Emerging roles for histone deacetylases in age-related muscle atrophy
Abstract
BACKGROUND: Skeletal muscle atrophy during aging, a process known as sarcopenia, is associated with muscle weakness, frailty, and the loss of independence in older adults. The mechanisms contributing to sarcopenia are not totally understood, but muscle fiber loss due to apoptosis, reduced stimulation of anabolic pathways, activation of catabolic pathways, denervation, and altered metabolism have been observed in muscle from old rodents and humans.
OBJECTIVE: Recently, histone deacetylases (HDACs) have been implicated in muscle atrophy and dysfunction due to denervation, muscular dystrophy, and disuse, and HDACs play key roles in regulating metabolism in skeletal muscle. In this review, we will discuss the role of HDACs in muscle atrophy and the potential of HDAC inhibitors for the treatment of sarcopenia.
CONCLUSIONS: Several HDAC isoforms are potential targets for intervention in sarcopenia. Inhibition of HDAC1 prevents muscle atrophy due to nutrient deprivation. HDAC3 regulates metabolism in skeletal muscle and may inhibit oxidative metabolism during aging. HDAC4 and HDAC5 have been implicated in muscle atrophy due to denervation, a process implicated in sarcopenia. HDAC inhibitors are already in use in the clinic, and there is promise in targeting HDACs for the treatment of sarcopenia.
1Introduction
The age-related loss of muscle mass, or sarcopenia, is a debilitating condition affecting older adults. Sarcopenia is a major contributor to frailty and is associated with a number of co-morbidities, including bone loss, infection, and metabolic disease, and can lead to falls and placement in nursing homes [1, 2]. Muscle strength is inversely correlated with all-cause mortality in men and women, suggesting sarcopenia is a biomarker for health in older adults [3–6]. Sarcopenia is characterized by molecular, histological, and functional alterations to skeletal muscle, peripheral nerves, and neuromuscular junctions [7]. While the causes of these changes are not fully understood, potential mechanisms include lifestyle factors, like lack of exercise, or pathologies, like denervation and altered metabolism. No pharmaceutical treatments exist for sarcopenia, and the only generally accepted intervention is physical exercise and resistance training, which can induce muscle hypertrophy in elderly men and women [8–10]. Furthermore, hormonal therapy and nutritional supplements are currently under investigation. This review will discuss the emerging role of histone deacetylases (HDACs) in skeletal muscle disease and the implications for the treatment of sarcopenia.
2Sarcopenia
The loss of muscle mass is relatively constant during aging; approximately 1% of lean mass is lost each year after age 50 [11, 12]. Muscle strength declines at approximately three times this rate [13], suggesting the loss of muscle mass is not the only contributing factor to weakness during aging, and other factors, including peripheral nerve and excitation-contraction dysfunction and inflammation, may contribute to the loss of strength during aging. Some researchers use the term “dynapenia” to describe the loss of strength with age [14], and in fact strength may be more important for the maintenance of healthspan during aging. In this review, we will use the term “sarcopenia” to describe the various pathologies associated with skeletal muscle during aging, including atrophy, reduced strength, metabolic changes, inflammation, stem cell dysfunction, and denervation, as HDACs have potential roles in many of these processes. Sarcopenia is not technically a disease according to the International Classification of Diseases and is poorly defined in the clinic (one definition is muscle weakness two standard deviations from the average of a young reference group [15], and another definition by the European Working Group on Sarcopenia in Older People defines sarcopenia as low muscle mass with poor function, as set by age- and gender-specific standards [16]). The lack of a clinical definition of sarcopenia has hampered sarcopenia research and the development of treatments for sarcopenia.
Sarcopenia can be defined using muscle mass, muscle strength, or other performance measurements, and the prevalence of sarcopenia depends on the method for determining muscle mass or performance and the population examined. In one study, sarcopenia was estimated to affect approximately 23% of women and 27% of men aged 64–92 years of age [17]. In a longitudinal study, loss of muscle mass and strength was greater in men than in women [18]. The percent of sarcopenic individuals clearly increases in the eighth and ninth decade of life [19, 20]; muscle performance in 80 year olds is approximately half that of peak performance [21]. Elimination of sarcopenia would have enormous social and economic benefits. Sarcopenia is a major factor in the functional decline and disability due to aging, and most frail older adults are also sarcopenic [16]. Care for frail individuals is difficult because of the number of systems affected and increased social needs, and care often falls to family members and institutions. In 2000, sarcopenia was estimated to account for $18.5 billion in healthcare costs in the United States [22], and a 10% reduction in the incidence of sarcopenia would save the U.S. economy $1.1 billion according to a paper published in 2004 [23]. This number has undoubtedly risen in the decade since, and healthcare costs due to sarcopenia will continue to rise as the proportion of people over the age of 65 increases in the United States.
It is not known what causes sarcopenia, although a number of mechanisms have been proposed. The loss of muscle mass could be due to lifestyle factors like the lack of exercise or nutritional deficiencies, i.e. low protein or vitamin D intake [24]. A single serving of protein does increase protein synthesis in older adults [25, 26]; however, it is not clear if protein supplementation prevents sarcopenia, and even active adults experience muscle loss during aging [27–29]. Additionally, genetics contribute to muscle strength and likely plays a role in sarcopenia. Genetic factors account for 30% –85% of the variance in muscle strength, depending on the group studied and endpoints measured [30–32]. While genetic factors also contribute to the age-related decline in muscle strength [33, 34], the genetic contributions to muscle strength declines during aging, with the effects of environment becoming more important to muscle strength in older adults [35].
Muscle strength [4, 6, 36] and mass [37] correlate with all-cause mortality, suggesting that basic aging processes are an underlying cause of sarcopenia. Several signaling pathways related to aging have also been implicated in muscle atrophy, including growth hormone and insulin-like growth factor signaling through mTOR (mammalian target of rapamycin), Akt (protein kinase B), and FOXO (Forkhead box O) and inflammatory signaling through NF-κB (nuclear factor kappa-light-chain-enhancer of activated B cells) and TNF-α (tumor necrosis factor alpha) [38–46]. Alterations in these signaling pathways can lead to activation of the major degradation processes, including the ubiquitin proteasome system, autophagy, necrosis, and apoptosis, or the inhibition of protein synthesis, potentially causing muscle atrophy, although the effect of aging on these processes in skeletal muscle is complex [47–56]. There are conflicting reports on the effect of age on stem cell numbers, but the regenerative capacity of muscle stem cells is clearly reduced in older adults and aged mice compared to young controls, which is at least partly due to the aged niche and altered expression of myogenic regulatory transcription factors [57–62]. Transplantation of stem cells from aged mice rejuvenated ex vivo can restore function to muscle in aged mice, although depletion of stem cells during aging does not lead to sarcopenia per se [63, 64]. Thus, pathogenic processes like the imbalance between protein synthesis and protein degradation, endocrine dysfunction, denervation, loss of muscle fibers by apoptosis, stem cells dysfunction, and altered metabolism are possible mechanisms that cause sarcopenia.
3Histone deacetylases
Chromatin is a highly organized complex of DNA and histone proteins, and its structure is associated with transcriptional activity. Tightly packaged chromatin, or heterochromatin, is associated with repression of transcriptional activity, and open chromatin, or euchromatin, is associated with transcriptional activation. The nucleosome is the basic unit of chromatin and is formed from DNA and four histone proteins: 146 base pairs of DNA circumscribe an octamer composed of two copies each of histone H3 and histone H4 and two copies of the histone H2A/H2B dimer. Each histone has an N-terminal tail that protrudes from the nucleosome and is subject to a number of post-translational modifications, including acetylation, methylation, ubiquitination, and phosphorylation. These post-translational modifications are responsible for recruiting DNA binding proteins and can affect histone-DNA interactions. Acetylation is a well-described histone modification that masks the positive charge of lysine, reducing the attraction between negatively charged DNA and positively charged histones. Thus, increased levels of histone acetylation are associated with euchromatin and increased transcriptional activity [65]. At the same time, bromodomain-containing proteins can bind acetyllysine modifications and can activate or inhibit transcription [66]. Protein acetylation is regulated by acetyl-CoA availability and cellular pH in addition to enzymatic regulation [67].
Protein acetylation is regulated by two classes of enzymes: histone acetyltransferases (HATs) and HDACs. These enzymes are also called to lysine acetyltransferases and lysine deacetylases, reflecting the fact that several thousand non-histone proteins are acetylated, including the majority of metabolic enzymes as well as muscle contractile proteins, including several myosin heavy chain proteins, tropomyosin 2, and nebulin [68]. Histone acetyltransferases are broadly divided into three families: GCN5-related acetyltransferases, the MYST [MOZ (monocytic leukemia zinc finger), Ybf2, Sas2 (something about silencing 2), and Tip60 (Tat interactive protein, 60 kDa)] family, and the p300/CBP [cAMP response element-binding protein (CREB) binding protein] family. Mammalian HDACs are divided into four classes based on homology to Saccharomyces cerevisiae HDACs [69]. Class I HDACs include HDAC1–3 and HDAC8 and are related to yeast rpd3 (reduced potassium dependency 3); class II HDACs include HDAC4–7 and HDAC9–10 and are related to yeast hda1 (histone deacetylase 1); class III HDACs are the NAD+ (nicotinamide adenine dinucleotide)-dependent sirtuins, Sirt1–7 (silent mating type information regulation 2 homolog 1–7), and are related to yeast sir2 (silent information regulator 2); HDAC11 is the sole member of class IV. Eukaryotic species typically have representative but a varying number of members from each class. Sirtuins are extensively studied in the field of aging, and their role in muscle atrophy has been reviewed elsewhere [70, 71]. Sirtuin activation is associated with improved metabolism in skeletal muscle during aging. For example, Sirt1 activates PGC-1α (peroxisome proliferator-activated receptor gamma coactivator 1-alpha) [72], which is known to protect against sarcopenia [56], and supplementation with the sirtuin cofactor NAD+ protects against age-related metabolic changes in skeletal muscle in a Sirt1-dependent manner [73]. While Sirt1 inhibits myoblast differentiation [74], it is not known if this is relevant to muscle stem cell dysfunction during aging. This review will focus on the class I and II HDACs and their potential as targets for interventions in sarcopenia.
Class I, II, and IV HDACs catalyze the Zn2 +-dependent hydrolysis of acetyllysine to lysine and acetate. These classical HDACs are emerging targets for metabolic disease [75], cancer [76], and neurodegeneration [77]. Class I HDACs are ubiquitously expressed and localized in the nucleus and are found in transcriptional corepressor complexes with Sin3 (switch independent transcription regulator family member A), NuRD (nucleosome-remodeling complex), and CoREST (repressor element-1 silencing transcription factor corepressor 1) [78]. These enzymes inhibit many transcription factors, including Sp1 (specificity protein 1), p53, and retinoblastoma protein. Class II HDACs are further subdivided into class IIa and class IIb. Class IIa HDACs shuttle between the nucleus and cytoplasm and are involved in signal transduction pathways [78]. Class IIa HDACs are phosphorylated by a number of kinases, including protein kinase D1 (PKD) [79], protein kinase C [80], salt inducible kinase 1 (SIK1) [81], AMP-activated protein kinase (AMPK) [82], and calcium/calmodulin-dependent kinase II (CaMK II) [83], in the nucleus, resulting in the exposure of a nuclear export signal, binding by 14-3-3 proteins, and export from the nucleus. The class IIb HDAC6 and HDAC10 are mostly cytoplasmic and acetylate tubulin and proteins involved in autophagy [84]. Little is known about HDAC11, the sole class IV HDAC.
4The role of histone deacetylases in skeletal muscle development and maintenance
4.1Histone deacetylases in muscle development
Skeletal muscle is formed by the differentiation and fusion of muscle precursor cells. During this process, the expression of hundreds of genes is altered, including muscle structural and remodeling genes. HDACs have emerged as crucial regulators of muscle development, and this topic has been reviewed extensively elsewhere [85] and will only be briefly discussed in this review. Myogenesis is coordinated by a family of basic helix-loop-helix (bHLH) transcription factors that include myoblast determination protein (MyoD), myogenic factor 5 (Myf-5), and myogenin, which are regulated by class I and class II HDACs. The activity of these bHLH transcription factors is enhanced by myocyte enhancer factor 2 (MEF2), which is in turn regulated by the class IIa HDACs. The class IIa HDACs, namely HDAC4, HDAC5, HDAC7, and HDAC9, have been implicated in myogenesis via the regulation of MEF2 [86–88]. These HDACs interact with MEF2 [89] and inhibit the differentiation of myoblasts [90, 91]. While class IIa HDACs repress MEF2, this repression is independent of the deacetylase domain, suggesting that the effects of HDACs are due to the recruitment of other co-repressors or exclusion of transcriptional activators. In addition, the class I HDAC HDAC1 regulates the activity of MyoD and inhibits myoblast differentiation [92, 93].
The inhibition of muscle differentiation by HDACs can be overcome by histone acetyltransferases or pharmacological HDAC inhibitors. The HATs p300 and glucocorticoid receptor interacting protein can displace HDACs bound to MEF2 and activate myoblast differentiation [94, 95]. Similarly, the HATs p300/CBP and p300/CBP-associated factor (PCAF) regulate MyoD-induced gene transcription [96]. Pharmacological inhibition of HDACs can similarly modulate myoblast differentiation, which is induced by low-serum conditions. When HDAC inhibitors are included in low-serum differentiation media, primary human myoblasts and rodent the C2C12 myoblast cell line are unable to differentiate. However, differentiation is enhanced in myoblasts treated with HDAC inhibitors while they are in growth media and prior to the induction of differentiation [97]. The roles of HDACs in muscle stem cells in mature animals are not as well defined. Aging affects muscle stem cell differentiation and proliferation [reviewed in [98]], which are key processes regulated by HDACs, suggesting that HDAC inhibitors may have therapeutic use in regenerative therapies for sarcopenia.
4.2Histone deacetylases in neurogenic muscle atrophy
Neurogenic muscle atrophy results from the death and dysfunction of motor neurons or alterations of neuronal transmission at the synapse between the motor neuron and muscle fiber, which is called the neuromuscular junction. This form of muscle atrophy can result from peripheral nerve injury, toxins that affect motor neurons, and inherited and acquired diseases like amyotrophic lateral sclerosis and Lambert-Eaton syndrome. HDAC4 is specifically upregulated in animal models and human diseases resulting from neurogenic muscle atrophy [99]. HDAC4 and HDAC5 regulate the transcription factor myogenin to execute the muscle atrophy program after loss of innervation by motor neurons [100]. In response to denervation, HDAC4 travels from the neuromuscular junction to the nucleus and activates myogenin, leading to the activation of the ubiquitin-proteasome system. Phosphorylation of HDAC4 leads to the exposure of a nuclear export sequence, binding of HDAC4 to a 14-3-3 protein, and export from the nucleus [101]. Mice lacking HDAC4 or HDAC5 are protected from muscle atrophy in response to denervation [100]. These class IIa HDACs repress Dach2 (dachshund homolog 2), which negatively regulates myogenin-dependent activation of the ubiquitin-proteasome system, including the E3 ligases muscle-specific RING finger-1 (MuRF1) and atrogin-1 [100].
Another key mediator of denervation-induced muscle atrophy is FOXO3a, whose activity is regulated by acetylation. Increasing acetylation of FOXO3a by activating HATs and inhibiting HDACs inhibits its transcriptional activity and can protect against muscle atrophy due to denervation [102]. HDACs are extensively regulate and are regulated by microRNAs during denervation and other skeletal muscle diseases [103–107], adding another layer of complexity and therapeutic option for targeting HDACs in sarcopenia. Inhibitors of HDACs have shown promise in several preclinical models of neurogenic muscle atrophy, including amyotrophic lateral sclerosis [108], spinal muscular atrophy [109–111], and peripheral nerve injury [112].
4.3Histone deacetylases in metabolism and exercise
Muscle atrophy due to denervation is associated with changes in skeletal muscle fiber type. Following motor neuron death and denervation, motor neurons adjacent to the denervated muscle fiber re-innervate by axonal sprouting. Because muscle fiber type is determined by motor neuron innervation, this cycle of denervation and re-innervation results in grouping of muscle fibers of the same fiber type. Class IIa HDACs and MEF2 regulate this specification of muscle fiber type. In type I fibers, HDAC4 is selectively degraded, resulting in activation of genes involved in oxidative metabolism and the formation of type I oxidative fibers [113]. Overexpression of HDAC4 also enhances glycolysis in skeletal muscle and is required for the reduction in glycolytic enzymes that occur after denervation [114]. Defects in oxidative metabolism are also observed when HDAC5 is not responsive to phosphorylation [115], suggesting that inhibition of HDACs could increase oxidative metabolism in skeletal muscle.
HDACs also play a role in the metabolic adaptations that occur during exercise. After 60 minutes of cycling, histone acetylation is increased at histone H3 lysine 36 in skeletal muscle, which is associated with transcriptional activation [116]. This change is associated with the phosphorylation HDAC4 and HDAC5 by PKD and their export from the nucleus, resulting in elevated MEF2 activity and increased expression of glucose transporter type 4 (GLUT4) [82, 115, 116]. Another class IIa HDAC kinase, AMPK, is known to activate the expression of GLUT4 in skeletal muscle by phosphorylating HDAC5 [82]. The class I HDACs also play a critical role in skeletal muscle metabolism. Mice lacking both HDAC1 and HDAC2 experience a myopathy characterized by muscle degeneration, mitochondrial dysfunction, inhibition of autophagy, and increased oxidative metabolism [117]. In this regard, there is a clear difference between the total loss of HDACs, as occurs in genetic models, and pharmacological inhibitors of HDACs, which only reduce activity and maintain the key ability of HDACs to form complexes with other proteins. Pharmacological inhibitors of HDACs increase the expression of GLUT4 in skeletal muscle [118], and HDAC inhibitors have shown promise in models of obesity and diabetes, preventing insulin resistance and reducing circulating triglycerides and cholesterol [119, 120], although these effects may be due to the roles of HDACs in liver and adipose tissue [121–123].
4.4Histone deacetylases in muscular dystrophy
Muscular dystrophies are a group of skeletal muscle diseases characterized by muscle wasting and weakness. The prototypical muscular dystrophy, Duchenne muscular dystrophy, is caused by mutations in the dystrophin gene, which encodes a structural protein that connects the muscle cytoskeleton with the extracellular matrix. HDAC2 is elevated in muscle from dystrophin-deficient (MDX) mice, and class-I-specific HDAC inhibitors and siRNA targeting HDAC2 are protective in MDX mice [124]. Increased activity of CREB in skeletal muscle leads to a condition resembling muscular dystrophy, and expression of the class II HDAC kinase SIK1 prevents this dysfunction [81], suggesting class II HDACs may also play a role in muscular dystrophy. HDAC inhibitors improve morphological and functional deficits in MDX mice, including increasing muscle fiber size and reducing fibrosis and inflammation [125, 126]. HDAC inhibitors increase muscle fiber size through the production of follistatin, an antagonist of myostatin [97]. Myostatin is a member of the TGF-β (transforming growth factor beta) family that, when mutated, results in muscular individuals. The ability of HDAC inhibitors to reduce fibrosis in models of muscular dystrophy has been linked to effects on fibro/adipogenic progenitors (FAPs) residing in skeletal muscle. In diseased conditions, FAPs contribute to the formation of intramuscular fat and fibrosis. However, FAPs are also able to promote muscle regeneration by enhancing muscle stem cell differentiation [127] and increasing clearance of damaged muscle debris [128]. In mouse models of muscular dystrophy, HDAC inhibitors enhance the ability of FAPs to stimulate muscle stem cells during early stages of the disease [129].
4.5Histone deacetylases in muscle atrophy due to disuse and poor nutrition
Similar to denervation-induced muscle atrophy, myogenin is activated in skeletal muscle in response to fasting or nutrient deprivation, but under these conditions HDAC1 activates myogenin. Reduction in the level of HDAC1 or inhibition of its activity prevents muscle atrophy after nutrient deprivation [130]. The non-specific HDAC inhibitor trichostatin A (TSA) prevents muscle atrophy in a rodent model of disuse atrophy [131]. TSA reduces oxidative stress and prevents the expression of MuRF1 after hindlimb unloading in rodents. This effect was found to be due to the inhibition of HDAC1 using class-specific HDAC inhibitors [130]. In a cast immobilization model of disuse atrophy, class I HDAC inhibitors prevented muscle atrophy and the expression of ubiquitin-proteasomal and autophagic genes, while class II HDAC inhibitors had no effect. HDAC1 was subsequently identified as cause of muscle atrophy in these models using dominant-negative HDAC variants [130]. These studies demonstrate the power of pharmacological and genetic manipulation of HDACs to uncover the isoforms that contribute to specific skeletal muscle diseases.
4.6Potential role of histone deacetylases in sarcopenia
One potential contributor to sarcopenia is deterioration of the motor unit [132], which is a motor neuron and all the muscle fibers it innervates and the neuromuscular junction. Loss of motor neuron activity and alterations at neuromuscular junctions are key events in the loss of muscle mass during aging [133–136], even though motor neuron numbers in the spinal cord do not decline with age in mice [137]. We found that HDAC4 is elevated in muscle from old mice, consistent with a role of denervation in sarcopenia [138]. Even though denervation is apparent at 26 months of age in C57Bl/6 mice, we did not observe phosphorylated HDAC4 in skeletal muscle from old mice. Thus, similar to studies on muscular dystrophy [81] and metabolism [82], targeting class II HDAC kinases is a promising approach that may avoid the potential side effects of a non-specific HDAC inhibitor. Another result of the cycle of denervation and reinnervation is the increase in size of the motor unit [139] and reduced acetylcholine receptor numbers and firing release [140], and these effects are likely a major contributor to the loss of muscle strength during aging. Regression analysis revealed that only 13% of the variation in muscle strength is explained by muscle mass in older adults [141] and that muscle fiber cross sectional area has a similarly small correlation with strength [142], suggesting that other factors, like innervation by motor neurons, are critical for the loss of strength during aging [143]. Thus, further research is needed on the role of HDACs in motor neurons and neuromuscular junctions.
Muscle from sarcopenic individuals and patients with muscular dystrophy share some phenotypes, including increased fibrosis and neuromuscular junction fragmentation [144]. However, the decline in motor units observed during sarcopenia is not seen in muscular dystrophy, and there are no reports of alterations in the dystrophin-associated protein complex during aging. Thus, there is little evidence that sarcopenia is a form a muscular dystrophy. However, studies on HDACs and muscular dystrophy demonstrate an important principle for the potential role of HDACs in sarcopenia; namely, that specific HDAC isoforms are implicated in different diseases of skeletal muscle. Further research is needed using targeted genetic and pharmacological approaches to determine if specific HDAC isoforms contribute to sarcopenia.
We and others have shown a loss of structural and physiological integrity in motor neurons and at neuromuscular junctions during aging and have implicated oxidative stress in these processes [137, 145–147]. We recently showed that the HDAC inhibitor butyrate reduces oxidative damage and increases antioxidant activity in muscle from old mice [138]. A compound related to butyrate, the ketone body beta-hydroxybutyrate, is an endogenous histone deacetylases inhibitor that is upregulated by dietary restriction and protects against oxidative stress in mice [148]. Beta-hydroxybutyrate and another HDAC inhibitor, TSA, extend lifespan in Caenorhabditis elegans, and RNA interference of two C. elegans HDAC genes also increases lifespan [149, 150]. These effects likely occur through dietary restriction mechanisms [149, 150], suggesting that dietary restriction and HDAC inhibitors function through similar mechanisms. Together with data demonstrating that deletion of the model class I HDAC rpd3 extends lifespan in S. cerevisiae [151] and that HDAC inhibitors increase lifespan in Drosophila [152], it is possible that HDACs play a role in general aging processes.
Class IIa HDACs activate glycolysis in skeletal muscle [114, 115], and HDAC3, a class I HDAC, inhibits mitochondrial biogenesis [153]. Interestingly, dietary restriction, which preserves muscle mass during aging [154, 155], prevents the decline in oxidative metabolism in skeletal muscle from old mice [156], suggesting that dietary restriction might modulate histone deacetylases during aging. Shifts in body composition during aging may also contribute to sarcopenia, as fat mass is inversely correlated with muscle strength and mobility in the elderly [22, 157–159]. This may be due to deposition of fat in skeletal muscle during aging, which is associated with insulin resistance and low muscle strength in older adults [13, 160]. HDAC inhibitors increase oxidative metabolism and reduce intramuscular fat during aging, which may explain improvements in metabolism in old mice [138].
An imbalance between protein synthesis and degradation can result in muscle growth or atrophy, and sarcopenia may result from increased protein degradation [53, 161]. HDACs are critical mediators of catabolic pathways, including ubiquitin-proteasomal degradation and autophagy. Although a number of studies have investigated these pathways, it is still not clear if catabolism is increased during aging [50, 52–56, 162]. Reduced protein synthesis might also contribute to the net reduction in protein levels during aging. Mitochondrial protein synthesis rates are reduced by middle age, although no further decline is seen in the elderly [163], even though a further loss of muscle mass is reported. Another study found similar results with whole body protein synthesis rates as well as myosin heavy chain synthesis in skeletal muscle [164]. Importantly, the elderly are able to increase protein synthesis rates in response to amino acid intake [25]. Whether HDACs affect protein synthesis is an outstanding question.
Although the role of muscle stem cells in the development of sarcopenia has recently been called into question [63], stem cells remain a promising therapy for sarcopenia, but we need to know more about the role of HDACs in muscle stem cells in vivo to determine if HDAC inhibitors might play a role in regenerative therapies for sarcopenia. Similarly, while HDAC inhibitors inhibit myostatin by increasing follistatin, there is not a clear role for myostatin in the development of sarcopenia [165]. However, myostatin is a promising target for intervention [166] and is under investigation by a number of pharmaceutical companies for the treatment of sarcopenia and other skeletal muscle diseases [167].
5Conclusions and future directions
Many challenges remain for the development of HDAC inhibitors and other pharmacological agents to treat sarcopenia. There is no universal method to diagnose sarcopenia in an individual patient, and it is not clear what endpoints are most relevant for clinical trials. Broad goals of restoring function and preventing dependent living need to be distilled into clinically measurable endpoints like mortality and hospitalization rate. It is an exciting time for sarcopenia research, as new targets for intervention have recently been identified in rodent models, including the cytokine TWEAK (tumor necrosis factor-related weak inducer of apoptosis) [168, 169], TGF-beta inhibitors [170], activin [171], sirtuins [70], angiotensin receptors [172], GDF11 (growth differentiation factor 11) [173–175], NAD+ [176], and ATF4 (activating transcription factor 4) [177], along with continued research on myostatin [178], PGC-1α [56], and steroids [179].
Studies on HDACs in models of denervation, exercise, metabolism, muscular dystrophy, and disuse atrophy have given us a better understanding of the potential mechanisms underlying sarcopenia and may lead to successful intervention in sarcopenia (see Fig. 1). Further research is needed to determine if specific isoforms contribute to sarcopenia and the mechanisms by which HDAC inhibitors act to prevent muscle loss. Although the expression of most HDACs has not been reported to change during aging, HDACs function throughout the genome and affect non-histone protein function. Chromatin immunoprecipitation followed by DNA sequencing and analysis of the acetylome during aging may uncover additional HDAC-related targets for intervention in sarcopenia. HDAC inhibitors are relatively well studied in cancer treatment, and several HDAC inhibitors are already approved by the U.S. Food and Drug Administration. The increasing interest in HDAC inhibitors has led to the development of class-specific inhibitors, with isoform-specific inhibitors currently under development, which will help researchers uncover the role of HDACs in sarcopenia. Based on the numerous beneficial effects of HDAC inhibitors in skeletal muscle under pathological conditions, we believe that HDAC inhibitors are promising therapeutics for the treatment of sarcopenia and potentially age-related metabolic disease.
References
[1] | Gillick MR . Long-term care options for the frail elderly. Journal of the American Geriatrics Society. (1989) ;37: 12:1198–203. |
[2] | Visser M . Towards a definition of sarcopenia–results from epidemiologic studies. J Nutr Health Aging. (2009) ;13: 8:713–6. |
[3] | Ruiz J , Sui X , Lobelo F , Morrow J , Jackson A , Sjöström M , et al. Association between muscular strength and mortality in men: Prospective cohort study. BMJ. (2008) ;337. |
[4] | Newman A , Kupelian V , Visser M , Simonsick E , Goodpaster B , Kritchevsky S , et al. Strength, but not muscle mass, is associated with mortality in the health, aging and body composition study cohort. The Journals of Gerontology Series A: Biological Sciences and Medical Sciences. (2006) ;61: 1:72–7. |
[5] | Rantanen T , Volpato S , Luigi Ferrucci MD , Eino Heikkinen MD , Fried L , Guralnik J . Handgrip strength and cause-specific and total mortality in older disabled women: Exploring the mechanism. Journal of the American Geriatrics Society. (2003) ;51: 5:636–41. |
[6] | Metter J , Talbot L , Schrager M , Conwit R . Skeletal muscle strength as a predictor of all-cause mortality in healthy men. J Gerontol A Biol Sci Med Sci. (2002) ;57: 10:B359–65. |
[7] | Marcell T . Review article: Sarcopenia: Causes, consequences, and preventions. The Journals of Gerontology Series A: Biological Sciences and Medical Sciences. (2003) ;58: 10:M911–M6. |
[8] | Verdijk L , Gleeson B , Jonkers R , Meijer K , Savelberg H , Dendale P , et al. Skeletal muscle hypertrophy following resistance training is accompanied by a fiber type–specific increase in satellite cell content in elderly men. The Journals of Gerontology Series A: Biological Sciences and Medical Sciences. (2009) ;64A: 3:332–9. |
[9] | Charette SL , McEvoy L , Pyka G , Snow-Harter C , Guido D , Wiswell RA , et al. Muscle hypertrophy response to resistance training in older women. Journal of Applied Physiology (Bethesda, Md: 1985). (1991) ;70: 5:1912–6. |
[10] | Burton L , Sumukadas D . Optimal management of sarcopenia. Clinical Interventions in Aging. (2010) ;5: , 217–28. |
[11] | Sehl ME , Yates FE . Kinetics of human aging: I. Rates of senescence between ages 30 and 70 years in healthy people. The Journals of Gerontology Series A, Biological Sciences and Medical Sciences. (2001) ;56: 5. |
[12] | Hughes V , Roubenoff R , Wood M , Frontera W , Evans W , Fiatarone Singh M . Anthropometric assessment of 10-y changes in body composition in the elderly. The American Journal of Clinical Nutrition. (2004) ;80: 2:475–82. |
[13] | Goodpaster B , Park S , Harris T , Kritchevsky S , Nevitt M , Schwartz A , et al. The loss of skeletal muscle strength, mass, and quality in older adults: The health, aging and body composition study. The Journals of Gerontology Series A: Biological Sciences and Medical Sciences. (2006) ;61: 10:1059–64. |
[14] | Clark B , Manini T . Sarcopenia=/=dynapenia. The Journals of Gerontology Series A, Biological Sciences and Medical Sciences. (2008) ;63: 8:829–34. |
[15] | Baumgartner RN , Koehler KM , Gallagher D , Romero L , Heymsfield SB , Ross RR , et al. Epidemiology of sarcopenia among the elderly in New Mexico. American Journal of Epidemiology. (1998) ;147: 8:755–63. |
[16] | Cruz-Jentoft A , Baeyens J , Bauer J , Boirie Y , Cederholm T , Landi F , et al. Sarcopenia: European consensus on definition and diagnosis. Age and Ageing. (2010) ;39: 4:412–23. |
[17] | Iannuzzi-Sucich M , Prestwood K , Kenny A . Prevalence of sarcopenia and predictors of skeletal muscle mass in healthy, older men and women. The Journals of Gerontology Series A, Biological Sciences and Medical Sciences. (2002) ;57: 12. |
[18] | Gallagher D , Visser M , De Meersman RE , Sepúlveda D , Baumgartner RN , Pierson RN , et al. Appendicular skeletal muscle mass: Effects of age, gender, and ethnicity. Journal of Applied Physiology (Bethesda, Md: 1985). (1997) ;83: 1:229–39. |
[19] | Baumgartner R , Koehler K , Gallagher D , Romero L , Heymsfield S , Ross R , et al. Epidemiology of sarcopenia among the elderly in New Mexico. American Journal of Epidemiology. (1998) ;147: 8:755–63. |
[20] | Castillo E , Goodman-Gruen D , Kritz-Silverstein D , Morton D , Wingard D , Barrett-Connor E . Sarcopenia in elderly men and women: The Rancho Bernardo study. American Journal of Preventive Medicine. (2003) ;25: 3:226–31. |
[21] | Faulkner J , Larkin L , Claflin D , Brooks S . Age-related changes in the structure and function of skeletal muscles. Clinical and Experimental Pharmacology & Physiology. (2007) ;34: 11:1091–6. |
[22] | Janssen I , Heymsfield S , Ross R . Low relative skeletal muscle mass (Sarcopenia) in older persons is associated with functional impairment and physical disability. Journal of the American Geriatrics Society. (2002) ;50: 5:889–96. |
[23] | Janssen I , Shepard D , Katzmarzyk P , Roubenoff R . The healthcare costs of sarcopenia in the United States. Journal of the American Geriatrics Society. (2004) ;52: 1:80–5. |
[24] | Visser M , Deeg D , Lips P . Low vitamin D and high parathyroid hormone levels as determinants of loss of muscle strength and muscle mass (sarcopenia): The Longitudinal Aging Study Amsterdam. The Journal of Clinical Endocrinology and Metabolism. (2003) ;88: 12:5766–72. |
[25] | Koopman R , Verdijk L , Manders R , Gijsen A , Gorselink M , Pijpers E , et al. Co-ingestion of protein and leucine stimulates muscle protein synthesis rates to the same extent in young and elderly lean men. The American Journal of Clinical Nutrition. (2006) ;84: 3:623–32. |
[26] | Symons B , Sheffield-Moore M , Wolfe R , Paddon-Jones D . A moderate serving of high-quality protein maximally stimulates skeletal muscle protein synthesis in young and elderly subjects. Journal of the American Dietetic Association. (2009) ;109: 9:1582–6. |
[27] | Kohrt WM , Malley MT , Dalsky GP , Holloszy JO . Body composition of healthy sedentary and trained, young and older men and women. Medicine and Science in Sports and Exercise. (1992) ;24: 7:832–7. |
[28] | Wiswell R , Hawkins S , Jaque V , Hyslop D , Constantino N , Tarpenning K , et al. Relationship between physiological loss, performance decrement, and age in master athletes. The Journals of Gerontology Series A: Biological Sciences and Medical Sciences. (2001) ;56: 10:M618–M26. |
[29] | Hawkins S , Wiswell R , Marcell T . Exercise and the master athlete— a model of successful aging? The Journals of Gerontology Series A: Biological Sciences and Medical Sciences, (2003) ;58: 11:M1009–M11. |
[30] | Reed T , Fabsitz RR , Selby JV , Carmelli D . Genetic influences and grip strength norms in the NHLBI twin study males aged 59-69. Annals of Human Biology. (1991) ;18: 5:425–32. |
[31] | Huygens W , Thomis MA , Peeters MW , Aerssens J , Vlietinck R , Beunen GP . Quantitative trait loci for human muscle strength: Linkage analysis of myostatin pathway genes. Physiological Genomics. (2005) ;22: 3:390–7. |
[32] | Karlsson J , Komi PV , Viitasalo JH . Muscle strength and muscle characteristics in monozygous and dizygous twins. Acta physiologica Scandinavica. (1979) ;106: 3:319–25. |
[33] | Zhai G , Stankovich J , Ding C , Scott F , Cicuttini F , Jones G . The genetic contribution to muscle strength, knee pain, cartilage volume, bone size, and radiographic osteoarthritis: A sibpair study. Arthritis and Rheumatism. (2004) ;50: 3:805–10. |
[34] | Tiainen K , Sipilä S , Alén M , Heikkinen E , Kaprio J , Koskenvuo M , et al. Shared genetic and environmental effects on strength and power in older female twins. Medicine and Science in Sports and Exercise. (2005) ;37: 1:72–8. |
[35] | Carmelli D , Reed T . Stability and change in genetic and environmental influences on hand-grip strength in older male twins. Journal of Applied Physiology. (2000) ;89: 5:1879–83. |
[36] | Rantanen T , Harris T , Leveille S , Visser M , Foley D , Masaki K , et al. Muscle strength and body mass index as long-term predictors of mortality in initially healthy men. The Journals of Gerontology Series A: Biological Sciences and Medical Sciences. (2000) ;55: 3:M168–73. |
[37] | Srikanthan P , Karlamangla A . Muscle mass index as a predictor of longevity in older adults. The American Journal of Medicine. (2014) ;127: 6:547–53. |
[38] | Sandri M , Barberi L , Bijlsma AY , Blaauw B , Dyar KA , Milan G , et al. Signalling pathways regulating muscle mass in ageing skeletal muscle. The role of the IGF1-Akt-mTOR-FoxO pathway. Biogerontology. (2013) ;14: 3:303–23. |
[39] | Luo L , Lu AM , Wang Y , Hong A , Chen Y , Hu J , et al. Chronic resistance training activates autophagy and reduces apoptosis of muscle cells by modulating IGF-1 and its receptors, Akt/mTOR and Akt/FOXO3a signaling in aged rats. Experimental Gerontology. (2013) ;48: 4:427–36. |
[40] | Velloso CP . Regulation of muscle mass by growth hormone and IGF-I. British Journal of Pharmacology. (2008) ;154: 3:557–68. |
[41] | Léger B , Cartoni R , Praz M , Lamon S , Dériaz O , Crettenand A , et al. Akt signalling through GSK-3β, mTOR and Foxo1 is involved in human skeletal muscle hypertrophy and atrophy. The Journal of Physiology. (2006) ;576: 3:923–33. |
[42] | Phillips T , Leeuwenburgh C . Muscle fiber specific apoptosis and TNF-alpha signaling in sarcopenia are attenuated by life-long calorie restriction. FASEB Journal. (2005) ;19: 6:668–70. |
[43] | Roubenoff R , Parise H , Payette H , Abad L , D’Agostino R , Jacques P , et al. Cytokines, insulin-like growth factor 1, sarcopenia, and mortality in very old community-dwelling men and women: The Framingham Heart Study. The American Journal of Medicine. (2003) ;115: 6:429–35. |
[44] | Tilstra J , Robinson A , Wang J , Gregg S , Clauson C , Reay D , et al. NF-κB inhibition delays DNA damage-induced senescence and aging in mice. The Journal of Clinical Investigation. (2012) ;122: 7:2601–12. |
[45] | Buford T , Cooke M , Manini T , Leeuwenburgh C , Willoughby D . Effects of age and sedentary lifestyle on skeletal muscle NF-kappaB signaling in men. The Journals of Gerontology Series A, Biological Sciences and Medical Sciences. (2010) ;65: 5:532–7. |
[46] | Merritt E , Stec M , Thalacker-Mercer A , Windham S , Cross J , Shelley D , et al. Heightened muscle inflammation susceptibility may impair regenerative capacity in aging humans. Journal of Applied Physiology. (2013) ;115: 6:937–48. |
[47] | Carnio S , LoVerso F , Baraibar M , Longa E , Khan M , Maffei M , et al. Autophagy impairment in muscle induces neuromuscular junction degeneration and precocious aging. Cell Reports. (2014) ;8: 5:1509–21. |
[48] | Marzetti E , Lawler J , Hiona A , Manini T , Seo A , Leeuwenburgh C . Modulation of age-induced apoptotic signaling and cellular remodeling by exercise and calorie restriction in skeletal muscle. Free Radical Biology and Medicine. (2008) ;44: 2:160–8. |
[49] | Combaret L , Dardevet D , Béchet D , Taillandier D , Mosoni L , Attaix D . Skeletal muscle proteolysis in aging. Current Opinion in Clinical Nutrition and Metabolic Care. (2009) ;12: 1:37–41. |
[50] | Altun M , Besche H , Overkleeft H , Piccirillo R , Edelmann M , Kessler B , et al. Muscle wasting in aged, sarcopenic rats is associated with enhanced activity of the ubiquitin proteasome pathway. Journal of Biological Chemistry. (2010) ;285: 51:39597–608. |
[51] | Cheema N , Herbst A , McKenzie D , Aiken J . Apoptosis and necrosis mediate skeletal muscle fiber loss in age-induced mitochondrial enzymatic abnormalities. Aging Cell. (2015) ;14: 6:1085–93. |
[52] | Edström E , Altun M , Hägglund M , Ulfhake B . Atrogin-1/MAFbx and MuRF1 are downregulated in aging-related loss of skeletal muscle. The Journals of Gerontology Series A: Biological Sciences and Medical Sciences. (2006) ;61: 7:663–74. |
[53] | Hepple R , Qin M , Nakamoto H , Goto S . Caloric restriction optimizes the proteasome pathway with aging in rat plantaris muscle: Implications for sarcopenia. American Journal of Physiology Regulatory, Integrative and Comparative Physiology. (2008) );295: 4. |
[54] | Hwee D , Baehr L , Philp A , Baar K , Bodine S . Maintenance of muscle mass and load-induced growth in Muscle RING Finger 1 null mice with age. Aging Cell. (2014) ;13: 1:92–101. |
[55] | Radák Z , Takahashi R , Kumiyama A , Nakamoto H , Ohno H , Ookawara T , et al. Effect of aging and late onset dietary restriction on antioxidant enzymes and proteasome activities, and protein carbonylation of rat skeletal muscle and tendon. Experimental Gerontology. (2002) ;37: 12:1423–30. |
[56] | Wenz T , Rossi S , Rotundo R , Spiegelman B , Moraes C . Increased muscle PGC-1α expression protects from sarcopenia and metabolic disease during aging. Proceedings of the National Academy of Sciences. (2009) ;106: 48:20405–10. |
[57] | Conboy I , Conboy M , Wagers A , Girma E , Weissman I , Rando T . Rejuvenation of aged progenitor cells by exposure to a young systemic environment. Nature. (2005) ;433: 7027:760–4. |
[58] | Sousa-Victor P , Gutarra S , Garcia-Prat L , Rodriguez-Ubreva J , Ortet L , Ruiz-Bonilla V , et al. Geriatric muscle stem cells switch reversible quiescence into senescence. Nature. (2014) ;506: 7488:316–21. |
[59] | Pietrangelo T , Puglielli C , Mancinelli R , Beccafico S , Fanò G , Fulle S . Molecular basis of the myogenic profile of aged human skeletal muscle satellite cells during differentiation. Experimental Gerontology. (2009) ;44: 8:523–31. |
[60] | Chargé S , Brack A , Hughes S . Aging-related satellite cell differentiation defect occurs prematurely after Ski-induced muscle hypertrophy. American Journal of Physiology Cell Physiology. (2002) );283: 4. |
[61] | Shefer G , Van de Mark D , Richardson J , Yablonka-Reuveni Z . Satellite-cell pool size does matter: Defining the myogenic potency of aging skeletal muscle. Developmental Biology. (2006) ;294: 1:50–66. |
[62] | Bigot A , Jacquemin V , Debacq-Chainiaux F , Butler-Browne G , Toussaint O , Furling D , et al. Replicative aging down-regulates the myogenic regulatory factors in human myoblasts. Biology of the Cell / Under the Auspices of the European Cell Biology Organization. (2008) ;100: 3:189–99. |
[63] | Fry C , Lee J , Mula J , Kirby T , Jackson J , Liu F , et al. Inducible depletion of satellite cells in adult, sedentary mice impairs muscle regenerative capacity without affecting sarcopenia. Nat Med. (2015) ;21: 1:76–80. |
[64] | Cosgrove B , Gilbert P , Porpiglia E , Mourkioti F , Lee S , Corbel S , et al. Rejuvenation of the muscle stem cell population restores strength to injured aged muscles. Nature Medicine. (2014) ;20: 3:255–64. |
[65] | Tse C , Sera T , Wolffe AP , Hansen JC . Disruption of higher-order folding by core histone acetylation dramatically enhances transcription of nucleosomal arrays by RNA polymerase III. Molecular and Cellular Biology. (1998) ;18: 8:4629–38. |
[66] | Owen D , Ornaghi P , Yang JC , Lowe N , Evans P , Ballario P , et al. The structural basis for the recognition of acetylated histone H4 by the bromodomain of histone acetyltransferase Gcn5The EMBO Journal (2000) ;19: 22:6141–9. |
[67] | McBrian M , Behbahan IS , Ferrari R , Su T , Huang T-W , Li K , et al. Histone acetylation regulates intracellular pH. Molecular Cell. (2013) ;49: 2:310–21. |
[68] | Lundby A , Lage K , Weinert B , Bekker-Jensen D , Secher A , Skovgaard T , et al. Proteomic analysis of lysine acetylation sites in rat tissues reveals organ specificity and subcellular patterns. Cell Reports. (2012) ;2: 2:419–31. |
[69] | Gregoretti I , Lee Y-M , Goodson H . Molecular evolution of the histone deacetylase family: Functional implications of phylogenetic analysis. Journal of Molecular Biology. (2004) ;338: 1:17–31. |
[70] | Pardo P , Boriek A . The physiological roles of Sirt1 in skeletal muscle. Aging. (2011) ;3: 4:430–7. |
[71] | Vinciguerra M , Fulco M , Ladurner A , Sartorelli V , Rosenthal N . SirT1 in muscle physiology and disease: Lessons from mouse models. Disease Models & Mechanisms. (2010) ;3: 5-6:298–303. |
[72] | Gerhart-Hines Z , Rodgers J , Bare O , Lerin C , Kim S-H , Mostoslavsky R , et al. Metabolic control of muscle mitochondrial function and fatty acid oxidation through SIRT1/PGC-1alpha. The EMBO Journal. (2007) ;26: 7:1913–23. |
[73] | Gomes A , Price N , Ling A , Moslehi J , Montgomery M , Rajman L , et al. Declining NAD+induces a pseudohypoxic state disrupting nuclear-mitochondrial communication during aging. Cell. (2013) ;155: 7:1624–38. |
[74] | Fulco M , Schiltz L , Iezzi S , King T , Zhao P , Kashiwaya Y , et al. Sir2 regulates skeletal muscle differentiation as a potential sensor of the redox state. Molecular Cell. (2003) ;12: 1:51–62. |
[75] | Christensen D , Dahllöf M , Lundh M , Rasmussen D , Nielsen M , Billestrup N , et al. Histone deacetylase (HDAC) inhibition as a novel treatment for diabetes mellitus. Molecular Medicine (Cambridge, Mass). (2011) ;17: 5-6:378–90. |
[76] | Ropero S , Esteller M . The role of histone deacetylases (HDACs) in human cancer. Molecular Oncology. (2007) ;1: 1:19–25. |
[77] | Dietz K , Casaccia P . HDAC inhibitors and neurodegeneration: At the edge between protection and damage. Pharmacological Research: The Official Journal of the Italian Pharmacological Society. (2010) ;62: 1:11–7. |
[78] | Dokmanovic M , Clarke C , Marks P . Histone deacetylase inhibitors: Overview and perspectives. Molecular Cancer Research. (2007) ;5: 10:981–9. |
[79] | Kim M-S , Fielitz J , McAnally J , Shelton J , Lemon D , McKinsey T , et al. Protein kinase D1 stimulates MEF2 activity in skeletal muscle and enhances muscle Performance. Mol Cell Biol. (2008) ;28: 11:3600–9. |
[80] | D’Andrea M , Pisaniello A , Serra C , Senni MI , Castaldi L , Molinaro M , et al. Protein kinase C theta co-operates with calcineurin in the activation of slow muscle genes in cultured myogenic cells. Journal of Cellular Physiology. (2006) ;207: 2:379–88. |
[81] | Berdeaux R , Goebel N , Banaszynski L , Takemori H , Wandless T , Shelton D , et al. SIK1 is a class II HDAC kinase that promotes survival of skeletal myocytes. Nature Medicine. (2007) ;13: 5:597–603. |
[82] | McGee S , van Denderen B , Howlett K , Mollica J , Schertzer J , Kemp B , et al. AMP-Activated protein kinase regulates GLUT4 transcription by phosphorylating histone deacetylase 5. Diabetes. (2008) ;57: 4:860–7. |
[83] | Backs J , Backs T , Bezprozvannaya S , McKinsey T , Olson E . Histone Deacetylase 5 Acquires Calcium/Calmodulin-Dependent Kinase II Responsiveness by Oligomerization with Histone Deacetylase 4. Mol Cell Biol. (2008) ;28: 10:3437–45. |
[84] | Lee J-Y , Koga H , Kawaguchi Y , Tang W , Wong E , Gao Y-S , et al. HDAC6 controls autophagosome maturation essential for ubiquitin-selective quality-control autophagy. EMBO J. advance online publication. (2010) ;5 969–80. |
[85] | Duby R , Olson E . Signaling pathways in skeletal muscle remodeling. Annual Review of Biochemistry. (2006) ;75: 1:19–37. |
[86] | Lu J , McKinsey TA , Nicol RL , Olson EN . Signal-dependent activation of the MEF2 transcription factor by dissociation from histone deacetylases. Proc Natl Acad Sci U S A. (2000) ;97: 8:4070–5. |
[87] | Miska EA , Karlsson C , Langley E , Nielsen SJ , Pines J , Kouzarides T . HDAC4 deacetylase associates with and represses the MEF2 transcription factor. The EMBO Journal. (1999) ;18: 18:5099–107. |
[88] | Wang AH , Bertos NR , Vezmar M , Pelletier N , Crosato M , Heng HH , et al. HDAC4, a human histone deacetylase related to yeast HDA1, is a transcriptional corepressor. Molecular and Cellular Biology. (1999) ;19: 11:7816–27. |
[89] | Lu J , McKinsey T , Zhang C-L , Olson E . Regulation of skeletal myogenesis by association of the MEF2 transcription factor with class II histone deacetylases. Molecular Cell. (2000) ;6: 2:233–44. |
[90] | Dressel U , Bailey P , Wang M , Downes M , Evans R , Muscat G . A Dynamic role for HDAC7 in MEF2-mediated muscle differentiation. Journal of Biological Chemistry. (2001) ;276: 20:17007–13. |
[91] | McKinsey T , Zhang C-L , Lu J , Olson E . Signal-dependent nuclear export of a histone deacetylase regulates muscle differentiation. Nature. (2000) ;408: 6808:106–11. |
[92] | Mal A , Sturniolo M , Schiltz RL , Ghosh MK , Harter ML . A role for histone deacetylase HDAC1 in modulating the transcriptional activity of MyoD: Inhibition of the myogenic program. The EMBO Journal. (2001) ;20: 7:1739–53. |
[93] | Sartorelli V , Puri PL , Hamamori Y , Ogryzko V , Chung G , Nakatani Y , et al. Acetylation of MyoD directed by PCAF is necessary for the execution of the muscle program. Molecular Cell. (1999) ;4: 5:725–34. |
[94] | Chen S , Feng B , George B , Chakrabarti R , Chen M , Chakrabarti S . Transcriptional coactivator p300 regulates glucose-induced gene expression in endothelial cells. American Journal of Physiology - Endocrinology and Metabolism. (2010) ;298: 1:E127–E37. |
[95] | Ma K , Chan J , Zhu G , Wu Z . Myocyte enhancer factor 2 acetylation by p300 enhances its DNA binding activity, transcriptional activity, and myogenic differentiation. Molecular and Cellular Biology. (2005) ;25: 9:3575–82. |
[96] | Puri PL , Sartorelli V , Yang XJ , Hamamori Y , Ogryzko VV , Howard BH , et al. Differential roles of p300 and PCAF acetyltransferases in muscle differentiation. Molecular Cell. (1997) ;1: 1:35–45. |
[97] | Iezzi S , Cossu G , Nervi C , Sartorelli V , Puri P . Stage-specific modulation of skeletal myogenesis by inhibitors of nuclear deacetylases. Proc Natl Acad Sci U S A. (2002) ;99: 11:7757–62. |
[98] | Sousa-Victor P , García-Prat L , Serrano A , Perdiguero E , Muñoz-Cánoves P . Muscle stem cell aging: Regulation and rejuvenation. Trends in Endocrinology & Metabolism. (2016) ;26: 6:287–96. |
[99] | Cohen T , Waddell D , Barrientos T , Lu Z , Feng G , Cox G , et al. The histone deacetylase HDAC4 connects neural activity to muscle transcriptional reprogramming. The Journal of Biological Chemistry. (2007) ;282: 46:33752–9. |
[100] | Moresi V , Williams A , Meadows E , Flynn J , Potthoff M , McAnally J , et al. Myogenin and class II HDACs control neurogenic muscle atrophy by inducing E3 ubiquitin ligases. Cell. (2010) ;143: 1:35–45. |
[101] | Wang AH , Kruhlak MJ , Wu J , Bertos NR , Vezmar M , Posner BI , et al. Regulation of histone deacetylase 4 by binding of 14-3-3 proteins. Molecular and Cellular Biology. (2000) ;20: 18:6904–12. |
[102] | Senf S , Sandesara P , Reed S , Judge A . p300 Acetyltransferase activity differentially regulates the localization and activity of the FOXO homologues in skeletal muscle. American Journal of Physiology Cell Physiology. (2011) );300: 6. |
[103] | Chen J-F , Mandel E , Thomson M , Wu Q , Callis T , Hammond S , et al. The role of microRNA-1 and microRNA-133 in skeletal muscle proliferation and differentiation. Nature Genetics. advanced online publication. (2005) ;2 228–33. |
[104] | Williams A , Valdez G , Moresi V , Qi X , McAnally J , Elliott J , et al. MicroRNA-206 delays ALS progression and promotes regeneration of neuromuscular synapses in mice. Science. (2009) ;326: 5959:1549–54. |
[105] | Winbanks C , Wang B , Beyer C , Koh P , White L , Kantharidis P , et al. TGF-beta regulates miR-206 and miR-29 to control myogenic differentiation through regulation of HDAC4. The Journal of Biological Chemistry. (2011) ;286: 16:13805–14. |
[106] | Nguyen-Tran D-H , Hait N , Sperber H , Qi J , Fischer K , Ieronimakis N , et al. Molecular mechanism of sphingosine-1-phosphate action in Duchenne muscular dystrophy. Disease Models and Mechanisms. (2014) ;7: 1:41–54. |
[107] | Liu N , Williams A , Maxeiner J , Bezprozvannaya S , Shelton J , Richardson J , et al. microRNA-206 promotes skeletal muscle regeneration and delays progression of Duchenne muscular dystrophy in mice. Journal of Clinical Investigation. (2012) ;122: 6:2054–65. |
[108] | Yoo Y-E , Ko C-P . Treatment with trichostatin A initiated after disease onset delays disease progression and increases survival in a mouse model of amyotrophic lateral sclerosis. Experimental Neurology. (2011) ;231: 1:147–59. |
[109] | Minamiyama M , Katsuno M , Adachi H , Waza M , Sang C , Kobayashi Y , et al. Sodium butyrate ameliorates phenotypic expression in a transgenic mouse model of spinal and bulbar muscular atrophy. Human Molecular Genetics. (2004) ;13: 11. |
[110] | Chang JG , Hsieh-Li HM , Jong YJ , Wang NM , Tsai CH , Li H . Treatment of spinal muscular atrophy by sodium butyrate. Proc Natl Acad Sci U S A. (2001) ;98: 17:9808–13. |
[111] | Bricceno K , Sampognaro P , Van Meerbeke J , Sumner C , Fischbeck K , Burnett B . Histone deacetylase inhibition suppresses myogenin-dependent atrogene activation in spinal muscular atrophy mice. Human Molecular Genetics. (2012) ;21: 20:4448–59. |
[112] | Walsh M , Bhattacharya A , Liu Y , Van Remmen H . Butyrate prevents muscle atrophy after sciatic nerve crush. Muscle & Nerve. (2015) . |
[113] | Potthoff M , Wu H , Arnold M , Shelton J , Backs J , McAnally J , et al. Histone deacetylase degradation and MEF2 activation promote the formation of slow-twitch myofibers. The Journal of Clinical Investigation. (2007) ;117: 9:2459–67. |
[114] | Tang H , Macpherson P , Marvin M , Meadows E , Klein W , Yang X-J , et al. A histone deacetylase 4/myogenin positive feedback loop coordinates denervation-dependent gene induction and suppression. Molecular Biology of the Cell. (2009) ;20: 4:1120–31. |
[115] | McGee S , Swinton C , Morrison S , Gaur V , Campbell D , Jorgensen S , et al. Compensatory regulation of HDAC5 in muscle maintains metabolic adaptive responses and metabolism in response to energetic stress. FASEB Journal. (2014) ;28: 8:3384–95. |
[116] | McGee S , Fairlie E , Garnham A , Hargreaves M . Exercise-induced histone modifications in human skeletal muscle. The Journal of Physiology. (2009) ;587: Pt 24:5951–8. |
[117] | Moresi V , Carrer M , Grueter C , Rifki O , Shelton J , Richardson J , et al. Histone deacetylases 1 and 2 regulate autophagy flux and skeletal muscle homeostasis in mice. Proceedings of the National Academy of Sciences of the United States of America. (2012) ;109: 5:1649–54. |
[118] | Raichur S , Teh SH , Ohwaki K , Gaur V , Long YC , Hargreaves M , et al. Histone deacetylase 5 regulates glucose uptake and insulin action in muscle cells. Journal of Molecular Endocrinology. (2012) ;49: 3:203–11. |
[119] | Sharma S , Taliyan R , Ramagiri S . Histone deacetylase inhibitor, trichostatin A, improves learning and memory in high-fat diet-induced cognitive deficits in mice. Journal of Molecular Neuroscience: MN. (2015) ;56: 1:1–11. |
[120] | Gao Z , Yin J , Zhang J , Ward R , Martin R , Lefevre M , et al. Butyrate improves insulin sensitivity and increases energy expenditure in mice. Diabetes. (2009) ;58: 7. |
[121] | Chatterjee T , Basford J , Yiew KH , Stepp D , Hui D , Weintraub N . Role of histone deacetylase 9 in regulating adipogenic differentiation and high fat diet-induced metabolic disease. Adipocyte. (2014) ;3: 4:333–8. |
[122] | Feng D , Liu T , Sun Z , Bugge A , Mullican S , Alenghat T , et al. A circadian rhythm orchestrated by histone deacetylase 3 controls hepatic lipid metabolism. Science. (2011) ;331: 6022:1315–9. |
[123] | Mihaylova M , Vasquez D , Ravnskjaer K , Denechaud P-D , Yu R , Alvarez J , et al. Class IIa histone deacetylases are hormone-activated regulators of FOXO and mammalian glucose homeostasis. Cell. (2011) ;145: 4:607–21. |
[124] | Colussi C , Mozzetta C , Gurtner A , Illi B , Rosati J , Straino S , et al. HDAC2 blockade by nitric oxide and histone deacetylase inhibitors reveals a common target in Duchenne muscular dystrophy treatment. Proceedings of the National Academy of Sciences. (2008) ;105: 49:19183–7. |
[125] | Consalvi S , Mozzetta C , Bettica P , Germani M , Fiorentini F , Del Bene F , et al. Preclinical studies in the mdx mouse model of duchenne muscular dystrophy with the histone deacetylase inhibitor givinostat. Molecular Medicine (Cambridge, Mass). (2013) ;19: , 79–87. |
[126] | Minetti G , Colussi C , Adami R , Serra C , Mozzetta C , Parente V , et al. Functional and morphological recovery of dystrophic muscles in mice treated with deacetylase inhibitors. Nat Med. (2006) .advanced online publication(10):1147–50. |
[127] | Joe A , Yi L , Natarajan A , Le Grand F , So L , Wang J , et al. Muscle injury activates resident fibro/adipogenic progenitors that facilitate myogenesis. Nature Cell Biology. (2010) ;12: 2:153–63. |
[128] | Heredia J , Mukundan L , Chen F , Mueller A , Deo R , Locksley R , et al. Type 2 innate signals stimulate fibro/adipogenic progenitors to facilitate muscle regeneration. Cell. (2013) ;153: 2:376–88. |
[129] | Mozzetta C , Consalvi S , Saccone V , Tierney M , Diamantini A , Mitchell K , et al. Fibroadipogenic progenitors mediate the ability of HDAC inhibitors to promote regeneration in dystrophic muscles of young, but not old Mdx mice. EMBO Molecular Medicine. (2013) ;5: 4:626–39. |
[130] | Beharry A , Sandesara P , Roberts B , Ferreira L , Senf S , Judge A . HDAC1 activates FoxO and is both sufficient and required for skeletal muscle atrophy. Journal of Cell Science. (2014) ;127: Pt 7:1441–53. |
[131] | Dupré-Aucouturier S , Castells J , Freyssenet D , Desplanches D . Trichostatin A, a histone deacetylase inhibitor, modulates unloaded-induced skeletal muscle atrophy. Journal of Applied Physiology. (2015) :jap.01031.2014. |
[132] | Lexell J . Evidence for nervous system degeneration with advancing age. The Journal of Nutrition. (1997) ;127: 5 Suppl:1011S–1013S. |
[133] | Lexell J , Taylor C , Sjöström M . What is the cause of the ageing atrophy? Journal of the Neurological Sciences (1988) ;84: 2-3:275–94. |
[134] | Campbell MJ , McComas AJ , Petito F . Physiological changes in ageing muscles. Journal of Neurology, Neurosurgery, and Psychiatry. (1973) ;36: 2:174–82. |
[135] | Edström L , Larsson L . Effects of age on contractile and enzyme-histochemical properties of fast- and slow-twitch single motor units in the rat. The Journal of Physiology. (1987) ;392: , 129–45. |
[136] | Jang Y , Liu Y , Hayworth C , Bhattacharya A , Lustgarten M , Muller F , et al. Dietary restriction attenuates age-associated muscle atrophy by lowering oxidative stress in mice even in complete absence of CuZnSOD. Aging Cell. (2012) ;11: 5:770–82. |
[137] | Chai RJ , Vukovic J , Dunlop S , Grounds M , Shavlakadze T . Striking denervation of neuromuscular junctions without lumbar motoneuron loss in geriatric mouse muscle. PloS One. (2011) ;6: 12. |
[138] | Walsh M , Bhattacharya A , Sataranatarajan K , Qaisar R , Sloane L , Rahman M , et al. The histone deacetylase inhibitor butyrate improves metabolism and reduces muscle atrophy during aging. Aging Cell. (2015) ;14: 6:957–70. |
[139] | Fling B , Knight C , Kamen G . Relationships between motor unit size and recruitment threshold in older adults: Implications for size principle. Experimental Brain Research. (2009) ;197: 2:125–33. |
[140] | Erim Z , Beg F , Burke D , de Luca C . Effects of Aging on Motor-Unit Control Properties. Journal of Neurophysiology. (1999) ;82: 5:2081–91. |
[141] | Chen L , Nelson D , Zhao Y , Cui Z , Johnston J . Relationship between muscle mass and muscle strength, and the impact of comorbidities: A population-based, cross-sectional study of older adults in the United States. BMC Geriatrics. (2013) ;13: 1:74. |
[142] | Maughan RJ , Watson JS , Weir J . Strength and cross-sectional area of human skeletal muscle. The Journal of Physiology. (1983) ;338: 1:37–49. |
[143] | Kadhiresan VA , Hassett CA , Faulkner JA . Properties of single motor units in medial gastrocnemius muscles of adult and old rats. The Journal of Physiology. (1996) ;493: Pt 2:543–52. |
[144] | Rudolf R , Khan MM , Labeit S , Deschenes M . Degeneration of neuromuscular junction in age and dystrophy. Frontiers in Aging Neuroscience. (2014) ;6. |
[145] | Opalach K , Rangaraju S , Madorsky I , Leeuwenburgh C , Notterpek L . Lifelong calorie restriction alleviates age-related oxidative damage in peripheral nerves. Rejuvenation Research. (2010) ;13: 1:65–74. |
[146] | Hamilton R , Bhattacharya A , Walsh M , Shi Y , Wei R , Zhang Y , et al. Elevated protein carbonylation, and misfolding in sciatic nerve from db/db and sod1–/– mice: Plausible link between oxidative stress and demyelination. PloS One. (2013) ;8: 6:e65725. |
[147] | Jang Y , Lustgarten M , Liu Y , Muller F , Bhattacharya A , Liang H , et al. Increased superoxide in vivo accelerates age-associated muscle atrophy through mitochondrial dysfunction and neuromuscular junction degeneration. FASEB J. (2010) ;24: 5:1376–90. |
[148] | Shimazu T , Hirschey M , Newman J , He W , Shirakawa K , Le Moan N , et al. Suppression of oxidative stress by β-hydroxybutyrate, an endogenous histone deacetylase inhibitor. Science (New York, NY). (2013) ;339: 6116:211–4. |
[149] | Edwards C , Canfield J , Copes N , Rehan M , Lipps D , Bradshaw P . D-beta-hydroxybutyrate extends lifespan in C. elegans. Aging. (2014) ;6: 8:621–44. |
[150] | Calvert S , Tacutu R , Sharifi S , Teixeira R , Ghosh P , de Magalhães J . A network pharmacology approach reveals new candidate caloric restriction mimetics in C. elegans. Aging Cell. (2015) ):n/a-n/a. |
[151] | Kim S , Benguria A , Lai CY , Jazwinski SM . Modulation of life-span by histone deacetylase genes in Saccharomyces cerevisiae. Molecular Biology of the Cell. (1999) ;10: 10:3125–36. |
[152] | Kang H-L , Benzer S , Min K-T . Life extension in Drosophila by feeding a drug. Proceedings of the National Academy of Sciences of the United States of America. (2002) ;99: 2:838–43. |
[153] | Galmozzi A , Mitro N , Ferrari A , Gers E , Gilardi F , Godio C , et al. Inhibition of class I histone deacetylases unveils a mitochondrial signature and enhances oxidative metabolism in skeletal muscle and adipose tissue. Diabetes. (2013) ;62: 3:732–42. |
[154] | Jang Y , Lustgarten M , Liu Y , Van Remmen H . Calorie Restriction (CR) Protects against oxidative stress-induced muscle atrophy by preserving mitochondrial function and muscle integrity even in the absence of antioxidant enzyme CuZnSOD. Free Radical Biology and Medicine. (2010) ;49: , S81. |
[155] | Colman R , Anderson R , Johnson S , Kastman E , Kosmatka K , Beasley M , et al. Caloric restriction delays disease onset and mortality in rhesus monkeys. Science. (2009) ;325: 5937:201–4. |
[156] | Lanza I , Zabielski P , Klaus K , Morse D , Heppelmann C , Bergen R , et al. Chronic caloric restriction preserves mitochondrial function in senescence without increasing mitochondrial biogenesis. Cell Metabolism. (2012) ;16: 6:777–88. |
[157] | Dufour A , Hannan M , Murabito J , Kiel D , McLean R . Sarcopenia definitions considering body size and fat mass are associated with mobility limitations: The Framingham Study. The Journals of Gerontology Series A, Biological Sciences and Medical Sciences. (2013) ;68: 2:168–74. |
[158] | Newman A , Haggerty C , Goodpaster B , Harris T , Kritchevsky S , Nevitt M , et al. Strength and muscle quality in a well-functioning cohort of older adults: The Health, Aging and Body Composition Study. Journal of the American Geriatrics Society. (2003) ;51: 3:323–30. |
[159] | Bouchard D , Héroux M , Janssen I . Association between muscle mass, leg strength, and fat mass with physical function in older adults: Influence of age and sex. Journal of Aging and Health. (2011) ;23: 2:313–28. |
[160] | Cree M , Newcomer B , Katsanos C , Sheffield-Moore M , Chinkes D , Aarsland A , et al. Intramuscular and liver triglycerides are increased in the elderly. The Journal of Clinical Endocrinology and Metabolism. (2004) ;89: 8:3864–71. |
[161] | Attaix D , Mosoni L , Dardevet D , Combaret L , Mirand PP , Grizard J . Altered responses in skeletal muscle protein turnover during aging in anabolic and catabolic periods. The International Journal of Biochemistry & Cell Biology. (2005) ;37: 10:1962–73. |
[162] | Husom A , Peters E , Kolling E , Fugere N , Thompson L , Ferrington D . Altered proteasome function and subunit composition in aged muscle. Archives of Biochemistry and Biophysics. (2004) ;421: 1:67–76. |
[163] | Rooyackers OE , Adey DB , Ades PA , Nair KS . Effect of age on in vivo rates of mitochondrial protein synthesis in human skeletal muscle. Proceedings of the National Academy of Sciences of the United States of America. (1996) ;93: 26:15364–9. |
[164] | Balagopal P , Rooyackers OE , Adey DB , Ades PA , Nair KS . Effects of aging on in vivo synthesis of skeletal muscle myosin heavy-chain and sarcoplasmic protein in humans. Am J Physiol. (1997) ;273: 4 Pt 1:E790–800. |
[165] | White T , LeBrasseur N . Myostatin and sarcopenia: Opportunities and challenges - a mini-review. Gerontology. (2014) ;60: 4:289–93. |
[166] | Siriett V , Platt L , Salerno M , Ling N , Kambadur R , Sharma M . Prolonged absence of myostatin reduces sarcopenia. J Cell Physiol. (2006) ;209: 3:866–73. |
[167] | Smith R , Lin B . Myostatin inhibitors as therapies for muscle wasting associated with cancer and other disorders. Current Opinion in Supportive and Palliative Care. (2013) ;7: 4:352–60. |
[168] | Johnston A , Murphy K , Jenkinson L , Laine D , Emmrich K , Faou P , et al. Targeting of Fn14 prevents cancer-induced cachexia and prolongs survival. Cell. (2015) ;162: 6:1365–78. |
[169] | Tajrishi M , Sato S , Shin J , Zheng T , Burkly L , Kumar A . The TWEAK-Fn14 dyad is involved in age-associated pathological changes in skeletal muscle. Biochemical and Biophysical Research Communications. (2014) ;446: 4:1219–24. |
[170] | Yousef H , Conboy M , Morgenthaler A , Schlesinger C , Bugaj L , Paliwal P , et al. Systemic attenuation of the TGF-β pathway by a single drug simultaneously rejuvenates hippocampal neurogenesis and myogenesis in the same old mammal. Oncotarget. (2015) ;6: 14:11959–78. |
[171] | Trendelenburg AU , Meyer A , Jacobi C , Feige J , Glass D . TAK-1/p38/nNFκB signaling inhibits myoblast differentiation by increasing levels of Activin A. Skeletal Muscle. (2012) ;2: 1. |
[172] | Yabumoto C , Akazawa H , Yamamoto R , Yano M , Kudo-Sakamoto Y , Sumida T , et al. Angiotensin II receptor blockade promotes repair of skeletal muscle through down-regulation of aging-promoting C1q expression. Scientific Reports. (2015) ;5: , 14453. |
[173] | Sinha M , Jang Y , Oh J , Khong D , Wu E , Manohar R , et al. Restoring systemic GDF11 levels reverses age-related dysfunction in mouse skeletal muscle. Science. (2014) ;344: 6184:649–52. |
[174] | Egerman M , Cadena S , Gilbert J , Meyer A , Nelson H , Swalley S , et al. GDF11 increases with age and inhibits skeletal muscle regeneration. Cell Metabolism. (2015) ;22: 1:164–74. |
[175] | Hinken A , Powers J , Luo G , Holt J , Billin A , Russell A . Lack of evidence for GDF11 as a rejuvenator of aged skeletal muscle satellite cells. Aging Cell. (2016) :n/a-n/a. |
[176] | Gomes A , Price N , Ling A , Moslehi J , Montgomery M , Rajman L , et al. Declining NAD(+) induces a pseudohypoxic state disrupting nuclear-mitochondrial communication during aging. Cell. (2013) ;155: 7:1624–38. |
[177] | Ebert S , Dyle M , Bullard S , Dierdorff J , Murry D , Fox D , et al. Identification and small molecule inhibition of an activating transcription factor 4 (ATF4)-dependent pathway to age-related skeletal muscle weakness and atrophy. The Journal of Biological Chemistry. (2015) ;290: 42:25497–511. |
[178] | Siriett V , Salerno MS , Berry C , Nicholas G , Bower R , Kambadur R , et al. Antagonism of myostatin enhances muscle regeneration during sarcopenia. Molecular Therapy: The Journal of the American Society of Gene Therapy. (2007) ;15: 8:1463–70. |
[179] | Morley J . Sarcopenia in the elderly. Family Practice. (2012) ;29: suppl 1:i44–i8. |
Figures and Tables
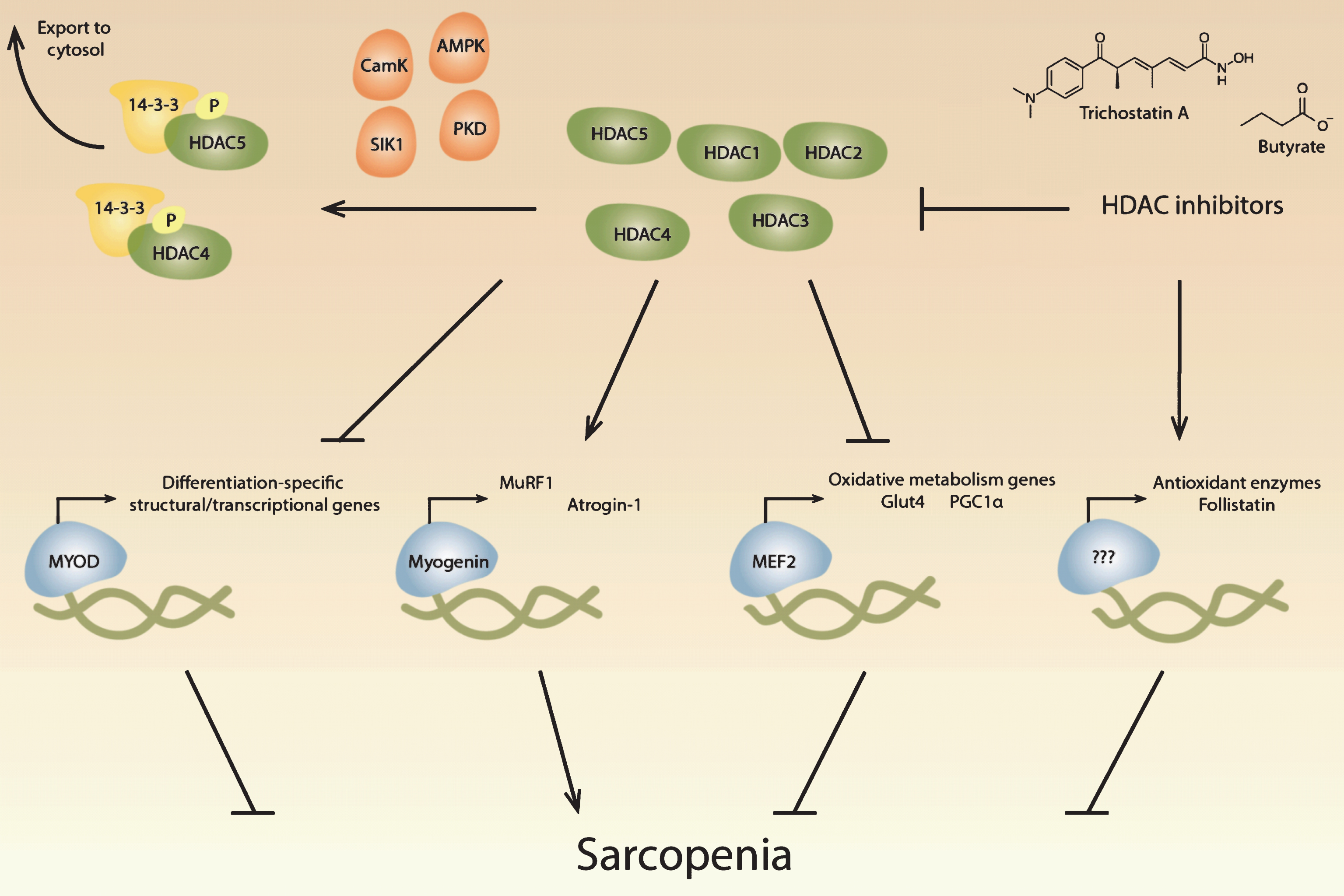
Fig.1
Potential roles of histone deacetylases in sarcopenia. Histone deacetylases (HDACs) regulate critical muscle-specific transcription factors (blue). Different HDAC isoforms (green) can affect the differentiation process, which is regulated by MyoD and other transcription factors, suggesting that HDACs may play a role in age-related changes in satellite cells. HDAC4 and HDAC5 activate myogenin-dependent gene transcription, which includes the ubiquitin proteolytic machinery, that may contribute to muscle atrophy during aging. HDACs also regulate Mef2, which regulates metabolic processes known to change during aging. HDAC inhibitors have potentially beneficial effects in skeletal muscle, including the upregulation of antioxidant enzymes and the increase of follistatin, which inhibits myostatin. HDACs can be inhibited pharmacologically with HDAC inhibitors, including trichostatin A and butyrate, and multiple signaling pathways converge on class II HDACs, resulting in their phosphorylation by kinases (orange) and transport out of the nucleus by 14-3-3 proteins (yellow).