
A central role for hypoxia-inducible factor (HIF)-2α in hepatic glucose homeostasis
Abstract
Hepatic glucose production is regulated by hormonal and dietary factors. At fasting, 80% of glucose released into the circulation is derived from the liver, among which gluconeogenesis accounts for 55% and the rest by glycogenolysis. Studies suggest a complex mechanism involved in the regulation of hepatic glucose metabolism during fasting and post-absorptive phase. Oxygen plays a key role in numerous metabolic pathways such as TCA cycle, gluconeogenesis, glycolysis and fatty acid oxidation. Oxygenation of the gastrointestinal tract including liver and intestine is dynamically regulated by changes in the blood flow and metabolic activity. Cellular adaptation to low oxygen is mediated by the transcription factors HIF-1α and HIF-2α. HIF-1α regulates glycolytic genes whereas HIF-2α is known to primarily regulate genes involved in cell proliferation and iron metabolism. This review focuses on the role of the oxygen sensing signaling in the regulation of hepatic glucose output with an emphasis on hypoxia inducible factor (HIF)-2α. Recent studies have established a metabolic role of HIF-2α in systemic glucose homeostasis. Understanding the HIF-2α dependent mechanism in hepatic metabolism will greatly enhance our potential to utilize the oxygen sensing mechanisms to treat metabolic diseases.
1Introduction
Glucose is the primary energy source required to produce ATP through aerobic and anaerobic respiration. Sustained low blood glucose (hypoglycemia) results in adverse and irreversible detrimental effects at cellular and organismal level. Conversely, chronic elevation in blood glucose (hyperglycemia) leads to type 2 diabetes and other metabolic diseases. Two major hormones, insulin and glucagon tightly regulate blood glucose levels. Insulin regulates systemic glucose homeostasis by increasing glucose uptake and utilization in insulin sensitive target tissues and decreasing hepatic gluconeogenesis. In contrast, glucagon acts on the liver to increase blood glucose levels through gluconeogenesis and glycogenolysis. Numerous mechanisms are proposed in the pathogenesis of insulin resistance that includes but not limited to dyslipidemia, inflammation, and unfolded protein response [1]. At the cellular level, insulin resistance is associated with a decrease in the expression of insulin receptor, insulin receptor substrate (IRS) phosphorylation and increase in inflammation. Insulin resistance either at peripheral level (liver, adipose tissue and skeletal muscle) or at central level (brain) is associated with decrease in the cellular response to insulin. There is also a complex crosstalk between the peripheral tissues and the brain in the predisposition and/or acceleration of insulin resistance, which leads to chronic elevation in blood glucose. Chronic hyperglycemia results in a compensatory increase in pancreatic insulin secretion, which if persists for a prolonged period, may lead to β-cell failure and type 2 diabetes [2–4].
Evolutionarily oxygen plays a central role in numerous key cellular processes. Intracellular oxygen levels vary significantly depending on the cell type, oxygen availability and metabolic activity. Recent studies have unraveled an integrative pathway between hepatic hypoxic response and systemic glucose regulation. This review will focus on the role of hepatic hypoxia in liver metabolism by regulating key signaling pathways involved in hepatic gluconeogenesis.
2Oxygen dynamics and oxygen sensing pathways in the liver
Oxygen dynamics in the liver is rapid compared to other tissues due to its high metabolic activity and poor supply of oxygenated blood from the portal vein [5, 6]. Approximately, a 50% drop in the blood oxygen tension was observed between the periportal to perivenous region, causing a steep oxygen gradient along the hepatic sinusoids of the liver [7]. Liver was once thought to be a homogenous tissue; however recent studies have determined the existence of zones having a distinct pattern of gene expression involved in hepatic glucose and lipid metabolism [8–12]. Oxygen gradient in addition to metabolic status (fasting versus fed), metabolites, and insulin/glucagon ratio vary significantly between the periportal and perivenous areas and have profound effect on the zonal function of normal as well as diseased liver [7, 9, 13–15]. Numerous signaling pathways have been implicated in the regulation of hepatic zonation such as transforming growth factor (TGF)-β, fibroblast growth factor (FGF), bone morphogenetic growth factor, Wnt/β-catenin signaling, hepatocyte growth factor and Hedgehog signaling [16]. Of these signaling pathways, Wnt/β-catenin signaling is considered to be the gatekeeper of hepatic zonation. Hypoxia is known to significantly enhance Wnt/β-catenin signaling by regulating the nuclear translocation of β-catenin, LEF-1 and TCF-1 proteins [17, 18]. The role of hypoxia in hepatic zonation needs further studies, but it is clear that the oxygen gradient along the liver sinusoids is critical for the metabolic zonation of liver from perivenous to periportalregion.
Cells adapt to acute decrease in cellular oxygen (physiological hypoxia) via oxygen sensitive transcription factor hypoxia inducible factor (HIF). HIF is a heterodimer of an oxygen-dependent α subunit, (HIF-1α, HIF-2α, or HIF-3α) and constitutively expressed β-subunit or Aryl hydrocarbon receptor nuclear translocator (Arnt) [19, 20]. Under normoxia, HIFα is hydroxylated by two enzymes, prolyl 4-hydroxylase domain-containing proteins (PHD)s and 2-oxoglutarate (2-OG)-dependent oxygenase factor inhibiting HIF-1α (FIH-1). Hydroxylated HIFs are subsequently bound by the E3 ubiquitin ligase von Hippel Lindau (VHL) protein which targets HIFα to ubiquitin-mediated degradation [21] (Fig. 1).
Fig.1
Regulation of HIF stability by oxygen. Under normoxic conditions, proline residues on HIFα subunit are hydroxylated by prolyl 4-hydroxylase domain-containing proteins (PHD). Hydroxylated HIF alpha bind the E3 ubiquitin ligase von Hippel Lindau tumor suppressor protein (VHL), which induces ubiquitin-mediated degradation of HIFα. Hypoxic conditions, decrease the ability of PHDs to hydroxylate HIFα subunit, resulting in stabilization and heterodimerization of HIFα with aryl hydrocarbon receptor nuclear translocator (ARNT). HIFα/ARNT complex binds to response elements (RCGTG) on HIF target genes and induces their expression.
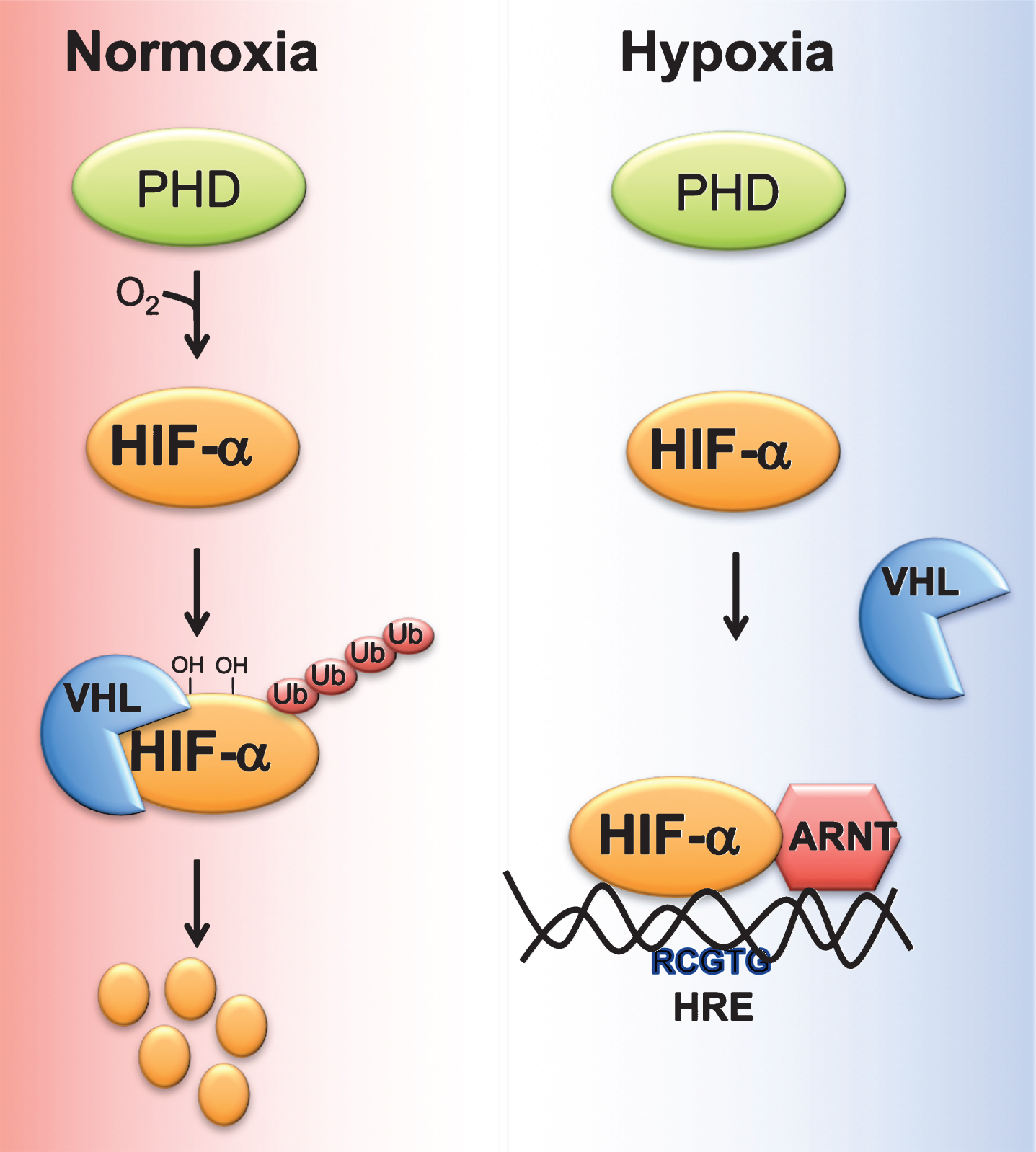
PHDs hydroxylate the proline residues present in the LXXLAP core sequence on HIFα subunits [22]. Four isoforms of PHDs have been characterized namely PHD1 (also known as egl nine homolog (EGLN)-2), PHD2 (also known as EGLN-1), PHD3 (also known as EGLN-3) and prolyl 4-hydroxylase transmembrane (P4H-TM) [23, 24]. PHDs vary in their tissue distribution and intracellular localization. Among the PHDs, PHD2 is abundantly expressed by all tissues [25]. P4H-TM is anchored to the ER membrane, whereas the other PHDs are localized in the cytoplasm. The C-terminal domain containing the hydroxylase activity is well conserved compared to the N-terminal domain. PHDs use oxygen and 2-OG as substrate, and iron and ascorbate as cofactors [26]. PHD activity is also regulated by metabolic intermediates. An increase in TCA metabolites such as fumarate, succinate and itaconate will alter PHD activity by competing with 2-OG [27–31]. Similarly, divalent metals such as nickel and cobalt inhibit PHD activity by decreasing the intracellular levels of ascorbate [29].
PHDs also exhibit substrate specificity. PHD1 and PHD3 predominantly regulate HIF-2α and PHD2 regulates HIF-1α [32–34]. PHDs sequentially hydroxylate the C-terminal proline residue followed by N-terminal proline on HIFs. The secondary structure was speculated to regulate the selectivity of the PHDs in determining the hydroxylation of C-terminal versus N-terminal proline residues on HIFs [35]. PHD1 and PHD3 display relatively less activity in hydroxylation of the N-terminal proline residue than PHD2. Interestingly N-terminal proline hydroxylation is more sensitive to hypoxia compared to C-terminal proline. Thus, it is speculated that the oxygen-independent but iron-dependent differential regulation of HIFs by PHDs may be conferred by their structural difference and proline selectivity [22]. Similarly, binding partners of HIF and PHDs also were shown to regulate PHD specificity. For instance, OS-9 increased ubiquitin mediated degradation of HIF-1α by forming ternary complex with PHD2 and PHD3 rather than to PHD1 [36, 37].
HIF bind to core HIF response elements (HRE) that compose of 5’-RCGTG-3’ present at the promoter of HIF target genes. At the R position “A” is preferred over the “G”, and “T” and “C” at the 5’ and 3’ position of the core response element, respectively. The promoters of HIF target genes also contain “CAC” at position +13 of core HIF response element [38, 39]. HIF-1α and HIF-2α bind to the same core HREs, but can regulate distinct target genes by currently unclear mechanisms. Typically, HIF-1α HREs are present closer to the transcription start site compared to HIF-2α. However, motifs closer to core response element could not be identified to determine their differential regulation of HIF target genes, suggesting the importance of epigenetic mechanisms to dictate target specificity [38, 39].
3Hypoxia and hepatic circadian metabolism
One of the important regulators of hepatic metabolism including gluconeogenesis is the well-coordinated circadian clock machinery. The suprachiasmatic nucleus (SCN) in the anterior hypothalamus orchestrates the circadian rhythmicity of various genes in response to light, and recent evidence suggests food to be an important regulator of circadian rhythmicity [40, 41]. Chronic activation of hypoxia signaling in liver disrupts circadian expression of genes through dysregulation of hepatic Rev-erbα signaling in a HIF-2α dependent manner [42]. In liver, 20% of protein is regulated by circadian clock at the transcriptional and/or at the post-translational level [43]. Circadian rhythms regulate the release of insulin and glucagon on a daily basis in response to food and central nervous system stimulation. Several mouse models with disruption in circadian genes develop glucose intolerance and insulin resistance suggesting a key role of rhythmicity in glucose homeostasis [44, 45]. Recently, Adamovich et al., demonstrated that circadian oscillations in oxygen tension in the blood and peripheral tissues are essential to synchronize clock genes in the liver [46]. In the muscle, HIF-1α directly binds to the circadian gene BMAL1 and induces the expression of glycolytic genes [47]. However, constitutive activation of hypoxia signaling disrupts rhythmicity of circadian genes resulting in various pathological diseases [42, 48]. Further investigation is required to understand the cross talk between circadian mechanisms and hypoxia signaling in hepatic homeostasis including gluconeogenesis.
4Hepatic HIF and insulin signaling
Insulin exerts its action by binding to the insulin receptor. Upon ligand binding, insulin receptor undergoes a conformational change resulting in the recruitment and binding of IRS family of proteins [49, 50]. The function of IRS-1 and IRS-2 are well characterized in the liver. Disruption of hepatic IRS-1 and/or IRS-2 results in glucose intolerance and insulin resistance [51–53]. IRS mediates insulin signaling by binding to phosphoinositide-3-kinase (PI3K), which generates PIP3 leading to the recruitment of AKT and 3-phosphoinositide-dependent protein kinase 1 (PDK1). PDK1 phosphorylates Thr-308 on AKT, which is followed by Ser-473 phosphorylation by mechanistic target of rapamycin 2 (mTORC2) [54, 55]. Downstream targets of AKT are essential in glucose metabolism, lipid metabolism, growth and survival of the cells. Importantly, AKT regulates genes involved in glucose and lipid metabolism by phosphorylating Forkhead box O (FOXO) family of protein resulting in their nuclear exclusion and degradation [54, 55].
In liver, HIF-1α regulates genes involved in glycolysis, inflammation and fatty acid oxidation. Hepatic HIF-2α regulates genes involved in fatty acid oxidation, iron metabolism, fibrosis and inflammation [42, 56]. Recent studies have unraveled a new role of HIF-2α in glucose homeostasis [34, 57, 58]. Taniguchi et al., have demonstrated that knockdown of PHD3 in the liver results in stabilization of HIF-2α [34]. The increase in hepatic HIF-2α but not HIF-1α, improves glucose tolerance and insulin sensitivity [34]. Disruption of hepatic PHD3 alone is sufficient to observe a significant decrease in the expression of gluconeogenic genes in the liver; however compound disruption of PHD3 with other PHDs resulted in hepatic steatosis, probably due to a robust increase in hepatic HIF-2α signaling [42, 56]. Mechanistically, HIF-2α improved insulin sensitivity by increasing the expression of IRS-2 [34] (Fig. 2). Disruption of IRS-2 mitigated the improvement in glucose tolerance in PHD3-/- mice. Similar conclusions were derived by Wei et al. [58]. Inhibition of vascular endothelial growth factor (VEGF)-b by pharmacological and genetic approaches decreased angiogenesis and hepatic vascular function, which led to cellular hypoxia and stabilization of HIF-2α [58–60]. The increase in hepatic hypoxia ameliorated dyslipidemia, decreased hepatic gluconeogenesis, improved glucose tolerance and hepatic insulin sensitivity in a HIF-2α-IRS-2 dependent manner [34, 57, 58](Fig. 2).
Fig.2
Regulation of hepatic gluconeogenesis by HIF-2. Liver hypoxia, refeeding, vascular endothelial growth factor (VEGF) inhibitors or prolyl 4 hydroxylase domain-containing protein (PHD) inhibitors stabilize hypoxia-inducible factor (HIF)-2α in the liver. HIF-2α improves insulin sensitivity by increasing insulin receptor substrate (IRS)-2 levels directly at transcriptional level or indirectly through transrepression by sterol regulatory binding protein (SREBP)-1c. IRS-2 decreases gluconeogenesis by enhancing AKT-mediated repression of gluconeogenic genes. In addition, HIF-2α attenuates postprandial glucagon signaling through ERK1/2-dependent increase in phosphodiesterase (PDE) mediated hydrolysis of intracellular cyclic AMP (cAMP), resulting in a decrease in protein kinase A (PKA)-mediated activation of cAMP response element-binding protein (CREB). Lastly, HIF-2α-mediated increase in systemic erythropoietin (EPO) levels inhibit gluconeogenesis through a STAT3-dependent mechanism.
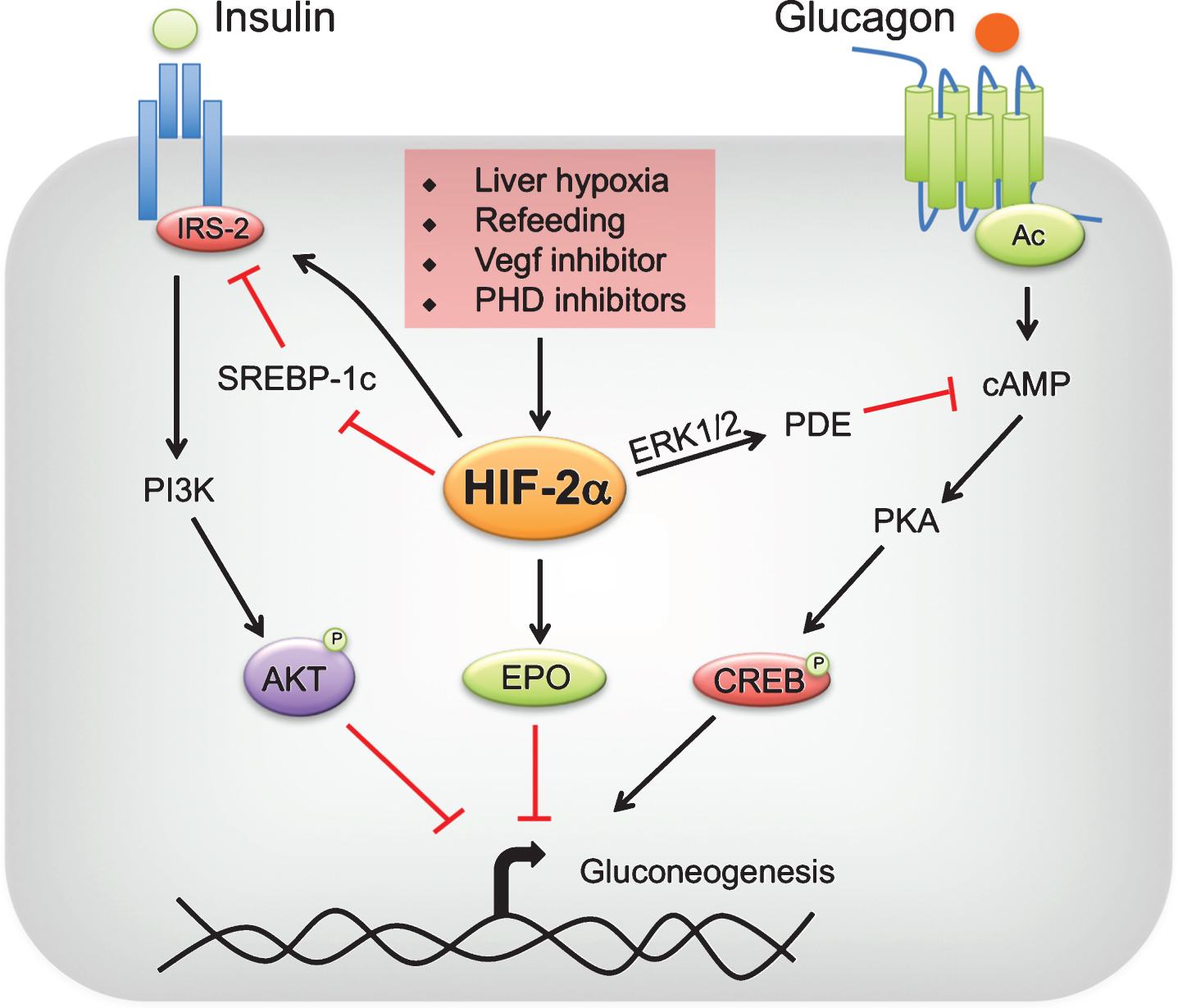
5Hypoxia and glucagon signaling in the regulation of glucose homeostasis
The counter regulatory hormone glucagon increases hepatic glucose output by inducing the expression of genes involved in gluconeogenesis such as phosphoenolpyruvate carboxykinase (Pepck) and glucose 6-phosphatase (G6pase). Glucagon binding to its receptor results in the activation of adenylyl cyclase, which increases intracellular cyclic AMP (cAMP) production from ATP. Protein kinase A (PKA) is a heterodimer that comprise of two regulatory and two catalytic subunit [61]. cAMP binding to the regulatory subunit of PKA results in the release of catalytic subunit which then phosphorylates cAMP response element-binding protein (CREB) [62, 63]. Additionally, glucagon dephosphorylates CREB regulated transcriptional co-activator 2 (CRTC2), and together this results in the expression of gluconeogenic target genes such as Pepck and G6pase [64, 65].
Although not the cause, aberrant levels of glucagon plays a key role in the progression of diabetes. Chronic elevation of glucagon increases the gluconeogenic capacity of insulin resistant livers, thus further aggravating disease progression [64–66]. Liver-specific glucagon receptor knockout mice are resistant to diet-induced hyperglycemia and ameliorates glucose intolerance in mouse model of type 1 diabetes [67, 68].
Upon refeeding glucagon signaling is ablated in concert with an increase in insulin action to maintain systemic glucose homeostasis and curb postprandial gluconeogenesis. Unlike insulin, glucagon levels are not rapidly regulated immediately after refeeding and remain at high levels [69]. This suggests a potential role of other pathways in the repression of glucagon signaling immediately after refeeding. Recent work demonstrates that refeeding results in induction of liver hypoxia and HIF-2α [57]. The increase in HIF-2α is critical to inhibit hepatic glucagon signaling during the acute postprandial state. Mouse models that overexpress HIF-2α but not HIF-1α specifically in hepatocytes improved glucose tolerance and decreased hepatic gluconeogenesis, independent of insulin sensitivity [57]. HIF-2α increased IRS-2 but not IRS-1 expression [34]. However, liver-specific IRS-1 but not IRS-2 knockout mice develops insulin resistance under refeeding conditions suggesting that an increase in hepatic HIF-2α-IRS-2 may not be a major contributor in the regulation postprandial glucose metabolism [52]. HIF-2α inhibited glucagon signaling by increasing the hydrolysis of cAMP via upregulating the activity of phosphodiesterases (PDEs). Mechanistically, HIF-2α induced upon refeeding activated ERK, a known activator of PDEs [57]. PDE abrogated glucagon signaling by decreasing intracellular cAMP levels. Inhibiting PDE4 in hepatocytes partially restored HIF-2α-mediated repression of hepatic glucagon signaling and gluconeogenesis (Fig. 2). Furthermore, activation of HIF-2α is sufficient to improve glucose tolerance in streptozotocin induced diabetes model [57]. Thus, HIF-2α plays a critical role in postprandial glucose homeostasis by regulation hepatic glucagon signaling.
6Hypoxia and mTOR signaling in the regulation of glucose homeostasis
Mechanistic target of rapamycin (mTOR) signaling plays an important role in responding to the cellular cues for protein synthesis and cell growth by regulating its downstream targets such as the eukaryotic initiation factor 4E-binding protein-1 (4EBP1) and ribosomal p70 S6 kinase (S6K) [70]. Dysregulation of mTOR signaling is associated with type 2 diabetes and metabolic diseases [71, 72]. In liver, mTOR signaling controls lipogenesis by regulating the expression of SREBP-1c [73]. The lipogenic effect of insulin in the liver occurs through the mTOR-mediated induction of SREPB-1c and a concomitant repression of hepatic FOXO1 signaling [74]. Inhibition of mTOR using rapamycin decreased insulin-stimulated SREBP-1c levels without any significant effect on insulin-mediated repression of gluconeogenic genes [75, 76]. Improvement in insulin sensitivity following acute treatment with rapamycin occurs through inhibition of S6 kinase mediated IRS-1 phosphorylation [77]; however chronic inhibition of mTOR results in insulin resistance [72, 78]. Interestingly, in diet-induced obesity, where the glucoregulatory effect of insulin is attenuated but not lipogenic effect, mTOR signaling has been proposed to uncouple lipogenesis from gluconeogenesis through SREBP-1c dependent mechanism [75, 76].
Transient increases in HIF-2α through inhibition of Vegf or PHD3 improves insulin sensitivity through upregulation of IRS-2 expression without any significant increase in hepatic triglyceride [58]. The induction of IRS-2 by HIF-2α is associated with a significant decrease in the expression of SREBP-1c, a known IRS-2 repressor [58]. However, the mechanism by which HIF-2α decreases SREBP-1c is not currently known. During hypoxia mTORC-mediated anabolic pathway is inhibited by increase in the expression of regulated in development and DNA damage 1 (REDD1) [79, 80]. It is not known if the cross talk between HIF-2α and mTORC signaling is involved in the regulation of SREBP-1c-IRS-2 expression. Therefore, further investigation on the cross talk between HIF-2α and mTORC signaling is essential to therapeutically target HIF-2α in the treatment of diabetes without the consequential effect of HIF-2α on steatosis.
7Hypoxia and erythropoietin (EPO) signaling in the regulation of glucose homeostasis
EPO is a glycopeptide hormone that induces erythropoiesis from precursor erythroid progenitor cells [81]. EPO is primarily secreted by the kidney and to some extent by the liver. Recombinant EPO is used in patients that undergo hemodialysis and in chronic kidney disease patients to treat anemia [82]. Chronic hypoxia resulted in a robust increase in EPO mRNA (∼1500 fold) levels in mice [83]. Using RNA interference technology and in vivo animal models, EPO was shown to be specifically regulated by HIF-2α but not HIF-1α. Overexpression of HIF-2α is sufficient to significantly increase the hepatic, renal and serum levels of EPO [84, 85].
Chronic EPO treatment in hemodialysis patients decreased serum glucose levels and improved parameters of insulin sensitivity [86–88]. Similarly, EPO improved systemic glucose homeostasis, insulin sensitivity and β-cell function in animal models. Recombinant EPO treatment increased AKT phosphorylation and decreased inflammation in livers of mice fed with a high fat diet [87, 89]. In skeletal muscle and adipose tissue, EPO significantly decreased inflammation and improved glucose uptake in these tissues [90, 91]. HIF-2α induced EPO provides yet another mechanism by which HIF-2α improves glucose homeostasis.
8Deleterious effects of chronic HIF-2α activation in liver
Transient activation of hypoxia signaling has an essential role in hepatic metabolism by regulating insulin and glucagon signaling (described above). Hypoxia signaling is also a cellular adaptation for survival to acute injury or insult, which primarily affects the intracellular oxygen levels. For instance, to prevent ischemic reperfusion (I/R) injury during surgical resection (liver) and transplantation (liver, kidney and heart), ischemic preconditioning is performed to decrease apoptosis, mitochondrial dysfunction and inflammation in the tissues [92, 93]. One of the key signaling pathways that is activated by ischemic preconditioning is HIF signaling [92–95]. Mouse models with tissue and cell type specific knockout have established a critical role of HIF signaling in protection against IR injury [92–95]. However, chronic intermittent exposure to hypoxia resulted in dysregulated lipid levels in mouse models of obesity [96, 97]. Several independent groups demonstrate that mouse models with hepatocyte specific activation of HIF exhibit dyslipidemia, elevated hepatic triglyceride and cholesterol levels [42, 56, 98]. Mechanistically, HIF-2α increases hepatic lipid accumulation through a decrease in PPARα activity and disruption in circadian regulation of cholesterol-bile synthesis [42, 56, 98]. Chronic activation of HIF-2α also results in a significant increase in inflammatory cytokines and infiltration of inflammatory cells in the liver. Moreover, numerous genes involved in liver fibrosis are significantly increased in the liver resulting in hepatic fibrosis [98]. Moreover, constitutive activation of HIF-2α also results in polycythemia due to increase in EPO-mediated erythropoiesis [84].
Hypoxia is a hallmark of cancer. An imbalance in the cell growth to tissue blood supply results in tumor hypoxia, leading to activation HIF target genes. Increased HIF-1α target gene such as Glut-1 in hepatocellular carcinoma (HCC) is needed for a switch to glycolytic metabolism. This is a critical survival advantage to cancer cells in the face of limited nutrient supply. However, the role of HIF-2α in cancer appears controversial. Report suggests that increase in HIF-2α in HCC results in cell death [99]. On the other hand, knock down of HIF-1α resulted in a compensatory increase in HIF-2α, which decreased caspase-3 activity leading to the progression of HCC [100]. Therefore, further investigation is required to delineate the role of HIF-1α and HIF-2α in HCC in order to target HIF signaling in metabolic diseases.
9PHD inhibitors and their use in diabetes
Numerous small molecule inhibitors of PHDs have been developed, tested, and are under clinical trial for use in the treatment of anemia in chronic kidney disease patients [101]. Competitive inhibitors of 2-OG such as dimethyloxaloylglycine (DMOG), FG-4497 and FG-4592 effectively inhibit PHDs and stabilize HIFs. PHD2 inhibitors are currently used in the treatment of anemia in chronic kidney diseases [102]. PHD2 treatment induces intestinal iron absorption and hepatic secretion of EPO, which increases RBC count through erythropoiesis [102]. Independent of its effect on erythropoiesis, EPO decreases hepatic gluconeogenesis and protects mice against diet-induced glucose intolerance and hepatic inflammation [103]. Further investigation is needed to assess whether treatment with PHD inhibitors will improve glucose homeostasis in chronic kidney disease patients through EPO dependent mechanism.
Chronic high levels of hepatic of HIF-2α result in steatohepatitis. However, achieving moderate increase in HIF-2α in liver is highly protective in mouse models of diabetes [57, 58]. PHD inhibitor FG-4497 ameliorates diet-induced glucose intolerance and hepatic steatosis, and decreases visceral adiposity in mice [104]. Modest stabilization of HIF-1α and HIF-2α by FG-4497 decreases the incidence of atherosclerosis by decreasing serum cholesterol levels, inflammation and by increasing autoantibodies against oxidized-LDL cholesterol [105]. Thus, PHD inhibitors that stabilize HIFs may have beneficial effects in improving metabolic homeostasis.
10Conclusion
Hepatic HIFα signaling is not only essential for cellular adaptation to hypoxia but also critical to maintain metabolic homeostasis. The role of HIF-2α in HCC needs further investigation, and it is evident that chronic high activation of HIF-2α leads to steatohepatitis and fibrosis. However, transient and/or moderate increase in HIF-2α is beneficial in decreasing hepatic glucagon signaling, increasing insulin sensitivity and improving hepatic lipid metabolism. Currently, PHD inhibitors are used in the treatment of anemia in chronic kidney disease patients. However, their beneficial role of HIF in regulating hepatic glucose homeostasis provides good rationale to investigate the use of HIF activators in the treatment of type 2 diabetes.
Conflict of interests
The authors declare no conflict of interest.
Acknowledgments
Y.S. is supported by grants from the NIH (CA148828 and DK095201) and S.K.R is supported by a grant from the NIH (K99DK110537).
References
[1] | Samuel VT , Shulman GI . Mechanisms for insulin resistance: Commonthreads and missing links. Cell. (2012) ;148: (5):852–71. |
[2] | Perry RJ , Samuel VT , Petersen KF , Shulman GI . The role of hepatic lipids in hepatic insulin resistance and type 2 diabetes. Nature. (2014) ;510: (7503)84–91. |
[3] | Roden M , Price TB , Perseghin G , Petersen KF , Rothman DL , Cline GW , et al. Mechanism of free fatty acid-induced insulin resistance inhumans. J Clin Invest. (1996) ;97: (12):2859–65. |
[4] | Kahn SE , Hull RL , Utzschneider KM . Mechanisms linking obesity to insulin resistance and type 2 diabetes. Nature. (2006) ;444: (7121):840–6. |
[5] | Cho CH , Park J , Nagrath D , Tilles AW , Berthiaume F , Toner M , et al. Oxygen uptake rates and liver-specific functions of hepatocyte and 3T3 fibroblast co-cultures. Biotechnol Bioeng. (2007) ;97: (1):188–99. |
[6] | Sato A , Kadokura K , Uchida H , Tsukada K . An in vitro hepaticzonation model with a continuous oxygen gradient in a microdevice. Biochem Biophys Res Commun. (2014) ;453: (4):767–71. |
[7] | Jungermann K , Kietzmann T . Oxygen: Modulator of metabolic zonation and disease of the liver. Hepatology. (2000) ;31: (2):255–60. |
[8] | Chen KS , Katz J . Zonation of glycogen and glucose syntheses, butnot glycolysis, in rat liver. Biochem J. (1988) ;255: (1):99–104. |
[9] | Guzman M , Bijleveld C , Geelen MJ . Flexibility of zonation of fatty acid oxidation in rat liver. Biochem J. (1995) ;311: (Pt 3):853–60. |
[10] | Hailfinger S , Jaworski M , Braeuning A , Buchmann A , Schwarz M . Zonal gene expression in murine liver: Lessons from tumors. Hepatology. (2006) ;43: (3):407–14. |
[11] | Katz NR , Fischer W , Giffhorn S . Distribution of enzymes of fattyacid and ketone body metabolism in periportal and perivenousrat-liver tissue. Eur J Biochem. (1983) ;135: (1):103–7. |
[12] | Wolfle D , Schmidt H , Jungermann K . Short-term modulation of glycogen metabolism, glycolysis and gluconeogenesis by physiological oxygen concentrations in hepatocyte cultures. Eur J Biochem. (1983) ;135: (3):405–12. |
[13] | Behari J , Li H , Liu S , Stefanovic-Racic M , Alonso L , O’Donnell CP , et al. beta-catenin links hepatic metabolic zonation with lipid metabolism and diet-induced obesity in mice. Am J Pathol. (2014) ;184: (12):3284–98. |
[14] | Gebhardt R . Metabolic zonation of the liver: Regulation and implications for liver function. Pharmacol Ther. (1992) ;53: (3):275–354. |
[15] | Kudryavtseva MV , Sakuta GA , Stein GI , Kudryavtsev BN . The metabolic zonation of glycogen synthesis in rat liver after fasting and refeeding. Tissue Cell. (1992) ;24: (1):31–5. |
[16] | Gebhardt R , Matz-Soja M . Liver zonation: Novel aspects of its regulation and its impact on homeostasis. World J Gastroenterol. (2014) ;20: (26):8491–504. |
[17] | Kietzmann T . Metabolic zonation of the liver: The oxygen gradient revisited. Redox Biol. (2017) ;11: :622–30. |
[18] | Mazumdar J , O’Brien WT , Johnson RS , LaManna JC , Chavez JC , Klein PS , et al. O2 regulates stem cells through Wnt/beta-catenin signalling. Nat Cell Biol. (2010) ;12: (10):1007–13. |
[19] | Huang LE , Gu J , Schau M , Bunn HF . Regulation of hypoxia-inducible factor 1alpha is mediated by an O2-dependent degradation domain via the ubiquitin-proteasome pathway. Proc Natl Acad Sci U S A. (1998) ;95: (14):7987–92. |
[20] | Wang GL , Jiang BH , Rue EA , Semenza GL . Hypoxia-inducible factor 1 is a basic-helix-loop-helix-PAS heterodimer regulated by cellular O2 tension. Proc Natl Acad Sci U S A. (1995) ;92: (12):5510–4. |
[21] | Ivan M , Kondo K , Yang H , Kim W , Valiando J , Ohh M , et al. HIFalpha targeted for VHL-mediated destruction by proline hydroxylation: Implications for O2 sensing. Science. (2001) ;292: (5516):464–8. |
[22] | Hirsila M , Koivunen P , Gunzler V , Kivirikko KI , Myllyharju J . Characterization of the human prolyl 4-hydroxylases that modify the hypoxia-inducible factor. J Biol Chem. (2003) ;278: (33):30772–80. |
[23] | Fong GH , Takeda K . Role and regulation of prolyl hydroxylase domain proteins. Cell Death Differ. (2008) ;15: (4):635–41. |
[24] | Koivunen P , Tiainen P , Hyvarinen J , Williams KE , Sormunen R , Klaus SJ , et al. An endoplasmic reticulum transmembrane prolyl 4-hydroxylase is induced by hypoxia and acts on hypoxia-inducible factor alpha. J Biol Chem. (2007) ;282: (42):30544–52. |
[25] | Berra E , Benizri E , Ginouves A , Volmat V , Roux D , Pouyssegur J . HIF prolyl-hydroxylase 2 is the key oxygen sensor setting low steady-state levels of HIF-1alpha in normoxia. EMBO J. (2003) ;22: (16):4082–90. |
[26] | Hagen T , Taylor CT , Lam F , Moncada S . Redistribution of intracellular oxygen in hypoxia by nitric oxide: Effect on HIF1alpha. Science. (2003) ;302: (5652):1975–8. |
[27] | Lampropoulou V , Sergushichev A , Bambouskova M , Nair S , Vincent EE , Loginicheva E , et al. Itaconate Links Inhibition of Succinate Dehydrogenase with Macrophage Metabolic Remodeling and Regulation of Inflammation. Cell Metab. (2016) ;24: (1):158–66. |
[28] | Mills EL , Kelly B , Logan A , Costa AS , Varma M , Bryant CE , et al. Succinate Dehydrogenase Supports Metabolic Repurposing of Mitochondria to Drive Inflammatory Macrophages. Cell. (2016) ;167: (2):457–70 e13. |
[29] | Salnikow K , Donald SP , Bruick RK , Zhitkovich A , Phang JM , Kasprzak KS . Depletion of intracellular ascorbate by the carcinogenic metals nickel and cobalt results in the induction of hypoxic stress. J Biol Chem. (2004) ;279: (39):40337–44. |
[30] | Selak MA , Armour SM , MacKenzie ED , Boulahbel H , Watson DG , Mansfield KD , et al. Succinate links TCA cycle dysfunction to oncogenesis by inhibiting HIF-alpha prolyl hydroxylase. Cancer Cell. (2005) ;7: (1):77–85. |
[31] | Tannahill GM , Curtis AM , Adamik J , Palsson-McDermott EM , McGettrick AF , Goel G , et al. Succinate is an inflammatory signal that induces IL-1beta through HIF-1alpha. Nature. (2013) ;496: (7444):238–42. |
[32] | Appelhoff RJ , Tian YM , Raval RR , Turley H , Harris AL , Pugh CW , et al. Differential function of the prolyl hydroxylases PHD1, PHD2,and PHD3 in the regulation of hypoxia-inducible factor. J BiolChem. (2004) ;279: (37):38458–65. |
[33] | Huang J , Zhao Q , Mooney SM , Lee FS . Sequence determinants in hypoxia-inducible factor-1alpha for hydroxylation by the prolyl hydroxylases PHD1, PHD2, and PHD3. J Biol Chem. (2002) ;277: (42):39792–800. |
[34] | Taniguchi CM , Finger EC , Krieg AJ , Wu C , Diep AN , LaGory EL , et al. Cross-talk between hypoxia and insulin signaling through Phd3regulates hepatic glucose and lipid metabolism and amelioratesdiabetes. Nat Med. (2013) ;19: (10):1325–30. |
[35] | Flashman E , Bagg EA , Chowdhury R , Mecinovic J , Loenarz C , McDonough MA , et al. Kinetic rationale for selectivity toward N- and C-terminal oxygen-dependent degradation domain substrates mediated by a loop region of hypoxia-inducible factor prolyl hydroxylases. J Biol Chem. (2008) ;283: (7):3808–15. |
[36] | Baek JH , Mahon PC , Oh J , Kelly B , Krishnamachary B , Pearson M , et al. OS-9 interacts with hypoxia-inducible factor 1alpha and prolylhydroxylases to promote oxygen-dependent degradation ofHIF-1alpha. Mol Cell. (2005) ;17: (4):503–12. |
[37] | McDonough MA , Li V , Flashman E , Chowdhury R , Mohr C , Lienard BM , et al. Cellular oxygen sensing: Crystal structure ofhypoxia-inducible factor prolyl hydroxylase (PHD2). Proc Natl AcadSci U S A. (2006) ;103: (26):9814–9. |
[38] | Mole DR , Blancher C , Copley RR , Pollard PJ , Gleadle JM , Ragoussis J , et al. Genome-wide association of hypoxia-inducible factor (HIF)-1alpha and HIF-2alpha DNA binding with expression profiling of hypoxia-inducible transcripts. J Biol Chem. (2009) ;284: (25):16767–75. |
[39] | Schodel J , Oikonomopoulos S , Ragoussis J , Pugh CW , Ratcliffe PJ , Mole DR . High-resolution genome-wide mapping of HIF-binding sites by ChIP-seq. Blood. (2011) ;117: (23):e207–17. |
[40] | Eckel-Mahan K , Sassone-Corsi P . Metabolism and the circadian clock converge. Physiol Rev. (2013) ;93: (1):107–35. |
[41] | Sujino M , Masumoto KH , Yamaguchi S , van der Horst GT , Okamura H , Inouye ST . Suprachiasmatic nucleus grafts restore circadian behavioral rhythms of genetically arrhythmic mice. Curr Biol. (2003) ;13: (8):664–8. |
[42] | Ramakrishnan SK , Taylor M , Qu A , Ahn SH , Suresh MV , Raghavendran K , et al. Loss of von Hippel-Lindau protein (VHL) increases systemic cholesterol levels through targeting hypoxia-inducible factor 2alpha and regulation of bile acid homeostasis. Mol Cell Biol. (2014) ;34: (7):1208–20. |
[43] | Reddy AB , Karp NA , Maywood ES , Sage EA , Deery M , O’Neill JS , et al. Circadian orchestration of the hepatic proteome. Curr Biol. (2006) ;16: (11):1107–15. |
[44] | Marcheva B , Ramsey KM , Buhr ED , Kobayashi Y , Su H , Ko CH , et al. Disruption of the clock components CLOCK and BMAL1 leads to hypoinsulinaemia and diabetes. Nature. (2010) ;466: (7306):627–31. |
[45] | Turek FW , Joshu C , Kohsaka A , Lin E , Ivanova G , McDearmon E , et al. Obesity and metabolic syndrome in circadian Clock mutant mice. Science. (2005) ;308: (5724):1043–5. |
[46] | Adamovich Y , Ladeuix B , Golik M , Koeners MP , Asher G . Rhythmic Oxygen Levels Reset Circadian Clocks through HIF1alpha. Cell Metab. (2017) ;25: (1):93–101. |
[47] | Peek CB , Levine DC , Cedernaes J , Taguchi A , Kobayashi Y , Tsai SJ , et al. Circadian Clock Interaction with HIF1alpha Mediates Oxygenic Metabolism and Anaerobic Glycolysis in Skeletal Muscle. Cell Metab. (2017) ;25: (1):86–92. |
[48] | Wu Y , Tang D , Liu N , Xiong W , Huang H , Li Y , et al. Reciprocal Regulation between the Circadian Clock and Hypoxia Signaling at the Genome Level in Mammals. Cell Metab. (2017) ;25: (1):73–85. |
[49] | Schaffer L . A model for insulin binding to the insulin receptor. Eur J Biochem. (1994) ;221: (3):1127–32. |
[50] | Baron V , Kaliman P , Gautier N , Van Obberghen E . The insulin receptor activation process involves localized conformational changes. J Biol Chem. (1992) ;267: (32):23290–4. |
[51] | Dong X , Park S , Lin X , Copps K , Yi X , White MF . Irs1 and Irs2signaling is essential for hepatic glucose homeostasis andsystemic growth. J Clin Invest. (2006) ;116: (1):101–14. |
[52] | Kubota N , Tobe K , Terauchi Y , Eto K , Yamauchi T , Suzuki R , et al. Disruption of insulin receptor substrate 2 causes type 2 diabetes because of liver insulin resistance and lack of compensatory beta-cell hyperplasia. Diabetes. (2000) ;49: (11):1880–9. |
[53] | Tamemoto H , Kadowaki T , Tobe K , Yagi T , Sakura H , Hayakawa T , et al. Insulin resistance and growth retardation in mice lacking insulin receptor substrate-1. Nature. (1994) ;372: (6502):182–6. |
[54] | Puigserver P , Rhee J , Donovan J , Walkey CJ , Yoon JC , Oriente F , et al. Insulin-regulated hepatic gluconeogenesis through FOXO1-PGC-1alpha interaction. Nature. (2003) ;423: (6939):550–5. |
[55] | Yang G , Murashige DS , Humphrey SJ , James DE . A Positive Feedback Loop between Akt and mTORC2 via SIN1 Phosphorylation. Cell Rep. (2015) ;12: (6):937–43. |
[56] | Rankin EB , Rha J , Selak MA , Unger TL , Keith B , Liu Q , et al. Hypoxia-inducible factor 2 regulates hepatic lipid metabolism. Mol Cell Biol. (2009) ;29: (16):4527–38. |
[57] | Ramakrishnan SK , Zhang H , Takahashi S , Centofanti B , Periyasamy S , Weisz K , et al. HIF2alpha Is an Essential Molecular Brake for Postprandial Hepatic Glucagon Response Independent of Insulin Signaling. Cell Metab. (2016) ;23: (3):505–16. |
[58] | Wei K , Piecewicz SM , McGinnis LM , Taniguchi CM , Wiegand SJ , Anderson K , et al. A liver Hif-2alpha-Irs2 pathway sensitizes hepatic insulin signaling and is modulated by Vegf inhibition. Nat Med. (2013) ;19: (10):1331–7. |
[59] | Dijkstra MH , Pirinen E , Huusko J , Kivela R , Schenkwein D , Alitalo K , et al. Lack of cardiac and high-fat diet induced metabolic phenotypes in two independent strains of Vegf-b knockout mice. Sci Rep. (2014) ;4: :6238. |
[60] | Wu LE , Meoli CC , Mangiafico SP , Fazakerley DJ , Cogger VC , Mohamad M , et al. Systemic VEGF-A neutralization ameliorates diet-induced metabolic dysfunction. Diabetes. (2014) ;63: (8):2656–67. |
[61] | Herberg FW , Taylor SS , Dostmann WR . Active site mutations define the pathway for the cooperative activation of cAMP-dependent protein kinase. Biochemistry. (1996) ;35: (9):2934–42. |
[62] | Sandoval DA , D’Alessio DA . Physiology of proglucagon peptides:Role of glucagon and GLP-1 in health and disease. Physiol Rev. (2015) ;95: (2):513–48. |
[63] | Manni S , Mauban JH , Ward CW , Bond M . Phosphorylation of the cAMP-dependent protein kinase (PKA) regulatory subunit modulates PKA-AKAP interaction, substrate phosphorylation, and calcium signaling in cardiac cells. J Biol Chem. (2008) ;283: (35):24145–54. |
[64] | D’Alessio D . The role of dysregulated glucagon secretion in type 2 diabetes. Diabetes Obes Metab. (2011) ;13: (Suppl 1):126–32. |
[65] | Koo SH , Flechner L , Qi L , Zhang X , Screaton RA , Jeffries S , et al. The CREB coactivator TORC2 is a key regulator of fasting glucosemetabolism. Nature. (2005) ;437: (7062):1109–11. |
[66] | Dunning BE , Gerich JE . The role of alpha-cell dysregulation in fasting and postprandial hyperglycemia in type 2 diabetes and therapeutic implications. Endocr Rev. (2007) ;28: (3):253–83. |
[67] | Conarello SL , Jiang G , Mu J , Li Z , Woods J , Zycband E , et al. Glucagon receptor knockout mice are resistant to diet-inducedobesity and streptozotocin-mediated beta cell loss andhyperglycaemia. Diabetologia. (2007) ;50: (1):142–50. |
[68] | Lee Y , Wang MY , Du XQ , Charron MJ , Unger RH . Glucagon receptor knockout prevents insulin-deficient type 1 diabetes in mice. Diabetes. (2011) ;60: (2):391–7. |
[69] | Ruiter M , La Fleur SE , van Heijningen C , van der Vliet J , Kalsbeek A , Buijs RM . The daily rhythm in plasma glucagon concentrations in the rat is modulated by the biological clock and by feeding behavior. Diabetes. (2003) ;52: (7):1709–15. |
[70] | Laplante M , Sabatini DM . mTOR signaling in growth control and disease. Cell. (2012) ;149: (2):274–93. |
[71] | Blagosklonny MV . TOR-centric view on insulin resistance and diabetic complications: Perspective for endocrinologists and gerontologists. Cell Death Dis. (2013) ;4: :e964. |
[72] | Verges B , Cariou B . mTOR inhibitors and diabetes. Diabetes Res Clin Pract. (2015) ;110: (2):101–8. |
[73] | Porstmann T , Santos CR , Griffiths B , Cully M , Wu M , Leevers S , et al. SREBP activity is regulated by mTORC1 and contributes to Akt-dependent cell growth. Cell Metab. (2008) ;8: (3):224–36. |
[74] | Titchenell PM , Quinn WJ , Lu M , Chu Q , Lu W , Li C , et al. Direct Hepatocyte Insulin Signaling Is Required for Lipogenesis but Is Dispensable for the Suppression of Glucose Production. Cell Metab. (2016) ;23: (6):1154–66. |
[75] | Laplante M , Sabatini DM . mTORC1 activates SREBP-1c and uncouples lipogenesis from gluconeogenesis. Proc Natl Acad Sci U S A. (2010) ;107: (8):3281–2. |
[76] | Li S , Brown MS , Goldstein JL . Bifurcation of insulin signaling pathway in rat liver: mTORC1 required for stimulation of lipogenesis, but not inhibition of gluconeogenesis. Proc Natl Acad Sci U S A. (2010) ;107: (8):3441–6. |
[77] | Zhang J , Gao Z , Yin J , Quon MJ , Ye J . S6K directly phosphorylates IRS-1 on Ser-270 to promote insulin resistance in response to TNF-(alpha) signaling through IKK2. J Biol Chem. (2008) ;283: (51):35375–82. |
[78] | Lamming DW , Ye L , Katajisto P , Goncalves MD , Saitoh M , Stevens DM , et al. Rapamycin-induced insulin resistance is mediated by mTORC2 loss and uncoupled from longevity. Science. (2012) ;335: (6076):1638–43. |
[79] | DeYoung MP , Horak P , Sofer A , Sgroi D , Ellisen LW . Hypoxia regulates TSC1/2-mTOR signaling and tumor suppression through REDD1-mediated 14-3-3 shuttling. Genes Dev. (2008) ;22: (2):239–51. |
[80] | Schneider A , Younis RH , Gutkind JS . Hypoxia-induced energy stress inhibits the mTOR pathway by activating an AMPK/REDD1 signaling axis in head and neck squamous cell carcinoma. Neoplasia. (2008) ;10: (11):1295–302. |
[81] | Schnoder TM , Arreba-Tutusaus P , Griehl I , Bullinger L , Buschbeck M , Lane SW , et al. Epo-induced erythroid maturation is dependent on Plcgamma1 signaling. Cell Death Differ. (2015) ;22: (6):974–85. |
[82] | Drueke TB , Locatelli F , Clyne N , Eckardt KU , Macdougall IC , Tsakiris D , et al. Normalization of hemoglobin level in patients with chronic kidney disease and anemia. N Engl J Med. (2006) ;355: (20):2071–84. |
[83] | Kapitsinou PP , Liu Q , Unger TL , Rha J , Davidoff O , Keith B , et al. Hepatic HIF-2 regulates erythropoietic responses to hypoxia in renal anemia. Blood. (2010) ;116: (16):3039–48. |
[84] | Rankin EB , Biju MP , Liu Q , Unger TL , Rha J , Johnson RS , et al. Hypoxia-inducible factor-2 (HIF-2) regulates hepatic erythropoietin in vivo. J Clin Invest. (2007) ;117: (4):1068–77. |
[85] | Warnecke C , Zaborowska Z , Kurreck J , Erdmann VA , Frei U , Wiesener M , et al. Differentiating the functional role of hypoxia-inducible factor (HIF)-1alpha and HIF-2alpha (EPAS-1) by the use of RNA interference: Erythropoietin is a HIF-2alpha target gene in Hep3B and Kelly cells. FASEB J. (2004) ;18: (12):1462–4. |
[86] | Borissova AM , Djambazova A , Todorov K , Dakovska L , Tankova T , Kirilov G . Effect of erythropoietin on the metabolic state and peripheral insulin sensitivity in diabetic patients on haemodialysis. Nephrol Dial Transplant. (1993) ;8: (1):93. |
[87] | Katz O , Stuible M , Golishevski N , Lifshitz L , Tremblay ML , Gassmann M , et al. Erythropoietin treatment leads to reduced blood glucose levels and body mass: Insights from murine models. J Endocrinol. (2010) ;205: (1):87–95. |
[88] | Spaia S , Pangalos M , Askepidis N , Pazarloglou M , Mavropoulou E , Theodoridis S , et al. Effect of short-term rHuEPO treatment on insulin resistance in haemodialysis patients. Nephron. (2000) ;84: (4):320–5. |
[89] | Ge Z , Zhang P , Hong T , Tang S , Meng R , Bi Y , et al. Erythropoietinalleviates hepatic insulin resistance via PPARgamma-dependent AKTactivation. Sci Rep. (2015) ;5: :17878. |
[90] | Caillaud C , Mechta M , Ainge H , Madsen AN , Ruell P , Mas E , et al. Chronic erythropoietin treatment improves diet-induced glucose intolerance in rats. J Endocrinol. (2015) ;225: (2):77–88. |
[91] | Alnaeeli M , Raaka BM , Gavrilova O , Teng R , Chanturiya T , Noguchi CT . Erythropoietin signaling: A novel regulator of white adipose tissue inflammation during diet-induced obesity. Diabetes. (2014) ;63: (7):2415–31. |
[92] | Cai Z , Luo W , Zhan H , Semenza GL . Hypoxia-inducible factor 1 is required for remote ischemic preconditioning of the heart. Proc Natl Acad Sci U S A. (2013) ;110: (43):17462–7. |
[93] | Zhuonan Z , Sen G , Zhipeng J , Maoyou Z , Linglan Y , Gangping W , et al. Hypoxia preconditioning induced HIF-1alpha promotes glucose metabolism and protects mitochondria in liver I/R injury. Clin Res Hepatol Gastroenterol. (2015) ;39: (5):610–9. |
[94] | Kapitsinou PP , Sano H , Michael M , Kobayashi H , Davidoff O , Bian A , et al. Endothelial HIF-2 mediates protection and recovery from ischemic kidney injury. J Clin Invest. (2014) ;124: (6):2396–409. |
[95] | Schneider M , Van Geyte K , Fraisl P , Kiss J , Aragones J , Mazzone M , et al. Loss or silencing of the PHD1 prolyl hydroxylase protectslivers of mice against ischemia/reperfusion injury. Gastroenterology. (2010) ;138: (3):1143–54 e1-2. |
[96] | Li J , Grigoryev DN , Ye SQ , Thorne L , Schwartz AR , Smith PL , et al. Chronic intermittent hypoxia upregulates genes of lipid biosynthesis in obese mice. J Appl Physiol(1985). (2005) ;99: (5):1643–8. |
[97] | Savransky V , Bevans S , Nanayakkara A , Li J , Smith PL , Torbenson MS , et al. Chronic intermittent hypoxia causes hepatitis in a mouse model of diet-induced fatty liver. Am J Physiol Gastrointest Liver Physiol. (2007) ;293: (4):G871–7. |
[98] | Qu A , Taylor M , Xue X , Matsubara T , Metzger D , Chambon P , et al. Hypoxia-inducible transcription factor 2alpha promotes steatohepatitis through augmenting lipid accumulation, inflammation, and fibrosis. Hepatology. (2011) ;54: (2):472–83. |
[99] | Sun HX , Xu Y , Yang XR , Wang WM , Bai H , Shi RY , et al. Hypoxia inducible factor 2 alpha inhibits hepatocellular carcinomagrowth through the transcription factor dimerization partner 3/E2F transcription factor 1-dependent apoptotic pathway. Hepatology. (2013) ;57: (3):1088–97. |
[100] | Menrad H , Werno C , Schmid T , Copanaki E , Deller T , Dehne N , et al. Roles of hypoxia-inducible factor-1alpha (HIF-1alpha) versus HIF-2alpha in the survival of hepatocellular tumor spheroids. Hepatology. (2010) ;51: (6):2183–92. |
[101] | Brigandi RA , Johnson B , Oei C , Westerman M , Olbina G , de Zoysa J , et al. A Novel Hypoxia-Inducible Factor-Prolyl Hydroxylase Inhibitor (GSK863) for Anemia in CKD: A 28-Day, Phase 2A Randomized Trial. Am J Kidney Dis. (2016) ;67: (6):861–71. |
[102] | Bernhardt WM , Wiesener MS , Scigalla P , Chou J , Schmieder RE , Gunzler V , et al. Inhibition of prolyl hydroxylases increases erythropoietin production in ESRD. J Am Soc Nephrol. (2010) ;21: (12):2151–6. |
[103] | Meng R , Zhu D , Bi Y , Yang D , Wang Y . Erythropoietin inhibits gluconeogenesis and inflammation in the liver and improves glucose intolerance in high-fat diet-fed mice. PLoS One. (2013) ;8: (1):e53557. |
[104] | Rahtu-Korpela L , Karsikas S , Horkko S , Blanco Sequeiros R , Lammentausta E , Makela KA , et al. HIF prolyl 4-hydroxylase-2inhibition improves glucose and lipid metabolism and protectsagainst obesity and metabolic dysfunction. Diabetes. (2014) ;63: (10):3324–33. |
[105] | Rahtu-Korpela L , Maatta J , Dimova EY , Horkko S , Gylling H , Walkinshaw G , et al. Hypoxia-Inducible Factor Prolyl 4-Hydroxylase-2 Inhibition Protects Against Development of Atherosclerosis. Arterioscler Thromb Vasc Biol. (2016) ;36: (4):608–17. |

