
Patient-Derived Renal Cell Carcinoma Xenografts Capture Tumor Genetic Profiles and Aggressive Behaviors
Abstract
BACKGROUND:
Patient-derived xenografts (PDX) have emerged as one of the most promising model systems to study cancer biology and to develop new antineoplastic drugs. Renal cell carcinoma (RCC) represents up to 90% of all kidney tumors, exhibits aggressive behavior, and has a propensity for metastasis. At diagnosis, 30% of patients with RCC have metastases, while up to 50% of those with localized disease treated with curative protocols experience recurrence.
OBJECTIVE:
This study aimed to establish an RCC PDX platform to identify novel clinical and molecular biomarkers of recurrence risk in order to facilitate precision medicine.
METHODS:
Tumor samples were obtained from surgical specimens of 87 RCC patients; fragments were implanted in immunodeficient NOD/SCID/gamma (NSG) mice. 17 Fragments were implanted subcutaneously in an initial group while a second group of 70 samples were implanted orthotopically in the subcapsular space.
RESULTS:
A total of 19 PDX developed only after orthotopic implantation, and included 15 cases of clear cell RCC subtype, 3 cases of papillary subtype, and 1 unclassifiable tumor. 1 PDX of clear cell RCC recapitulated the phenotype of vena caval tumor thrombus extension that had been diagnosed in the source patient. PDX characterization by immunohistochemistry and targeted sequencing indicated that all PDXs preserved RCC identity and major molecular alterations. Moreover, the capacity of tumor engraftment was a strong prognostic indicator for patients with locally advanced disease.
CONCLUSION:
Taken together, these results suggest that the orthotopic xenograft model of RCC represents a suitable tool to study RCC biology, identify biomarkers, and to test therapeutic candidates.
INTRODUCTION
Renal cell carcinoma (RCC) represents the most common kidney tumor, and exhibits aggressive behavior with a propensity for metastasis [1]. Worldwide, RCC is among the 10 most common malignancies, having caused an estimated 430,000 incident cases and 180,000 deaths during 2020 [2]. In a large Latin American cohort, the 5- and 10-year cancer-specific survival rates were 89.9% and 81.8%, respectively [3]. RCC comprises a group of morphologically and molecularly heterogeneous tumors that include several histological subtypes [4]. The most common are clear cell (ccRCC) and papillary (pRCC) RCCs [4, 5]. Formerly used classifications placed pRCC into two subgroups, papillary types 1 and 2, based upon morphological characteristics that are currently not recommended for histologic diagnosis [5]. The most recent studies show that the so-called type 1 pRCC includes a homogeneous group of tumors, with typical morphologic, immunohistochemical, and molecular features that do not occur in type 2 tumors, which are a heterogeneous group of neoplasms that include fumarate hydratase (FH)-deficient RCC, microphthalmia-associated transcription factor family translocation RCC, and other subtypes [6].
Most renal tumors are initially limited to the kidney. Surgery is the first treatment option [1]. However, despite significant improvements in surgical techniques and curative resection, hematogenous metastasis occurs in approximately 30% of patients [7]. The most frequent sites of distant metastases are lungs, bones, brain, adrenal glands, contralateral kidney, and liver [8]. In approximately 4% to 25% of patients with RCC, the primary tumor spreads by infiltrating the renal vein and inferior vena cava, and can extend to the right atrium, usually through the formation of a large tumor thrombus that portends a poor prognosis [9]. Systemic therapy is indicated for advanced and metastatic disease. Immune checkpoint inhibitor therapy (CTLA-4 plus PD-1 monoclonal antibodies) alone or in combination with anti-angiogenic tyrosine kinase inhibitors is the most effective therapeutic option; however, many patients do not respond to these treatments and also suffer from significant drug toxicities [10]. Recently, adjuvant pembrolizumab therapy significantly improved disease-free survival in patients at high risk for recurrence [11]. However, longer follow-up will be critical to assess the effect of pembrolizumab on overall survival [12]. Furthermore, more reliable biomarkers for recurrence risk should be identified to inform patient enrolment in clinical trials [13].
To overcome these uncertainties, experimental models that accurately capture the idiosyncrasies of RCC genetics, biochemistry, and pharmacology are essential to improve the predictive power of preclinical testing to determine the safety and efficacy of new therapeutic approaches. Consequently, patient-derived xenografts (PDX) are emerging as a relevant model system to translate new technology from the bench to the bedside [14, 15]. The most pronounced advantages of PDX arise from the fact that in addition to maintaining the 3D architecture of the original tumors, this model system may also capture systemic features that can only be recapitulated in vivo, such as metastasis [16, 17].
Herein, we present a comprehensive characterization of a cohort of RCC PDX. Morphologic and genetic tests confirmed that PDX preserved histology and intra- and intertumoral heterogeneity even after successive in vivo passages. Our results also demonstrated that in vivo tumor growth capacity is associated with a worse prognosis for patients with locally advanced disease.
MATERIAL AND METHODS
Patients
Patients with primary renal lesions were recruited after providing signed informed consent and undergoing partial or total nephrectomy. Tumor regions were identified in surgical specimens by a pathologist, and fresh fragments of up to 1 cm3 were separated and individually prepared for tumor implantation or DNA extraction and formalin fixation. Pathologic subtyping and staging were determined after tumor inoculation, occasionally resulting in the implantation of benign tumors, such as oncocytomas, renal cysts, and angiomyolipoma; and also RCCs other than ccRCC and pRCC, such as chromophobe RCC, which usually behave as indolent lesions. Such cases were excluded from the analysis, resulting in 87 patients with confirmed malignancies. Only one case of pT4 in which a biopsy specimen was used to inoculate NSG. This study was approved by Fundação Antonio Prudente, A.C. Camargo Cancer Center Institutional Review Board (process number 2201/16).
Animals
Male and female 2– 4 months old NOD. Cg-Prkdcscid Il2rgtm1Wjl mice, herein designated NOD/SCID/gamma or NSG mice, were obtained from the Jackson Laboratory (Bar Harbor, ME, USA). NSG colonies were maintained in the A.C. Camargo animal facility following the National Institutes of Health (USA) and institutional guidelines for animal welfare and experimental conduct [18]. This study was approved by the Animal Care and Use Committee of Fundação Antonio Prudente, A.C. Camargo Cancer Center (process number 075/16).
Tumor implantation
Ectopic: Tumor fragments of 1 to 3 mm were prepared from surgical specimen and implanted subcutaneously on the dorsum of NSG mice previously anesthetized with Ketamine/Xylasin cocktail. One to five fragments were implanted using a 11 G x 3-1/4” needle (Cadence Science) in each animal. Animals were monitored periodically for tumor growth for up to 15 months.
Orthotopic: Animals were anesthetized with Ketamine/Xylasin cocktail. The left lumbar region was trichotomized, followed by longitudinal lumbotomy of approximately 1 cm. Using small forceps, the left kidney was externalized and a transverse incision (2 mm) was made in the renal capsule. The capsule was dissected from the renal cortex with a blunt forceps, and 1 mm tumor fragments were gently positioned separately in subcapsular renal poles (2 to 4 fragment for each animal). The kidney was then returned to the abdominal cavity, and the lumbotomy incision was sutured. Animals were monitored to evaluate tumor growth using palpation. Monitoring was carried out up to 15 months. Animals that presented evidence of tumor growth were euthanized; tumors were removed and divided for reimplantation, cell culture, cryopreservation, DNA extraction, and histological evaluation.
Cryopreservation
We used two different protocols for cryopreservation: slow freezing and vitrification. Slow freezing is the conventional method used in living biobanks, and uses 10% dimethyl-sulfoxide (DMSO), 50% fetal bovine serum, and 40% culture medium. Cryotubes with tumor samples were transferred to freezing containers with slow cooling rates. Tubes were rapidly thawed at 37°C, and tumor fragments were washed twice in cold PBS and implanted. The vitrification method consisted of rapid freezing of tumor tissue in liquid nitrogen using a solution with basal medium, 0.5M sucrose + 10% fetal bovine serum (FBS). For thawing, successive washes were performed in three steps, first with a solution containing 80% DMEN, 20% FBS, and 0.5M sucrose for 45 min in orbital shaking at 4°C. In the second step, sucrose concentration was decreased to 0.25M. Fragments were incubated with sucrose-free medium during the third step [19].
Primary Cultures
Tumor fragments from PDX were minced into small pieces with the aid of a scalpel in 100 mm Petri dishes in cold Hank's balanced salt solution and centrifuged for two minutes at 1500 rpm. The pellet was resuspended in a mixture of 0.1% collagenase II or IV and 0.1% trypsin in orbital shaking (600 rpm) at 37°C for 90 min. Enzymatic activity was neutralized with Dulbecco's Modified Eagle Medium (DMEM) with high glucose plus 20% FBS. Tubes were centrifuged for 2 minutes at 1500 rpm, and pellets were resuspended in DMEM with high glucose. Cells were filtered through a 40-μm filter to remove non-dissociated cell clumps. The single cell suspension was then cultured in DMEM with 20% fetal bovine serum and 1% gentamicin on culture dishes in a modified atmosphere with 5% CO2 at 37°C.
Immunohistochemistry and immunofluorescence
Immunohistochemical staining was performed in the Ventana Discovery XT automated system (Ventana Medical Systems, Tucson, AZ, USA) using commercially available antibodies against PAX8 (clone MRQ-50-Ventana/Cell Marque), cytokeratin-7 (CK7, clone SP52-Ventana/Cell Marque), carbonic anhydrase IX (CAIX, clone EP161 Cell Marque), and alpha-methylacyl-CoA racemase (AMACR, clone 13H4, Cell Marque). Images were acquired by using a Aperio Imagescope (Leica Biosystems). Cultured cells were fixed for immunofluorescence with 4% paraformaldehyde for 20 min, washed twice with PBS, and incubated overnight with primary antibodies followed by incubation with fluorescent-conjugated AlexaFluor 488 antibodies. Images of cultured cells were obtained by using a Leica SP5 TCS SP II confocal microscope.
Targeted next-generation sequencing
Samples were provided by the A.C. Camargo Cancer Center Biobank. DNA extraction from fresh frozen or formalin-fixed paraffin-embedded patient tumors was performed in the QIASymphony platform (Qiagen, CA, EUA), following standard procedures. Purity and quantification were evaluated in the Nanodrop™ ND-1000 (Thermo Scientific, DE, USA) and the Qubit QUANT-IT dsDNA BR assay kit (Invitrogen Life Technologies, CA, USA), respectively. DNA integrity was determined in Agilent 4200 TapeStation (Agilent Technologies, CA, USA). DNA libraries were generated using QIAseq Targeted DNA Custom Panel and QIAseq 96-Index I Set A kit, following the manufacturer’s instructions and sequenced in the NextSeq 500 Illumina platform. The custom panel consisted of the complete coding regions of 21 frequently mutated genes in kidney cancer: VHL, ARID1A, KDM6A, PBRM1, SMARCA4, SMARCB1, BAP1, PCF11, NFE2L2, SETD2, AR, STAG2, KDM5 C, MET, CDKN2A, PTEN, FAT1, FH, TP53, NF2, and FLCN. This panel also included hotspot regions in MTOR, PIK3CA, NRAS, BRAF, EGFR, KRAS, and TCEB1.
To determine whether the mutational repertoires of patient tumors were present in the respective xenograft tumors, we performed either amplicon sequencing or sequencing of the custom gene panel in DNA extracted from PDXs at passage one. Amplicon libraries were generated using the Ion Plus Fragment Library kit (Thermo Fisher Scientific) according to the manufacturer’s protocol, and sequencing was performed on the Ion Proton platform (Thermo Fisher Scientific). Gene panel sequencing was performed as described for patients’ tumors. For the gene panel, variant calling and annotation were performed with GeneGlobe software (Qiagen) and Varseq software (Golden Helix), respectively. Variants were filtered based on variant allele frequency (>3%), variant coverage (100X), absence from leucocyte DNA pools (from 4 to 5 patients) and minor allele frequency <1%. For amplicon sequencing, visual inspection of base calling and absolute base frequencies annotation were performed using Integrative Genome Viewer (IGV) [20]. The mutation was considered present if the variant allele frequency was > 3%, with a minimum coverage depth of 500X.
Statistical analysis
Pathologic stage was assigned according to the 8th edition of the American Joint Committee on Cancer Staging Manual and histologic grade from International Society of Urological Pathology, which is currently recommended by the World Health Organization. Associations were assessed with χ2 tests or Fisher’s exact test to compare histopathologic features between patients whose tumors were successfully vs. unsuccessfully engrafted as PDX, or between orthotopic and ectopic implantation, with a significance level of 5%. The Kaplan– Meier method was used to evaluate metastasis-free survival and overall survival, with the log-rank test used for comparison. Statistical analysis was performed using the SPSS 25.0 software package (SPSS, Chicago, IL, USA).
RESULTS
The initial attempt to establish RCC PDX was conducted by using ectopic (subcutaneous) transplantation in the dorsal region of NSG mice. Seventeen patients with tumors of different stages and subtypes were recruited. Three patients developed post-transplantation lymphoproliferative disease unrelated to RCC (data not shown). No tumor growth was observed following ectopic implantation. PDX were obtained only following orthotopic implantation in the renal subcapsular space. Tumor fragments from 70 patients gave rise to 19 PDX consisting of 15 clear cell, three papillary, and one unclassifiable subtype. The overall take rate was 27% and the time to observe tumor growth varied from 1 to 13 months. All PDX were reinoculated in second and third passages with 100% of take rate and no alterations in tumor growth time.
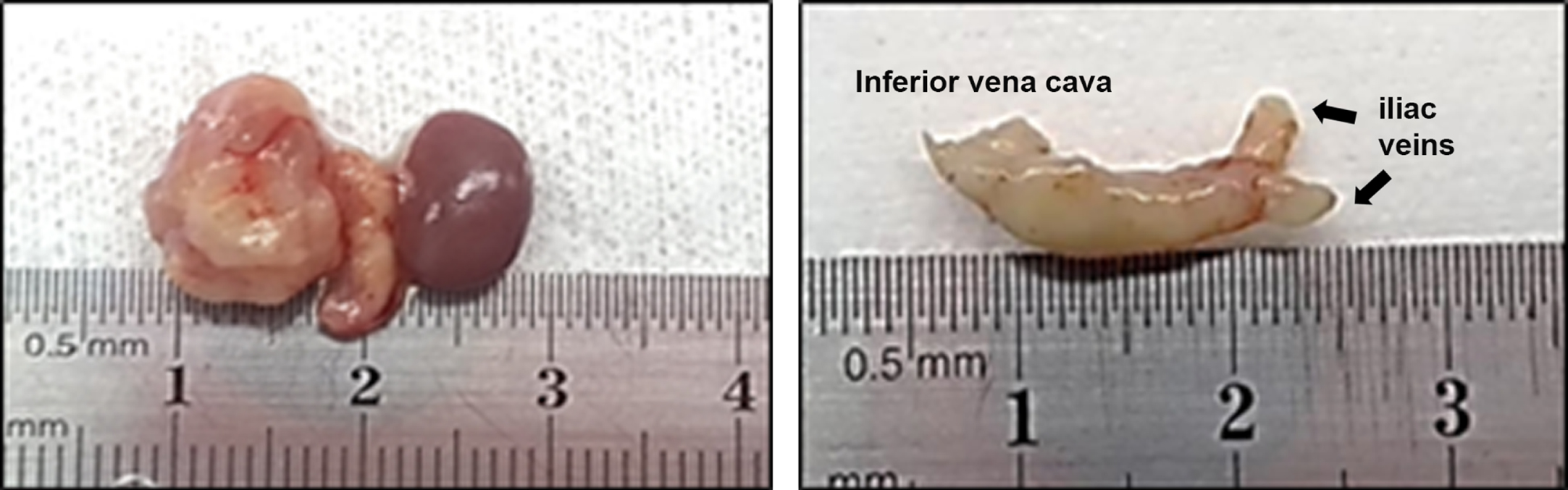
PDX take rate was unrelated to tumor size (p = 0.071). However, an association between tumor growth and take rate was suggested by increasing take rates of 18%, 36%, and 100% among pT1 (pT1a + pT1b), pT3a (invasion of perinephric fat and renal vasculature), and pT3b (invasion of renal vein and inferior vena cava) stage tumors, respectively (Table 1). Interestingly, the PDX derived from a patient with vena caval tumor thrombus extension (ID 55) resulted in the same phenotype (Fig. 1). In this specific case, we obtained ectopic implants after passage 1. We observed only one case of pT4, in which a biopsy was used to inoculate NSG mice, and no tumor growth was observed. Histologic grade and the presence of necrosis were unrelated to PDX take rate. However, the presence of either sarcomatoid or rhabdoid cells, which also characterize grade 4, was associated with PDX growth (p = 0.019 and p = 0.036, respectively).
Table 1
Clinical characteristics of patients and tumor implantation route influencing PDX take rate
| Orthotopic | Ectopic | ||||
| PDX engraftment | Yes | Total | P | Total | |
| N = 19 | N = 70 | N = 17 | |||
| Gender | Male | 12 | 48 | 0.76 | 10 |
| Female | 7 | 22 | 7 | ||
| Age | Mean | 59.5 | 56.9 | 63 | |
| Time to growth (months) | Mean | 6 | – | – | |
| Minimum | 1 | – | |||
| Maximum | 13 | – | |||
| Subtype | Clear cell | 15 | 55 | 1.00 | 13 |
| Papillary | 3 | 10 | 3 | ||
| MiT family translocation | 0 | 2 | 0 | ||
| Unclassifiable | 1 | 3 | 1 | ||
| Staging | pT1a | 5 | 24 | 0.09 | 11 |
| pT1b | 4 | 21 | 3 | ||
| pT2a | 0 | 2 | 0 | ||
| pT3a | 7 | 19 | 3 | ||
| pT3b | 3 | 3 | 0 | ||
| pT4 | 0 | 1 | 0 | ||
| Histologic grade | 1 | 0 | 3 | 0.49 | 0 |
| 2 | 6 | 24 | 6 | ||
| 3 | 7 | 29 | 8 | ||
| 4 | 6 | 14 | 3 | ||
| Sarcomatoid | Yes | 5 | 9 | 0.05 | 0 |
| No | 14 | 61 | 17 | ||
| Rhabdoid | Yes | 6 | 11 | 0.05 | 0 |
| No | 13 | 59 | 17 | ||
| Necrosis | Yes | 8 | 23 | 0.393 | 5 |
| No | 11 | 47 | 12 | ||
Fig. 1
Vena caval thrombosis in orthotopic PDX model. Left image shows nephrectomy with vena cava. Right image shows the isolated inferior vena cava with complete obstruction caused by tumor thrombus.
Demographic and clinical characteristics were similar between patients whose tumors were implanted ectopically or orthotopically. There were no differences in gender (p = 0.567), age (p = 0.106), subtype (p = 0.539), staging (p = 0.62), histologic grade (p = 1.000), the presence of necrosis (p = 1.000), or the presence of sarcomatoid or rhabdoid cells (p = 0.195 and 0.112, respectively) between the two groups. These findings did not identify any differences that could explain the absence of tumor growth in the ectopic cohort and strongly suggest that the site of implantation is a major determinant of tumor growth. All information is summarized in Table 1 and the individual compilation of clinical characteristics is available in Supplementary Table 1.
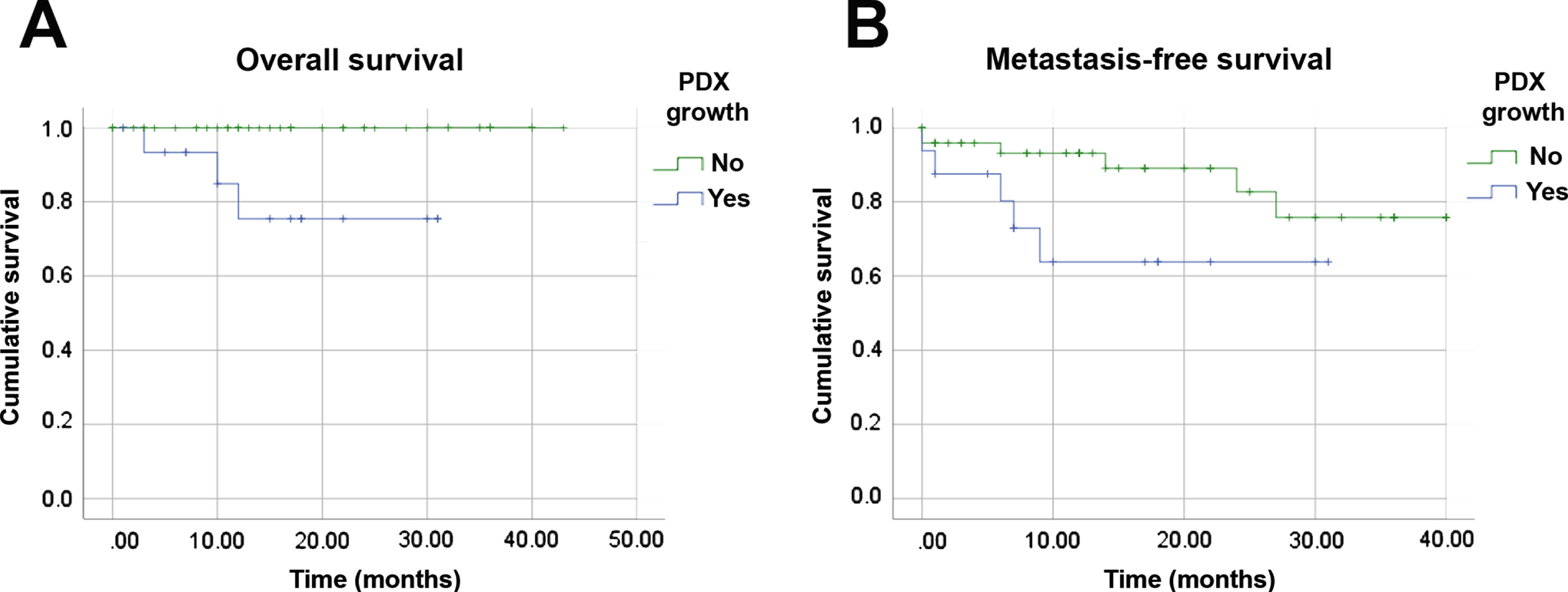
Due to the similarities between patients’ tumors and their derived xenografts, we investigated whether PDX growth capacity could represent a prognostic factor for RCC (Fig. 2). Patients whose tumor fragments engrafted experienced worse overall survival (OS) than those whose tumor fragments that did not engraft (p = 0.003). Our data also suggested a similar association with metastasis-free survival (MFS) (p = 0.063) that may be further evaluated in a larger cohort. Only one pT1 patient whose tumor yielded a PDX experienced disease progression (ID 43). All other patients who developed metastasis who died were at the pT3 stage. The median MFS for patients with successful PDX was 21.6 months (95% CI: 14.9, 28.3), compared to a median of 34.4 months (95% CI: 30.4, 38.5 p = 0.003) among PDX engraftment-negative patients. These findings suggest that tumor engraftment capacity can identity patients at increased risk of relapse or death. Furthermore, pT1 stage tumors with the ability to engraft could also facilitate the identification of risk factors related to the rare pT1 cases with disease progression.
Fig. 2
Tumor engraftment capacity is a prognostic factor in RCC. Kaplan-Meier analyses of overall (A) and metastasis-free (B) survivals of 70 RCC patients related to engraftment of orthotopic PDX. All histologic subtypes and clinical stages were included.
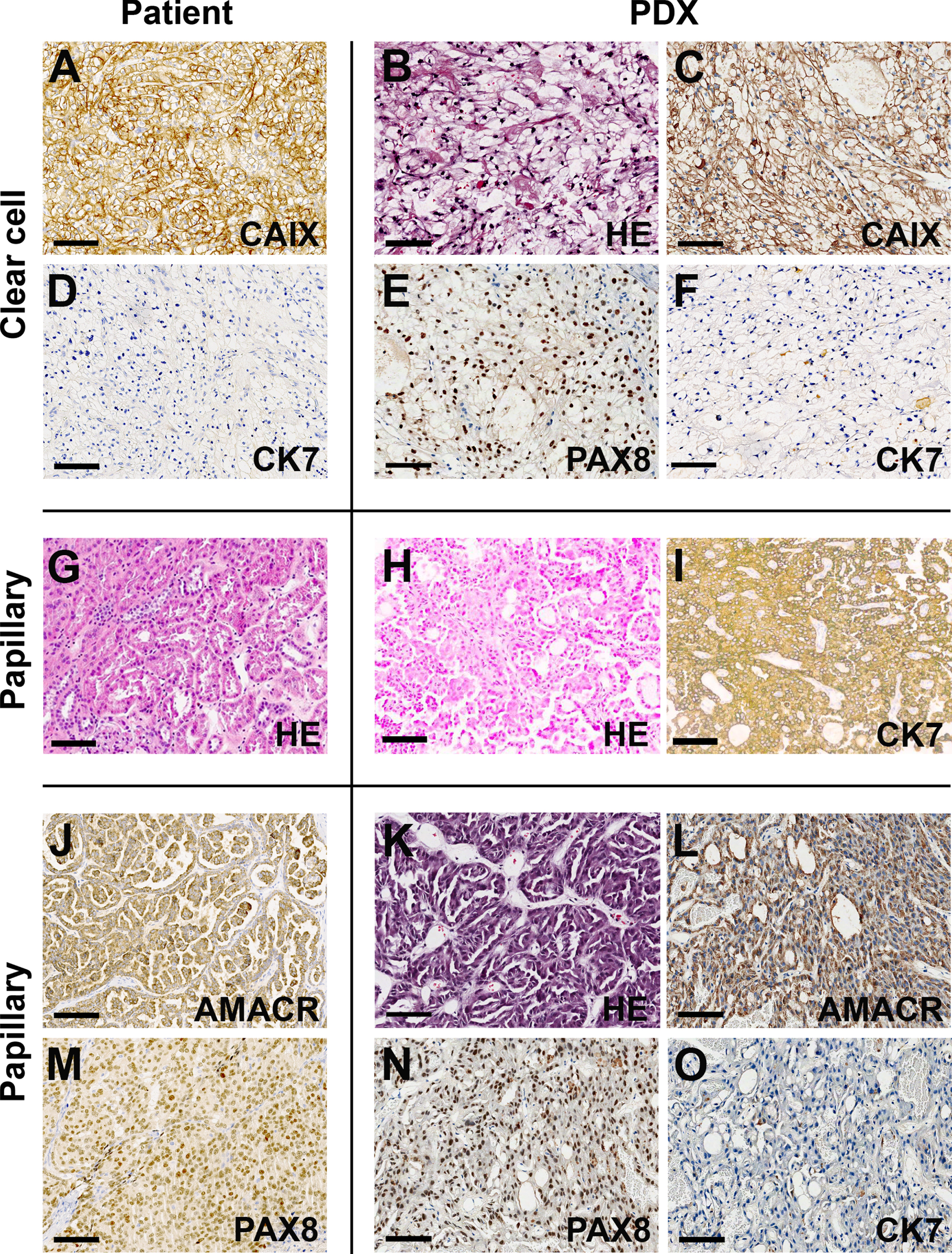
RCC PDX were characterized by immunohistochemistry and next-generation sequencing. Figure 3 shows representative images of PDX of clear cell and papillary subtypes. Carbonic Anhydrase IX (CAIX) and Paired Box 8 (PAX8) positivity associated with cytokeratin 7 (CK7) status were used to determine the clear cell phenotype. The combined use of Alpha-methylacyl-CoA racemase (AMACR) and CK7 was used to determine papillary RCCs. The similarity of tumor and PDX immunotypes was demonstrated in all cases.
Fig. 3
Patient-derived xenografts preserve the major morphologic characteristics and molecular markers of RCC. Representative images from clear cell (A-F) and papillary (G-O) cases comparing the presence of RCC markers in patient tumors (left panels) and their respective PDX (right panels). Calibration bars = 100μm. AMACR – Alpha-methylacyl-CoA racemase; CAIX – Carbonic anhydrase XI; CK7 – Cytokeratin 7; HE – hematoxylin and eosin; PAX8 – Paired box 8.
An essential aspect of PDX, in the case of RCC, is high-quality cryopreservation maintenance in a biobank. In our experience, slow freezing protocols, such as those used to cryopreserve primary cells, compromise PDX viability and impair tumor growth (data not shown). We currently use a vitrification method to cryopreserve PDX fragments that, after thawing and reimplantation, exhibit growth rates similar to those of the original tumors.
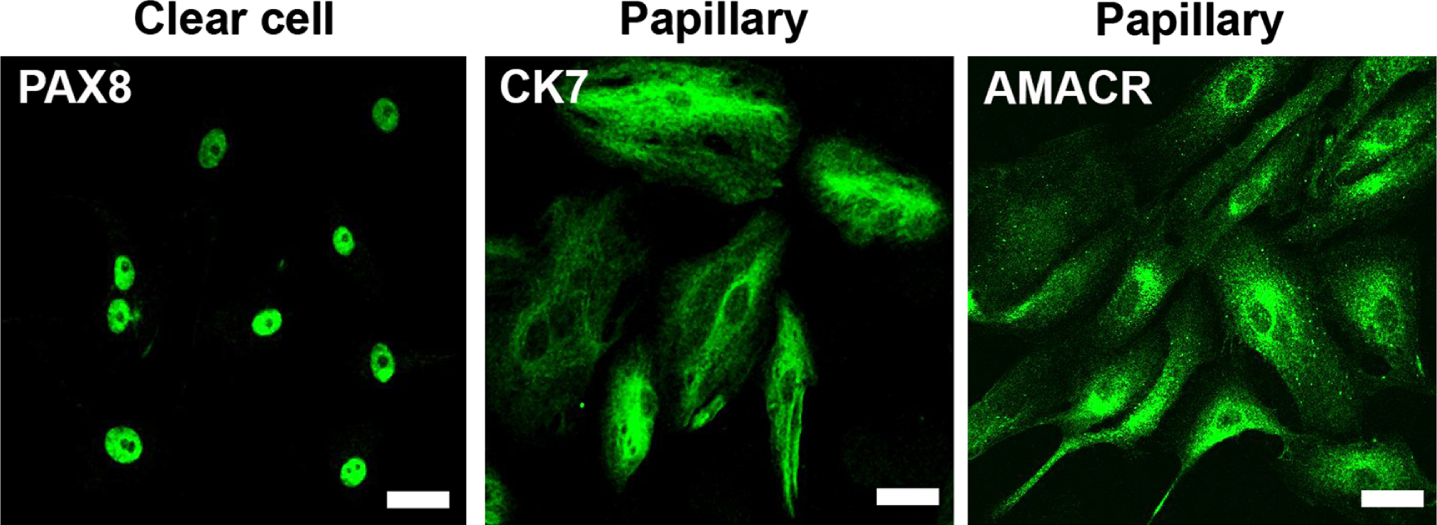
To complement the PDX platform with in vitro models, we derived early passage primary cultures from all PDX and performed immunofluorescence to demonstrate the fidelity of tumor cells to their subtypes (Fig. 4). The large majority of cultures presented a limited number of passages (4 to 6) before entering senescence. An immortalized lineage was generated by serial passaging in only one case (clear cell), and exhibited tumorigenic activity in NSG mice (data not shown).
Fig. 4
Primary cultures from PDX maintain RCC characteristics. Immunofluorescence of representative cell cultures of clear cell and papillary cases expressing PAX8, CK7 and AMACR, respectively. Calibration bars = 50μm.
To characterize the somatic tumor mutation repertoire of our PDX cohort, we used a customized target sequencing panel containing the most frequently mutated genes in RCC. From the total of 19 PDX, we compared the sequences of 13 patients’ tumor samples with their respective PDX. In most patients (92% - 11/13), at least one mutation in patients’ tumor was identified in the PDX (Table 2). No alterations were identified in only one case (ID 52). The most frequently mutated genes were VHL (50% - 6/12) and PBRM1 (41.7% - 5/12), followed by SETD2 (25% - 3/12), BAP1 and KDM5C (both 16.7% - 2/12), and ARID1A (8.3% - 1/12). Interestingly, most of the matching mutations (83.3% - 20/24) presented a higher variant allele frequency (VAF) in the PDX than in the respective tumors, with several PDX exhibiting VAFs compatible with loss of heterozygosity (VAFs > 65%).
Table 2
Somatic mutations identified in tumors from patients and PDX
| ID | Histology | pT Stage | Information Ref Seq | Patient Allele freq. (%) | PDX Allele freq. (%) | |||
| Gene | Effect | Variant codon | Variant protein | |||||
| 43 | Clear cell | pT1a | VHL | Missense | c.287A > C | p.Gln96Pro | 31.8% | 99.9% |
| PBRM1 | LoF | c.447 + 1 G > C | p.spl? | n.d | 99.9% | |||
| 48 | Clear cell | pT1a | VHL | Missense | c.349T > C | p.Trp117Arg | 17.3% | 0.0% |
| PBRM1 | LoF | c.457 C > T | p.Gln153Ter | 19.9% | 0.0% | |||
| 50 | Clear cell | pT1a | VHL | LoF | c.444delT | p.Phe148Leufs*11 | 46.1% | 86.1% |
| PBRM1 | LoF | c.3253 G > T | p.Glu1085Ter | 50.5% | 88.5% | |||
| AR | Missense | c.170T > A | p.Leu57Gln | 7.1% | 2.4% | |||
| 52 | Papillary | pT1a | – | – | – | – | – | |
| 59 | Papillary | pT1a | MET | Missense | c.3209T > C | p.Val1070Ala | 51.5% | 67.0% |
| SMARCA4 | Missense | c.3699 G > A | p.Met1233Ile | 37.8% | 51.6% | |||
| 27 | Clear cell | pT1b | BAP1 | Missense | c.517T > A | p.Tyr173Asn | 10.0% | 99.0% |
| CDKN2A | Missense | c.262 G > A | p.Glu88Lys | 2.9% | n.d. | |||
| TP53 | Missense | c.711 G > A | p.Met237Ile | 10.9% | 82.0% | |||
| TP53 | LoF | c.403delT | p.Cys135Alafs | 6.7% | 4.4% | |||
| 32 | Papillary | pT3a N1 | ARID1A | LoF | c.5904T > A | p.Cys1968Ter | 42.0% | 49.2% |
| ARID1A | LoF | c.6781delA | p.Ile2261Serfs | 42.8% | 46.0% | |||
| 35 | Clear cell | pT3a | VHL | LoF | c.344_345insC | p.Leu116Profs | 28.5% | 79.3% |
| PBRM1 | LoF | c.813 + 1 G > A | p.spl? | 31.8% | 98.0% | |||
| KDM5C | LoF | c.503delinsAG | p.Gln168Hisfs | 47.0% | 99.0% | |||
| 51 | Clear cell | pT3a | VHL | LoF | c.162_172delGGAGGCCGGGC | p.Glu55Alafs | 53.3% | 78.1% |
| SETD2 | LoF | c.4917 + 1 G > A | p.spl? | 54.2% | 71.9% | |||
| PBRM1 | LoF | c.3079 C > T | p.Arg1027Ter | 50.3% | 69.0% | |||
| 55 | Clear cell | pT3a | VHL | Missense | c.458T > C | p.Leu153Pro | 21.2% | 89.0% |
| BAP1 | Missense | c.38 G > T | p.Gly13Val | 19.8% | 92,1% | |||
| FAT1 | LoF | c.5178delC | p.Phe1727Leufs*9 | 16.6% | n.d. | |||
| 64 | Clear cell | pT3a | VHL | LoF | c.280 G > T | p.Glu94Ter | 39.6% (FF); 44.0% (FFPE) | 99.1% |
| PBRM1 | LoF | c.778-1 G > A | p.spl? | 33.5% (FF); 2.8% (FFPE) | n.d. | |||
| SETD2 | LoF | c.1265 C > G | p.Ser422Ter | 11.0% (FF); 1.7% (FFPE) | n.d. | |||
| PBRM1 | LoF | c.1063A > T | p.Lys355Ter | 0.0% (FF); 42.0% (FFPE) | 100.0% | |||
| 53 | Clear cell | pT3b | SETD2 | LoF | c.6837dupT | p.Val2280Cysfs*89 | 31.1% | 68.1% |
| KDM5C | Missense | c.1883_1903del | p.Gln628_Arg634del | 26.9% | 57.1% | |||
| 68 | Clear cell | pT3b | TP53 | Missense | c.511 G > A | p.Glu171Lys | 32.8% | 99.7% |
Of the ten sequenced ccRCC cases, seven had VHL mutations, two had BAP1 mutations, and one had concomitant VHL and BAP1 mutations (ID 55). Two ccRCC patients who died had both VHL and PBRM1 mutations, while the third death was of a pRCC patient with an ARID1A mutation. One pRCC case (ID 59) had an MET mutation, while the other case no displayed no alterations. We performed panel sequencing of two areas of one patient tumor (ID 64). Three mutations in VHL, PBRM1 (c.778-1 G > A) and SETD2 were identified in both regions of the tumor, but with striking differences in VAF for the PBRM1 mutations. Additionally, one PBRM1 mutation (c.1063A > T) was identified only in one lesion, and with a high VAF (42%). In the PDX, only the VHL and PBRM1 (c.1063A > T) mutations were identified. This case illustrates the intratumor molecular heterogeneity of ccRCC tumors.
DISCUSSION
Herein, we report a workflow to generate patient-derived RCC xenografts with high take rates. PDX tumors preserved the major biological features of the respective patient tumors, including histological and molecular markers and the potential to disseminate in a venous tumor thrombus. Tumor engraftment indicated a poor patient prognosis.
Low-passage patient-derived tumor models are currently the best tools to translate knowledge from basic cancer research to clinical practice [21]. These models may be established in vitro as patient-derived organoids (PDO) and in vivo as PDX. Both models have their pros and cons, and may be used as complementary methods. While PDO are generated without animals and preserve the tumor microenvironment (including immune infiltration) and tumor heterogeneity, this model system is limited by its inability to recapitulate clinical features that entail whole organs, multiple organ systems, and distant anatomic sites; such as metastasis [16, 22] or systemic dissemination as observed in one case from our cohort. Consequently, PDX models are still necessary to fully understand tumorigenesis and also to evaluate antineoplastic drug candidates [23, 24]. The restoration of the immune systems of immunodeficient mice with human immune cells (humanized models) brings PDX even closer to human tumors [25, 26].
We obtained PDX only from fresh human tumor tissue using the orthotopic route, suggesting that the organ microenvironment is necessary for tumor growth [17, 27]. Only in one case were we able to obtain a secondary ectopic implant, but this case was unique in terms of aggressiveness, with prominent sarcomatoid and rhabdoid features, perinephric fat invasion, and extensive tumor dissemination through the vena cava. These features are biomarkers for tumor relapse [28– 31] and are important to guide follow-up, but frequently fail to predict outcomes [32]. Consequently, the development of animal models that recapitulate the micro- and macroscopic characteristics of RCC is imperative. The 27% take rate observed in our cohort is higher than those of previous studies [33, 34]. Our cohort included less aggressive tumors, such as pT1 stage and histologic grade 2 lesions, indicating that our PDX cohort was not biased by tumors with greater potential for growth in NSG mice. These findings also have prognostic value, since the capacity to generate tumors in immunodeficient mice indicated poor overall and metastasis-free survivals. Similar results were reported in other studies of RCC [33] and also of head and neck [35], mesothelioma [36], melanoma [37], breast [38, 39], lung [40] and pancreatic [41] cancers. Our results could also facilitate the identification of biomarkers to guide adjuvant therapy of pT3 cases. The use of immune checkpoint inhibitors as adjuvant therapy was recently reported [11], but the combination of immune checkpoint inhibitors with tyrosine kinase inhibitors to improve disease-free survival remains a clinical challenge [42]. Thus, the discovery of more reliable biomarkers is urgently needed to enable patients and their oncologists to adhere to this therapeutic modality. Novel biomarkers of high recurrence risk could be sought in patients with increased capacity to generate PDX.
An important aspect observed in our cohort was the cryopreservation of PDX tissue. Cryopreservation methods that are efficient in maintaining cellular viability are essential to establish a PDX biorepository that maintains primary tumor characteristics, especially those related to tumor heterogeneity. Many studies have used methods similar to those used for in vitro cell preservation, with the use of cryopreservation agents, such as DMSO [27,43]. In our lab conditions, this method is ineffective, and limited to very aggressive tumors. Instead, we used vitrification that utilized rapid freezing of tumor tissue (in the presence of cryopreservation agents) in liquid nitrogen [44]. The primary advantage of this technique is the solidification of living cells in the form of glass, which obviates the formation of ice crystals that are cytotoxic during freezing and thawing [45]. The use of slow or rapid freezing may be have pros and cons in different tumor types. For example, slow-freezing of prostate PDX is recommended over rapid-freezing [46]. The eventual differences in cryopreservation methods should be better characterized for kidney tissue and other urologic cancers.
Another point of intense discussion is murine-specific intratumoral evolution that can occur during PDX development [47, 48]. Our data demonstrate that the majority of PDX carry the same mutations found in the respective patient’s tumor; however, higher variant allele frequencies (VAFs) are found in 83% of PDX, which is compatible with loss of heterozygosity (LoH) [47]. Nonetheless, it is important to note that the molecular characterizations of our PDX cohort could also be influenced by multi-region heterogeneity [32, 49]. In advanced cases treated with total nephrectomy, regional differences of molecular alteration can also cause discrepancies in PDX VAFs. A recent study demonstrated that copy number alteration evolution during PDX engraftment and passaging is similar to the variation observed in different samples from the same patient, preserving molecular fidelity to the patient’s tumor, thus favoring the use of PDX as a preclinical drug testing tool [48].
As demonstrated in multiple studies, PDX from RCC are predictive models for therapeutic responses to anti-angiogenic agents, mTOR (mammalian Target of Rapamycin) inhibitors [33, 34, 50, 51] and new anti-tumoral agents, such as HIF-2 inhibitors [52, 53]. Together, these findings support the use of PDX for a variety of studies, because the modeling of aspects of RCC basic tumor biology that still elude other translational initiatives will culminate in patient benefits through either the development of new therapeutic strategies or the use of these models as precision medicine tools.
FUNDING
This work was supported by Sao Paulo State Research Support Foundation (FAPESP, grant numbers 2014/50943-1, 2015/50428-2 and 2018/25541-8), National Council for Scientific and Technological Development (CNPq, grant number 465682/2014-6) and Programa Nacional de Apoio à Atenção Oncológica (PRONON). Fellowships from Higher Education Improvement Coordination (CAPES) to A.O.B, E.C.E, A.I and I.T.M. FAPESP fellowship to E.C.E. PNL was supported by NCI Cancer Center Support Grant P30 CA093373.
AUTHOR CONTRIBUTIONS
A.O.B performed the experimental mouse work, contributed to data interpretation and manuscript writing. E.C.E and A.I performed cell culture experiments and manuscript writing. S.M.B conducted pathological analysis, contributed to data interpretation and manuscript writing. H.D contributed to mouse work. I.W.C contributed to pathological analysis and manuscript writing. G.T.T coordinated genetic analysis and contributed to manuscript writing. I.T.M. performed genetic analysis. D.M.C contributed in data interpretation and manuscript writing. P.N.L developed the hypothesis and contributed to manuscript writing. S.C.Z developed the hypothesis, interpreted the data and contributed to manuscript writing. V.R.M developed the hypothesis, interpreted the data and contributed to manuscript writing. T.G.S developed the hypothesis, led the project, interpreted the data and wrote the manuscript.
COMPETING INTERESTS
AOB, ECE, AI, SMB, HD, IWC, GTT, ITM, DMC, PNL, SCZ, VRM, TGS declare no competing interests. Dr. Primo N. Lara, Jr. is co-Editor-in – Chief and Dr. Stenio C. Zequi is an Editorial Board Member of this journal, but they were not involved in the peer-review process of this paper, nor had access to any information regarding its peer-review.
ACKNOWLEDGMENTS
We are thankful to Dr. Marilene Lopes and Dr. José Augusto Rinck for critically reading the manuscript and helpful discussions.
SUPPLEMENTARY MATERIAL
[1] The supplementary material is available in the electronic version of this article: https://dx.doi.org/10.3233/KCA-210011.
REFERENCES
[1] | Capitanio U , Montorsi F . Renal cancer. The Lancet. (2016) ;387: (10021):894–906. |
[2] | Sung H , Ferlay J , Siegel RL , Laversanne M , Soerjomataram I , Jemal A , et al. Global Cancer Statistics GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA: A Cancer Journal for Clinicians. (2021) ;71: (3):209–49. |
[3] | Zequi S de C , Mourão TC , de Oliveira MM , Curado MP , Gueglio G , da Costa WH , et al. Predictors of Survival Outcomes in Non-Metastatic Renal Cell Carcinoma in Latin America and Spain: A Multicentric Analysis. Kidney Cancer.(2019) ;3: (4):253–61. |
[4] | Moch H , Cubilla AL , Humphrey PA , Reuter VE , Ulbright TM . The WHO Classification of Tumours of the Urinary System and Male Genital Organs— Part A: Renal, Penile, and Testicular Tumours. European Urology. (2016) ;70: (1):93–105. |
[5] | Trpkov K , Hes O , Williamson SR , Adeniran AJ , Agaimy A , Alaghehbandan R , et al. New developments in existing WHO entities and evolving molecular concepts: The Genitourinary Pathology Society (GUPS) update on renal neoplasia. Modern Pathology. (2021) ;34: (7):1392–424. |
[6] | Linehan WM , Ricketts CJ . The Cancer Genome Atlas of renal cell carcinoma: findings and clinical implications. Nature Reviews Urology. (2019) ;16: (9):539–52. |
[7] | Siegel RL , Miller KD , Jemal A . Cancer statistics, 2018. CA: A Cancer Journal for Clinicians. (2018) ;68: (1):7–30. |
[8] | Bianchi M , Sun M , Jeldres C , Shariat SF , Trinh Q-D , Briganti A , et al. Distribution of metastatic sites in renal cell carcinoma: a population-based analysis. Annals of oncology: official journal of the European Society for Medical Oncology / ESMO. (2012) ;23: (4):973–80. |
[9] | Reese AC , Whitson JM , Meng M V . Natural history of untreated renal cell carcinoma with venous tumor thrombus. Urologic Oncology. (2013) ;31: (7):1305–9. |
[10] | Motzer RJ , Banchereau R , Hamidi H , Powles T , McDermott D , Atkins MB , et al. Molecular Subsets in Renal Cancer Determine Outcome to Checkpoint and Angiogenesis Blockade. Cancer Cell. (2020) ;38: (6):803–817.e4. |
[11] | Choueiri TK , Tomczak P , Park SH , Venugopal B , Ferguson T , Chang Y-H , et al. Adjuvant Pembrolizumab after Nephrectomy in Renal-Cell Carcinoma. New England Journal of Medicine. (2021) ;385: (8):683–94. |
[12] | McKay RR . The Promise of Adjuvant Immunotherapy in Renal-Cell Carcinoma. New England Journal of Medicine. (2021) ;385: (8):756–8. |
[13] | Gul A , Rini BI . Adjuvant therapy in renal cell carcinoma. Cancer. (2019) ;125: (17):2935–44. |
[14] | Byrne AT , Alférez DG , Amant F , Annibali D , Arribas J , Biankin A V , et al. Interrogating open issues in cancer precision medicine with patient-derived xenografts. Nature Reviews Cancer. (2017) ;17: (4):254–68. |
[15] | Sun H , Cao S , Mashl RJ , Mo C-K , Zaccaria S , Wendl MC , et al. Comprehensive characterization of 536 patient-derived xenograft models prioritizes candidates for targeted treatment. Nature Communications. (2021) ;12: (1):5086. |
[16] | Grisanzio C , Seeley A , Chang M , Collins M , Di Napoli A , Cheng S-C , et al. Orthotopic xenografts of RCC retain histological, immunophenotypic and genetic features of tumours in patients. The Journal of Pathology. (2011) ;225: (2):212–21. |
[17] | Hoffman RM . Patient-derived orthotopic xenografts: better mimic of metastasis than subcutaneous xenografts. Nature Reviews Cancer. (2015) ;15: (8):451–2. |
[18] | Brown MJ , Pearson PT , Tomson FN . Guidelines for animal surgery in research and teaching. AVMA Panel on Animal Surgery in Research and Teaching, and the ASLAP (American Society of Laboratory Animal Practitioners). American Journal of Veterinary Research. (1993) ;54: (9):1544–59. |
[19] | Lunardi FO , de Aguiar FLN , Apolloni LB , Duarte ABG , de Sá NAR , Leal ÉSS , et al. Sheep Isolated Secondary Follicles Are Able to Produce Metaphase II Oocytes After Vitrification and Long-Term In Vitro Growth. Biopreservation and Biobanking. (2017) ;15: (4):321–31. |
[20] | Robinson JT , Thorvaldsdóttir H , Winckler W , Guttman M , Lander ES , Getz G , et al. Integrative genomics viewer. Nature Biotechnology. (2011) ;29: (1):24–6. |
[21] | Bleijs M , Wetering M , Clevers H , Drost J . Xenograft and organoid model systems in cancer research. The EMBO Journal. (2019) ;38: (15). |
[22] | Metildi CA , Kaushal S , Hoffman RM , Bouvet M . In vivo serial selection of human pancreatic cancer cells in orthotopic mouse models produces high metastatic variants irrespective of Kras status. The Journal of Surgical Research. (2013) ;184: (1):290–8. |
[23] | Malaney P , Nicosia SV , Davé V . One mouse, one patient paradigm: New avatars of personalized cancer therapy. Cancer Letters. (2014) ;344: (1):1–12. |
[24] | Aparicio S , Hidalgo M , Kung AL . Examining the utility of patient-derived xenograft mouse models. Nature Reviews Cancer. (2015) ;15: (5):311–6. |
[25] | Chen Q , Wang J , Liu WN , Zhao Y . Cancer Immunotherapies and Humanized Mouse Drug Testing Platforms. Translational Oncology. (2019) ;12: (7):987–95. |
[26] | Wang M , Yao L , Cheng M , Cai D , Martinek J , Pan C , et al. Humanized mice in studying efficacy and mechanisms of PD-1-targeted cancer immunotherapy. The FASEB Journal. (2018) ;32: (3):1537–49. |
[27] | Pavía-Jiménez A , Tcheuyap VT , Brugarolas J . Establishing a human renal cell carcinoma tumorgraft platform for preclinical drug testing. Nature Protocols. (2014) ;9: (8):1848–59. |
[28] | Bakouny Z , Braun DA , Shukla SA , Pan W , Gao X , Hou Y , et al. Integrative molecular characterization of sarcomatoid and rhabdoid renal cell carcinoma. Nature Communications. (2021) ;12: (1):808. |
[29] | da Costa WH , Moniz RR , da Cunha IW , Fonseca FP , Guimaraes GC , de Cássio Zequi S . Impact of renal vein invasion and fat invasion in pT3a renal cell carcinoma. BJU International. (2012) ;109: (4):544–8. |
[30] | Zhang BY , Thompson RH , Lohse CM , Leibovich BC , Boorjian SA , Cheville JC , et al. A novel prognostic model for patients with sarcomatoid renal cell carcinoma. BJU International. (2015) ;115: (3):405–11. |
[31] | Leibovich BC , Lohse CM , Cheville JC , Potretzke TA , Tsivian M , Shah PH , et al. Renal Cell Carcinoma with Inferior Vena Cava Extension: Can Classification Be Optimized to Predict Perioperative Outcomes? Kidney Cancer. (2020) ;4: (2):111–5. |
[32] | Turajlic S , Xu H , Litchfield K , Rowan A , Horswell S , Chambers T , et al. Deterministic Evolutionary Trajectories Influence Primary Tumor Growth: TRACERx Renal. Cell. (2018) ;173: (3):595–610.e11. |
[33] | Sivanand S , Peña-Llopis S , Zhao H , Kucejova B , Spence P , Pavia-Jimenez A , et al. A validated tumorgraft model reveals activity of dovitinib against renal cell carcinoma. Science Translational Medicine. (2012) ;4: (137):137ra75. |
[34] | Lang H , Béraud C , Bethry A , Danilin S , Lindner V , Coquard C , et al. Establishment of a large panel of patient-derived preclinical models of human renal cell carcinoma. Oncotarget. (2016) ;7: (37):59336–59. |
[35] | Karamboulas C , Bruce JP , Hope AJ , Meens J , Huang SH , Erdmann N , et al. Patient-Derived Xenografts for Prognostication and Personalized Treatment for Head and Neck Squamous Cell Carcinoma. Cell Reports. (2018) ;25: (5):1318–1331.e4. |
[36] | Wu L , Allo G , John T , Li M , Tagawa T , Opitz I , et al. Patient-Derived Xenograft Establishment from Human Malignant Pleural Mesothelioma. Clinical Cancer Research. (2017) ;23: (4):1060–7. |
[37] | Némati F , Sastre-Garau X , Laurent C , Couturier J , Mariani P , Desjardins L , et al. Establishment and Characterization of a Panel of Human Uveal Melanoma Xenografts Derived from Primary and/or Metastatic Tumors. Clinical Cancer Research. (2010) ;16: (8):2352–62. |
[38] | DeRose YS , Wang G , Lin Y-C , Bernard PS , Buys SS , Ebbert MTW , et al. Tumor grafts derived from women with breast cancer authentically reflect tumor pathology, growth, metastasis and disease outcomes. Nature Medicine. (2011) ;17: (11):1514–20. |
[39] | McAuliffe PF , Evans KW , Akcakanat A , Chen K , Zheng X , Zhao H , et al. Ability to Generate Patient-Derived Breast Cancer Xenografts Is Enhanced in Chemoresistant Disease and Predicts Poor Patient Outcomes. Coleman WB, editor. PLOS ONE. (2015) ;10: (9):e0136851. |
[40] | John T , Kohler D , Pintilie M , Yanagawa N , Pham N-A , Li M , et al. The Ability to Form Primary Tumor Xenografts Is Predictive of Increased Risk of Disease Recurrence in Early-Stage Non-Small Cell Lung Cancer. Clinical Cancer Research. (2011) ;17: (1):134–41. |
[41] | Garrido-Laguna I , Uson M , Rajeshkumar NV , Tan AC , de Oliveira E , Karikari C , et al. Tumor Engraftment in Nude Mice and Enrichment in Stroma- Related Gene Pathways Predict Poor Survival and Resistance to Gemcitabine in Patients with Pancreatic Cancer. Clinical Cancer Research. (2011) ;17: (17):5793–800. |
[42] | Kazarian AG , Chawla NS , Muddasani R , Pal SK . Adjuvant Therapy in Renal Cell Carcinoma: Current Status and Future Directions. Kidney Cancer. (2021) ;5: (1):9–20. |
[43] | Kimple RJ , Harari PM , Torres AD , Yang RZ , Soriano BJ , Yu M , et al. Development and Characterization of HPV-Positive and HPV-Negative Head and Neck Squamous Cell Carcinoma Tumorgrafts. Clinical Cancer Research. (2013) ;19: (4):855–64. |
[44] | Vajta G , Holm P , Kuwayama M , Booth PJ , Jacobsen H , Greve T , et al. Open pulled straw (OPS) vitrification: A new way to reduce cryoinjuries of bovine ova and embryos. Molecular Reproduction and Development. (1998) ;51: (1):53–8. |
[45] | Wietcovsky L , Til D , Salvador RA , Amaral NLL , Senn AP , Amaral VLL . Ovarian tissue vitrification and heterotopic autologous transplantation in prepubertal Wistar rats. JBRA Assisted Reproduction. 2018; |
[46] | Porter LH , Lawrence MG , Wang H , Clark AK , Bakshi A , Obinata D , et al. Establishing a cryopreservation protocol for patient-derived xenografts of prostate cancer. The Prostate. (2019) ;79: (11):1326–37. |
[47] | Ben-David U , Ha G , Tseng Y-Y , Greenwald NF , Oh C , Shih J , et al. Patient-derived xenografts undergo mouse-specific tumor evolution. Nature Genetics. (2017) ;49: (11):1567–75. |
[48] | Woo XY , Giordano J , Srivastava A , Zhao Z-M , Lloyd MW , de Bruijn R , et al. Conservation of copy number profiles during engraftment and passaging of patient-derived cancer xenografts. Nature Genetics. (2021) ;53: (1):86–99. |
[49] | Gerlinger M , Rowan AJ , Horswell S , Larkin J , Endesfelder D , Gronroos E , et al. Intratumor Heterogeneity and Branched Evolution Revealed by Multiregion Sequencing. New England Journal of Medicine. (2012) ;366: (10):883–92. |
[50] | Dong Y , Manley BJ , Becerra MF , Redzematovic A , Casuscelli J , Tennenbaum DM , et al. Tumor Xenografts of Human Clear Cell Renal Cell Carcinoma But Not Corresponding Cell Lines Recapitulate Clinical Response to Sunitinib: Feasibility of Using Biopsy Samples. European Urology Focus. (2017) ;3: (6):590–8. |
[51] | Jiménez-Valerio G , Martínez-Lozano M , Bassani N , Vidal A , Ochoa-de-Olza M , Suárez C , et al. Resistance to Antiangiogenic Therapies by Metabolic Symbiosis in Renal Cell Carcinoma PDX Models and Patients. Cell Reports. (2016) ;15: (6):1134–43. |
[52] | Cho H , Du X , Rizzi JP , Liberzon E , Chakraborty AA , Gao W , et al. On-target efficacy of a HIF-2α antagonist in preclinical kidney cancer models. Nature. (2016) ;539: (7627):107–11. |
[53] | Chen W , Hill H , Christie A , Kim MS , Holloman E , Pavia-Jimenez A , et al. Targeting renal cell carcinoma with a HIF-2 antagonist. Nature. (2016) ;539: (7627)112–7. |


