
Hereditary Renal Cell Carcinoma
Abstract
BACKGROUND:
Hereditary renal cell carcinoma (RCC) is a complex and rapidly evolving topic as there is a growing body of literature regarding inherited syndromes and mutations associated with an increased risk of RCC.
OBJECTIVES:
We sought to systematically review 13 hereditary syndromes associated with RCC; von Hippel-Lindau Disease associated RCC (VHLRCC), BAP-1 associated clear cell RCC (BAPccRCC), Familial non-von Hippel Lindau clear cell RCC (FccRCC), Tuberous Sclerosis Complex associated RCC (TSCRCC), Birt-Hogg-Dub
RESULTS:
Hereditary RCC is generally associated with an early age of onset, multifocal and/or bilateral lesions, and aggressive disease course. VHLRCC, BAPccRCC, FccRCC, and certain mutations resulting in SDHRCC are associated with clear cell RCC (ccRCC). HPRCC is associated with Type 1 papillary RCC. HLRCC is associated with type 2 papillary RCC. BHDRCC is associated with Chromophobe RCC. TSCRCC, PHTSRCC, MiTFtRCC, TFEBRCC, ADPKDRCC, certain SDHRCC and ALKRCC have variable histology.
CONCLUSIONS:
There has been tremendous advancement in our understanding of the pathophysiology of hereditary RCC. Ongoing research will refine our understanding of hereditary RCC and its therapeutic targets.
INTRODUCTION
Hereditary forms of renal cell carcinoma (RCC) comprise up to 5% of RCC cases. In patients presenting with metastatic RCC (mRCC) and uncommon RCC variants, the prevalence of a germline mutation was estimated at 16% and 20%, respectively [1]. General features common to most hereditary forms of RCC are early age of onset, multifocal and/or bilateral lesions, and aggressive disease course. There are a growing number of cancer syndromes and predisposing mutations without identified syndromes that have been connected to RCC. Mammalian target of rapamycin (mTOR) and hypoxia-inducible factor (HIF)-2 alpha (HIF-2α) are an established link in the phosphoinositide 3-kinase (PI3K)-protein kinase B (AKT)-mTOR-HIF-erythropoietin (EPO) axis that plays a vital role in hereditary RCC (Fig. 1) [2–4]. The treatment landscape for clear cell RCC (ccRCC) has evolved rapidly over the last few years. In August, 2021, the Food and Drug Association (FDA) approved the HIF-2α inhibitor belzutifan for cancers associated with von Hippel Lindau Disease (VHL), including VHLRCC [5]. Table 1 outlines the key characteristics of heritable RCC and potential therapeutic targets and clinical trials.
Fig. 1
Summary hereditary RCCs via common pathway of PI3K-AKT-mTOR-EPO. Abbreviations: Phosphoinositide 3-Kinase (PI3K), Protein Kinase B (AKT), Mammalian Target of Rapamycin (mTOR), Hypoxia-Inducible Factor Alpha (HIFα), Tricarboxylic Acid (TCA), Erythropoetin (EPO), Tyrosine Kinase Met/Hepatocyte Growth Factor Receptor (MET), Phosphatase and Tensin Homolog (PTEN), Anaplastic Lymphoma Kinase (ALK), Folliculin (FLCN), Ubiquitin Carboxyl-terminal Hydrolase BAP1 (BAP1), Polycystin 1 (PC1), Fumarate Hydratase (FH), Succinate Dehydrogenase (SDH), Tuberous Sclerosis Complex-1 and 2 (TSC1 and TSC2), other Chromosome 3 alterations (Chr3), E3- Von Hipple Lindau protein (E3pVHL), Melanocyte Inducing Transcription Factor (MiTF). Image created using BioRender.com.
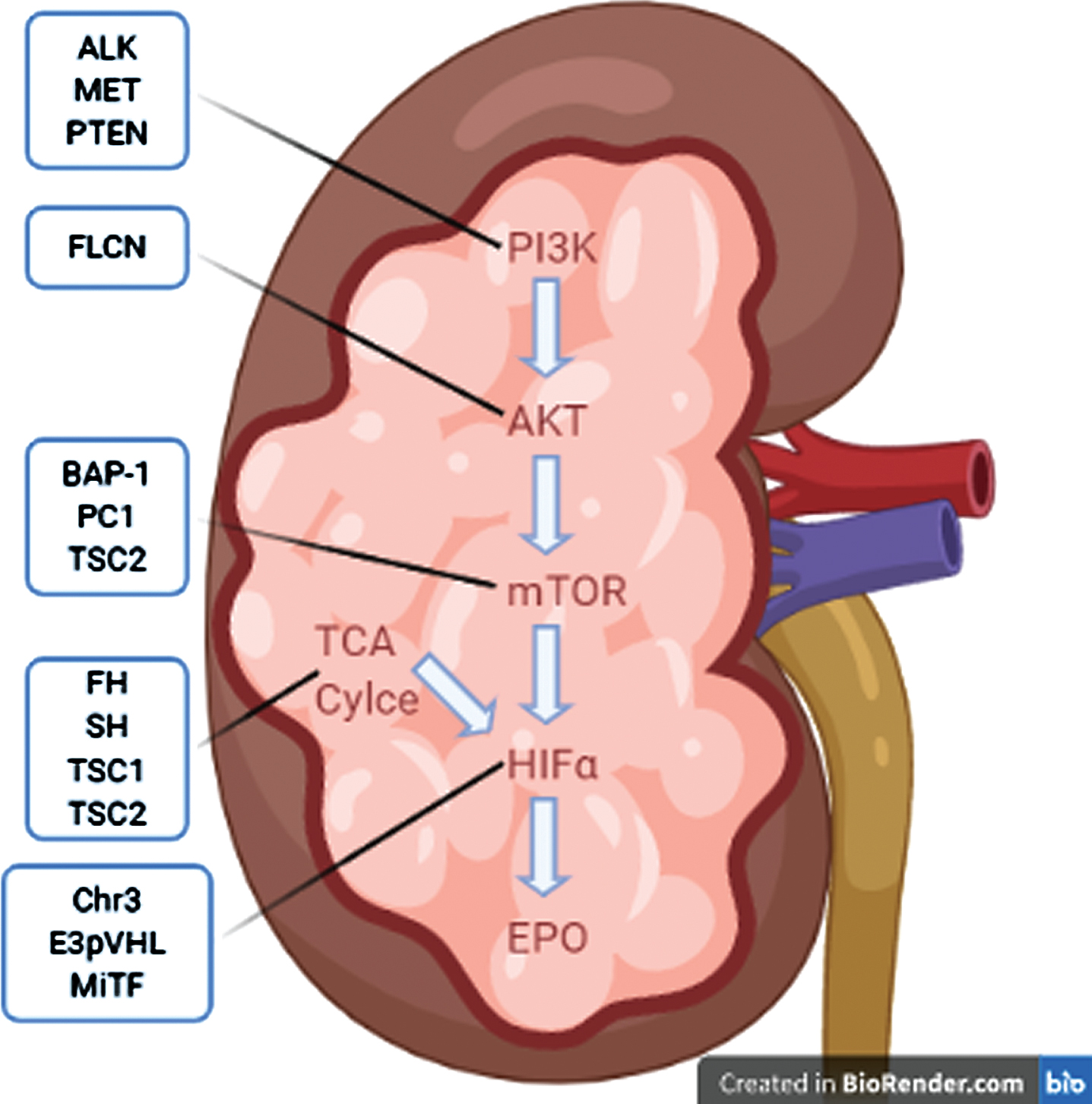
Table 1
Summary of hereditary RCC with gene, loci based on OMIM database, typical histology, therapeutic targets, and ongoing clincial trials
| Syndrome | Gene | Chromosome OMIM location [126] | Histology | Potential therapeutic target | Related clinical trials |
| vHLRCC | VHL | 3p25.3 | Clear cell | HIF | NCT04195750 |
| NCT03634540 | |||||
| NCT03108066 | |||||
| PARP | NCT04068831 | ||||
| Hsp90 | NCT00088374 | ||||
| BAPcRCC | BAP-1 | 3p21.1 | Clear cell | PARP | NCT03207347 |
| NCT03786796 | |||||
| Radiation? | |||||
| FcRCC | Various | Other chromosome 3 translocations | Clear cell | Depends on specific mutation | |
| TSRCC | TSC1 | 9q34.13 | Various including tubulopapillary and chromophobe-like, also RAML | mTOR, HIF | NCT02201212 |
| TSC2 | 16p13.3 | ||||
| BHD | FLCN | 17q11.2 | Oncocytic/chromophobe and chromophobe | mTOR | |
| PHTS | PTEN | 10q23.31 | Papillary and chromophobe | DNA repair | NCT03207347 |
| mTOR | NCT02145559 | ||||
| MiTRCC | TFE3 | Xp11.23 | Tubulocystic, varied | NCT03595124 | |
| TFEB | 6p21.1 | ||||
| 6p | TFEB | Amplification/other rearrangement | |||
| ADPKD | PKD1 | 16p13.3 | Various, particularly sarcomatoid and papillary | VEGF | |
| PKD2 | 4q22.1 | ||||
| HLRCC | FH | 1q43 | Type II papillary, tubulocystic | VEGF, EGFR | NCT01130519 |
| NCT02495103 | |||||
| NCT04068831 | |||||
| SDHRCC | SDHB | 1p36.13 | Various, giant mitochondria present as cytoplasmic granules | VEGF, EGFR, DNMT, PARP | NCT02495103 |
| SDHC | 1q23.3 | NCT03165721 | |||
| SDHD | 11q23.1 | NCT04068831 | |||
| NCT02071862 | |||||
| NCT03635892 | |||||
| HPRCC | MET | 7q31.2 | Type I papillary | MET | NCT02019693 |
| NCT00726323 | |||||
| ALK | ALK | 2p23.2, various ALK fusions | Varied | ALK | NCT01524926 |
| NCT02568267 |
Herein, we review hereditary RCC; von Hippel-Lindau Disease associated RCC (VHLRCC), BAP-1 associated clear cell RCC (BAPccRCC), Familial non-von Hippel-Lindau clear cell RCC (FccRCC), Tuberous Sclerosis Complex associated RCC (TSCRCC), Birt-Hogg-Dub
CLEAR CELL PREDOMINANT HERITABLE RCCs
von Hippel-Lindau disease associated RCC (VHLRCC)
VHL is AD with an incidence of approximately 1 in 30,000. VHL is characterized by mutations of VHL, found at loci 3p25, and a predisposition to various tumors including hemangioblastoma of the central nervous system and retina, pheochromocytoma, and RCC [6]. The VHL gene expresses the protein VHL (pVHL), which forms a complex with elongin B, elongin C, Rbx1 and cullin 2, thus forming the pVHL-E3 ubiquitin ligase complex (pVHLE3) [7–10]. This complex then ubiquitinates HIF-α, leading to its degradation [8, 11–15].
RCC develops in about 25–45% of patients with VHL, with age of onset during 2nd to 4th decade of life and by age 60, penetrance is ∼70% [16]. VHLRCC is typically ccRCC, multifocal and bilateral [17].
Understanding of the pVHLE3-HIFα-EPO pathway ushered in the use of multi-kinase targeted therapies which have transformed the therapeutic landscape in ccRCC [18]. Multi-kinase inhibitors alone and in combination with checkpoint inhibitors form the backbone of ccRCC treatment.
HIF-2α is a critical therapeutic target in ccRCC, and the advent of HIF-2α inhibitors has further revolutionized the treatment for RCC. Belzutifan (MK-6482), a HIF-2α inhibitor, showed activity in a phase II trial in patients with localized VHLRCC in which 53 out of 61 patients had response [5]. In another phase I/II trial in advanced RCC with 1 prior line of treatment, belzutifan resulted in tumor response in 35 of 52 patients and was recently granted FDA approval [19].
In VHLRCC patients, sunitinib did not show efficacy for hemangioblastomas, but pazopanib demonstrated response in VHL lesions other than VHLRCC [20, 21].
BAP-1 associated clear cell RCC (BAPccRCC)
BAPccRCC is an inheritable form of ccRCC associated with BAP-1 mutation; loss of BAP-1 has been demonstrated to indirectly upregulate mTOR [22]. In sporadic ccRCC, a common pathway in tumorigenesis involves an initial mutation to VHL followed by a mutation to BAP-1 or PBRM1 followed by a third mutation of the remaining wildtype BAP-1 or PBRM1, a gene that encodes SWI/SNF chromatin remodeling complex found in 41% of ccRCC [23, 24]. Inherited BAPccRCC was found in 83 unrelated patients with unexplained familial RCC [22]. Tumors with BAP-1 and PBRMI loss were associated with rhabdoid features [25]. BAP-1 and PBRM1 mutations may define subtypes of ccRCC with poorer prognosis for BAP-1 subgroup [23]. BAP-1 loss in in vitro models sensitized cells to ionizing radiation and poly adenosine diphosphate-ribose polymerase (PARP) inhibitors are being studied in ongoing trials (NCT03786796, NCT03207347) [25].
Familial non-von Hippel-Lindau clear cell RCC (FccRCC)
FccRCC is AD and characterized by inherited ccRCC without evidence of VHL [26, 27]. Mutations to various genes via chromosome 3 translocations, excluding those to VHL, MET or CUL2, can account for these cases [28]. In a review of 60 kindreds with at least 2 patients with ccRCC, there were 145 affected patients with familial RCC without another syndrome or mutation [28]. The mean age of onset was 53 years, which is younger than sporadic cases but older than VHLRCC; 14/145 individuals had multiple tumors at diagnosis or developed a second primary and 17/145 patients developed an additional benign or malignant neoplasm, but there was no evidence to suggest inherited susceptibility.
NON-CLEAR CELL PREDOMINANT HERITABLE RCCs
Tuberous sclerosis complex (TSC) associated RCC (TSCRCC)
TSCRCC is an AD disease characterized by seizures, cognitive impairment, hamartomas in various organs and RCC [29]. Mutations of TSC1 and TSC2, loci 9q34 and 16p13 respectively, result in this syndrome [30, 31]. TSC1 encodes Hamartin and TSC2 encodes Tuberin, which heterodimerize to produce a complex that ultimately inhibits the mTOR pathway [32, 33]. The incidence of TSCRCC is similar to the incidence of RCC in the general population, except with earlier age of onset and pathological heterogeneity [34, 35]. Renal Angiomyolipoma (RAML) and atypical RAML are also associated with TSC [36, 37].
Due to signaling through mTOR and HIF, mTOR and HIF inhibitors are potential therapies. Everolimus resulted in durable response in TSC2 mutant RCC [38]. In the EXIST-2 trial, a phase 3 trial of 118 patients with at least one 3 cm or larger angiomyolipoma and a diagnosis of tuberous sclerosis or sporadic lymphangioleiomyomatosis treated with everolimus or placebo, everolimus demonstrated 42% response rate in angiomyolipomas [39]. Four year follow up of EXIST-2 with median duration of everolimus exposure of 46.9 months demonstrated a renal lesion reduction in 97% of patients [40]. There is an ongoing phase II clinical trial in solid tumor patients with mutant TSC1 or TSC2 using everolimus (NCT02201212).
Birt-Hogg-Dubé syndrome associated RCC (BHDRCC)
BHD is as an AD syndrome characterized by fibrofolliculomas of the skin, spontaneous pneumothorax, and increased risk of RCC [41–44]. The FLCN gene, loci 17p11.2 [45], encodes for the protein FLCN that functions to regulate cell metabolism in conjunction with Folliculin-Interacting Protein (FNIP) 1 and 2. This regulation occurs through the AMPK energy sensing, mTOR proliferation cascade, and Ppargc1a mitochondrial oxidation [46–52]. In a review of 89 individuals from 51 families with BHD mutations, 34% of individuals had RCC, with median age of onset around 50 years [53]. The predominant histologies are Hybrid oncocytic/chromophobe tumors (HOCT) and chromophobe RCC [41, 54]. These tumors are generally slow growing and can be managed surgically [55]. Everolimus showed response as 6th line therapy in a patient with BHD-RCC [56].
PTEN hamartoma tumor syndrome associated RCC (PHTSRCC)
PHTS encompasses AD conditions caused by germline mutation of PTEN, including Cowden Syndrome [57] and Bannayan-Riley-Ruvalcaba Syndrome [58]. PTEN, loci 10q23, encodes for the protein Phosphatase and tensin homolog [59, 60], which blocks PIP3 dependent PI3K activation of AKT [61–64]. In a series of 219 patients with a germ line PTEN mutation, 9 were found to have RCC, with 6 being papillary and 2 being chromophobe [65]. RECORD-3 was a phase IIb clinical trial comparing everolimus to sunitinib in patients with advanced RCC that did not meet its primary outcome [66]. However, subsequent analysis of based on PTEN expression found longer PFS (10.5 months vs 5.3 months, p < 0.001) in patients with PTEN loss vs not, respectively, for those treated with everolimus. There was no significant difference in PFS (10.3 months vs 10.9 months, p = 0.475) based on PTEN expression in those treated with sunitinib [67]. This evidence suggests that decreased PTEN expression, as would be seen in PHTS, would derive more benefit from everolimus compared to sunitinib.
Microphthalmia-associated transcription family (MiTF)translocation RCC (MiTFtRCC)
MiTFtRCC is caused by AD germline mutation of one of the MiTF transcription factors, which are: MITF, TFE3 loci Xp11.23, TFEB loci 6p21.1, and TFEC [68–72]. There are 2 further subgroups of MiTFtRCC defined by the presence of rearrangement of TFE3 via Xp11 and TFEB via t(6;11) [73]. TFE3 and TFEB function in the regulation of autophagy induction in response to DNA damage and starvation [74]. MITF mutation MiE318 has been linked to RCC and melanoma, with mutation carriers having a 5 times higher risk of melanoma, RCC or both [75]. Xp11 translocation RCC causes 20–40% of childhood RCC and 1–4% of adult RCC with age of onset around 50 years [76, 77]. The presence of ASPSCR1-TFE3 fusion has a poorer prognosis than other fusions [78]. These tumors have cells with clear to eosinophilic cytoplasm, often mixing features of clear cell and papillary RCC [79]. t(6;11) RCC is quite rare with an age of onset of 30 years and poor prognosis [79–81]. These RCCs usually have weak expression of epithelial markers on IHC, sometimes express melanoma markers, and may not respond well to VEGF targeted therapy, so a misclassification as clear cell may lead to unexpectedly poor outcomes [54]. There is an ongoing phase II trial to evaluate the efficacy of axitinib plus nivolumab in patients with TFE/translocation RCC (NCT03595124).
RCC with chromosome 6p (TFEB) amplification (TFEBRCC)
TFEBRCC is related to MiTFtRCC because it has an amplification or rearrangement of the TFEB gene other than t(6;11). The unique features to this category are age of onset about 65 years old, areas of high grade epithelioid cells with pseudo or true papillary histology, and more aggressive course [82–84]. These patients have a more aggressive course, including more likely to present with advanced disease and to metastasize [82]. The series by Gupta et al demonstrated 24 cases with associated amplification of VEGFA with 11 (46%) of patients having metastasis and death from RCC [84].
Autosomal dominant polycystic kidney disease (ADPKD) associated RCC (ADPKDRCC)
The association of ADPKD and RCC has been a controversial topic over the years. ADPKD is caused by mutations of PKD1 at loci 16p13.3, which encodes for the transmembrane protein polycystin-1, and PKD2 at loci 4q21.1, which encodes for the transmembrane cation channel polycystin-2, with polycystin-1 regulating the function of polycystin-2. Approximately 85% of ADPKD cases are due to mutant PKD1 and 15% due to mutant PKD2 [85]. ADPKD is characterized by cysts of the kidneys, liver, pancreas, seminal vesicles, and meninges and connective tissue disorders affecting heart and vasculature [85–87]. Mutations of PKD1 are associated with more renal cysts and onset of renal failure about 20 years earlier [88, 89].
The RCC found in patients with ADPKD has unique characteristics. In a case series of 25 cases of RCC in ADPKD (3 new cases from the Mayo Clinic and 22 cases from literature review), there was no increased risk of RCC in those with ADPKD compared to historical controls. However, the following characteristics were more common in ADPKD than the general population of RCC patients: presence of a triad of fever, flank pain, weight loss, concurrent bilateral or multifocal tumors, and sarcomatoid histology [90, 91]. A more contemporary retrospective review of patients with ADPKD included 240 pts with a median age of 54 years that underwent nephrectomy and 5% were incidentally found to have RCC with the majority being papillary histology [92]. A review of renal transplant patients in the US of 10,626 patients with PKD, including ADPKD, found an increased risk of RCC compared to the general population but did not find ADPKD to be a risk factor for developing RCC [93].
One potentially confounding factor in ADPKDRCC is hemodialysis and its impact on the incidence of RCC. It is known that the majority of patients requiring chronic hemodialysis, regardless of the etiology, eventually develop cystic kidney disease. The presence of this acquired cystic kidney disease increases their risk of developing RCC by 3–6 times over historical general United States population risk from the Mayo Clinic [94]. It has also been shown that depending on the types of renal injuries, patients may have a predilection to a specific type of RCC [95]. A retrospective analysis of patients with ADPKD who did not have end stage renal disease in Taiwan demonstrated a higher incidence of liver, colon and renal cancers in those with ADPKD as compared to age matched controls [96]. This supports the theory that ADPKD, not just associated hemodialysis, increases the risk of RCC. Although the mechanism is thought to be mediated via the mTOR pathway, clinical studies have not yet demonstrated efficacy of mTOR inhibitors for treatment. It is possible that promising preclinical dual mTOR/PI3K inhibitors will demonstrate efficacy in the treatment of ADPKD and/or ADPKDRCC[97].
Hereditary leiomyomatosis associated RCC (HLRCC)
HLRCC is an AD disease characterized by uterine and skin leiomyomas and RCC [98]. Mutations of Fumarate Hydratase (FH) gene, found at loci 1q42-q44, account for these cases. The enzyme fumarate hydratase catalyzes the conversion of fumarate to malate in the Krebs cycle [99]. In a study of 21 families with HLRCC, renal tumors occurred in 13/21 families and while various histologies were seen, tumors had amphophilic cytoplasm and large nuclei with large inclusion-like nucleoli, making them similar to type II papillary RCC [98].
The RCC cells of HLRCC depended on glucose for adenosine triphosphate generation via aerobic glycolysis, known as the Warburg effect, due to a defect in the Krebs cycle. Increased oxidative stress and/or increased fumarate lead to increased levels of HIF, which results in increased VEGF [100]. The AVATAR trial (NCT01130519) is a phase II study in HLRCC or sporadic papillary RCC using a combination of bevacizumab and erlotinib and demonstrated median PFS of 21.1 months in the HLRCC cohort and 8.7 months in the sporadic cohort [101]. Another clinical trial (NCT02495103) using vandetanib and metformin in HLRCC or SDH associated RCC or sporadic papillary RCC has completed and results are pending. Vandetanib, an inhibitor of vascular endothelial growth factor receptor (VEGFR), epidermal growth factor receptor (EGFR), and rearranged during transfection (RET), is used to treat medullary thyroid cancer [102]. A retrospective review of 10 HLRCC patients in Korea showed clinical benefit with bevacizumab plus erlotinib in 9 patients with PFS 13.3 months and overall survival of 14.1 months [103].
Succinate dehydrogenase RCC (SDHRCC)
SDHRCC is an AD disease characterized by RCC with or without pheochromocytoma and paraganglioma [104, 105]. The 4 genes that encode for mitochondrial SDH are SDHA at loci 5p15 [106], SDHB at loci 1p36 [107], SDHC at loci 1q21 [108], SDHD at 11q23 [104] and this syndrome is known to occur in all except SDHA. When SDH is dysfunctional, succinate accumulates in the mitochondria and is transported into the cytosol where it inhibits HIFα prolyl hydroxylases which leads to stabilization and activation of HIFα [109]. SDHRCC has been described in patients with mutation of SDHB, SDHC, and SDHD. SHDBRCC was first described in 2004 in a kindred study of 2 families in which there was RCC diagnosed with age of onset < 30 years old and variable histology with solid, clear cell, and cells with granular eosinophilic cytoplasm [110]. In this study, the prevalence of RCC in SDHB mutation carriers was 5–10%. The unique histology of SDHBRCC includes nests of cuboidal tumor cells with granular or bubbly cytoplasm and prominent cytoplasmic inclusions that correspond to giant mitochondria [111, 112]. SDHCRCC and SDHDRCC typically have a later age of onset, in the 40’s, and clear cell histology as compared to SDHCRCC [113].
In SDHRCC, as in HLRCC, treatments that interact with cellular metabolism may be of value. There is on ongoing trial of pan VEFG-inhibitor, vandetanib, plus metformin in patients with HLRCC or sporadic papillary carcinoma (NCT02495103). There is also a phase II trial of guadecitabine, a DNA methyl transferase inhibitor in patients with SDHRCC (NCT03165721). There is a trial of talazoparib, a PARP inhibitor [114] and avelumab, an anti PD-L1 antibody [115], in patients with VHL, FH or SDH deficiency (NCT04068831). The compound CB-839, a glutaminase inhibitor is also under investigation in a trial including SDHRCC patients (NCT02071862). A phase II trial is underway to evaluate cabozantinib, a VEGF-inhibitor, and nivolumab, a PD-1 inhibitor, in patients with non-clear cell RCC including SHDRCC and HLRCC (NCT03635892).
Hereditary papillary RCC (HPRCC)
HPRCC is an ADRCC disease characterized by familial papillary RCC. Mutations in the MET gene, found at loci 7q31-34 [116], account for this disease. HPRCC tumors demonstrate type I papillary architecture, first described in a kindred study of 3 families with hereditary papillary RCC [117]. Age of onset is variable. In a study of 129 sporadic papillary tumors, MET mutations were found in only 17 cases, demonstrating that non hereditary papillary RCC is likely driven by other genetic alterations [118].
Due to the role of MET in HPRCC and chromosome 7 trisomy in sporadic papillary RCC, MET inhibitors have the potential to make a significant advancement in treatment. There are 2 clinical trials with MET inhibitors in papillary RCC (NCT00726323, NCT02019693). A phase II study of foretinib, inhibitor of MET, VEGF, RON, AXL, and Tie-2, in patients with papillary RCC included 10 patients with germline MET mutation and found that response rates were higher in patients with MET mutations compared to those without [19].
ALK-rearrangement RCC (ALKRCC)
ALKRCC is characterized by various Anaplastic Lymphoma Kinase (ALK) fusion proteins, including VCL-ALK in patients with sickle cell trait [119, 120], TPM3-ALK [121], STRN-ALK [122]. ALK, loci 2p23.2, is a receptor tyrosine kinase of the insulin superfamily [123, 124]. With various fusion partners, ALK has been reported in many malignancies and is a relatively new entity in RCC. These cases have varied histopathological features and comprises < 1% of RCC [125]. Due to the presence of ALK fusion, this group of RCC may be responsive to ALK inhibitors. CREATE is an ongoing phase II basket study with crizotinib in solid tumors with activating alterations to ALK or MET (NCT01524926). Another phase II basket trial is testing entrectinib in solid tumors with NTRK1/2/3, ROS1, or ALK gene fusion that is ongoing as well (NCT02568267).
CONCLUSION
The identification of cases of RCC that shared unusual characteristics such as early age of onset, other syndrome features, atypical histologies, and focal versus multifocal presentation led to identification of genes with oncogenic potential. This has revolutionized our understanding of RCC and more patients with inherited susceptibility to RCC are being identified as our knowledge expands. The next step is connecting the mutant genes to their aberrant gene products and related pathways. This will provide understanding that can not only be applied to RCC but also across scientific specialties to further our understanding of the various pathways. The final step is to discover targeted therapeutics tailored to the specific aberrant pathway(s) in each patient’s cancer. Ongoing clinical trials with known targeted therapeutics along with ongoing drug discovery are critical to our continued advancement and goal of personalized medicine in future.
ACKNOWLEDGMENTS
The authors have no acknowledgements.
FUNDING
The authors report no funding
AUTHOR CONTRIBUTIONS
Conceptualization, S.J.D. and S.G; writing—original draft preparation, S.J.D.; writing—review and editing, S.J.D. and S.G.; visualization, S.J.D.; supervision, S.G.; project administration, S.G. All authors have read and agreed to the published version of the manuscript.
CONFLICT OF INTEREST
Scott J. Dawsey and Shilpa Gupta have no conflict of interest to report.
REFERENCES
[1] | Carlo MI , Mukherjee S , Mandelker D , Vijai J , Kemel Y , Zhang L , et al. Prevalence of germline mutations in cancer susceptibility genes in patients with advanced renal cell carcinoma. JAMA Oncol. (2018) ;4: (9):1228–35. |
[2] | Miyazawa M , Yasuda M , Fujita M , Hirabayashi K , Hirasawa T , Kajiwara H , et al. Granulosa cell tumor with activated mTOR-HIF-1alpha-VEGF pathway. J Obstet Gynaecol Res. (2010) ;36: (2):448–53. |
[3] | Chen H , Xiong T , Qu Y , Zhao F , Ferriero D , Mu D . mTOR activates hypoxia-inducible factor-1alpha and inhibits neuronal apoptosis in the developing rat brain during the early phase after hypoxia-ischemia. Neurosci Lett. (2012) ;507: (2):118–23. |
[4] | Shackelford DB , Vasquez DS , Corbeil J , Wu S , Leblanc M , Wu CL , et al. mTOR and HIF-1alpha-mediated tumor metabolism in an LKB1 mouse model of Peutz-Jeghers syndrome. Proc Natl Acad Sci U S A. (2009) ;106: (27):11137–42. |
[5] | Jonasch E , Donskov F , Iliopoulos O , Rathmell WK , Narayan V , Maughan BL , et al. Phase II study of the oral HIF-2α inhibitor MK-for Von Hippel-Lindau disease–associated renal cell carcinoma. Journal of Clinical Oncology. (2020) ;38: (15_suppl):5003-. |
[6] | Latif F , Tory K , Gnarra J , Yao M , Duh FM , Orcutt ML , et al. Identification of the von Hippel-Lindau disease tumor suppressor gene. Science. (1993) ;260: (5112):1317–20. |
[7] | Stebbins CE , Kaelin WG Jr , Pavletich NP . Structure of the VHL-ElonginC-ElonginB complex: implications for VHL tumor suppressor function. Science. (1999) ;284: (5413):455–61. |
[8] | Ohh M , Park CW , Ivan M , Hoffman MA , Kim TY , Huang LE , et al. Ubiquitination of hypoxia-inducible factor requires direct binding to the beta-domain of the von Hippel-Lindau protein. Nat Cell Biol. (2000) ;2: (7):423–7. |
[9] | Kaelin WG . The von Hippel-Lindau tumor suppressor protein: roles in cancer and oxygen sensing. Cold Spring Harb Symp Quant Biol. (2005) ;70: :159–66. |
[10] | Tanimoto K , Makino Y , Pereira T , Poellinger L . Mechanism of regulation of the hypoxia-inducible factor-1 alpha by the von Hippel-Lindau tumor suppressor protein. EMBO J. (2000) ;19: (16):4298–309. |
[11] | Ohh M . Ubiquitin pathway in VHL cancer syndrome. Neoplasia. (2006) ;8: (8):623–9. |
[12] | Maxwell PH , Wiesener MS , Chang GW , Clifford SC , Vaux EC , Cockman ME , et al. The tumour suppressor protein VHL targets hypoxia-inducible factors for oxygen-dependent proteolysis. Nature. (1999) ;399: (6733):271–5. |
[13] | Jaakkola P , Mole DR , Tian YM , Wilson MI , Gielbert J , Gaskell SJ , et al. Targeting of HIF-alpha to the von Hippel-Lindau ubiquitylation complex by O2-regulated prolyl hydroxylation. Science. 292. (2001) ;292: (5516):468–72. |
[14] | Salceda S , Caro J . Hypoxia-inducible factor 1alpha (HIF-1alpha) protein is rapidly degraded by the ubiquitin-proteasome system under normoxic conditions. Its stabilization by hypoxia depends on redox-induced changes. J Biol Chem. (1997) ;272: (36):22642–7. |
[15] | Huang LE , Gu J , Schau M , Bunn HF . Regulation of hypoxia-inducible factor 1alpha is mediated by an O2-dependent degradation domain via the ubiquitin-proteasome pathway. Proc Natl Acad Sci U S A. (1998) ;95: (14):7987–92. |
[16] | Nordstrom-O’Brien M , van der Luijt RB , van Rooijen E , van den Ouweland AM , Majoor-Krakauer DF , Lolkema MP , et al. Genetic analysis of von Hippel-Lindau disease. Hum Mutat. (2010) ;31: (5):521–37. |
[17] | Poston CD , Jaffe GS , Lubensky IA , Solomon D , Zbar B , Linehan WM , et al. Characterization of the renal pathology of a familial form of renal cell carcinoma associated with von Hippel-Lindau disease: clinical and molecular genetic implications. J Urol. (1995) ;153: (1):22–6. |
[18] | Gkolfinopoulos S , Psyrri A , Bamias A . Clear-cell renal cell carcinoma - A comprehensive review of agents used in the contemporary management of advanced/metastatic disease. Oncol Rev. (2021) ;15: (1):530. |
[19] | Choueiri TK , Plimack ER , Bauer TM , Merchan JR , Papadopoulos KP , McDermott DF , et al. Phase I/II study of the oral HIF-2 α inhibitor MK-in patients with advanced clear cell renal cell carcinoma (RCC). Journal of Clinical Oncology. (2020) ;38: (6_suppl):611-. |
[20] | Jonasch E , McCutcheon IE , Waguespack SG , Wen S , Davis DW , Smith LA , et al. Pilot trial of sunitinib therapy in patients with von Hippel-Lindau disease. Ann Oncol. (2011) ;22: (12):2661–6. |
[21] | Jonasch E , McCutcheon IE , Gombos DS , Ahrar K , Perrier ND , Liu D , et al. Pazopanib in patients with von Hippel-Lindau disease: a single-arm, single-centre, phase 2 trial. Lancet Oncol. (2018) ;19: (10):1351–9. |
[22] | Farley MN , Schmidt LS , Mester JL , Pena-Llopis S , Pavia-Jimenez A , Christie A , et al. A novel germline mutation in BAP1 predisposes to familial clear-cell renal cell carcinoma. Mol Cancer Res. (2013) ;11: (9):1061–71. |
[23] | Kapur P , Pena-Llopis S , Christie A , Zhrebker L , Pavia-Jimenez A , Rathmell WK , et al. Effects on survival of BAP1 and PBRM1 mutations in sporadic clear-cell renal-cell carcinoma: a retrospective analysis with independent validation. Lancet Oncol. (2013) ;14: (2):159–67. |
[24] | Varela I , Tarpey P , Raine K , Huang D , Ong CK , Stephens P , et al. Exome sequencing identifies frequent mutation of the SWI/SNF complex gene PBRM1 in renal carcinoma. Nature. (2011) ;469: (7331):539–42. |
[25] | Pena-Llopis S , Vega-Rubin-de-Celis S , Liao A , Leng N , Pavia-Jimenez A , Wang S , et al. BAP1 loss defines a new class of renal cell carcinoma. Nat Genet.. (2012) ;44: (7):751–9. |
[26] | Teh BT , Giraud S , Sari NF , Hii SI , Bergerat JP , Larsson C , et al. Familial non-VHL non-papillary clear-cell renal cancer. Lancet. (1997) ;349: (9055):848–9. |
[27] | Woodward ER , Clifford SC , Astuti D , Affara NA , Maher ER . Familial clear cell renal cell carcinoma (FCRC): clinical features and mutation analysis of the VHL, MET, and CUL2 candidate genes. J Med Genet. (2000) ;37: (5):348–53. |
[28] | Woodward ER , Ricketts C , Killick P , Gad S , Morris MR , Kavalier F , et al. Familial non-VHL clear cell (conventional) renal cell carcinoma: clinical features, segregation analysis, and mutation analysis of FLCN. Clin Cancer Res. (2008) ;14: (18):5925–30. |
[29] | Eker R . Familial renal adenomas in Wistar rats; a preliminary report. Acta Pathol Microbiol Scand. (1954) ;34: (6):554–62. |
[30] | van Slegtenhorst M , de Hoogt R , Hermans C , Nellist M , Janssen B , Verhoef S , et al. Identification of the tuberous sclerosis gene TSC1 on chromosome 9q34. Science. (1997) ;277: (5327):805–8. |
[31] | European Chromosome 16 Tuberous Sclerosis C. Identification and characterization of the tuberous sclerosis gene on chromosome 16. Cell. (1993) ;75: (7):1305–15. |
[32] | Manning BD , Tee AR , Logsdon MN , Blenis J , Cantley LC . Identification of the tuberous sclerosis complex-2 tumor suppressor gene product tuberin as a target of the phosphoinositide 3-kinase/akt pathway. Mol Cell. . (2002) ;10: (1):151–62. |
[33] | Dan HC , Sun M , Yang L , Feldman RI , Sui XM , Ou CC , et al. Phosphatidylinositol 3-kinase/Akt pathway regulates tuberous sclerosis tumor suppressor complex by phosphorylation of tuberin. J Biol Chem. (2002) ;277: (38):35364–70. |
[34] | Crino PB , Nathanson KL , Henske EP . The tuberous sclerosis complex. N Engl J Med. (2006) ;355: (13):1345–56. |
[35] | Bjornsson J , Short MP , Kwiatkowski DJ , Henske EP . Tuberous sclerosis-associated renal cell carcinoma. Clinical, pathological, and genetic features. Am J Pathol. (1996) ;149: (4):1201–8. |
[36] | Lane BR , Aydin H , Danforth TL , Zhou M , Remer EM , Novick AC , et al. Clinical correlates of renal angiomyolipoma subtypes in 209 patients: classic, fat poor, tuberous sclerosis associated and epithelioid. J Urol. . (2008) ;180: (3):836–43. |
[37] | Delgado R , de Leon Bojorge B , Albores-Saavedra J . Atypical angiomyolipoma of the kidney: a distinct morphologic variant that is easily confused with a variety of malignant neoplasms. Cancer. (1998) ;83: (8):1581–92. |
[38] | Alsidawi S , Kasi PM . Exceptional response to everolimus in a novel tuberous sclerosis complex-2 mutation-associated metastatic renal-cell carcinoma. Cold Spring Harb Mol Case Stud. (2018) ;4: (2). |
[39] | Bissler JJ , Kingswood JC , Radzikowska E , Zonnenberg BA , Frost M , Belousova E , et al. Everolimus for angiomyolipoma associated with tuberous sclerosis complex or sporadic lymphangioleiomyomatosis (EXIST-2): a multicentre, randomised, double-blind, placebo-controlled trial. Lancet. (2013) ;381: (9869):817–24. |
[40] | Bissler JJ , Kingswood JC , Radzikowska E , Zonnenberg BA , Belousova E , Frost MD , et al. Everolimus long-term use in patients with tuberous sclerosis complex: Four-year update of the EXIST-2 study. PloS one. (2017) ;12: (8):e0180939. |
[41] | Pavlovich CP , Walther MM , Eyler RA , Hewitt SM , Zbar B , Linehan WM , et al. Renal tumors in the Birt-Hogg-Dube syndrome. Am J Surg Pathol. (2002) ;26: (12):1542–52. |
[42] | Toro JR , Glenn G , Duray P , Darling T , Weirich G , Zbar B , et al. Birt-Hogg-Dube syndrome: a novel marker of kidney neoplasia. Arch Dermatol. (1999) ;135: (10):1195–202. |
[43] | Roth JS , Rabinowitz AD , Benson M , Grossman ME . Bilateral renal cell carcinoma in the Birt-Hogg-Dube syndrome. J Am Acad Dermatol. (1993) ;29: (6):1055–6. |
[44] | Birt AR , Hogg GR , Dube WJ . Hereditary multiple fibrofolliculomas with trichodiscomas and acrochordons. Arch Dermatol. (1977) ;113: (12):1674–7. |
[45] | Schmidt LS , Warren MB , Nickerson ML , Weirich G , Matrosova V , Toro JR , et al. Birt-Hogg-Dube syndrome, a genodermatosis associated with spontaneous pneumothorax and kidney neoplasia, maps to chromosome 17p11. 2. Am J Hum Genet. (2001) ;69: (4):876–82. |
[46] | Hasumi H , Baba M , Hasumi Y , Huang Y , Oh H , Hughes RM , et al. Regulation of mitochondrial oxidative metabolism by tumor suppressor FLCN. J Natl Cancer Inst. (2012) ;104: (22):1750–64. |
[47] | Hasumi H , Baba M , Hasumi Y , Lang M , Huang Y , Oh HF , et al. Folliculin-interacting proteins Fnip1 and Fnip2 play critical roles in kidney tumor suppression in cooperation with Flcn. Proc Natl Acad Sci U S A. (2015) ;112: (13):E1624–31. |
[48] | Hasumi H , Baba M , Hong SB , Hasumi Y , Huang Y , Yao M , et al. Identification and characterization of a novel folliculin-interacting protein FNIP2. Gene. (2008) ;415: (1-2):60–7. |
[49] | Hasumi Y , Baba M , Ajima R , Hasumi H , Valera VA , Klein ME , et al. Homozygous loss of BHD causes early embryonic lethality and kidney tumor development with activation of mTORC1 and mTORC2. Proc Natl Acad Sci U S A. (2009) ;106: (44):18722–7. |
[50] | Hasumi Y , Baba M , Hasumi H , Huang Y , Lang M , Reindorf R , et al. Folliculin (Flcn) inactivation leads to murine cardiac hypertrophy through mTORC1 deregulation. Hum Mol Genet. (2014) ;23: (21):5706–19. |
[51] | Baba M , Furihata M , Hong SB , Tessarollo L , Haines DC , Southon E , et al. Kidney-targeted Birt-Hogg-Dube gene inactivation in a mouse model: Erk1/2 and Akt-mTOR activation, cell hyperproliferation, and polycystic kidneys. J Natl Cancer Inst. (2008) ;100: (2):140–54. |
[52] | Baba M , Hong SB , Sharma N , Warren MB , Nickerson ML , Iwamatsu A , et al. Folliculin encoded by the BHD gene interacts with a binding protein, FNIP1, and AMPK, and is involved in AMPK and mTOR signaling. Proc Natl Acad Sci U S A. (2006) ;103: (42):15552–7. |
[53] | Toro JR , Wei MH , Glenn GM , Weinreich M , Toure O , Vocke C , et al. BHD mutations, clinical and molecular genetic investigations of Birt-Hogg-Dube syndrome: a new series of 50 families and a review of published reports. J Med Genet. (2008) ;45: (6):321–31. |
[54] | Warren AY , Harrison D . WHO/ISUP classification, grading and pathological staging of renal cell carcinoma: standards and controversies. World J Urol. (2018) ;36: (12):1913–26. |
[55] | Hasumi H , Baba M , Hasumi Y , Furuya M , Yao M . Birt-Hogg-Dube syndrome: Clinical and molecular aspects of recently identified kidney cancer syndrome. Int J Urol. (2016) ;23: (3):204–10. |
[56] | Nakamura M , Yao M , Sano F , Sakata R , Tatenuma T , Makiyama K , et al. [A case of metastatic renal cell carcinoma associated with Birt-Hogg-Dube syndrome treated with molecular-targeting agents]. Hinyokika Kiyo. (2013) ;59: (8):503–6. |
[57] | Starink TM , van der Veen JP , Arwert F , de Waal LP , de Lange GG , Gille JJ , et al. The Cowden syndrome: a clinical and genetic study in 21 patients. Clin Genet. (1986) ;29: (3):222–33. |
[58] | Hendriks YM , Verhallen JT , van der Smagt JJ , Kant SG , Hilhorst Y , Hoefsloot L , et al. Bannayan-Riley-Ruvalcaba syndrome: further delineation of the phenotype and management of PTEN mutation-positive cases. Familial cancer. (2003) ;2: (2):79–85. |
[59] | Marsh DJ , Kum JB , Lunetta KL , Bennett MJ , Gorlin RJ , Ahmed SF , et al. PTEN mutation spectrum and genotype-phenotype correlations in Bannayan-Riley-Ruvalcaba syndrome suggest a single entity with Cowden syndrome. Hum Mol Genet. (1999) ;8: (8):1461–72. |
[60] | Zbuk KM , Eng C . Cancer phenomics: RET and PTEN as illustrative models. Nat Rev Cancer. (2007) ;7: (1):35–45. |
[61] | Li DM , Sun H . TEP1, encoded by a candidate tumor suppressor locus, is a novel protein tyrosine phosphatase regulated by transforming growth factor beta. Cancer Res. (1997) ;57: (11):2124–9. |
[62] | Myers MP , Pass I , Batty IH , Van der Kaay J , Stolarov JP , Hemmings BA , et al. The lipid phosphatase activity of PTEN is critical for its tumor supressor function. Proc Natl Acad Sci U S A. (1998) ;95: (23):13513–8. |
[63] | Maehama T , Dixon JE . The tumor suppressor, PTEN/MMAC1, dephosphorylates the lipid second messenger, phosphatidylinositol 3,4,5-trisphosphate. J Biol Chem. (1998) ;273: (22):13375–8. |
[64] | Stambolic V , Suzuki A , de la Pompa JL , Brothers GM , Mirtsos C , Sasaki T , et al. Negative regulation of PKB/Akt-dependent cell survival by the tumor suppressor PTEN. Cell. (1998) ;95: (1):29–39. |
[65] | Mester JL , Zhou M , Prescott N , Eng C . Papillary renal cell carcinoma is associated with PTEN hamartoma tumor syndrome. Urology. (1187) ;79: (5):e1–7. |
[66] | Motzer RJ , Barrios CH , Kim TM , Falcon S , Cosgriff T , Harker WG , et al. Phase II randomized trial comparing sequential first-line everolimus and second-line sunitinib versus first-line sunitinib and second-line everolimus in patients with metastatic renal cell carcinoma. J Clin Oncol. (2014) ;32: (25):2765–72. |
[67] | Voss MH , Chen D , Reising A , Marker M , Shi J , Xu J , et al. PTEN expression, not mutation status in TSC1, TSC2, or mTOR, correlates with the outcome on everolimus in patients with renal cell carcinoma treated on the randomized RECORD-3 trial. Clin Cancer Res. (2019) ;25: (2):506–14. |
[68] | Carr CS , Sharp PA . A helix-loop-helix protein related to the immunoglobulin E box-binding proteins. Mol Cell Biol. (1990) ;10: (8):4384–8. |
[69] | Hodgkinson CA , Moore KJ , Nakayama A , Steingrimsson E , Copeland NG , Jenkins NA , et al. Mutations at the mouse microphthalmia locus are associated with defects in a gene encoding a novel basic-helix-loop-helix-zipper protein. Cell. (1993) ;74: (2):395–404. |
[70] | Zhao GQ , Zhao Q , Zhou X , Mattei MG , de Crombrugghe B . TFEC, a basic helix-loop-helix protein, forms heterodimers with TFE3 and inhibits TFE3-dependent transcription activation. Mol Cell Biol. (1993) ;13: (8):4505–12. |
[71] | Beckmann H , Su LK , Kadesch T . TFE a helix-loop-helix protein that activates transcription through the immunoglobulin enhancer muE3 motif. Genes Dev. (1990) ;4: (2):167–79. |
[72] | Henthorn PS , Stewart CC , Kadesch T , Puck JM . The gene encoding human TFE3, a transcription factor that binds the immunoglobulin heavy-chain enhancer, maps to Xp11. 22. Genomics. (1991) ;11: (2):374–8. |
[73] | Moch H , Cubilla AL , Humphrey PA , Reuter VE , Ulbright TM . The WHO classification of tumours of the urinary system and male genital organs-Part A: renal, penile, and testicular tumours. Eur Urol. (2016) ;70: (1):93–105. |
[74] | Brady OA , Jeong E , Martina JA , Pirooznia M , Tunc I , Puertollano R . The transcription factors TFE3 and TFEB amplify p53 dependent transcriptional programs in response to DNA damage. Elife. 2018;7. |
[75] | Bertolotto C , Lesueur F , Giuliano S , Strub T , de Lichy M , Bille K , et al. A SUMOylation-defective MITF germline mutation predisposes to melanoma and renal carcinoma. Nature. (2011) ;480: (7375):94–8. |
[76] | Kmetec A , Jeruc J . Xp 11. 2 translocation renal carcinoma in young adults; recently classified distinct subtype. Radiol Oncol. (2014) ;48: (2):197–202. |
[77] | Sukov WR , Hodge JC , Lohse CM , Leibovich BC , Thompson RH , Pearce KE , et al. TFE3 rearrangements in adult renal cell carcinoma: clinical and pathologic features with outcome in a large series of consecutively treated patients. Am J Surg Pathol. (2012) ;36: (5):663–70. |
[78] | Ellis CL , Eble JN , Subhawong AP , Martignoni G , Zhong M , Ladanyi M , et al. Clinical heterogeneity of Xp11 translocation renal cell carcinoma: impact of fusion subtype, age, and stage. Mod Pathol. (2014) ;27: (6):875–86. |
[79] | Inamura K , Fujiwara M , Togashi Y , Nomura K , Mukai H , Fujii Y , et al. Diverse fusion patterns and heterogeneous clinicopathologic features of renal cell carcinoma with t(6;11) translocation. Am J Surg Pathol. (2012) ;36: (1):35–42. |
[80] | Argani P , Yonescu R , Morsberger L , Morris K , Netto GJ , Smith N , et al. Molecular confirmation of t(6;11)(p21;q12) renal cell carcinoma in archival paraffin-embedded material using a break-apart TFEB FISH assay expands its clinicopathologic spectrum. Am J Surg Pathol. (2012) ;36: (10):1516–26. |
[81] | Smith NE , Illei PB , Allaf M , Gonzalez N , Morris K , Hicks J , et al. t(6;11) renal cell carcinoma (RCC): expanded immunohistochemical profile emphasizing novel RCC markers and report of 10 new genetically confirmed cases. Am J Surg Pathol. (2014) ;38: (5):604–14. |
[82] | Argani P , Reuter VE , Zhang L , Sung YS , Ning Y , Epstein JI , et al. TFEB-amplified renal cell carcinomas: an aggressive molecular subset demonstrating variable melanocytic marker expression and morphologic heterogeneity. Am J Surg Pathol. (2016) ;40: (11):1484–95. |
[83] | Williamson SR , Grignon DJ , Cheng L , Favazza L , Gondim DD , Carskadon S , et al. Renal cell carcinoma with chromosome 6p amplification including the TFEB gene: a novel mechanism of tumor pathogenesis? Am J Surg Pathol. (2017) ;41: (3):287–98. |
[84] | Gupta S , Johnson SH , Vasmatzis G , Porath B , Rustin JG , Rao P , et al. TFEB-VEGFA (6p21. 1) co-amplified renal cell carcinoma: a distinct entity with potential implications for clinical management. Mod Pathol. (2017) ;30: (7):998–1012. |
[85] | Niemczyk M , Niemczyk S , Paczek L . Autosomal dominant polycystic kidney disease and transplantation. Ann Transplant. (2009) ;14: (4):86–90. |
[86] | Grantham JJ , Torres VE , Chapman AB , Guay-Woodford LM , Bae KT , King BF Jr , et al. Volume progression in polycystic kidney disease. N Engl J Med.. (2006) ;354: (20):2122–30. |
[87] | Torres VE , Harris PC , Pirson Y . Autosomal dominant polycystic kidney disease. Lancet. (2007) ;369: (9569):1287–301. |
[88] | Harris PC , Bae KT , Rossetti S , Torres VE , Grantham JJ , Chapman AB , et al. Cyst number but not the rate of cystic growth is associated with the mutated gene in autosomal dominant polycystic kidney disease. J Am Soc Nephrol. (2006) ;17: (11):3013–9. |
[89] | Hateboer N , v Dijk MA , Bogdanova N , Coto E , Saggar-Malik AK , San Millan JL , et al. Comparison of phenotypes of polycystic kidney disease types 1 and 2. European PKD1-PKD2 Study GrouLancet. (1999) ;353: (9147):103–7. |
[90] | Keith DS , Torres VE , King BF , Zincki H , Farrow GM . Renal cell carcinoma in autosomal dominant polycystic kidney disease. J Am Soc Nephrol. (1994) ;4: (9):1661–9. |
[91] | McNichols DW , Segura JW , DeWeerd JH . Renal cell carcinoma: long-term survival and late recurrence. J Urol. (1981) ;126: (1):17–23. |
[92] | Jilg CA , Drendel V , Bacher J , Pisarski P , Neeff H , Drognitz O , et al. Autosomal dominant polycystic kidney disease: prevalence of renal neoplasias in surgical kidney specimens. Nephron Clin Pract. (2013) ;123: (1-2):13–21. |
[93] | Karami S , Yanik EL , Moore LE , Pfeiffer RM , Copeland G , Gonsalves L , et al. Risk of renal cell carcinoma among kidney transplant recipients in the United States. Am J Transplant. (2016) ;16: (12):3479–89. |
[94] | Levine E , Slusher SL , Grantham JJ , Wetzel LH . Natural history of acquired renal cystic disease in dialysis patients: a prospective longitudinal CT study. AJR American journal of roentgenology. (1991) ;156: (3):501–6. |
[95] | Peired AJ , Lazzeri E , Guzzi F , Anders H-J , Romagnani P . From kidney injury to kidney cancer. Kidney International. (2021) ;100: (1):55–66. |
[96] | Yu T-M , Chuang Y-W , Yu M-C , Chen C-H , Yang C-K , Huang S-T , et al. Risk of cancer in patients with polycystic kidney disease: a propensity-score matched analysis of a nationwide, population-based cohort study. The Lancet Oncology. (2016) ;17: (10):1419–25. |
[97] | Liu Y , Pejchinovski M , Wang X , Fu X , Castelletti D , Watnick TJ , et al. Dual mTOR/PI3K inhibition limits PI3K-dependent pathways activated upon mTOR inhibition in autosomal dominant polycystic kidney disease. Scientific Reports. (2018) ;8: (1):5584. |
[98] | Wei MH , Toure O , Glenn GM , Pithukpakorn M , Neckers L , Stolle C , et al. Novel mutations in FH and expansion of the spectrum of phenotypes expressed in families with hereditary leiomyomatosis and renal cell cancer. J Med Genet. (2006) ;43: (1):18–27. |
[99] | Toro JR , Nickerson ML , Wei MH , Warren MB , Glenn GM , Turner ML , et al. Mutations in the fumarate hydratase gene cause hereditary leiomyomatosis and renal cell cancer in families in North America. Am J Hum Genet. (2003) ;73: (1):95–106. |
[100] | Linehan WM , Rouault TA . Molecular pathways: Fumarate hydratase-deficient kidney cancer–targeting the Warburg effect in cancer. Clin Cancer Res. (2013) ;19: (13):3345–52. |
[101] | Srinivasan R , Gurram S , Al Harthy M , Singer EA , Sidana A , Shuch BM , et al. Results from a phase II study of bevacizumab and erlotinib in subjects with advanced hereditary leiomyomatosis and renal cell cancer (HLRCC) or sporadic papillary renal cell cancer. Journal of Clinical Oncology. (2020) ;38: (15_suppl):5004. |
[102] | Wells SA Jr , Robinson BG , Gagel RF , Dralle H , Fagin JA , Santoro M , et al. Vandetanib in patients with locally advanced or metastatic medullary thyroid cancer: a randomized, double-blind phase III trial. J Clin Oncol. (2012) ;30: (2):134–41. |
[103] | Choi Y , Keam B , Kim M , Yoon S , Kim D , Choi JG , et al. Bevacizumab plus erlotinib combination therapy for advanced hereditary leiomyomatosis and renal cell carcinoma-associated renal cell carcinoma: a multicenter retrospective analysis in Korean patients. Cancer Res Treat. (2019) ;51: (4):1549–56. |
[104] | Baysal BE , Ferrell RE , Willett-Brozick JE , Lawrence EC , Myssiorek D , Bosch A , et al. Mutations in SDHD, a mitochondrial complex II gene, in hereditary paraganglioma. Science. (2000) ;287: (5454):848–51. |
[105] | Baysal BE . Hereditary paraganglioma targets diverse paraganglia. J Med Genet. (2002) ;39: (9):617–22. |
[106] | Burnichon N , Brière JJ , Libé R , Vescovo L , Rivière J , Tissier F , et al. SDHA is a tumor suppressor gene causing paraganglioma. Hum Mol Genet. (2010) ;19: (15):3011–20. |
[107] | Astuti D , Latif F , Dallol A , Dahia PL , Douglas F , George E , et al. Gene mutations in the succinate dehydrogenase subunit SDHB cause susceptibility to familial pheochromocytoma and to familial paraganglioma. Am J Hum Genet. (2001) ;69: (1):49–54. |
[108] | Niemann S , Muller U . Mutations in SDHC cause autosomal dominant paraganglioma, type 3. Nat Genet. (2000) ;26: (3):268–70. |
[109] | Selak MA , Armour SM , MacKenzie ED , Boulahbel H , Watson DG , Mansfield KD , et al. Succinate links TCA cycle dysfunction to oncogenesis by inhibiting HIF-alpha prolyl hydroxylase. Cancer Cell. (2005) ;7: (1):77–85. |
[110] | Vanharanta S , Buchta M , McWhinney SR , Virta SK , Peczkowska M , Morrison CD , et al. Early-onset renal cell carcinoma as a novel extraparaganglial component of SDHB-associated heritable paraganglioma. Am J Hum Genet. (2004) ;74: (1):153–9. |
[111] | Gill AJ , Pachter NS , Chou A , Young B , Clarkson A , Tucker KM , et al. Renal tumors associated with germline SDHB mutation show distinctive morphology. Am J Surg Pathol. (2011) ;35: (10):1578–85. |
[112] | Gill AJ , Pachter NS , Clarkson A , Tucker KM , Winship IM , Benn DE , et al. Renal tumors and hereditary pheochromocytoma-paraganglioma syndrome type 4. N Engl J Med. (2011) ;364: (9):885–6. |
[113] | Ricketts CJ , Shuch B , Vocke CD , Metwalli AR , Bratslavsky G , Middelton L , et al. Succinate dehydrogenase kidney cancer: an aggressive example of the Warburg effect in cancer. J Urol. (2012) ;188: (6):2063–71. |
[114] | de Bono J , Ramanathan RK , Mina L , Chugh R , Glaspy J , Rafii S , et al. Phase I, dose-escalation, two-part trial of the PARP inhibitor talazoparib in patients with advanced germline BRCA1/2 mutations and selected sporadic cancers. Cancer Discov. (2017) ;7: (6):620–9. |
[115] | Heery CR , O’Sullivan-Coyne G , Madan RA , Cordes L , Rajan A , Rauckhorst M , et al. Avelumab for metastatic or locally advanced previously treated solid tumours (JAVELIN Solid Tumor): a phase 1a, multicohort, dose-escalation trial. Lancet Oncol. (2017) ;18: (5):587–98. |
[116] | Schmidt L , Duh FM , Chen F , Kishida T , Glenn G , Choyke P , et al. Germline and somatic mutations in the tyrosine kinase domain of the MET proto-oncogene in papillary renal carcinomas. Nat Genet. (1997) ;16: (1):68–73. |
[117] | Zbar B , Tory K , Merino M , Schmidt L , Glenn G , Choyke P , et al. Hereditary papillary renal cell carcinoma. J Urol. (1994) ;151: (3):561–6. |
[118] | Schmidt L , Junker K , Nakaigawa N , Kinjerski T , Weirich G , Miller M , et al. Novel mutations of the MET proto-oncogene in papillary renal carcinomas. Oncogene. (1999) ;18: (14):2343–50. |
[119] | Debelenko LV , Raimondi SC , Daw N , Shivakumar BR , Huang D , Nelson M , et al. Renal cell carcinoma with novel VCL-ALK fusion: new representative of ALK-associated tumor spectrum. Mod Pathol. (2011) ;24: (3):430–42. |
[120] | Marino-Enriquez A , Ou WB , Weldon CB , Fletcher JA , Perez-Atayde AR . ALK rearrangement in sickle cell trait-associated renal medullary carcinoma. Genes Chromosomes Cancer. (2011) ;50: (3):146–53. |
[121] | Sugawara E , Togashi Y , Kuroda N , Sakata S , Hatano S , Asaka R , et al. Identification of anaplastic lymphoma kinase fusions in renal cancer: large-scale immunohistochemical screening by the intercalated antibody-enhanced polymer method. Cancer. (2012) ;118: (18):4427–36. |
[122] | Kusano H , Togashi Y , Akiba J , Moriya F , Baba K , Matsuzaki N , et al. Two cases of renal cell carcinoma harboring a novel STRN-ALK fusion gene. Am J Surg Pathol. (2016) ;40: (6):761–9. |
[123] | Pulford K , Lamant L , Morris SW , Butler LH , Wood KM , Stroud D , et al. Detection of anaplastic lymphoma kinase (ALK) and nucleolar protein nucleophosmin (NPM)-ALK proteins in normal and neoplastic cells with the monoclonal antibody ALK1. Blood. (1997) ;89: (4):1394–404. |
[124] | Fischer P , Nacheva E , Mason DY , Sherrington PD , Hoyle C , Hayhoe FG , et al. A Ki-1 (CD30)-positive human cell line (Karpas 299) established from a high-grade non-Hodgkin’s lymphoma, showing a 2;5 translocation and rearrangement of the T-cell receptor beta-chain gene. Blood. (1988) ;72: (1):234–40. |
[125] | Sukov WR , Hodge JC , Lohse CM , Akre MK , Leibovich BC , Thompson RH , et al. ALK alterations in adult renal cell carcinoma: frequency, clinicopathologic features and outcome in a large series of consecutively treated patients. Mod Pathol. (2012) ;25: (11):1516–25. |
[126] | Online Mendelian Inheritance in Man, OMIM® [online database]. Baltimore, MD:McKusick-Nathans Institute of Genetic Medicine, Johns Hopkins University 2022 [Available from: https://omim.org/. |


