N-3 polyunsaturated fatty acid and neuroinflammation in aging and Alzheimer’s disease
Abstract
The innate immune system of the brain is mainly composed of microglial cells, which play a key role in the maintenance of synapses and the protection of neurons against noxious agents or lesions owing to their phagocytic activity. In the healthy brain, microglia are highly motile and strongly interact with neurons either by physical contact, induction of oxidative stress or through specific mediators, such as chemokines and cytokines. In response to inflammatory insult however, microglial cells get activated and produce inflammatory cytokines. The action of cytokines on specific receptors expressed in the brain triggers the development of sickness behavior and altered cognitive and emotional processes. The effects are acute and reversible as normal behavior is restored once the synthesis of inflammatory brain cytokines returns to baseline after a few hours. However, in pathological situations, these cytokines may reach toxic levels and have irreversible consequences such as neuronal death, as observed in neurodegenerative disorders such as Alzheimer’s disease. Omega-3 (n-3) polyunsaturated fatty acids (PUFAs) are essential nutrients and fundamental components of neuronal and glial cell membranes. They accumulate in the brain during the perinatal period in a dietary supply-dependent fashion. Their brain levels may diminish with age, but can be increased by diets enriched in n-3 PUFAs. Changes in the immune profile have been associated with n-3 PUFAs intake in humans and animal models. Therefore, the increasing exposure of the population to diets low in n-3 PUFAs could contribute to the deleterious effects of the chronic activation of microglia in the brain.
1Introduction
The central nervous system (CNS) has long been considered as an immunoprivileged organ. Indeed, in physiological conditions, the blood brain barrier (BBB), owing to its tight junctions, considerably restricts the entry of immune cells, notably lymphocytes, into the brain. Research in neuroimmunology has shown that the brain possesses its own line of defense, activated by immune stimuli, and closely linked to the peripheral immune system. Although not as isolated as first thought, there are important distinctions between the peripheral and the central immune systems. Inflammatory cytokines, which are important mediators of the communication within the immune system, also act in the brain where they can activate microglial cells and astrocytes that in turn, can produce cytokines, chemokines, complement proteins and nitric oxide [1–3]. In physiological conditions, the synthesis of brain immune mediators is finely regulated, allowing a rapid return to basal levels, without leading either to BBB failure or cerebral lesion. However, when synthesized in large amounts or in a chronic fashion, these factors may have toxic effects on neurons, resulting in substantial neuronal dysfunction that can lead to cell death [4–7]. During aging, chronic microglial reactivity and production of low levels of inflammatory cytokines occurs and can produce alterations of neuronal functions [8]. Indeed, this age-associated inflammation, characterized by the increased production of brain cytokines, increases the vulnerability of the aging brain to immune stimuli, and the risk of developing delirium and/or neurodegenerative disorders with an inflammatory component, such as Alzheimer’s disease (AD) [9–11]. Accordingly, clinical and epidemiological studies have suggested a possible association between the systemic expression levels of inflammatory cytokines and the incidence of functional/behavioral alterations (cognitive or mood disorders) in psychiatric and elderly subjects [12–15]. In this context, limiting the development of chronic neuroinflammation could protect the brain against neurodegenerative disorders. This could be achieved with the diet, a modifiable environmental factor to which each individual is exposed throughout his life.
Increasing attention has been paid to omega-3 (n-3) and omega-6 (n-6) polyunsaturated fatty acids (PUFAs). These micronutrients are essential since their precursors cannot be synthesized de novo by the organism. Moreover, their synthesis is extremely limited in most mammals (less than 5% of the precursors are converted) [16]. Increasing data in animal models as well as in humans suggest that dietary PUFA exert immunomodulatory effects [17–20]. Indeed, n-3 long chain PUFAs form the basis of lipid derivatives (neuroprotectins and resolvins) with anti-inflammatory properties [21–26]. Moreover, they are precursors of eicosanoids including leukotrienes, prostaglandins and thromboxanes, which are important modulators of the inflammatory response. However, they have lower biological potency than eicosanoids derived from n-6 PUFAs such as arachidonic acid [27, 28]. The brain is extremely rich in PUFAs, and the accumulation of PUFAs in brain tissues first takes place during the perinatal period in proportions which are dependent on maternal dietary levels [29, 30]. Conversely, the levels of brain PUFA has been reported to diminish with aging, although this decrease can be prevented or corrected by appropriate nutritional strategies [31]. Since the Industrial Revolution, we have observed a decrease in energy expenditure related mainly to sedentary lifestyle combined with an increase in the consumption of high energy foods characterized by elevated levels of sugar, saturated fats, and n-6 PUFAs, poor in n-3 PUFAs, vitamins, proteins and micronutriments [32, 33]. The resulting imbalance in the n-6:n-3 ratio from this dramatic modification in the dietary intake is currently estimated to 10:1 to 20:1 in Western diets, whereas the current recommended ratio is 1:1 to 2:1 [32, 33]. With the known immunomodulation resulting from n-3 PUFAs intake, this imbalance could therefore contribute to prime the brain to the deleterious effects of inflammatory cytokines, and eventually to the development of neurodegenerative and/or neurobehavioral disorders.
2The brain innate immune system (BIIS)
In the periphery, tissue injuries caused by trauma or pathogens induce a rapid local inflammatory response involving local cells and characterized by the synthesis and release of proinflammatory factors, among which cytokines and chemokines, followed by systemic recruitment of immune cells. The purpose of this local response is to eliminate pathogens and to promote tissue repair. However, the failure of resolving the insult and dysregulated injury may lead to chronic inflammation, which is toxic for the tissue, ultimately resulting in cell death. In addition, peripheral inflammation can also influence brain immunity [1]. However, it appears that the steps involved in the cerebral immune response are distinct from those of the periphery. Therefore, the term “neuroinflammation” is broadly used to discriminate the brain from the peripheral inflammation. “Neuroinflammation” describes the brain inflammatory response involving not only peripheral immune cells influx into the brain but also the specialized response of brain innate immune system (BIIS) [34].
Microglial cells are the main cells of the BIIS. They are the parenchymal resident macrophages of the brain where they act as a first line of defense(phagocytosis, antigen presentation, cell recruitment and secretion of cytokines) [35, 36]. They account for 5 to 20% of the non-neuronal glial cells, depending on the brain structures analyzed. Microglia are distinct from the brain macrophages found in the meninges, choroid plexus, and perivascular space, owing to their different developmental origin. Indeed, recent data highlight that microglia derives from macrophages produced by primitive hematopoiesis in the yolk sac [37–40] while brain macrophages derive from myeloid precursor in the bone marrow [41, 42]. Microglia precursors colonize the CNS during the embryonic and fetal phases of development [43]. Interestingly, an increase of CD11b+/F4/80+ microglia occurs in the post-natal brain of rodents [37]. However, recent evidence suggests that this increase in microglial cell number is not induced by the recruitment of blood-derived myeloid precursors but instead results from the proliferation of resident microglial cells [39, 44]. Recent studies provide new insights into the development of the microglial population from yolk sac progenitors during embryogenesis. In contrast with macrophages from other tissues, microglia persist throughout the entire life of an individual [42]. There is a growing interest to characterize microglia cells profile and signature. Very recent data reveals that microglia express a decreased number of mRNA types as compared to tissue macrophages [45] and display a specific mRNA signature that is dependent on TGFβ [46].
Microglia are particularly sensitive to changes in their microenvironment and readily become activated in response to infection, trauma or disease [47]. In healthy brain, the microglia can phagocyte apoptotic neurons and debris and reduce neuroinflammation which, in turn is beneficial to viable neurons [48]. However, in inflammatory or pathologic situations, the phagocytic adaptor protein MFG-E8 is released by microglia and binds to phosphatidylserine (PS) exposed on apoptotic neurons. This activates neuronal phagocytosis by microglia via the vitronectin receptor [49]. Annexin A1, another eat-me signal released by microglia, serves as a bridge with PS on dying neuron, helping the microglia to discriminate between apoptotic neurons and healthy neurons [50]. Recently, new data revealed that activated microglia in inflammatory state appears to lose their ability to discriminate between apoptotic and viable neurons, resulting in phagocytosis of diseased as well as healthy neurons [51].
In the adult brain, microglial cells have a ramified morphology when quiescent, and ameboid morphology when activated. Ramified microglia generally display less phagocytic activity and weakly express ligands and receptors involved in macrophage function. Disseminated throughout the brain parenchyma, they use their processes to receive signals such as danger-associated molecular patterns (DAMP) and pathogen-associated molecular patterns (PAMP) from their microenvironment, which reveal the existence of an endogenous pathological signal or the presence of a pathogen, respectively. In order to do this, microglial cells express a set of pattern recognition receptors (PRRs) including the Toll-like receptors (TLRs) that allow the recognition of PAMPs, such as the bacterial endotoxin [52, 53], and DAMPS, such as misfolded proteins [54], and promote brain inflammatory reaction [55]. The activation of PRRs by PAMPS and DAMPs induces a signaling cascade leading to the secretion of cytokines and chemokines. The microglial cells further coordinate the inflammatory response via the expression of membrane receptors for inflammatory cytokines interleukin (IL)-1β, tumor necrosis factor (TNF)α and IL-6 and several chemokines. In vivo, IL-1β, TNFα and IL-6 are produced by microglia in response to peripheral immune stimuli such as bacterial endotoxin lipopolysaccharide (LPS) [56].
The BIIS response promotes clearance of pathogens, toxic cellular debris and apoptotic cells and therefore protects the brain. Indeed, a selective ablation of proliferating microglia exacerbates brain damage in adult and in neonatal hypoxic ischemic injury models [57]. However, under some circumstances, the sustained expression of inflammatory factors such as cytokines can lead to neurodegeneration [7, 58]. The BIIS response is therefore a double-edged sword depending on a fine balance between protective and detrimental effects that needs to be tightly controlled. Real-time in vivo imaging technology has revealed that both in health and disease, microglia exist in an active state but their role in the brain depends on the broad phenotype spectrum they can adopt [59]. Microglia phenotypes, so called polarization, could be crucial in the protective or detrimental role of PRR-activated BIIS response toward neurons. According to what was described for macrophages, it has been suggested that activated M1 cells have cytotoxic properties, M2a are involved in repair and regeneration, M2b promote immunomodulation, while M2c have an acquired-deactivating phenotype [60] (Fig. 1). In vivo, microglia express proinflammatory cytokines associated with a M1 phenotype (IL-1, IL-6, IL-12 and TNFα) in response to an immune stimulus. Recent evidence indicates that neurons exert some control on microglia activity [61]. The extent of neuroinflammation therefore depends on the bi-directional interactions between neurons and microglia. Recruitment and activation of microglial cells require well-organized reciprocal communication between these two cell types [36, 62]. As a result, neurons release ON or OFF signals to regulate the activation of microglia. On the one hand, OFF signals (CD200, CX3CL1, CD47, CD55 and HMGB1) are produced by healthy neurons to keep microglia in their surveillance mode. On the other hand, damaged neurons express inducible ON signals (chemokines, purine and glutamate) to activate microglia and phagocytosis [36]. Interestingly, such neuron-glia interaction is impaired in the aged brain leading to amplified and prolonged microglial activation and production of proinflammatory cytokines [63, 64].
3BIIS in the aging brain and neurodegenerative diseases
3.1Neuroinflammation, the aged brain and Alzheimer’s disease
Aging is associated with senescence of microglia, impaired microglia phagocytic activity and low-grade neuroinflammation [65], which is characterized by a higher expression of proinflammatory cytokines IL-1β, IL-6 and TNFα to the detriment of anti-inflammatory factors such as IL-10 and IL-4. This state is called inflammaging at the periphery and in the brain [66]. The overproduction of proinflammatory cytokines in the absence of infection or injury in the aged brain could be linked to the impairment of microglial activity. Indeed, microglia number and activity increases during normal aging [67]. These aged cells, in addition to producing proinflammatory cytokines, contain lipofuscine granules, and decreased processes complexity, a morphological change found in activated microglia [47, 68]. In addition, microglia in the aged brain express higher levels of CD86, major histocompatibility complex II (MHC II), TLR and CR3/CD11b which are markers of activation [69, 70]. Senescent microglia display reduced phagocytic activities of beta-amyloid in aged transgenic mice which could be due to their M1 phenotype [71]. Mechanisms involved in increased microglia activation in the aged brain are not fully understood, however as CD200 and CX3CR1 expression is impaired it could be that neuron-glia interactions are disturbed [67, 72].
The involvement of microglia in the communication of systemic inflammatory signals to neurons has been recently reviewed [73]. One of the most important new knowledge is that microglia become more susceptible to inflammatory stimuli when they are primed by pathological insults contributing to the progression of neurodegenerative diseases such as Alzheimer’s Disease (AD) [73, 74]. The characteristics of primed microglia remain to be determined. However, increased number of microglia, together with changes in morphology and increased expression of cell surface antigens such as CD68, complement receptors (CR3) and/or MHC have been consistently reported in priming context as first reported in prion disease [75]. Priming phenomenon has been observed in aging [76], after the administration of proinflammatory endotoxins [77, 78] and in animal models of AD and Parkinson disease [7, 79]. Interestingly, aging microglia express a specific sensome (defined as proteins sensing microbes) that could confer them a higher vulnerability to inflammatory stimuli [70]. Indeed, the concept of microglia priming provides new view of how systemic inflammation could contribute to the progression of neurodegenerative diseases, through increased production of inflammatory cytokines that could damage neurons and/or loss of neuroprotective properties.
The first evidence of an inflammatory response in AD comes from case-control studies showing immune anomalies in blood or CSF from living individuals or cerebral tissue collected post-mortem. For example, plasma cytokine profiles, key components of the complement system and immune cells are altered early in the disease [80–83]. High levels of pro and anti-inflammatory factors, and increased PRR and chemokines expression are found in the brain of AD patients [4, 84]. Activated microglia in the brain of AD patients have been detected by (R)-[3H]PK11195 PET binding to peripheral benzodiazepine receptors [85]. A second set of evidence comes from epidemiology. Indeed, chronic non-steroidal anti-inflammatory drugs (NSAID) use has been associated to lower risk of AD [86, 87]. Finally, data generated in animal models suggest a causal link between brain amyloid or tau pathologies and the triggering of an immune response [5, 88, 89], although this view is also disputed [90]. In transgenic APP/PS1 or 3xTg-AD mice, reports suggest that microglia activation starts between 6 and 12 months, as assessed with CD45, Iba1 and/or F4/80 IHC [91–94]. Therefore, despite conflicting data, it appears clear that changes in parameters involved in central or peripheral inflammation are associated with AD.
However, the role of inflammation or the immune system in AD pathogenesis is still a matter of debate as both beneficial and adverse effects of inflammation have been reported [95–101]. Recent data highlighted that microglia, because of its impaired activity in the AD brain, cannot phagocytocize Aβ that therefore accumulates [102, 103]. In turn, Aβ accumulation activates microglia in a chronic proinflammatory state that contributes to the disease progression and, ultimately cognitive decline [4]. Higher levels of IL-1 have been implicated in both the initiation and progression of neuropathological changes [104]. Accordingly, overexpression of IL-1 in the AD brain has been linked to an increased microglial activity, frequently associated with amyloid plaques [105]. In addition, brain from Tg2576 mice (a model of AD) exhibits significant increases in IL-1 expression in comparison to healthy animals [106]. Overexpresssion of IL-1β in the 3xTg-AD mouse model of AD suggest that amyloid and tau pathology are differentially regulated, with a reduction in amyloid deposit but an exacerbation of Tau hyperphosphorylation [107]. Such effects could involve the fractalkine pathway as the expression of its receptor CX3CR1 have been reported to be associated with either a decrease [108] or an activation of Aβ clearance [109–112] and increase Tau phosphorylation [113]. Therefore, further studies are needed to precise the role of the fractalkine pathway in these processes.Globally, a crucial issue that remains unclear is whether the immune response observed in AD is a compensatory mechanism with a neuroprotective effect, part of a vicious circle leading to neuronal death and cognitive decline, or both [72, 103, 114, 115]. Answers to this question will determine how neuroinflammation can be “tamed” for the purpose of developing therapeutic interventions.
In the last decades, trials aiming at modulating brain inflammation in AD patients have been dis- appointing [72, 116–118]. This included assays with NSAID, steroid anti-inflammatory (prednisone) or with immunomodulators such as TNF antagonists and polyclonal or monoclonal immunoglobulins [72, 118, 119]. However, consistent with epidemiology data [86], it remains likely that the modulation of inflammation is more suitable for a preventive intervention [120]. Unfortunately, no such preventive study has been completed yet, the ADAPT study on COX-1 and COX-2 inhibitors having been ended abruptly due to unsuspected side effects [72, 121]. Nevertheless, further data analysis from the ADAPT study do not provide a clear answer to the hypothesis that celecoxib or naproxen exert a preventive action against AD [122, 123]. Because the role of BIIS in the development of AD is complex, other strategies aiming at optimizing microglia activity, rather than just blocking inflammatory factors synthesis in the brain could be more beneficial [4, 72].
4Polyunsaturated fatty acids and neuroinflammation
4.1N-3 PUFAs: A brief overview
PUFAs of the n-3 or n-6 families are essential nutrients, as the precursors of these two series (linoleic acid (18:2n-6, LA) and α-linolenic acid (18:3n-3, ALA)) cannot be generated de novo in mammals, they have to be provided by the diet. They are respectively metabolized by a series of elongation and desaturation steps into arachidonic acid (20:4 n-6, AA) and eicosapentaenoic acid (20:5 n-3, EPA) and docosahexaenoic acid (22:6 n-3, DHA) (Fig. 2). These long chain PUFAs are incorporated into cell membranes as phospholipids. The liver is the main site of conversion of LA and ALA into long chain PUFAs, although other organs such as the brain also express the necessary elongases and desaturases [124]. Since the two series of PUFAs compete for the use of the enzymes necessary for their biosynthesis, and because the conversion into long chain is extremely limited (less than 5% of the precursors is converted), their supply by the diet is of particular importance. Foods that were previously consumed by humans were obtained from hunting and fishing and were relatively rich in n-3 long chain PUFAs. Since the Industrial Revolution, the ratio of n-6:n-3 PUFAs in the diet has increased from 1 to almost 20 in industrialized countries like the United States, leading to a suboptimal dietary consumption of n-3 PUFAs [33, 125].
The dietary deficiency in n-3 PUFAs is associated with a significant decrease in DHA in the brain, and could thus promote neuroinflammatory processes and the subsequent development of inflammation-related CNS disorders [8]. However, the effect of n-3 long chain PUFA supplementation to generate a favorable inflammatory marker profile is currently subject to debate. In populations with a high n-3 long chain PUFA dietary intake due to elevated fish consumption, such as Greenland Inuits, the incidence of ischemic heart and autoimmune diseases (such as psoriasis, asthma or multiple sclerosis) is low [126, 127], but inflammatory markers such as YKL-40 and hsCRP are rather found to be increased [128]. While some clinical studies have reported anti-inflammatory effects of n-3 long chain PUFAs administered in the context of chronic and autoimmune inflammatory disorders, other reports fail to reproduce these findings (for review [28]).
4.2N-3 PUFAs and inflammatory mechanisms
Attenuation of the inflammatory response is one of the most frequently cited mechanisms of action of n-3 PUFAs, despite limited convincing evidence of such an action in cerebral tissues until recently [129]. As stated above, a high n-3 PUFA intake decreases AA content and at the same time, increases DHA in the brain. Since n-3 PUFAs compete with AA as substrates for cyclooxygenase (COX) and lipoxygenases (LOX), increased n-3 PUFA concentration is expected to reduce the production of the more potent inflammatory eicosanoids derived from AA [25, 130–132]. Therefore, food rich in n-3 PUFA such as fish or walnut have been found to limit the inflammatory response in preclinical studies [133–136].
More specifically, n-3 PUFAs also decrease the production of various important inflammatory cytokines such as TNFα, IL-1, and IL-6 [20, 130, 131, 137]. Indeed, DHA decreases the expression of brain inflammatory markers following systemic LPS administration [138], brain ischemia-reperfusion [20, 139] and spinal cord injury [140]. However it remains difficult to gauge the direct effect of DHA on BIIS, since the primary CNS injury is also attenuated in these studies. High-fat intake in n-3 PUFA-deprived animals induces a rise of GFAP, a marker of astrogliosis, in mouse brains [141]. Recently, we have demonstrated thatin vitro, in murine microglia induced by LPS, the production of IL-1β and TNFα by is strongly inhibited by DHA through its effect on LPS signaling pathway nuclear factor-κB [137]. In vivo, chronic dietary n-3 PUFA deficiency significantly increases the production and release of IL-6 and TNFα in the blood [142]. In addition, mice exposed throughout life to a diet devoid of n-3 PUFAs display lower brain DHA and higher LPS-induced IL-6 levels in the plasma and the hippocampus [138]. In parallel, n-3 PUFAs have been shown to decrease the levels of COX-2 in vitro [143] and in vivo [20, 144].
The recent discovery of a novel family of endogenously generated autacoids, namely resolvins and protectins, with potent anti-inflammatory and proresolving activities offer a better understanding of the mechanisms responsible for the protective effect of DHA in the brain [25, 145]. In particular, resolvin D1 (RvD1), which originates from DHA via lipoxygenases, promotes the resolution of inflammation and has been detected in the brain [21]. Very interestingly, DHA and RvD1 promotes macrophage polarization toward a M2 state in obese mice adipose tissue associated to a decrease of pro-inflammatory cytokines and an increase of anti-inflammatory cytokines [146, 147]. In a model of LPS injection, Orr et al (2013) [129] showed that DHA exerts its anti-inflammatory effects in the brain, via its conversion into resolvins. Neuroprotectin D1 (NPD1), a DHA derived docosanoid has also been detected in the brain where it could exert anti-inflammatory and protective activities [23]. Chronic infusions of DHA or NPD1 in the brain significantly decreased neuroinflammatory processes triggered by a middle cerebral artery occlusion [139]. NPD1 even had a more potent effect than DHA [139, 148]. However, it remains to be demonstrated that NPD1 is the intermediary of the anti-inflammatory effect of DHA in the brain.
Experiments conducted in animal models have highlighted brain DHA as a potent mediator of the protective effects of dietary n-3 PUFAs. Low dietary intake of n-3 PUFA decreases DHA levels in the animal brain [149–151]. As a result, emotional behavior (depressive-like symptoms and anxiety) as well as learning and memory are impaired as shown by others and us [152–155]. On the other hand, positive effects of diets enriched in DHA on learning and memory have been demonstrated in laboratory animals [156–160].
4.3N-3 PUFAs and age-related neuroinflammation
As previously mentioned, PUFAs represent potent immunomodulatory agents. During aging, the levels and the turn-over rate of brain PUFAs decrease, particularly in the hippocampus, cortex, striatum and hypothalamus [161–164]. Brain levels of DHA and AA diminish in aging rats with alterations in cognition and in long-term potentiation (LTP) in the hippocampus [162]. In senescence-accelerated mouse (SAMP8), a spontaneous model of accelerated aging, DHA levels decrease, whereas lipid peroxidation increases with age including that of DHA [165, 166]. In addition, the conversion of the precursors LA and ALA into their long chain derivatives becomes less efficient. The activity of the Δ6 desaturase decreases with age in the liver and the brain [167, 168]. Phospholipid synthesis pathways are also altered with age, thus reducing the incorporation of PUFAs into membranes [169]. The combination and interaction of these different alterations associated with aging contributes to a reduction in the level of DHA, i.e. a reduction in the index of membrane fluidity, in the brain of elderly people. In animals, aging was found to be associated with a decrease in the membrane content of AA in the hippocampus together with an attenuation of LTP that can be reestablished by a diet containing AA [170].
With aging, IL-6 expression is increased in the cortex of both n-3 deficient and n-3 adequate CD1 mice while IL-10 expression is decreased with no effect of long term ALA deficient or enriched diet [155]. Conversely, short-term exposure to dietary EPA reduces IL-1-induced spatial memory deficit and anxiolytic behavior [171, 172] and improves LPS and Aβ-induced inhibition of LTP in both adult and aged rats [173]. The expression of markers of microglial activation (CD68, MHCII and CD11b) increases with age in animals, as does the number of microglia in the brain of humans, attesting of the occurrence of age-related neuroinflammation [174]. Microglial cell reactivity is involved in the age-dependent increase in the production of inflammatory cytokines, as demonstrated by the inhibition of inflammatory cytokine overexpression by minocycline in aged rats [69]. In rats, the age-related activation of the microglia, production of IL-1β and alterations in hippocampal LTP are attenuated by EPA [175, 176]. Importantly, a 2-month fish-oil dietary supply increases DHA in the brain, prevents proinflammatory cytokine expression and astrocyte morphology changes in the hippocampus and restored spatial memory deficits and c-Fos-associated activation in the hippocampus of aged mice [31]. These data support the idea of the importance of DHA dietary supply in aged mammals.
To detect an anti-inflammatory action in neurodegenerative diseases, one must work with an animal model displaying an easily quantifiable inflammatory response, which is not always the case in AD models [90]. In contrast, animal model of stroke offers an easier opportunity to directly probe this mechanism of action. To visualize the effects of DHA on stroke-induced neuroinflammation, we recently used the TLR2-fluc-GFP transgenic mice exposed to either (i) a control diet; (ii) a diet depleted in n-3 polyunsaturated fatty acid (PUFA) (iii) a diet enriched in n-3 long chain PUFA (0.7 g kg−1 day−1, with DHA:EPA ratio of 4:1) during 3 months [20]. Real-time biophotonic/bioluminescence imaging of the TLR2 response was performed before and after middle cerebral artery occlusion (MCAO), whereas cytokines concentrations and stroke area analyses were performed 3 and 7 days after MCAO, respectively. We have observed that 3 months of DHA/EPA treatment abolished the TLR2 response after ischemic injury, while increasing the brain n-3:n-6 PUFA ratio, preventing microglial activation, reducing the ischemic lesion size and increasing concentrations of the anti-apoptotic molecule Bcl-2 in the brain [20]. Additional analysis further revealed a significant decrease in the levels of COX-2 and IL-1beta , but not other pro-inflammatory cytokines [20]. The result of this study argues for the use of n-3 long chain PUFA in preventing the initiation of TLR2-dependent signaling cascade, which lays upstream of the main pathways leading to a neuroinflammatory response.
In epidemiological and observational studies, a higher level of blood n-3 PUFAs is associated with lower proinflammatory cytokine production [177–180]. In a cohort of elderly subjects, depressive individuals with an elevated plasma n-6:n-3 ratio were found to exhibit higher levels of TNFα and of IL-6 [180]. F2-isoprostane, a marker of oxidative stress, and telomere length, an indicator of immune cell aging, are both decreased in the blood of subjects supplemented with EPA/DHA [181]. Additionally, n-3 PUFA supplementation in elderly subjects reduced the levels of inflammatory cytokines produced by blood leukocytes stimulated in vitro [182]. The production of prostaglandin E2 by monocytes is inversely correlated to the EPA content of leukocytes obtained from aged subjects after the consumption of dietary complements containing different doses of EPA [183]. To the extent that the level of peripheral cytokines can reflect that of brain cytokines, these results would suggest that dietary n-3 PUFAs modulate neuroinflammation and associated neurobehavioral effects in elderly individuals [8] (Fig. 3).
Epidemiological studies further highlight the importance of n-3 PUFA levels in the development of age-linked neurodegenerative disorders. Indeed, decreases in plasma and brain DHA levels have been shown in patients with AD. These results, however, remain controversial, since other studies have demonstrated an increase or an absence of variation in brain DHA levels in similar populations. Nonetheless, the risk of dementia was found to be augmented in elderly subjects presenting low levels of circulating EPA [184]. In addition, regular consumption of diets rich in n-3 PUFA, such as the Mediterranean diet, appears to contribute to a decrease in the risk of depression and/or dementia in the elderly [185, 186]. Using a mouse model of AD, the Tg2576 mouse, dietary supply of DHA reduces both the formation of amyloid plaques or the accumulation of caspase-cleaved products, while protecting against the loss of synaptic markers [187–189]. However, the administration of DHA-containing dietary supplements to patients with AD or mild cognitive impairment has not yielded conclusive results [190]. Only in vitro studies suggested a potential beneficial effect of DHA mediators. Indeed, it was shown in vitro that RvD1 promotes Aβ phagocytosis [191] and that NPD1 downregulates inflammatory signaling and amyloidogenic APP cleavage [192]. Data from preclinical and clinical studies all indicate that the effect of DHA differs according to apolipoprotein E (APOE) genotype [193–196]. A reduced brain uptake has been suggested as a mechanism underlying the lack of benefit of DHA in APOE4 carriers [193, 197]. However, APOE4 could simply counteract any effect of DHA by its known aggravating impact on peripheral immune and inflammatory responses, including microglial activation [198, 199]. Therefore, although preclinical studies support an effect of DHA in the prevention and / or treatment of age-related diseases, further clinical trials adapted to APOE genotype remain required to determine the best approach for an optimal therapeutic effect.
5Conclusion
Growing evidence highlight an association between inflammation in cerebral tissues and mood and cognitive disorders during infection, aging and neurodegenerative disorders. Whether neuroinflammation processes are a cause or a consequence of the underlying disease and its symptoms is still debated. However, the current state of knowledge strongly points to potential disease modification value of treating neuroinflammation in the preclinical stages of neurodegenerative diseases. More specifically, the impact of DHA, either from the diet or from dietary-supplementation, as a preventive therapeutic strategy needs to be investigated.
Acknowledgments
This work has been supported by FRM, Région Aquitaine, Société française de Nutrition and INRA (SL). The work of FC is supported by grants from the Canadian Institutes of Health Research (CIHR), the Alzheimer Society Canada, the Parkinson Society Canada and the Fonds de la recherche en santé du Québec (FRQ-S)
References
[1] | Dantzer R(1998) Cytokines and sickness behaviorAnn N Y Acad Sci840: 586590 |
[2] | Czirr E, Wyss-Coray T(2012) The immunology of neurodegenerationJ Clin Invest122: 11561163 |
[3] | Wilson CJ, Finch CE, Cohen HJ(2002) Cytokines and cognition-the case for a head-to-toe inflammatory paradigmJ Am Geriatr Soc50: 20412056 |
[4] | Lampron A, Elali A, Rivest S(2013) Innate immunity in the CNS: Redefining the relationship between the CNS and Its environmentNeuron78: 214232 |
[5] | Wyss-Coray T(2006) Inflammation in Alzheimer disease: Driving force, bystander or beneficial response?Nat Med12: 10051015 |
[6] | Hurley LL, Tizabi Y(2013) Neuroinflammation, neurodegeneration, and depressionNeurotox Res23: 131144 |
[7] | Perry VH, Nicoll JA, Holmes C(2010) Microglia in neurodegenerative diseaseNat Rev Neurol6: 193201 |
[8] | Laye S(2010) Polyunsaturated fatty acids, neuroinflammation and well beingProstaglandins Leukot Essent Fatty Acids82: 295303 |
[9] | Perry VH, Newman TA, Cunningham C(2003) The impact of systemic infection on the progression of neurodegenerative diseaseNat Rev Neurosci4: 103112 |
[10] | Norden DM, Godbout JP(2013) Review: Microglia of the aged brain: Primed to be activated and resistant to regulationNeuropathol Appl Neurobiol39: 1934 |
[11] | Mosher KI, Wyss-Coray T(2014) Microglial dysfunction in brain aging and Alzheimer’s diseaseBiocem Pharmacol88: 594604 |
[12] | Capuron L(2011) Chronic low-grade inflammation in elderly persons is associated with altered tryptophan and tyrosine metabolism: Role in neuropsychiatric symptomsBiol Psychiatry70: 175182 |
[13] | Capuron L(2009) Vitamin E status and quality of life in the elderly: Influence of inflammatory processesBr J Nutr102: 13901394 |
[14] | Kreisl WC(2013) A genetic polymorphism for translocator protein 18 kDa affects both in vitro and in vivo radioligand binding in human brain to this putative biomarker of neuroinflammationJ Cereb Blood Flow Metab33: 5358 |
[15] | Bettcher BM(2012) C-reactive protein is related to memory and medial temporal brain volume in older adultsBrain Behav Immun26: 103108 |
[16] | Plourde M, Cunnane SC(2007) Extremely limited synthesis of long chain polyunsaturates in adults: Implications for their dietary essentiality and use as supplementsAppl Physiol Nutr Metab32: 619634 |
[17] | Mizota T(2009) Effect of dietary fatty acid composition on Th1/Th2 polarization in lymphocytesJPEN J Parenter Enteral Nutr33: 390396 |
[18] | Gopinath R, Yelliboina S, Singh M, Prasad VB(2013) Impact of supplementing preoperative intravenous omega 3 Fatty acids in fish oil on immunomodulation in elderly patients undergoing hip surgeryIndian J Surg75: 478484 |
[19] | Sierra S, Lara-Villoslada F, Comalada M, Olivares M, Xaus J(2006) Dietary fish oil n-3 fatty acids increase regulatory cytokine production and exert anti-inflammatory effects in two murine models of inflammationLipids41: 11151125 |
[20] | Lalancette-Hebert M(2011) Accumulation of dietary docosahexaenoic acid in the brain attenuates acute immune response and development of postischemic neuronal damageStroke42: 29032909 |
[21] | Hong S(2007) Resolvin D1, protectin D1, and related docosahexaenoic acid-derived products: Analysis via electrospray/low energy tandem mass spectrometry based on spectra and fragmentation mechanismsJ Am Soc Mass Spectrom18: 128144 |
[22] | Hong S, Lu Y(2013) Omega-3 fatty acid-derived resolvins and protectins in inflammation resolution and leukocyte functions: Targeting novel lipid mediator pathways in mitigation of acute kidney injuryFront Immunol4: 13 |
[23] | Bazan NG(2009) Neuroprotectin D1-mediated anti-inflammatory and survival signaling in stroke, retinal degenerations, and Alzheimer’s diseaseJ Lipid Res50: SupplS400S405 |
[24] | Bazan NG, Calandria JM, Serhan CN(2010) Rescue and repair during photoreceptor cell renewal mediated by docosahexaenoic acid-derived neuroprotectin D1J Lipid Res51: 20182031 |
[25] | Bannenberg G, Serhan CN(1801) Specialized pro-resolving lipid mediators in the inflammatory response: An updateBiochim Biophys Acta12601273 |
[26] | Dalli J(2013) Resolvin D3 and aspirin-triggered resolvin D3 are potent immunoresolventsChem Biol20: 188201 |
[27] | Calder PC(2008) Polyunsaturated fatty acids, inflammatory processes and inflammatory bowel diseasesMol Nutr Food Res52: 885897 |
[28] | Calder PC(2013) Omega-3 polyunsaturated fatty acids and inflammatory processes: Nutrition or pharmacology?Br J Clin Pharmacol75: 645662 |
[29] | Bowen RA, Clandinin MT(2005) Maternal dietary 6n-3 is more effective than 3n-3 in increasing the 6n-3 content in phospholipids of glial cells from neonatal rat brainBr J Nutr93: 601611 |
[30] | Destaillats F(2010) Differential effect of maternal diet supplementation with alpha-Linolenic adcid or n-3 long-chain polyunsaturated fatty acids on glial cell phosphatidylethanolamine and phosphatidylserine fatty acid profile in neonate rat brainsNutr Metab (Lond)7: 2 |
[31] | Labrousse VF(2012) Short-term long chain omega3 diet protects from neuroinflammatory processes and memory impairment in aged micePLoS Onee7: 36861 |
[32] | Simopoulos AP(2001) n-3 fatty acids and human health: Defining strategies for public policyLipids36: SupplS83S99 |
[33] | Simopoulos AP(2011) Evolutionary aspects of diet: The omega-6/omega-3 ratio and the brainMol Neurobiol44: 203215 |
[34] | Glass CK, Saijo K, Winner B, Marchetto MC, Gage FH(2010) Mechanisms underlying inflammation in neurodegenerationCell140: 918934 |
[35] | Town T, Nikolic V, Tan J(2005) The microglial “activation” continuum: From innate to adaptive responsesJ Neuroinflammation2: 24 |
[36] | Biber K, Neumann H, Inoue K, Boddeke HW(2007) Neuronal ‘On’ and ‘Off’ signals control microgliaTrends Neurosci 30: 596602 |
[37] | Alliot F, Godin I, Pessac B(1999) Microglia derive from progenitors, originating from the yolk sac, and which proliferate in the brainBrain Res Dev Brain Res117: 145152 |
[38] | Herbomel P, Thisse B, Thisse C(2001) Zebrafish early macrophages colonize cephalic mesenchyme and developing brain, retina, and epidermis through a M-CSF receptor-dependent invasive processDev Biol238: 27488 |
[39] | Ginhoux F(2010) Fate mapping analysis reveals that adult microglia derive from primitive macrophagesScience 330: 841845 |
[40] | Kierdorf K, Katzmarski N, Haas CA, Prinz M(2013) Bone marrow cell recruitment to the brain in the absence of irradiation or parabiosis biasPLoS Onee8: 58544 |
[41] | Saijo K, Glass CK(2011) Microglial cell origin and phenotypes in health and diseaseNat Rev Immunol11: 775787 |
[42] | Prinz M, Priller J(2014) Microglia and brain macrophages in the molecular age: From origin to neuropsychiatric diseaseNat Rev Neurosci15: 300312 |
[43] | Rezaie P, Male D(2002) Mesoglia & microglia–a historical review of the concept of mononuclear phagocytes within the central nervous systemJ Hist Neurosci11: 325374 |
[44] | Schulz C(2012) A lineage of myeloid cells independent of Myb and hematopoietic stem cellsScience336: 8690 |
[45] | Gautier EL(2012) Gene-expression profiles and transcriptional regulatory pathways that underlie the identity and diversity of mouse tissue macrophagesNat Immunol 13: 11181128 |
[46] | Butovsky O(2014) Identification of a unique TGF-beta-dependent molecular and functional signature in microgliaNat Neurosci17: 131143 |
[47] | Hanisch UK, Kettenmann H(2007) Microglia: Active sensor and versatile effector cells in the normal and pathologic brainNat Neurosci10: 13871394 |
[48] | Neumann J(2008) Microglia cells protect neurons by direct engulfment of invading neutrophil granulocytes: A new mechanism of CNS immune privilegeJ Neurosci28: 59655975 |
[49] | Neher JJ(2011) Inhibition of microglial phagocytosis is sufficient to prevent inflammatory neuronal deathJ Immunol186: 49734983 |
[50] | McArthur S(2010) Annexin A A central player in the anti-inflammatory and neuroprotective role of microgliaJ Immunol185: 63176328 |
[51] | Brown GC, Neher JJ(2014) Microglial phagocytosis of live neuronsNat Rev Neurosci15: 209216 |
[52] | Beutler B (2004) Inferences, questions and possibilities in Toll-like receptor signallingNature430: 257263 |
[53] | Mariathasan S, Monack DM(2007) Inflammasome adaptors and sensors: Intracellular regulators of infection and inflammationNat Rev Immunol7: 3140 |
[54] | Drouin-Ouellet J, Cicchetti F(2012) Inflammation and neurodegeneration: The story ‘retolled’Trends Pharmacol Sci33: 542551 |
[55] | Zhang X, Mosser DM(2008) Macrophage activation by endogenous danger signalsJ Pathol214: 161178 |
[56] | Dantzer R, O’Connor JC, Freund GG, Johnson RW, Kelley KW(2008) From inflammation to sickness and depression: When the immune system subjugates the brainNat Rev Neurosci9: 4656 |
[57] | Lalancette-Hebert M, Gowing G, Simard A, Weng YC, Kriz J(2007) Selective ablation of proliferating microglial cells exacerbates ischemic injury in the brainJ Neurosci27: 25962605 |
[58] | Ransohoff RM, Perry VH(2009) Microglial physiology: Unique stimuli, specialized responsesAnnu Rev Immunol 27: 119145 |
[59] | Cherry JD, Olschowka JA, O’Banion MK(2014) Are “resting” microglia more “m2”? Front Immunol5: 594 |
[60] | David S, Kroner A(2011) Repertoire of microglial and macrophage responses after spinal cord injuryNat Rev Neurosci 12: 388399 |
[61] | Zhan Y(2014) Deficient neuron-microglia signaling results in impaired functional brain connectivity and social behaviorNat Neurosci17: 400406 |
[62] | Neumann H(2001) Control of glial immune function by neuronsGlia36: 191199 |
[63] | Streit WJ(2006) Microglial senescence: Does the brain’s immune system have an expiration date?Trends Neurosci29: 506510 |
[64] | Cox FF, Carney D, Miller AM, Lynch MA(2012) CD200 fusion protein decreases microglial activation in the hippocampus of aged ratsBrain Behav Immun26: 789796 |
[65] | Streit WJ, Xue QS(2014) Human CNS immune senescence and neurodegenerationCurr Opin Immunol29C: 9396 |
[66] | Franceschi C(2000) Inflamm-aging. An evolutionary perspective on immunosenescenceAnn N Y Acad Sci 908: 244254 |
[67] | Dilger RN, Johnson RW(2008) Aging, microglial cell priming, and the discordant central inflammatory response to signals from the peripheral immune systemJ Leukoc Biol84: 932939 |
[68] | Tremblay ME(2011) The role of microglia in the healthy brainJ Neurosci31: 160641609 |
[69] | Griffin R(2006) The age-related attenuation in long-term potentiation is associated with microglial activationJ Neurochem99: 12631272 |
[70] | Hickman SE(2013) The microglial sensome revealed by direct RNA sequencingNat Neurosci16: 18961905 |
[71] | Heneka MT, O’Banion MK, Terwel D, Kummer MP(2010) Neuroinflammatory processes in Alzheimer’s diseaseJ Neural Transm117: 919947 |
[72] | Wyss-Coray T, Rogers J(2012) Inflammation in Alzheimer disease-a brief review of the basic science and clinical literatureCold Spring Harb Perspect Meda2: 006346 |
[73] | Perry VH, Holmes C(2014) Microglial priming in neurodegenerative diseaseNat Rev Neurol10: 217224 |
[74] | Perry VH, Cunningham C, Holmes C(2007) Systemic infections and inflammation affect chronic neurodegenerationNat Rev Immunol7: 161167 |
[75] | Cunningham C, Wilcockson DC, Campion S, Lunnon K, Perry VH(2005) Central and systemic endotoxin challenges exacerbate the local inflammatory response and increase neuronal death during chronic neurodegenerationJ Neurosci25: 92759284 |
[76] | Norden DM, Muccigrosso MM, Godbout JP(2014) Microglial Priming and Enhanced Reactivity to Secondary Insult in Aging, and Traumatic CNS injury, and Neurodegenerative DiseaseNeuropharmacology10.028, Epub ahead of print |
[77] | Cunningham C(2009) Systemic inflammation induces acute behavioral and cognitive changes and accelerates neurodegenerative diseaseBiol Psychiatry65: 304312 |
[78] | Field R, Campion S, Warren C, Murray C, Cunningham C(2010) Systemic challenge with the TLR3 agonist poly I:C induces amplified IFNalpha/beta and IL-1beta responses in the diseased brain and exacerbates chronic neurodegenerationBrain Behav Immun24: 9961007 |
[79] | Cunningham C(2013) Microglia and neurodegeneration: The role of systemic inflammationGlia61: 7190 |
[80] | Ray S(2007) Classification and prediction of clinical Alzheimer’s diagnosis based on plasma signaling proteinsNat Med13: 13591362 |
[81] | Galimberti D(2008) Intrathecal levels of IL-6, IL-11 and LIF in Alzheimer’s disease and frontotemporal lobar degenerationJ Neurol255: 539544(2008) |
[82] | Bennett S(2012) Plasma levels of complement 4a protein are increased in Alzheimer’s diseaseAlzheimer Dis Assoc Disord26: 329334 |
[83] | Thambisetty M(2011) Plasma biomarkers of brain atrophy in Alzheimer’s diseasePLoS Onee6: 28527 |
[84] | Sokolova A(2009) Monocyte chemoattractant protein-1 plays a dominant role in the chronic inflammation observed in Alzheimer’s diseaseBrain Pathol19: 392398 |
[85] | Edison P(2008) Microglia, amyloid, and cognition in Alzheimer’s disease: An [11C](R)PK1115-PET and [11C]PIB-PET studyNeurobiol Dis32: 412419 |
[86] | int’ Veld BA(2001) Nonsteroidal antiinflammatory drugs and the risk of Alzheimer’s diseaseN Engl J Med345: 15151521 |
[87] | McGeer PL, Rogers J, McGeer EG(2006) Inflammation, anti-inflammatory agents and Alzheimer disease: The last 12 yearsJ Alzheimers Dis9: 271276 |
[88] | Schwab C, Klegeris A, McGeer PL(1802) Inflammation in transgenic mouse models of neurodegenerative disordersBiochim Biophys Acta889902 |
[89] | Akiyama H(2000) Inflammation and Alzheimer’s diseaseNeurobiol Aging21: 383421 |
[90] | Quinn J(2003) Inflammation and cerebral amyloidosis are disconnected in an animal model of Alzheimer’s diseaseJ Neuroimmunol137: 3241 |
[91] | Janelsins MC(2005) Early correlation of microglial activation with enhanced tumor necrosis factor-alpha and monocyte chemoattractant protein-1 expression specifically within the entorhinal cortex of triple transgenic Alzheimer’s disease miceJ Neuroinflammation2: 23 |
[92] | Kitazawa M, Oddo S, Yamasaki TR, Green KN, LaFerla FM(2005) Lipopolysaccharide-induced inflammation exacerbates tau pathology by a cyclin-dependent kinase 5-mediated pathway in a transgenic model of Alzheimer’s diseaseJ Neurosci25: 88438853 |
[93] | Poisnel G(2012) PET imaging with [18F]AV-45 in an APP/PS1-21 murine model of amyloid plaque depositionNeurobiol Aging33: 25612571 |
[94] | Ferretti MT, Bruno MA, Ducatenzeiler A, Klein WL, Cuello AC(2012) Intracellular Abeta-oligomers and early inflammation in a model of Alzheimer’s diseaseNeurobiol Aging33: 13291342 |
[95] | Boissonneault V(2009) Powerful beneficial effects of macrophage colony-stimulating factor on beta-amyloid deposition and cognitive impairment in Alzheimer’s diseaseBrain132: 10781092 |
[96] | Michaud JP(2013) Toll-like receptor 4 stimulation with the detoxified ligand monophosphoryl lipid A improves Alzheimer’s disease-related pathologyProc Natl Acad Sci U S A110: 19411946 |
[97] | Yang Y-M, Shang D-S, Zhao W-D, Fang W-G, Chen Y-H(2013) Microglial TNF-α-Dependent Elevation of MHC Class I Expression on Brain Endothelium Induced by Amyloid-Beta Promotes T Cell Transendothelial MigrationNeurochemical research38: 22952304 |
[98] | Lee MH(2010) TGF-beta induces TIAF1 self-aggregation via type II receptor-independent signaling that leads to generation of amyloid beta plaques in Alzheimer’s diseaseCell Death Dise1: 110 |
[99] | Giuliani F, Vernay A, Leuba G, Schenk F(2009) Decreased behavioral impairments in an Alzheimer mice model by interfering with TNF-alpha metabolismBrain Res Bull80: 302308 |
[100] | McAlpine FE(2009) Inhibition of soluble TNF signaling in a mouse model of Alzheimer’s disease prevents pre-plaque amyloid-associated neuropathologyNeurobiol Dis34: 163177 |
[101] | Chakrabarty P(2012) Hippocampal expression of murine IL-4 results in exacerbation of amyloid depositionMol Neurodegener7: 36 |
[102] | Krabbe G(2013) Functional impairment of microglia coincides with Beta-amyloid deposition in mice with Alzheimer-like pathologyPLoS Onee8: 60921 |
[103] | Rivest S(2009) Regulation of innate immune responses in the brainNat Rev Immunol9: 429439 |
[104] | Rothwell NJ, Luheshi GN (2000) Trends Neurosci 23: 618 625 Interleukin 1 in the braBiology, pathology and therapeutic target |
[105] | Griffin WS, Sheng JG, Roberts GW, Mrak RE(1995) Interleukin-1 expression in different plaque types in Alzheimer’s disease: Significance in plaque evolutionJ Neuropathol Exp Neurol54: 276281 |
[106] | Apelt J, Schliebs R(2001) Beta-amyloid-induced glial expression of both pro- and anti-inflammatory cytokines in cerebral cortex of aged transgenic Tgmice with Alzheimer plaque pathologyBrain Res894: 2130 |
[107] | Ghosh S(2013) Sustained interleukin-1beta overexpression exacerbates tau pathology despite reduced amyloid burden in an Alzheimer’s mouse modelJ Neurosci33: 50535064 |
[108] | Cho SH(2011) CX3CR1 protein signaling modulates microglial activation and protects against plaque-independent cognitive deficits in a mouse model of Alzheimer diseaseJ Biol Chem286: 3271332722 |
[109] | St-Amour I(2014) IVIg protects the 3xTg-AD mouse model of Alzheimer’s disease from memory deficit and Abeta pathologyJ Neuroinflammation11: 54 |
[110] | Liu Z, Condello C, Schain A, Harb R, Grutzendler J(2010) CX3CR1 in microglia regulates brain amyloid deposition through selective protofibrillar amyloid-beta phagocytosisJ Neurosci30: 1709117101 |
[111] | Lee S(2010) CX3CR1 deficiency alters microglial activation and reduces beta-amyloid deposition in two Alzheimer’s disease mouse modelsAm J Pathol177: 25492562 |
[112] | Fuhrmann M(2010) Microglial Cx3cr1 knockout prevents neuron loss in a mouse model of Alzheimer’s diseaseNat Neurosci13: 411413 |
[113] | Bhaskar K(2010) Regulation of tau pathology by the microglial fractalkine receptorNeuron68: 1931 |
[114] | Hoozemans JJ, Rozemuller AJ, van Haastert ES, Eikelenboom P, van Gool WA(2011) Neuroinflammation in Alzheimer’s disease wanes with ageJ Neuroinflammation8: 171 |
[115] | Querfurth HW, LaFerla FM(2010) Alzheimer’s diseaseN Engl J Med362: 329344 |
[116] | Aisen PS(2000) A randomized controlled trial of prednisone in Alzheimer’s disease. Alzheimer’s Disease Cooperative StudyNeurology54: 588593 |
[117] | Breitner JC(1995) Delayed onset of Alzheimer’s disease with nonsteroidal anti-inflammatory and histamine H2 blocking drugsNeurobiol Aging16: 523530 |
[118] | von Bernhardi R(2010) Immunotherapy in Alzheimer’s disease: Where do we stand? Where should we go?J Alzheimers Dis19: 405421 |
[119] | Butchart J, Holmes C(2012) Systemic and central immunity in Alzheimer’s disease: Therapeutic implicationsCNS Neurosci Ther18: 6476 |
[120] | Cummings JL(2007) Treatment of Alzheimer’s disease: The role of symptomatic agents in an era of disease-modifying therapiesRev Neurol Dis4: 5762 |
[121] | Meinert CL, McCaffrey LD, Breitner JC(2009) Alzheimer’s Disease Anti-inflammatory Prevention Trial: Design, methods, and baseline resultsAlzheimers Dement5: 93104 |
[122] | Breitner JC(2011) Extended results of the Alzheimer’s disease anti-inflammatory prevention trialAlzheimers Dement7: 402411 |
[123] | Group A. D. A. P. T. R.(2013) Results of a follow-up study to the randomized Alzheimer’s Disease Anti-inflammatory Prevention Trial (ADAPT)Alzheimers Dement9: 714723 |
[124] | Cunnane SC(1994) Antioxidants, free radicals and PUFAProstaglandins Leukot Essent Fatty Acids50: 363364 |
[125] | Simopoulos AP(2006) Evolutionary aspects of diet, the omega-6/omega-3 ratio and genetic variation: Nutritional implications for chronic diseasesBiomed Pharmacother60: 502507 |
[126] | Bang HO, Dyerberg J, Sinclair HM(1980) The composition of the Eskimo food in north western GreenlandAm J Clin Nutr33: 26572661 |
[127] | Kromann N, Green A(1980) Epidemiological studies in the Upernavik district, GreenlandIncidence of some chronic diseases -Acta Med Scand208: 401406 |
[128] | Schaebel LH, Vestergaard H, Laurberg P, Rathcke CN, Andersen S(2013) Intake of traditional Inuit diet vary in parallel with inflammation as estimated from YKL-40 and hsCRP in Inuit and non-Inuit in GreenlandAtherosclerosis228: 496501 |
[129] | Orr SK, Trepanier MO, Bazinet RP(2013) n-3 Polyunsaturated fatty acids in animal models with neuroinflammationProstaglandins Leukot Essent Fatty Acids88: 97103 |
[130] | Jump DB(2002) Dietary polyunsaturated fatty acids and regulation of gene transcriptionCurr Opin Lipidol13: 155164 |
[131] | Calder PC(2005) Polyunsaturated fatty acids and inflammationBiochem Soc Trans33: 423427 |
[132] | Chapkin RS, Kim W, Lupton JR, McMurray DN(2009) Dietary docosahexaenoic and eicosapentaenoic acid: Emerging mediators of inflammationProstaglandins Leukot Essent Fatty Acids81: 187191 |
[133] | Willis LM, Shukitt-Hale B, Joseph JA(2009) Modulation of cognition and behavior in aged animals: Role for antioxidant- and essential fatty acid-rich plant foodsAm J Clin Nutr89: 1602S1606S |
[134] | Willis LM, Shukitt-Hale B, Cheng V, Joseph JA(2009) Dose-dependent effects of walnuts on motor and cognitive function in aged ratsBr J Nutr101: 11401144 |
[135] | Carey AN, Fisher DR, Joseph JA, Shukitt-Hale B(2013) The ability of walnut extract and fatty acids to protect against the deleterious effects of oxidative stress and inflammation in hippocampal cellsNutr Neurosci16: 1320 |
[136] | Poulose SM, Bielinski DF, Shukitt-Hale B(2013) Walnut diet reduces accumulation of polyubiquitinated proteins and inflammation in the brain of aged ratsJ Nutr Biochem 24: 912919 |
[137] | De Smedt-Peyrusse V(2008) Docosahexaenoic acid prevents lipopolysaccharide-induced cytokine production in microglial cells by inhibiting lipopolysaccharide receptor presentation but not its membrane subdomain localizationJ Neurochem105: 296307 |
[138] | Mingam R(2008) Uncoupling of interleukin-6 from its signalling pathway by dietary n-3-polyunsaturated fatty acid deprivation alters sickness behaviour in miceEur J Neurosci28: 18771886 |
[139] | Marcheselli VL(2003) Novel docosanoids inhibit brain ischemia-reperfusion-mediated leukocyte infiltration and pro-inflammatory gene expressionJ Biol Chem278: 4380743817 |
[140] | Huang WL(2007) A combination of intravenous and dietary docosahexaenoic acid significantly improves outcome after spinal cord injuryBrain130: 30043019 |
[141] | Julien C(2010) High-fat diet aggravates amyloid-beta and tau pathologies in the 3xTg-AD mouse modelNeurobiol Aging31: 15161531 |
[142] | McNamara RK(2010) Omega-3 fatty acid deficiency increases constitutive pro-inflammatory cytokine production in rats: Relationship with central serotonin turnoverProstaglandins Leukot Essent Fatty Acids83: 185191 |
[143] | Curtis CL(2000) n-3 fatty acids specifically modulate catabolic factors involved in articular cartilage degradationJ Biol Chem275: 721724 |
[144] | Michael-Titus AT(2007) Omega-3 fatty acids and neurological injuryProstaglandins Leukot Essent Fatty Acids 77: 295300 |
[145] | Serhan CN, Chiang N, Van Dyke TE(2008) Resolving inflammation: Dual anti-inflammatory and pro-resolution lipid mediatorsNat Rev Immunol8: 349361 |
[146] | Claria J, Gonzalez-Periz A, Lopez-Vicario C, Rius B, Titos E(2011) New insights into the role of macrophages in adipose tissue inflammation and Fatty liver disease: Modulation by endogenous omega-3 Fatty Acid-derived lipid mediatorsFront Immunol2: 49 |
[147] | Titos E(2011) Resolvin D1 and its precursor docosahexaenoic acid promote resolution of adipose tissue inflammation by eliciting macrophage polarization toward an M2-like phenotypeJ Immunol187: 54085418 |
[148] | Lukiw WJ, Bazan NG(2008) Docosahexaenoic acid and the aging brainJ Nutr138: 25102514 |
[149] | Calder PC(2007) Immunomodulation by omega-3 fatty acidsProstaglandins Leukot Essent Fatty Acids77: 327335 |
[150] | Carrie I, Guesnet P, Bourre JM, Frances H(2000) Diets containing long-chain n-3 polyunsaturated fatty acids affect behaviour differently during development than ageing in miceBr J Nutr83: 439447 |
[151] | Connor WE, Neuringer M, Lin DS(1990) Dietary effects on brain fatty acid composition: The reversibility of n-3 fatty acid deficiency and turnover of docosahexaenoic acid in the brain, erythrocytes, and plasma of rhesus monkeysJ Lipid Res31: 237247 |
[152] | Fedorova I, Salem NJ(2006) Omega-3 fatty acids and rodent behaviorProstaglandins Leukot Essent Fatty Acids75: 271289 |
[153] | Lafourcade M(2011) Nutritional omega-3 deficiency abolishes endocannabinoid-mediated neuronal functionsNat Neurosci14: 345350 |
[154] | Larrieu T, Madore C, Joffre C, Laye S(2012) Nutritional n-3 polyunsaturated fatty acids deficiency alters cannabinoid receptor signaling pathway in the brain and associated anxiety-like behavior in miceJ Physiol Biochem68: 671681 |
[155] | Moranis A(2012) Long term adequate n-3 polyunsaturated fatty acid diet protects from depressive-like behavior but not from working memory disruption and brain cytokine expression in aged miceBrain Behav Immun26: 721731 |
[156] | Gamoh S, Hashimoto M, Hossain S, Masumura S(2001) Chronic administration of docosahexaenoic acid improves the performance of radial arm maze task in aged ratsClin Exp Pharmacol Physiol28: 266270 |
[157] | Carrie I(2002) Docosahexaenoic acid-rich phospholipid supplementation: Effect on behavior, learning ability, and retinal function in control and n-3 polyunsaturated fatty acid deficient old miceNutr Neurosci5: 4352 |
[158] | Gamoh S(1999) Chronic administration of docosahexaenoic acid improves reference memory-related learning ability in young ratsNeuroscience93: 237241 |
[159] | Yehuda S, Rabinovitz S, Mostofsky DI(1999) Treatment with a polyunsaturated fatty acid prevents deleterious effects of Ro4-Eur J Pharmacol365: 2734 |
[160] | Arsenault D, Julien C, Tremblay C, Calon F(2011) DHA improves cognition and prevents dysfunction of entorhinal cortex neurons in 3xTg-AD micePLoS Onee6: 17397 |
[161] | Lopez GH, Ilincheta de Boschero MG, Castagnet PI, Giusto NM(1995) Age-associated changes in the content and fatty acid composition of brain glycerophospholipidsComp Biochem Physiol B Biochem Mol Biol112: 331343 |
[162] | Favreliere S(2003) DHA-enriched phospholipid diets modulate age-related alterations in rat hippocampusNeurobiol Aging24: 233243 |
[163] | Little SJ, Lynch MA, Manku M, Nicolaou A(2007) Docosahexaenoic acid-induced changes in phospholipids in cortex of young and aged rats: A lipidomic analysisProstaglandins Leukot Essent Fatty Acids77: 155162 |
[164] | McNamara RK, Liu Y, Jandacek R, Rider T, Tso P(2008) The aging human orbitofrontal cortex: Decreasing polyunsaturated fatty acid composition and associated increases in lipogenic gene expression and stearoyl-CoA desaturase activityProstaglandins Leukot Essent Fatty Acids78: 293304 |
[165] | Petursdottir AL, Farr SA, Morley JE, Banks WA, Skuladottir GV(2007) Lipid peroxidation in brain during aging in the senescence-accelerated mouse (SAM)Neurobiol Aging28: 11701178 |
[166] | Petursdottir AL, Farr SA, Morley JE, Banks WA, Skuladottir GV(2008) Effect of dietary n-3 polyunsaturated fatty acids on brain lipid fatty acid composition, learning ability, and memory of senescence-accelerated mouseJ Gerontol A Biol Sci Med Sci63: 11531160 |
[167] | Bourre JM, Piciotti M(1992) Delta-6 desaturation of alpha-linolenic acid in brain and liver during development and aging in the mouseNeurosci Lett141: 6568 |
[168] | Kumar VB(1999) Changes in membrane fatty acids and delta-9 desaturase in senescence accelerated (SAMP8) mouse hippocampus with agingLife Sci65: 16571662 |
[169] | Ilincheta de Boschero MG, Roque ME, Salvador GA, Giusto NM(2000) Alternative pathways for phospholipid synthesis in different brain areas during agingExp Gerontol35: 653668 |
[170] | McGahon BM, Martin DS, Horrobin DF, Lynch MA(1999) Age-related changes in synaptic function: Analysis of the effect of dietary supplementation with omega-3 fatty acidsNeuroscience94: 305314 |
[171] | Song C, Leonard BE, Horrobin DF(2004) Dietary ethyl-eicosapentaenoic acid but not soybean oil reverses central interleukin-1-induced changes in behavior, corticosterone and immune response in ratsStress7: 4354 |
[172] | Song C, Manku MS, Horrobin DF(2008) Long-chain polyunsaturated fatty acids modulate interleukin-1beta-induced changes in behavior, monoaminergic neurotransmitters, and brain inflammation in ratsJ Nutr138: 954963 |
[173] | Minogue AM, Lynch AM, Loane DJ, Herron CE, Lynch MA(2007) Modulation of amyloid-beta-induced and age-associated changes in rat hippocampus by eicosapentaenoic acidJ Neurochem103: 914926 |
[174] | Godbout JP(2005) Exaggerated neuroinflammation and sickness behavior in aged mice following activation of the peripheral innate immune systemFASEB J19: 13291331 |
[175] | Lynch AM(2007) Eicosapentaenoic acid confers neuroprotection in the amyloid-beta challenged aged hippocampusNeurobiol Aging28: 845855 |
[176] | Martin DS, Spencer P, Horrobin DF, Lynch MA(2002) Long-term potentiation in aged rats is restored when the age-related decrease in polyunsaturated fatty acid concentration is reversedProstaglandins Leukot Essent Fatty Acids67: 121130 |
[177] | Farzaneh-Far R, Harris WS, Garg S, Na B, Whooley MA(2009) Inverse association of erythrocyte n-3 fatty acid levels with inflammatory biomarkers in patients with stable coronary artery disease: The Heart and Soul StudyAtherosclerosis205: 538543 |
[178] | Ferrucci L(2006) Relationship of plasma polyunsaturated fatty acids to circulating inflammatory markersJ Clin Endocrinol Metab91: 439446 |
[179] | Kalogeropoulos N(2010) Unsaturated fatty acids are inversely associated and n-6/n-3 ratios are positively related to inflammation and coagulation markers in plasma of apparently healthy adultsClin Chim Acta411: 584591 |
[180] | Kiecolt-Glaser JK(2007) Depressive symptoms, omega-omega-3 fatty acids, and inflammation in older adultsPsychosom Med69: 217224 |
[181] | Kiecolt-Glaser JK(2013) Omega-3 fatty acids, oxidative stress, and leukocyte telomere length: A randomized controlled trialBrain Behav Immun28: 1624 |
[182] | Meydani SN(1994) Oral (n-3) fatty acid supplementation suppresses cytokine production and lymphocyte proliferation: Comparison between young and older womenJ Nutr 1121: 547555 |
[183] | Rees D(2006) Dose-related effects of eicosapentaenoic acid on innate immune function in healthy humans: A comparison of young and older menAm J Clin Nutr83: 331342 |
[184] | Samieri C(2011) omega-3 fatty acids and cognitive decline: Modulation by ApoEepsilon4 allele and depressionNeurobiol Aging32: 2317e13-2317.e22. |
[185] | Feart C(2008) Plasma eicosapentaenoic acid is inversely associated with severity of depressive symptomatology in the elderly: Data from the Bordeaux sample of the Three-City StudyAm J Clin Nutr87: 11561162 |
[186] | Feart C(2011) Adherence to a Mediterranean diet and plasma fatty acids: Data from the Bordeaux sample of the Three-City studyBr J Nutr106: 149158 |
[187] | Lim GP(2005) A diet enriched with the omega-3 fatty acid docosahexaenoic acid reduces amyloid burden in an aged Alzheimer mouse modelJ Neurosci25: 30323040 |
[188] | Calon F(2004) Docosahexaenoic acid protects from dendritic pathology in an Alzheimer’s disease mouse modelNeuron43: 633645 |
[189] | Calon F(2005) Dietary n-3 polyunsaturated fatty acid depletion activates caspases and decreases NMDA receptors in the brain of a transgenic mouse model of Alzheimer’s diseaseEur J Neurosci22: 617626 |
[190] | Calon F, Cole G(2007) Neuroprotective action of omega-3 polyunsaturated fatty acids against neurodegenerative diseases: Evidence from animal studiesProstaglandins Leukot Essent Fatty Acids77: 287293 |
[191] | Mizwicki MT(2013) 1alpha,25-dihydroxyvitamin D3 and resolvin D1 retune the balance between amyloid-beta phagocytosis and inflammation in Alzheimer’s disease patientsJ Alzheimers Dis34: 155170 |
[192] | Zhao Y(2011) Docosahexaenoic acid-derived neuroprotectin D1 induces neuronal survival via secretase- and PPARgamma-mediated mechanisms in Alzheimer’s disease modelsPLoS Onee6: 15816 |
[193] | Salem NJ, Vandal M, Calon F(2015) The benefit of docosahexaenoic acid for the adult brain in aging and dementiaProstaglandins Leukot Essent Fatty Acids92: 1522 |
[194] | Frautschy SA, Cole GM(2011) What was lost in translation in the DHA trial is whom you should intend to treatAlzheimers Res Ther3: 2 |
[195] | Quinn JF(2010) Docosahexaenoic acid supplementation and cognitive decline in Alzheimer disease: A randomized trialJAMA304: 19031911 |
[196] | Barberger-Gateau P, Samieri C, Feart C, Plourde M(2011) Dietary omega 3 polyunsaturated fatty acids and Alzheimer’s disease: Interaction with apolipoprotein E genotypeCurr Alzheimer Res8: 479491 |
[197] | Vandal M(2014) Reduction in DHA transport to the brain of mice expressing human APOE4 compared to APOE2J Neurochem129: 516526 |
[198] | Egensperger R, Kosel S, von Eitzen U, Graeber MB(1998) Microglial activation in Alzheimer disease: Association with APOE genotypeBrain Pathol8: 439447 |
[199] | Li X, Montine KS, Keene CD, Montine TJ(2015) Different mechanisms of apolipoprotein E isoform-dependent modulation of prostaglandin E2 production and triggering receptor expressed on myeloid cells 2 (TREM2) expression after innate immune activation of microgliaFASEB J14-262683Epub ahead of print |
Figures and Tables
Fig.1
Microglia phenotype plasticity. Microglia can adopt different phenotypes: M1 (classical activation), M2a (alternative activation), M2b (immunoregulatory) and M2c (acquired-deactivation). According to their phenotype, microglia cells express different clusters of differentiation (CD) such as CD86 or CD206, or type-II proteins of major histocompatibility complex (MHC) and secrete different cytokines and chemokines. CCL: chemokine (C-C motif) ligand; IFN: interferon; IL: interleukine; LPS: lipopolysaccharide; TGF: transforming growth factor; TNF: tumor necrosis factor; Ym1: YKL-40, chitinase 3-like 3.
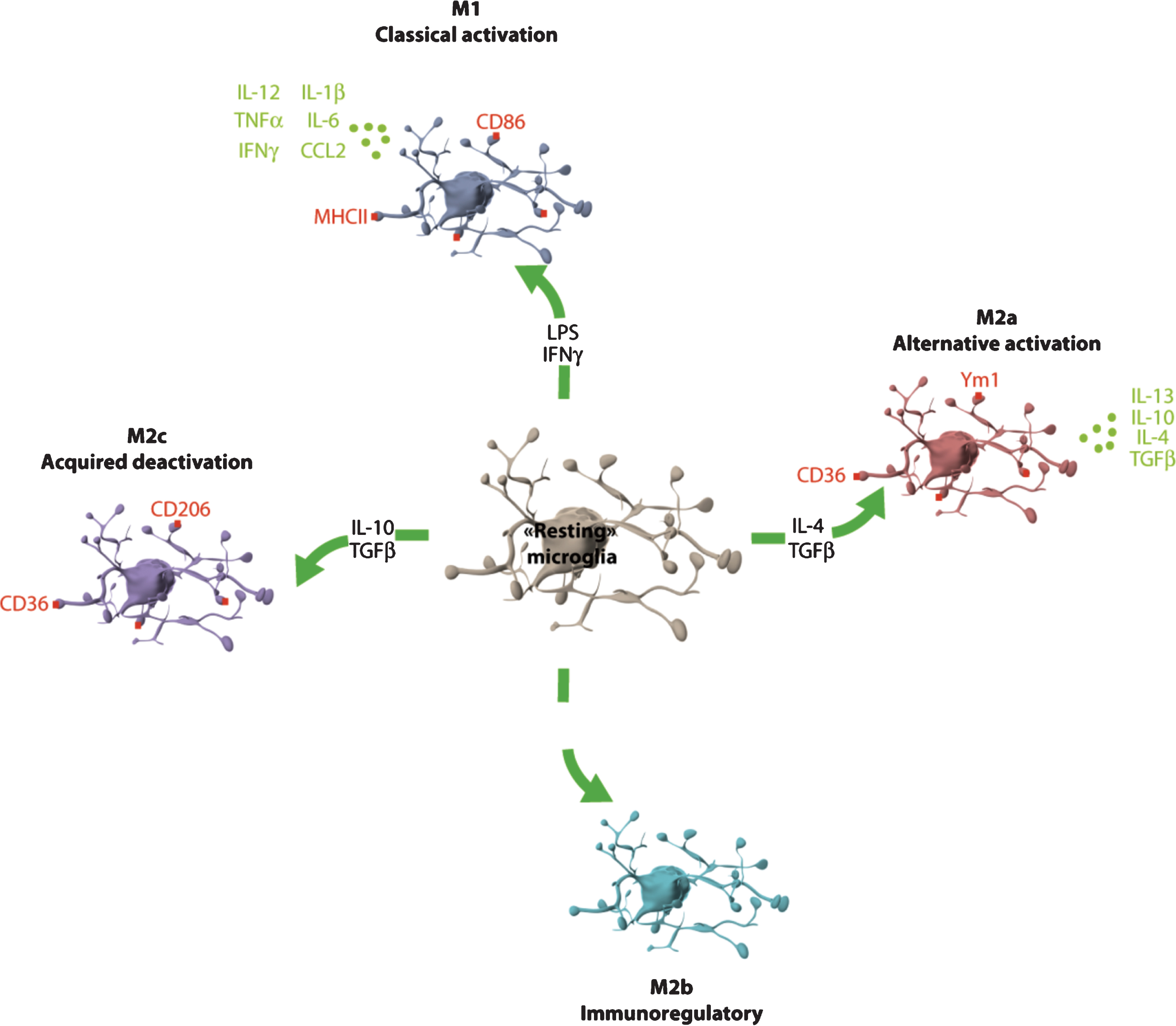
Fig.2
Elongation and desaturation of n-6 and n-3 PUFA and pathways in active eicosanoid metabolism from arachidonic acid, eicosapentaenoic acid and docosahexaenoic acid. n-6 and n-3 essential fatty acids precursors are linoleic acid (LA) and α-linolenic acid (ALA). These precursors are metabolized into arachidonic acid (AA) and eicosapentaenoic (EPA) and docosahexaenoic acid (DHA) respectively. AA is metabolized into derivatives that belong to the eicosanoid family, series 2 and 4. EPA and DHA metabolic derivatives belong to the eicosanoid family, series 3 and 5, resolving family (series D and E) and neuroprotectins. Tx: thromboxane; PG: prostaglandines; COX-2: cycloxygenase 2; LT: leukotrienes; LOX: lipoxygenase; Lx: lipoxin; NPD1: neuroprotectin D1.
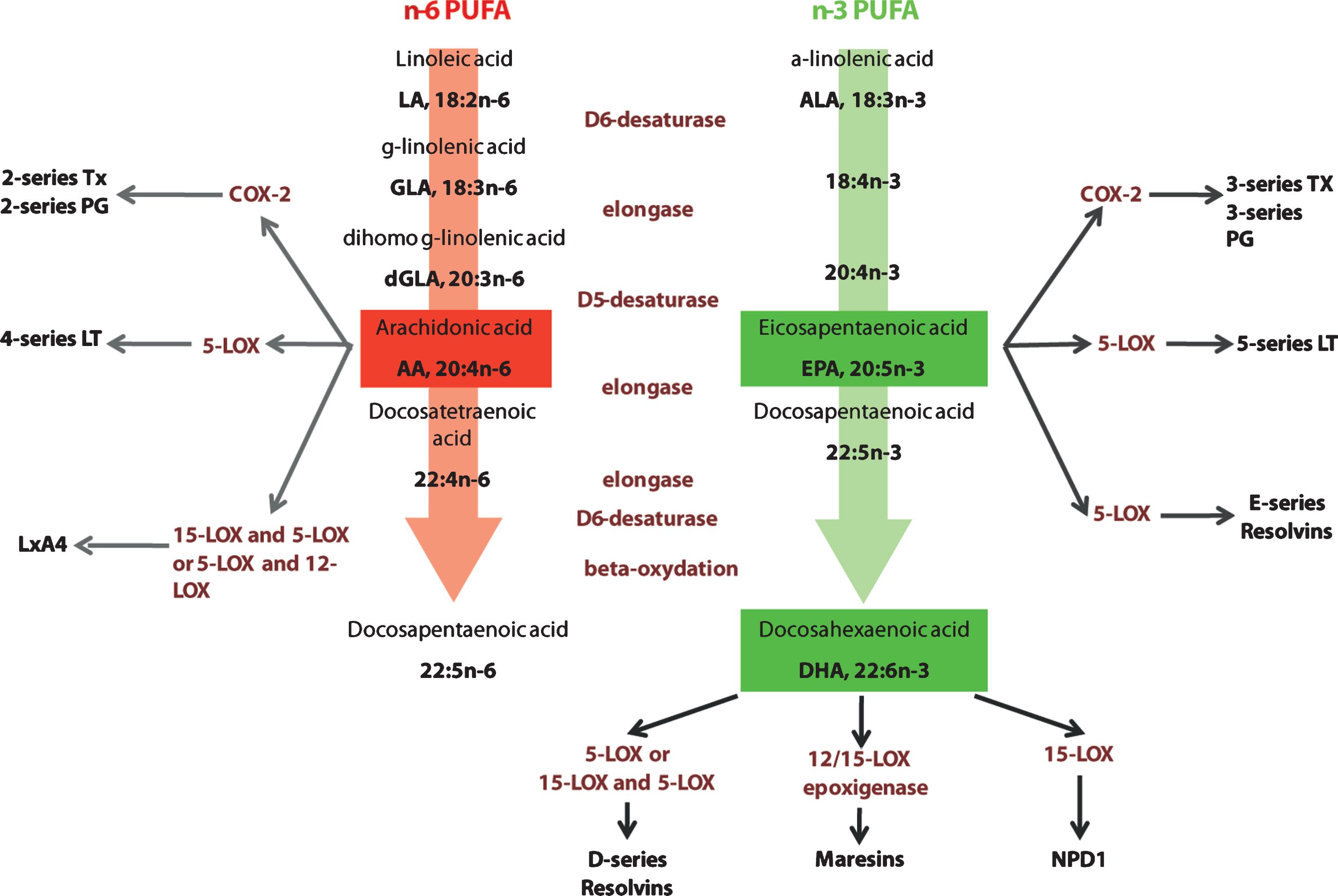
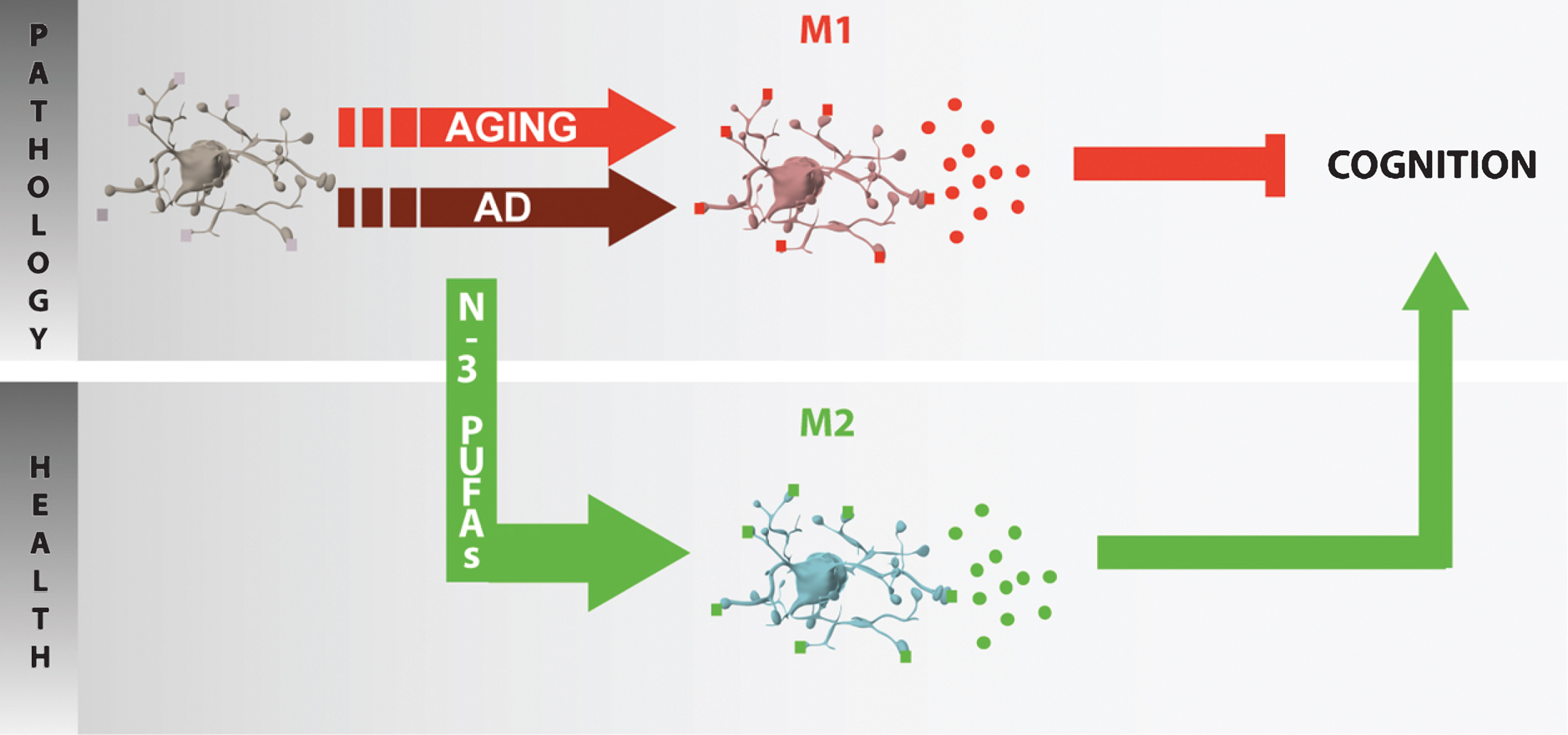
Fig.3
Potential role of n-3 PUFA in inflammaging. In the aged or diseased brain (AD neuropathology), microglia are primed and polarized into various phenotypes (e.g. M1 in aging) and secrete pro-inflammatory cytokines that could play a role in cognitive impairment. The protective effect of n-3 PUFAs toward cognitive deficit in aging or neurodegeneration could be linked to the promotion of an anti-inflammatory M2 phenotype.