
Quantum processing of cytidine derivatives and evaluating their in silico interactions with the COVID-19 main protease
Abstract
This work was performed by the importance of exploring possible medications for COVID-19 pandemic. In this regard, cytidine (Cyd) derivatives were investigated to reach a point to see their benefit of employing for the purpose. Each of halogenated models of Cyd including CydF, CydCl, CydBr, and CydI were investigated in addition to the original CydH model. Density functional theory (DFT) based quantum processing were performed to obtain stabilized structures in addition to evaluation of frontier molecular orbitals features. Next, molecular docking (MD) simulations were performed to reach a point of formations of interacting ligand-target complexes. Among the investigated models CydH and CydI were working better than other model for reaching the purpose of this work, in which the derived CydI model was indeed the ligand with the highest suitability for formation of ligand-target complexes. As a consequence, such ligands of original and halogenated Cyd models might work for inhibition of main protease (MPro) enzyme of COVID-19 based on the obtained meaningful vales for complex strengths in addition interacting with the amino acids of active site. More precisely, the CydI model could be proposed as promising ligand for showing the inhibitory effects towards the MPro target of COVID-19.
1Introduction
Since the end of 2019, the world has been shocked and seriously affected by the appearance of coronavirus disease (COVID-19) [1–3]. After about two years, the problem has not been solved yet and so many people all around the world have been infected by this mysterious virus [4–6]. To this point, considerable efforts have been done to innovate pharmaceutical compounds for prevention or medication of infection by COVID-19; however, the results have not been certain yet and investigation are still under developments [7–9]. Moreover, several procedures were employed for detection of such COVID-19 in the infected people [10–12]. Among various medical protocols of vaccination or medication, several attempts have been devoted to find inhibitors for the main protease (MPro) enzyme of coronavirus preventing the infection development [13–15]. To this aim, available drugs have been extensively examined for inhibiting the enzyme activity in addition to developing new pharmaceutical compounds for achieving a possible solution for this serious problem [16–18]. By complexity of performing investigations on biological systems and the COVID-19 related studies, employing the in silico approach could help to provide insightful information for approaching to a brighter point about this really unknown disease [19–21]. The current work was performed to examine potency of cytidine for inhibiting the activity of MPro COVID-19 employing the computer-based in silico approach. In recent years, several computer-based tools have been developed to solve the problems in science and technology, especially those related to the living systems [22–24].
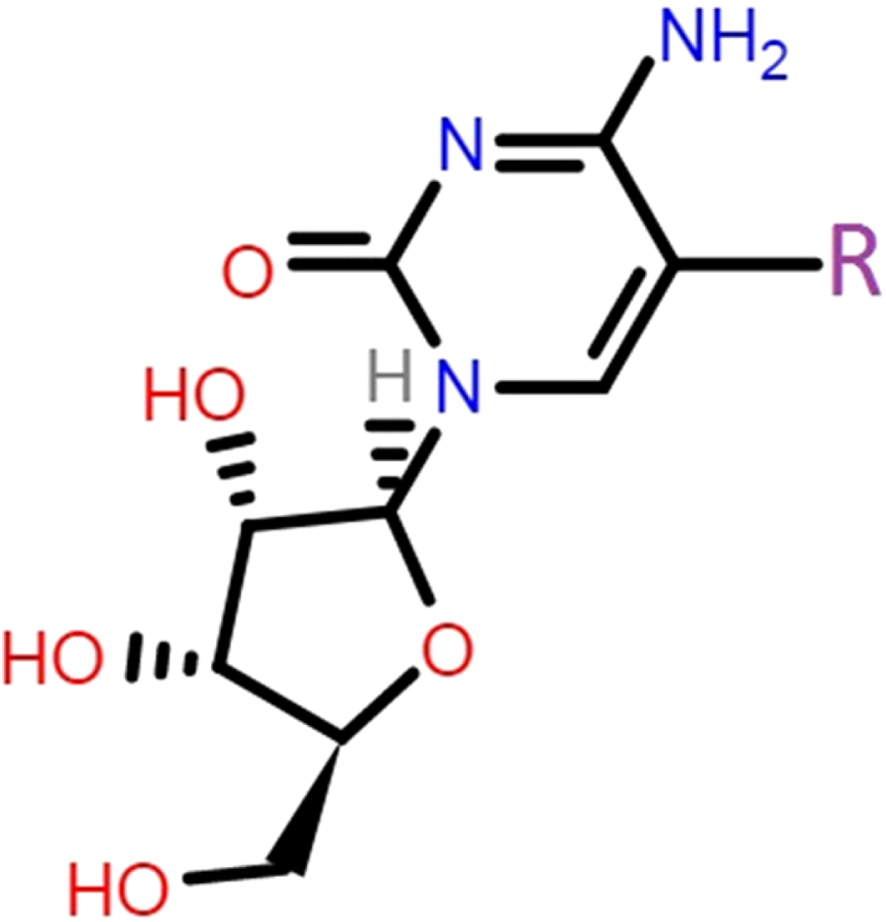
Cytidine (Cyd) is a nucleoside derivative of cytosine nucleobase, which showed several features in medical applications [25–27]. Indeed, considerable attempts have been devoted to explore medical functions of nucleobases derivatives in living systems after the pioneering characterization work of Watson and Crick [28–30]. To this point, Cyd has been seen as a pharmaceutical compound for enzymatic activity inhibitions by earlier works [31–33]. Hence, the hypothesis of MPro inhibition by Cyd was investigated in this work to show potency of this already known compound towards medication of COVID-19 patients. In addition to the original nucleobases, halogenated derivatives have been always important for showing specific functions, in which such halogenated derivatives of Cyd were also examined in this work to approach the purpose. As a consequence, molecular models of Cyd in the role of ligand, and molecular model of MPro in the role of target, were considered in this work for analyzing their corresponding features. To do this, quantum processing of ligands were done employing density functional theory (DFT) approach to obtain theoretically meaningful ligand compounds for participating in interactions with the MPro target. Accordingly, molecular docking (MD) simulations were performed to reach the interacting ligand-target complexes by evaluating the interaction strength in addition to identifying the surrounding amino acids around the centralized ligand. It is important to mention here that existence of hydrogen bond and van der Waals interactions could both help to formation of interacting ligand-target complexes [34]. Table 1 and Fig. 1 represent the ligands specifications and interacting features for the models to examine the hypothesis of MPro inhibition by assistance of the investigated Cyd derivatives. It is worth to mention that the result of current work could provide knowledge about existence of possible inhibitors for the medication of COVID-19 patients.
Table 1
Ligands specifications
 | |||||||
| Ligand | R | HOMO Ev | LUMO eV | EG eV | H eV | S eV–1 | DM Debye |
| CydH | H | –6.36 | –0.99 | 5.37 | 2.68 | 0.37 | 4.88 |
| CydF | F | –6.32 | –1.21 | 5.11 | 2.56 | 0.39 | 3.99 |
| CydCl | Cl | –6.42 | –1.29 | 5.13 | 2.56 | 0.39 | 3.68 |
| CydBr | Br | –6.38 | –1.27 | 5.11 | 2.56 | 0.39 | 3.79 |
| CydI | I | –6.28 | –1.36 | 4.92 | 2.46 | 0.41 | 4.01 |
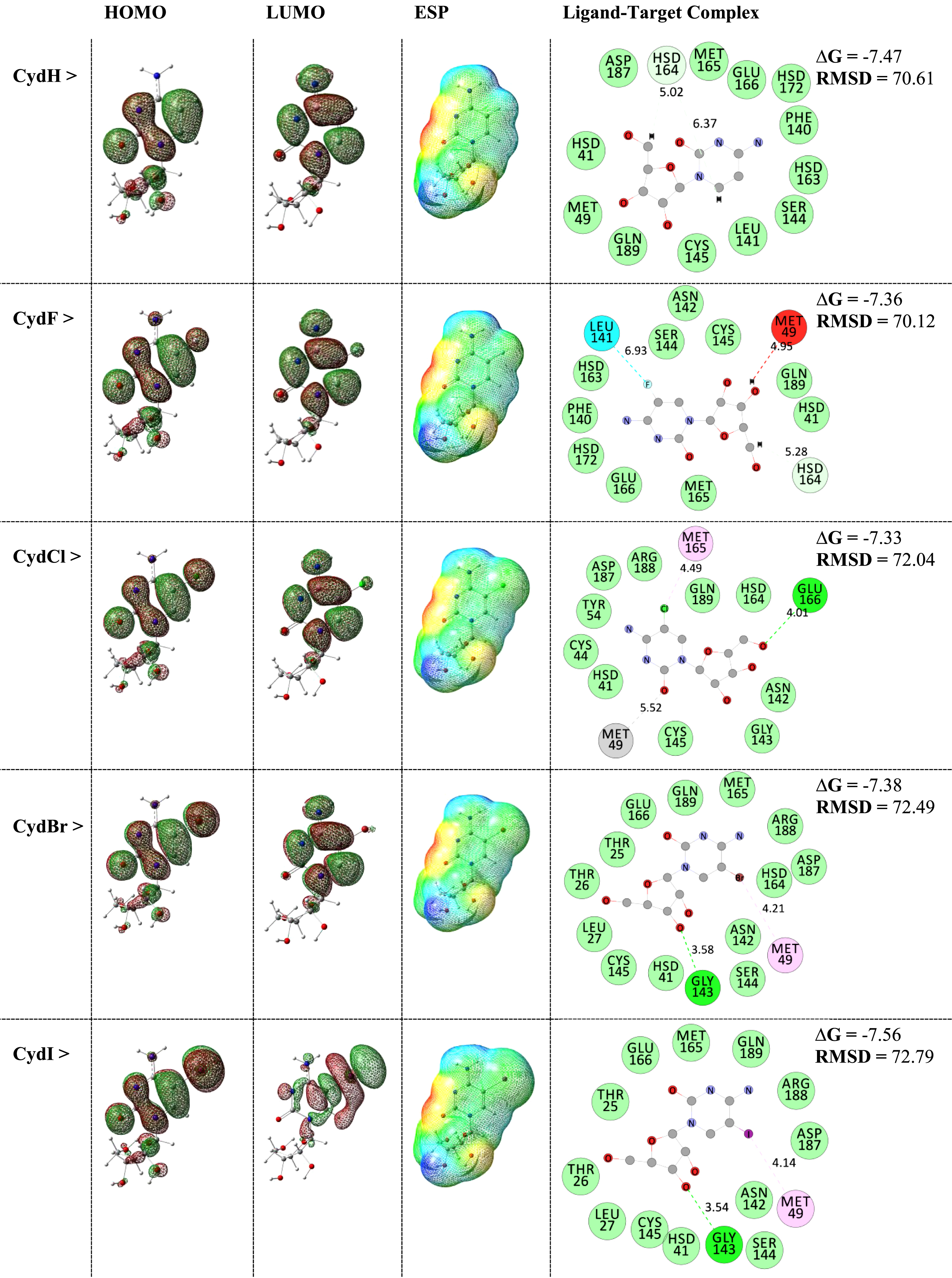
Fig. 1
HOMO-LUMO distribution patterns and ESP surfaces of ligands and interacting ligand-target complexes.
2Materials and methods
Within this work, the original models of Cyd in addition to halogenated derivatives were considered as the ligands, in which their specifications were listed in Table 1. Earlier works indicated that the carbon atom number five of pyrimidine nucleobases is an important atomic site to be functionalized with other substances such as halogen atoms [35]. Accordingly, such benefit was employed in this work to functionalize cytosine counterpart of Cyd by each of F, Cl, Br, and I halogenated atoms. As a consequence, four derivatives were added to the original Cyd to provide ligands of this work including CydH, CydF, CydCl, CydBr, and CydI. Quantum processing were performed to optimize the molecular models of each of ligand structures employing DFT approach as implemented in the Gaussian program [36]. By doing this step, stabilized models with theoretically meaningful geometries were obtained for further investigating their own features in addition to their contribution to interaction with the MPro enzyme target of COVID-19. The 3D macromolecular model of MPro (6lu7) was obtained from the Protein Data Bank (PDB) [37] to be considered for the target model of interacting complex formations with each of Cyd ligands. The accurate MD simulations were performed by submitting each of provided ligand and target structures to the Swiss Dock web server [38] for evaluating interacting ligand-target complexes in a defined 70*70*70 grid box. As a consequence, required features were evaluated for describing each of ligand structures in addition to the obtained interacting ligand-target complexes. Returning to Table 1, frontier molecular orbital quantities were evaluated for the ligands including energy levels of the highest occupied and the lowest unoccupied molecular orbitals (HOMO and LUMO), energy differences of HOMO and LUMO levels in terms of energy gap (EG), chemical hardness and softness (H and S), and dipole moment (DM). Moreover, representations of HOMO-LUMO distribution patterns and electrostatic potential (ESP) surfaces of the ligands were visualized in Fig. 1 besides the evaluated interacting ligand-target complex systems. Values of ΔG (kcal/mol) and RMSD implied for energetic strengths of interactions and conformational fluctuations of ligands from the start point up to the end point of MD simulations. As a consequence, required information were provided for discussing the hypothesis of employing Cyd ligands for inhibition of the MPro of COVID-19.
3Results and discussion
The hypothesis of inhibition of MPro of COVID-19 by assistance of Cyd derivatives was examined in this work employing the in silico approach. To this aim, five models of Cyd derivatives were obtained by halogenation of the atomic site number five of main cytosine group to evaluate CydF, CydCl, CydBr, and CydI derivatives in addition the original CydH compound. As described in Table 1, the models were variated by the attached R group, in which each of F, Cl, Br, and I atoms were substituted instead of the H atom of original Cyd. As a consequence, the models structures were provided to be included in the DFT based quantum processing to approach to the minimized energy structures. Doing this step, five theoretically meaningful ligands were obtained for further analyzing their features besides examining their contributions to interactions with the MPro target. For analyzing the ligand structures, values of HOMO and LUMO were evaluated to show their tendency of participating in electron transferring processes. Indeed, such molecular orbitals features could reactivity of a molecule for contributing to interactions with other substances [39]. HOMO implies for the highest occupied molecular orbital with electrons, in which its occupation means that how a molecule could participate in electron donating process. On the other hand, LUMO implies for the lowest unoccupied molecular orbital without electrons, in which its vacancy means that how a molecule could participate in electron accepting process. In this regard, their energy levels are important because of providing appropriate levels of electron transferring. Moreover, energy differences of HOMO and LUMO of a molecule in terms of energy gap (EG) imply for energy distances for internal electron transitions, as an important feature for electron conductivity processes. These points are important because of the detection role of an inhibitor for detecting the correct target prior to contributing to interactions with it, in which concepts of biosensors could be considered for such purpose. Therefore, analyzing such frontier molecular orbitals features could show insightful information about the characteristic properties of investigated Cyd derivatives.
As could be seen in Table 1, each of HOMO and LUMO levels detected effects of halogenation by changes of values among the models. With the exception of CydCl model, HOMO levels for all other halogenated models moved to higher level in comparison with the original CydH model. However, LUMO levels of all halogenated derivatives were moved to lower levels in comparison with the original CydH model. Among the models, the characteristic HOMO-LUMO achievements were obtained for CydI model with the highest HOMO level and the lowest LUMO level among all halogenated derivatives. Such characteristic structural feature could mean that another characteristic activity could be expected for such ligand model. Indeed, structure-activity relationship (SAR) is an important concept in the field of drug design for innovating structures with promising activity in living systems [40]. As a consequence, structural features could lead to desired activity for approaching the purposes of detection ad medication in living systems. Comparing values of EG shows shorter energy distances for the halogenated Cyd derivatives in comparison with the original CydH model, in which that of CydI was placed at the shortest distance. Accordingly, evaluated values of chemical hardness and softness (H and S) from the obtained HOMO and LUMO levels could show lower hardness and higher softness for CydI model for contributing to reactions/interactions with other substances. Among the models, H and S of CydF, CydCl, and CydBr were almost close to those of CydH; however, CydI was indeed distinguished in the models. Lead optimization is another important concept in drug design meaning how to modify a known structure to find better features regarding the desired purposes [41], in which such concept was explored in the current work by modifying CydH model to reach a more efficient compound as could be seen by the obtained features of CydI model. By the changes of functionalized groups and total structural configurations, values of dipole moment (DM) could show such fluctuations for electric charge distributions of the models. Although an atomic substitution was done for the ligand models, but changes of DM were notable affirming the importance of knowing structural features and their corresponding electric properties to show evaluate appropriate models. As a consequence of performing quantum processing on Cyd ligands, halogenations brought new features for the models with promising roles for the obtained CydI model.
As could be seen in Fig. 1, the evaluated HOMO-LUMO distribution patterns and ESP surfaces by performing quantum processing and the evaluated interacting ligand-target complexes by performing MD simulations were visualized and represented. Within these results, it could be found that the shapes of HOMO-LUMO patterns were almost similar for CydH, CydF, CydCl, and CydBr models but different from the already distinguished CydI model. Moreover, careful comparison of the ESP surfaces could show that the occupied space of was increased by additional of halogen atoms, in which increasing the size of atomic radius of halogen atom leaded to expanding the model surface. Red, yellow, green, light blue, and blue colors of ESP surfaces imply for localizing negative, slightly negative, neutral, slightly positive, and positive charges at the surface. In this case, the models could be defined for participating in interactions with other substances. Returning again to values of HOMO-LUMO, those of CydI model were in characteristic mode with repeating such characteristic feature for distribution patterns. Indeed, different type of localization of HOMO-LUMO might be an advantage of molecule for better contributing to the external interactions, in which such feature could be expected for the CydI model better than the other models. In this regard, the obtained interacting ligand-target complexes could show that such halogenation changed strength of obtained complexes. Based on the obtained ΔG values, strengths of all complex models were meaningful in accordance with the obtained energies showing total advantage of investigating such Cyd derivatives for possible inhibition of the MPro of COVID-19. Moreover, each of CydH and CydI models showed even better features of interactions in comparisons with the other models. The surrounding amino acids of centralized ligand were all in the active site region of enzyme, in which various types of interactions were seen for ligand-target complexes. In addition to existence of conventional hydrogen bond (HB) interactions, non-HB interactions also play important roles for formations of biomolecular complexes as shown for the investigated models. Another impact of halogenation was seen by increasing RMSD for the ligand counterpart to reach the relaxed conformation towards the target. Indeed, such conformational changes could help each of ligand and target structures to fine the best positions of complex formations. As a consequence, performing halogenated mode of lead optimization for Cyd yielded CydI by distinguished features in single-standing mode and complex formation. Hence, this model could be proposed for further investigating regarding innovation of inhibitors for the MPro of COVID-19.
4Conclusion
DFT based quantum processing and MD simulations were performed to provide required results for analyzing Cyd models for the purpose of MPro inhibiting of COVID-19. In this regard, the results indicated that the stabilized models of halogenated Cyd could be achievable by performing DFT calculations and their frontier molecular orbital features could provide information about their characteristic features. To this point, quantities of HOMO-LUMO and corresponding distribution patterns all showed significant features for the models distinguishing CydI model for approaching the purpose. Chemical hardness and softness were more suitable for CydI model in comparison with other models, in which complex formation of this ligand structure was still at the most suitability. Analyzing HOMO-LUMO distribution patterns showed changes of such orbital localization for CydI model in comparison with the other models implying for the promising features of CydI model. As a concluding result of this work, lead optimization of Cyd model yielded a better model of CydI derivative with appropriate features to be considered for investigating MPro inhibitors of COVID-19.
Acknowledgments
The support of this work by the research council of Isfahan University of Medical Sciences under grant number 299126 is acknowledged.
References
[1] | Velavan T.P. and Meyer C.G. , The COVID-19 epidemic, Tropical Medicine & International Health 25: ((2020) ), 278–80. |
[2] | Ozkendir O.M. , Askar M. and Kocer N.E. , Influence of the epidemic COVID-19: an outlook on health, business and scientific studies, Lab-in-Silico 1: ((2020) ), 26–30. |
[3] | Arshizadeh S. , Gorgani S.H. , Taheri P. , Givgol M. , Shahrokhi S. and Abdalisousan A. , The impact of COVID-19 on oil supply in the short term, Advanced Journal of Science and Engineering 2: ((2021) ), 120–35. |
[4] | Lazarus J.V. , Ratzan S.C. , Palayew A. , Gostin L.O. , Larson H.J. , Rabin K. , Kimball S. and El-Mohandes A. , A global survey of potential acceptance of a COVID-19 vaccine, Nature Medicine 27: ((2021) ), 225–8. |
[5] | Vandenberg O. , Martiny D. , Rochas O. , van Belkum A. and Kozlakidis Z. , Considerations for diagnostic COVID-19 tests, Nature Reviews Microbiology 19: ((2021) ), 171–83. |
[6] | Brodin P. , Immune determinants of COVID-19 disease presentation and severity, Nature Medicine 27: ((2021) ), 28–33. |
[7] | Rahman M.T. and Idid S.Z. , Can Zn be a critical element in COVID-19 treatment? Biological Trace Element Research 199: ((2021) ), 550–8. |
[8] | Liu H. , Liu J. , Hou S. , Tao T. and Han J. , Perception consistency ultrasound image super-resolution via self-supervised CycleGAN, Neural Computing and Applications. 2021: in press. |
[9] | Dabbous H.M. , Abd-Elsalam S. , El-Sayed M.H. , Sherief A.F. , Ebeid F.F. , Abd El Ghafar M.S. , Soliman S. , Elbahnasawy M. , Badawi R. and Tageldin M.A. , Efficacy of favipiravir in COVID-19 treatment: a multi-center randomized study, Archives of Virology 166: (202), 949–54. |
[10] | Yang Y. , Liu J. and Zhou X. , A CRISPR-based and post-amplification coupled SARS-CoV-2 detection with a portable evanescent wave biosensor, Biosensors and Bioelectronics 190: ((2021) ), 113418. |
[11] | Xue C. , You J. , Zhang H. , Xiong S. , Yin T. and Huang Q. , Capacity of myofibrillar protein to adsorb characteristic fishy-odor compounds: Effects of concentration, temperature, ionic strength, pH and yeast glucan addition, Food Chemistry 363: ((2021) ), 130304. |
[12] | Li X. , Dong Z.Q. , Yu P. , Wang L.P. , Niu X.D. , Yamaguchi H. and Li D.C. , Effect of self-assembly on fluorescence in magnetic multiphase flows and its application on the novel detection for COVID-19, Physics of Fluids 33: ((2021) ), 042004. |
[13] | Khan S.L. , Siddiqui F.A. , Jain S.P. and Sonwane G.M. , Discovery of potential inhibitors of SARS-CoV-2 (COVID-19) Main Protease (Mpro) from Nigella Sativa (black seed) by molecular docking study, Coronaviruses 2: ((2021) ), 384–402. |
[14] | Daoud S. , Alabed S.J. and Dahabiyeh L.A. , Identification of potential COVID-19 main protease inhibitors using structure-based pharmacophore approach, molecular docking and repurposing studies, Acta Pharmaceutica 71: ((2021) ), 163–74. |
[15] | Ashjaee Y. and Zandi H. , Molecular analysis of 5-COR derivatives of uracil and evaluating their affinity against the MPro target of COVID-19, Advanced Journal of Science and Engineering 2: ((2021) ), 79–85. |
[16] | Kumar D. , Chandel V. , Raj S. and Rathi B. , In silico identification of potent FDA approved drugs against Coronavirus COVID-19 main protease: A drug repurposing approach, Chemical Biology Letters 7: ((2020) ), 166–75. |
[17] | Khalid H. , Hussain R. and Hafeez A. , Virtual screening of piperidine based small molecules against COVID-19, Lab-in-Silico 1: ((2020) ), 50–5. |
[18] | Mirzaei M. , Harismah K. , Da’i M. , Salarrezaei E. and Roshandel Z. , Screening efficacy of available HIV protease inhibitors on COVID-19 protease, Journal Military Medicine 22: ((2020) ), 100–7. |
[19] | Shah B. , Modi P. and Sagar S.R. , In silico studies on therapeutic agents for COVID-19: drug repurposing approach, Life Sciences 252: ((2020) ), 117652. |
[20] | Joshi T. , Joshi T. , Sharma P. , Mathpal S. , Pundir H. , Bhatt V. and Chandra S. , In silico screening of natural compounds against COVID-19 by targeting Mpro and ACE2 using molecular docking, European Review for Medical and Pharmacological Sciences 24: ((2020) ), 4529–36. |
[21] | Wang Y. , Wu M. , Tan X. and Gu J. , Insights into the reaction mechanism between phosphacyclopropenylidene and methyleneimine: a theoretical study, Main Group Chemistry 19: ((2020) ), 237–44. |
[22] | Wang X.F. , Gao P. , Liu Y.F. , Li H.F. and Lu F. , Predicting thermophilic proteins by machine learning, Current Bioinformatics 15: ((2020) ), 493–502. |
[23] | Zou Q. , Xing P. , Wei L. and Liu B. , Gene2vec: gene subsequence embedding for prediction of mammalian N6-methyladenosine sites from Mrna, RNA 25: ((2019) ), 205–18. |
[24] | Xu Q. , Zeng Y. , Tang W. , Peng W. , Xia T. , Li Z. , Teng F. , Li W. and Guo J. , Multi-task joint learning model for segmenting and classifying tongue images using a deep neural network, IEEE Journal of Biomedical and Health Informatics 24: ((2020) ), 2481–9. |
[25] | Narayanasamy R.K. , Rada P. , Zdrha A. , van Ranst M. , Neyts J. , Tachezy J. , Cytidine nucleoside analog is an effective antiviral drug against Trichomonasvirus, Journal of Microbiology, Immunology and Infection. 2021; in press. |
[26] | Rana K.M. , Maowa J. , Alam A. , Dey S. , Hosen A. , Hasan I. , Fujii Y. , Ozeki Y. , Kawsar S.M. , In silico DFT study, molecular docking, and ADMET predictions of cytidine analogs with antimicrobial and anticancer properties, In Silico Pharmacology 9: ((2021) ), 1–24. |
[27] | Yaghoobi R. , Mirzaei M. , Computational analyses of cytidine and aza-cytidine molecular structures, Lab-in-Silico. 1: ((2020) ), 21–6. |
[28] | Wong X.K. , Yeong K.Y. , From Nucleic Acids to Drug Discovery: Nucleobases as Emerging Templates for Drug Candidates. Current Medicinal Chemistry. 2021; in press. |
[29] | Niu M. , Lin Y. , Zou Q. , sgRNACNN: identifying sgRNA on-target activity in four crops using ensembles of convolutional neural networks, Plant Molecular Biology 105: ((2021) ), 483–95. |
[30] | Dolečelová E. , Klejch T. , Špaček P. , Slapničková M. , Guddat L. , Hocková D. , Zíková A. , A cyclic nucleoside phosphonateswith adenine nucleobase inhibit Trypanosoma brucei adeninephosphoribosyltransferase in vitro, Scientific Reports 11: ((2021) ), 1–27. |
[31] | Chua G.N. , Wassarman K.L. , Sun H. , Alp J.A. , Jarczyk E.I. , Kuzio N.J. , Bennett M.J. , Malachowsky B.G. , Kruse M. , Kennedy A.J. , Cytosine-based TET enzyme inhibitors, ACS Medicinal Chemistry Letters 10: ((2019) ), 180–5. |
[32] | Ghamsari P.A. , Samadizadeh M. , Mirzaei M. , Halogenated derivatives of cytidine: structural analysis and binding affinity, Journal of Theoretical and Computational Chemistry 19: ((2020) ), 2050033. |
[33] | Crane C. , Hirsch A.E. , Alphey M. , Sgraja T. , Lauw S. , Illarionova V. , Rohdich F. , Eisenreich W. , Hunter W. , Bacher A. , Diederich F. , Synthesis and characterization of cytidine derivatives that inhibit the kinase IspE of the non-mevalonate pathway for isoprenoid biosynthesis, ChemMedChem 3: ((2008) ), 91. |
[34] | Hayakawa D. , Sawada N. , Watanabe Y. , Gouda H. , A molecular interaction field describing nonconventional intermolecular interactions and its application to protein–ligand interaction prediction, Journal of Molecular Graphics and Modelling 96: ((2020) ), 107515. |
[35] | Puffer B. , Kreutz C. , Rieder U. , Ebert M.O. , Konrat R. , Micura R. , 5-Fluoro pyrimidines: labels to probe DNA and RNA secondary structures by 1D 19 F NMR spectroscopy, Nucleic Acids Research 37: ((2009) ), 7728–40. |
[36] | Frisch M.J. , Trucks G.W. , Schlegel H.B. , Scuseria G.E. , Robb M.A. , Cheeseman J.R. , Scalmani G. , Barone V. , et al. Gaussian 09. Gaussian, Inc., Wallingford, CT (2009) . |
[37] | Goodsell D.S. , Zardecki C. , Di Costanzo L. , Duarte J.M. , Hudson B.P. , Persikova I. , Segura J. , Shao C. , Voigt M. , Westbrook J.D. and Young J.Y. , RCSB Protein Data Bank: Enabling biomedical research and drug discovery, Protein Science 29: ((2020) ), 52–65. |
[38] | Patil N.S. , Rohane S.H. , Organization of Swiss Dock: In study of computational and molecular docking study, Asian Journal of Research in Chemistry 14: ((2021) ), 145–8. |
[39] | Huang Y. , Rong C. , Zhang R. , Liu S. , Evaluating frontier orbital energy and HOMO/LUMO gap with descriptors from density functional reactivity theory, Journal of Molecular Modeling 23: ((2017) ), 1–2. |
[40] | Rahman M.M. , Saha T. , Islam K.J. , Suman R.H. , Biswas S. , Rahat E.U. , Hossen M.R. , Islam R. , Hossain M.N. , Mamun A.A. , Khan M. , Virtual screening, molecular dynamics and structure–activity relationship studies to identify potent approved drugs for Covid-19 treatment, Journal of Biomolecular Structure and Dynamics 39: ((2021) ), 6231–41. |
[41] | de Souza Neto L.R. , Moreira-Filho J.T. , Neves B.J. , Maidana R.L. , Guimarães A.C. , Furnham N. , Andrade C.H. , Silva F.P. Jr., Insilico strategies to support fragment-to-lead optimization in drugdiscovery, Frontiers in Chemistry 8: ((2020) ), 93. |

