
Cell-free Circulating Tumor DNA (ctDNA) in Metastatic Renal Cell Carcinoma (mRCC): Current Knowledge and Potential Uses
Abstract
Historically, tumor biopsies and clinical laboratory testing have been the gold standard for diagnosis and prognosis in metastatic renal cell carcinoma (mRCC). Genomic profiling in mRCC has traditionally been performed on tumor tissue; however, challenges and limitations in obtaining tissue biopsies led to the discovery of alternative biological specimens, namely circulating cell-free DNA (cfDNA). Rapidly evolving technologies, with increased sensitivity and specificity, have been used to query cfDNA in the clinical research setting. These investigations are rapidly establishing cfDNA and liquid biopsies as valuable complementary specimens to the gold standard, and in some instances surpassing these with unique insight into the contemporary genomic landscape and tumor heterogeneity. In this review, we will discuss recent research into the prognostic, diagnostic, and predictive utility of liquid biopsies in mRCC. We will explore their potential role in precision treatment of mRCC and conclude with what is needed in order to translate them to clinical practice.
INTRODUCTION
The treatment landscape for metastatic renal cell carcinoma (mRCC) is evolving at a staggering pace. Currently, the therapeutic armamentarium for mRCC includes multi-targeted tyrosine kinase inhibitors (TKIs), VEGF targeted therapies, combined immune checkpoint inhibitors (ICIs), and combinations of VEGF targeted therapy and immune checkpoint inhibitors [1–7]. Since many of these agents were approved over a similar time period, there are few direct head-to-head comparisons of these agents. With minimal head-to-head clinical trial data to guide treatment selection, predictive biomarkers capable of optimizing individual outcomes are needed in mRCC. Tissue biopsies are routinely used to diagnose patients with mRCC, and they are also the primary source of tumor cells for molecular profiling by next-generation sequencing (NGS). NGS of tumor tissue has improved our understanding of the biology and genomic landscape of mRCC [8].
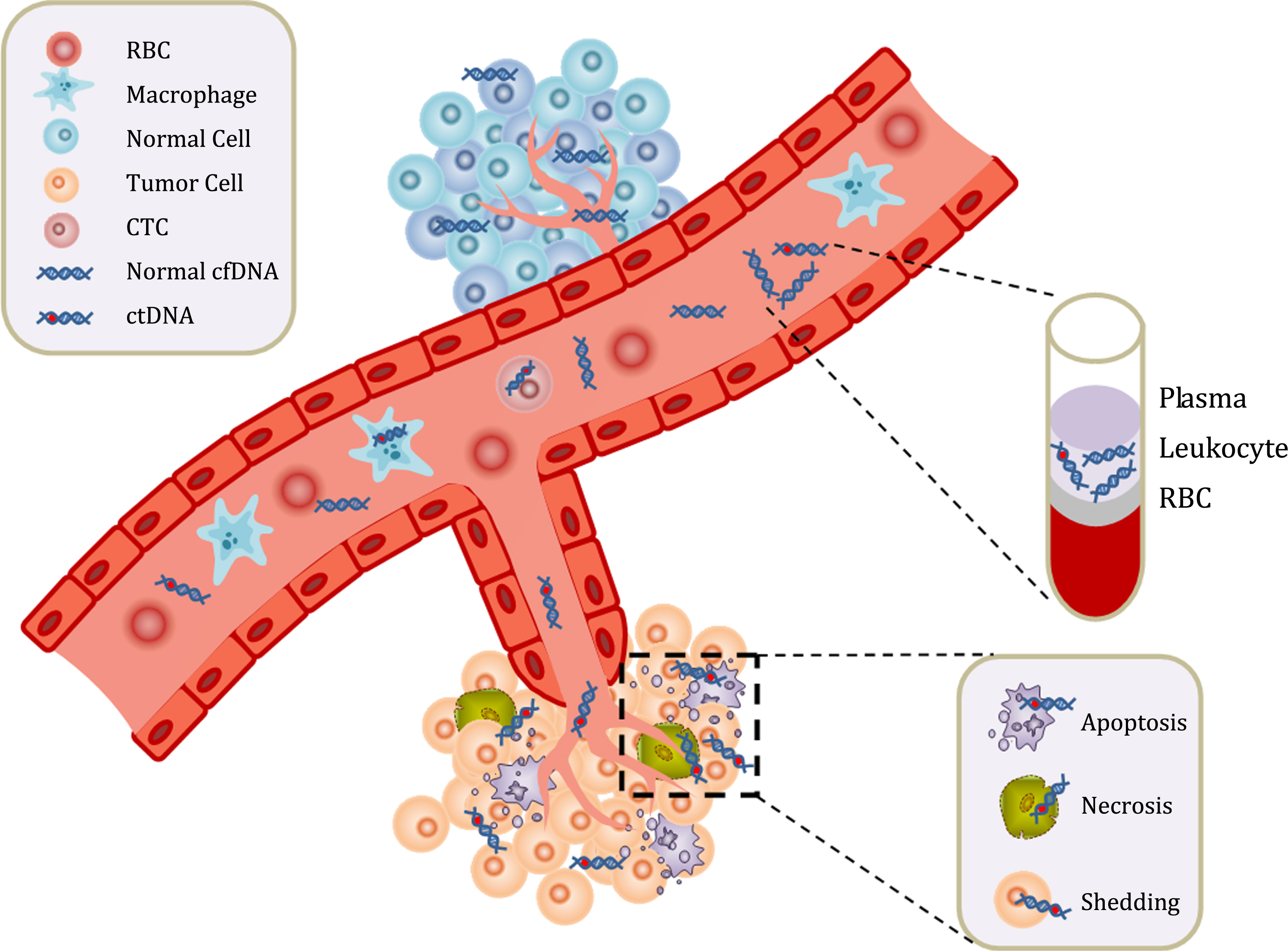
To date, in mRCC, the majority of NGS studies were retrospective and utilized archived tumor tissue from the initial nephrectomy, which may not accurately reflect the contemporary genomic profile of metastatic disease. For patients with mRCC, analysis of cell-free circulating tumor DNA (ctDNA), hereafter referred to as liquid biopsy, is a promising addition to the diagnostic, predictive, and prognostic tools currently utilized in the clinical setting. A thorough discussion on liquid biopsies is beyond the scope of this review, but we recommend readers refer to a recent publication by Corcoran and Chabner for more details [9]. Current dogma predicts that cancer cells undergoing apoptosis and necrosis release ctDNA into circulation, which then combines with plasma cell-free circulating DNA (cfDNA) from normal cells (Fig. 1) [10]. Liquid biopsy is a minimally invasive blood-based test that is easy to repeat, may offer insight into tumor heterogeneity, and provides contemporary monitoring of tumor evolution [11]. Furthermore, metastases in mRCC are often solitary and in anatomic sites that are difficult to biopsy, such as bone or brain. In this review, we will discuss the promising clinical utility of liquid biopsies for diagnosis, prognosis, and to guide precision treatment of mRCC patients.
Fig.1
Circulating cell-free DNA (cfDNA) and circulating tumor DNA (ctDNA) are found in serum and plasma fractions from blood. The mechanism of ctDNA release is unknown, though apoptosis, necrosis, and active shedding from tumor cells has been hypothesized. Once ctDNA is isolated, it can be quantitated and analyzed for genomic alterations.
ANALYTICAL CHARACTERIZATION OF LIQUID BIOPSY IN mRCC
Nuclease-resistant cell-free DNA (cfDNA), typically associated to nucleosomes, corresponds to double-stranded DNA fragments ranging in length between 150–200 base pairs. The abundance, degree of fragmentation (genomic and mitochondrial), and methylation of tumor-suppressor genes has been investigated in cfDNA from RCC patients.
Analysis of cfDNA levels and fragmentation in mRCC
In spite of significant variability in cfDNA levels between patients, a study of 92 RCC patients and 41 healthy controls reported increasing levels between healthy controls and tumor, as well as with increasing Fuhrman nuclear grade [12]. Moreover, cfDNA levels were lower in healthy controls compared to cT1aN0M0 RCC patients, and higher in patients with lymphovascular invasion (LVI) [12]. Additionally, the median fragment size of plasma cfDNA was shorter in RCC patients compared to healthy controls, and also shorter with higher Fuhrman grade (3/4) and positive LVI [12]. Yamamoto and colleagues also noted that cfDNA fragment size increased following removal of the primary tumor, and that shorter fragment size and higher plasma cfDNA levels were associated with shorter PFS [12]. These findings suggest that cfDNA levels and median fragment size may have diagnostic and prognostic utility for patients with all stages of RCC.
Analysis of mitochondrial cfDNA fragments in mRCC
A different report investigated whether genomic and mitochondrial cfDNA fragments may have diagnostic and prognostic potential [13]. CfDNA was extracted from 40 healthy controls and 229 RCC patients (145 with localized disease and 84 metastatic). Results from this study indicated two mitochondrial fragments of 65 and 175bp were able to distinguish metastatic from localized and healthy control cfDNA with high confidence (p < 0.0001). In contrast, only 1 out of 5 genomic cfDNA fragments (APP fragment 3, 306bp) was capable of distinguishing between RCC patients, who had lower levels, and healthy controls. The results reported by Lu and colleagues suggests that combinations of genomic and mitochondrial cfDNA fragments may help with early diagnosis of metastatic disease, which could be especially valuable in clinical trials evaluating adjuvant therapy for RCC [13].
Analysis of cfDNA levels and tumor suppressor gene methylation in mRCC
Earlier detection of RCC at lower clinical staging improves the probability of cure. With this goal in mind, Skrypkina and colleagues investigated whether cfDNA levels and methylation of specific tumor suppressor genes could be useful in RCC [14]. Their results, in agreement with those from previous reports, showed that cfDNA concentration in plasma was higher in RCC patients compared to healthy controls. In addition, they noted that methylation of the tumor suppressor genes RASSF1 (62.9%), FHIT (55.6%), and APC (51.9%) in cfDNA had the highest specificity for distinguishing patients with RCC from healthy controls. Finally, the authors utilized receiver-operating characteristics analysis to investigate the diagnostic potential of combining cfDNA level with the methylation status of the three tumor-suppressor genes (RASSF1A, FHIT, and APC). The ROC results indicate the highest achievable sensitivity/specificity (100%) for RCC diagnosis in the localized setting requires a combination of the cfDNA level and methylation status of all three genes. A significant limiting factor in this study was the number of RCC patients and controls investigated, 27 and 15 respectively. Due to potential significance of these results, validation of their findings in large, prospective studies is needed.
GENOMIC LANDSCAPE OF mRCC BY LIQUID BIOPSY
In order for liquid biopsy to be of clinical utility, targeted panels must include genes with common genomic alterations (GAs) found in patients with mRCC. While currently there are no predictive biomarkers for mRCC, a group of commonly altered genes that are of interest includes VHL, PBRM1, SETD2, BAP1, KDM5C, mTOR, TSC1, and PTEN [15–17]. To date, there is only one large study that reported the frequency of GAs in mRCC by NGS of a liquid biopsy [18]. In that study, Pal and colleagues described the genomic landscape in 220 patients with mixed histology mRCC who underwent testing using a commercial platform, Guardant360. Among 69 patients with clear cell mRCC, 32% had an alteration in VHL and 30% had an alteration in TP53. ARID1A (18%), NF1 (22%), and EGFR (13%) were also commonly altered in this cohort. The commercial gene panel used did not include PBRM1, SETD2, BAP1, or KDM5C, so the presence of GAs in these genes in ctDNA was not determined.
The frequency of GAs detected in ctDNA from mRCC patients differs from reports on NGS of tumor tissue. Multiple, large studies of tumor tissue NGS have been reported in patients with all stages of RCC, yet there is only one tissue NGS study that focused on patients with mRCC [19]. The gold standard for reporting the genomic landscape in tumor tissue across all stages and histologies of RCC is The Cancer Genome Atlas (TCGA) study [8, 20]. In the most recent report from TCGA of 488 patients with clear cell RCC, GAs in VHL were more common than in the study by Pal, et al (55% vs. 32%), while TP53 alterations were less prevalent (3% vs. 30%) (Table 1). While the TCGA cohort includes patients with all histologies of RCC, only 11% had clear cell mRCC. Thus, one could anticipate that the frequency of GAs would differ between the two studies. However, even if one were to consider only clear cell mRCC patients (n = 76) from the TCGA cohort, the frequency of GAs in VHL and TP53 would remain mostly the same (50% and 4% respectively). It is also important to note that >80% of ccRCC patients in the TCGA cohort had loss of chromosomal arm 3p where the VHL gene is located. The commercial platform, Guardant360, does not detect large deletions; therefore, loss of the VHL gene in the Pal et al cohort would go undetected. In contrast, a post-hoc analysis of the RECORD-3 trial performed NGS of tumor tissue on 220 patients with mRCC [19]. The prevalence of GAs in VHL and TP53 were 75% and 8% respectively (Table 1). A limitation of both, the TCGA and RECORD-3 studies on patients with mRCC, was that the predominant source of tissue came from primary tumor nephrectomies, thus these may not reflect the evolving genomic landscape and heterogeneity of the metastatic setting. Additionally, it is possible that low levels of ctDNA, possibly due to therapy, could result in alterations being below the limit of detection. Large, carefully controlled studies, comparing the genomic landscape assessed by NGS of tumor tissue and ctDNA are needed in order to fully understand the discrepancies in GAs detected by either platform.
Table 1
Frequency of Genomic Alterations in Clear-Cell mRCC by ctDNA and Tissue Next-Generation Sequencing
| Gene | ctDNA clear-cell (n = 69) | TCGA clear-cell (n = 488) | RECORD-3 (n = 220) |
| TP53 | 30% | 3% | 8% |
| VHL | 32% | 55% | 75% |
| PBRM1 | N/A* | 38% | 46% |
| SETD2 | N/A* | 13% | 30% |
| KDM5C | N/A* | 7% | 15% |
CtDNA = circulating tumor DNA, TCGA = The Cancer Genome Atlas, n = number, N/A = not available, * = not tested by commercially available ctDNA platform.
DIAGNOSTIC YIELD AND TIMING OF LIQUID BIOPSY IN mRCC
Currently, liquid biopsies are primarily utilized to enroll patients in biomarker-guided clinical trials; however, as their use becomes more prevalent, it’s important to understand the optimal timing for their collection in mRCC. In the largest study to date, 79% of 220 patients with mRCC had detectable ctDNA [18]. Furthermore, it appears that patients with higher tumor burden shed more ctDNA. In a study of 36 patients with mRCC, patients with detectable ctDNA had higher disease volume as determined by the sum of the longest diameter of measurable lesions on CT imaging compared to patients without detectable ctDNA (8.81 cm vs. 4.49 cm, p = 0.04) [21].
LIQUID BIOPSIES AND THERAPEUTIC EVOLUTION
It is also important to know how the genomic landscape of mRCC evolves after systemic treatment. In the same study of 220 patients with mRCC, Pal and colleagues showed that the genomic landscape for mRCC evolves after systemic treatment [18]. In a comparison of post-first-line and post-salvage-line treatment of mRCC, there was a significant difference in the frequency of GAs in TP53 (24% vs. 49%, p = 0.02) and NF1 (3% vs. 20%, p = 0.01). Furthermore, a non-significant difference was observed in the frequency of GAs in VHL (18% vs. 29%, p = 0.3) and PIK3CA (5% vs. 10%, p = 0.3). The evidence from these studies suggests that liquid biopsies will be most informative in those patients with mRCC who are progressing on systemic treatment or have high tumor burden.
LIQUID BIOPSIES AND TUMOR HETEROGENEITY
In a seminal paper by Gerlinger and colleagues, mRCC was shown to have significant intratumor heterogeneity, as 63–69% of somatic alterations were not detectable in every region biopsied [22]. Some have hypothesized that liquid biopsy will more accurately reflect the genomic landscape of mRCC by capturing tumor heterogeneity. In a hypothesis-generating study of 19 patients with mRCC, we performed matched tumor tissue and liquid biopsy NGS to assess concordance, and whether a liquid biopsy was capable of detecting tumor heterogeneity [23]. When controlling for genomic segments tested by both platforms, we noted a low concordance (8.6%) in detected GAs between platforms, this may suggest that liquid biopsies are more capable of detecting tumor heterogeneity. However, this study had significant methodological limitations. Primarily, this study was comparing two different commercial platforms, Guardant360 and FoundationOne. Multiple studies across cancer types have compared tissue NGS using FoundationOne and liquid biopsy NGS by Guardant360, and have reported low concordance between the two testing platforms [24–27]. The limitations of these comparisons include: the different pipelines used to call GAs, GAs with low variant allele frequencies, the timing between tests, the number of interventional therapies, and whether a patient was on therapy at the time of testing [28, 29]. While larger studies comparing tissue NGS and liquid biopsy in mRCC are needed, the available data supports the continued investigation of liquid biopsies as a tool for the contemporary, non-invasive, assessment of the genomic landscape in mRCC patients.
POTENTIAL ROLE OF LIQUID BIOPSY IN PRECISION TREATMENT OF mRCC
Biomarkers predictive of response to treatment are not routinely used in clinical practice for mRCC; however, there are some potential opportunities where liquid biopsies can inform precision treatment of mRCC. ICIs are approved in the first-line and salvage-line setting for mRCC [6, 30]. In contrast to non-small cell lung cancer (NSCLC) and other malignancies, PD-L1 staining has not been a reliable predictor of response for patients with mRCC [31]. Furthermore, PD-L1 staining requires tumor tissue, and metastases in mRCC are often difficult to access. Tumor mutational burden (TMB) is an alternative test that can be assessed by liquid biopsy and may be a better predictor of response to ICIs. Across all malignancies, RCC has the highest number of insertions and deletions, which can result in a high neo-antigen load [32]. TMB is a surrogate for overall neo-antigen load in a tumor [33, 34]. Multiple studies across a variety of malignancies, including mRCC, have demonstrated that high tissue TMB is predictive of response to ICIs [35, 36]. A study using data from the POPLAR and OAK trials of atezolizumab in NSCLC showed that blood-based TMB can also be used to predict response to ICIs [37]. Recently, Guardant Health has updated their cfDNA assay, Guardant360, to include TMB reporting. Furthermore, a study of 69 patients with diverse solid tumors, including mRCC, found that “hypermutated ctDNA”, defined as ≥6 GAs, was predictive of improved response to ICIs [38]. While prospective studies are needed, TMB and hypermutated ctDNA in liquid biopsies are promising biomarkers of response to ICIs for patients with mRCC.
While there are no predictive biomarkers for VEGF targeted therapies, liquid biopsy NGS can be used to guide enrollment of patients with mRCC in biomarker-guided clinical trials. Currently, there are multiple biomarker-guided clinical trials open to patients with mRCC. While clear cell is the most common histologic subtype of mRCC, 15–20% of patients will have non-clear cell disease, including papillary mRCC, which are associated with inferior outcomes [39, 40]. Genomic profiling has shown that MET dysregulation is common in papillary mRCC, and a phase 2 clinical trial of savolitinib, a selective MET tyrosine kinase inhibitor, showed promising activity in MET-positive, papillary mRCC [41, 42]. Currently, there are two phase 3 clinical trials evaluating MET inhibitors in patients with papillary mRCC, SWOG 1500 and SAVOIR, and liquid biopsy NGS is capable of detecting MET alterations. Moving forward, gene targeted liquid biopsies may become an ideal companion test for future biomarker-driven clinical trials in mRCC.
CONCLUSION
Due to advances in many technologies and increased sensitivity, the use of liquid biopsies is becoming increasingly prevalent across many different cancers. While there are currently no indications to use liquid biopsy in routine management of mRCC, there are many promising avenues currently under investigation. Based on multiple studies, analysis of cfDNA concentration, fragment size, and methylation status may aid in early diagnosis of localized or metastatic RCC and help with prognostication. While the majority of mRCC patients appear to shed ctDNA, the yield of liquid biopsy NGS is higher in patients with increased tumor burden and in those whose disease is progressing. Additionally, liquid biopsy NGS in mRCC has shown that the genomic landscape evolves after systemic treatment and is capable of detecting tumor heterogeneity. In order for liquid biopsy to achieve its full clinical potential, standardization of pre-analytical and analytical procedures, terminology, cross-platform validation, and sufficiently powered prospective studies are needed.
ACKNOWLEDGMENTS
CONFLICT-OF-INTEREST STATEMENT
Neeraj Agarwal reports consultancy to Pfizer, Novartis, Merck, Genentech, Eisai, Exelixis, Clovis, EMD Serono, BMS, Astra Zeneca, Foundation One, Astellas, Ely Lilly, Bayer, Argos, Medivation, Clovis, and Nektar; and research funding to my institution on my behalf from Active Biotech, Astra Zeneca, Bavarian Nordic, BMS, Calithera, Celldex, Eisai, Exelixis, Genetech, Glaxosmithkline, Immunomedics, Janssen, Medivation, Merck, New link Genetics, Novartis, Pfizer, Prometheus, Rexahn, Sanofi, Takeda, and Tracon. Benjamin L. Maughan reports advisory board for Janssen Oncology, Exelixis, Peloton Therapeutics, and TEMPUS. Andrew W. Hahn and Roberto H. Nussenzveig have no conflicts to report.
REFERENCES
[1] | Choueiri TK , Halabi S , Sanford BL , et al. Cabozantinib Versus Sunitinib As Initial Targeted Therapy for Patients With Metastatic Renal Cell Carcinoma of Poor or Intermediate Risk: The Alliance A031203 CABOSUN Trial. J Clin Oncol. (2017) ;35: :591–7. |
[2] | Motzer RJ , Hutson TE , Glen H , et al. Lenvatinib, everolimus, and the combination in patients with metastatic renal cell carcinoma: A randomised, phase 2, open-label, multicentre trial. Lancet Oncol. (2015) ;16: :1473–82. |
[3] | Motzer RJ , Hutson TE , Tomczak P , et al. Sunitinib versus interferon alfa in metastatic renal-cell carcinoma. N Engl J Med. (2007) ;356: :115–24. |
[4] | Sternberg CN , Davis ID , Mardiak J , et al. Pazopanib in locally advanced or metastatic renal cell carcinoma: Results of a randomized phase III trial. J Clin Oncol. (2010) ;28: :1061–8. |
[5] | Escudier B , Bellmunt J , Negrier S , et al. Phase III trial of bevacizumab plus interferon alfa-2a in patients with metastatic renal cell carcinoma (AVOREN): Final analysis of overall survival. J Clin Oncol. (2010) ;28: :2144–50. |
[6] | Motzer RJ , Tannir NM , McDermott DF , et al. Nivolumab plus Ipilimumab versus Sunitinib in Advanced Renal-Cell Carcinoma. N Engl J Med (2018) ;378: (14):1277–90. |
[7] | Motzer RJ , Powles T , Atkins MB , et al. IMmotion A Randomized Phase III Study of Atezolizumab Plus Bevacizumab vs Sunitinib in Untreated Metastatic Renal Cell Carcinoma (mRCC). J Clin Oncol. (2018) ;36: :Abstr578. |
[8] | Ricketts CJ , De Cubas AA , Fan H , et al. The Cancer Genome Atlas Comprehensive Molecular Characterization of Renal Cell Carcinoma. Cell reports. (2018) ;23: :313–26.e5. |
[9] | Corcoran RB , Chabner BA . Application of Cell-free DNA Analysis to Cancer Treatment. N Engl J Med. (2018) ;379: :1754–65. |
[10] | Husain H , Velculescu VE . Cancer DNA in the Circulation: The Liquid Biopsy. JAMA. (2017) ;318: :1272–4. |
[11] | Wan JCM , Massie C , Garcia-Corbacho J , et al. Liquid biopsies come of age: Towards implementation of circulating tumour DNA. Nat Rev Cancer. (2017) ;17: :223–38. |
[12] | Yamamoto Y , Uemura M , Nakano K , et al. Increased level and fragmentation of plasma circulating cell-free DNA are diagnostic and prognostic markers for renal cell carcinoma. Oncotarget. (2018) ;9: :20467–75. |
[13] | Lu H , Busch J , Jung M , et al. Diagnostic and prognostic potential of circulating cell-free genomic and mitochondrial DNA fragments in clear cell renal cell carcinoma patients. Clin Chim Acta. (2016) ;452: :109–19. |
[14] | Skrypkina I , Tsyba L , Onyshchenko K , et al. Concentration and Methylation of Cell-Free DNA from Blood Plasma as Diagnostic Markers of Renal Cancer. Dis Markers. (2016) ;2016: :3693096. |
[15] | Rini BI , Campbell SC , Escudier B . Renal cell carcinoma. Lancet. (2009) ;373: :1119–32. |
[16] | Hakimi AA , Ostrovnaya I , Reva B , et al. Adverse outcomes in clear cell renal cell carcinoma with mutations of 3p21 epigenetic regulators BAP1 and SETD A report by MSKCC and the KIRC TCGA research network. Clin Cancer Res. (2013) ;19: :3259–67. |
[17] | Kapur P , Pena-Llopis S , Christie A , et al. Effects on survival of BAP1 and PBRM1 mutations in sporadic clear-cell renal-cell carcinoma: A retrospective analysis with independent validation. Lancet Oncol. (2013) ;14: :159–67. |
[18] | Pal SK , Sonpavde G , Agarwal N , et al. Evolution of Circulating Tumor DNA Profile from First-line to Subsequent Therapy in Metastatic Renal Cell Carcinoma. Eur Urol (2017) ;72: (4):557–64. |
[19] | Hsieh JJ , Chen D , Wang PI , et al. Genomic Biomarkers of a Randomized Trial Comparing First-line Everolimus and Sunitinib in Patients with Metastatic Renal Cell Carcinoma. Eur Urol. (2017) ;71: :405–14. |
[20] | Comprehensive molecular characterization of clear cell renal cell carcinoma. Nature. (2013) ;499: :43–9. |
[21] | Maia MC , Bergerot PG , Dizman N , et al. Association of Circulating Tumor DNA (ctDNA) Detection in Metastatic Renal Cell Carcinoma (mRCC) with Tumor Burden. Kidney Cancer. (2017) ;1: :65–70. |
[22] | Gerlinger M , Rowan AJ , Horswell S , et al. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N Engl J Med. (2012) ;366: :883–92. |
[23] | Hahn AW , Gill DM , Maughan B , et al. Correlation of genomic alterations assessed by next-generation sequencing (NGS) of tumor tissue DNA and circulating tumor DNA (ctDNA) in metastatic renal cell carcinoma (mRCC): Potential clinical implications. Oncotarget. (2017) ;8: :33614–20. |
[24] | Kuderer NM , Burton KA , Blau S , et al. Comparison of 2 Commercially Available Next-Generation Sequencing Platforms in Oncology. JAMA oncology. (2017) ;3: :996–8. |
[25] | Chae YK , Davis AA , Carneiro BA , et al. Concordance between genomic alterations assessed by next-generation sequencing in tumor tissue or circulating cell-free DNA. Oncotarget. (2016) ;7: :65364–73. |
[26] | Chae YK , Davis AA , Jain S , et al. Concordance of Genomic Alterations by Next-Generation Sequencing in Tumor Tissue versus Circulating Tumor DNA in Breast Cancer. Mol Cancer Ther. (2017) ;16: :1412–20. |
[27] | Barata P , Koshkin V , Funchain P , et al. Next-generation sequencing of cell-free circulating tumor DNA and tumor tissue in patients with advanced urothelial cancer: A pilot assessment of concordance. Ann Oncol (2017) ;28: (10):2458–63. |
[28] | Hahn AW NR , Pal SK , Agarwal N . Blood and tissue-based tumor genomics: A battle royale or match made in heaven? Ann Oncol. (2017) ;28: :2333–5. |
[29] | Agarwal N , Lanman RB , Pal SK . Regarding the Congruence Between 2 Circulating Tumor DNA Sequencing Assays. JAMA oncology. (2018) ;4: :1429–30. |
[30] | Motzer RJ , Escudier B , McDermott DF , et al. Nivolumab versus Everolimus in Advanced Renal-Cell Carcinoma. N Engl J Med. (2015) ;373: :1803–13. |
[31] | Zhu J , Armstrong AJ , Friedlander TW , et al. Biomarkers of immunotherapy in urothelial and renal cell carcinoma: PD-L1, tumor mutational burden, and beyond. Journal for immunotherapy of cancer. (2018) ;6: :4. |
[32] | Turajlic S , Litchfield K , Xu H , et al. Insertion-and-deletion-derived tumour-specific neoantigens and the immunogenic phenotype: A pan-cancer analysis. Lancet Oncol. (2017) ;18: :1009–21. |
[33] | Rizvi NA , Hellmann MD , Snyder A , et al. Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science. (2015) ;348: :124–8. |
[34] | Rooney MS , Shukla SA , Wu CJ , Getz G , Hacohen N . Molecular and genetic properties of tumors associated with local immune cytolytic activity. Cell. (2015) ;160: :48–61. |
[35] | Goodman AM , Kato S , Bazhenova L , et al. Tumor Mutational Burden as an Independent Predictor of Response to Immunotherapy in Diverse Cancers. Mol Cancer Ther. (2017) ;16: :2598–608. |
[36] | Rosenberg JE , Hoffman-Censits J , Powles T , et al. Atezolizumab in patients with locally advanced and metastatic urothelial carcinoma who have progressed following treatment with platinum-based chemotherapy: A single-arm, multicentre, phase 2 trial. Lancet. (2016) ;387: :1909–20. |
[37] | Gandara DR , Paul SM , Kowanetz M , et al. Blood-based tumor mutational burden as a predictor of clinical benefit in non-small-cell lung cancer patients treated with atezolizumab. Nat Med. (2018) ;24: :1441–8. |
[38] | Khagi Y , Goodman AM , Daniels GA , et al. Hypermutated Circulating Tumor DNA: Correlation with Response to Checkpoint Inhibitor-Based Immunotherapy. Clin Cancer Res. (2017) ;23: :5729–36. |
[39] | Shuch B , Amin A , Armstrong AJ , et al. Understanding pathologic variants of renal cell carcinoma: Distilling therapeutic opportunities from biologic complexity. Eur Urol. (2015) ;67: :85–97. |
[40] | Salgia M , Adashek J , Bergerot P , Pal SK . Non-Clear Cell Renal Cell Carcinoma: Current Management and Best Practice. Kidney Cancer. (2017) ;1: :99–105. |
[41] | Choueiri TK , Plimack E , Arkenau HT , et al. Biomarker-Based Phase II Trial of Savolitinib in Patients With Advanced Papillary Renal Cell Cancer. J Clin Oncol. (2017) ;35: :2993–3001. |
[42] | Shuch B , Hahn AW , Agarwal N . Current Treatment Landscape of Advanced Papillary Renal Cancer. J Clin Oncol. (2017) ;35: :2981–3. |


