Exploration of genetics commonness between bladder cancer and breast cancer based on a silcio analysis on disease subtypes
Abstract
BACKGROUND AND OBJECTIVE:
Muscle-invasive bladder cancers (MIBCs) are heterogeneous cancers and can be grouped into basal-like and luminal subtypes that are highly reminiscent of those found in breast cancer. Like basal-like breast cancers, basal-like MIBCs are associated with advanced stage and metastatic disease. However, the biological and clinical significance of molecular subtypes of MIBCs remain unclear. Therefore, we implemented a serious of bioinformatics methods to explore genetic similarities between bladder and breast cancers.
METHODS AND RESULTS:
In the current study, by the application of multiple levels data analysis including random forest analysis, PPI and transcription factor regulation network construction, Gene Ontology (GO) and KEGG pathway enrichment analysis, we explored the genetics commonness between MIBC and breast cancers from the molecular heterogeneity based on the disease subtypes.
CONCLUSIONS:
Our study identified some basal-related and luminal-related genes shared by two cancers. These studies can help shed light on the potential relationships between MIBC and breast cancer as a whole.
1.Introduction
Bladder cancer is a malignant tumor that occurs in the bladder epithelium and cause the high mortality. Bladder cancer can be divided into non-muscle invasive bladder cancer (NMIBC) and muscle invasive bladder cancer (MIBC) based on invasion into the muscularis propria of the bladder [1]. Recent studies based on the integrated genomic and protein analysis have better defined molecular subtypes of MIBCs, termed basal-like and luminal, with differences in clinical outcome [2]. Basal-like MIBCs are enriched with squamous features, with a keratinized/squamous phenotype, and showed a high level of sarcomatoid differentiation, whereas luminal MIBCs showed papillary histopathological features [3]. Compared with luminal MIBCs, basal-like MIBCs have more invasive and metastatic disease, and associated with shorter disease-specific survival as well as overall survival [2]. Therefore, the identification of MIBC subtype-specific biomarkers will help provide biological value for the MIBC clinical trials and therapy strategies.
Figure 1.
The flowchart of our work, which includes (1) identification of bladder cancer and breast cancer subtypes-related DEGs, (2) GO and KEGG enrichment analysis for the identified DEGs, (3) random forest analysis using overlapping genes shared by two cancers (4) PPI network and TF-regulation network construction based on subtype special genes involved in the defined four groups.
Recently, more and more evidences showed that intrinsic subtypes of MIBC are remarkably similar to the intrinsic subtypes of breast cancer [4, 5]. Breast cancer is heterogeneous disease and can be grouped into five molecular subtypes, including claudin-low, basal-like, luminal-A, luminal-B, and HER2 enrichment, based on shared gene expression patterns [6, 7, 8]. Meanwhile, basal-like and luminal-A are two major breast cancer subtypes which have shown significant differences in terms of clinical outcome, prognosis and response to treatment. When compared with the molecular profile of several tumor types, basal-like MIBC is very similar to basal-like breast cancer, whereas luminal MIBC is similar to luminal-A breast cancer [5]. Their similar molecular profiles reflect in their similar functional roles. For example, a great amount of basal-like and luminal breast cancer specific gene signatures were enriched on the corresponding MIBC subtypes, including bona fide luminal breast cancer pathways, such as GATA3 and estrogen receptor signaling in the luminal MIBC subtype [5]. Previous studies have established that high level of
In this study, we revealed the molecular commonalities between intrinsic subtypes of MIBC and breast cancer, including basal-like and luminal (for breast cancer is luminal-A) subtypes by the application of bioinformatics method. First, the MIBC and breast cancer datasets were downloaded from published works and public database, respectively. Then, we identified differentially expressed genes (DEGs) of both cancers. Second, we used DEGs identified in first step to perform GO and KEGG enrichment analyses to explore their potential functions, and consequently found some overlapped terms shared by two cancers. Third, we selected overlapping genes extracted from the MIBC and breast cancer DEGs to perform random forest analysis. We constructed molecular subtype-related classifiers and extract some important cancer subtypes-related genes by random forest analysis. Finally, we divided the common genes shared by two cancers into four groups: all basal-related genes for two cancers, all luminal-related genes for two cancers, basal-related genes for bladder cancer whereas luminal-related for breast cancer, luminal-related genes for bladder cancer whereas basal-related for breast cancer, respectively. For these four groups of genes, we explored their relationships in protein-protein interaction (PPI) networks and transcription factor (TF) regulatory networks. The workflow of our works was shown in Fig. 1, and the detail was provided in the next section.
2.Materials and methods
2.1Data source and data preprocessing
In the current study, we performed the analysis for a set of bladder cancer subtype samples extracted from a MIBC dataset [13]. The original MIBC dataset was already normalized using a robust multichip averaging method. The dataset contained 1000 MIBC subtype related genes with high standard deviation. Twenty-two basal samples and 132 luminal samples were included in this dataset [13]. We used independent samples t-test by BRB-Array Tools to filter statistically significant DEGs that distinguish basal-like and luminal bladder cancer subtypes. In this method, repeated permutations of the data are used to determine if the expression of any gene is significant related to the phenotype. To get more information,
2.2GO and KEGG enrichment analysis
In order to see whether the identified DEGs of both cancers can focus on some commonness function, we performed Gene Ontology (GO) and KEGG pathway enrichment analysis using DAVID web tool (http://david.abcc.ncifcrf.gov/). A GO term (or a KEGG pathway) with a p-value of less that 0.01 was considered to be significant. We did not perform the multiple test correction to avoid a loss of true-positive results.
2.3Random forest analysis
To further explore relationships between MIBC and breast cancer in the view of data, we analyze those DEGs shared by two cancers using machine learning method. In this analysis, we selected the common DEGs shared by two cancers, and then applied Random forests (RF) method to construct molecular subtype related classifiers and extract the important cancer subtype related genes. RF is an ensemble classifier that consists of many decision trees and each tree depends on the values of a random vector sampled independently [15]. The method to obtain subtype-related feature gene G is permuting the expression value of gene G of out-of bag according to random forest algorithm. If gene G is a good predictor, then it will appear in a large number of split trees. We used Mean Decrease Gini (MDG) to evaluate whether gene G is a feature gene or not. MDG was the total decrease in node impurities measured by the Gini index from splitting on the variable, averaged over all trees. It provided possible ways to quantify which genes contribute most to classification accuracy. Greater MDG will indicate that the degree of impurity arising from category could be reduced farthest by gene G, and thus suggests an important feature gene [16]. The program was implemented with the randomForest package of R software.
2.4Exploration of the potential regulation relationships of common subtype-related genes shared by MIBC and breast cancer
Among the common DEGs shared by two cancers, we defined two kinds of gene: basal-related gene when it shows higher expression in basal-like samples than in luminal (luminal-A), and luminal-related gene when it shows higher expression in luminal (luminal-A) samples than in basal-like, respectively. Under these conditions, we further divided these genes into four groups: all basal-related genes for two cancers, all luminal-related genes for two cancers, basal-related genes for MIBC whereas luminal-related for breast cancer, luminal-related genes for MIBC whereas basal-related for breast cancer, respectively. Then, we explored the potential regulation relationships of common subtype-related genes shared by MIBC and breast cancer.
2.4.1Exploration of the interaction in PPI network of subtype-related genes
For four genes groups, we explored their relationships in protein-protein interaction (PPI) networks. In the practice, the PPI network is the basic method for systematically displaying protein-protein interaction information. In PPI network, the protein as a node, the interaction between two proteins as an edge, and the proteome as a whole is mapped to a system network. In this analysis, we use String database (http://string.org) to get the protein-protein pairs. Then, the Cytoscape software (version 3.4.0; http://cytoscape.org/) was used to visualize the PPI networks and to further explore the association between genes.
2.4.2Exploration of transcription factor regulatory network of subtype-related genes
The transcription factor can bind to the nucleotide sequence specific upstream of the gene, thereby regulating the expression of the gene. Gene transcription regulation network can be constructed by gene transcriptional regulation data. For four groups of genes, we explored their transcription factor regulation networks. In this analysis, we selected TRANSFAC database (Release 2016) (http://www.biobase-international.com/product/transcription-factor-binding-sites) which is a manually curated database of eukaryotic transcription factors, their genomic binding sites and DNA binding profiles. The gene transcriptional regulatory network describes the relationship between transcription factors and their target genes, which can be represented by directed graphs. We used Cytoscape software to visualize these networks.
3.Results
3.1Identification of DEGs for bladder cancer and breast cancer
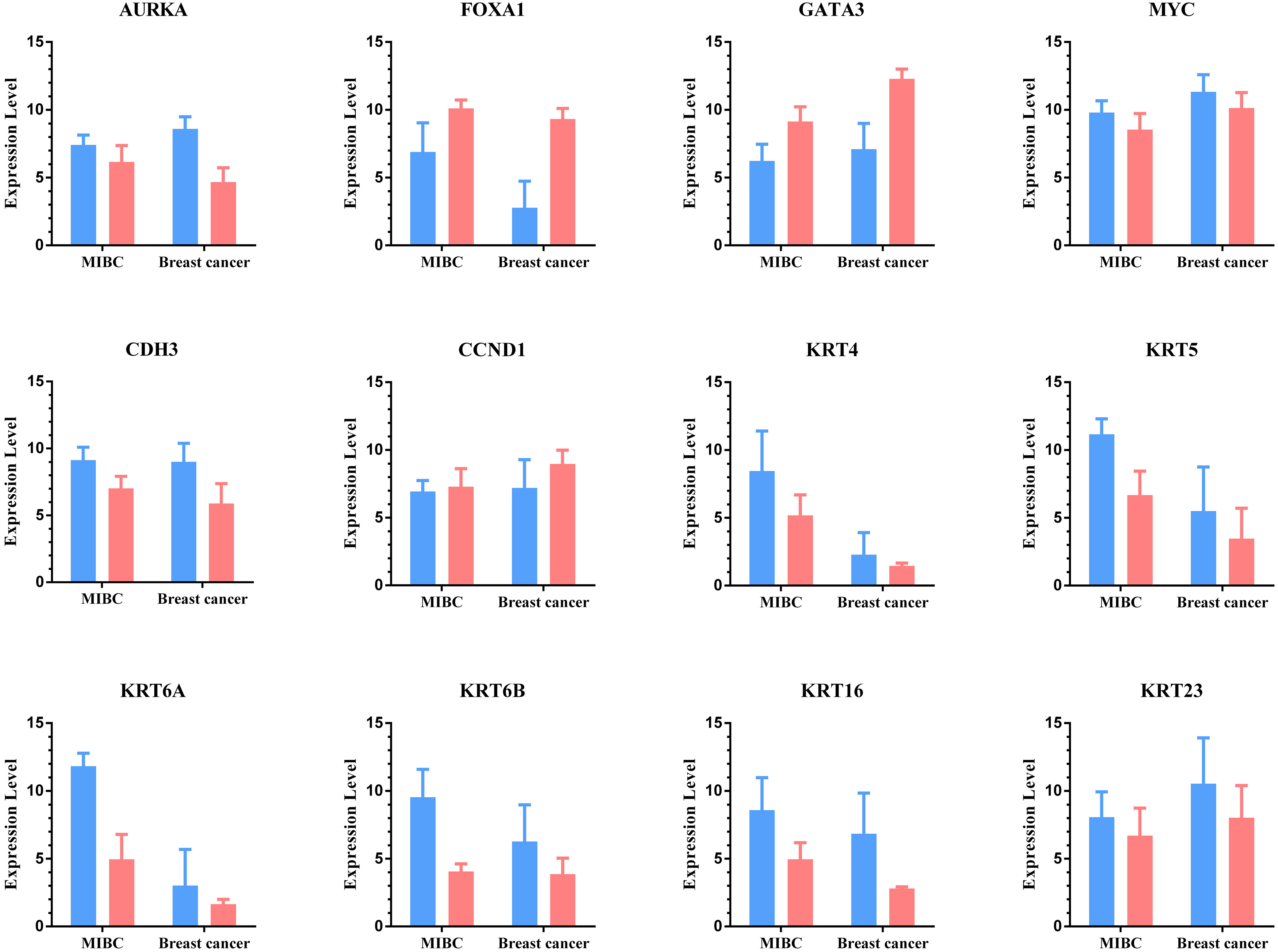
The classic t-test in the BRB-Array Tools and Limma package of R were used to analyze the gene expression profiles of MIBC and breast cancer, and identified 741 MIBC subtype-related DEGs and 9946 breast cancer subtype-related DEGs. We described some important basal-like and luminal genes in both cancers in the Supplementary Fig. 1. We observed 9 genes overexpressed in basal-like samples whereas 3 genes overexpressed in luminal samples. Among these genes, KRT6A, KRT5 and CDH3 are reported direct DeltaNp63 transcriptional targets and can restore the expression of the basal kertains [2, 17]. Luminal subtype MIBC show features similar to those of Luminal-A breast cancer, with high mRNA expression in luminal samples than in basal-like samples, such as FOXA1 and GATA3 [8, 11, 12, 18].
3.2GO and KEGG enrichment analysis
We performed GO and KEGG enrichment analysis for the identified DEGs of both cancers in the first step using DAVID web tool. We used all 741 DEGs to perform enrichment analysis for bladder cancer. Notably, for breast cancer, because the number of DEGs was too great to analyze by online tool owing to overload, we chose a subset of genes (2677 DEGs) which are high significant (screening threshold:
Figure 2.
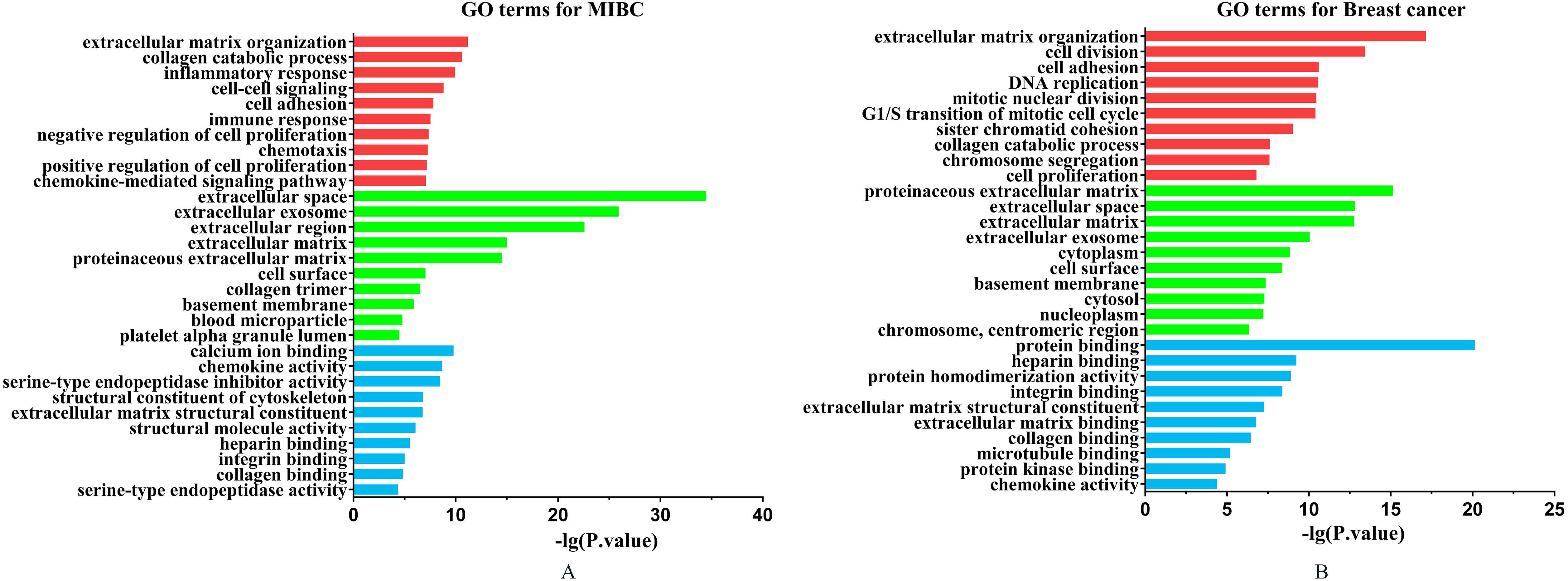
The GO analysis results for two cancers. The GO terms included biological process (red bars), cellular component (green bars) and molecular function (blue bars). The length of bar is positive correlation with significant level. (A) GO analysis results for MIBC. (B) GO analysis results for breast cancer.
3.2.1GO enrichment analysis
We listed the top ten significant GO terms for MIBC and breast cancer from Cellular Component (CC), Biological Process (BP) and Molecular Function (MF), respectively (see Fig. 2). From the GO enrichment analysis results, we found those common DEGs shared by MIBC and breast cancer focus on some significant functions. The most significant GO terms shared by both cancers were extracellular matrix organization. Extracellular matrix (ECM) is defined as a complex mixture of various proteins that provides structural and mechanical support for cells and tissues, and has an important role in the regulation of gene expression, cell division, survival, shape, and movement [19]. The altered ECM produced by the oncogene-exposed or transformed by carcinogen may play an important role in determining the ability of human bladder epithelial tumor to proliferate, relapse, or progress [20]. For breast cancer, a great amount of studies indicated that the ECM may be involved in various processes of breast tumors growth [21]. Some studies demonstrated that the ECM components were more heterogeneously at basal-like tumors when compared with the luminal subtype [22]. Interestingly, in our study we found that most of the GO terms shared by MIBC and breast cancer were related to extracellular, such as extracellular space, extracellular exosome, ECM structural constituent and so on.
Figure 3.
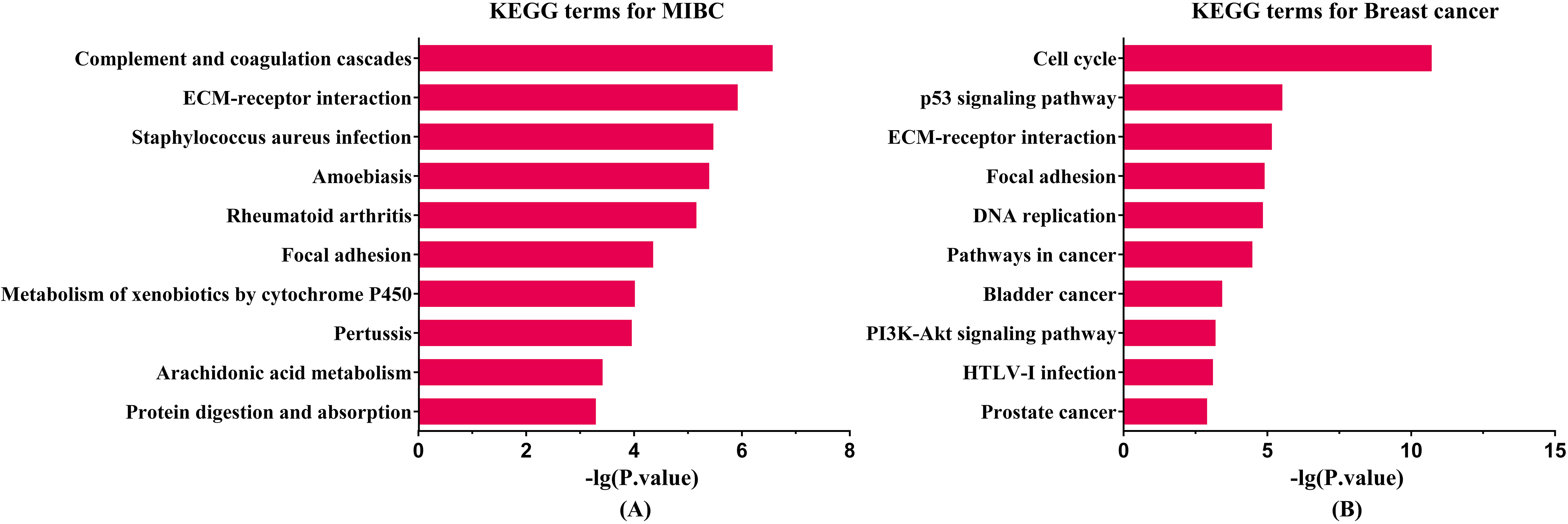
KEGG enrichment analysis results for two cancers. The length of bar is positive correlation with significant level. (A) Enriched KEGG pathway for MIBC. (B) Enriched KEGG pathway for breast cancer.
Furthermore, we found some other co-enrichment GO terms of two kinds of cancers, such as collagen catabolic process, cell adhesion, integrin binding and chemokine activity, and all of these biochemical mechanisms play important roles in the development of the tumors.
3.2.2KEGG enrichment analysis
We listed the top ten significant KEGG pathways of MIBC and breast cancer (see Fig. 3). The KEGG enrichment analysis results showed that the shared pathway by both cancers were ECM-receptor interaction and focal adhesion. Previous study reported that ECM molecules play an important role in the development of invasion, progression and metastasis in bladder cancer [20, 23]. The ECM in MIBCs may be a potential therapeutic target for treatment [23]. In the case of breast cancer, ECM is necessary for normal functional differentiation of mammary epithelia [21]. The altered ECM proteins may lead to different phenotypes of breast cancer. Moreover, an increasing number of researches indicated that the extracellular matrix and extracellular matrix receptors may be involved in controlling most of the successive stages of breast tumors, from appearance to progression and metastasis [21]. Focal adhesion kinase (FAK) is an intracellular non-receptor tyrosine kinase. FAK is a vital symbolic component that is activated by many stimuli, acting as a biosensor or an integrator to control cell motility [24]. FAK is an important regulator of bladder cancer cell invasion and migration, and it may become a potential therapeutic target [25]. For breast cancer, FAK may play a role in breast cancer by regulating breast stem cells (MaSCs) and breast cancer stem cells (MaCSCs) [24]. Emerging data suggested that FAK can be an effective therapeutic target in breast tumors, particularly in highly invasive triple-negative breast cancer (also terms basal-like breast cancer) [26]. Interestingly, we found that the amount of breast cancer related genes were enriched on bladder cancer and prostate cancer pathway.
3.3Random forest analysis
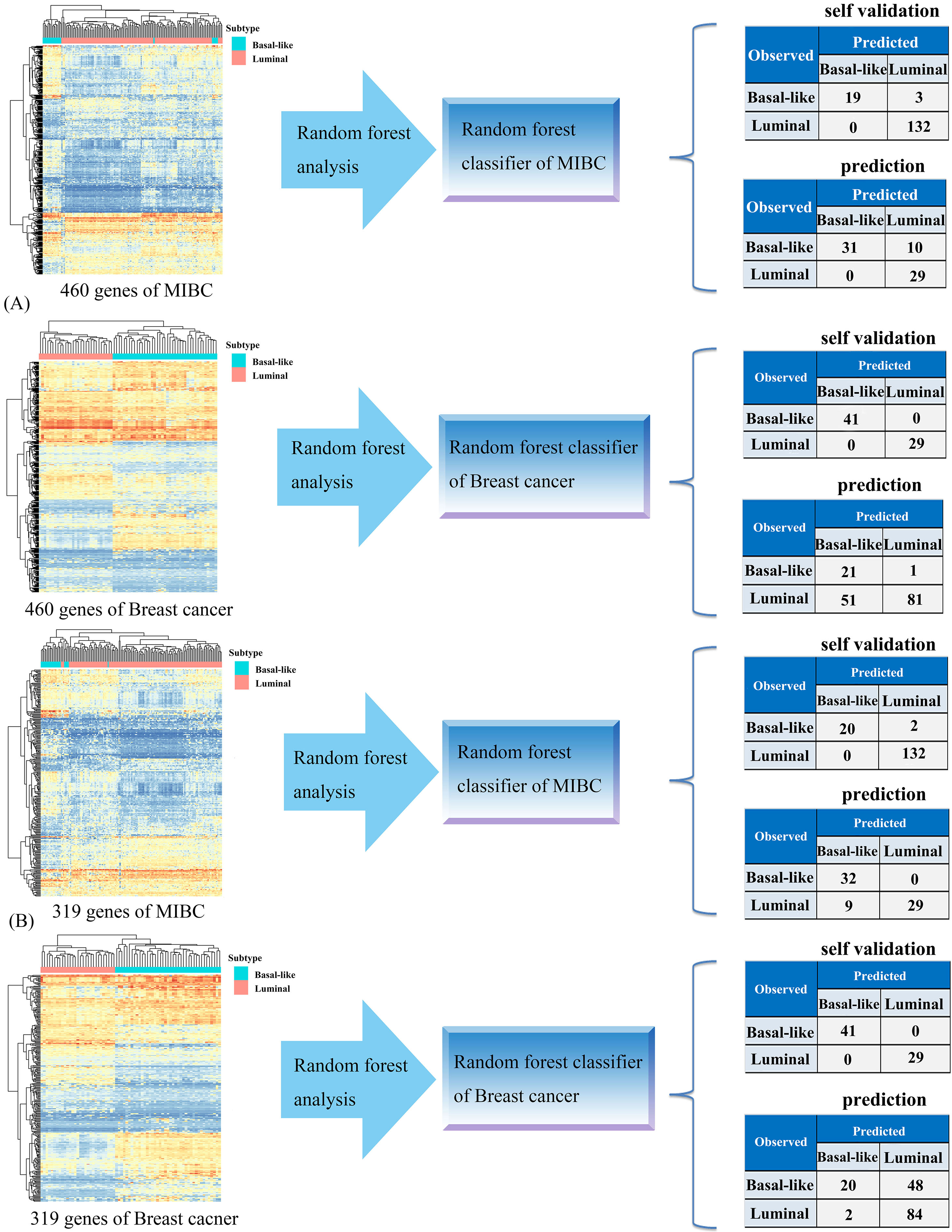
We next sought to define a set of genes that could accurately classify MIBC and breast cancer samples into the basal-like and luminal intrinsic subtypes. We extracted 460 overlapping genes shared by MIBC and breast cancer from DEGs, and then used these genes to construct MIBC type classifier and breast cancer type classifier. Two types of classifier can predict subtypes of two cancers.
3.3.1Classification accuracy
The classification result showed that the MIBC and breast cancer subtype classifiers have powerful classification performance when 460 overlapping genes were used as predictor variables to classify samples. The classification accuracy of these two type classifiers were up to 98.1% and 100%, respectively. Importantly, when classified breast cancer subtype samples with MIBC type classifier, the classification accuracy is 85.7% (see Fig. 4A), indicating that the gene expression patterns that distinguish basal-like and luminal MIBC reflect the mRNA expression patterns that defined the intrinsic subtypes of breast cancer. However, the accuracy of breast cancer type classifier was only 66.2% when predict MIBC subtype samples. The reason of this case may be that we didn’t further select the subset genes with high fold change to create random forest models because
of the limitation of available datasets. For this, we selected a gene subset with higher
fold change (
Figure 4.
Generation of the cancer subtype classifiers. (A) Random forest analysis was performed using 460 overlapping genes. Each classifier generated by one kind of cancer predicted the other kind of cancer. (B) Random forest analysis was performed using 319 overlapping genes extracted from 460 DEGs according to the cutoff of
3.3.2Identification of important cancer subtype-related genes based on MDG
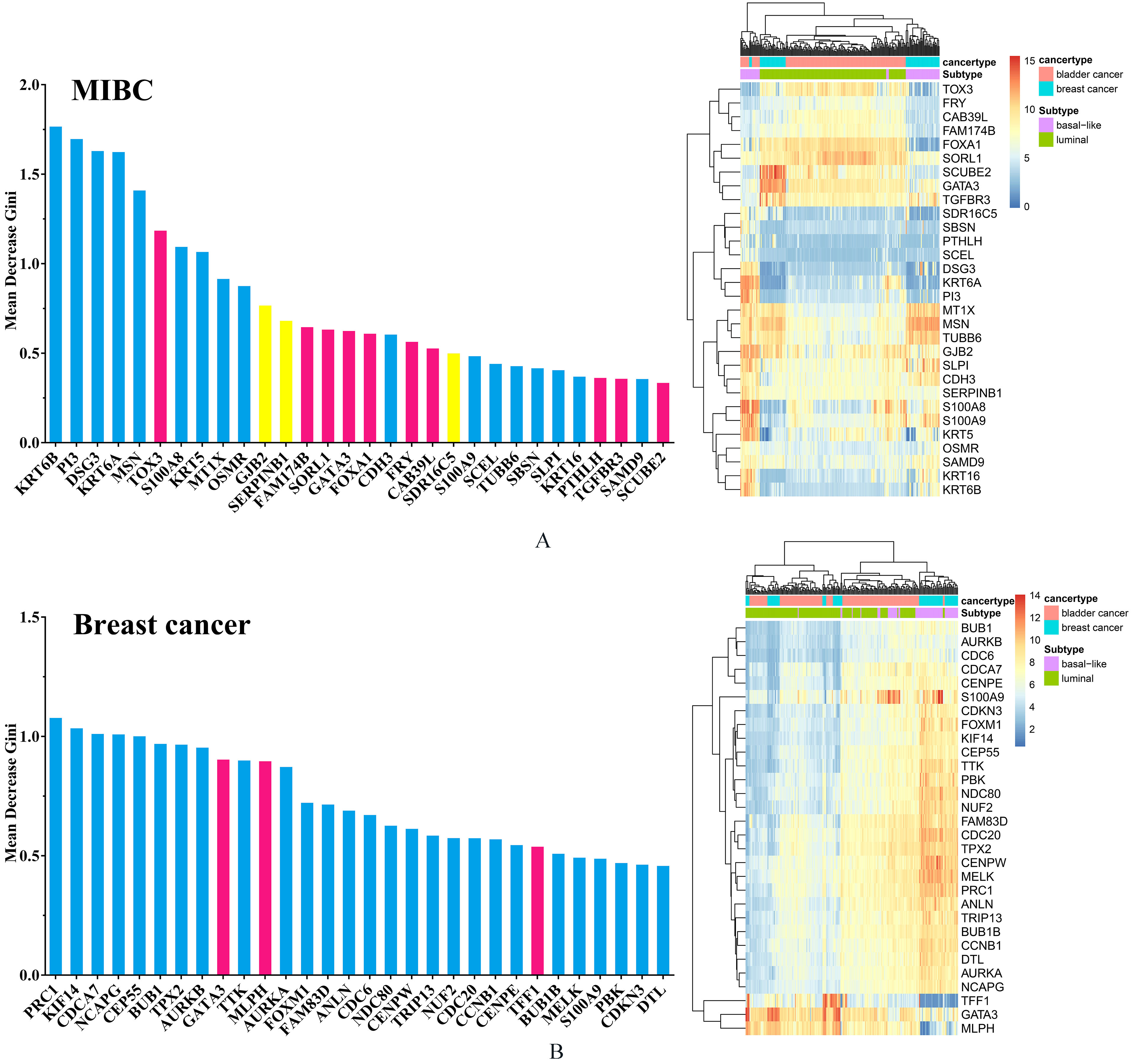
Specially, it is often interest to know which of genes are important in classification in Random Forests program. In this analysis, we used MDG to measure the distinguishing ability of genes. Greater MDG will indicate that the degree of impurity arising from category could be reduced farthest by a gene, and thus suggests an important gene. We listed the top 30 genes for MIBC and breast cancer respectively according to their MDG on Supplementary Fig. 2.
For MIBC genes, 17 genes were all basal-related genes whereas 9 genes were all luminal-related genes. Meanwhile, as a basal-related biomarker, KRT6B has the highest MDG. In the case of breast cancer, 26 genes were all basal-related genes whereas 4 genes were all luminal-related genes. Among these genes, PRC1 had the highest MDG. Among the top 30 MDG for each of cancers, there were some overlapped genes shared by both cancers, such as GATA3 and S100A9. The genomic gains of GATA3 were associated with the urothelial differentiation component. GATA3 is important for the proliferation of luminal breast tumors and maybe play the same role in bladder tumors presenting a molecular urothelial differentiation [5, 13, 18, 27]. The overexpression of S100A9 significantly correlated with tumor progression and worse outcome in the patients with bladder cancer, prostate cancer and breast cancer [28].
3.4Exploration of the potential regulation relationships of common subtype-related genes shared by MIBC and breast cancer
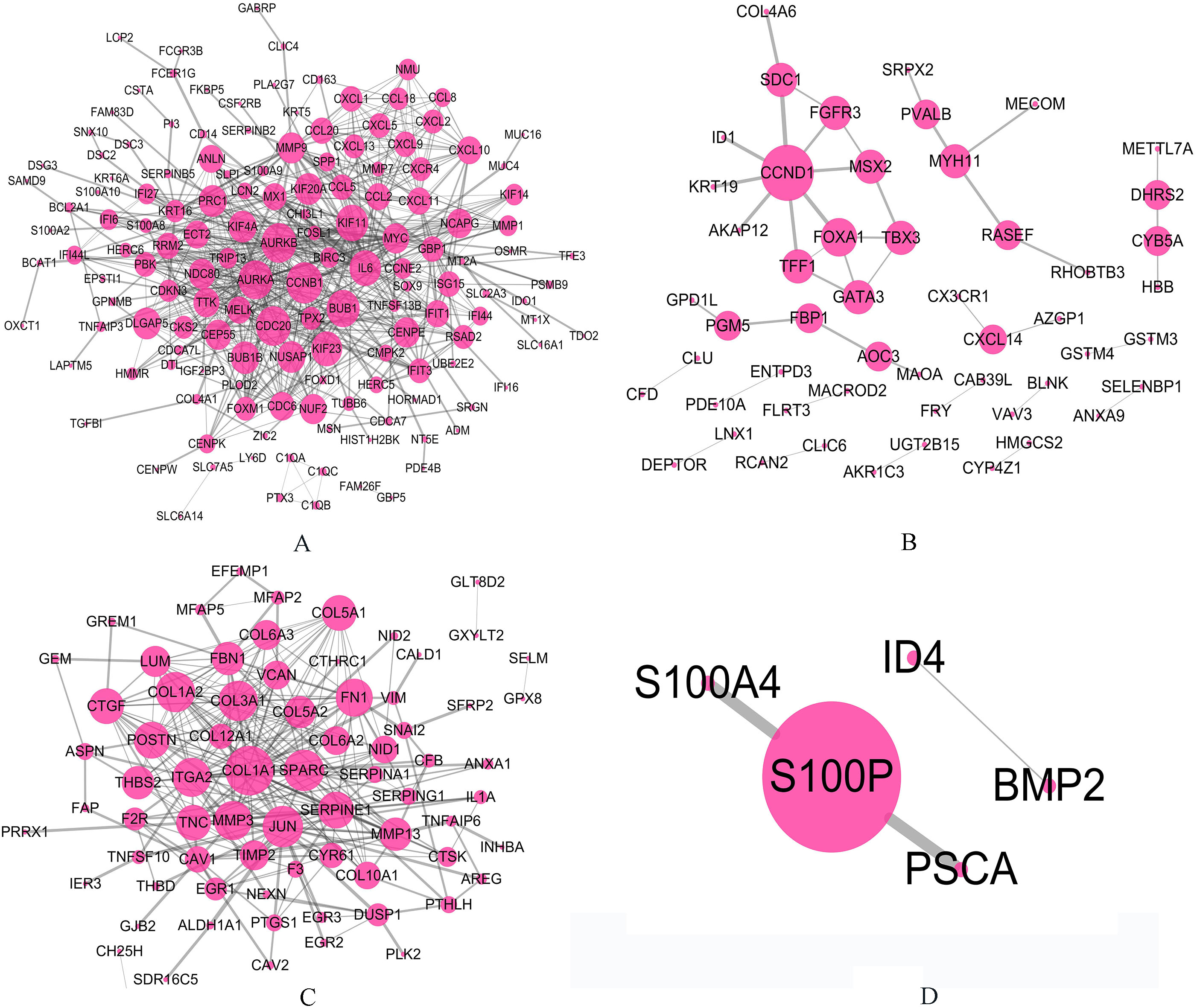
We divided 460 overlapping DEGs into four groups: all basal-related genes for two cancers (189 genes), all luminal-related genes for two cancers (130 genes), basal-related genes for bladder cancer whereas luminal-related for breast cancer (103 genes), luminal-related genes for bladder cancer whereas basal-related for breast cancer (38 genes), respectively. Then, we performed network analysis to explore the potential regulation relationships among these genes.
Figure 5.
The relationships for genes involved in four groups respectively based on PPI network. The larger circle indicates the gene with higher degree. (A) All basal-related genes for two cancers. (B) All luminal-related genes for two cancers. (C) Basal-related genes for MIBC whereas luminal-related for breast cancer. (D) Luminal-related genes for MIBC whereas basal-related for breast cancer.
3.4.1Exploration of the interaction in PPI network of subtype-related genes
The relationships for genes involved in four groups respectively based on PPI network was shown in the Fig. 5. We found that some important genes have higher degree than other genes in PPI networks. As we know, basal-like subtype in both cancers is susceptible to have more aggressive and metastatic disease at the time of initial diagnosis and is associated with shorter disease specificity and overall survival. In basal-related group, genes with high degree were CCNB, CDC20, AURKA, AURKB, BUB1, KIF11, IL6 and KIF23.
Among these genes, CCNB1 had the highest degree (degree
In the luminal-related group, we identified 9 important genes (CCND1, FOXA1, TFF1, GATA3, SDC1, TBX3, MSX2, FGFR3, and MYH11). We know that the luminal subtype is not as intrinsically aggressive as basal-like subtypes in both cancers. The gene with the highest degree in luminal-related group was CCND1 (degree
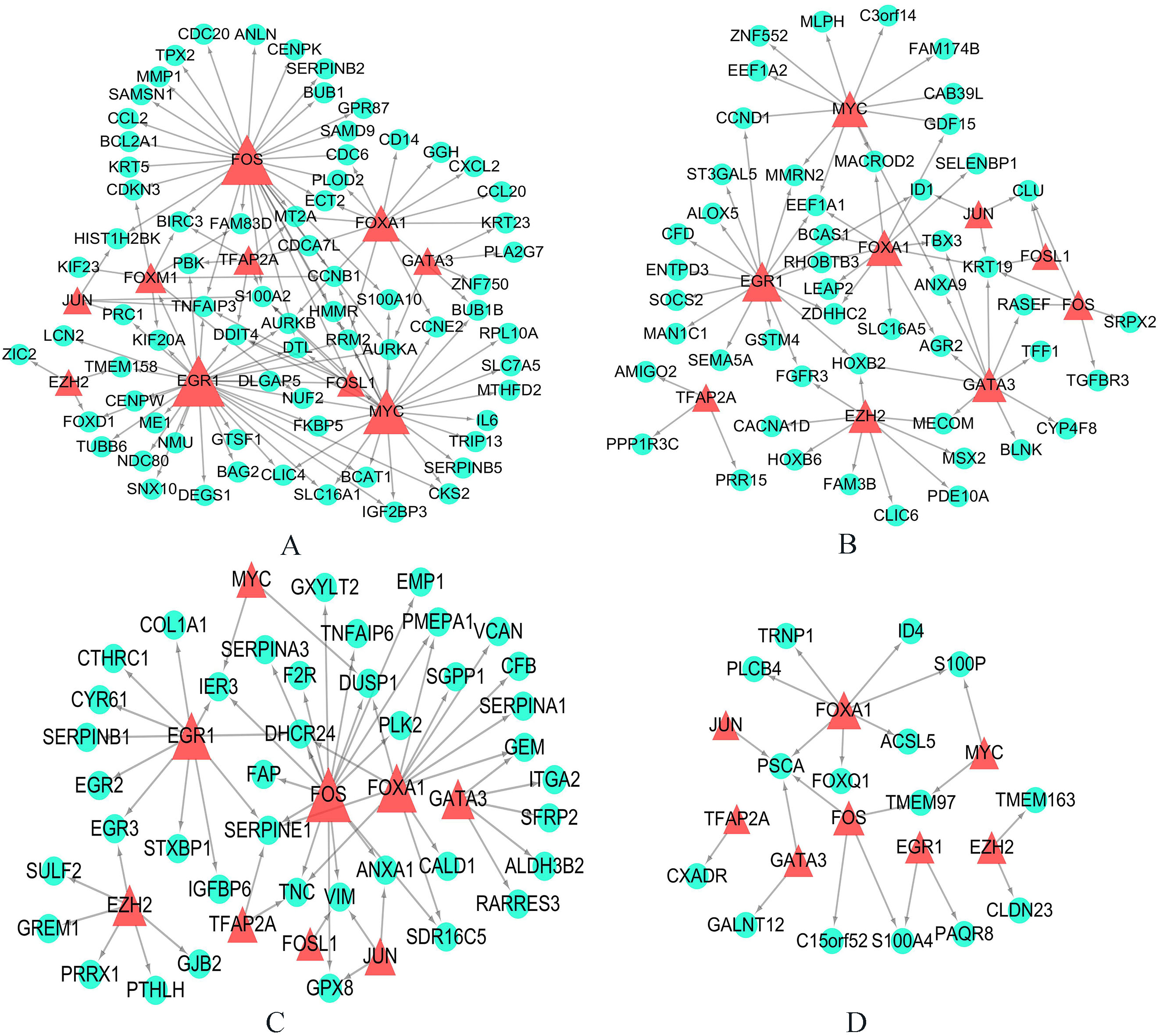
3.4.2Exploration of transcription factor regulatory network of subtype-related genes
The transcription factor regulatory relationships based on TRANSFAC database were shown in the Fig. 6. In this study, we found some transcription factors regulate genes involved in different groups with different ways, such as FOXA1, FOSL1, FOXM1, GATA3, TFAP2A, JUN, EGR1, MYC and so on. Among these genes, all basal-related transcription factors for two cancers included FOSL1, FOXM1, MYC, whereas all luminal-related transcription factors for two cancers included FOXA1 and GATA3. These transcription factors play important roles in regulate progression of carcinogenesis. The FOXM1 transcription factor plays a key role in regulating cell proliferation, differentiation, and transformation [42]. Overexpression of FOXM1 is associated with a variety of invasive solid carcinomas, including bladder cancer and breast cancer [42]. The MYC gene encodes about 60 kD of nuclear phosphoric acid protein with DNA binding activity [43]. Abnormal regulation of MYC can lead to phenotypic transformation, abnormal cell cycle control and genomic instability [43]. Our study, and others, found that key transcription factors FOXA1 and GATA3 were up regulated in the luminal subtypes of both kinds of cancers, and both transcription factors have important roles in luminal epithelial differentiation [2, 18, 40].
Figure 6.
The transcription factor regulatory networks for genes involved in four groups respectively based on TRANSFAC database. The green circles indicate genes in each group, and the red triangles indicate transcription factors which regulate these genes. (A) All basal-related genes for two cancers. (B) All luminal-related genes for two cancers. (C) Basal-related genes for MIBC whereas luminal-related for breast cancer. (D) Luminal-related genes for MIBC whereas basal-related for breast cancer.
4.Discussion
MIBC is extremely aggressive disease which cause a high mortality, and the traditional treatment of its therapeutic effect is limited [2]. For an early diagnosis and appropriate targeted treatment, studies about molecular characterization diseases phenotypes and prediction of novel biomarkers are essential. MIBC tumors can be distinguished into basal-like and luminal subtypes which remarkably similar to breast cancer subtypes in molecular features and clinical outcomes [4]. In the past few years, bioinformatics research on breast cancer subtypes has yielded great results, and there is a very mature mechanism of molecular subtype classification and therapeutic options [11]. Therefore, given the potential similarities between MIBC and breast cancer at the molecular level, the abundant breast cancer knowledge base may be utilized to help make progress more rapidly in the clinical management of MIBC.
Here, we applied bioinformatics method to analyze MIBC and breast cancer mRNA datasets, and consequently found some genetics commonness between MIBC and breast cancer subtypes. According to enrichment analysis, we found that the main biological processes based on GO focus on ECM related terms. Moreover, the KEGG enrichment analyses showed that DEGs of both cancers were significantly enriched on ECM-receptor interaction pathway. It is obvious that ECM and ECM-receptor status are altered in tumors with profound functional implications which are of value for cancer diagnosis, prognosis and therapy [20]. Bergamaschi et al. demonstrated that primary breast cancer can be classified based on the differential expression of a set of ECM-related genes, and this classification has implications for clinical outcome [19]. We believe that the association study between ECM-related genes and bladder cancer subtypes can help understand MIBC pathology from molecular levels. It is no doubt that explore the relationships between ECM related genes and MIBC subtypes is extraordinary helpful in molecular subtypes classification and target therapy.
We created gene classifiers which accurately discriminate intrinsic cancer subtypes based on the common DEGs shared by two cancers. By random forest analysis, we constructed two types classifier: one is MIBC type classifier which trained by MIBC mRNA expression and the other is breast cancer type classifier which trained by breast cancer mRNA expression respectively. Subsequently, we predicted breast cancer subtype samples by MIBC type classifier and MIBC subtype samples by breast cancer type classifier. Furthermore, different subtypes of bladder cancer and breast cancer can be identified by a similar basic regulatory system, even if the phenotype and tissue origin may differ. We further identify the gene co-regulation relation in basal-like and luminal subtypes. We divided those DEGs shared by two cancers into four groups and then performed biological network analysis for each group. We identified some hub genes that play important roles in basal-like and luminal tumor development. In addition, we identified some transcription factors which regulate genes involved in four groups, such as FOS, FOXA1, GATA3, TFAP2A, FOXM1, JUN, EZH2, EGR1, FOSL1 and MYC. Abnormities in these transcription factors causing gene expression abnormities play important roles in human cancers including bladder and breast cancer.
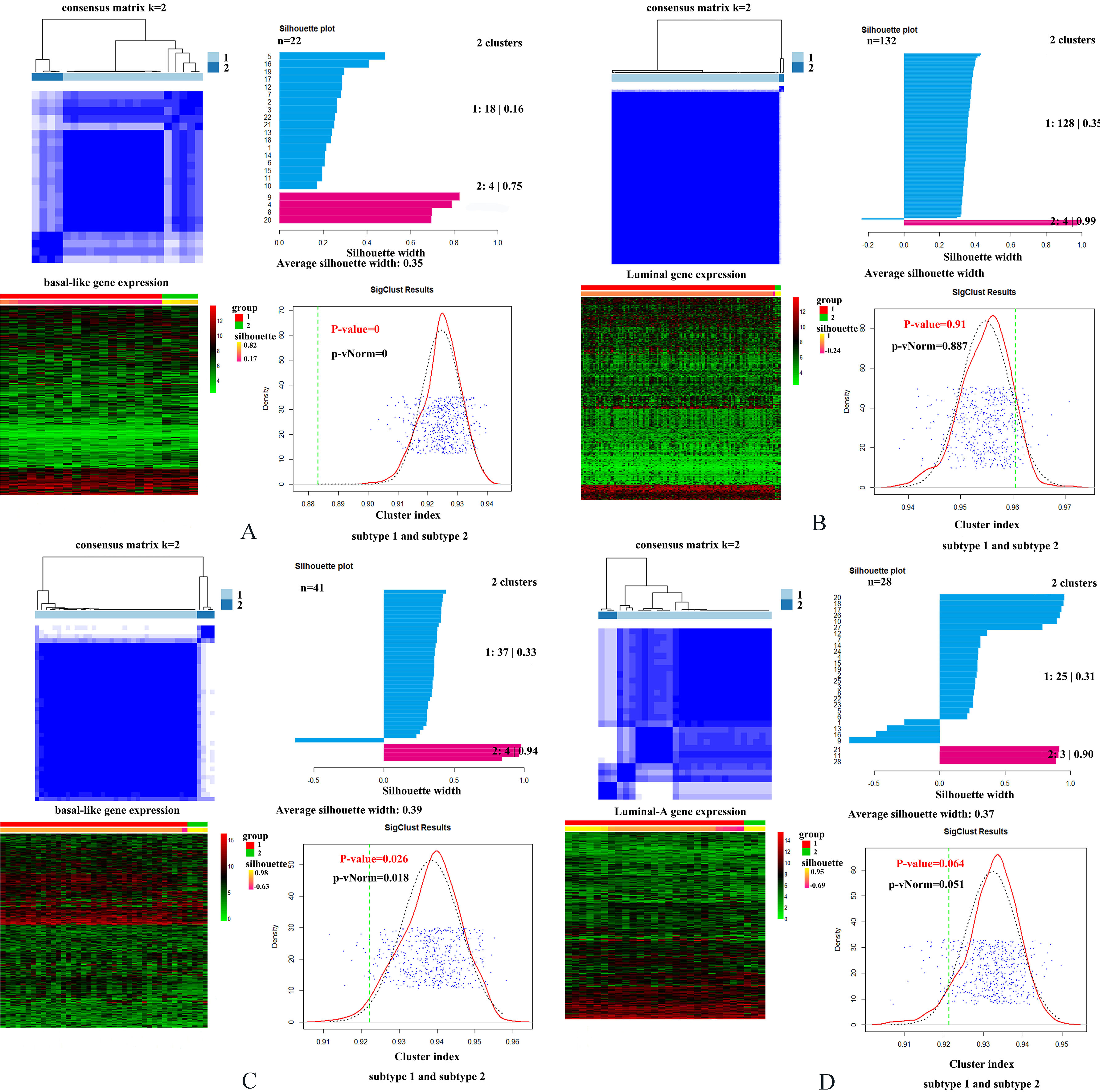
Specially, we performed added exploratory study to see whether existing potential interior difference between two MIBC molecular subtypes as well as breast cancer subtypes. To find the potential interior difference between two molecular subtypes for each cancer, we performed cluster analysis on basal-like samples and luminal samples based on consensus cluster method, respectively. For bladder cancer, we used original 1000 genes to implement cluster analysis. For breast cancer, for the purpose of improving cluster efficiency, we selected equal numbers of genes (1000 genes) with highest standard deviation extracted from 9946 breast cancer DEGs as input to perform the analysis. CancerSubtypes package of R software (http://www.r-project.org) was used to implement this analysis. In this study, we divided each kind of cancer into two subtypes: basal-like subtype and luminal subtype. Accordingly, four types of samples were included: basal-like MIBC samples, basal-like breast cancer samples, luminal MIBC samples and luminal-A breast cancer samples. Then, for each type of samples, we clustered samples into two clusters using consensus cluster method, and calculated the corresponding silhouette scores. Silhouette score indicates the consistency within clusters of data. A higher silhouette score indicates the greater similarity within the samples involved in the same cluster. Finally, for two obtained clusters of each type of samples, we implemented paired comparison between of them. For basal-like subtype in two cancers, the paired comparison between two clusters were all significant (
It also should be pointed out the limitations of our analysis. In the present study, we only selected the limited datasets to perform a series of analyses. This may cause the inadequate conclusions and generate bias in some cases. Therefore the identified candidate genes need to be validated for further experimentation. Specifically, the lack of MIBC and breast cancer subtype expression profiling datasets causes the limitations in the data analysis and these results need to be approved in the future studies when more bladder cancer and breast cancer subtype expression profiling datasets are available.
In summary, like the basal-like and luminal subtypes of breast cancer have distinct and reciprocal gene expression profiles as well as large differences in clinical characteristics, the basal-like and luminal subtypes of MIBC also reflect many different aspects of urothelial physiological development. In this study, we use bioinformatics methods to analyze mRNA expression datasets of MIBC and breast cancer and explored the genetics commonness between MIBC and breast cancer. We constructed random forest classifiers that distinguish cancer subtypes based on gene expression profiles. The genes contributing greatly to classify tumor subtypes may be important potential biomarkers for MIBC and breast cancer development and progression, and eventually become candidates for therapeutic targeting.
Acknowledgments
This work is supported by Beijing Natural Science Foundation (Grant No. 7142015). This study is also funded by the Special Subject of Capital General Medicine Research (17QK15), and the foundation-clinical cooperation project of Capital Medical University (16JL58 and 17JL54).
Conflict of interest
None to report.
References
[1] | Kaufman DS, Shipley WU, Feldman AS. Bladder cancer. Lancet. (2009) ; 374: : 239-49. |
[2] | Choi W, Porten S, Kim S, Willis D, Plimack ER, Hoffman-Censits J, et al. Identification of Distinct Basal and Luminal Subtypes of Muscle-Invasive Bladder Cancer with Different Sensitivities to Frontline Chemotherapy. Cancer Cell. (2014) ; 25: : 152-65. |
[3] | Lindgren D, Sjödahl G, Lauss M, Staaf J, Chebil G, Lövgren K, et al. Integrated Genomic and Gene Expression Profiling Identifies Two Major Genomic Circuits in Urothelial Carcinoma. PLOS ONE. (2012) ; 7: : e38863. |
[4] | Mcconkey DJ, Choi W, Dinney CP. New insights into subtypes of invasive bladder cancer: considerations of the clinician. European Urology. (2014) ; 66: : 609-10. |
[5] | Damrauer JS, Hoadley KA, Chism DD, Fan C, Tiganelli CJ, Wobker SE, et al. Intrinsic subtypes of high-grade bladder cancer reflect the hallmarks of breast cancer biology. Proceedings of the National Academy of Sciences of the United States of America. (2014) ; 111: : 3110-5. |
[6] | Zhu S, Reggi M, Pergamenschikov A, Børresendale AL, Eisen M, Sørlie T, et al. Molecular portraits of human breast tumours. Nature. (2000) ; 490: : 61. |
[7] | Prat A, Ellis MJ, Perou CM. Practical implications of gene-expression-based assays for breast oncologists. Nature Reviews Clinical Oncology. (2012) ; 9: : 48-57. |
[8] | The Cancer Genome Atlas Research N. Comprehensive Molecular Characterization of Urothelial Bladder Carcinoma. Nature. (2014) ; 507: : 315-22. |
[9] | Carroll DK, Carroll JS, Leong CO, Cheng F, Brown M, Mills AA, et al. p63 regulates an adhesion programme and cell survival in epithelial cells. Nature Cell Biology. (2006) ; 8: : 551-61. |
[10] | Cheung KJ, Gabrielson E, Werb Z, Ewald AJ. Collective invasion in breast cancer requires a conserved basal epithelial program. Cell. (2013) ; 155: : 1639-51. |
[11] | Network CGA. Comprehensive molecular portraits of human breast tumours. Nature. (2012) ; 490: : 61. |
[12] | Guo CC, Dadhania V, Zhang L, Majewski T, Bondaruk J, Sykulski M, et al. Gene Expression Profile of the Clinically Aggressive Micropapillary Variant of Bladder Cancer. European Urology. (2016) ; 70: : 611-20. |
[13] | Biton A, BernardPierrot I, Lou Y, et al. Independent Component Analysis Uncovers the Landscape of the Bladder Tumor Transcriptome and Reveals Insights into Luminal and Basal Subtypes. Cell Reports. (2014) ; 9: : 1235-45. |
[14] | Gruosso T, Mieulet V, Cardon M, Bourachot B, Kieffer Y, Devun F, et al. Chronic oxidative stress promotes H2AX protein degradation and enhances chemosensitivity in breast cancer patients. EMBO Molecular Medicine. (2016) . |
[15] | Breiman L. Random Forests. Machine Learning. (2001) ; 45: : 5-32. |
[16] | Hua L, Li DG, Lin H, Li L, Li X, Liu ZC. The correlation of gene expression and co-regulated gene patterns in characteristic KEGG pathways. Journal of Theoretical Biology. (2010) ; 266: : 242-9. |
[17] | Romano RA, Ortt K, Birkaya B, Smalley K, Sinha S. An Active Role of the ΔN Isoform of p63 in Regulating Basal Keratin Genes K5 and K14 and Directing Epidermal Cell Fate. Plos One. (2009) ; 4: : e5623. |
[18] | Martin-Doyle W, Kwiatkowski DJ. Molecular Biology of Bladder Cancer. Hematology/Oncology Clinics of North America. (2015) ; 29: : 191-203. |
[19] | Bergamaschi A, Tagliabue E, Sørlie T, Naume B, Triulzi T, Orlandi R, et al. Extracellular matrix signature identifies breast cancer subgroups with different clinical outcome. Journal of Pathology. (2008) ; 214: : 357-67. |
[20] | Gordon JN, Shu WP, Schlussel RN, Droller MJ, Liu BC. Altered extracellular matrices influence cellular processes and nuclear matrix organizations of overlying human bladder urothelial cells. Cancer Research. (1993) ; 53: : 4971-7. |
[21] | Lochter A, Bissell MJ. Involvement of extracellular matrix constituents in breast cancer. Seminars in Cancer Biology. (1995) ; 6: : 165-73. |
[22] | Acerbi I, Cassereau L, Dean I, Shi Q, Au A, Park C, et al. Human breast cancer invasion and aggression correlates with ECM stiffening and immune cell infiltration. Integrative Biology Quantitative Biosciences from Nano to Macro. (2015) ; 7: : 1120-34. |
[23] | Brunner A, Tzankov A. The role of structural extracellular matrix proteins in urothelial bladder cancer (review). Biomark Insights. (2007) ; 2: : 418-27. |
[24] | Luo M, Guan JL. Focal adhesion kinase: a prominent determinant in breast cancer initiation, progression and metastasis. Cancer Letters. (2010) ; 289: : 127-39. |
[25] | Kong DB, Chen F, Sima N. Focal adhesion kinases crucially regulate TGFβ-induced migration and invasion of bladder cancer cells via Src kinase and E-cadherin. Oncotargets & Therapy. (2017) : 1783-92. |
[26] | Golubovskaya VM, Ylagan L, Miller A, Hughes M, Wilson J, Wang D, et al. High focal adhesion kinase expression in breast carcinoma is associated with lymphovascular invasion and triple-negative phenotype. Bmc Cancer. (2014) ; 14: : 769. |
[27] | Choi W, Czerniak B, Ochoa A, Su X, Siefkerradtke A, Dinney C, et al. Intrinsic basal and luminal subtypes of muscle-invasive bladder cancer. Nature Reviews Urology. (2014) ; 11: : 400-10. |
[28] | Narumi K, Miyakawa R, Ueda R, Hashimoto H, Yamamoto Y, Yoshida T, et al. Proinflammatory Proteins S100A8/S100A9 Activate NK Cells via Interaction with RAGE. Journal of Immunology. (2015) ; 194: : 5539. |
[29] | Song Y, Zhao C, Dong L, Fu M, Xue L, Huang Z, et al. Overexpression of cyclin B1 in human esophageal squamous cell carcinoma cells induces tumor cell invasive growth and metastasis. Carcinogenesis. (2008) ; 29: : 307. |
[30] | Choi JW, Kim Y, Lee JH, Kim YS. High expression of spindle assembly checkpoint proteins CDC20 and MAD2 is associated with poor prognosis in urothelial bladder cancer. Virchows Archiv. (2013) ; 463: : 681-7. |
[31] | Krishnamurthy S. Cdc20 and securin overexpression predict short-term breast cancer survival. British Journal of Cancer. (2014) ; 110: : 2905-13. |
[32] | De MM, Shariat SF, Hofbauer SL, Lucca I, Taus C, Wiener HG, et al. Aurora A Kinase as a diagnostic urinary marker for urothelial bladder cancer. World Journal of Urology. (2015) ; 33: : 105-10. |
[33] | Opyrchal M, Khoury T, Boland P, Galanis E, Haddad TC, D’Assoro AB. Aurora Kinase Inhibitors in Breast Cancer Treatment. Toxicon. (2015) ; 38: : 1491-503. |
[34] | Yamamoto Y, Matsuyama H, Chochi Y, Okuda M, Kawauchi S, Inoue R, et al. Overexpression of BUBR1 is associated with chromosomal instability in bladder cancer. Cancer Genetics & Cytogenetics. (2007) ; 174: : 42-7. |
[35] | Han JY, Han YK, Park GY, Kim SD, Kim JS, Jo WS, et al. Bub1 is required for maintaining cancer stem cells in breast cancer cell lines. Scientific Reports. (2015) ; 5: : 15993. |
[36] | Chen MF, Lin PY, Wu CF, Chen WC, Wu CT. IL-6 Expression Regulates Tumorigenicity and Correlates with Prognosis in Bladder Cancer. Plos One. (2013) ; 8: : e61901-e. |
[37] | Dethlefsen C, Højfeldt G, Hojman P. The role of intratumoral and systemic IL-6 in breast cancer. Breast Cancer Research & Treatment. (2013) ; 138: : 657-64. |
[38] | Tobin NP, Sims AH, Lundgren KL, Lehn S, Landberg G. Cyclin D1, Id1 and EMT in breast cancer. BMC Cancer,11,1(2011-09-28). (2011) ; 11: : 1-14. |
[39] | Seiler R, Thalmann GN, Rotzer D, Perren A, Fleischmann A. CCND1/CyclinD1 status in metastasizing bladder cancer: a prognosticator and predictor of chemotherapeutic response. Modern Pathology An Official Journal of the United States & Canadian Academy of Pathology Inc. (2014) ; 27: : 87. |
[40] | Warrick JI, Walter V, Yamashita H, Chung E, Shuman L, Amponsa VO, et al. FOXA1, GATA3 and PPAR? Cooperate to Drive Luminal Subtype in Bladder Cancer: A Molecular Analysis of Established Human Cell Lines. Scientific Reports. (2016) ; 6: : 38531. |
[41] | Szarvas T, Reis H, Kramer G, Shariat SF, Vom DF, Tschirdewahn S, et al. Enhanced stromal syndecan-1 expression is an independent risk factor for poor survival in bladder cancer. Human Pathology. (2014) ; 45: : 674-82. |
[42] | Liu D, Zhang Z, Kong CZ. High FOXM1 expression was associated with bladder carcinogenesis. Tumour Biology the Journal of the International Society for Oncodevelopmental Biology & Medicine. (2013) ; 34: : 1131-8. |
[43] | Lipponen PK. Expression of c-myc protein is related to cell proliferation and expression of growth factor receptors in transitional cell bladder cancer. Journal of Pathology. (1995) ; 175: : 203-10. |
Appendices
Supplementary material
Supplementary Figure 1.
Expression of some overlapped genes shared by two cancers. The blue bar represent basal-like subtype whereas the pink bar represent luminal (luminal-A) subtype.
Supplementary Figure 2.
(A) The top 30 MIBC subtype-related genes with the highest MDG. Among these genes, blue bars indicate all basal-related genes whereas red bars indicate all luminal-related genes and the yellow bar indicate basal-related genes for bladder cancer but luminal-related genes for breast cancer. The right sub-graph of (A) indicates the heatmap of top 30 MIBC subtype-related genes with the highest MDG in two cancers. (B) The top 30 breast cancer subtype-related genes with the highest MDG. Among these genes, blue bars indicate all basal-related genes whereas red bars indicate all luminal-related genes. The right sub-graph of (B) indicates the heatmap of top 30 breast cancer subtype-related genes with the highest MDG in two cancers.
Supplementary Figure 3.
Cluster analyses for two subtypes of MIBC and breast cancer. (A) Cluster analysis for MIBC basal-like subtype. (B) Cluster analysis for MIBC luminal subtype. (C) Cluster analysis for breast cancer basal-like subtype. (D) Cluster analysis for breast cancer luminal-A subtype. Meanwhile, four small graphs involved in each sub-graph indicate consensus matrix for subtype, silhouette plot for two clusters, heatmap for cluster analysis, and the significance of paired comparison between two clusters respectively.