
Comparative study of molecular docking, structural, electronic, vibrational and hydrogen bonding interactions on 4-hydroxy benzo hydrazide (4HBH) and its newly designed derivative [(E)–N′-((1H-Pyrrol-2-YL)methylene) –4-hydroxy benzo hydrazide and its isomers (I, II and III)] (potential inhibitors) for COVID-19 protease
Abstract
Studies have shown that hydrazides and thier derivatives are used for pharmaceutical and medicinal purposes. At present, the whole world is suffering for COVID-19 virus. There are some vaccines or medicines available to treat this disease all over the world. Today the one fourth of the world’s population is under lockdown condition. In this scenario, scientists from the whole world are doing different types of research on this disease. Being a molecular modeller, this inspires us to design new types of species (may be drugs) which may be capable for COVID-19 Protease. In the present effort, we have performed docking studies of title compounds with COVID-19 protein (6LU7) for anti-COVID-19 activity. A comparative quantum chemical calculations of molecular geometries (bond lengths and bond angles) of 4-Hydroxy Benzo Hydrazide (4HBH) and its newly designed derivatve [(E)-N′-((1H-Pyrrol-2-YL)Methylene) –4-Hydroxy Benzo Hydrazide and its isomers (I, II and III)] in the ground state have also been carried out due to its biological importance and compared with the similer type of compound found in literature i.e. benzohydrazide. The optimized geometry and wavenumber of the vibrational bands of the molecules have been calculated by density functional theory (DFT) using Becke’s three-parameters hybrid functional (B3LYP/CAM-B3LYP) with 6–311G (d, p) as the basis set. Vibrational wavenumbers are compared with the observed FT-IR and FTRaman spectra of 4-Hydroxy Benzo Hydrazide. TDDFT calculations are also done on the same level of theory and a theoretical UV-vis spectrum of title molecules are also drawn. HOMO-LUMO analysis has been done to describe the way
the molecule interacts with other species. Natural bond orbitals (NBO) analysis has been carried out to inspect the intra- and inter- molecular hydrogen-bonding, conjugative and hyper conjugative interactions and their second order stabilization energy. Nonlinear optical (NLO) analysis has been performed to study the non-linear optical properties of the molecule by computing the first hyperpolarizability. The variation of thermodynamic properties with temperature has been studied. QATIM analysis shows that hydrogen bonding occurs in 4HBH, isomer II and III respectively.
1Introduction
The COVID-19 virus from the family of corona emerged in December 2019 in china and then spread rapidly worldwide, particularly to China, Japan, South Korea, Italy, Spain, USA and India etc. As of march 31, 2020, a total of 803,313 confirmed cases of coronavirus disease 2019 (COVID-19) and 39033 deaths have been reported all over the world [1, 2]. Scientists are trying to find vaccines and drugs to treat this disease. In the absence of any medicine or vaccine, some antivirals including interferon α (IFN-α), lopinavir/ritonavir and chloroquine phosphate have been used for tentative treatment of COVID-19 [3]. It is seen that hydrazides have different biological activities [4–6]. Studies have shown that hydrazides and its derivatives are used for pharmaceutical and medicinal purposes [7]. Hydrazides have been known to be associated with anti-bacterial, antifungal, anthelmintic and anticonvulsant activities [8–11]. Abdulaziz et al. [12] have studied the comparative study of structures of benzohydroxamic acid (BHA) and benzohydrazide (BH). Suresh et al. [13] have done spectroscopics investigations of 2,4-dihydroxy-N′-(4-methoxybenzylidene)benzohydrazide by with experimental and theoretical aspects. A paper entitled “FTIR, FT-Raman and UV–Vis spectra of the Schiff base compound (E)-N′-(4-methoxybenzylidene) benzohydrazide (MBBH)” is also reported by Saleem et al. [14]. p-Hydroxy benzohydrazide moiety and its analogues are suitable parent compounds upon which variety of biological activities are reported such as antitumor [15], antianginal [16], antitubercular [17], antihypertensive [18] and antibacterial [19]. Suresh et al. [20] have investigated the experimental and theoretical vibrational modes of 2,4-dihydroxy-N′-(4-methoxybenzylidene)benzohydrazide. Arjunan et al. [21] have assigned and analysed the vibrational modes of benzohydrazide (BH) by using FTIR and FT-Raman spectral data. Marta Sánchez-Lozano et al. [22] have prepared the rhenium (I) carbonyl bromide complex, [ReBr(CO)3(HL)], from 2,4-dihydroxybenzaldehyde and 4 hydroxybenzoic acid hydrazide (HL). Bharty et al. [23] have synthesised two new compounds N′ [bis(methylsulfanyl) methylene]-2-hydroxybenzohydrazide {Hbmshb(1)} and N′-(4 methoxy benzoyl)-hydrazine carbodithioic acid ethyl ester {H2mbhce(2)}.
As a part of our on-going research [24–28], we have reported a camparative quantum chemical study on (E)-N′-((1H-Pyrrol-2-YL)Methylene) –4-Hydroxy Benzo Hydrazide isomers and 4-Hydroxy Benzo Hydrazide in the ground state by using combination of DFT /B3LYP/CAM-B3LYP theory and 6–311G (d, p) as the basis set and compared with the similer type of compound found in literature i.e. benzohydrazide [29]. This DFT study on these compounds most likely help researchers to understand some modification in chemical reaction such as oxidation/reduction which generate new binding reactive sites. We have also performed molecular docking of title compounds. In this paper NLO properties of these compounds are reported for the first time which helps to explore its various NLO applications [30, 31].
1.1Experimental details and computational methods
Geometry optimization have been done using Gaussian 03 [32] and Gauss View molecular visualization program pakages [33]. The molecular structure of the title compounds in ground state (in gas phase) were optimized by DFT/B3LYP [34, 35] and CAM-B3LYP [36] methods with 6–311 G (d, p) as the basis set. B3LYP, the most attractive and well known DFT functional, uses Becke’s 3 parameter exchange correlation functional which embrace 3 parameters to mix in the exact Hartree–Fock (HF) exchange correlation and Lee Yang and Parr (LYP) correlation functional that refurbish dynamic electron correlation. The CAM-B3LYP functional laid down by Tawada et al. consolidates the long-range correction and the hybrid qualities of B3LYP. We have used density functional theory with B3LYP and CAM-B3LYP as the basis sets. As B3LYP is most successful functional so far while CAM-B3LYP combines the hybrid and long range correlation. CAM-B3LYP performs well for charge transfer excitations which B3LYP underestimates enormously. So we have used these both basis sets for our calculation. These optimized geometry was used in the vibrational frequency calculations. Harmonic modes were multiplied by the factor 0.9614 for DFT/B3LYP and 0.9624 for DFT/ CAM-B3LYP to scale the frequencies [37]. The calculated vibrational frequencies and their corresponding assignments were investigated in detail by potential energy distribution (PED) analysis using VEDA 4 program [38]. The Chem Craft program [39] was used to evaluate the theoretical IR spectrum. Also the components of the electric moments such as total dipole moment (μ), mean polarizability < α>and total first hyperpolarizability (β) [40, 41] have been calculated and discussed at B3LYP and CAM-B3LYP functionals. Lastly in this study, the molecular docking of the title molecules and 6LU7 protein has been investigated by Swissdock online server.
2Results and discussion
2.1Geometry optimization
Alternate positions of enaminone group attached to 4-Hydroxy Benzo Hydrazide have been studied, but finally we get these three optimized structures of (E)-N′-((1H-Pyrrol-2-YL)Methylene) –4-Hydroxy Benzo Hydrazide. Optimized parameters of 4-Hydroxy Benzo Hydrazide derivatives (three isomers of (E)-N′-((1H-Pyrrol-2-YL)Methylene) –4-Hydroxy Benzo Hydrazide), calculated by B3LYP/CAM-B3LYP methods with 6–311 G (d, p) as the basis set are listed in supplementary Table 1, which are in accordance with the atom numbering scheme as shown in Fig. 1 also with the possible optimized structure of 4-Hydroxy Benzo Hydrazide. Local minimum energies are –778.7766 a.u., –778.7750 a.u., –778.7777 a.u. and –531.5123 for B3LYP while –778.3938 a.u., –778.3919, –778.3950, –531.3831 for CAM-B3LYP methods for I, II, III and 4HBH respectively given in Table 1. Optimized structures of 4-HBH, I, II and III have C1 point group symmetry. In I, II and III, Both rings are not in the same plane as the molecules are twisted from the middle chain. C–C bond distances are found to be in the range of 1.388–1.499 A°, 1.383–1.466 A° & 1.383–1.466 A° for B3LYP and 1.382–1.495 A°, 1.378–1.466 A° & 1.378–1.467 A° for CAM-B3LYP respectively for isomers I, II and III. For C–O, these values are 1.361 A°, 1.345–1.362 A° & 1.337–1.361 A° for B3LYP and 1.355 A°, 1.337–1.356 A° & 1.329–1.355 A° for CAM-B3LYP respectively. In case of C–H bond distances, they lie in the range of 1.083–1.096 A°, 1.078–1.086 A° & 1.078–1.087 A° for B3LY P and 1.078–1.085 A°, 1.078–1.090 A° & 1.078–1.087 A° for CAM-B3LYP, respectively. The C–C bond distance of literature compound [29] i.e. BH lies in the range 1.391–1.40A while the ring C–H bond distances were found 1.085A. The longer C–C bond distance in title compound shows that there is no delocalization of the nitrogen lone pair of electrons towards the ring. Planes of the benzene ring and the planar hydrazide group of 4-Hydroxy Benzo Hydrazide are inclined at 24.5° with respect to each other. Except amino and hydrazide hydrogens, molecule is essentially planar as evidenced by the torsion angles C6–C1–C7–O8, N10–N9–C7–C1 and N10–N9–C7–O8 of –23.3°, –23.2° and 158.5°. Here we have seen that calculated bond lengths by B3LYP method are very closer to literature [29]. This shows the supremacy of B3LYP method over CAM-B3LYP.
Fig. 1
Model Molecular Structure of 4-HBH and three isomers of title compound.
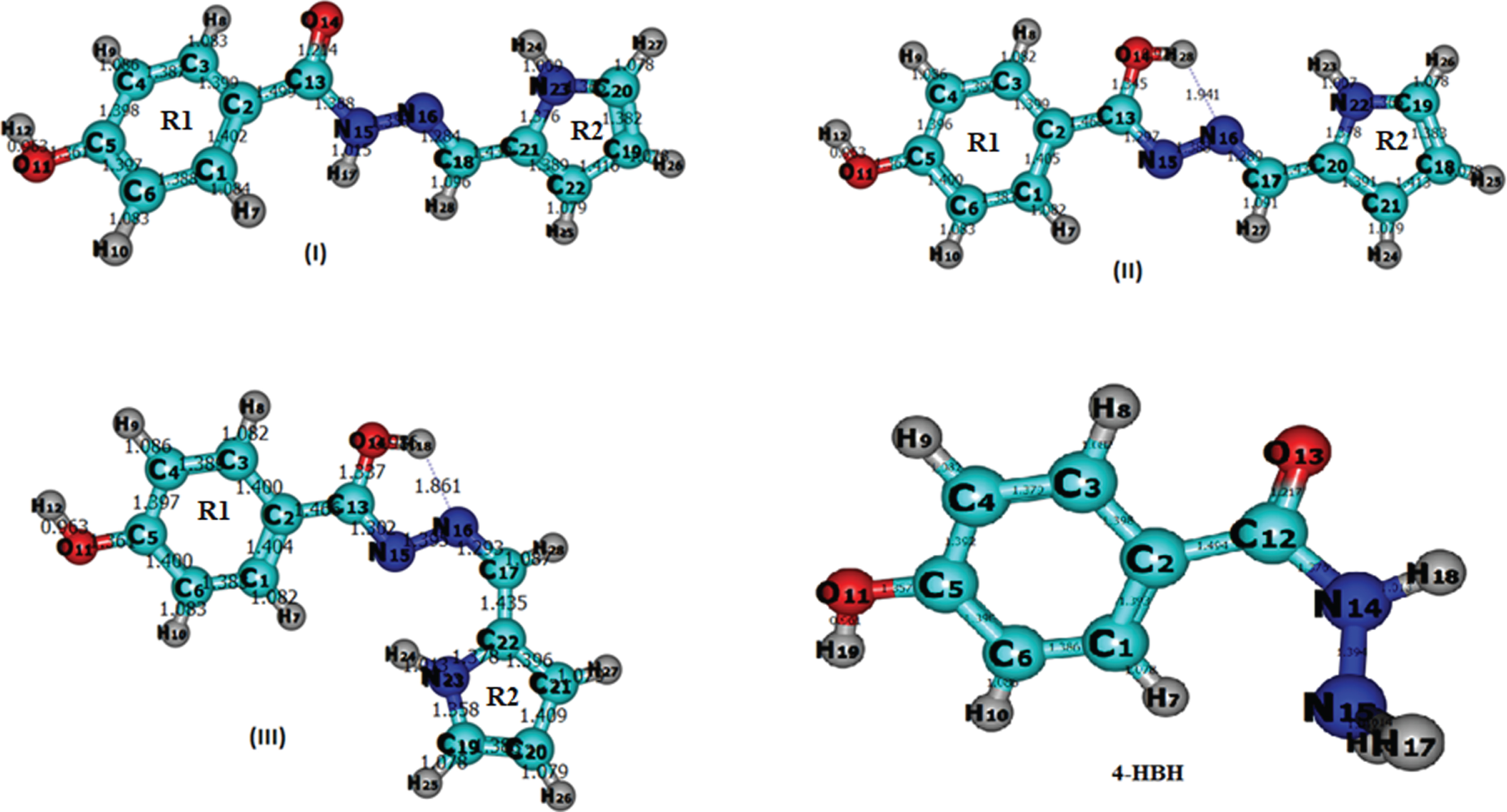
Table 1
Comparative energy (in a.u.) of 4HBH and the three Isomers of title compound at 6–311 G (d,p) level
| Functional | Isomer 1 | Isomer 2 | Isomer 3 | 4HBH |
| B3LYP | –778.7766 | –778.7750 | –778.7777 | –531.5123 |
| CAM-B3LYP | –778.3938 | –778.3919 | –778.3950 | –531.3831 |
2.2Vibrational analysis
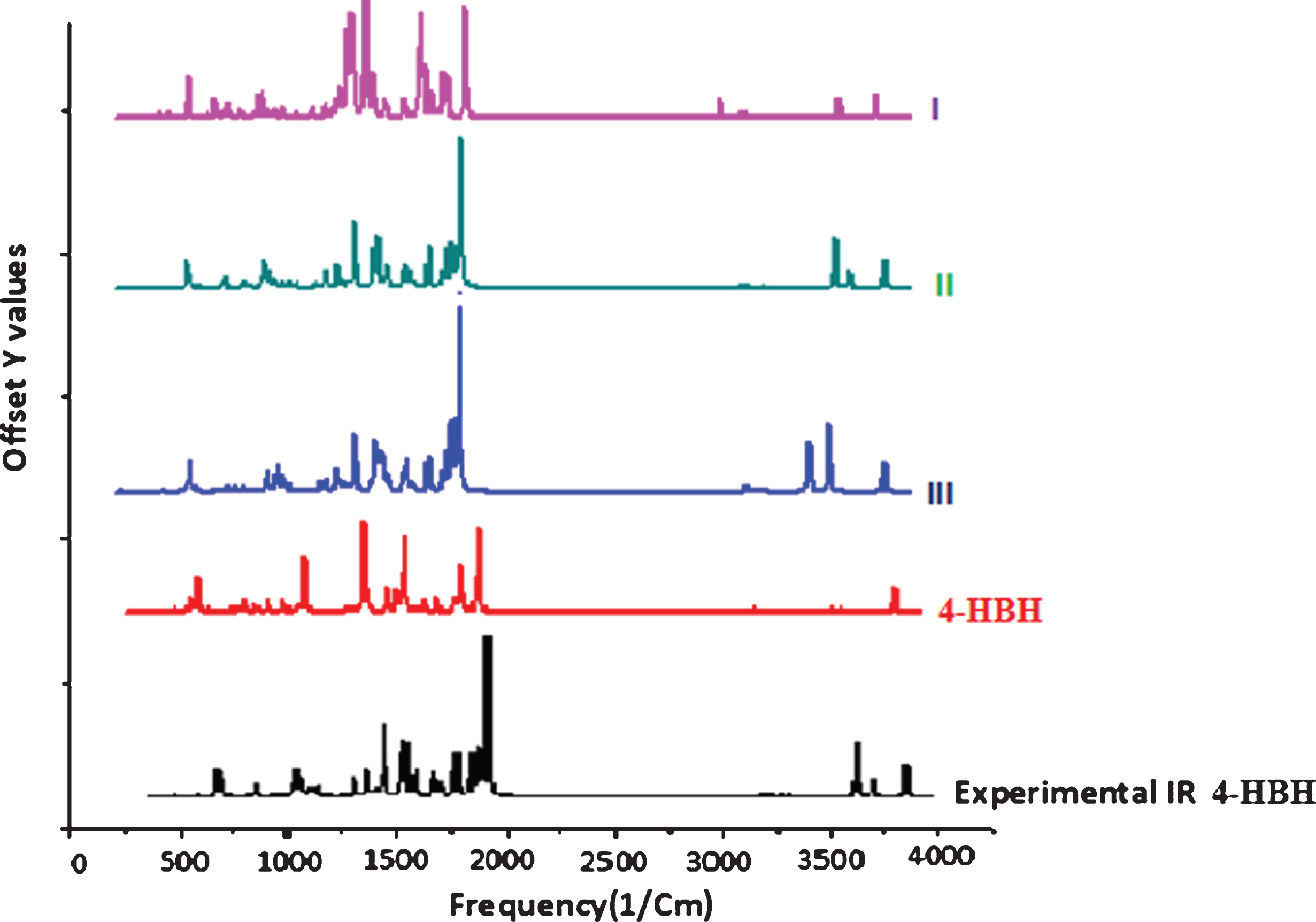
The 4-HBH contains 19 while its derivatives (I, II and III) have 28 atoms and they have 51 and 78 normal modes of vibration, respectively. All the fundamental vibrations are active IR. The harmonic vibrational frequencies are calculated at B3LYP/CAM-B3LYP and 6–311 G (d, p) level and experimental frequencies {FTIR and FTRaman- supplementary Fig. 1 (a & b)} have been recored for 4-Hydroxy Benzo Hydrazide only. No experimental FTIR spectrum for 4-Hydroxy Benzo Hydrazide derivatives is available for comparison so the theoretical spectrum (supplementary Figure 2) will be a suitable path for experimental researchers. Vibrational assignments of the normal modes of I, II and III are discussed in Table 2, 3 and 4. A comparison of all IR spectrum is shown in Fig. 2. Vibrational assignments are based on the observation of the animated modes in GaussView and discussed below.
Table 2
Comparison of the calculated Vibrational spectra with assignments of I by B3LYP/CAM-B3LYP/6-311 G (d, p) methods
| B3LYP | CAM-B3LYP | Vibrational Assignment | ||
| Scaled Freq. | IR intensity | Scaled Freq. | IR intensity | |
| 483 | 42.8554 | 477 | 51.6086 | βout (15N–17H) (43%) |
| 490 | 23.387 | 497 | 13.6821 | R1[βout (C–C–C) +βout(C–H)] +βout (15N–17H) (21%) |
| 526 | 22.3151 | 533 | 22.0461 | R1[βout (C–C–C) +βout(C–H)] |
| 549 | 48.2771 | 573 | 50.1374 | βout (23N–28H) (69%) |
| 606 | 32.765 | 640 | 32.0031 | Ring R1 Breathing |
| 651 | 48.3251 | 689 | 51.2412 | βout (11O–12H) (24%)] + R1[τ (C–C–C–C) (21%)] |
| 704 | 91.7578 | 722 | 94.6277 | βout (19C–26H) (36%) +βout (20C–27H) (29%) |
| 739 | 26.5499 | 756 | 32.411 | R1[βout (C–C–C) +βout(C–H)] +βout(13C–14O) (17%) |
| 773 | 21.6174 | 799 | 16.4497 | R2[βout (C–C–C) (31%) +βout(C–H) (18%)] |
| 816 | 31.1816 | 835 | 32.7719 | R1[βout (C–C–C) (28%) +βout(C–H) (32%)] |
| 857 | 12.2726 | 877 | 10.6485 | R2[β (C–C–C) (61%) +β (C–H) (21%)] |
| 881 | 23.8969 | 895 | 29.036 | Ring R1&R2 Breathing |
| 908 | 11.2773 | 933 | 12.1061 | β (18C–28H) (81%) |
| 952 | 37.713 | 966 | 32.4391 | R2[β(C–N–C) (15%) +β (C–C–C) (21%) +β(C–H) (31%)] |
| 1012 | 52.1344 | 1016 | 54.2541 | β (19C–26H) (26%) +β (22C–25H) (19%) |
| 1037 | 12.3927 | 1060 | 10.0571 | R2[β(C–N–C) +β (C–C–C) +β(C–H)] |
| 1066 | 44.9576 | 1079 | 53.4016 | R1[β (C–C) (24%) +β(C–H)](31%) +β(11O–12H) (14%) |
| 1083 | 73.279 | 1092 | 51.5572 | β (23N–24H) (21%) +β (20C–27H) (15%) |
| 1094 | 39.0411 | 1104 | 24.8774 | β (15N–17H) (13%) +υ(15N–116N) (17%) |
| + R1[β (C–C) +β(C–H)] +β(11O–12H) (14%) | ||||
| 1123 | 190.888 | 1147 | 23.2847 | R1 β (C–C) (39%) +β(C–H) (32%)] +β(11O–12H) (22%) |
| 1142 | 302.9124 | 1152 | 460.2024 | β(11O–12H) (27%)] +β (4C–9H) (15%) |
| 1150 | 123.5665 | 1156 | 69.9927 | υ (13C–2C) (25%) +β(15N–17H) (19%) |
| + R1[β (C–C) +β(C–H)] +β(11O–12H) (12%) | ||||
| 1208 | 281.8507 | 1227 | 36.7442 | R2[β(C–N–C) (17%)] +β (C–C–C) (24%) |
| +β(C–H) (13%)] +β(18C–28H) (11%) | ||||
| 1216 | 82.5483 | 1238 | 350.4964 | R1 β (C–C) (33%) +β(C–H) (21%)] +β(11O–12H) (12%) |
| 1247 | 107.1672 | 1270 | 145.3033 | R2[β(C–N–C) (29%)] +β (C–C–C) (37%) |
| +β(C–H) (24%)] +β(18C–28H) (14%) | ||||
| 1261 | 24.8574 | 1273 | 25.0607 | R2[β(C–N–C) +β (C–C–C) +β(C–H)] |
| 1300 | 39.717 | 1315 | 70.9183 | β (28H–18C) (29%) +β(15N–17H) (23%) |
| +β(22C–25H) (17%) | ||||
| 1316 | 36.8194 | 1318 | 8.9768 | R1[υ (C–C) (35%) +β(C–H) (32%) +β(11O–12H) (21%) |
| 1395 | 17.8486 | 1419 | 26.3573 | υ (23N–21C) (45%) +β(23N–24H) (32%) |
| 1396 | 34.6263 | 1426 | 10.801 | R2[β(C–N–C) (17%)] +β (C–C–C) (37%) +β(C–H) (34%)] |
| 1405 | 20.4706 | 1429 | 31.4119 | R1[υ (C–C) (25%) +β(C–H) (32%)] +β(11O–12H) (12%) |
| 1423 | 10.0616 | 1449 | 17.464 | υ (20N–22C) (27%)] + R2[υ (C–C) (32%) |
| +β (C–H) (19%)] +β (15N–17C) (13%) | ||||
| 1475 | 345.1893 | 1497 | 449.4911 | R1[υ (C–C) (23%) +β(C–H) (32%)] +β (15N–17C) (11%) |
| 1492 | 119.7245 | 1568 | 107.6335 | β (15N–17C) (15%) +β (18C–28H) (11%) |
| + R1[υ (C–C) (23%) +β(C–H) (21%)] | ||||
| 1533 | 65.0114 | 1588 | 79.4396 | R2[β (C–N–C) (29%)] + R2[υ(C–C) (37%) |
| 1561 | 17.644 | 1599 | 13.4733 | R1[υ (C–C) (33%) +β(C–H) (41%)] +β(11O–12H) (11%) |
| 1587 | 127.5757 | 1625 | 155.7993 | R1[υ (C–C) (36%) +β(C–H) (49%)] |
| 1608 | 98.9418 | 1670 | 50.815 | υ(16N–18C) (49%) +υ(21C–18C) (32%) |
| 1693 | 278.3323 | 1741 | 288.0971 | υ(14O–13C) (89%) |
| 2916 | 45.9041 | 2948 | 41.8969 | υ(28H–18C) (83%) |
| 3026 | 23.5403 | 3051 | 17.7611 | υ(9H–4C) (73%) |
| 3041 | 11.8602 | 3065 | 10.3821 | υ(7H–1C) (43%) +υ (10H–5C) (48%) |
| 3372 | 7.5641 | 3423 | 9.2716 | υ(15N–17H) (89%) |
| 3497 | 70.3324 | 3519 | 84.7768 | υ(23N–24H) (99%) |
| 3674 | 91.5524 | 3716 | 103.5788 | υ(11O–12H)[100%] |
Table 3
Comparison of the calculated Vibrational spectra with assignmentsof II by B3LYP/ CAM-B3LYP/6-311 G (d, p) methods
| B3LYP | CAM-B3LYP | Vibrational Assignment | ||
| Scaled Frequency | IR intensity | Scaled Frequency | IR intensity | |
| 350 | 118.4313 | 363 | 124.9219 | βout (11O–12H) (54%) |
| 362 | 18.2986 | 382 | 17.439 | R1[[τ(C–C–C–C) (11%) |
| +τ (2C–13C–14O–28H) (17%)] | ||||
| +τ (15N–16N–17C–28H) (21%) | ||||
| 512 | 58.9415 | 555 | 57.4514 | βout (22N–23H) (46%) |
| 612 | 24.4195 | 645 | 22.456 | Ring R1 Breathing |
| 643 | 7.477 | 681 | 10.6966 | βout (14O–28H) (26%)] + R1[τ (C–C–C–C) (31%)] |
| 697 | 27.2591 | 740 | 40.1221 | βout (14O–28H) (24%)] + R1[τ (C–C–C–C) (21%)] |
| 706 | 96.6931 | 754 | 91.9246 | R2[βout (C–H) (35%)] |
| 732 | 78.2065 | 780 | 82.918 | R1[[τ(C–C–C–C) (23%)] |
| + [τ (2C–13C–14O–28H) (19%) | ||||
| 764 | 22.8306 | 806 | 25.7312 | R2[β (C–H) (29%) +β (C–C) (31%)] |
| 783 | 8.1018 | 841 | 14.281 | βout (21C–24H) (18%) +βout (18C–25H) (24%) |
| +βout (19C–26H) (42%) | ||||
| 797 | 19.4004 | 844 | 17.6158 | R1[β(C–C–C) (38%)] |
| 821 | 31.1122 | 873 | 32.7204 | βout (1C–7H) (28%) +βout (5C–10H) (32%) |
| 941 | 10.26 | 1004 | 11.8064 | β(10C–26H) (35%) +β (18C–25H) (39%) |
| 949 | 16.8708 | 1007 | 10.2561 | β(17C–27H) (47%) |
| 998 | 43.4569 | 1061 | 86.0462 | β(21C–24H) (38%) +β (18C–25H) (42%) |
| 1015 | 55.8184 | 1068 | 17.2139 | β(21C–24H) (27%) +υ(15N–16N) (16%) |
| +β(17C–27H) (22%) | ||||
| 1065 | 76.0312 | 1123 | 88.908 | R2[β (C–H) (47%) +β (N–H) (29%)] |
| 1075 | 31.006 | 1133 | 22.399 | R1[β (C–H) (61%)] |
| 1104 | 14.0347 | 1173 | 18.7888 | R1[β(C–C) (27%) +β(C–H) (39%)] |
| +υ(15N–16N) (17%) | ||||
| 1142 | 150.386 | 1198 | 26.7802 | R1[β (C–H) (71%)] |
| 1149 | 172.7663 | 1204 | 253.243 | β(11O–12H) (89%)] |
| 1233 | 129.7987 | 1299 | 135.9068 | β(14O–28H) (77%)] |
| 1246 | 162.2252 | 1322 | 176.7406 | R1[β (C–C–C) (19%) +β(C–H) (22%)] |
| +β(17C–27H) (11%) + R2[β(C–H) (23%)] | ||||
| 1258 | 28.083 | 1322 | 11.4024 | R2[β (C–H) (23%)] + R2[β(N–H) (11%)] |
| +β(17C–27H) (21%) | ||||
| 1278 | 16.0233 | 1339 | 10.6391 | R1[β (C–C–C) (10%)] + R1[β(C–H) (21%)] |
| 1292 | 26.6546 | 1365 | 57.2207 | R1[β (17C–27H) (57%)] + R2[β(C–H) (17%) |
| 1316 | 69.5266 | 1371 | 65.1115 | R1[β (C–H) (37%)] + R1[β(C–C) (32%) |
| + R2[β (11O–12H) (26%)] | ||||
| 1375 | 81.3562 | 1456 | 116.5657 | υ (13C–14O) (29%) +β(14O–28H) (16%) |
| + R1[β (C–H) (13%) | ||||
| 1390 | 24.9431 | 1484 | 13.6506 | υ(20C–22N)[31%] +υ(19C–22N)[29%] |
| +β (22N–28H)19% | ||||
| 1404 | 29.623 | 1485 | 44.6845 | R2[β (C–C) (31%)] + R2[β(C–H) (27%) |
| +υ(20C–22N)[15%] | ||||
| 1415 | 11.7508 | 1496 | 11.7704 | R1[β (C–H) (21%)] + R1[β(C–C) (12%) |
| + R2[β (N–H) (11%)] + R2[β(C–H) (17%) | ||||
| 1423 | 16.6892 | 1508 | 25.2429 | R2[β (C–N–C) (39%)] + [υ(C–O) (31%) |
| 1486 | 211.4073 | 1577 | 189.6982 | R1[β (C–H) (35%)] + R1[υ(C–C) (43%) |
| +υ (15C–13N) (17%) | ||||
| 1544 | 156.9155 | 1658 | 149.6037 | R1[β (C–H) (43%)] + R1[υ(C–C) (37%) |
| +β(13N–14O–28H) (16%) +υ (15C–13N) (13%) | ||||
| 1571 | 43.281 | 1694 | 147.1628 | R1[β (C–H) (50%)] + R1[υ(C–C) (42%) |
| 1589 | 131.7016 | 1696 | 97.5399 | υ(17C–16N) (17%) +υ (15C–13N) (21%) |
| +β(13N–14O–28H) (27%) + R1[υ(C–C) (32%) | ||||
| 1612 | 707.6691 | 1736 | 803.3336 | υ(17C–16N) (37%) +υ (15C–13N) (26%) |
| +β(13N–14O–28H) (29%) | ||||
| 2981 | 14.1793 | 3136 | 9.894 | R1[υ(17C–27H) (24%)] |
| 3026 | 28.8138 | 3177 | 22.1502 | R1[υ(9H–4C) (41%) +υ (8H–3C) (58%)] |
| 3425 | 143.2864 | 3622 | 186.5282 | υ(14O–28H) (98%) |
| 3522 | 47.8866 | 3695 | 58.2308 | υ(22N–23H) (99%) |
| 3673 | 103.6369 | 3870 | 112.2172 | υ(11O–12H)[100%] |
Table 4
Comparison of the calculated Vibrational spectra with assignmentsof III by B3LYP/ CAM-B3LYP/6-311 G (d, p) methods
| B3LYP | CAM-B3LYP | Vibrational Assignment | ||
| Scaled Frequency | IR intensity | Scaled Frequency | IR intensity | |
| 367 | 108.2927 | 377 | 109.2724 | βout (11O–12H) (54%) |
| 385 | 10.8785 | 407 | 11.7887 | R1[[τ(C–C–C–C) (11%) |
| +τ (2C–13C–14O–28H) (17%)] | ||||
| +τ(15N–16N–17C–28H) (21%) | ||||
| 531 | 22.4624 | 567 | 22.3186 | βout (22N–23H) (46%) |
| 571 | 21.3294 | 602 | 19.5022 | Ring R1&R2 Breathing |
| 587 | 10.87 | 626 | 12.3398 | R2[[τ(C–N–C–C) +τ (H–C–C–H)] |
| 610 | 10.4678 | 642 | 10.523 | Ring R1 Breathing |
| 647 | 1.7592 | 688 | 1.6907 | βout (14O–28H) (26%)] + R1[τ (C–C–C–C) (31%)] |
| 686 | 3.3197 | 730 | 5.3781 | R2[βout (C–H) (35%)] |
| 717 | 37.3949 | 763 | 37.4237 | R2[βout (C–H) (35%)] |
| 722 | 56.7497 | 769 | 60.1092 | R1[[τ(C–C–C–C) (23%)] |
| + [τ (2C–13C–14O–28H) (19%) | ||||
| 767 | 33.4405 | 809 | 34.1047 | R2[β (C–H) (29%) +β (C–C) (31%)] |
| 774 | 99.9665 | 821 | 123.2933 | βout (21C–24H) (18%) +βout (18C–25H) (24%) |
| +βout (19C–26H) (42%) | ||||
| 791 | 56.0716 | 848 | 40.1394 | R1[β(C–C–C) (38%)] |
| 805 | 12.3208 | 852 | 15.9248 | β(15N–16N–17C) (24%) |
| + R2[β(C–N–C) +β(C–C–C) +β(C–H)] | ||||
| 816 | 24.8227 | 869 | 23.7864 | βout (1C–7H) (28%) +βout (5C–10H) (32%) |
| 851 | 0.0005 | 909 | 10.9717 | R1[β(C–C–C) +β(C–H)] |
| + R2[β(C–N–C) +β(C–C–C) +β(C–H)] | ||||
| 942 | 18.9666 | 1002 | 1.6787 | β(10C–26H) (35%) +β(18C–25H) (39%) |
| 965 | 28.5874 | 1036 | 32.1442 | β(17C–27H) (47%) |
| 985 | 0.9315 | 1040 | 12.0086 | β(21C–24H) (38%) +β(18C–25H) (42%) |
| 1017 | 47.1457 | 1064 | 49.7147 | β(21C–24H) (27%) +υ(15N–16N) (16%) |
| +β(17C–27H) (22%) | ||||
| 1059 | 87.0593 | 1117 | 100.0931 | R2[β (C–H) (47%) +β (N–H) (29%)] |
| 1079 | 35.875 | 1134 | 28.1347 | R1[β (C–H) (61%)] |
| 1096 | 37.8923 | 1153 | 25.7973 | β(20C–26H) (37%) +β(21C–27H) (41%) |
| 1103 | 5.7146 | 1171 | 21.5208 | R1[β(C–C) (27%) +β(C–H) (39%)] |
| +υ(15N–16N) (17%) | ||||
| 1143 | 133.6493 | 1199 | 22.9502 | R1[β (C–H) (71%)] |
| 1151 | 166.1405 | 1205 | 236.3207 | β(11O–12H) (89%)] |
| 1217 | 20.1759 | 1282 | 23.0342 | β(14O–28H) (77%)] |
| 1237 | 190.1962 | 1308 | 183.314 | R1[β (C–C–C) (19%) +β(C–H) (22%)] |
| +β(17C–27H) (11%) + R2[β(C–H) (23%)] | ||||
| 1249 | 142.0432 | 1325 | 144.2228 | R2[β (C–H) (23%)] + R2[β(N–H) (11%)] |
| +β(17C–27H) (21%) | ||||
| 1270 | 67.8627 | 1335 | 28.8497 | R1[β (C–C–C) (10%)] + R1[β(C–H) (21%)] |
| 1279 | 88.7216 | 1344 | 145.3576 | R1[β (17C–27H) (57%)] + R2[β(C–H) (17%) |
| 1314 | 34.5646 | 1370 | 56.246 | R1[β (17C–27H) (57%)] + R2[β(C–H) (17%) |
| 1318 | 32.6606 | 1388 | 29.7065 | R1[β (C–H) (37%)] + R1[β(C–C) (32%) |
| + R2[β (11O–12H) (26%)] | ||||
| 1374 | 66.1348 | 1455 | 85.4573 | υ (13C–14O) (29%) +β(14O-28H) (16%) |
| + R1[β (C–H) (13%) | ||||
| 1383 | 55.1456 | 1467 | 108.9683 | υ(20C–22N)[31%] +υ(19C–22N)[29%] |
| +β (22N–28H)19% | ||||
| 1388 | 34.3131 | 1480 | 3.8144 | R2[β (C–C) (31%)] + R2[β(C–H) (27%) |
| s+υ(20C–22N)[15%] | ||||
| 1415 | 10.8257 | 1496 | 20.0693 | R1[β (C–H) (21%)] + R1[β(C–C) (12%) |
| + R2[β (N–H) (11%)] + R2[β(C–H) (17%) | ||||
| 1428 | 8.8952 | 1513 | 10.8952 | R2[β (C–N–C) (39%)] +υ(C–O) (31%) |
| 1484 | 201.5601 | 1576 | 185.4923 | R1[β (C–H) (35%)] + R1[υ(C–C) (43%) |
| +υ (15C–13N) (17%) | ||||
| 1545 | 186.5887 | 1657 | 167.0252 | R1[β (C–H) (43%)] + R1[υ(C–C) (37%) |
| +β(13N–14O–28H) (16%) +υ (15C–13N) (13%) | ||||
| 1574 | 42.6826 | 1694 | 120.2316 | R1[β (C–H) (50%)] + R1[υ(C–C) (42%) |
| 1589 | 117.7402 | 1696 | 251.6079 | υ(17C–16N) (17%) +υ (15C–13N) (21%) |
| +β(13N–14O–28H) (27%) + R1[υ(C–C) (32%) | ||||
| 1605 | 723.5927 | 1730 | 696.6936 | υ(17C–16N) (37%) +υ (15C–13N) (26%) |
| +β(13N–14O–28H) (29%) | ||||
| 3020 | 12.2277 | 3171 | 10.2649 | R1[υ(17C–27H) (24%)] |
| 3027 | 25.2308 | 3179 | 19.3602 | R1[υ(9H–4C) (41%) +υ (8H–3C) (58%)] |
| 3298 | 189.0629 | 3492 | 220.0088 | υ(14O–28H) (98%) |
| 3420 | 222.3406 | 3592 | 271.9228 | υ(22N–23H) (99%) |
| 3674 | 103.9801 | 3870 | 114.1538 | υ(11O–12H)[100%] |
Fig. 2
Comparative IR spectra of all compounds.
2.3(O–H) and (C–H) vibrations
The aromatic C–H stretching vibrations lie in the range 3100–3000 cm-1 [42]. The C–H stretching vibrations for 4-Hydroxy Benzo Hydrazide are observed at 3081(Raman) and 3012 (IR) cm–1 which are in good agreement with the aromatic C–H stretching frequencies, observed at 3062, 3047, 3060 and 3080 cm–1 of benzene and its derivatives [43]. The aromatic C–H present in the benzene ring of BH are seen as medium to strong bands in IR at 3062, 3050 and 3027 cm–1 [29]. In accordance with the bending vibrations of benzene [44] the peaks seen at 1090, 1049 cm–1 (IR) and 1046 cm–1 (Raman) in 4-Hydroxy Benzo Hydrazide spectra are attributed to the C–H in-plane bending vibrations. The ring C–H out of plane bending vibrations of 4-Hydroxy Benzo Hydrazide are seen in the infrared spectrum at 965, 956, 941 and 777 cm–1 [43]. The stretching frequency of O–H is identified at 3428 cm–1 in IR spectrum while it at 3430 cm–1 in calculated data. The O–H in-plane bending mode is identified at 1257 cm-1 in the IR and at 1255 cm-1 in Raman spectra whereas the out of plane bending mode is identified at 631 cm-1 in the IR and at 641 cm-1 in Raman spectra which are in good agreement with calculated data. Experimental frequencies nearly matches with the the calculated frequencies of B3LYP as campared to CAM-B3LYP method. In vibrational assignments, the C–H and O–H stretching vibrations are in the same range for all the derivatives (I, II, III) discussed in Table 2, 3 and 4.
2.4(C–C) and (N–H) vibrations
The aromatic ring carbon–carbon stretching modes of benzene and its derivatives are noticed in the range of 1650–1200 cm–1 [45, 46]. Strong to medium lines as observed in the IR spectrum of 4-HBH at 1591, 1539, 1512, 1467, 1340 and 1282 cm–1 are described to the C–C stretching modes. Strong bands in IR at 1607 cm–1 and at 1612 cm–1 in Raman are assigned to the –NH2 deformation mode of 4-Hydroxy Benzo Hydrazide while in literature, The aromatic ring carbon–carbon stretchingmodes are also expected in the range from 1650 to 1200 cm–1. Benzene has two degenerate modes at 1596 cm–1 and 1485 cm–1. Similarly the frequency of two non-degenerate modes observed at 1310 cm–1 and 995 cm–1 in benzene [43]. The N–H stretching vibration of hydrazide group appears at 3280 cm-1 in IR and at 3256 cm-1 in Raman spectra. The N–H stretching vibrations are normally viewed in the region 3300–3600 cm–1. For isomer I, the N–H stretching vibration is calculated at 3372 cm–1 while it is 3522 and 3420 cm–1 for isomer II and III respectivelly. In lower region (below 800 cm–1), torsional vibration of C=C–N–H are also seen in the assignment of all isomers (I, II & III). For isomer I, the C–C stretching vibrations are calculated at 1492 (1568) cm–1 while it is at 1486 (1577) and 1484 (1576) cm–1 [B3LYP(CAM-B3LYP)] for isomer II and III respectivelly.
2.5Other modes of vibration
In 4-Hydroxy Benzo Hydrazide, the ring breathing mode is calculated at 602 cm–1 which is in good agreement with the experimental data, that is, 605 cm–1, while in all the derivatives (I,II, III), ring breathing modes are at 606 (640), 612 (645), and 610 (642) cm–1 having appropriate IR intensity. As expected, torsion modes along with wagging modes appear in the lower frequency range. For 4-Hydroxy Benzo Hydrazide, strong torsion mode of C–C–C–C is at 564 cm–1 in calculated spectrum which matches well with the experimental one, that is, 570 cm–1,while strong torsion modes of C–C–C–C are at 651, 697 and 647 cm–1 in calculated spectrum for isomer I, II and III respectively. A very strong stretching vibration of C=O is found at 1693 (1741) cm–1 for isomer I while C–O vibrations are at 1423 cm–1 and 1430 cm–1 in calculated spectra for isomer II and III. In literature [29], the frequency of the carbonyl stretching vibration is absorbed at 1661 cm–1 in the infrared spectrum, the reason being that the double bond character of the C=O group is less due to the nitrogen lone pair electron being delocalized towards the carbonyl end. There are some frequencies in lower region having appreciable IR intensity. Furthermore, the study of low frequency vibrations is of great significance, because it gives information on weak intermolecular interactions, which take place in enzyme reactions [47]. Knowledge of low frequency mode is also essential for the interpretation of the effect of electromagnetic radiation on biological systems [48]. The full interpretation of vibrational spectra of 4-Hydroxy Benzo Hydrazide and its derivatives is in good agreement with the literature [49]. The aim of vibrational analysis is to acquire direct information on lower and higher frequency vibrations of such 4-Hydroxy Benzo Hydrazide and its derivatives. No experimental FTIR spectrum is available for comparison for derivatives of 4-Hydroxy Benzo Hydrazide so it will provide a suitable path for experimental researchers.
2.6Electronic properties and TDDFT analysis
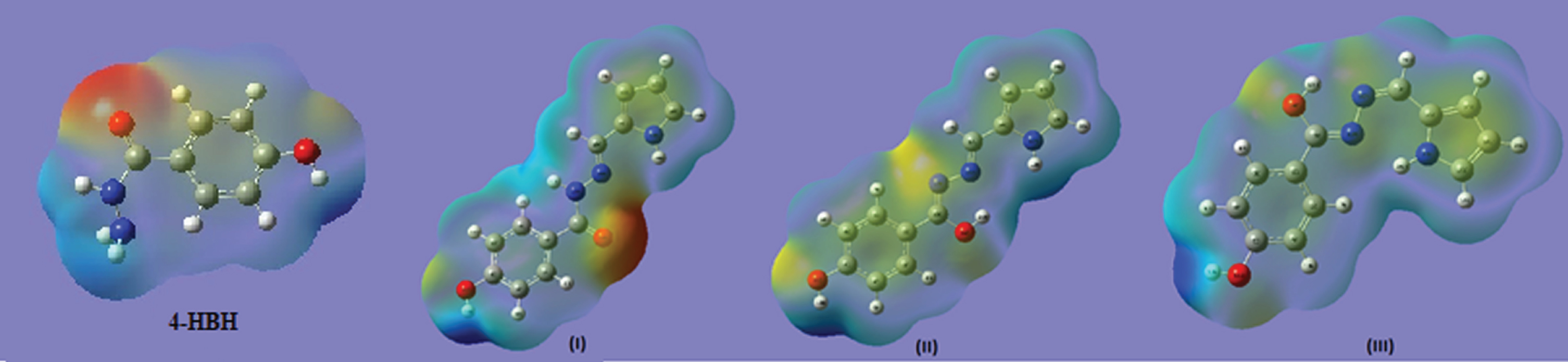
Frontier orbital energy gap, that is, the gap between HOMO and LUMO, shows the interaction of that molecule with other species. Frontier orbital energy gap helps to differentiate the chemical reactivity of the molecules [50, 51]. In case of 4-Hydroxy Benzo Hydrazide and its derivatives (I, II nad III), frontier orbital energy gap is 5.35792, 2.03075, 3.62059 and 3.79059 eV, respectively for B3LYP method, and is given in Table 5 with CAM-B3LYP method too. In literature [29], the frontier orbital energy gap of benzo hydrazide is nearly 5.6052 eV, So it can be concluded that isomer I is the most reactive compound among all. The HOMOs and LUMOs are seen to be localized on molecules as a whole for 4-Hydroxy Benzo Hydrazide, isomers II and III while for isomer I, the LUMOs are seen to be localized on molecules as a whole but HOMOs are seen over enaminone group only. Molecular electrostatic potential maps are very useful three dimensional diagrams of molecules. They enable us to visualize the charge distributions of molecules and charge related properties of molecules. They also allow us to visualize the size and shape of molecules. In organic chemistry, electrostatic potential maps are invaluable in predicting the behaviour of complex molecules [52]. The pictures of HOMO, LUMO and electrostatic potential (MESP) for 4-HBH and derivatives (I, II & III) are shown in Fig. 3 & 4 respectivelly. DOS plot (Supplementary Figure 3) shows the features of the Molecular Orbital in a specific energy selection and population analysis per orbital. It shows interaction of anti-bonding or bonding characters among two orbitals. The edge is calculated by the degree of negative (anti-bonding interaction) and positive (bonding interaction) overlap for a specific MO. It illustrates anti-bonding charm of both the frontier orbitals. Charge localization over HOMO, LUMO as well as for affiliated MO are verified with the plot.
Table 5
HOMO-LUMO orbital energies (eV) and their energy band gap, dipole moment (D) of the three Isomers of title compound computed at B3LYP and CAM-B3LYP/6-311 G (d, p) level
| Parameters | B3LYP | CAM-B3LYP | ||||||
| 4HBH | Isomer I | Isomer II | Isomer III | 4HBH | Isomer I | Isomer II | Isomer III | |
| ELUMO | –0.04106 | –0.09246 | –0.05607 | –0.06032 | –0.00399 | –0.00762 | –0.01428 | –0.01794 |
| EHOMO | –0.23877 | –0.16712 | –0.18918 | –0.19968 | –0.27888 | –0.25269 | –0.23629 | –0.24845 |
| EGAP | 0.19771 | 0.07466 | 0.13311 | 0.13936 | 0.27489 | 0.24607 | 0.22201 | 0.23051 |
| EGAP (eV) | 5.35792 | 2.03075 | 3.62059 | 3.79059 | 7.44952 | 6.66590 | 6.01647 | 6.24682 |
| Dipole moment (μ) | 5.02854 | 2.6453 | 2.2634 | 2.1035 | 5.0889 | 2.8119 | 2.2895 | 2.1053 |
Fig. 3
LUMO-HOMO orbital Plots of 4-HBH and three isomers of title compound.
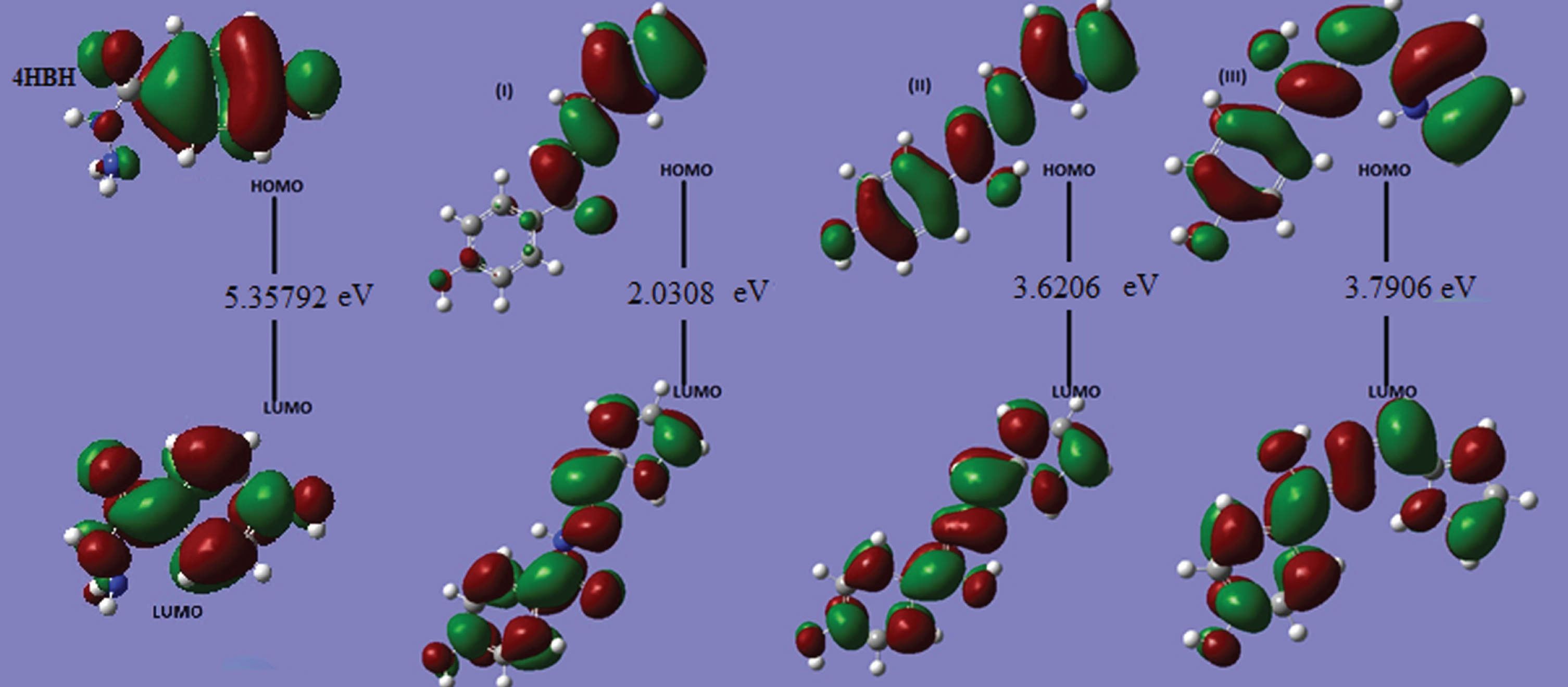
Fig. 4
MESP surfaces of 4-HBH and three isomers of title compound.
TDDFT method is an important tool for studying the nature of the transitions and UV spectrum of the title compound. TDDFT of title molecules are calculated by using combination of DFT/B3LYP and 6–311G(d, p) as the basis set. The calculated high oscillatory strength of electronic transitions are listed in Table 6 and shown in supplementary Figure 4. A prominent peak occurs at 236 nm, 323 nm, 330 nm and 335 nm in 4-Hydroxy Benzo Hydrazide (4HBH) and its newly designed derivatve I, II, III respectively. In 4HBH prominent peaks occurs due to transition of electron from HOMO-1⟶LUMO, HOMO⟶LUMO, HOMO-2⟶LUMO, HOMO-2⟶LUMO + 1, HOMO-1⟶LUMO + 1 with 11%, 62%, 5% 3% and 4% contribution respectively. In I, II & III isomers, prominent peaks occurs due to electronic transitions occurs from HOMO⟶LUMO with contribution 96% 100% 99% respectively. In I and III isomer, a bumf occurs at 286nm and 279 with their corresponding transition from HOMO⟶LUMO + 1(93%), HOMO⟶LUMO + 2(4%) and HOMO⟶LUMO + 1(91%), HOMO-4⟶LUMO (5%) respectively. On basis of assignment, these transition originated due to np⟶Ry*.
Table 6
The calculated electronic transitions: E (eV), oscillatory strength (f),λmax (nm) using TD–DFT/B3LYP/6–311G (d, p)
| S.N. | Electronic Transitions | E (eV) | Oscillatory Strength (f) | Calculated (λmax) | % Contribution | Assignment |
| Isomer I | ||||||
| 1 | H⟶L | 3.84 | 0.723 | 323 | 96 | np⟶Ry* |
| 2 | H⟶L + 1 | 4.33 | 0.040 | 286 | 93 | np⟶Ry* |
| H⟶L + 2 | 4 | |||||
| 3 | H-3⟶L | 4.43 | 0.037 | 280 | 59 | np⟶Ry* |
| H-1⟶L | 31 | |||||
| H-3⟶L + 2 | 2 | |||||
| Isomer II | ||||||
| 1 | H⟶L | 3.76 | 1.219 | 330 | 100 | np⟶Ry* |
| 2 | H-2⟶L | 4.07 | 0.001 | 305 | 99 | np⟶Ry* |
| 3 | H-1⟶L | 4.54 | 0.011 | 273 | 38 | np⟶Ry* |
| H⟶L + 1 | 52 | |||||
| H-4⟶L | 2 | |||||
| H⟶L + 2 | 3 | |||||
| Isomer III | ||||||
| 1 | H⟶L | – | 0.6475 | 335 | 99 | np⟶Ry* |
| 2 | H-2⟶L | – | 0.0014 | 306 | 92 | np⟶Ry* |
| H-1⟶L | 5 | |||||
| 3 | H⟶L + 1 | – | 0.0393 | 279 | 91 | np⟶Ry* |
| H-4⟶L | 5 | |||||
| 4-Hydroxy-Benzo-Hydrazide | ||||||
| 1 | H-2⟶L | 4.83 | 0.0161 | 257 | 17 | np⟶Ry* |
| H-1⟶L | 56 | |||||
| H⟶L | 13 | |||||
| H-3⟶L | 5 | |||||
| 2 | H-2⟶L | 5.07 | 0.0114 | 245 | 30 | np⟶Ry* |
| H⟶L + 1 | 56 | |||||
| H-3⟶L | 6 | |||||
| H-1⟶L | 6 | |||||
| 3 | H-1⟶L | 5.25 | 0.2595 | 236 | 11 | np⟶Ry* |
| H⟶L | 62 | |||||
| H-2⟶L | 5 | |||||
| H-2⟶L + 1 | 3 | |||||
| H-1⟶L + 1 | 4 | |||||
2.7Optical, dipole moment and thermo-dynamical
Dipole moment (μ), polarizability < α>and total first static hyperpolarizability β [53, 54] can be expressed in terms of x, y, z components and are given by following equations 1, 2 and 3-
(1)
(2)
(3)
The β components of Gaussian output are reported in atomic units (where 1 a.u. = 8.3693*10–33 e.s.u.). The calculated dipole moments (Table 5) for 4-Hydroxy Benzo Hydrazide and its derivatives are 5.02854, 2.6453, 2.2634 and 2.1035 Debye respectively. So, 4-HBH is a better solvent among them all. A greater contribution of αzz (lesser of αxx) is seen in all compounds that means these compounds are elongated more towards Z direction and is more contracted in the X direction. βxxx, βxxy and βyyy contribute lager part of hyperpolarizability in all the molecules. This shows that X-axis, XY plane and Y axis are more optically active in these directions. The values of hyperpolarizability indicate a possible use of these compounds in electro-optical applications. Supplementary Table 2 shows the values of polarizability and hyperpolarizability of 4-Hydroxy Benzo Hydrazide and its derivatives. Internal thermal energy (E), constant volume heat capacity Cv, and entropy S, calculated at B3LYP/CAM-B3LYP/6-311G (d, p) level, are listed in supplementary Table 3. We know that the conduction band is almost empty at the room temperature, so electronic contribution in total energy is negligible. Thermodynamic properties show that the vibrational motion plays an important role as compared to other motions.
2.8NBO analysis
Natural bond analysis is an important tool for studying intermolecular and intramolecular interaction and charge transfer and conjugate interaction in molecular system [55]. The strength of interaction depend on second order perturbation energy E(2). The larger value of E(2) mean stronger interaction however lower E(2) value means weak interaction. The strength of delocalization interaction or second order energy For acceptor NBO (j) and, donor NBO (i), is related with second order energy lowering equation 4 as [56, 57]
(4)
Here, qi is the population of donor orbital or donor orbital occupancy, ɛi, ɛj are orbital energies (diagonal elements) of acceptor and donor NBO orbital’s respectively, Fij is the off–diagonal Fock or Kohn–Sham matrix element between i and j NBO orbitals. NBO analyses of all compounds are calculated by using same level of theory are given in supplementary Tables 4, 5, 6 and 7.
Table 7
Calculated ɛHOMO, ɛLUMO, energy band gap (ɛLUMO –ɛHOMO), chemical potential (μ), electronegativity (χ), global hardness (η), global softness (S), and global electrophilicity index (ω) forI, II and III at B3LYP/6-311 G (d, p) level
| Folder | ɛH | ɛL | ɛH - ɛL | χ | μ | η | S | ω |
| I | –0.16712 | –0.09246 | –0.07466 | 0.12979 | –0.12979 | 0.03733 | 13.39405 | 3.47682 |
| II | –0.18918 | –0.05607 | –0.13311 | 0.12262 | –0.12262 | 0.06655 | 7.51258 | 1.84238 |
| III | –0.19968 | –0.06032 | –0.13936 | 0.13000 | –0.13000 | 0.06968 | 7.17566 | 1.86567 |
| 4HBH | –0.23877 | –0.04106 | –0.19771 | 0.13991 | –0.13991 | 0.09885 | 5.05791 | 1.41530 |
In NBO analysis of 4-Hydroxy Benzo Hydrazide and its derivatives, significant interactions formed by orbital overlap between Lp(1)N/Lp(1)O, σ(N–N), π(C–C) and π*(C–C), σ* (C–C)/π*(C–N) which stabilizes the system by inter molecular charge transfer. In 4-HBH, significant contribution occurs in between Lp(1)N14⟶π*(C12 - O13), Lp(2)O13⟶σ*(C12–N13)/σ*(C12–C2), Lp(2)O11⟶π*(C5–C6), π(C1–C2)⟶π*(C3–C4)/π*(C5–C6)/π*(C12–O13), which stabilize the molecule by 38 kcalmol–1, 23 kcalmol–1/18 kcalmol–1, 30 kcalmol–1 23 kcalmol–1/16 kcalmol–1/17 kcalmol–1 respectively. In isomer I of 4-HBH, most significant contribution occurs in between π(C1–C6) ⟶π* (C3–C4)/ π*(C5–C4), π(C4–C5) ⟶π*(C1–C6)/ π*(C2–C3), which stabilizes the molecules II and III by 17 kcalmol–1/24 kcalmol–1, 15 kcalmol–1/24 kcalmol–1 and 17 kcalmol–1/24 kcal mol–1,15 kcalmol–1/23 kcalmol–1 respectively. In isomer II, two significant contribution came from lp(1)N15⟶π*(C13–N15), lp(1)O11⟶π*(C4–C5) which stabilizes isomer II by 44 kcalmol–1, 30 kcalmol–1 respectively however in isomer III, two significant interaction occurs due to lp(1)N14⟶π*(C13–N15), lp(2)O11⟶π*(C4–C5) which stabilizes isomer III by 48 and 30 kcalmol–1 respectively. In isomer I, another significant contribution occurs due to orbital interaction between π(C1–C6) ⟶π*(C2–C3)/ π*(C4–C5), π(C4–C5) ⟶π*(C1–C6)/ π*(C2–C3), lp(1)N15⟶π*(C13–N15), lp(1)O11⟶π*(C4–C5) which stabilizes by 17 kcalmol–1/25 kcalmol–1, 16 kcalmol–1/25 kcalmol–1, 44 kcalmol–1, 30 kcalmol–1 respectively. NBO analysis shows that most significant interaction occurs due to moment of π–electron cloud from donor to accepter which develops polarity of these isomers. The moment of π–electron clouds are responsible for the NLO activity these isomers.
2.9Global reactivity descriptors
Global reactivity descriptors are described as –
All these parameters for all compounds have been listed in Table 7.
According to these parameters, the chemical reactivity varies with the structural configuration of molecules. Chemical hardness (softness) value of compound (I) is lesser (greater) among all the molecules. Thus, compound (I) is found to be more reactive than all whereas, compound 4-HBH is less reactive. Compound 4-HBH possesses higher electronegativity (lower electrophilicity index) among them all. Correlations have been found between electrophilicity of various chemical compounds and reaction rates in biochemical systems.
2.10Molecular docking
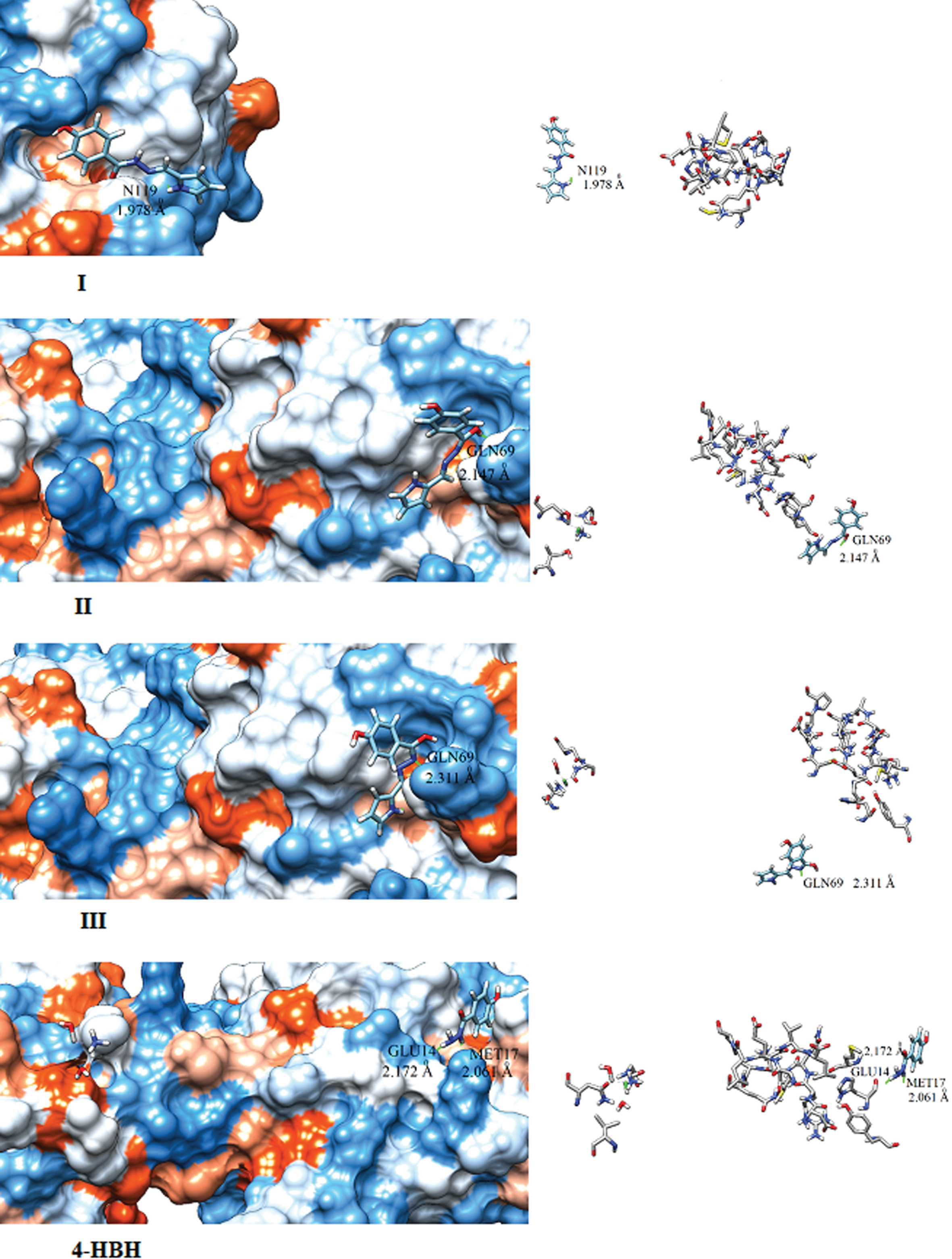
Hydrazides are well known pharmaceutical compound and show better activity against for anti-bacterial, antifungal, anticonvulsant activities In this segment, we calculate the biolocal activities of 4-HBH and its isomers (I, II, III). For this we first calculate their electro-topological indices like LogP,LogS by using ALOGPS 2.1 software [63]. This Program was developed by Tetko et.al. [64–66]. The calculated value of LogP is largest for isomer I however 4HBH have lowest Log P value. This means transportation through cell membranes varies according to I > III > II > 4-HBH. The value of Log S for 4HBH and its derivatives lies in between –2.78 to –1.33 however 85% drugs having Log S value lie in between –1 to –5.36. The calculated values of Log S are favouring the permeability for 4HBH and its isomers into cell are better through membranes. Several Biological activities of 4HBH and its derivatives are calculated by PASS software. By using molecular mechanics PASS predict 900 pharmacological effects e.g. mutagenicity, teratogenicity and embryo toxicity. The Biological activities are calculated by PASS more than 46,000 drugs whose biological activities are determined experimentally are selected as training set and 85% results predicted by PASS software are correct [67]. In Table-8, we have compared calculated biological activities of 4-HBH and its isomers. All (4HBH and its isomers) show good biological activity against Anti-tuberculosis and Antivirals. Now a day, COVID 19 is a serious disaster however a number of studies are being carried out on antiviral and Anti-tuberculosis drugs to kill COVID 19 virus. In this way, our study has ability to become good drug for COVID19. To design new COVID 19 drug, first priority is to identify targets which, when inhibited, can kill the effected cells. In February 2020 researchers have identified COVID 19 protease and named as 6LU7. We have performed docking by using Swissdock online server [68] by using PDB file of 6LU7 protease [69]. Docking of 6LU7 protease with 4HBH and its isomers are blindly followed over whole molecules. The binding strength are describe by full fitness score (FF) and binding affinity. The more appropriate binding site between a ligand and its receptor is shown by highest negative FF score. The calculated FF, binding affinity, binding distance and binding site are listed in Table 9. The binding affinities (ΔG) obtained from docking of 6LU7 with I, II, III and 4HBH are –6.21 kcal/mol, –6.58 kcal/mol, –6.51 kcal/mol and –6.37 kcal/mol, respectively. The isomer-I, binds with polar Asparagine (N119) residue by distance 1.978 A0 however isomers II and III bind with polar amino acid Glutamine (GLN69) residue with distances 2.147 A0 and 3.310 A0 respectively. The 4HBH binds with two residues amphipathic amino acid Methionine (MET17), and polar amino acid Glutamine (GLU14) with distance 2.061 A0 and 2.172 A0 respectively. Docking picture of I, II, III & 4HBH with 6LU7 protease are shown in Fig. 5. This study only provides a path to experimental researchers to designed new COVID 19 drug. This study is only based on molecular modelling and docking, not on clinical trials, as we did not consider its side effects and toxicity.
Table 8
Biological activity of 4HBH and its Isomers predicted by PASS program
| Biological Activity | I | II | III | 4HBH | ||||
| Pa | Pi | Pa | Pi | Pa | Pi | Pa | Pi | |
| Antituberculosic | 0.659 | 0.005 | 0.704 | 0.009 | 0.753 | 0.003 | 0.768 | 0.003 |
| Antiviral | 0.445 | 0.024 | 0.505 | 0.019 | 0.519 | 0.041 | 0.526 | 0.040 |
Table 9
Electrotopological indices like log Pand log S of I, II, III and 4HBH compounds
| Parameter | I | II | III | 4HBH |
| LogP | 2.37 | 0.40 | 1.14 | 0.12 |
| LogS | –2.78 | –2.08 | –2.65 | –1.33 |
| FF(a.u) | –1176.25 | –1189.49 | –1183.29 | –1170.09 |
| ΔG(kcal/mol) | –6.21 | –6.58 | –6.51 | –6.37 |
| Binding length(A0) | 1.978 A0 | 2.147 A0 | 3.310 A0 | 2.061 A0, 2.172 A0 |
| Binding Residue | N119 | GLN69 | GLN69 | MET17, GLU14 |
Fig. 5
Docking pictures of 4-HBH and I, II and III compounds.
2.11AIM analysis
At bond critical point (BCP) for any molecular system QTAIM [70], is important method to analyze the H–bonding and other interactions in terms of topological parameters. For nonbonding interactions [71] the electron density (ρH …A) should lies within 0.002–0.040 a.u. ranges and Laplacian (▿2ρBCP) should varries in between 0.024–0.139 a.u. Based on this criteria, no H-bonding appears in isomer I however one H-bonding appears in 4HBH (N15-H7), isomer II (N16-H28) and two in isomer III (N15-H24, N16-H18).The AIM pictures of these compounds with BCP point(green color) are plotted in Fig. 6. The calculated topological parameters corresponding H-bonding intreactions for 4HBH and its derivatives e.g. electron density, ρ(r), Laplacian
Fig. 6
AIM picture of III, II and 4HBH (Green dots represent BCP, Red dots represent ring critical).
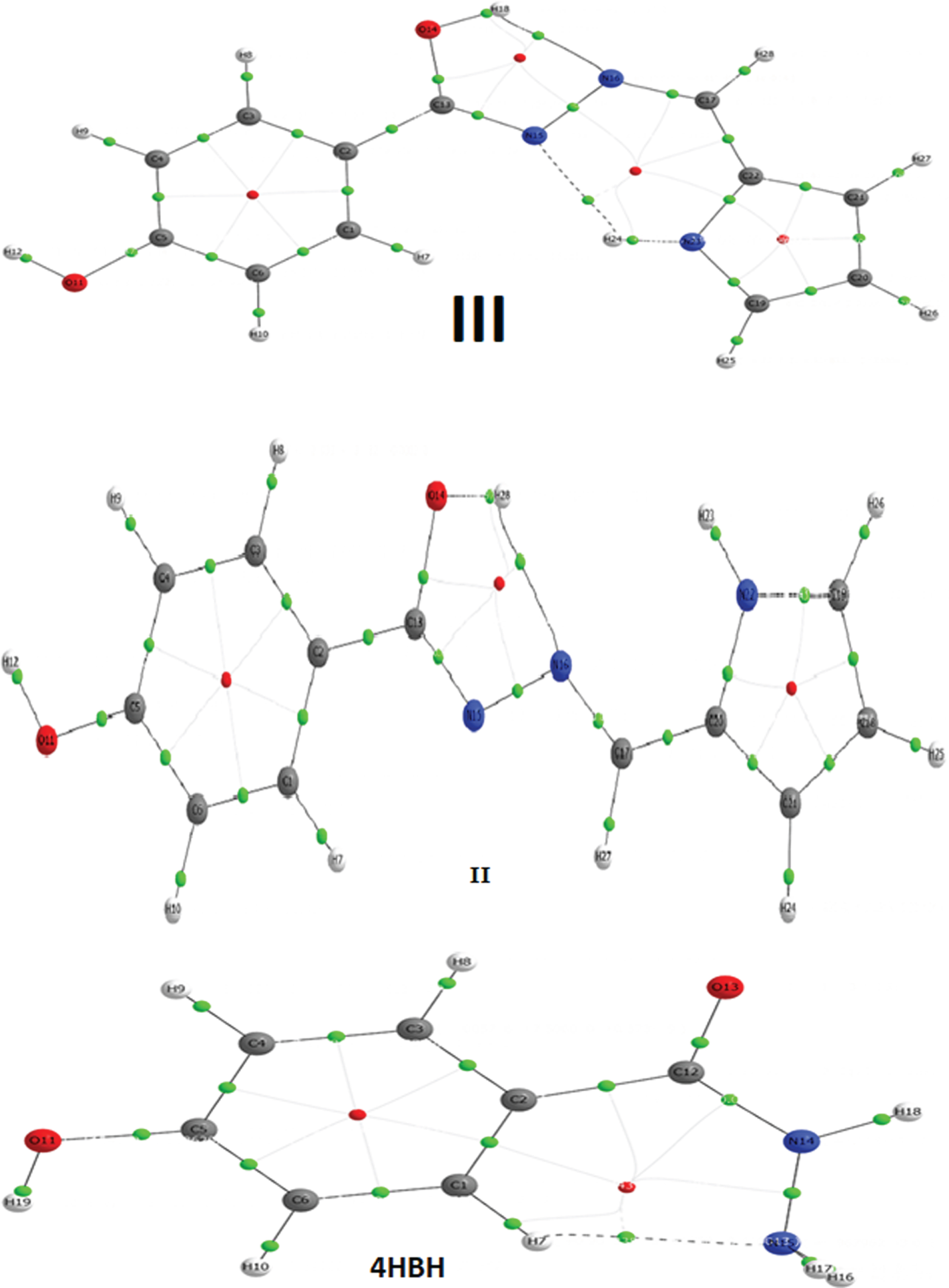
Table 10
Topological parameters for bonds of interacting atoms: electron density (ρBCP), Laplacian of electron density (∇2BCP), kinetic electron energy density (G BCP), potential electron energy density (VBCP), total electron energy density (HBCP), estimated interaction energy (Eint) at bond critical point (BCP)
| Species | Bond | ρbcp | ▿2ρbcp | Eigen value ▿2ρbcp | GBCP | VBCP |
| HBCP | Eint (kcal/mol) | |||
| λ1 | λ2 | λ3 |
| |||||||||
| Benzo | N15–H7 | 0.0176 | 0.0657 | –0.0318 | –0.2198 | 0.1620 | 0.196 | 0.0151 | –0.0140 | 0.927 | 0.0013 | 4.379 |
| Benzo-1 | – | – | – | – | – | – | – | – | – | – | – | – |
| Benzo-2 | N16–H28 | 0.0333 | 0.1174 | –0.0464 | –0.3213 | 0.1958 | 0.237 | 0.0319 | –0.0345 | 1.082 | –0.0026 | 10.818 |
| Benzo-3 | N15–H24 | 0.0249 | 0.0884 | –0.0289 | –0.0278 | 0.1451 | 0.199 | 0.0199 | –0.0178 | 0.894 | 0.0022 | 5.572 |
| N16–H18 | 0.0397 | 0.1254 | –0.0551 | –0.0492 | 0.2298 | 0.240 | 0.0337 | –0.0361 | 1.071 | –0.0024 | 11.326 | |
In energetically stable molecular system, the Laplacian
3Conclusion
We have designed and performed the DFT based calculations on 4-Hydroxy Benzo Hydrazide (4-HBH) and its derivatives (isomers I, II and III) with B3LYP/6-311 G (d, p) basis set. As 4-HBH and its derivatives (isomers I, II and III) are found to be potential pharmaceutically active, so we have focused our discussion mainly on vibrational, electronic and docking studies. On the basis of our calculations, we can conclude that, the addition of five membered ring to make new structures in 4-HBH structure tends to affect structural parameters of 4-HBH due to steric effects. The addition of five membered ring tends to increase the reactivity of molecules. Compound I is found to be more reactive than others due to its small energy gap between HOMO and LUMO. Chemical hardness (softness) value of compound I is lesser (greater) among all. Thus the compound (I) is found to be more reactive than all supported by hardness/ softness values too. The vibrational properties are also affected remarkably with the addition of five membered ring. In the absence of experimental IR spectra, we have reported theoretical vibrational spectra of all these compounds as this data is beneficial for the experimental researchers in near future. Therefore, the reported information should be reliable for future investigations. QATIM analysis shows that hydrogen bonding occurs in 4HBH, isomer II and III respectively. The most favourable binding occurs in isomer II by 6LU7 protease however most unfavourable binding occurs in between 4HBH and 6LU7 protease. From the above discussions in molecular docking section, we can say that 4-HBH and its derivatives (I, II, & III) may have good potential for treatment of COVID19. Therefore, we suggest quick clinical trials with 4-HBH and its derivatives (I, II, & III). Further studies are still in progress.
Acknowledgment
*This research is dedicated to all the volunteers from all over the world, who are serving against the COVID-19.
References
[1] | Update on the prevalence and control of novel coronavirus-induced pneumonia as of 24:00 on February 21. http://www.nhc.gov.cn/xcs/yqtb/202002/543cc508978a48d2b9322bdc83daa6fd.shtml (accessed February 23, 2020). |
[2] | http://aajtak.indiatoday.in (31, March 2020). |
[3] | Guidelines for the Prevention, Diagnosis, and Treatment of Novel Coronavirus-induced Pneumonia, The 6th ed. http://www.nhc.gov.cn/yzygj/s7653p/202002/8334a8326dd94d329df351d7da8aefc2/files/b218cfeb1bc54639af227f922bf6b817.pdf (accessed February 23, 2020). |
[4] | Ashiq U. , Ara R. , Mahroof–Tahir M. , Maqsood Z.T. , Khan K.M. , Khan S.N. , Siddiqui H and Choudhary M.I. , Chem Biodivers 5: (1) ((2008) ), 82. |
[5] | Ara R. , Ashiq U. , Mahroof-Tahir M. , Maqsood Z.T. , Khan K.M. , Lodhi M.A. and Choudhary M.I. , Chem Biodivers 4: ((2007) ), 58. |
[6] | Maqsood Z.T. , Khan K.M. , Ashiq U. , Jamal R.A. , Chohan Z.H. , Mahroof– Tahir M. and Supuran C.T. , J Enz Inhib Med Chem 21: ((2006) ), 37. |
[7] | Markley K.S. , Fatty Acids, their Chemistry, Properties and Uses, InterScience Publishers, New York, (1964) . pp. 1604. |
[8] | Mir I. , Siddiqui M.T. and Comrie A. , Tetrahedron 26: ((1970) ), 5235. |
[9] | Degener E. , Scheinplug H. and Schemelzer H.G. , Brit Patent No.: 1035474, 1967. |
[10] | Cavier R. and Rips R. , J Med Chem 8: ((1965) ), 706. |
[11] | Raval D.A. and Toliwal S.D. , J Oil Tech Assoc India 26: ((1994) ), 27. |
[12] | Al-Saadi Abdulaziz A. , Journal of Molecular Structure 1023: ((2012) ), 115. |
[13] | Suresh D.M. , Sajan D. , Diao Yun-Peng , Nemec I. , Hubert Joe I. and Bena Jothy V. , Spectrochimica Acta Part A: Molecular and Biomolecular Spectroscopy 110: ((2013) ), 157. |
[14] | Saleem H. , Subashchandrabose S. , Ramesh Babu N. and Syed Ali Padusha M. , Spectrochimica Acta Part A: Molecular and Biomolecular Spectroscopy 143: ((2015) ), 230. |
[15] | Vijayaraj K.K. , Narayana B. , Ashalatha B.V. , Suchetha Kumari N. and Sarojini B.K. , Eur J Med Chem 42: ((2007) ), 425. |
[16] | Abadi A.H. , Eissa A.A.H. and Hassan G.S. , Chem Pharm Bull 51: ((2003) ), 838. |
[17] | Bonde C.G. and Gaikwad N.J. , Bioorg Med Chem 12: ((2004) ), 2151. |
[18] | Bhandari S.V. , Patil A.A. , Sarkate V. , Gore S.G. and Bothra K.G. , Bioorg Med Chem 17: ((2009) ), 390. |
[19] | Sugano K. , Hamada H. , Machida M. and Ushio H. , J Biomol Screen 6: ((2001) ), 189. |
[20] | Suresh D.M. , Sajan D. , Diao Yun-Peng , Nemec I. , Hubert Joe I. and Bena Jothy V. , Spectrochimica Acta Part A: Molecular and Biomolecular Spectroscopy 110: ((2013) ), 157. |
[21] | Arjunan V. , Rani T. , Mythili C.V. and Mohan S. , Spectrochimica Acta Part A 79: ((2011) ), 486. |
[22] | Sánchez-Lozano Marta , Vázquez-López Ezequiel M. , HermidaRamón José M. and Estévez Carlos M. , Polyhedron 30: ((2011) ), 953. |
[23] | Bharty M.K. , Dani R.K. , Kushawaha S.K. , Om Prakash, Ranjan K. Singh , Sharma V.K. , Kharwarc R.N. and Singh N.K. , Spectrochimica Acta Part A: Molecular and Biomolecular Spectroscopy 145: ((2015) ), 98. |
[24] | Dwivedi A. , Srivastava A.K. and Bajpai A. , Spectrochimicaacta part A: molecular and biomoleculat spectroscopy 149: ((2015) ), 343. |
[25] | Dwivedi A. , Pandey A.K. and Misra N. , Spectroscopy: An International Journal 27: (3) ((2012) ), 155. |
[26] | Dwivedi A. , Pandey A.K. and Misra N. , Spectroscopy: An International Journal 26: ((2011) ), 367. |
[27] | Pandey A.K. , Dwivedi A. and Misra N. , Spectroscopy: An International Journal, Volume 2013 (2013): Article ID 937915, pp. 11. |
[28] | Dwivedi A. and Kumar A. , Polycyclic Aromatic Compounds, 2019, https://doi.org/10.1080/10406638.2019.1591466. |
[29] | Arjunan V. , et al., Spectrochimica Acta Part A 79: ((2011) ), 486–496. |
[30] | Gupta S.S. , Marchno A. , Pradhan R.D. , Desai C.F. and Melikechi J. , J Appl Phys 89: ((2001) ), 4939. |
[31] | Karna S.P. , J Phys Chem A 104: ((2000) ), 4671. |
[32] | Frisch M.J. , Trucks G.W. , Schlegel H.B. , et al., Gaussian 03. Wallingford (CT): Gaussian Inc.; 2003. |
[33] | Frisch E. , Hratchian H.P. , Dennington R.D. II, et al., Gaussian Inc., GaussView. 2003. |
[34] | Becke A.D. , J Chem Phys 98: ((1993) ), 5648. |
[35] | Lee C. , Yang W. and Parr R.G. , Phys Rev B 37: ((1988) ), 785. |
[36] | Tawada Y. , Tsuneda T. , Yanagisawa S. , et al., J Chem Phys 120: ((2004) ), 8425. |
[37] | Ar. H. , Ozpozan T. , Büyükmumcu Z. , et al., J Mol Struct 1122: ((2016) ), 48. |
[38] | Jamroz M.H. , “Vibrational Energy Distribution Analysis; VEDA4 Program,”, SpectroChimica Acta A 114: ((2013) ), 220. |
[39] | |
[40] | Kleinman D.A. , Phys Rev 126: ((1962) ), 1977. |
[41] | Pipek J. and Mezey P.Z. , J Chem Phys 90: ((1989) ), 4916. |
[42] | Mahmoudi G. , Rostamnia S. , Zaragoza G. , Brito I. , Cisterna J. and Cardenas A. , J Chil Chem Soc 64: (3), 2019. |
[43] | Bellamy L.J. , The Infrared Spectra of Complex Molecules, third ed., Wiley, New York, (1975) . |
[44] | Silverstein R.M. , Bassler G.C. and Morrill T.C. , Spectrometric Identification of Organic Compounds fifth ed., John Wiley & Sons, Inc., New York, (1981) . |
[45] | Arjunan V. , Mohan S. , Subramanian S. and Thimme B. , Gowda, Spectrochim Acta 60A: ((2004) ), 114. |
[46] | Varsanyi G. , Assignments for Vibrational Spectra of Seven Hundred Benzene Derivatives, vol. I, Adam Hilger, London, 1974. |
[47] | Chou K.C. , Biophysical Journal 45: (5) ((1984) ), 881. |
[48] | Frohlich H. , Biological Coherence and Response to External Stimuli, Springer, Berlin, Germany, (1988) . |
[49] | Arjunan V. , Jayaprakash A. , Carthigyan K. , Periandy S. and Mohan S. , Spectro chim acta A 108: ((2013) ), 100–114. |
[50] | Gutowski M. and Chalasinski G. , J Chem Phys 98: ((1993) ), 4540. |
[51] | Bose S.C. , Saleem H. , Erdogdu Y. , Rajarajan G. and Thanikachalam V. , Spectrochim. Acta part A 82: ((2011) ), 260. |
[52] | Murray J.S. and Sen K. , Molecular electrostatic potentials, Concepts and Applications Elsevier Amsterdam (1996) . |
[53] | Kleinman D.A. , Phys, Rev B 126: ((1977) ), 1962. |
[54] | Pipek J. and Mezey P.J. , J Chem Phys 90: ((1989) ), 4916. |
[55] | Erdogdu Y. , Unsalan O. and Gulluoglu M.T. , “FT-Raman, FT-IR spectral and DFT studies on 6, 8- dichloroflavone and 6, 8-dibromoflavone”, J Raman Spectrosc 41: ((2010) ), 820. |
[56] | Erdogdu Y. , Unsalan O. , Amalanathan M. and Hubert J.I. , J Mol Struct 980: ((2010) ), 24. |
[57] | Gonohe N. , Abe H. , Mikami N. and Ito M. , J Phys Chem 89: ((1985) ), 3642. |
[58] | Flippin L.A. , Gallagher D.W. and Jalali-Araghi K. , J Org Chem 54: ((1989) ), 1430. |
[59] | Parr R.G. , Szentpály L.V. and Liu S. , J Am Chem Soc 121: ((1999) ), 1922. |
[60] | Chattaraj P.K. and Giri S. , J Phys Chem A 111: ((2007) ), 11116. |
[61] | Padmanabhan J. , Parthasarathi Subramaniaan R. and Chattaraj P.K. , J Phys Chem A 111: ((2007) ), 1358. |
[62] | Ayers P.W. and Parr R.G. , J Am ChemSoc 122: ((2000) ), 2010. |
[63] | Jorgensen W.L. and Duffy E.M. , Bioorg Med Chem Lett 10: ((2000) ), 1155. |
[64] | Huuskonen J.J. , Livingstone D.J. and Tetko I.V. , J Chem Inf Comput Sci 40: (4) ((2000) ), 947. |
[65] | Tetk I.V. , Tanchuk V.Y. , Kasheva T.A. and Villa A.E. , J Chem Inf Comput Sci 41: ((2001) ), 1488. |
[66] | Tetk I.V. , Tanchuk V.Y. , Kasheva T.A. and Villa A.E. , J Chem Inf Comput Sci 41: ((2001) ), 1407–21. |
[67] | Tetko I.V. , Tanchuk V.Y. , Kasheva T.N. and Villa A.E.P. , J Chem Inf Comput Sci 41: ((2001) ), 246. |
[68] | |
[69] | http://www.rcsb.org/structure/6LU7 (accessed on 14 March 2020). |
[70] | Matta I.F. and Boyd R.J. , Wiley-VCH Verlag Gmbh, 2007. |
[71] | Koch U. and Popelier P. , J Phys Chem A 99: ((1995) ), 9747–9754. |
[72] | Bader R.F.W. , Atoms in Molecules: A Quantum Theory (2nd edn) (1990) (Oxford: New York, NY. |
[73] | Cremer D. , Kraka E. , Croat Chem Acta 57: (( 1984) ), 1259, 5B. D. Cremer, E. Kraka, Angew. Chem., Int Ed Engl 23 (1984), 627. doi:10.1002/ANIE.198406271 |
[74] | Bone R.G.A. and Bader R.F.W. , J Phys Chem A 100: ((1996) ), 10892. doi: 10.1021/JP953512M,. |
[75] | Bobrov M.F. , Popova G.V. and Tsirelson V.G. , Russ J Phys Chem 80: ((2006) ), 584. doi: 10.1134/S0036024406040182 |
[76] | Macchi P. and Sironi A. , Coord Chem Rev (2003), 238–239, 382. |
[77] | Espinosa E. , Alkorta I. , Elguero J. and Molins E. , J Chem Phys 117: ((2002) ), 5529. |
[78] | Bader R.F.W. , Atoms in Molecules: A Quantum Theory (2nd edn) n1990 (Oxford: New York, NY. |
[79] | Bader R.F.W. and Esse'n H. , J Chem Phys 80: ((1984) ), 1943. doi: 10.1063/1.446956. |
[80] | Jenkins S. and Morrison I. , Chem Phys Lett 317: ((2000) ), 97. |
[81] | Espinosa E. , Molins E. and Lecomte C. , Chem Phys Lett 285: ((1998) ), 170–173. |
[82] | Rozas I. , Alkorta I. and Elguero J. , J Am Chem Soc 122: ((2000) ), 11154–11161. |


