
Impact of Aspirin and Non-Aspirin Nonsteroidal Anti-Inflammatory Drugs on Outcomes in Patients with Metastatic Renal Cell Carcinoma
Abstract
Background:
Non-steroidal anti-inflammatory drugs (NSAIDs) have demonstrated an anti-tumorigenic effect in several cancers. However, their use is associated with an increased risk in renal cell carcinoma (RCC) and their effect has not been assessed in patients with metastatic disease.
Objective:
We investigated the impact of NSAIDs on survival outcomes in patients with metastatic RCC (mRCC).
Methods:
We conducted a pooled retrospective analysis of 4,736 mRCC patients treated on phase II and III clinical trials. Patients were categorized as: aspirin (ASA) only users, non-ASA NSAIDs only users, ASA and non-ASA NSAIDs users, and NSAIDs non-users. The primary endpoint was overall survival (OS). Progression free survival (PFS), overall response rate (ORR) and adverse events (AEs) were secondary endpoints. OS and PFS were estimated using the Kaplan–Meier method and were assessed using multivariate Cox regression analysis.
Results:
We identified 457 (10%) ASA only users, 639 (13%) non-ASA NSAIDs only users, 61 (1%) ASA and non-ASA NSAIDS users, and 3579 (76%) NSAIDs non-users. OS and PFS were significantly worse in non-ASA NSAIDs users compared to the NSAIDs non-users (OS hazard ratio (HR): 1.47, p < 0.0001, median 11.6 versus 21.1 months; PFS HR: 1.29, p < 0.0001, median 4.6 versus 7.4 months). There was no difference in survival in ASA users or ASA and non-ASA NSAIDs users compared to NSAIDs non-users.
Conclusions:
Our analysis demonstrates that NSAIDs do not confer a survival advantage in mRCC patients. Further studies are warranted to elucidate the interaction of NSAIDS with targeted therapy in mRCC.
INTRODUCTION
Non-steroidal anti-inflammatory drugs (NSAIDs) are among the most commonly utilized medications in the U.S., with an estimated use of 17% in 2002 [1]. These agents inhibit cyclooxygenase (COX) activity, which leads to the suppression of prostaglandin synthesis, and ultimately decreased inflammation. Some NSAIDs, such as ibuprofen, reversibly inhibit both Cox-1 and Cox-2 isoforms, while others selectively inhibit Cox-2. Additionally, aspirin (ASA) is unique in that it irreversibly inhibits Cox-1 and Cox-2 [2].
NSAID use is primarily indicated for the treatment of pain and inflammation as well as the prevention of cardiovascular disease [3]. Recently, there has been increasing interest in the role of NSAIDs as anti-tumorigenic drugs, particularly in colorectal cancer (CRC) [3, 4]. Epidemiological data demonstrate that NSAIDs use, including long-term ASA use, decreases incidence, metastasis and mortality risk in several cancers [5–10]. NSAIDs are thought to exert their antitumor activity mainly by inhibiting COX-2, although recent evidence suggests additional COX-independent mechanisms [11–14]. Several pre-clinical studies demonstrated the anti-tumor efficacy in vitro and in vivo [15–18]. NSAIDs inhibited formation of early malignant lesions and caused regression of tumors in rodent models of CRC [19–22]. Additionally, clinical trials have shown a modest benefit of NSAIDs in preventing the recurrence of colorectal adenomas [4, 23].
In RCC, limited studies have evaluated the role of ASA and non-ASA NSAIDs in patients with localized disease and none have characterized outcomes for patients with metastatic RCC (mRCC). Non-ASA NSAIDs, unlike ASA, have been implicated as a risk factor for RCC development [24, 25]. Elucidation of the impact of ASA/NSAIDs in patient with mRCC is relevant to optimizing the evolving treatment landscape for patients with metastatic disease. Therefore, we investigated the impact of ASA and non-ASA NSAIDs on overall survival (OS), progression free survival (PFS) and objective response rate (ORR) in a large clinical trials database of mRCC patients treated with targeted therapies (TT).
PATIENTS AND METHODS
Study design
We conducted a retrospective analysis of mRCC patients treated on phase II (NCT00054886, NCT00077974, NCT00267748, NCT00338884, NCT00137423, NCT00835978) and phase III (NCT00083889, NCT00065468, NCT000678392, NCT00474786, NCT00631371, NCT00920816) clinical trials sponsored by Pfizer. Eligible patients had a diagnosis of mRCC and available concomitant medication data.
Baseline demographic, clinical, and laboratory data were collected. Patients receiving ASA and non-ASA NSAIDs at baseline were defined as users. Patients were grouped into four cohorts: ASA only users, non-ASA NSAIDs only users, ASA and non-ASA NSAIDs users, and NSAIDs non-users (reference). Informed consent was obtained from all patients participating in this study.
Treatment outcomes
The primary endpoint was OS, defined as the time from randomization for randomized studies or from initiation of therapy for non-randomized studies to death from any cause. PFS, ORR and adverse events (AEs) were secondary endpoints. PFS was defined as the time from randomization for randomized studies or from initiation of therapy for non-randomized studies to date of progression or death from any cause, whichever came first. Response was assessed using the Response Evaluation Criteria in Solid Tumors (RECIST) version 1.0. Treatment-associated AEs were defined according to the Common Terminology Criteria for AEs, version 3.0.
Statistical analyses
OS and PFS were estimated using the Kaplan–Meier method and were assessed using multivariate Cox regression analysis, adjusting for age, gender, race, Eastern Cooperative Oncology group (ECOG) performance status, histology, International mRCC Database Consortium (IMDC) risk factors, prior nephrectomy, prior therapy, sites of metastasis, baseline hypertension, baseline diabetes, and baseline angiotensin system inhibitor (ASI) use. OS, PFS and ORR were evaluated in the total cohort and in the following subsets: type of therapy (vascular endothelial growth factor TT vs. mammalian target of rapamycin (mTOR) TT, vs. interferon-alpha (IFN-α) therapy) and line of therapy (first vs. second line). Serious grade ≥3 AEs occurring in >3% of patients and selected AEs of interest representing overlapping toxicities of TT and NSAIDs of any grade were summarized. All p values were two sided. Statistical analyses were conducted using SAS.
RESULTS
Baseline characteristics
Overall, 4,736 patients were included in the analysis of which 457 (10%) were defined as ASA users, 639 (13%) as non-ASA NSAIDs users, 61 (1%) as ASA and non-ASA NSAIDS users, and 3579 (76%) as NSAIDs non-users. The majority of patients were <65 years of age (n = 3258, 69%), male (n = 3363, 71%) with good performance status (n = 4653, 98%) (Table 1). Some baseline characteristics differed among the four cohorts, particularly, age, Asian race, region, IMDC risk group, bone metastasis and ASI use.
Table 1
Baseline patient and disease characteristics
| ASA users (N = 457) | Non-ASA NSAIDs users (N = 639) | ASA and non-ASA NSAIDs users (N = 61) | NSAIDs non-users (N = 3579) | Total Cohort (N = 4736) | |
| N (%) | N (%) | N (%) | N (%) | N (%) | |
| Age at initiation of therapy | |||||
| Median (min, max) | 65 (28,87) | 57 (22,85) | 66 (43, 80) | 59 (18,91) | 59 (18,91) |
| <65 years | 215 (47%) | 478 (75%) | 28 (46%) | 2537 (71%) | 3258 (69%) |
| ≥65 years | 242 (53%) | 161 (25%) | 33 (54%) | 1042 (29%) | 1478 (31%) |
| Sex | |||||
| Male | 350 (77%) | 428 (67%) | 47 (77%) | 2538 (71%) | 3363 (71%) |
| Female | 107 (23%) | 211 (33%) | 14 (23%) | 1041 (29%) | 1373 (29%) |
| Race | |||||
| White | 396 (87%) | 516 (81%) | 57 (93%) | 2695 (75.3%) | 3664 (77%) |
| Black | 11 (2%) | 9 (1%) | 0 | 56 (2%) | 76 (2%) |
| Asian | 36 (8%) | 91 (14%) | 3 (5%) | 624 (17%) | 754 (16%) |
| Other | 13 (3%) | 21 (4%) | 1 (2%) | 167 (6%) | 202 (5%) |
| Region | |||||
| United States | 237 (52%) | 172 (27%) | 26 (43%) | 887 (25%) | 1322 (28%) |
| Non-United States | 220 (48%) | 467 (73%) | 35 (57%) | 2692 (75%) | 3414 (72%) |
| ECOG PS | |||||
| 0 | 259 (57%) | 224 (35%) | 28 (46%) | 1984 (55%) | 2495 (53%) |
| 1 | 187 (41%) | 399 (62%) | 31 (51%) | 1541 (43%) | 2158 (46%) |
| 2 | 5 (1%) | 15 (2.3%) | 3 (3.3%) | 38 (1%) | 60 (1.3%) |
| Unknown | 6 (1.3%) | 1 (0.1%) | 0 | 16 (0.4%) | 23 (0.5%) |
| IMDC risk group | |||||
| Favorable | 66 (14%) | 48 (8%) | 4 (7%) | 528 (15%) | 646 (14%) |
| Intermediate | 221 (48%) | 251 (39%) | 24 (39%) | 1510 (42%) | 2006 (42%) |
| Poor | 81 (18%) | 218 (34%) | 21 (34%) | 823 (23%) | 1143 (24%) |
| Unknown | 89 (20%) | 122 (19%) | 12 (20%) | 718 (20%) | 941 (20%) |
| Baseline metastatic site | |||||
| Lung | 340 (74%) | 490 (77%) | 54 (89%) | 2745 (77%) | 3629 (77%) |
| Bone | 110 (24%) | 277 (43%) | 24 (39%) | 890 (25%) | 1301 (27%) |
| Liver | 121 (26%) | 198 (31%) | 16 (26%) | 903 (25%) | 1238 (26%) |
| Prior nephrectomy | |||||
| Yes | 359 (79%) | 443 (69%) | 41 (67%) | 2482 (69%) | 3325 (70%) |
| No | 84 (18%) | 144 (23%) | 14 (23%) | 965 (27%) | 1207 (26%) |
| Unknown | 14 (3%) | 52 (8%) | 6 (10%) | 132 (4%) | 204 (4%) |
| Prior therapy | |||||
| Any prior therapy | 145 (32%) | 176 (28%) | 22 (36%) | 1230 (34%) | 1573 (33%) |
| Cytokine therapy | 54 (12%) | 63 (10%) | 3 (5%) | 551 (15%) | 671 (14%) |
| VEGF therapya | 49 (11%) | 57 (9%) | 5 (8%) | 462 (13%) | 573 (12%) |
| Diabetes | |||||
| Yes | 124 (27%) | 61 (10%) | 16 (26%) | 4443 (12%) | 644 (14%) |
| No | 333 (73%) | 578 (90%) | 45 (74%) | 3136 (88%) | 4092 (86%) |
| ASI use | |||||
| Yes | 253 (55%) | 165 (26%) | 34 (56%) | 1035 (29%) | 1487 (31%) |
| No | 204 (45%) | 474 (74%) | 27 (44%) | 2544 (71%) | 3249 (69%) |
| Statin use | |||||
| Yes | 185 (40%) | 42 (7%) | 27 (44%) | 257 (7%) | 511 (11%) |
| No | 272 (60%) | 596 (93%) | 34 (56%) | 3322 (93%) | 4225 (89%) |
| Metformin use | |||||
| Yes | 46 (10%) | 23 (4%) | 5 (8%) | 144 (4%) | 218 (5%) |
| No | 411 (90%) | 616 (96%) | 56 (92%) | 3425 (96%) | 4518 (95%) |
Abbreviations: ASA = Aspirin; NSAIDs = Non-Steroidal Anti-Inflammatory Drugs; ECOG = Eastern Cooperative Oncology Group; PS = Performance Status; IMDC = International Metastatic RCC Database Consortium; VEGF = Vascular Endothelial Growth Factor; ASI = Angiotensin System Inhibitor. aVEGF therapy includes bevacizumab and VEGF tyrosine kinase inhibitors.
Treatment exposure
The majority of patients had undergone prior nephrectomy (n = 3325, 70%) and were naïve to systemic therapy (n = 3163, 67%) prior to clinical trial enrollment. Patients were treated with VEGF TT (n = 3511, 74%), mTOR TT (n = 665, 14%), and IFN-α (n = 560, 12%). Dose reductions (n = 1484, 31.3%) and treatment discontinuations (n = 675, 14.3%) due to AEs were similar across the cohorts.
Impact of ASA and non-ASA NSAIDS use on OS and PFS
For the overall cohort, OS was significantly worse in non-ASA NSAIDs users compared to the NSAIDs non-users (HR: 1.47, 95% confidence interval (CI) (1.31–1.65), p < 0.0001, median OS: 11.6 months and 21 months, respectively) (Table 2, Fig. 1). Consistent with OS, PFS was also significantly worse in non-ASA NSAIDs users when compared to non-NSAIDs users (HR: 1.29, (95% CI 1.16–1.44), p < 0.0001, median PFS: 4.6 and 7.4 months, respectively) (Table 2). However, there was no statistically significant difference in the PFS of ASA users and ASA and non-ASA NSAIDs users when compared to the PFS of NSAIDs non-users.
Table 2
Impact of NSAIDs use on OS and PFS
| N | OS | PFS | |||||
| Median (mo) | HRa (95% CI) | P | Median (mo) | HRa (95% CI) | P | ||
| Overall cohort (n = 4736) | |||||||
| ASA users | 457 | 23.6 | 1.00 (0.86–1.18) | 0.9567 | 8.0 | 1.07 (0.93–1.22) | 0.3515 |
| Non-ASA NSAIDs users | 639 | 11.6 | 1.47 (1.31–1.65) | < 0.0001 | 4.6 | 1.29 (1.16–1.44) | < 0.0001 |
| ASA and non-ASA NSAIDs users | 61 | 15.2 | 1.43 (0.99–2.06) | 0.0515 | 6.0 | 1.18 (0.86–1.63) | 0.3137 |
| NSAIDs non-users | 3579 | 21.1 | 7.4 | ||||
| Stratification by type of therapy (n = 4736)b | |||||||
| VEGF-TT (n = 3511) | |||||||
| ASA users | 340 | 26.5 | 1.05 (0.86–1.27) | 0.6572 | 8.6 | 1.06 (0.90–1.24) | 0.4772 |
| Non-ASA NSAIDs users | 369 | 41.1 | 1.58 (1.36–1.84) | < 0.0001 | 6.2 | 1.28 (1.11–1.47) | 0.0006 |
| ASA and non-ASA NSAIDs users | 32 | 19.1 | 1.43 (0.86–2.37) | 0.1672 | 7.0 | 1.34 (0.89–2.03) | 0.1607 |
| NSAIDs non-users | 2770 | 23.9 | 8.5 | ||||
| mTOR-TT (n = 665) | |||||||
| ASA users | 56 | 17.1 | 0.81 (0.53–1.23) | 0.3210 | 5.5 | 0.97 (0.67–1.41) | 0.8730 |
| Non-ASA NSAIDs users | 151 | 7.6 | 1.38 (1.09–1.76) | 0.0080 | 3.8 | 1.29 (1.02–1.63) | 0.0364 |
| ASA and non-ASA NSAIDs users | 13 | 11.5 | 0.92 (0.37–2.26) | 0.8516 | 5.9 | 0.70 (0.32–1.52) | 0.3671 |
| NSAIDs non-users | 445 | 11.6 | 4.2 | ||||
| IFN-α Therapy (n = 560) | |||||||
| ASA users | 61 | 14.9 | 1.07 (0.70–1.63 | 0.7648 | 3.9 | 1.16 (0.78–1.73) | 0.4686 |
| Non-ASA NSAIDs users | 119 | 12.1 | 1.43 (1.04–1.96) | 0.0262 | 3.5 | 1.13 (0.83–1.52) | 0.4433 |
| ASA and non-ASA NSAIDs users | 16 | 10.5 | 1.80 (0.90–3.62) | 0.0993 | 4.3 | 1.10 (0.51–2.33) | 0.8131 |
| NSAIDs non-users | 364 | 17.3 | 3.7 | ||||
| Stratification by line of therapy (n = 4736)b | |||||||
| First-line therapy (n = 3041) | |||||||
| ASA users | 317 | 23.5 | 1.06 (0.87–1.28) | 0.5805 | 8.1 | 1.03 (0.88–1.22) | 0.7015 |
| Non-ASA NSAIDs users | 458 | 11.9 | 1.59 (1.38–1.82) | < 0.0001 | 4.6 | 1.36 (1.19–1.55) | < 0.0001 |
| ASA and non-ASA NSAIDs users | 50 | 17.7 | 1.46 (0.96–2.23) | 0.0788 | 7.1 | 1.11 (0.76–1.61) | 0.5964 |
| NSAIDs non-users | 2216 | 22.8 | 8.3 | ||||
| Second-line therapy (n = 1695) | |||||||
| ASA users | 140 | 22.9 | 0.91 (0.68–1.22) | 0.5395 | 7.4 | 1.15 (0.91–1.45) | 0.2551 |
| Non-ASA NSAIDs users | 181 | 11.0 | 1.28 (1.04–1.58) | 0.0218 | 4.7 | 1.14 (0.94–1.39) | 0.1901 |
| ASA and non-ASA NSAIDs users | 11 | 10.1 | 2.02 (0.98–4.19) | 0.0568 | 4.6 | 2.06 (1.07–3.97) | 0.0302 |
| NSAIDs non-users | 1363 | 17.5 | 6.5 | ||||
Abbreviations: OS = Overall Survival; PFS = Progression Free Survival; mo = months; HR = Hazard Ratio; CI = Confidence Interval; ASA = Aspirin; NSAIDs = Non-Steroidal Anti-Inflammatory Drugs; VEGF = Vascular Endothelial Growth Factor; mTOR = Mechanistic Target of Rapamycin; IFN-α=Interferon alpha. aHR of NSAIDs users to NSAIDs non-users from multivariate analysis, adjusted age, gender, race, ECOG PS, histology, IMDC risk groups, prior nephrectomy, prior therapy, sites of metastasis, baseline hypertension, baseline diabetes, baseline statin use and baseline ASI use. bVEGF therapy users were categorized at patients receiving sunitinib, sorafenib, axitinib, bevacizumab, or bevacizumab and IFN-α. mTOR therapy users were categorized as patients receiving temsirolimus, temsirolimus and IFN-α, or temsirolimus and bevacizumab. P-values are from two-sided log rank test. Bolded p-values are statistically significant.
Fig.1
Kaplan Meier estimates of OS of ASA users, non-ASA NSAIDs users, ASA and non-ASA NSAIDs users and non-NSAIDs non-users in A) the overall cohort, B) patients receiving first therapy, C) patients receiving second line therapy, D) patients receiving VEGF therapy and E) patients receiving mTOR-TT.
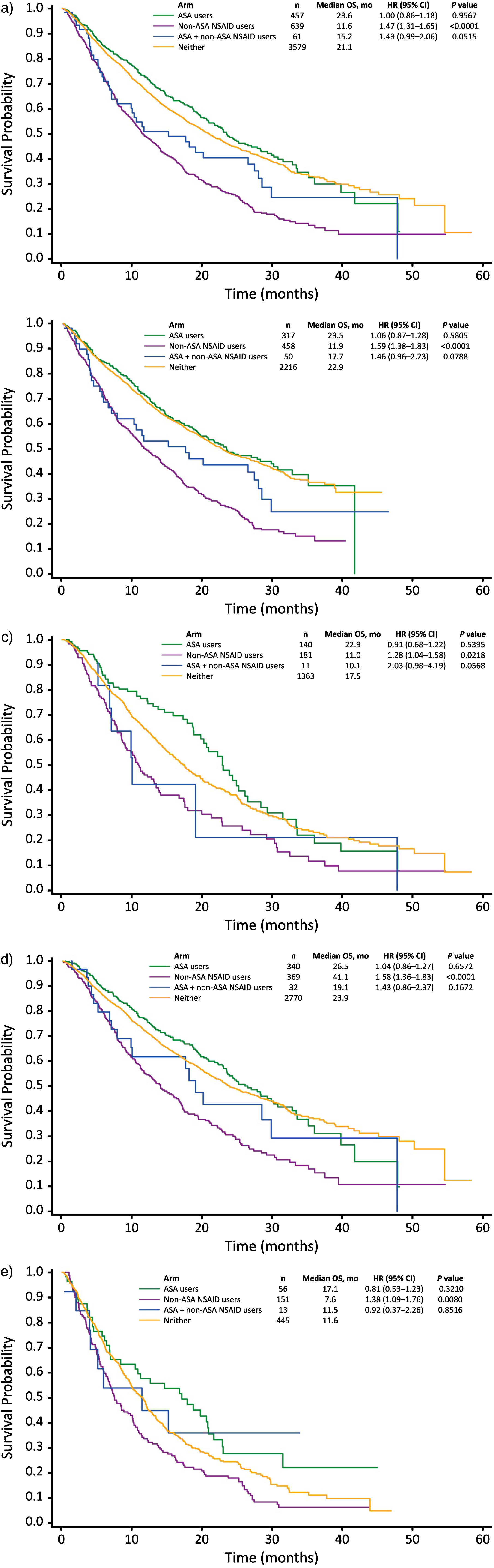
Similarly, OS was significantly shorter in non-ASA NSAIDs users compared to the NSAIDs non-users when stratified by line (first or second line) and type of therapy (VEGF-TT, mTOR-TT and IFN- α) (Table 2). PFS was lower the in non-ASA NSAIDs users compared to the NSAIDs non-users in the first line setting and in all types of therapy.
Impact of ASA and Non-ASA NSAIDS use on ORR
In the total cohort, ORR was 28% in ASA users, 26% in NSAIDs non-users, 20% in ASA and non-ASA NSAIDs users and 16% in non-ASA NSAIDs users. The majority of the patients had stable disease (n = 2403, 50.7%) followed by partial responses (n = 1127, 23.8%). The number of complete responses (CR) were low (n = 33, 0.7%).
Adverse events
Any grade AEs were similar between the cohorts and included fatigue, diarrhea and nausea (Table 3). The most common grade ≥3 AEs were fatigue (n = 735, 16%), hypertension (n = 420, 9%), anemia (n = 363, 8%), hand-foot syndrome (n = 267, 6%), diarrhea (rn = 256, 5%), and dyspnea (n = 193, 4%) (Table 3).
Table 3
Adverse events
| ASA users (N = 457) | Non-ASA NSAIDs users (N = 639) | ASA and non-ASA NSAIDs users (N = 61) | NSAIDs non-users (N = 3579) | Total Cohort (N = 4736) | |
| N (%) | N (%) | N (%) | N (%) | N (%) | |
| Selected adverse events (any grade) | |||||
| Fatigue | 346 (76%) | 428 (70%) | 48 (79%) | 2166 (61%) | 2988 (63%) |
| Diarrhea | 249 (54%) | 235 (37%) | 35 (57%) | 1560 (44%) | 2079 (44%) |
| Nausea | 201 (44%) | 257 (40%) | 27 (44%) | 1118 (31%) | 1603 (34%) |
| Hypertension | 124 (27%) | 132 (21%) | 10 (16%) | 1006 (28%) | 1272 (27%) |
| Anemia | 93 (20%) | 162 (25%) | 17 (28%) | 618 (17%) | 890 (19%) |
| Back pain | 81 (17%) | 115 (18%) | 10 (16%) | 550 (15%) | 756 (16%) |
| Arthralgia | 87 (19%) | 111 (17%) | 18 (30%) | 524 (15%) | 740 (16%) |
| Abdominal pain | 70 (15%) | 88 (14%) | 13 (21%) | 452 (13%) | 623 (13%) |
| Epistaxis | 91 (20%) | 70 (11%) | 10 (17%) | 449 (13%) | 620 (13%) |
| Dyspepsia | 68 (15%) | 67 (10%) | 12 (20%) | 395 (11%) | 542 (11%) |
| Thrombocytopenia | 46 (10%) | 63 (10%) | 10 (16%) | 329 (9%) | 448 (9%) |
| Renal Failure | 7 (1%) | 13 (2%) | 2 (3%) | 45 (1%) | 67 (1%) |
| Most frequent grade 3–5 adverse events | |||||
| (observed in >3% of patients) | |||||
| Fatigue | 83 (18%) | 111 (18%) | 16 (26%) | 525 (15%) | 735 (16%) |
| Hypertension | 49 (11%) | 53 (8%) | 3 (5%) | 315 (9%) | 420 (9%) |
| Anemia | 28 (6%) | 81 (13%) | 7 (12%) | 247 (7%) | 367 (8%) |
| Hand-foot syndrome | 30 (7%) | 25 (4%) | 2 (3%) | 210 (6%) | 267 (6%) |
| Diarrhea | 32 (7%) | 28 (4%) | 4 (7%) | 192 (5%) | 256 (5%) |
| Dyspnea | 20 (4%) | 36 (6%) | 8 (13%) | 129 (4%) | 193 (4%) |
| Neutropenia | 15 (3%) | 24 (4%) | 0 | 147 (4%) | |
| Proteinuria | 15 (3%) | 17 (3%) | 1 (2%) | 126 (4%) | |
| Decreased appetite | 12 (3%) | 35 (5%) | 3 (5%) | 91 (3%) |
DISCUSSION
Our study is the largest study to date evaluating the impact of ASA and non-ASA NSAIDS on survival outcomes in mRCC patients. It utilizes a large clinical trial database of 4,736 mRCC patients treated with a wide range of systemic treatments in the era of targeted therapy. Our data show that the use of non-ASA NSAIDs in mRCC patients is associated with shorter OS and PFS compared to the non-use of NSAIDs. We also demonstrate that there is no difference in the survival outcomes (OS and PFS) in ASA users or ASA and non-ASA NSAIDs users compared to NSAIDs non-users.
In the era of drug repurposing, ASA and non-ASA NSAIDs have been extensively investigated in cancer. However, these studies are limited in RCC, and no study has investigated the impact of these agents in the metastatic setting. In RCC, studies have reported on the association of ASA and non-ASA NSAIDs and the incidence risk [24–33]. A meta-analysis of 20 observational studies including 8,420 kidney cancer cases found that non-ASA NSAIDs use was associated with a higher incidence of RCC [25]. The association was stronger when non-ASA NSAIDs were used at higher doses and for longer periods of time. However, ASA was not associated with an increased risk of kidney cancer, although the analysis was limited by study heterogeneity. Similarly, Cho and colleagues examined the impact of NSAIDs on RCC risk in 126,568 subjects prospectively followed for 16–20 years as part of the Nurses’ Health Study and the Health Professionals Follow-up Study, longer use of non-ASA NSAIDs was associated with an increased risk of RCC while ASA was not [24]. A large prospective cohort study of 298,468 men and women evaluated the impact of NSAID in RCC and demonstrated an association between NSAID use and RCC risk only when stratified by age [29].
The anti-tumorigenic mechanism of NSAIDs has been largely attributed to their COX inhibitory activity, although COX-independent mechanisms have been suggested as well. They inhibit tumor growth by inducing apoptosis of tumor cells and inhibiting the Wnt/β-catenin signaling pathway [34]. In RCC, COX-2 expression is present in the majority of the tumors and correlates with worse stage, grade, and microvessel density and poorer survival [35–38].
COX-2 inhibition has been investigated in pre-clinical studies and subsequently in clinical trials due to its promising effects in vivo. In a phase II trial of celecoxib (Selective COX-2 inhibitor) and IFN-α in 25 mRCC patients, the addition of celecoxib did not increase the response rate and the time to progression compared to IFN-α alone [39].
The doses used for management of pain and inflammation may be suboptimal for achieving tumor growth and COX-inhibition, given that doses required for tumor growth inhibition are higher than those need for COX-inhibition [34]. Although the combination of COX-inhibitors and VEGF-TKIs has not been investigated in clinical trials, the combination was found to inhibit tumor growth in preclinical models where COX-2 expression was associated with hypoxia and sunitinib resistance [40]. In our study, NSAIDs did not confer a survival advantage in mRCC, which could be explained by their kidney-specific toxicity profile and their potency.
Unfortunately, the four groups were not balanced in terms of baseline and disease characteristics. However, we adjusted for known prognostic variables in our multivariate analysis including performance status, IMDC risk group, presence of bone and liver metastases, and ASI and statin use. In particular, non-ASA NSAIDs users had a higher percentage of patients with poor prognostic features including IMDC poor risk disease, presence of bone and liver metastases, and lower percentage of ASI and statin use. Given that non-ASA NSAIDs are a form of analgesic, this could reflect that patients using non-ASA NSAIDs have more symptomatic disease compared to patients using ASA for preventative reasons. Additionally, NSAIDs use is associated with an increase in blood pressure, decrease in renal function and risk of chronic renal failure. This is particularly worrisome in RCC patients who have underwent nephrectomy and are more susceptible to the nephrotoxic effects of NSAIDs. In our study, we did not adjust for baseline creatinine, as a surrogate for kidney function, however, non-ASA NSAID users did not have a higher rate of hypertension or proteinuria when compared to the NSAIDs non-users. They also had similar rates of dose reductions and treatment discontinuations due to AEs. Additionally, Charlson comorbidity scores were not available.
Since the database was not specifically designed for the purpose of this study, duration, dosage or constituents of the drugs used were not captured. Treatment with these agents was at the discretion of the treating physician, including indications, dosages and durations of use. It is also worth mentioning that non-ASA NSAIDs users and NSAIDs non-users were mostly treated outside the US. In our previous meta-analysis of the impact of the NSAIDs on the risk of kidney cancer, ASA was associated with a higher incidence risk in non-US countries but not in the US [25]. Additionally, the number of patients using both ASA and non-ASA NSAIDs was small (n = 61). Hence, these results should be interpreted cautiously as hypothesis-generating rather than definitive and they need to be validated in larger prospective studies. Finally, our patient population may not be representative of the “real-world” population as all patients were enrolled on clinical trials. Though a multiple targeted therapies were included in this analysis, we did not investigate the association with newer agents such as cabozantinib or immunotherapy.
In conclusion, NSAIDs do not confer a survival advantage in mRCC patients, with non-ASA NSAIDs being associated with worse survival outcomes when compared to NSAIDs non-users. Additional studies are warranted to investigate the association of NSAIDS with new agents such as cabozantinib and nivolumab. Additionally, our data are thought-provoking and warrant validation. Pre-clinical studies investigating the interaction of NSAIDS with targeted therapy and immunotherapy in RCC are warranted to corroborate our results and highlight the mechanism of action underlying our observations.
CONFLICT OF INTEREST
Rana R. McKay has received institutional research funding from Pfizer and Bayer. Toni K. Choueiri has received institutional research funding from Pfizer and has an advisory role at Pfizer, Novartis, GlaxoSmithKline, Genentech, Merck and Bayer and Onyx. Xun Lin is an employee at Pfizer. Ronit Simantov was a former employee at Pfizer. The remaining authors have no disclosures.
ACKNOWLEDGMENTS
This work was supported by Pfizer, Inc. Additionally, this research was supported in part by the Dana-Farber/Harvard Cancer Center Kidney SPORE, and the Trust Family, Michael Brigham, and Loker Pinard Funds for Kidney Cancer Research at Dana-Farber Cancer Institute for Toni K. Choueiri.
REFERENCES
[1] | Kaufman DW , Kelly JP , Rosenberg L , Anderson TE , Mitchell AA . Recent patterns of medication use in the ambulatory adult population of the United States: The Slone survey. JAMA. (2002) ;287: (3):337–44. |
[2] | Berg J , Christoph T , Widerna M , Bodenteich A . Isoenzyme-specific cyclooxygenase inhibitors: A whole cell assay system using the human erythroleukemic cell line HEL and the human monocytic cell line Mono Mac 6. J Pharmacol Toxicol Methods. (1997) ;37: (4):179–86. |
[3] | Bibbins-Domingo K , Force USPST. Aspirin use for the primary prevention of cardiovascular disease and colorectal cancer: U.S. preventive services task force recommendation statement. Ann Intern Med. (2016) ;164: (12):836–45. |
[4] | Giardiello FM , Hamilton SR , Krush AJ , Piantadosi S , Hylind LM , Celano P , et al. Treatment of colonic and rectal adenomas with sulindac in familial adenomatous polyposis. N Engl J Med. (1993) ;328: (18):1313–6. |
[5] | Muscat JE , Chen SQ , Richie JP Jr , Altorki NK , Citron M , Olson S , et al. Risk of lung carcinoma among users of nonsteroidal antiinflammatory drugs. Cancer. (2003) ;97: (7):1732–6. |
[6] | Rothwell PM , Wilson M , Elwin CE , Norrving B , Algra A , Warlow CP , et al. Long-term effect of aspirin on colorectal cancer incidence and mortality: 20-year follow-up of five randomised trials. Lancet. (2010) ;376: (9754):1741–50. |
[7] | Rothwell PM , Wilson M , Price JF , Belch JF , Meade TW , Mehta Z . Effect of daily aspirin on risk of cancer metastasis: A study of incident cancers during randomised controlled trials. Lancet. (2012) ;379: (9826):1591–601. |
[8] | Ruder EH , Laiyemo AO , Graubard BI , Hollenbeck AR , Schatzkin A , Cross AJ . Non-steroidal anti-inflammatory drugs and colorectal cancer risk in a large, prospective cohort. Am J Gastroenterol. (2011) ;106: (7):1340–50. |
[9] | Takkouche B , Regueira-Mendez C , Etminan M . Breast cancer and use of nonsteroidal anti-inflammatory drugs: A meta-analysis. J Natl Cancer Inst. (2008) ;100: (20):1439–47. |
[10] | Vidal AC , Howard LE , Moreira DM , Castro-Santamaria R , Andriole GL , Freedland SJ . Aspirin, NSAIDs, and risk of prostate cancer: Results from the REDUCE study. Clin Cancer Res. (2015) ;21: (4):756–62. |
[11] | Elder DJ , Halton DE , Hague A , Paraskeva C . Induction of apoptotic cell death in human colorectal carcinoma cell lines by a cyclooxygenase-2 (COX-2)-selective nonsteroidal anti-inflammatory drug: Independence from COX-2 protein expression. Clin Cancer Res. (1997) ;3: (10):1679–83. |
[12] | Janssen A , Maier TJ , Schiffmann S , Coste O , Seegel M , Geisslinger G , et al. Evidence of COX-2 independent induction of apoptosis and cell cycle block in human colon carcinoma cells after S- or R-ibuprofen treatment. Eur J Pharmacol. (2006) ;540: (1-3):24–33. |
[13] | Piazza GA , Rahm AK , Finn TS , Fryer BH , Li H , Stoumen AL , et al. Apoptosis primarily accounts for the growth-inhibitory properties of sulindac metabolites and involves a mechanism that is independent of cyclooxygenase inhibition, cell cycle arrest, and p53 induction. Cancer Res. (1997) ;57: (12):2452–9. |
[14] | Vogt T , McClelland M , Jung B , Popova S , Bogenrieder T , Becker B , et al. Progression and NSAID-induced apoptosis in malignant melanomas are independent of cyclooxygenase II. Melanoma Res. (2001) ;11: (6):587–99. |
[15] | Jacoby RF , Seibert K , Cole CE , Kelloff G , Lubet RA . The cyclooxygenase-2 inhibitor celecoxib is a potent preventive and therapeutic agent in the min mouse model of adenomatous polyposis. Cancer Res. (2000) ;60: (18):5040–4. |
[16] | Kawamori T , Rao CV , Seibert K , Reddy BS . Chemopreventive activity of celecoxib, a specific cyclooxygenase-2 inhibitor, against colon carcinogenesis. Cancer Res. (1998) ;58: (3):409–12. |
[17] | Narisawa T , Sato M , Tani M , Kudo T , Takahashi T , Goto A . Inhibition of development of methylnitrosourea-induced rat colon tumors by indomethacin treatment. Cancer Res. (1981) ;41: (5):1954–7. |
[18] | Rao CV , Rivenson A , Simi B , Zang E , Kelloff G , Steele V , et al. Chemoprevention of colon carcinogenesis by sulindac, a nonsteroidal anti-inflammatory agent. Cancer Res. (1995) ;55: (7):1464–72. |
[19] | Reddy BS , Hirose Y , Lubet R , Steele V , Kelloff G , Paulson S , et al. Chemoprevention of colon cancer by specific cyclooxygenase-2 inhibitor, celecoxib, administered during different stages of carcinogenesis. Cancer Res. (2000) ;60: (2):293–7. |
[20] | Reddy BS , Maruyama H , Kelloff G . Dose-related inhibition of colon carcinogenesis by dietary piroxicam, a nonsteroidal antiinflammatory drug, during different stages of rat colon tumor development. Cancer Res. (1987) ;47: (20):5340–6. |
[21] | Steele VE , Rao CV , Zhang Y , Patlolla J , Boring D , Kopelovich L , et al. Chemopreventive efficacy of naproxen and nitric oxide-naproxen in rodent models of colon, urinary bladder, and mammary cancers. Cancer Prev Res (Phila). (2009) ;2: (11):951–6. |
[22] | Takahashi M , Fukutake M , Yokota S , Ishida K , Wakabayashi K , Sugimura T . Suppression of azoxymethane-induced aberrant crypt foci in rat colon by nimesulide, a selective inhibitor of cyclooxygenase 2. J Cancer Res Clin Oncol. (1996) ;122: (4):219–22. |
[23] | Steinbach G , Lynch PM , Phillips RK , Wallace MH , Hawk E , Gordon GB , et al. The effect of celecoxib, a cyclooxygenase-2 inhibitor, in familial adenomatous polyposis. N Engl J Med. (2000) ;342: (26):1946–52. |
[24] | Cho E , Curhan G , Hankinson SE , Kantoff P , Atkins MB , Stampfer M , et al. Prospective evaluation of analgesic use and risk of renal cell cancer. Arch Intern Med. (2011) ;171: (16):1487–93. |
[25] | Choueiri TK , Je Y , Cho E . Analgesic use and the risk of kidney cancer: A meta-analysis of epidemiologic studies. Int J Cancer. (2014) ;134: (2):384–96. |
[26] | Chow WH , McLaughlin JK , Linet MS , Niwa S , Mandel JS . Use of analgesics and risk of renal cell cancer. Int J Cancer. (1994) ;59: (4):467–70. |
[27] | Friis S , Sorensen HT , McLaughlin JK , Johnsen SP , Blot WJ , Olsen JH . A population-based cohort study of the risk of colorectal and other cancers among users of low-dose aspirin. Br J Cancer. (2003) ;88: (5):684–8. |
[28] | Gago-Dominguez M , Yuan JM , Castelao JE , Ross RK , Yu MC . Regular use of analgesics is a risk factor for renal cell carcinoma. Br J Cancer. (1999) ;81: (3):542–8. |
[29] | Jacobs EJ , Thun MJ , Bain EB , Rodriguez C , Henley SJ , Calle EE . A large cohort study of long-term daily use of adult-strength aspirin and cancer incidence. J Natl Cancer Inst. (2007) ;99: (8):608–15. |
[30] | Liu W , Park Y , Purdue MP , Giovannucci E , Cho E . A large cohort study of nonsteroidal anti-inflammatory drugs and renal cell carcinoma incidence in the National Institutes of Health-AARP Diet and Health Study. Cancer Causes Control. (2013) ;24: (10):1865–73. |
[31] | McCredie M , Pommer W , McLaughlin JK , Stewart JH , Lindblad P , Mandel JS , et al. International renal-cell cancer study. II. Analgesics. Int J Cancer. (1995) ;60: (3):345–9. |
[32] | McLaughlin JK , Blot WJ , Mehl ES , Fraumeni JF Jr . Relation of analgesic use to renal cancer: Population-based findings. Natl Cancer Inst Monogr. (1985) ;69: :217–22. |
[33] | Paganini-Hill A , Chao A , Ross RK , Henderson BE . Aspirin use and chronic diseases: A cohort study of the elderly. BMJ. (1989) ;299: (6710):1247–50. |
[34] | Gurpinar E , Grizzle WE , Piazza GA . NSAIDs inhibit tumorigenesis, but how? Clin Cancer Res. (2014) ;20: (5):1104–13. |
[35] | Li JF , Chu YW , Wang GM , Zhu TY , Rong RM , Hou J , et al. The prognostic value of peritumoral regulatory T cells and its correlation with intratumoral cyclooxygenase-2 expression in clear cell renal cell carcinoma. BJU Int. (2009) ;103: (3):399–405. |
[36] | Miyata Y , Koga S , Takehara K , Kanetake H , Kanda S . Expression of thrombospondin-derived 4N1K peptide-containing proteins in renal cell carcinoma tissues is associated with a decrease in tumor growth and angiogenesis. Clin Cancer Res. (2003) ;9: (5):1734–40. |
[37] | Sozen S , Gurocak S , Erdem O , Acar C , Kordan Y , Akyol G , et al. Cyclooxygenase-2 expression: Does it have a probable role in tumorigenesis mechanisms of renal cell carcinoma? Int Urol Nephrol. (2008) ;40: (2):295–301. |
[38] | Tuna B , Yorukoglu K , Gurel D , Mungan U , Kirkali Z . Significance of COX-2 expression in human renal cell carcinoma. Urology. (2004) ;64: (6):1116–20. |
[39] | Rini BI , Weinberg V , Dunlap S , Elchinoff A , Yu N , Bok R , et al. Maximal COX-2 immunostaining and clinical response to celecoxib and interferon alpha therapy in metastatic renal cell carcinoma. Cancer. (2006) ;106: (3):566–75. |
[40] | Wang X , Zhang L , O’Neill A , Bahamon B , Alsop DC , Mier JW , et al. Cox-2 inhibition enhances the activity of sunitinib in human renal cell carcinoma xenografts. Br J Cancer. (2013) ;108: (2):319–26. |


