
The ‘Achilles Heel’ of Metabolism in Renal Cell Carcinoma: Glutaminase Inhibition as a Rational Treatment Strategy
Abstract
An important hallmark of cancer is ‘metabolic reprogramming’ or the rewiring of cellular metabolism to support rapid cell proliferation [1–5]. Metabolic reprogramming through oncometabolite-mediated transformation or activation of oncogenes in renal cell carcinoma (RCC) globally impacts energy production as well as glucose and glutamine utilization in RCC cells, which can promote dependence on glutamine supply to support cell growth and proliferation [6, 7]. Novel inhibitors of glutaminase, a key enzyme in glutamine metabolism, target glutamine addiction as a viable treatment strategy in metastatic RCC (mRCC). Here, we review glutamine metabolic pathways and how changes in cellular glutamine utilization enable the progression of RCC. This overview provides scientific rationale for targeting this pathway in patients with mRCC. We will summarize the current understanding of cellular and molecular mechanisms underlying anti-tumor efficacy of glutaminase inhibitors in RCC, provide an overview of clinical efforts targeting glutaminase in mRCC, and review approaches for identifying biomarkers for patient stratification and detecting therapeutic response early on in patients treated with this novel class of anti-cancer drug. Ultimately, results of ongoing clinical trials will demonstrate whether glutaminase inhibition can be a worthy addition to the current armamentarium of drugs used for patients with mRCC.
INTRODUCTION
Renal cell carcinoma (RCC) most commonly occurs due to loss of the von-Hippel Lindau (VHL) tumor suppressor, which leads to stabilization of the transcription factor hypoxia-inducible factor (HIF). Constitutive HIF signaling results in extensive metabolic reprogramming, including a shift towards increased glutamine utilization, which suggests that RCC may be particularly susceptible to interfering with glutamine metabolism. Therefore, a review of glutamine metabolic pathways in RCC was undertaken. Glutaminase is a mitochondrial enzyme that converts glutamine to glutamate. Here we review the role of glutamine in cellular metabolism, preclinical evidence of addiction of RCC cells to glutamine and glutaminase activity, and inhibitors of glutaminase as a novel strategy for the treatment of RCC.
GLUTAMINE - A CELL’S ‘MULTI-PURPOSE FUEL’
Glutamine is a non-essential amino acid that is synthesized and metabolized by all cells in the body. It participates in a wide array of physiological functions throughout the human body, not simply in cancer. This fact will be especially important for later discussions of early failed attempts at targeting the glutamine pathway for anti-cancer treatment. Hence, we will first review the role of glutamine in organismal and cellular physiology.
Glutamine is the most abundant amino acid in the plasma, where it serves as a universal carrier for the inter-organ transport of carbon and nitrogen - in essence, functioning as a ‘wildcard’ amino acid for the human body. The majority of glutamine is generated de novo by skeletal muscle, adipocytes, and the lungs, which maintain organism-wide glutamine homeostasis [8, 9]. When glutamine demand exceeds the biosynthetic capacity of the body such as during wound repair or sepsis, glutamine becomes an essential amino acid [10, 11].
In cells, it is used as ‘fuel’ for the biosynthesis of other amino acids, metabolites, nucleotides, lipids, proteins, and for generating energy in the form of adenosine triphosphate (ATP) [12–17]. Hence, rapidly dividing cells typically use the largest quantities of glutamine due to the high demand for the building blocks of macromolecules and for energy, including epithelial cells of the small intestine (enterocytes), immune cells (e.g. activated lymphocytes), and ultimately, cancer cells [18, 19]. If intracellular de novo synthesis is inadequate to meet the cellular demand for glutamine, then it can be imported into the cytoplasm via glutamine transporters of the solute carrier (SLC) family (in particular SLC1A5, see Fig. 1), macropinocytosis (the uptake of large vacuoles of extracellular fluid by endocytosis), or even released through the intracellular breakdown of macromolecules (autophagy) [20–22].
Fig.1
Cellular Uptake Routes and Intracellular Utilization of Glutamine. Glutamine (yellow) is either synthesized by cells de novo, taken up through the solute carrier 1A5 (SLC1A5), or derived from the intracellular breakdown of macromolecules (autophagy). Glutamine and its metabolic product, glutamate (orange), can be exported from cells in exchange for other essential amino acids (EAAs). Intracellularly, they are involved in a wide range of metabolic pathways that serve to generate other amino acids and glutathione (GSH) as well as precursors for the biosynthesis of nucleotides and reducing equivalents in the form of nicotinamide adenine dinucleotide (NADH) and NADH phosphate (NADPH) for generating energy and lipids. Most of glutamine is utilized by mitochondria, where the enzyme glutaminase 1 (GLS1, red) or glutaminase 2 (GLS2, not shown) convert glutamine into glutamate, which in the form of alpha-ketoglutarate (aKG) can enter the tricarboxylic acid cycle (TCA cycle). GLS1 is the target of glutaminase inhibitors such as BPTES, 968, and CB-839.
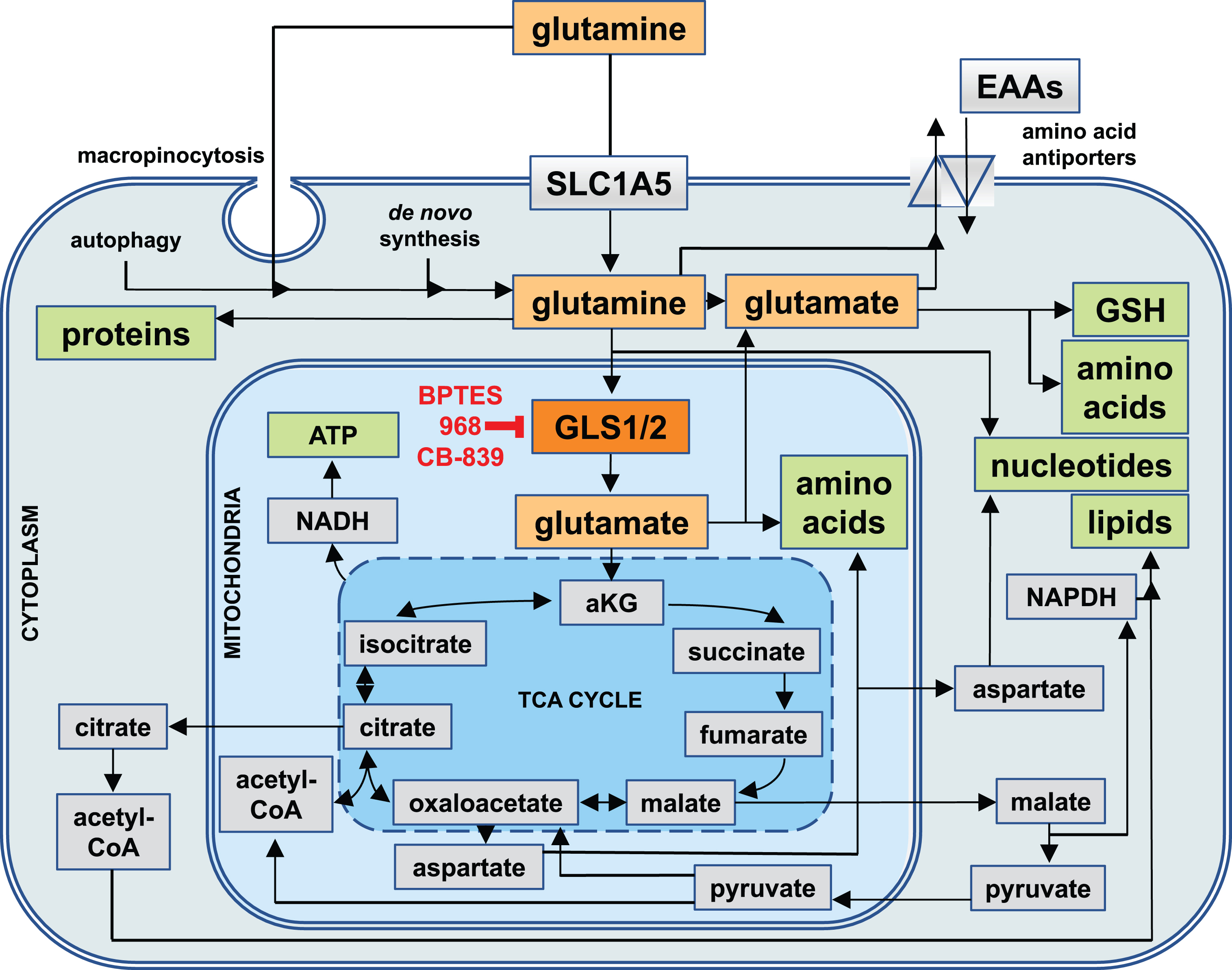
The intracellular fate of glutamine can be classified into two major categories: cytoplasmic and mitochondrial (see Fig. 1). In the cytoplasm, glutamine can be used for either protein and nucleotide biosynthesis, or for export by antiporters where it serves as a ‘currency’ in exchange for other amino acids. Moreover, cytoplasmic glutamine-derived glutamate is an important precursor for many amino acids, hexosamines (precursors for the glycosylation of signaling proteins), and glutathione - a major reducing agent for radical oxygen species (ROS) [12]. In the mitochondria, glutaminase 1 and 2 (GLS1 and GLS2) convert glutamine to glutamate and ammonia. Glutamate then becomes a primary commodity for amino acid biosynthesis or is converted into alpha-ketoglutarate (aKG), which enters the tricarboxylic acid (TCA) cycle - a central hub in cellular metabolism that can interconvert many precursors of biosynthetic reactions (see Fig. 1). Through the TCA cycle, glutamate provides precursors for the biosynthesis of amino acids, nucleotides, lipids, and reducing equivalents in the form of NADH (required for the synthesis of ATP by oxidative phosphorylation), and NADPH (needed for lipid and nucleotide biosynthesis, and the regeneration of cytoplasmic glutathione). Lastly, glutamine-derived glutamate can ‘fuel’ the TCA cycle by generating acetyl-CoA via malate and pyruvate.
Hence, glutamine is a major source of carbon, nitrogen, and electrons for virtually all cellular metabolites, macromolecules, and energy.
GLUTAMINE- AND GLUTAMINASE-ADDICTION IN RCC - WHEN GLUCOSE CAN’T DO THE JOB
In this section, we will review the molecular mechanisms proposed for the dependence on extracellular glutamine and glutaminase activity in RCC. This overview ultimately provides therapeutic rationale for targeting this specific metabolic pathway.
Increased glutamine uptake and elevated expression of glutaminase have been recognized as hallmarks of proliferating tumor cells in vitro and in vivo since the 1950s [23–30]. Subsequent studies in RCC cells confirmed that glutamine is consumed at high rates in vitro [31–33].
For the most common subtype of kidney cancer, clear cell RCC (ccRCC), tumors are consistently reported to have higher levels of glutamine and glutamate compared to normal kidney tissue in addition to increased expression of glutamine importers such as SLC1A5 [34–43]. Early glutamine deprivation studies demonstrated that some cancer cell lines are dependent on glutamine even under glucose-replete conditions [44]. Additional studies involving genetic perturbations further demonstrated that many tumors, including RCC, are dependent on glutaminase activity, thus implying that addiction to glutamine is a consequence of the increased need for glutamate [32, 45–56].
Glutaminase, the mitochondrial enzyme that converts glutamine to glutamate, exists as two isoenzymes, GLS1 and GLS2, encoded by the genes, GLS1 and GLS2 [57]. GLS1 has two splice variants, kidney-type glutaminase (KGA) and a shorter, more active variant, glutaminase C (GAC) [58, 59]. Both splice variants are widely expressed across tissues with especially the GAC variant frequently expressed at higher levels in tumor cells compared to corresponding normal cells [41, 48, 49, 51, 52, 59–68]. Interestingly, in most ccRCC tumors, expression levels of GLS1 seem not to be significantly changed, though expression of the more active GAC variant of GLS1 is slightly increased relative to the KGA variant in ccRCC cell lines [12, 31–33, 69]. GLS2 is predominantly found in the liver, brain, and pancreas and, like GLS1, has not been reported to be elevated in RCC [61].
The fact that glucose and glutamine are both abundant resources for cellular metabolism under normal in vitro culture conditions, and that both ‘fuel’ the same metabolic pathways through the TCA cycle, raises the question: why do RCC and other cancer cells become dependent on glutamine in the presence of glucose? The oncogenic transcription factor HIF, which is often activated in cancer cells by hypoxia in poorly perfused regions of solid tumors or by the activity of other oncogenes, plays a central role in this phenomenon.
In ccRCC, loss of VHL and fructose-1,6-bisphosphatase 1 (FBP1) tumor suppressor functions - fundamental features of this subtype of RCC - results in increased HIF activity in virtually all cancer cells, independently of hypoxia [70–72]. HIF reprograms cellular metabolism and increases glucose uptake and glycolysis via increased expression of glucose transporters and glycolytic enzymes. Moreover, HIF also shifts the TCA cycle from predominant glucose utilization to a predominantly glutamine-fuelled system in vitro and in vivo [31–33, 52, 73–77]. In normal cells, glucose-derived carbons enter the mitochondria via pyruvate, which drives the TCA cycle in a ‘clockwise’ fashion (see Fig. 1). However, even though ccRCC cells show increased glycolysis, increased HIF activity leads to inhibition of pyruvate dehydrogenase (PDH) and pyruvate carboxylase (PC) activity, thus resulting in drastically decreased entry of glucose-derived carbon into the TCA cycle [37, 39, 78, 79]. In consequence, these cells ‘fuel’ the TCA cycle primarily in a ‘counterclockwise’ direction by glutamine-derived glutamate via reductive carboxylation of aKG to isocitrate (see Fig. 1). Glutamine-addicted cancer cells operate the TCA cycle in this ‘reverse’ direction to generate citrate and malate for lipid biosynthesis, and oxaloacetate for nucleotide biosynthesis, while deriving most of their energy from glycolysis [31, 33, 74–77]. HIF activation in ccRCC is both necessary and sufficient for increased glutamine utilization and dependence, both by limiting the ability of glucose to ‘fuel’ the TCA cycle and by reprogramming glutamine metabolism to provide the requisite macromolecules needed for sustaining the increased needs of rapidly dividing cells [31, 32, 77].
The degree of metabolic reprogramming and glutamine dependence in ccRCC increases as tumors become more advanced and aggressive and accumulate genetic alterations in other oncogenes and tumor suppressor genes, for example in phosphoinositide 3-kinase (PI3K), c-MYC, or p53 [35, 39–41, 47, 48, 52, 80–89].
Interestingly, HIF activation does not seem to influence the expression or protein levels of glutamine transporters or GLS1 [32, 90, 91]. This suggests that even normal cells already have the capacity to use glutamine as the major ‘fuel’ for the TCA cycle, yet prefer to use glucose. Cancer cells in which HIF is activated, by contrast, lose the ability to use glucose as major TCA cycle ‘fuel’, thus developing a particular dependence on glutamine. This may explain why interfering with glutamine utilization via glutaminase inhibition negatively impacts cancer cells preferentially compared to normal cells.
Metabolic programming and glutamine dependence in non-clear cell RCC are less well studied. While genetic alterations have been clearly described for papillary type 1 and chromophobe RCC, the metabolic landscapes have not yet been characterized [92–97].
In hereditary leiomyomatosis and renal cell cancer (HLRCC) syndrome, where patients develop kidney tumors with papillary type 2 histology, RCC cells lack a functional enzyme in the TCA cycle, fumarate hydratase (FH), which converts fumarate to malate (see Fig. 1) [98]. FH deficiency causes an accumulation of fumarate, which stabilizes HIF protein [99, 100]. In vitro, these cells demonstrate increased glycolytic activity and reliance on both glucose and glutamine for cell proliferation and survival [74, 101–104].
Similarly, in familial and sporadic forms of succinate dehydrogenase (SDH) deficient RCC, there is a lack of the functional enzyme, SDH, which converts succinate to fumarate in the TCA cycle. Similar to FH-deficient cells, SDH-deficient cells also demonstrate increased HIF activity as well as increased glycolysis and glutamine utilization [105–112].
Finally, a c-MYC driven mouse model of collecting duct RCC (cdRCC), a particularly aggressive form of RCC arising from the epithelial cells of the renal collecting duct system, demonstrated upregulated glutamine utilization [41]. The c-MYC pathway is also activated in a subset of ccRCC tumors and in some rare forms of RCC caused by chromosomal translocations [35, 113]. Thus, as in ccRCC, glutaminase inhibition may be a viable drug target even for some non-clear cell RCC.
In summary, extensive metabolic reprogramming is a common theme in all forms of RCC. Glutamine-derived glutamate plays a central role in the TCA cycle to generate sufficient amounts of intermediates required for the biosynthesis of lipids, nucleotides, and other amino acids, as well as NADPH for maintaining redox homeostasis. The limited ability of ccRCC cells - and any cancer cells with activated HIF - to use glucose to drive the TCA cycle provides a plausible explanation for increased glutamine dependence in ccRCC and other cancers, as this metabolic strategy provides alternative carbon and nitrogen sources for generating macromolecules [114–116].
ANTI-TUMOR EFFICACY OF GLUTAMINASE INHIBITION IN RCC: PRECLINICAL DATA AND MOLECULAR MECHANISMS
Early studies demonstrated that RCC cells are dependent on glutaminase activity, suggesting that glutamine is required for maintaining intracellular glutamate supply [32, 45–56]. While it was believed for a long time that glutamine-addicted cancer cells require this glutamate for generating enough energy for proliferation and survival, more recent work suggests that ATP production does not seem to be the major limiting factor for cancer cell proliferation [5]. Rather, glutamine-derived glutamate plays a central role in the TCA cycle to generate enough intermediates required for the biosynthesis of lipids, nucleotides, other amino acids, and NADPH. Which of these intermediates become limiting, leading to impaired cellular functions, in glutamine-addicted cancer cells upon glutaminase inhibition? The development of selective glutaminase inhibitors over the last decade not only revived the interest in glutamine metabolism research in cancer, but also helped develop a better understanding of this central question of glutamine dependence in RCC and other cancers and have been used to study the role of glutaminase as a therapeutic target:
1) Bis-2-(5-phenylacetimido-1,3,4,thiadiazol-2-yl)ethyl sulfide (BPTES) inhibits both the KGA and GAC isoforms of GLS1 by stabilizing the tetramer form of the enzyme in an inactive conformation, but does not inhibit GLS2 [117–120].
2) The bromo-benzophenanthridinone compound 968, a GLS1 inhibitor thought to preferentially target the GAC isoform, binds to the monomeric form of the enzyme, thereby locking GLS1 in its inactive state [121]. Inhibition of GLS2 by 968 has not been investigated [122].
3) CB-839, a newer compound with structural similarity to BPTES, but higher potency and improved bioavailability, inhibits both isoforms of GLS1 (KGA and GAC), but not GLS2 [123].
Glutaminase inhibitors have anti-proliferative activity in vitro and in vivo in mice in a wide range of cancers including RCC [32, 33, 41, 49, 52, 53, 55, 64, 67, 68, 123–139]. In vitro, the effects of glutaminase inhibition generally mimic those of glutamine deprivation, suggesting that the mitochondrial fates of glutamine are most critical for cell proliferation and survival. However, these effects vary with cancer type and cell line being studied: in some cases cells become quiescent, whereas in others cells undergo apoptosis [32, 33]. Okazaki et al. investigated the molecular mechanism how GLS1 inhibition impairs proliferation of VHL-deficient RCC cells and found nucleotide biosynthesis and redox homeostasis to be the limiting functions that are mediated by GLS1 in RCC cells [33]. Inhibition of GLS1 by BPTES or CB-839 impaired the ability of VHL-deficient RCC cell to generate aspartate, required for the biosynthesis of pyrimidine nucleotides. GLS1 inhibition in these cells leads to DNA replication stress, more frequent DNA double-strand breaks (DSBs), increased ROS levels and decreased glutathione levels. Furthermore, Sun et al. showed that the growth defect of cancer cells upon glutamine depletion could be rescued by the addition of lipids or citrate [77]. Hence, in vitro, anti-proliferative effects of GLS1 inhibition in VHL-deficient RCC cells do not result from impaired energy production. Rather, the limited supply of TCA cycle intermediates as building blocks for the biosynthesis of nucleotides, lipids, and glutathione appear to be the limiting factors.
In vivo, tumor xenograft studies showed that treatment with glutaminase inhibitors results in reduced tumor growth in a number of cancer types, including RCC [32, 33, 41, 49, 52, 67, 68, 123, 136, 138]. Interestingly, glutaminase inhibitors can have additive or even synergistic effects with other anti-cancer drugs. For example, in an RCC xenograft model, CB-839 showed increased efficacy when combined with the poly(ADP-ribose) polymerase (PARP) inhibitor olaparib [33]. In mouse xenografts of the RCC cell line Caki-1, tumor growth was inhibited by treatment with CB-839 alone, but the combination with either cabozantinib or everolimus inhibited tumor growth more effectively than either agent alone (Calithera Biosciences, Inc., unpublished data, see ref. [140, 141]). In the syngeneic CT26 colon carcinoma mouse model, the addition of CB-839 to PD-L1 inhibition increased the number of complete tumor regressions compared to either single agent (Calithera Biosciences, Inc., unpublished data, see ref. [142]). Similarly, in a syngeneic B16 melanoma model, the combination of CB-839 with PD-L1 inhibition more effectively inhibits tumor growth than PD-L1 inhibition alone (Calithera Biosciences, Inc., unpublished data, see ref. [143]). Taken together, preclinical data suggest that CB-839 can have anti-tumor activity in RCC and other cancer types as a single agent and in combination with other agents.
TARGETING GLUTAMINE METABOLISM IN PATIENTS
Targeting glutamine metabolism can be exploited clinically. Notably, some FDA-approved drugs show off-target effects on glutamine utilization [89]. For example, bacterial L-asparaginases are used to treat acute lymphoblastic leukemia (ALL) and can metabolize both asparagine and glutamine and thereby deplete plasma glutamine levels [144]. Sorafenib, an FDA-approved multi-tyrosine kinase inhibitor (TKI) for advanced RCC, has recently been reported to also inhibit the cystine/glutamate antiporter SLC7A11/xCT [145]. Nevertheless, the main mechanism of efficacy of sorafenib and other TKIs is mostly attributed to inhibition of receptor tyrosine kinases.
Rather than relying on off-target effects, attempts to target the glutamine/glutaminase pathway more specifically began as early as the 1980s. Glutamine mimetics such as 6-diazo-5-oxo-L-norleucine (DON), azaserine, and acivicin were developed to competitively bind to glutamine transporters, thereby inhibiting cellular glutamine uptake [146]. Acivicin was tested in a clinical trial for metastatic RCC [147]. These drugs showed high efficacy in preclinical experiments, but turned out to be less potent in patients - acivicin had an objective response rate of 4% in RCC - and were highly toxic to the brain, bone marrow, and gastrointestinal tract [148]. These wide-ranging adverse effects can be understood in the context of glutamine’s crucial role in neurotransmitter biogenesis and in the metabolism of rapidly dividing normal cells [17]. The limited efficacy and severe toxicities of these drugs rendered glutamine metabolism a sub-optimal target in cancer therapy for a long time.
The refined knowledge about the importance of glutaminase in cellular glutamine utilization and about the increased dependence of cancer cells on GLS1 revived the interest in targeting glutamine metabolism and led to the development of selective glutaminase inhibitors for cancer therapy. However, the poor solubility of the GLS1 inhibitors BPTES and 968 in aqueous solution and their limited potency have hampered their translation into clinical applications. The availability of structural information about these drugs in complex with GLS1 has led to efforts to rationally evolve BPTES into more potent and stable analogues with better solubility. For example, the BPTES analog UPGL00004 is a more potent inhibitor of the the GAC variant of GLS1 than BPTES and inhibits breast cancer growth in mice when combined with the anti-vascular endothelial growth factor (VEGF) antibody bevacizumab [149, 150]. Other approaches explored delivering the drug by nanoparticles, which improved pharmacokinetics and efficacy of BPTES [151]. In parallel to GLS inhibitors, glutamate dehydrogenase (GLUD) inhibitors have been developed. GLUD catalyzes the reaction that converts glutamate to aKG and ammonia, downstream of GLS. The GLUD inhibitor epigallocatechin gallate (EGCG) is currently being evaluated as adjuvant therapy for colorectal cancer in a phase I clinical trial [152].
Of the next generation GLS inhibitors, CB-839, a newer compound with improved bioavailability, is the only agent that has reached clinical trials so far. It is being tested as single agent or in combination with established therapies in hematologic tumors and in a wide range of solid tumors including RCC. Early data from the phase I trials assessing the safety, pharmacokinetics, and pharmacodynamics indicate that CB-839 is tolerable as a single agent in patients with mRCC and other cancers [153]. Monotherapy with CB-839 in 21 patients with RCC resulted in one partial response (with duration of 356 days) and stable disease in 52% of patients. Moreover, CB-839 also appears to be well tolerated in combination with other drugs, with some patients experiencing tumor stabilization and clinical efficacy [153]. The combination of the mTOR inhibitor everolimus and CB-839 showed a disease control rate of 92% in 24 patients with RCC (with a median of two prior lines of therapy). In 12 patients with advanced RCC treated with the TKI cabozantinib and CB-839 (with a median of 3 prior lines of therapy), overall response rate was 40%, with 100% disease control rate. Whether CB-839 might have clinical efficacy for RCC in randomized trials is currently being tested (see Table 1). Lastly, novel GLS1 inhibitors are in development and also selective GLS2 inhibitors have been reported [154–156]. Glutaminase inhibitors thus represent a new class of drugs to clinically target cancer cell glutamine metabolism with promising early results in the management of mRCC.
Table 1
Ongoing Clinical Trials of Glutaminase Inhibitor CB-839 in Solid Tumors
| clinicaltrials.gov Identifier | Title | Phase |
| NCT02071862 | Study of the Glutaminase Inhibitor CB-839 in Solid Tumors | Phase I (ref. [153]) |
| NCT02771626 | Study of CB-839 in Combination With Nivolumab in Patients With Melanoma, ccRCC and NSCLC | Phase I/II |
| NCT03163667 | Study of CB-839 With Everolimus vs. Placebo With Everolimus in Patients With RCC (ENTRATA Trial) | Randomized Phase II |
| NCT03428217 | Study of CB-839 With Cabozantinib vs. Placebo With Cabozantinib in Patients With Metastatic RCC (CANTATA Trial) | Randomized Phase II |
BIOMARKERS FOR SENSITIVITY AND RESPONSE TO GLUTAMINASE INHIBITORS IN RCC
The early clinical results with CB-839 and the increasing number of novel glutaminase inhibitors and drug combinations on the horizon pose an opportunity and need for establishing biomarkers for patient stratification, therapeutic response, and resistance in parallel with developing novel glutaminase inhibitors. Developing assays for the activity of GLS1 protein as the actual drug target of GLS1 inhibitors will be vital to understand GLS1 regulation in RCC and to develop biomarkers for stratifying patients. Interestingly, beyond transcriptional regulation, GLS1 is regulated at virtually every post-transcriptional level (see Table 2). Gene expression analysis alone may therefore provide only limited information on the activation status of GLS1 and hence sensitivity to GLS1 inhibitors. The findings that GLS1 inhibition in RCC cells leads to depletion of the intracellular glutamate pool, impaired synthesis of TCA cycle intermediates and thereby to increased DNA replication stress, more frequent DNA double-strand breaks, impaired glutathione synthesis, and increased ROS levels, suggests that markers in these cellular pathways may be explored as biomarkers for response to glutaminase inhibitors in RCC patients [171].
Table 2
Modes of Regulation of GLS1
| Mode of GLS1 regulation | Effect | Reference |
| Transcription | STAT1 induces expression of GLS1 | [157] |
| Pre-mRNA splicing | RNA-binding proteins regulate GLS1 alternative splicing | [56, 69, 158, 159] |
| mRNA stability | GLS1 mRNA contains a pH-responsive stability element | [160] |
| Protein translation | c-MYC and NF-κB activity induce translation of the KGA isoform of GLS1 by inhibiting expression of the translational inhibitory microRNAs 23a and 23b | [161] |
| Post-translational modification | the GAC isoform of GLS1 is activated in cells transformed by diffuse B-cell lymphoma protein (Dbl; a GEF for Rho GTPases), likely by phosphorylation; | [49] |
| the KGA isoform of GLS1 is activated, likely through phosphorylation, in response to EGF stimulation through the RAF-MEK-ERK signaling pathway; | [162] | |
| GLS1 is inactivated through desuccinylation by Sirtuin 5, which is overexpressed in some lung cancers | [163,164] | |
| Protein localization | in neurons, the KGA isoform of GLS1 localizes to neurite terminals or mitochondria depending on the activity of BCL2/adenovirus E1B 19 kd-interacting protein (BNIP) family members | [165,166] |
| Enzymatic activity | GLS1 is activated by inorganic phosphate in vitro, with the GAC isoform of GLS1 showing the strongest increase in activity; glutamate, but not ammonia, inhibits GLS1 | [8, 51, 167] |
| Protein degradation | the KGA isoform of GLS1 is ubiquitinated by the anaphase-promoting complex (APC)-CDH1 E3 ubiquitin ligase complex and subsequently degraded; | [168, 169] |
| the GAC isoform of GLS1 is degraded by the LON protease upon diphenylarsinic acid (DPAA) treatment | [170] |
Two complementary approaches will be critical to establish and validate such biomarkers:
First, serial measurements of these candidate biomarkers in preclinical in vivo models will be needed. In human RCC tumors, multi-region metabolic profiling has revealed a remarkable level of spatial heterogeneity of metabolic phenotypes, which can not be modeled sufficiently in vitro. Moreover, increased glucose uptake by tumor cells limits glucose availability for tumor-infiltrating lymphocytes in mouse models and this metabolic competition causes T cell exhaustion [172, 173]. It will be important to study whether this also applies to glutamine metabolism in RCC to assess how the interactions of tumor cells with their microenvironment influence response and resistance to glutaminase inhibitors. Thus, in vivo model systems of RCC such as patient-derived xenografts that better reflect the complex metabolic landscape and microenvironment of solid tumors are needed to understand the mechanisms of response and resistance to glutaminase inhibitors in patients [174–178].
Second, in order to validate preclinical biomarkers, serial measurements in patients before and during glutaminase inhibitor therapy will be needed to correlate these biomarkers with clinical outcomes. A wide range of technologies is available to facilitate molecular measurements in patients: mass spectrometry may be useful to measure changes in metabolite abundance in biopsies from tumors treated with glutaminase inhibitors, for example, the depletion of glutamate and accumulation of glutamine [34, 39–42, 179]. Complementary nanotechnologies to allow minimally invasive serial sampling of patients during treatment have the potential to facilitate measurements of protein abundance and activity of cellular signaling pathways in scant clinical tissue specimens and in blood (e.g. circulating tumor cells, circulating RNA or proteins; refs. [180–184] and our own unpublished data). Serial measurements may be a crucial approach to find the best strategy to measure response and resistance early on during therapy, so that patients who do not benefit from glutaminase inhibitors can quickly move on to a different, more effective treatment.
In vivo, novel metabolic imaging using positron emission tomography (PET) tracers of glutamine metabolism have been developed: PET probes [18F](2S,4R)4-fluoroglutamine, [11C]glutamine, and (4 S)-4-(3-[18F]Fluoropropyl)-L-Glutamate (18F-FSPG) allow for glutamine and glutamate tracing in patients and have been tested preclinically and clinically to detect sites of increased uptake of these amino acids in multiple cancers including RCC [138, 185–195]. Since glutaminase inhibition depletes the intracellular glutamate pool, it is possible that tumors in which glutaminase activity is effectively inhibited may also show compensatory increased glutamate uptake. Studies to evaluate whether glutamine or glutamate PET tracers may be useful to detect tumors with increased activity of glutamine and glutamate metabolic pathways are underway. Future work will determine the optimal strategy of combining imaging, genetic, gene expression, protein, and/or metabolic biomarkers to identify the patient population with the highest likelihood to benefit from glutaminase inhibitor treatment.
CONCLUSION
Metabolic reprogramming in RCC changes both glucose and glutamine utilization and renders RCC cells dependent on exogenous glutamine supply. Glutamine is converted to glutamate in mitochondria by the enzyme glutaminase and serves to generate TCA cycle intermediates, which are the building blocks for the biosynthesis of amino acids, lipids, nucleotides, and antioxidants. The addiction of RCC cells to glutamine is an ‘Achilles heel’ in RCC metabolism. Glutaminase inhibitors, which lead to reduced cell proliferation in vitro and in vivo, may be a way to strike at this ‘Achilles heel’. Early data from clinical trials using the glutaminase inhibitor CB-839 alone or in combination with other drugs to treat metastatic RCC suggest that this novel class of drugs is well tolerated and able to control the disease in some cases. Yet, biomarkers are needed to identify patients who derive the most benefit from glutaminase inhibitors and for detecting therapeutic response and resistance early on during treatment. The combination of in vivo models for metastatic RCC, serial measurements in these models and in patients, novel imaging probes, and nanotechnologies to interrogate molecular biomarkers in scant clinical tissue specimens and in blood will help identify and validate such biomarkers.
CONFLICT OF INTEREST
AF is the site principal investigator of the glutaminase inhibitor CB-839 clinical trials NCT02771626, NCT03163667, and NCT03428217, and receives salary support from Calithera Biosciences, Inc.
AF is founder of Molecular Decisions, Inc.
ACKNOWLEDGMENTS
AF is supported by the Conquer Cancer Foundation, the NIH/NCI (grant 5U54CA199075), and Calithera Biosciences, Inc.
VC is supported by and NIH/NCI T32 grant and a Stanford University Translational Research and Applied Medicine (TRAM) award.
REFERENCES
[1] | Hanahan D , Weinberg RA . Hallmarks of cancer: The next generation. Cell. (2011) ;144: :646–674. |
[2] | Yang M , Soga T , Pollard PJ . Oncometabolites: Linking altered metabolism with cancer. J Clin Invest. (2013) ;123: :3652–8. |
[3] | DeBerardinis RJ , Chandel NS . Fundamentals of cancer metabolism. Sci Adv. (2016) ;2: :e1600200. |
[4] | Pavlova NN , Thompson CB . The Emerging Hallmarks of Cancer Metabolism. Cell Metab. (2016) ;23: :27–47. |
[5] | Vander Heiden MG , DeBerardinis RJ . Understanding the Intersections between Metabolism and Cancer Biology. Cell. (2017) ;168: :657–69. |
[6] | Wettersten HI , Aboud OA , Lara PN Jr , Weiss RH . Metabolic reprogramming in clear cell renal cell carcinoma. Nat Rev Nephrol. (2017) ;13: :410–19. |
[7] | Linehan WM , Srinivasan R , Schmidt LS . The genetic basis of kidney cancer: A metabolic disease. Nat Rev Urol. Nature Publishing Group. (2010) ;7: :277. |
[8] | Curthoys NP , Watford M . Regulation of glutaminase activity and glutamine metabolism. Annu Rev Nutr. (1995) ;15: :133–59. |
[9] | Mayers JR , Vander Heiden MG . Famine versus feast: Understanding the metabolism of tumors in vivo. Trends Biochem Sci. (2015) ;40: :130–140. |
[10] | Lacey JM , Wilmore DW . Is glutamine a conditionally essential amino acid? Nutr Rev. (1990) ;48: :297–309. |
[11] | Karinch AM , Pan M , Lin CM , Strange R , Souba WW . Glutamine metabolism in sepsis and infection. J Nutr. (2001) ;131: :2535S–8S; discussion 2550S-1S. |
[12] | DeBerardinis RJ , Cheng T . Q’s next: The diverse functions of glutamine in metabolism, cell biology and cancer. Oncogene. (2010) ;29: :313–24. |
[13] | Petersen MC , Vatner DF , Shulman GI . Regulation of hepatic glucose metabolism in health and disease. Nat Rev Endocrinol. (2017) ; doi: 10.1038/nrendo.2017.80 |
[14] | Gerich JE , Meyer C , Woerle HJ , Stumvoll M . Renal gluconeogenesis: Its importance in human glucose homeostasis. Diabetes Care. (2001) ;24: :382–91. |
[15] | Weiner ID , Mitch WE , Sands JM . Urea and Ammonia Metabolism and the Control of Renal Nitrogen Excretion. Clin J Am Soc Nephrol. (2015) ;10: :1444–58. |
[16] | Hamm LL , Nakhoul N , Hering-Smith KS . Acid-Base Homeostasis. Clin J Am Soc Nephrol. (2015) ;10: :2232–42. |
[17] | Albrecht J , Sidoryk-Węgrzynowicz M , Zielińska M , Aschner M . Roles of glutamine in neurotransmission. Neuron Glia Biol. (2010) ;6: :263–76. |
[18] | Kim M-H , Kim H . The Roles of Glutamine in the Intestine and Its Implication in Intestinal Diseases. Int J Mol Sci. 18. (2017) ;18. doi: 10.3390/ijms18051051 |
[19] | Calder PC , Newsholme P . Glutamine and the immune system. Nutrition and Immune Function. mprc.ajums.ac.ir; 2002; Available: http://mprc.ajums.ac.ir/_nrc/documents/Nutrition%20and%20Immune%20Function.pdf#page=123 |
[20] | Pochini L , Scalise M , Galluccio M , Indiveri C . Membrane transporters for the special amino acid glutamine: Structure/function relationships and relevance to human health. Front Chem. (2014) ;2: :61. |
[21] | Michalopoulou E , Bulusu V , Kamphorst JJ . Metabolic scavenging by cancer cells: When the going gets tough, the tough keep eating. Br J Cancer. (2016) ;115: :635–40. |
[22] | Villar VH , Merhi F , Djavaheri-Mergny M , Durán RV . Glutaminolysis and autophagy in cancer. Autophagy. (2015) ;11: :1198–208. |
[23] | Eagle H . Nutrition needs of mammalian cells in tissue culture. Science. (1955) ;122: :501–14. |
[24] | Kvamme E , Svenneby G . Effect of anaerobiosis and addition of keto acids on glutamine utilization by Ehrlich ascites-tumor cells. Biochim Biophys Acta. (1960) ;42: :187–8. |
[25] | Knox WE , Horowitz ML , Friedell GH . The proportionality of glutaminase content to growth rate and morphology of rat neoplasms. Cancer Res. (1969) ;29: :669–80. |
[26] | Linder-Horowitz M , Knox WE , Morris HP . Glutaminase activities and growth rates of rat hepatomas. Cancer Res. (1969) ;29: :1195–9. |
[27] | Kovacevic Z , McGivan JD . Mitochondrial metabolism of glutamine and glutamate and its physiological significance. Physiol Rev. (1983) ;63: :547–605. |
[28] | Matsuno T , Hirai H . Glutamine synthetase and glutaminase activities in various hepatoma cells. Biochem Int. (1989) ;19: :219–25. |
[29] | Matsuno T , Goto I . Glutaminase and glutamine synthetase activities in human cirrhotic liver and hepatocellular carcinoma. Cancer Res. (1992) ;52: :1192–4. |
[30] | Aledo JC , Segura JA , Medina MA , Alonso FJ , Núñez de Castro I , Márquez J . Phosphate-activated glutaminase expression during tumor development. FEBS Lett. (1994) ;341: :39–42. |
[31] | Metallo CM , Gameiro PA , Bell EL , Mattaini KR , Yang J , Hiller K , et al. Reductive glutamine metabolism by IDH1 mediates lipogenesis under hypoxia. Nature. (2011) ;481: :380–4. |
[32] | Gameiro PA , Yang J , Metelo AM , Pérez-Carro R , Baker R , Wang Z , et al. In vivo HIF-mediated reductive carboxylation is regulated by citrate levels and sensitizes VHL-deficient cells to glutamine deprivation. Cell Metab. (2013) ;17: :372–85. |
[33] | Okazaki A , Gameiro PA , Christodoulou D , Laviollette L , Schneider M , Chaves F , et al. Glutaminase and poly(ADP-ribose) polymerase inhibitors suppress pyrimidine synthesis and VHL-deficient renal cancers. J Clin Invest. (2017) ;127: :1631–45. |
[34] | Perroud B , Ishimaru T , Borowsky AD , Weiss RH . Grade-dependent proteomics characterization of kidney cancer. Mol Cell Proteomics. (2009) ;8: :971–85. |
[35] | Cancer Genome Atlas Research Network. Comprehensive molecular characterization of clear cell renal cell carcinoma. Nature. (2013) ;499: :43–9. |
[36] | Gatto F , Nookaew I , Nielsen J . Chromosome 3p loss of heterozygosity is associated with a unique metabolic network in clear cell renal carcinoma. Proc Natl Acad Sci U S A. (2014) ;111: :E866–75. |
[37] | Sanders E , Diehl S . Analysis and interpretation of transcriptomic data obtained from extended Warburg effect genes in patients with clear cell renal cell carcinoma. Oncoscience. (2015) ;2: :151–86. |
[38] | Minton DR , Fu L , Chen Q , Robinson BD , Gross SS , Nanus DM , et al. Analyses of the transcriptome and metabolome demonstrate that HIF1α mediates altered tumor metabolism in clear cell renal cell carcinoma. PLoS One. (2015) ;10: :e0120649. |
[39] | Wettersten HI , Hakimi AA , Morin D , Bianchi C , Johnstone ME , Donohoe DR , et al. Grade-Dependent Metabolic Reprogramming in Kidney Cancer Revealed by Combined Proteomics and Metabolomics Analysis. Cancer Res. (2015) ;75: :2541–52. |
[40] | Hakimi AA , Reznik E , Lee C-H , Creighton CJ , Brannon AR , Luna A , et al. An Integrated Metabolic Atlas of Clear Cell Renal Cell Carcinoma. Cancer Cell. (2016) ;29: :104–116. |
[41] | Shroff EH , Eberlin LS , Dang VM , Gouw AM , Gabay M , Adam SJ , et al. MYC oncogene overexpression drives renal cell carcinoma in a mouse model through glutamine metabolism. Proc Natl Acad Sci U S A. (2015) ;112: :6539–44. |
[42] | Okegawa T , Morimoto M , Nishizawa S , Kitazawa S , Honda K , Araki H , et al. Intratumor Heterogeneity in Primary Kidney Cancer Revealed by Metabolic Profiling of Multiple Spatially Separated Samples within Tumors. EBioMedicine. (2017) ;19: :31–8. |
[43] | van der Mijn JC , Panka DJ , Geissler AK , Verheul HM , Mier JW . Novel drugs that target the metabolic reprogramming in renal cell cancer. Cancer Metab. (2016) ;4: :14. |
[44] | Fuchs BC , Bode BP . Stressing out over survival: Glutamine as an apoptotic modulator. J Surg Res. (2006) ;131: :26–40. |
[45] | Lobo C , Ruiz-Bellido MA , Aledo JC , Márquez J , Núñez De Castro I , Alonso FJ . Inhibition of glutaminase expression by antisense mRNA decreases growth and tumourigenicity of tumour cells. Biochem J. (2000) ;348: (Pt 2):257–61. |
[46] | Lora J , Alonso FJ , Segura JA , Lobo C , Márquez J , Matés JM . Antisense glutaminase inhibition decreases glutathione antioxidant capacity and increases apoptosis in Ehrlich ascitic tumour cells. Eur J Biochem. (2004) ;271: :4298–306. |
[47] | Wise DR , DeBerardinis RJ , Mancuso A , Sayed N , Zhang X-Y , Pfeiffer HK , et al. Myc regulates a transcriptional program that stimulates mitochondrial glutaminolysis and leads to glutamine addiction. Proc Natl Acad Sci U S A. (2008) ;105: :18782–7. |
[48] | Gao P , Tchernyshyov I , Chang T-C , Lee Y-S , Kita K , Ochi T , et al. c-Myc suppression of miR-23a/b enhances mitochondrial glutaminase expression and glutamine metabolism. Nature. (2009) ;458: :762–5. |
[49] | Wang J-B , Erickson JW , Fuji R , Ramachandran S , Gao P , Dinavahi R , et al. Targeting mitochondrial glutaminase activity inhibits oncogenic transformation. Cancer Cell. (2010) ;18: :207–19. |
[50] | van den Heuvel APJ , Jing J , Wooster RF , Bachman KE . Analysis of glutamine dependency in non-small cell lung cancer: GLS1 splice variant GAC is essential for cancer cell growth. Cancer Biol Ther. (2012) ;13: :1185–94. |
[51] | Cassago A , Ferreira APS , Ferreira IM , Fornezari C , Gomes ERM , Greene KS , et al. Mitochondrial localization and structure-based phosphate activation mechanism of Glutaminase C with implications for cancer metabolism. Proc Natl Acad Sci U S A. (2012) ;109: :1092–7. |
[52] | Le A , Lane AN , Hamaker M , Bose S , Gouw A , Barbi J , et al. Glucose-independent glutamine metabolism via TCA cycling for proliferation and survival in B cells. Cell Metab. (2012) ;15: :110–21. |
[53] | Huang W , Choi W , Chen Y , Zhang Q , Deng H , He W , et al. A proposed role for glutamine in cancer cell growth through acid resistance. Cell Res. (2013) ;23: :724–7. |
[54] | Timmerman LA , Holton T , Yuneva M , Louie RJ , Padró M , Daemen A , et al. Glutamine sensitivity analysis identifies the xCT antiporter as a common triple-negative breast tumor therapeutic target. Cancer Cell. (2013) ;24: :450–65. |
[55] | Jacque N , Ronchetti AM , Larrue C , Meunier G , Birsen R , Willems L , et al. Targeting glutaminolysis has antileukemic activity in acute myeloid leukemia and synergizes with BCL-2 inhibition. Blood. (2015) ;126: :1346–56. |
[56] | Redis RS , Vela LE , Lu W , Ferreira de Oliveira J , Ivan C , Rodriguez-Aguayo C , et al. Allele-Specific Reprogramming of Cancer Metabolism by the Long Non-coding RNA CCAT2. Mol Cell. (2016) ;61: :520–34. |
[57] | Katt WP , Lukey MJ , Cerione RA . A tale of two glutaminases: Homologous enzymes with distinct roles in tumorigenesis. Future Med Chem. (2017) ;9: :223–43. |
[58] | Shapiro RA , Farrell L , Srinivasan M , Curthoys NP . Isolation, characterization, and in vitro expression of a cDNA that encodes the kidney isoenzyme of the mitochondrial glutaminase. J Biol Chem. (1991) ;266: :18792–6. |
[59] | Elgadi KM , Meguid RA , Qian M , Souba WW , Abcouwer SF . Cloning and analysis of unique human glutaminase isoforms generated by tissue-specific alternative splicing. Physiol Genomics. (1999) ;1: :51–62. |
[60] | Gómez-Fabre PM , Aledo JC , Del Castillo-Olivares A , Alonso FJ , Núñez De Castro I , Campos JA , et al. Molecular cloning, sequencing and expression studies of the human breast cancer cell glutaminase. Biochem J. (2000) ;345: (Pt 2):365–75. |
[61] | Aledo JC , Gómez-Fabre PM , Olalla L , Márquez J . Identification of two human glutaminase loci and tissue-specific expression of the two related genes. Mamm Genome. (2000) ;11: :1107–10. |
[62] | Turner A , McGivan JD . Glutaminase isoform expression in cell lines derived from human colorectal adenomas and carcinomas. Biochem J. (2003) ;370: :403–8. |
[63] | Szeliga M , Matyja E , Obara M , Grajkowska W , Czernicki T , Albrecht J . Relative expression of mRNAS coding for glutaminase isoforms in CNS tissues and CNS tumors. Neurochem Res. (2008) ;33: :808–13. |
[64] | Yuneva MO , Fan TWM , Allen TD , Higashi RM , Ferraris DV , Tsukamoto T , et al. The metabolic profile of tumors depends on both the responsible genetic lesion and tissue type. Cell Metab. (2012) ;15: :157–70. |
[65] | Huang F , Zhang Q , Ma H , Lv Q , Zhang T . Expression of glutaminase is upregulated in colorectal cancer and of clinical significance. Int J Clin Exp Pathol. (2014) ;7: :1093–100. |
[66] | Yang L , Moss T , Mangala LS , Marini J , Zhao H , Wahlig S , et al. Metabolic shifts toward glutamine regulate tumor growth, invasion and bioenergetics in ovarian cancer. Mol Syst Biol. (2014) ;10: :728. |
[67] | Tanaka K , Sasayama T , Irino Y , Takata K , Nagashima H , Satoh N , et al. Compensatory glutamine metabolism promotes glioblastoma resistance to mTOR inhibitor treatment. J Clin Invest. (2015) ;125: :1591–602. |
[68] | Xiang Y , Stine ZE , Xia J , Lu Y , O’Connor RS , Altman BJ , et al. Targeted inhibition of tumor-specific glutaminase diminishes cell-autonomous tumorigenesis. J Clin Invest. (2015) ;125: :2293–306. |
[69] | Xia Z , Donehower LA , Cooper TA , Neilson JR , Wheeler DA , Wagner EJ , et al. Dynamic analyses of alternative polyadenylation from RNA-seq reveal a 3’-UTR landscape across seven tumour types. Nat Commun. (2014) ;5: :5274. |
[70] | Kaelin WG Jr . Molecular basis of the VHL hereditary cancer syndrome. Nat Rev Cancer. (2002) ;2: :673–82. |
[71] | Li B , Qiu B , Lee DSM , Walton ZE , Ochocki JD , Mathew LK , et al. Fructose-1,6-bisphosphatase opposes renal carcinoma progression. Nature. (2014) ;513: :251–5. |
[72] | LaGory EL , Giaccia AJ . The ever-expanding role of HIF in tumour and stromal biology. Nat Cell Biol. (2016) ;18: :356–65. |
[73] | Semenza GL . HIF-1: Upstream and downstream of cancer metabolism. Curr Opin Genet Dev. (2010) ;20: :51–6. |
[74] | Mullen AR , Wheaton WW , Jin ES , Chen P-H , Sullivan LB , Cheng T , et al. Reductive carboxylation supports growth in tumour cells with defective mitochondria. Nature. (2011) ;481: :385–8. |
[75] | Wise DR , Ward PS , Shay JES , Cross JR , Gruber JJ , Sachdeva UM , et al. Hypoxia promotes isocitrate dehydrogenase-dependent carboxylation of α-ketoglutarate to citrate to support cell growth and viability. Proc Natl Acad Sci U S A. (2011) ;108: :19611–6. |
[76] | Filipp FV , Scott DA , Ronai ZA , Osterman AL , Smith JW . Reverse TCA cycle flux through isocitrate dehydrogenases 1 and 2 is required for lipogenesis in hypoxic melanoma cells. Pigment Cell Melanoma Res. (2012) ;25: :375–83. |
[77] | Sun RC , Denko NC . Hypoxic regulation of glutamine metabolism through HIF1 and SIAH2 supports lipid synthesis that is necessary for tumor growth. Cell Metab. (2014) ;19: :285–92. |
[78] | Kim J-W , Tchernyshyov I , Semenza GL , Dang CV . HIF-1-mediated expression of pyruvate dehydrogenase kinase: A metabolic switch required for cellular adaptation to hypoxia. Cell Metab. (2006) ;3: :177–85. |
[79] | Papandreou I , Cairns RA , Fontana L , Lim AL , Denko NC . HIF-1 mediates adaptation to hypoxia by actively downregulating mitochondrial oxygen consumption. Cell Metab. (2006) ;3: :187–97. |
[80] | Ramanathan A , Wang C , Schreiber SL . Perturbational profiling of a cell-line model of tumorigenesis by using metabolic measurements. Proc Natl Acad Sci U S A. (2005) ;102: :5992–7. |
[81] | Yuneva M , Zamboni N , Oefner P , Sachidanandam R , Lazebnik Y . Deficiency in glutamine but not glucose induces MYC-dependent apoptosis in human cells. J Cell Biol. (2007) ;178: :93–105. |
[82] | Tang S-W , Chang W-H , Su Y-C , Chen Y-C , Lai Y-H , Wu P-T , et al. MYC pathway is activated in clear cell renal cell carcinoma and essential for proliferation of clear cell renal cell carcinoma cells. Cancer Lett. (2009) ;273: :35–43. |
[83] | Csibi A , Lee G , Yoon S-O , Tong H , Ilter D , Elia I , et al. The mTORC1/S6K1 pathway regulates glutamine metabolism through the eIF4B-dependent control of c-Myc translation. Curr Biol. (2014) ;24: :2274–80. |
[84] | Hao Y , Samuels Y , Li Q , Krokowski D , Guan B-J , Wang C , et al. Oncogenic PIK3CA mutations reprogram glutamine metabolism in colorectal cancer. Nat Commun. (2016) ;7: :11971. |
[85] | Cox AG , Hwang KL , Brown KK , Evason K , Beltz S , Tsomides A , et al. Yap reprograms glutamine metabolism to increase nucleotide biosynthesis and enable liver growth. Nat Cell Biol. (2016) ;18: :886–96. |
[86] | Lau C-HE , Tredwell GD , Ellis JK , Lam EW-F , Keun HC . Metabolomic characterisation of the effects of oncogenic PIK3CA transformation in a breast epithelial cell line. Sci Rep. (2017) ;7: :46079. |
[87] | Guo H , German P , Bai S , Barnes S , Guo W , Qi X , et al. The PI3K/AKT Pathway and Renal Cell Carcinoma. J Genet Genomics. (2015) ;42: :343–53. |
[88] | Lukey MJ , Wilson KF , Cerione RA . Therapeutic strategies impacting cancer cell glutamine metabolism. Future Med Chem. (2013) ;5: :1685–700. |
[89] | Altman BJ , Stine ZE , Dang CV . From Krebs to clinic: Glutamine metabolism to cancer therapy. Nat Rev Cancer. (2016) ;16: :773. |
[90] | Stegen S , van Gastel N , Eelen G , Ghesquière B , D’Anna F , Thienpont B , et al. HIF-1α Promotes Glutamine-Mediated Redox Homeostasis and Glycogen-Dependent Bioenergetics to Support Postimplantation Bone Cell Survival. Cell Metab. (2016) ;23: :265–79. |
[91] | Munksgaard Thorén M , Vaapil M , Staaf J , Planck M , Johansson ME , Mohlin S , et al. Myc-induced glutaminolysis bypasses HIF-driven glycolysis in hypoxic small cell lung carcinoma cells. Oncotarget. (2017) ;8: :48983–95. |
[92] | Cancer Genome Atlas Research Network, Linehan WM , Spellman PT , Ricketts CJ , Creighton CJ , Fei SS , et al. Comprehensive Molecular Characterization of Papillary Renal-Cell Carcinoma. N Engl J Med. (2016) ;374: :135–45. |
[93] | Davis CF , Ricketts CJ , Wang M , Yang L , Cherniack AD , Shen H , et al. The somatic genomic landscape of chromophobe renal cell carcinoma. Cancer Cell. (2014) ;26: :319–30. |
[94] | Rathmell KW , Chen F , Creighton CJ . Genomics of chromophobe renal cell carcinoma: Implications from a rare tumor for pan-cancer studies. Oncoscience. (2015) ;2: :81–90. |
[95] | Durinck S , Stawiski EW , Pavía-Jiménez A , Modrusan Z , Kapur P , Jaiswal BS , et al. Spectrum of diverse genomic alterations define non-clear cell renal carcinoma subtypes. Nat Genet. (2015) ;47: :13–21. |
[96] | Klomp JA , Petillo D , Niemi NM , Dykema KJ , Chen J , Yang XJ , et al. Birt-Hogg-Dubé renal tumors are genetically distinct from other renal neoplasias and are associated with up-regulation of mitochondrial gene expression. BMC Med Genomics. (2010) ;3: :59. |
[97] | Sudarshan S , Karam JA , Brugarolas J , Thompson RH , Uzzo R , Rini B , et al. Metabolism of kidney cancer: From the lab to clinical practice. Eur Urol. (2013) ;63: :244–51. |
[98] | Tomlinson IPM , Alam NA , Rowan AJ , Barclay E , Jaeger EEM , Kelsell D , et al. Germline mutations in FH predispose to dominantly inherited uterine fibroids, skin leiomyomata and papillary renal cell cancer. Nat Genet. (2002) ;30: :406–10. |
[99] | Isaacs JS , Jung YJ , Mole DR , Lee S , Torres-Cabala C , Chung Y-L , et al. HIF overexpression correlates with biallelic loss of fumarate hydratase in renal cancer: Novel role of fumarate in regulation of HIF stability. Cancer Cell. (2005) ;8: :143–53. |
[100] | Linehan WM , Rouault TA . Molecular pathways: Fumarate hydratase-deficient kidney cancer–targeting the Warburg effect in cancer. Clin Cancer Res. (2013) ;19: :3345–52. |
[101] | Sudarshan S , Sourbier C , Kong H-S , Block K , Valera Romero VA , Yang Y , et al. Fumarate hydratase deficiency in renal cancer induces glycolytic addiction and hypoxia-inducible transcription factor 1alpha stabilization by glucose-dependent generation of reactive oxygen species. Mol Cell Biol. (2009) ;29: :4080–90. |
[102] | O’Flaherty L , Adam J , Heather LC , Zhdanov AV , Chung Y-L , Miranda MX , et al. Dysregulation of hypoxia pathways in fumarate hydratase-deficient cells is independent of defective mitochondrial metabolism. Hum Mol Genet. (2010) ;19: :3844–51. |
[103] | Tong W-H , Sourbier C , Kovtunovych G , Jeong SY , Vira M , Ghosh M , et al. The glycolytic shift in fumarate-hydratase-deficient kidney cancer lowers AMPK levels, increases anabolic propensities and lowers cellular iron levels. Cancer Cell. (2011) ;20: :315–27. |
[104] | Yang Y , Valera V , Sourbier C , Vocke CD , Wei M , Pike L , et al. A novel fumarate hydratase-deficient HLRCC kidney cancer cell line, UOK A model of the Warburg effect in cancer. Cancer Genet. (2012) ;205: :377–90. |
[105] | Selak MA , Armour SM , MacKenzie ED , Boulahbel H , Watson DG , Mansfield KD , et al. Succinate links TCA cycle dysfunction to oncogenesis by inhibiting HIF-alpha prolyl hydroxylase. Cancer Cell. (2005) ;7: :77–85. |
[106] | Guzy RD , Sharma B , Bell E , Chandel NS , Schumacker PT . Loss of the SdhB, but Not the SdhA, subunit of complex II triggers reactive oxygen species-dependent hypoxia-inducible factor activation and tumorigenesis. Mol Cell Biol. (2008) ;28: :718–31. |
[107] | Cardaci S , Zheng L , MacKay G , van den Broek NJF , MacKenzie ED , Nixon C , et al. Pyruvate carboxylation enables growth of SDH-deficient cells by supporting aspartate biosynthesis. Nat Cell Biol. (2015) ;17: :1317–26. |
[108] | Lussey-Lepoutre C , Hollinshead KER , Ludwig C , Menara M , Morin A , Castro-Vega L-J , et al. Loss of succinate dehydrogenase activity results in dependency on pyruvate carboxylation for cellular anabolism. Nat Commun. (2015) ;6: :8784. |
[109] | Niemann S , Müller U . Mutations in SDHC cause autosomal dominant paraganglioma, type 3. Nat Genet. (2000) ;26: :268–70. |
[110] | Astuti D , Latif F , Dallol A , Dahia PL , Douglas F , George E , et al. Gene mutations in the succinate dehydrogenase subunit SDHB cause susceptibility to familial pheochromocytoma and to familial paraganglioma. Am J Hum Genet. (2001) ;69: :49–54. |
[111] | Baysal BE , Ferrell RE , Willett-Brozick JE , Lawrence EC , Myssiorek D , Bosch A , et al. Mutations in SDHD, a mitochondrial complex II gene, in hereditary paraganglioma. Science. (2000) ;287: :848–51. |
[112] | Killian JK , Kim SY , Miettinen M , Smith C , Merino M , Tsokos M , et al. Succinate dehydrogenase mutation underlies global epigenomic divergence in gastrointestinal stromal tumor. Cancer Discov. (2013) ;3: :648–57. |
[113] | Drabkin HA , Bradley C , Hart I , Bleskan J , Li FP , Patterson D . Translocation of c-myc in the hereditary renal cell carcinoma associated with a t(3;8)(p14.2;q24.13) chromosomal translocation. Proc Natl Acad Sci U S A. (1985) ;82: :6980–4. |
[114] | Medina MA , Sánchez-Jiménez F , Márquez J , Rodríguez Quesada A , Núñez de Castro I . Relevance of glutamine metabolism to tumor cell growth. Mol Cell Biochem. (1992) ;113: :1–15. |
[115] | Souba WW . Glutamine and cancer. Ann Surg. (1993) ;218: :715–28. |
[116] | Deberardinis RJ , Sayed N , Ditsworth D , Thompson CB . Brick by brick: Metabolism and tumor cell growth. Curr Opin Genet Dev. (2008) ;18: :54–61. |
[117] | Robinson MM , McBryant SJ , Tsukamoto T , Rojas C , Ferraris DV , Hamilton SK , et al. Novel mechanism of inhibition of rat kidney-type glutaminase by bis-2-(5-phenylacetamido-1,2,4-thiadiazol-2-yl)ethyl sulfide (BPTES). Biochem J. (2007) ;406: :407–14. |
[118] | DeLaBarre B , Gross S , Fang C , Gao Y , Jha A , Jiang F , et al. Full-length human glutaminase in complex with an allosteric inhibitor. Biochemistry. (2011) ;50: :10764–70. |
[119] | Hartwick EW , Curthoys NP . BPTES inhibition of hGA124-551, a truncated form of human kidney-type glutaminase. J Enzyme Inhib Med Chem. Taylor & Francis. (2012) ;27: :861–7. |
[120] | Ferreira APS , Cassago A , Gonçalves K de A , Dias MM , Adamoski D , Ascenção CFR , et al. Active glutaminase C self-assembles into a supratetrameric oligomer that can be disrupted by an allosteric inhibitor. J Biol Chem. (2013) ;288: :28009–20. |
[121] | Stalnecker CA , Ulrich SM , Li Y , Ramachandran S , McBrayer MK , DeBerardinis RJ , et al. Mechanism by which a recently discovered allosteric inhibitor blocks glutamine metabolism in transformed cells. Proc Natl Acad Sci U S A. (2015) ;112: :394–9. |
[122] | Katt WP , Cerione RA . Glutaminase regulation in cancer cells: A druggable chain of events. Drug Discov Today. (2014) ;19: :450–7. |
[123] | Gross MI , Demo SD , Dennison JB , Chen L , Chernov-Rogan T , Goyal B , et al. Antitumor activity of the glutaminase inhibitor CB-839 in triple-negative breast cancer. Mol Cancer Ther. (2014) ;13: :890–901. |
[124] | Seltzer MJ , Bennett BD , Joshi AD , Gao P , Thomas AG , Ferraris DV , et al. Inhibition of glutaminase preferentially slows growth of glioma cells with mutant IDH1. Cancer Res. (2010) ;70: :8981–7. |
[125] | Katt WP , Ramachandran S , Erickson JW , Cerione RA . Dibenzophenanthridines as inhibitors of glutaminase C and cancer cell proliferation. Mol Cancer Ther. (2012) ;11: :1269–78. |
[126] | Simpson NE , Tryndyak VP , Pogribna M , Beland FA , Pogribny IP . Modifying metabolically sensitive histone marks by inhibiting glutamine metabolism affects gene expression and alters cancer cell phenotype. Epigenetics. (2012) ;7: :1413–20. |
[127] | Son J , Lyssiotis CA , Ying H , Wang X , Hua S , Ligorio M , et al. Glutamine supports pancreatic cancer growth through a KRAS-regulated metabolic pathway. Nature. (2013) ;496: :101–5. |
[128] | Qie S , Chu C , Li W , Wang C , Sang N . ErbB2 activation upregulates glutaminase 1 expression which promotes breast cancer cell proliferation. J Cell Biochem. (2014) ;115: :498–509. |
[129] | Emadi A , Jun SA , Tsukamoto T , Fathi AT , Minden MD , Dang CV . Inhibition of glutaminase selectively suppresses the growth of primary acute myeloid leukemia cells with IDH mutations. Exp Hematol. (2014) ;42: :247–51. |
[130] | Richard SM , Martinez Marignac VL . Sensitization to oxaliplatin in HCT116 and HT29 cell lines by metformin and ribavirin and differences in response to mitochondrial glutaminase inhibition. J Cancer Res Ther. (2015) ;11: :336–40. |
[131] | Sontakke P , Koczula KM , Jaques J , Wierenga ATJ , Brouwers-Vos AZ , Pruis M , et al. Hypoxia-Like Signatures Induced by BCR-ABL Potentially Alter the Glutamine Uptake for Maintaining Oxidative Phosphorylation. PLoS One. (2016) ;11: :e0153226. |
[132] | Hudson CD , Savadelis A , Nagaraj AB , Joseph P , Avril S , DiFeo A , et al. Altered glutamine metabolism in platinum resistant ovarian cancer. Oncotarget. (2016) ;7: :41637–49. |
[133] | Xie C , Jin J , Bao X , Zhan W-H , Han T-Y , Gan M , et al. Inhibition of mitochondrial glutaminase activity reverses acquired erlotinib resistance in non-small cell lung cancer. Oncotarget. (2016) ;7: :610–21. |
[134] | Ye X , Zhou Q , Matsumoto Y , Moriyama M , Kageyama S , Komatsu M , et al. Inhibition of Glutaminolysis Inhibits Cell Growth via Down-regulating Mtorc1 Signaling in Lung Squamous Cell Carcinoma. Anticancer Res. (2016) ;36: :6021–9. |
[135] | Matre P , Velez J , Jacamo R , Qi Y , Su X , Cai T , et al. Inhibiting glutaminase in acute myeloid leukemia: Metabolic dependency of selected AML subtypes. Oncotarget. (2016) ;7: :79722–35. |
[136] | Rajeshkumar NV , Yabuuchi S , Pai S , De Oliveira E , Kamphorst JJ , Rabinowitz JD , et al. Treatment of pancreatic cancer patient-derived xenograft panel with metabolic inhibitors reveals efficacy of phenformin. Clin Cancer Res. (2017) ; doi: 10.1158/1078-0432.CCR-17-1115 |
[137] | Han T , Guo M , Zhang T , Gan M , Xie C , Wang J-B . A novel glutaminase inhibitor-968 inhibits the migration and proliferation of non-small cell lung cancer cells by targeting EGFR/ERK signaling pathway. Oncotarget. (2017) ;8: :28063–73. |
[138] | Momcilovic M , Bailey ST , Lee JT , Fishbein MC , Magyar C , Braas D , et al. Targeted Inhibition of EGFR and Glutaminase Induces Metabolic Crisis in EGFR Mutant Lung Cancer. Cell Rep. (2017) ;18: :601–10. |
[139] | Thompson RM , Dytfeld D , Reyes L , Robinson RM , Smith B , Manevich Y , et al. Glutaminase inhibitor CB-839 synergizes with carfilzomib in resistant multiple myeloma cells. Oncotarget. (2017) ;8: :35863–76. |
[140] | Emberley E , Bennett M , Chen J , Gross M , Huang T , Li W , et al. CB-839, a Selective Glutaminase Inhibitor, has Anti-Tumor Activity in Renal Cell Carcinoma and Synergizes with Cabozantinib and Everolimus. Keystone Symposia, Tumor Metabolism: Mechanisms and Targets, Whistler Canada. (2017) . Available: https://www.calithera.com/wp-content/uploads/2017/12/03.2017-Keystone-poster-Emberley-2017.pdf |
[141] | Parlati F . Anti-Tumor Activity of Glutaminase Inhibitor CB-839 in Solid Tumor Malignancies (oral presentation). Keystone Symposium: Tumor Metabolism. 2018; Available: https://www.calithera.com/wp-content/uploads/2018/02/Keystone-Metabolism-2018-parlati-final.pdf (accessed 09/03/2018) |
[142] | Meric-Bernstam F , Gordon M , Tykodi S , Lam E , Vaishampayan U , Chaves J , et al. CX-839-004: A phase 1/2 study of CB-839, a first-in-class glutaminase inhibitor, combined with nivolumab in patients with advanced Melanoma (MEL), Renal Cell Carcinoma (RCC), or Non-Small Cell Lung Cancer (NSCLC) (oral presentation). Society for Immunotherapy of Cancer Annual Meeting. (2017) ; Available: https://www.calithera.com/wp-content/uploads/2017/12/CX-839-004-SITC-11-11-2017-FMB-FINAL-for-PDF.pdf (accessed 09/03/2018) |
[143] | MacKinnon A , Chen J , Gross M , Marguier G , Shwonek P , Sotirovska N , et al. Targeting Tumor Glutamine Metabolism with CB-839 Enhances the Efficacy of Immune Checkpoint Inhibitors (poster presentation). Society for Immunotherapy of Cancer Annual Meeting. 2016; Available: https://www.calithera.com/wp-content/uploads/2017/12/1st-post.pdf (accessed 09/03/2018) |
[144] | Avramis VI . Asparaginases: Biochemical pharmacology and modes of drug resistance. Anticancer Res. (2012) ;32: :2423–37. |
[145] | Dahlmanns M , Yakubov E , Chen D , Sehm T , Rauh M , Savaskan N , et al. Chemotherapeutic xCT inhibitors sorafenib and erastin unraveled with the synaptic optogenetic function analysis tool. Cell Death Discovery. Nature Publishing Group. (2017) ;3: :17030. |
[146] | Ahluwalia GS , Grem JL , Hao Z , Cooney DA . Metabolism and action of amino acid analog anti-cancer agents. Pharmacol Ther. (1990) ;46: :243–71. |
[147] | Elson PJ , Kvols LK , Vogl SE , Glover DJ , Hahn RG , Trump DL , et al. Phase II trials of 5-day vinblastine infusion (NSC 49842), L-alanosine (NSC 153353), acivicin (NSC 163501), and aminothiadiazole (NSC 4728) in patients with recurrent or metastatic renal cell carcinoma. Invest New Drugs. (1988) ;6: :97–103. |
[148] | Mueller C , Al-Batran S , Jaeger E , Schmidt B , Bausch M , Unger C , et al. A phase IIa study of PEGylated glutaminase (PEG-PGA) plus 6-diazo-5-oxo-L-norleucine (DON) in patients with advanced refractory solid tumors. J Clin Orthod. American Society of Clinical Oncology. (2008) ;26: :2533. |
[149] | Shukla K , Ferraris DV , Thomas AG , Stathis M , Duvall B , Delahanty G , et al. Design, synthesis, and pharmacological evaluation of bis-2-(5-phenylacetamido-1,2,4-thiadiazol-2-yl)ethyl sulfide 3 (BPTES) analogs as glutaminase inhibitors. J Med Chem. (2012) ;55: :10551–63. |
[150] | Huang Q , Stalnecker C , Zhang C , McDermott LA , Iyer P , O’Neill J , et al. Characterization of the interactions of potent allosteric inhibitors with glutaminase C, a key enzyme in cancer cell glutamine metabolism. J Biol Chem. (2018) ;293: :3535–45. |
[151] | Elgogary A , Xu Q , Poore B , Alt J , Zimmermann SC , Zhao L , et al. Combination therapy with BPTES nanoparticles and metformin targets the metabolic heterogeneity of pancreatic cancer. Proc Natl Acad Sci U S A. (2016) ;113: :E5328–36. |
[152] | Choi Y-K , Park K-G . Targeting Glutamine Metabolism for Cancer Treatment. Biomol Ther. (2018) ;26: :19–28. |
[153] | Meric-Bernstam F , Tannir N , Harding J , Voss M , Mier J , DeMichele A , et al. Phase 1 study of CB-839, a small molecule inhibitor of glutaminase, in combination with everolimus in patients (pts) with clear cell and papillary renal cell cancer (RCC). Eur J Cancer. (2016) ;69: :S12–S13. |
[154] | McDermott LA , Iyer P , Vernetti L , Rimer S , Sun J , Boby M , et al. Design and evaluation of novel glutaminase inhibitors. Bioorg Med Chem. (2016) ;24: :1819–39. |
[155] | Ramachandran S , Pan CQ , Zimmermann SC , Duvall B , Tsukamoto T , Low BC , et al. Structural basis for exploring the allosteric inhibition of human kidney type glutaminase. Oncotarget. (2016) ;7: :57943–54. |
[156] | Lee Y-Z , Yang C-W , Chang H-Y , Hsu H-Y , Chen I-S , Chang H-S , et al. Discovery of selective inhibitors of Glutaminase-2, which inhibit mTORC1, activate autophagy and inhibit proliferation in cancer cells. Oncotarget. (2014) ;5: :6087–101. |
[157] | Zhao L , Huang Y , Tian C , Taylor L , Curthoys N , Wang Y , et al. Interferon-α regulates glutaminase 1 promoter through STAT1 phosphorylation: Relevance to HIV-1 associated neurocognitive disorders. PLoS One. (2012) ;7: :e32995. |
[158] | Ince-Dunn G , Okano HJ , Jensen KB , Park W-Y , Zhong R , Ule J , et al. Neuronal Elav-like (Hu) proteins regulate RNA splicing and abundance to control glutamate levels and neuronal excitability. Neuron. (2012) ;75: :1067–80. |
[159] | Masamha CP , Xia Z , Yang J , Albrecht TR , Li M , Shyu A-B , et al. CFIm25 links alternative polyadenylation to glioblastoma tumour suppression. Nature. (2014) ;510: :412–6. |
[160] | Hansen WR , Barsic-Tress N , Taylor L , Curthoys NP . The 39-nontranslated region of rat renal glutaminase mRNA contains a pH-responsive stability element. American Journal of Physiology-Renal Physiology. Am Physiological Soc. (1996) ;271: :F126–F131. |
[161] | Rathore MG , Saumet A , Rossi J-F , de Bettignies C , Tempé D , Lecellier C-H , et al. The NF-κB member p65 controls glutamine metabolism through miR-23a. Int J Biochem Cell Biol. (2012) ;44: :1448–56. |
[162] | Thangavelu K , Pan CQ , Karlberg T , Balaji G , Uttamchandani M , Suresh V , et al. Structural basis for the allosteric inhibitory mechanism of human kidney-type glutaminase (KGA) and its regulation by Raf-Mek-Erk signaling in cancer cell metabolism. Proc Natl Acad Sci U S A. (2012) ;109: :7705–10. |
[163] | Polletta L , Vernucci E , Carnevale I , Arcangeli T , Rotili D , Palmerio S , et al. SIRT5 regulation of ammonia-induced autophagy and mitophagy. Autophagy. (2015) ;11: :253–70. |
[164] | Lu W , Zuo Y , Feng Y , Zhang M . SIRT5 facilitates cancer cell growth and drug resistance in non-small cell lung cancer. Tumour Biol. (2014) ;35: :10699–705. |
[165] | Buschdorf JP , Li Chew L , Zhang B , Cao Q , Liang F-Y , Liou Y-C , et al. Brain-specific BNIP-2-homology protein Caytaxin relocalises glutaminase to neurite terminals and reduces glutamate levels. J Cell Sci. (2006) ;119: :3337–50. |
[166] | Boulay A-C , Burbassi S , Lorenzo H-K , Loew D , Ezan P , Giaume C , et al. Bmcc1s interacts with the phosphate-activated glutaminase in the brain. Biochimie. (2013) ;95: :799–807. |
[167] | Krebs HA . Metabolism of amino-acids: The synthesis of glutamine from glutamic acid and ammonia, and the enzymic hydrolysis of glutamine in animal tissues. Biochem J. (1935) ;29: :1951–69. |
[168] | Colombo SL , Palacios-Callender M , Frakich N , De Leon J , Schmitt CA , Boorn L , et al. Anaphase-promoting complex/cyclosome-Cdh1 coordinates glycolysis and glutaminolysis with transition to S phase in human T lymphocytes. Proc Natl Acad Sci U S A. (2010) ;107: :18868–73. |
[169] | Colombo SL , Palacios-Callender M , Frakich N , Carcamo S , Kovacs I , Tudzarova S , et al. Molecular basis for the differential use of glucose and glutamine in cell proliferation as revealed by synchronized HeLa cells. Proc Natl Acad Sci U S A. (2011) ;108: :21069–74. |
[170] | Kita K , Suzuki T , Ochi T . Diphenylarsinic acid promotes degradation of glutaminase C by mitochondrial Lon protease. J Biol Chem. (2012) ;287: :18163–72. |
[171] | Viswanathan VS , Ryan MJ , Dhruv HD , Gill S , Eichhoff OM , Seashore-Ludlow B , et al. Dependency of a therapy-resistant state of cancer cells on a lipid peroxidase pathway. Nature. (2017) ;547: :453–7. |
[172] | Ho P-C , Bihuniak JD , Macintyre AN , Staron M , Liu X , Amezquita R , et al. Phosphoenolpyruvate Is a Metabolic Checkpoint of Anti-tumor T Cell Responses. Cell. (2015) ;162: :1217–28. |
[173] | Chang C-H , Qiu J , O’Sullivan D , Buck MD , Noguchi T , Curtis JD , et al. Metabolic Competition in the Tumor Microenvironment Is a Driver of Cancer Progression. Cell. (2015) ;162: :1229–41. |
[174] | Thong AE , Zhao H , Ingels A , Valta MP , Nolley R , Santos J , et al. Tissue slice grafts of human renal cell carcinoma: An authentic preclinical model with high engraftment rate and metastatic potential. Urol Oncol. (2014) ;32: :43.e23–30. |
[175] | Ingels A , Zhao H , Thong AE , Saar M , Valta MP , Nolley R , et al. Preclinical trial of a new dual mTOR inhibitor, MLN0128, Nusing renal cell carcinoma tumorgrafts. Int J Cancer. (2014) ;134: :2322–9. |
[176] | Valta MP , Zhao H , Ingels A , Thong AE , Nolley R , Saar M , et al. Development of a realistic in vivo bone metastasis model of human renal cell carcinoma. Clin Exp Metastasis. (2014) ;31: :573–84. |
[177] | Zhao H , Nolley R , Chan AMW , Rankin EB , Peehl DM . Cabozantinib inhibits tumor growth and metastasis of a patient-derived xenograft model of papillary renal cell carcinoma with MET mutation. Cancer Biol Ther. (2016) ;0. |
[178] | Inoue T , Terada N , Kobayashi T , Ogawa O . Patient-derived xenografts as in vivo models for research in urological malignancies. Nat Rev Urol. (2017) ;14: :267–83. |
[179] | Banerjee S , Zare RN , Tibshirani RJ , Kunder CA , Nolley R , Fan R , et al. Diagnosis of prostate cancer by desorption electrospray ionization mass spectrometric imaging of small metabolites and lipids. Proc Natl Acad Sci U S A. (2017) ;114: :3334–9. |
[180] | Fan AC , Deb-Basu D , Orban MW , Gotlib JR , Natkunam Y , O’Neill R , et al. Nanofluidic proteomic assay for serial analysis of oncoprotein activation in clinical specimens. Nat Med. (2009) ;15: :566–71. |
[181] | Seetharam M , Fan AC , Tran M , Xu L , Renschler JP , Felsher DW , et al. Treatment of higher risk myelodysplastic syndrome patients unresponsive to hypomethylating agents with ON 01910.Na. Leuk Res. (2012) ;36: :98–103. |
[182] | DeChristopher BA , Fan AC , Felsher DW , Wender PA . “Picolog,” a synthetically-available bryostatin analog, inhibits growth of MYC-induced lymphoma in vivo. Oncotarget. (2012) ;3: :58–66. |
[183] | Fan AC , O’Rourke JJ , Praharaj DR , Felsher DW . Real-time nanoscale proteomic analysis of the novel multi-kinase pathway inhibitor rigosertib to measure the response to treatment of cancer. Expert Opin Investig Drugs. (2013) ;22: :1495–509. |
[184] | Liu S , Tian Z , Zhang L , Hou S , Hu S , Wu J , et al. Combined cell surface carbonic anhydrase 9 and CD147 antigens enable high-efficiency capture of circulating tumor cells in clear cell renal cell carcinoma patients. Oncotarget. (2016) ;7: :59877–91. |
[185] | Lieberman BP , Ploessl K , Wang L , Qu W , Zha Z , Wise DR , et al. PET imaging of glutaminolysis in tumors by 18F-(2S, 4R) 4-fluoroglutamine. J Nucl Med. Soc Nuclear Med. (2011) ;52: :1947–55. |
[186] | Venneti S , Dunphy MP , Zhang H , Pitter KL , Zanzonico P , Campos C , et al. Glutamine-based PET imaging facilitates enhanced metabolic evaluation of gliomas in vivo. Sci Transl Med. (2015) ;7: :274ra17. |
[187] | Hassanein M , Hight MR , Buck JR , Tantawy MN , Nickels ML , Hoeksema MD , et al. Preclinical Evaluation of 4-[18F]Fluoroglutamine PET to Assess ASCT2 Expression in Lung Cancer. Mol Imaging Biol. (2016) ;18: :18–23. |
[188] | Zhou R , Pantel AR , Li S , Lieberman BP , Ploessl K , Choi H , et al. [(18)F](2S,4R)4-Fluoroglutamine PET Detects Glutamine Pool Size Changes in Triple-Negative Breast Cancer in Response to Glutaminase Inhibition. Cancer Res. (2017) ;77: :1476–84. |
[189] | Abu Aboud O , Habib SL , Trott J , Stewart B , Liang S , Chaudhari AJ , et al. Glutamine Addiction in Kidney Cancer Suppresses Oxidative Stress and Can Be Exploited for Real-Time Imaging. Cancer Res. (2017) ;77: :6746–58. |
[190] | Koglin N , Mueller A , Berndt M , Schmitt-Willich H , Toschi L , Stephens AW , et al. Specific PET Imaging of xC- Transporter Activity Using a 18F-Labeled Glutamate Derivative Reveals a Dominant Pathway in Tumor Metabolism. Clin Cancer Res. American Association for Cancer Research. (2011) ;17: :6000–11. |
[191] | Baek S , Choi C-M , Ahn SH , Lee JW , Gong G , Ryu J-S , et al. Exploratory clinical trial of (4S)-4-(3-[18F] fluoropropyl)-l-glutamate for imaging xC- transporter using positron emission tomography in patients with non–small cell lung or breast cancer. Clin Cancer Res. AACR. (2012) ;18: :5427–37. |
[192] | Baek S , Mueller A , Lim Y-S , Lee HC , Lee Y-J , Gong G , et al. (4S)-4-(3-18F-fluoropropyl)-L-glutamate for imaging of xC transporter activity in hepatocellular carcinoma using PET: Preclinical and exploratory clinical studies. J Nucl Med. (2013) ;54: :117–23. |
[193] | Mittra ES , Koglin N , Mosci C , Kumar M , Hoehne A , Keu KV , et al. Pilot Preclinical and Clinical Evaluation of (4S)-4-(3-[18F]Fluoropropyl)-L-Glutamate (18F-FSPG) for PET/CT Imaging of Intracranial Malignancies. PLoS One. (2016) ;11: :e0148628. |
[194] | Kavanaugh G , Williams J , Morris AS , Nickels ML , Walker R , Koglin N , et al. Utility of [18F]FSPG PET to Image Hepatocellular Carcinoma: First Clinical Evaluation in a US Population. Mol Imaging Biol. (2016) ;18: :924–34. |
[195] | Park S , Hatami N , Rutledge O , Koglin N , Loo B , Fan A , et al. Pilot study of 18F-FSPG vs18F-FDG PET imaging for response assessment in cancer. J Nucl Med. (2017) ;58: :118. |


