January 2002 laying down the general principles and requirements of food law, establishing the European Food Safety Authority and laying down procedures in matters of food safety. Official Journal of the European Communities, L 31, (February 01, (2002) ). |
[6] | EU Medical Device Regulation: Regulation (EC) No 1223/2009 of the European Parliament and of the Council of 30 November 2009 on cosmetic products. Official Journal of the European Union, L 342, (December 22, (2009) ). |
[7] | International Organization for Standardization. ISO 13485:2016. Medical devices – Quality management systems – Requirements for regulatory purposes. Geneva: ISO; (2016) . |
[8] | International Electrotechnical Commission. IEC 60601. Medical electrical equipment – all parts. Geneva: IEC; (2021) . |
[9] | International Organization for Standardization. ISO 14117:2019. Active implantable medical devices – Electromagnetic compatibility – EMC test protocols for implantable cardiac pacemakers, implantable cardioverter defibrillators and cardiac resynchronization devices. Geneva: ISO; (2019) . |
[10] | International Organization for Standardization. ISO 10993-18:2020. Biological evaluation of medical devices – Part 18: Chemical characterization of medical device materials within a risk management process. Geneva: ISO; (2020) . |
[11] | International Organization for Standardization. ISO 16142-1:2016. Medical devices – Recognized essential principles of safety and performance of medical devices – Part 1: General essential principles and additional specific essential principles for all non-IVD medical devices and guidance on the selection of standards. Geneva: ISO; (2016) . |
[12] | CE marking [Internet]. Brussels: European Comission; [Cited 2021 Oct 10]. Available from: https://ec.europa.eu/growth/single-market/ce-marking_hr. |
[13] | Notified bodies [Internet]. Brussels: European Commission; [Cited 2021 Oct 10]. Available from: https://ec.europa.eu/growth/single-market/goods/building-blocks/notified-bodies_en. |
[14] | Food and Drug Administration (FDA) [Internet]. Silver Spring (MA): Food and Drug Administration; [Cited 2021 Oct 11]. Available from: https://www.fda.gov/. |
[15] | UL (Underwriters Laboratories) marking [Internet]. Northbrook (IL): Underwriter Laboratories; [Cited 2021 Oct 11]. Available from: https://marks.ul.com/about/. |
[16] | Samadi F. Master thesis: “Regulatory requirements of Medical Devices in MENA countries”. Bonn. der Mathematisch-Naturwissenschaftlichen Fakultät der Rheinischen Friedrich-Wilhelms-Universität Bonn. (2015) . |
[17] | Medical device marking in China, CCC (China Compulsory Certification) [Internet]. Beijing: China Compulsory Certification; c2012 [Cited 2021 Oct 11]. Available from: http://www.ccc-cn.org/en/AboutCCC.html. |
[18] | Medical device marking in Japan, Product Safety Electrical Appliance & Material marking [Internet]. Tokyo: Japan Quality Assurance Organization; c2021 [Cited 2021 Oct 12]. Available from: https://www.jqa.jp/english/safety/service/mandatory/pse/. |
[19] | Medical device labelling obligations [Internet]. Canberra: Therapeutc Goods Administration; c2021 [Cited 2021 Oct 12]. Available from: https://www.tga.gov.au/medical-device-labelling-obligations. |
[20] | Guidance Regulating medical devices in the UK [Internet]. London: Medicines and Healthcare Products Regulatory Agency (MHRA); c2020 [Cited 2021 Oct 12]. Available from: https://www.gov.uk/guidance/regulating-medical-devices-in-the-uk#contents. |
[21] | Health Canada Guidance Document: Guidance for the Labelling of Medical Devices, not including in vitro diagnostic devices – Appendices for the Labelling of Soft, Decorative, Contact Lenses and Menstrual Tampons. Health Canada Guidance Document, (June 18, (2015) ). |
[22] | Effective post-market surveillance: Understanding and conducting vigilance and post-market clinical follow-up [Pamphlet]. London (UK): BSI Standards Ltd.; (2014) . |
[23] | Associated Press. Medical devices for pain, other conditions have caused more than 80,000 deaths since 2008. Associated Press. (2018) Nov 25. Available at: https://www.statnews.com/2018/11/25/medical-devices-pain-other-conditions-more-than-80000-deaths-since-2008/. |
[24] | Page MJ, McKenzie JE, Bossuyt PM, Boutron I, Hoffmann TC, Mulrow CD, Shamseer L, Tetzlaff JM, Akl EA, Brennan SE, Chou R. The PRISMA 2020 statement: an updated guideline for reporting systematic reviews. Bmj. (2021) Mar 29; 372. |
[25] | Medline [Internet]. Bethesda (MD): National Library of Medicine (US); [updated 2021 Oct 15; cited 2021 Oct 15]. Available from: https://medline.gov/. |
[26] | EBSCO Host [Internet]. Ipswich (MA): EBSCO Industries; [updated 2021 Oct 15; cited 2021 Oct 15]. Available from: https://search.ebscohost.com/Login.aspx. |
[27] | PubMed [Internet]. Bethesda (MD): National Library of Medicine (US); [updated 2021 Oct 15; cited 2021 Oct 15]. Available from: https://pubmed.ncbi.nlm.nih.gov/. |
[28] | Badnjević A, Pokvić LG, Džemić Z, Bičić F. Risks of emergency use authorizations for medical products during outbreak situations: A COVID-19 case study. BioMedical Engineering OnLine. (2020) Dec; 19: (1): 1-4. |
[29] | Schwendimann R, Blatter C, Dhaini S, Simon M, Ausserhofer D. The occurrence, types, consequences and preventability of in-hospital adverse events – a scoping review. BMC Health Services Research. (2018) ; 18: (1): 1-13. |
[30] | Giardina C, Cutroneo PM, Mocciaro E, Russo GT, Mandraffino G, Basile G, Arcoraci V, et al. Adverse drug reactions in hospitalized patients: Results of the FORWARD (facilitation of reporting in hospital ward) study. Frontiers in Pharmacology. (2018) ; 9: : 350. |
[31] | White SK, Walters AN. Assessing risk by analogy: A case study of us medical device risk management policy. Health, Risk & Society. (2018) Nov 17; 20: (7-8): 358-78. |
[32] | Principles of Conformity Assessment for Medical Devices [Internet]. Global Harmonization Task Force (GHTF); c2000 [Cited 2021 October 14]. Available from: http://www.imdrf.org/docs/ghtf/final/sg1/technical-docs/ghtf-sg1-n40-2006-guidance-ca-principles-060626.pdf. |
[33] | Imgrf.org [Internet]. The International Medical Device Regulators Forum (IMDRF); [Cited 2021 Oct 13] Available from: http://www.imdrf.org/ghtf/ghtf-archives.asp. |
[34] | Guidance for post-market surveillance and market surveillance of medical devices, including in vitro diagnostics. Genewa (SW): World Health Organisation (WHO); (2020) . |
[35] | Al-Surimi K, Househ M, Almohandis E, Alshagathrh F. Establishing a national medical device registry in Saudi Arabia: lessons learned and future work. In Enabling Health Informatics Applications. IOS Press. (2015) . 23-26. |
[36] | Bisdas T, Bohan P, Lescan M, Zeebregts CJ, Tessarek J, van Herwaarden J, et al. Research methodology and practical issues relating to the conduct of a medical device registry. Clinical Trials. (2019) Oct; 16: (5): 490-501. |
[37] | Kramer DB, Parasidis E. Informed consent and compulsory medical device registries: ethics and opportunities. Journal of Medical Ethics. (2021) Feb 19. |
[38] | Medical Devices Regulation (MDR): Regulation (EU) 2017/745 of the European Parliament and of the Council of 5 April 2017 on medical devices, amending Directive 2001/83/EC, Regulation (EC) No 178/2002 and Regulation (EC) No 1223/2009 and repealing Council Directives 90/385/EEC and 93/42/EEC. Official Journal of the European Union, L 117, (May 5, (2017) ). |
[39] | CFR – Code of Federal Regulations Title 21 [Internet]. Silver Spring (MA): United States Food and Drug Administration (FDA), c2021 [Cited 14 Oct 2021]. Available from: https://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfcfr/CFRSearch.cfm?CFRPart=820. |
[40] | Federal Food, Drug, and Cosmetic Act, Section 522 [Internet]. Silver Spring (MA): United States Food and Drug Administration (FDA), c2021 [Cited 14 Oct 2021]. Available from: https://www.regulations.gov/docket/FDA-2011-D-0514. |
[41] | Decree 4 CFDA: Provisions for Medical Device Registration [Internet]. Beijing: National Medicinal Product Administration, c2021 [Cited 14 Oct 2021]. Available from: https://chinameddevice.com/resources3/nmpa-cfda-regulations-3/. |
[42] | Supervision and Administration of Medical Devices as State Council Order Number 739 [Internet]. Toledo: North American Science Associates (NAMSA), c.2021 [Cited 15 Oct 2021]. Available from: https://namsa.com/china-state-council-releases-order-739/. |
[43] | Pharmaceuticals and Medical Devices Act (PMD Act) of November 2015 [Internet]. Tokyo: Pharmaceutical and Medical Device Agency (PMDA), c2021 [Cited 15 Oct 2021]. Available from: https://www.pmda.go.jp/english/. |
[44] | Ministerial Ordinance No. 169 [Internet]. Tokyo: Ministry of Health, Labour and Welfare (MHLW), c2021 [Cited 15 Oct 2021]. Available from: https://www.emergobyul.com/file/3286/download?token=n19oSH_g. |
[45] | Therapeutic Goods Act (TGA) 1989: Compilation No. 72. Federal Register of Legislation, Statutory rules No. 21, (January 22, (2019) ). |
[46] | Therapeutic Goods (Medical Devices) Regulation 200: Compilation No. 32. Federal Register of Legislation, Statutory rules No. 236, (July 12, (2017) ). |
[47] | The UK Medical Device Regulations 2002. UK Statutory Instruments No. 618, (May 20, (2002) ). |
[48] | Medical Device Ordinance (MedDO) [Internet]. Genewa: The Swiss Federal Council [Cited 16 Oct 2021]. Available from: https://www.fedlex.admin.ch/eli/cc/2020/552/en. |
[49] | Gurbeta L, Badnjević A, Kurta E. EVerlab: software tool for medical device safety and performance inspection management. In International Conference on Medical and Biological Engineering. Springer, Cham. (2019) May 16. pp. 429-435. |
[50] | Gurbeta L, Sejdinović D, Alić B, Abd El-Ilah L, Badnjević A, Žunić E. Software solution for tracking inspection processes of medical devices from legal metrology system. In XIV Mediterranean Conference on Medical and Biological Engineering and Computing 2016. Springer, Cham. (2016) . pp. 957-961. |
[51] | Gurbeta L, Badnjević A, Žunić E, Pinjo N, Ljumić F. Software package for tracking status of inspection dates and reports of medical devices in healthcare institutions of Bosnia and Herzegovina. In 2015 XXV International Conference on Information, Communication and Automation Technologies (ICAT). IEEE. (2015) Oct 29. pp. 1-5. |
[52] | Fraser AG, Byrne RA, Kautzner J, Butchart EG, Szymański P, Leggeri I, et al. Implementing the new European Regulations on medical devices – clinical responsibilities for evidence-based practice: A report from the Regulatory Affairs Committee of the European Society of Cardiology. European Heart Journal. (2020) Jul 14; 41: (27): 2589-96. |
[53] | Campbell B. Regulation and safe adoption of new medical devices and procedures. British Medical Bulletin. (2013) Sep 1; 107: (1). |
[54] | Kavanagh KT, Brown RE Jr, Kraman SS, Calderon LE, Kavanagh SP. Reporter’s occupation and source of adverse device event reports contained in the FDA’s MAUDE database. Patient Related Outcome Measures. (2019) ; 10: : 205. |
[55] | Kumar A, Matheny ME, Ho KK, Yeh RW, Piemonte TC, Waldman H, et al. The data extraction and longitudinal trend analysis network study of distributed automated postmarket cardiovascular device safety surveillance. Circulation: Cardiovascular Quality and Outcomes. (2015) Jan; 8: (1): 38-46. |
[56] | Ross JS, Blount KL, Ritchie JD, Hodshon B, Krumholz HM. Post-market clinical research conducted by medical device manufacturers: A cross-sectional survey. Medical Devices (Auckland, NZ). (2015) ; 8: : 241. |
[57] | Tarricone R, Callea G, Ogorevc M, Prevolnik Rupel V. Improving the methods for the economic evaluation of medical devices. Health Economics. (2017) Feb; 26: : 70-92. |
[58] | Kramer DB, Baker M, Ransford B, Molina-Markham A, Stewart Q, Fu K, et al. Security and privacy qualities of medical devices: An analysis of FDA postmarket surveillance. PloS One. (2012) Jul 19; 7: (7): e40200. |
[59] | Badnjevic A, Gurbeta L, Jimenez ER, Iadanza E. Testing of mechanical ventilators and infant incubators in healthcare institutions. Technology and Health Care. (2017) Jan 1; 25: (2): 237-50. |
[60] | Gurbeta L, Dzemic Z, Bego T, Sejdic E, Badnjevic A. Testing of anesthesia machines and defibrillators in healthcare institutions. Journal of Medical Systems. (2017) Sep; 41: (9): 1-0. |
[61] | Gurbeta L, Alic B, Dzemic Z, Badnjevic A. Testing of dialysis machines in healthcare institutions in Bosnia and Herzegovina. In EMBEC & NBC 2017. Springer, Singapore. (2017) Jun 11. pp. 470-473. |
[62] | Gurbeta L, Alic B, Dzemic Z, Badnjevic A. Testing of infusion pumps in healthcare institutions in Bosnia and Herzegovina. In EMBEC & NBC 2017. Springer, Singapore. (2017) Jun 11. pp. 390-393. |
[63] | Gurbeta L, Džemic Z, Badnjevic A. Establishing traceability chain of infusion and perfusor pumps using legal metrology procedures in Bosnia and Herzegovina. In World Congress on Medical Physics and Biomedical Engineering 2018. Springer, Singapore. (2019) . pp. 45-49. |
[64] | Badnjević A, Cifrek M, Magjarević R, Džemić Z, eds. Inspection of medical devices: for regulatory purposes. Springer Singapore; (2018) . |
[65] | Natsume J, Numaguchi A, Ohno A, Mizuno M, Takahashi Y, Okumura A, Noda M, et al. Death review of children receiving medical care at home. Pediatric Research. (2022) ; 91: (5): 1286-1289. |
[66] | Porte PJ, Smits M, Verweij LM, de Bruijne MC, van der Vleuten CPM, Wagner C. The Incidence and Nature of Adverse Medical Device Events in Dutch Hospitals. (2021) . |
[67] | Ryan M, Myers AM, Yustein AS. Reporting Device-Associated Death Events to the FDA. JAMA Internal Medicine. (2022) ; 182: (4): 462-462. |
[68] | China State Council Releases Order 739, Creating New Market Access Opportunities for Global Medical Device and IVD Manufacturers |
[69] | Yang X, Mu R, Fan Y, Wang C, Li D. The current situation and development of medical device testing institutes in China. Expert Review of Medical Devices. (2017) Apr 3; 14: (4): 263-9. |
[70] | Japan MHLW & PMDA Medical Device and Pharmaceutical Regulations [Internet]. Bethesda (MD): Pacific Bridge Medical, c2021 [Cited 16 Oct 2021]. Available from: https://www.pacificbridgemedical.com/regulation/japan-medical-device-pharmaceutical-regulations/. |
[71] | Outline of Post-marketing Safety Measures [Internet]. Tokyo: Pharmaceutical and Medical Device Agency (PMDA), c2021 [Cited 16 Oct 2021]. Available from: https://www.pmda.go.jp/english/safety/outline/0001.html. |
[72] | oiml.org [Internet]. Paris: International organization of Legal Metrology (OIML), c2021 [Cited 16 Oct 2021]. Available from: https://www.oiml.org/en/structure/members/memberslist_view?varMember=1. |
[73] | International Organization for Standardization. ISO/IEC 17020:2012 – Conformity assessment – Requirements for the operation of various types of bodies performing inspection. Geneva: ISO; (2012) . |
[74] | International Organization for Standardization. ISO/IEC 17025:2017 – General requirements for the competence of testing and calibration laboratories. Geneva: ISO; (2017) . |
[75] | Kovačević Ž, Pokvić LG, Spahić L, Badnjević A. Prediction of medical device performance using machine learning techniques: Infant incubator case study. Health and Technology. (2020) Jan; 10: (1): 151-5. |
[76] | Hrvat F, Spahić L, Pokvić LG, Badnjević A. Artificial Intelligence for prediction of medical device performance: Infusion and perfusor pumps case study. In 9th Mediterranean Conference on Embedded Computing MECO. (2020) . |
[77] | Hadžić L, Fazlić A, Hasanić O, Kudić N, Spahić L. Expert System for Performance Prediction of Anesthesia Machines. In International Conference on Medical and Biological Engineering. Springer, Cham. (2019) May 16. pp. 671-679. |
[78] | Badnjević A, Pokvić LG, Hasičić M, Bandić L, Mašetić Z, Kovačević Ž, et al. Evidence-based clinical engineering: Machine learning algorithms for prediction of defibrillator performance. Biomedical Signal Processing and Control. (2019) Sep 1; 54: : 101629. |
[79] | Gurbeta L, Badnjević A. Inspection process of medical devices in healthcare institutions: Software solution. Health and Technology. (2017) Mar; 7: (1): 109-17. |
[80] | Gurbeta L, Badnjevic A, Dzemic Z, Jimenez ER, Jakupovic A. Testing of therapeutic ultrasound in healthcare institutions in Bosnia and Herzegovina. In 2nd EAI International Conference on Future Access Enablers of Ubiquitous and Intelligent Infrastructures. (2016) Oct. pp. 24-25. |
[81] | Badnjević A, Cifrek M, Magjarević R, Džemić Z. Inspection of Medical Devices. Series in Biomedical Engineering. (2017) Oct. |
[82] | IEEE Standards Association. IEEE P2727 Working Group: MEDICAL DEVICES WITH MEASURING FUNCTIONS WORKING GROUP [Internet]. New York: Institute of Electrical and Electronic Engineers, c2021 [Cited 17 Oct 2021]. Available from: https://sagroups.ieee.org/2727/. |
[83] | P2727.1 – Standard for Conformity Assessment Testing of Cardiac Defibrillators for Legal Metrology Purposes [Internet]. New York: Institute of Electrical and Electronic Engineers, c2021 [Cited 17 Oct 2021]. Available from: https://standards.ieee.org/project/2727_1.html. |
[84] | Pane J, Francisca RD, Verhamme KM, Orozco M, Viroux H, Rebollo I, et al. EU postmarket surveillance plans for medical devices. Pharmacoepidemiology and Drug Safety. (2019) Sep; 28: (9): 1155-65. |
[85] | Fraser AG, Nelissen RG, Kjærsgaard-Andersen P, Szymański P, Melvin T, Piscoi P, CORE–MD Investigators (see Appendix). Improved clinical investigation and evaluation of high-risk medical devices: The rationale and objectives of CORE–MD (Coordinating Research and Evidence for Medical Devices). EFORT Open Reviews. (2021) Oct; 6: (10): 839-49. |
[86] | Iwaishi C, Iwasaki K. A Comprehensive Analysis of Postmarket Surveillance Study Orders: Device Characteristics, Study Statuses, Outcomes, and Potential Contributions. Therapeutic Innovation & Regulatory Science. (2020) Jul; 54: (4): 953-63. |
[87] | Lim RR. Model competencies in regulatory therapeutic product assessment: Health Canada’s good review guiding principles as a reviewing community’s code of intellectual conduct. Pharmacoepidemiology and Drug Safety. (2007) Aug; 16: (8): 933-41. |
[88] | Peña C, Li K, Felten R, Ogden N, Melkerson M. An example of US Food and Drug Administration evice regulation: Medical devices indicated for use in acute ischemic stroke. Stroke. (2007) Jun 1; 38: (6): 1988-92. |
[89] | Randall H. Post-marketing surveillance and vigilance for medical devices. Drug Safety. (2001) Oct; 24: (12): 869-72. |
[90] | Rome BN, Kramer DB, Kesselheim AS. Approval of high-risk medical devices in the US: Implications for clinical cardiology. Current Cardiology Reports. (2014) Jun 1; 16: (6): 489. |
[91] | Ross JS, Blount KL, Ritchie JD, Hodshon B, Krumholz HM. Post-market clinical research conducted by medical device manufacturers: A cross-sectional survey. Medical Devices (Auckland, NZ). (2015) ; 8: : 241. |


