
Parallel (Randomized) Phase II Evaluation of Tivantinib (ARQ197) and Tivantinib in Combination with Erlotinib in Papillary Renal Cell Carcinoma: SWOG S1107
Abstract
Background:
Papillary renal cell carcinoma (pRCC) is associated with EGFR expression and activation of MET signaling pathway. A randomized multicenter parallel two-stage phase II trial of MET inhibitor tivantinib alone or in combination with EGFR inhibitor erlotinib was conducted in patients with pRCC.
Methods:
Patients with advanced pRCC and 0-1 prior systemic therapy were randomly assigned to tivantinib 360 mg BID (Arm 1) or tivantinib 360 mg BID plus erlotinib 150 mg daily (Arm 2). Target max accrual was 70 patients (35 per arm) with interim analysis planned after enrollment of 20 patients per arm.
Results:
Six % of patients had type 1 pRCC, 42% had type 2, and 52% had no subtype assigned. The study was closed after the first stage when both arms yielded RR of 0%. Median progression free survival (PFS) was 2.0 and 3.9 months, and OS was 10.3 and 11.3 months in Arms 1 and 2 respectively. Treatment was well tolerated. Exome of tumor tissue from 16 patients were successfully sequenced using Agilent SureSelect probes. Only 1 of 16 samples harbored MET mutation. Other mutations associated primarily with type 2 pRCC were noted and included CDKN2A, PBRM1, SETD2, KDM6A, FAT1 and NF2.
Conclusions:
Tivantinib - either alone or in combination with erlotinib has no clinical activity in patients with advanced pRCC. The S1107 cohort had a low proportion of patients with MET alterations. MET remains a reasonable therapeutic target in pRCC, but selection of patient subsets exhibiting MET activation may be required to better benefit from therapy with MET inhibitors.
INTRODUCTION
The remarkable progress in the management of advanced kidney cancer over the last decade focused primarily on the most common histologic type - clear cell carcinoma (ccRCC) [1]. Less prevalent histologic subtypes have been traditionally categorized as “non-clear cell RCC” (nccRCC) and have been under-represented in drug development efforts. The most common of the nccRCC is papillary renal cell carcinoma (pRCC) accounting for approximately 15% of all kidney cancer cases [2]. Morphologically pRCC is divided into 2 subsets. Type 1 tumors exhibit small cells with scanty pale cytoplasm arranged in a single layer on the basement membrane of papillary cores. They frequently express cytokeratin 7. In contrast, type 2 tumors have cells that are larger with pseudostratified nuclei and usually display voluminous eosinophilic cytoplasm [3]. The analysis of data from available clinical trials suggests that the benefit of currently available RCC agents targeting vascular endothelial growth factor (VEGF) and mammalian target of rapamycin (MTOR) pathways although present in pRCC, is inferior to that observed in clear cell histology perhaps reflecting different underlying pathophysiology and lesser dependency on VEGF pathway signaling [3].
The biological significance of MET pathway in pRCC and other tumor types has been well recognized. The c-MET (MET) receptor tyrosine kinase (RTK) and its ligand hepatocyte growth factor (HGF) play important role in the processes of cancer cell migration, proliferation, metastasis and apoptosis [4]. Enhanced HGF/MET signaling has been observed in variety of tumor types including pRCC, ccRCC, bladder, prostate, lung and hepatocellular carcinoma [5]. Activating germline mutations in the MET tyrosine kinase (TK) domain are present in the majority of patients with hereditary pRCC. More common sporadic pRCCs exhibit activating MET mutation only in about 10–20% of cases but other mechanisms of MET activation including amplification of the MET gene locus resulting in MET overexpression are common [6].
Even though other molecular characteristics of pRCC are quite diverse, the recognition that MET is an important component of the pathophysiology of pRCC, both in hereditary and sporadic cases, provides a potential target for novel therapeutic agents. In preclinical models, MET inhibitors showed activity against several hereditary papillary renal cell carcinoma-related mutations and tumor xenografts [7]. The Cancer Genome Atlas (TCGA) molecular characterization of pRCC revealed significant heterogeneity of this entity with type 1 pRCC exhibiting higher rate of MET alterations as compared to type 2 pRCC [8]. In emerging clinical experience (Supplementary Table S1), a phase II study of the dual MET and VEGF receptor 2 (VEGFR2) inhibitor foretinib (XL880) in patients with pRCC revealed modest activity including response rate (RR) of 13.5% and a median progression free survival (PFS) of 9.3 months. Interestingly the presence of germline MET mutation (noted in 7% of patients) was associated with a 50% response rate. Multiple other measures of somatic MET pathway activation were noted, including gain of chromosome 7 (27%), somatic MET mutations (7%), and MET amplification (3%) [9]. Selective MET inhibitor savolitinib (AZD6094) demonstrated modest activity in a biomarker-based trial in pts with pRCC but the benefit seemed to be restricted to approximately 50% of pts who exhibited some form of MET aberration, in contrast the RR in pts without MET abnormalities was 0% and PFS was 1.4 months [10]. The epidermal growth factor receptor (EGFR) pathway has also been explored as a potential therapeutic target in pRCC. Preclinical RCC data had revealed that absence of von Hippel-Lindau (VHL) mutations (as typically observed in pRCC) is associated with greater activity of EGFR inhibitors against RCC [11]. EGFR TK inhibitor erlotinib was evaluated by the SWOG in a phase II trial of patients with metastatic pRCC [12]. Overall RR was 10% and median overall survival was reported to be an encouraging 26.9 months. The results of this study demonstrated clinical activity of erlotinib in patients with pRCC and provided support for further evaluation of erlotinib either alone or in combination with therapies targeting other relevant pathways (such as MET).
To support the concept of dual EGFR and MET inhibition in pRCC, our group tested the combination of erlotinib and tivantinib (ARQ-197) which respectively inhibit EGFR and MET in the ACHN renal papillary cancer cell line [3]. The combined treatment out-performed either individual agent, and was additive to synergistic at the effective doses tested.
Tivantinib (formerly ARQ 197; ArQule, Woburn, MA; Daiichi Sankyo, Tokyo, Japan) was initially developed as a non–adenosine triphosphate–competitive small molecule MET inhibitor first identified as a proapoptotic agent in a cell-based screen and subsequently found to have selectivity for MET [13]. The drug was reported to stabilize the inactive conformation of MET, disrupting downstream signaling, and demonstrated antiproliferative activity in multiple cancer models. Tivantinib has been studied in phase I and II trials as monotherapy and in Phase I, II and III trials in combination with erlotinib; it demonstrated a relatively mild toxicity profile with dose-limiting toxicity of neutropenia, and ability to be combined with erlotinib at full doses [14–16]. Based on the clinical data demonstrating activity of erlotinib in pRCC, the importance of MET signaling and preclinical studies indicating activity of MET inhibitors alone and in combination with EGFR inhibitor in pRCC SWOG conducted a parallel randomized phase 2 study (S1107) evaluating tivantinib (ARQ-197) alone and in combination with erlotinib in pRCC.
METHODS
Patients
Patients were recruited from institutions in the United States. Eligible patients had histologically or cytologically confirmed metastatic or locally advanced pRCC based on local pathology review. No central pathology review was performed. Patients may have received up to one prior systemic therapy for metastatic pRCC. Patients were required to have radiographically measurable disease per Response Evaluation Criteria in Solid Tumors (RECIST) 1.0 guidelines [17]. Patients with history of brain metastases who were asymptomatic and had not received steroid therapy in the 14 days prior to registration were eligible.
Adequate hematologic and hepatic function and Zubrod performance status of 0–2 were required. Patients with a known history of specific corneal diseases, or known to be HIV-positive and receiving combination anti-retroviral therapy, were not eligible. Tumor blocks or unstained archival tumor tissue slides for molecular analysis were collected with patient consent.
Study Design and Treatment
In this open-label randomized parallel two-stage phase II study, patients were assigned to receive oral tivantinib (360 mg twice daily) or tivantinib (360 mg twice daily) plus erlotinib 150 mg daily. Treatment was administered continuously, although divided into 28-day cycles, until progressive disease or unacceptable toxicity. Patient randomization was stratified based on prior systemic therapy for metastatic disease: none versus one.
End Points and Statistical Considerations
The primary end point of the trial was to assess the RECIST response rate (RR) (confirmed complete and partial response) of patients with locally advanced or metastatic papillary renal cell carcinoma treated with either tivantinib or tivantinib combined with erlotinib. The secondary end points included the assessment of the progression free survival (PFS), overall survival (OS) and safety and tolerability of tivantinib alone and in combination with erlotinib. Tissue specimens were evaluated for the expression of biomarkers such as c-MET and EGFR and correlated with clinical outcomes. Tumor status was assessed locally with computed tomography (CT) or magnetic resonance imaging (MRI) every 8 weeks (2 cycles).
The study employed a two-stage design. A RR of≥30% was to be considered a promising outcome. Target accrual was 35 per arm with first stage response assessment after the first 20 patients. If 0–1 responses per arm were noted this would constitute lack of evidence of sufficient activity, and that arm would close to accrual. If the arm showed adequate activity in the first stage it would be expanded until a total of 35 eligible patients per arm were accrued. If the total number of patients out of 35 who respond was 8 + that would be consider worthy of further investigation. The design for each arm had a significance level (probability of falsely declaring that an agent with 10% RR is worthy of further investigation) of 2% and a power (probability of correctly declaring that an agent with RR of 30% is worthy of further investigation) of 87%.
Adverse events were classified using the National Cancer Institute’s Common Terminology Criteria for Adverse Events (version 4.0) on day 1 of each cycle.
Molecular Analyses
The QIAamp DNA FFPE Tissue kit (Qiagen, Germantown, MD; Cat. 56404) was used to extract DNA from FFPE tissues. The protocol included an overnight lysis at 56°C and incubation at 90°C for 10 minutes. We used a shorter incubation time based on the tissue size to avoid extensive fragmentation of DNA. To increase the DNA yield, the elution of DNA was performed twice from the same column with 15ul of EB buffer each time. The quality of DNA was evaluated with Nano drop for purity, Qubit for dsDNA concentration and QPCR for estimation of amplifiable DNA content.
Illumina sequencing library preparation and sequencing on Hiseq2500
250 ng of QC passed FFPE genomic DNA was fragmented by using Covaris S220 (Covaris, Woburn, MA) with the 200bp peak setting. The fragmented DNA was end-repaired and ligated to Illumina adaptor oligonucleotides with Kapa Hyper Prep Kit (KAPA Biosystems, Wilmington, MA; Cat.KK8504). Ligation products were purified and amplified with a 7 cycle of PCR. The enriched PCR products were subject to the exome capture procedure using the SureSelecXT Human All Exon V6 + COMSIC kit (Agilent, Technologies, Santa Clara, CA; Cat 5190-9307) according to manufacturer’s protocols. The captured products were further amplified with an 8-cycle of PCR and the purified products were used for cluster generation by using cBot cluster generation system with HiSeq PE Cluster Kit V4 (Illumina, San Diego, CA; Cat PE-401-4001). Sequencing run was performed in the paired end mode of 101 cycles of read1, 7 cycles of index read and 101 cycles of read2 using HiSeq2500 platform with HiSeq SBS Kit V4 (Illumina, San Diego, CA; Cat.FC-401-4003). Real-time analysis (RTA) V1.18.66 software was used to process the image analysis and base calling.
Sequencing Data Analysis
Sequencing data were aligned to hg19 genome using Novoalign with default settings. Only reads aligned to unique genomic location was retained. The single point mutations and small indels were identified using GATK HaplotypeCaller, with the criteria of 1) minimum depth of coverage > = 10; 2) minimum reads supporting variants > = 3; and 3) minimum variant frequency > = 15%. The exonic variants that are nonsynonymous, indels (frameshift and nonframeshit), stopgain and stoploss are deemed functional. The variants are further filtered to retain the ones that are most likely relevant to tumor based on the following criteria: 1) variant frequency <2% or not present in esp6500 database; 2) variant frequency <2% or not present in 1000 genome project; 3) not present in dbSNP v138; 4) not present in EXAC database or frequency <2%. Germ line copy number variants (CNV) were identified using CoNifer, and virtual somatic copy number variants were identified using custom R scripts and Bioconductor package “DNAcopy”. Pathway analysis was performed using “ReactomeFIVIZ” in Cytoscape. Genes with mutations in at least 2 samples were uploaded to ReactomeFIVIZ to generate the network based on their interactions defined in ReactomeFI database. The pathways enrichment analysis was performed and the ones with FDR less than 0.05 were considered significantly enriched in the network. FDR is adjusted p value, which represents the level of enrichment of each pathway in the uploaded gene list compared to the genome background.
Study Oversight
The clinical study was designed by the Principal Investigator with input from co-investigators. Data collection occurred in a monitored fashion and was maintained by the SWOG Statistical and Data Management Center (SDMC). The data analysis was performed at the SWOG SDMC. Local Institutional Review Boards approved the study at all participating sites. Signed informed consent was obtained from all patients enrolled onto the study. The study was registered at clinicaltrials.gov before any patient entry (NCT01688973).
RESULTS
Patients
Patients were accrued by participating SWOG and Eastern Cooperative Group (ECOG) institutions. The study was activated on 8/20/2012 and permanently closed on 5/29/2014. Data for this report are based on a database freeze as of June 20, 2017. Robust accrual resulted in 55 patients registered by the time of interim closure. Fifty patients were eligible (25 in each arm). The study population had a median age of 63 years and was primarily made of white (76%) males (68%) who did not receive prior therapy for metastatic disease (66%). 78% of patients underwent prior nephrectomy. Histologic subset (type 1 or 2) of pRCC was not determined in majority of patients (52%) (Table 1).
Table 1
Patient Characteristics of Eligible Randomized Patients, By Arm
| ARQ 197 | ARQ 197 + Erlotinib | |||
| (n = 25) | (n = 25) | |||
| AGE | ||||
| Median | 62.1 | 63.6 | ||
| Minimum | 20.3 | 22.8 | ||
| Maximum | 76.1 | 81.9 | ||
| SEX | ||||
| Males | 19 | 76% | 15 | 60% |
| Females | 6 | 24% | 10 | 40% |
| HISPANIC | ||||
| Yes | 1 | 4% | 1 | 4% |
| No | 23 | 92% | 21 | 84% |
| Unknown | 1 | 4% | 3 | 12% |
| RACE | ||||
| White | 19 | 76% | 19 | 76% |
| Black | 6 | 24% | 5 | 20% |
| Unknown | 0 | 0% | 1 | 4% |
| HISTOLOGIC GRADE | ||||
| Unknown | 11 | 44% | 11 | 44% |
| 1 | 0 | 0% | 0 | 0% |
| 2 | 3 | 12% | 5 | 20% |
| 3 | 7 | 28% | 6 | 24% |
| 4 | 4 | 16% | 3 | 12% |
| HISTOLOGIC SUBSET | ||||
| Pure papillary | 20 | 80% | 23 | 92% |
| Mixed histology | 5 | 20% | 2 | 8% |
| HISTOLOGIC TYPE | ||||
| Not Assigned | 12 | 48% | 14 | 56% |
| Type 1 | 2 | 8% | 1 | 4% |
| Type 2 | 11 | 44% | 10 | 40% |
| PRIOR NEPHRECTOMY | ||||
| No | 4 | 16% | 7 | 28% |
| Yes | 21 | 84% | 18 | 72% |
| PRIOR SYSTEMIC THERAPY | ||||
| None | 16 | 64% | 17 | 68% |
| One | 9 | 36% | 8 | 32% |
| PERFORMANCE STATUS | ||||
| 0 | 12 | 48% | 9 | 36% |
| 1 | 11 | 44% | 13 | 52% |
| 2 | 2 | 8% | 3 | 12% |
RR, PFS and OS
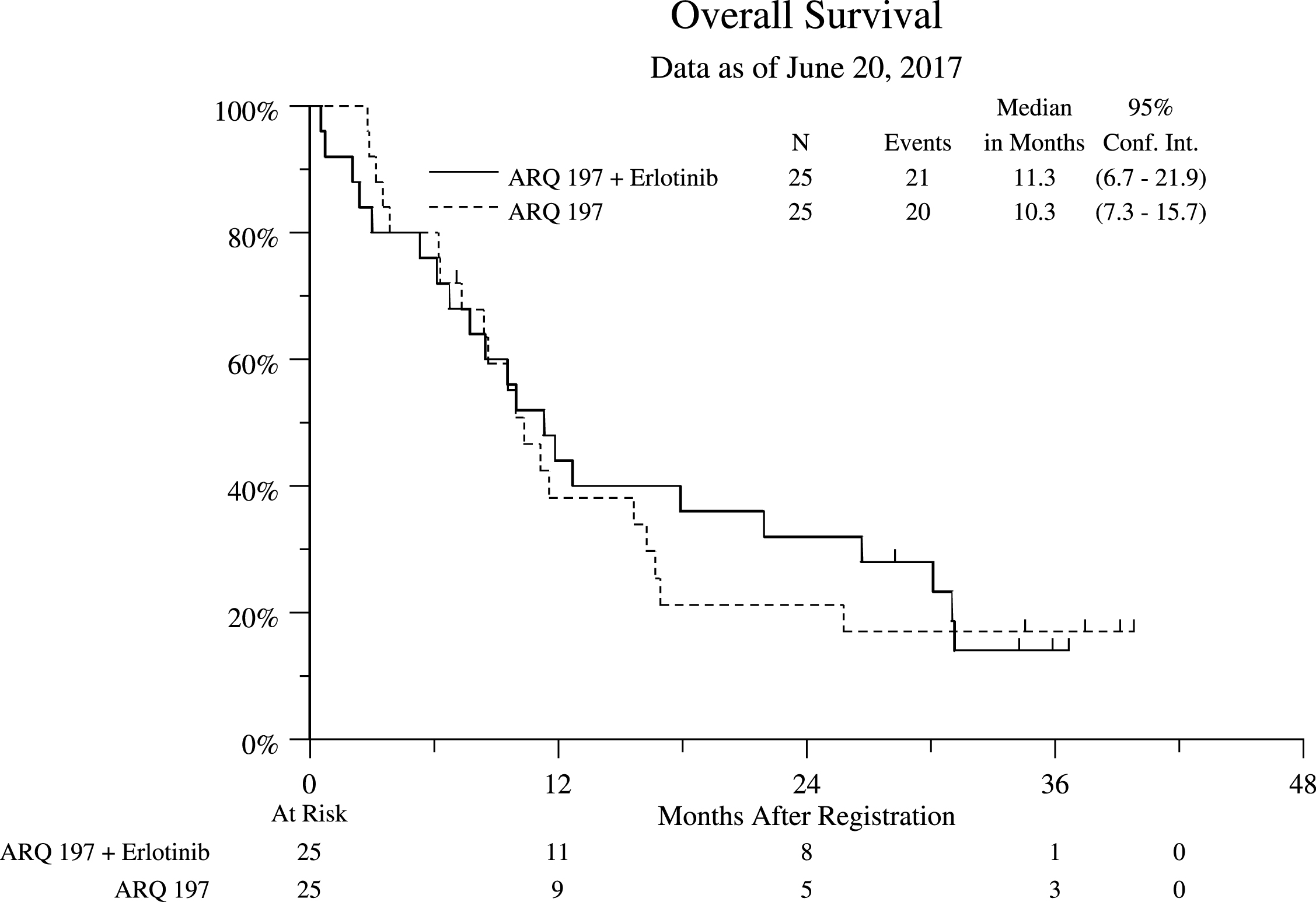
Both arms yielded RR of 0%. Median progression free survival (PFS) was 2.0 (95% CI 1.8, 3.0) and 3.9 (95% CI 1.8, 7.3) months, and OS was 10.3 (95% CI 7.3, 15.7) and 11.3 (95% CI 6.7, 21.9) months in Arms 1 and 2 respectively (Figs. 1 and 2).
Fig.1
Progression Free Survival (PFS) Stratified by Treatment Arm for All Eligible Patients.
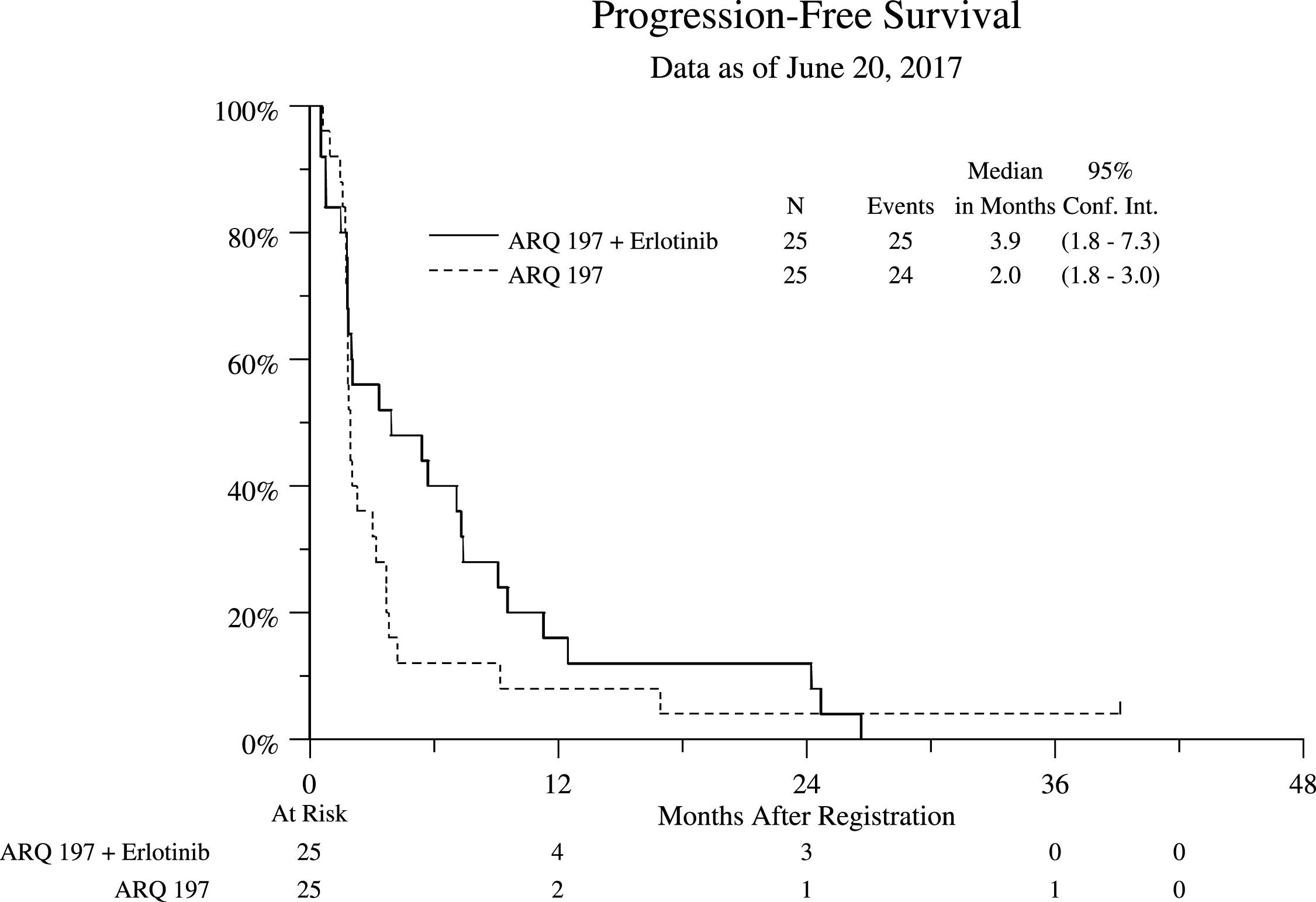
Fig.2
Overall Survival (OS) Stratified by Treatment Arm for All Eligible Patients.
Toxicity
In general, treatment was well tolerated. As is common with erlotinib therapy low-grade rash, nausea, fatigue and diarrhea were common in Arm 2 (tivantinib plus erlotinib). Anemia was the most common in Arm 1 (Tivantinib alone). (Supplementary Table S2).
Variant and Copy Number Analysis
Tissue specimens were collected from 35 patients. In the majority of cases they represented primary tumor samples from nephrectomy. Exome of 16 patients were successfully sequenced using Agilent SureSelect probes. The mean coverage of target regions range from 45x to 91x. The single point mutations and small indels were identified with filtering steps outlined in the method section. These filtering resulted in 143 – 289 variants identified in these samples. It is interesting to note that most of these mutations are only identified in one patient, indicating the high diversity of variants with this patient cohort (Supplementary Figure S1). The genes affected with at least 4 cases are shown in Fig. 3, with IGSF3 being affected in 15 out of 16 patients. This gene was found to be mutated in only 1.4% of 280 cases in TCGA project and is not known to be linked to cancer. The significance of majority of mutations observed in our cohort is not well understood [18]. MUC2 and MUC4 mutations detected in our samples are also frequently mutated in TCGA cohort, and they have been shown to be associated with various types of cancers [19]. Only 1 of 16 samples harbored MET mutation located in tyrosine kinase domain (K1198I). Other mutations associated primarily with type 2 pRCC were noted and included CDKN2A, PBRM1, SETD2, KDM6A, FAT1, NF2 and CUL. (Table 2) No EGFR and FH mutations were detected. To examine the functional consequence of the mutations, we applied network analysis with the 306 genes mutated in at least 2 cases using CytoScape and Reactome FI database (Supplementary Table S3). The most enriched pathways include WNT signaling pathway, cadherin signaling pathway, and Mitotic G2-G2/M phase pathway. These pathways have been shown to be linked to tumorigenesis [20], suggesting other potential therapeutic targets might be worth exploring in these patients.
Fig.3
The most frequently mutated genes.
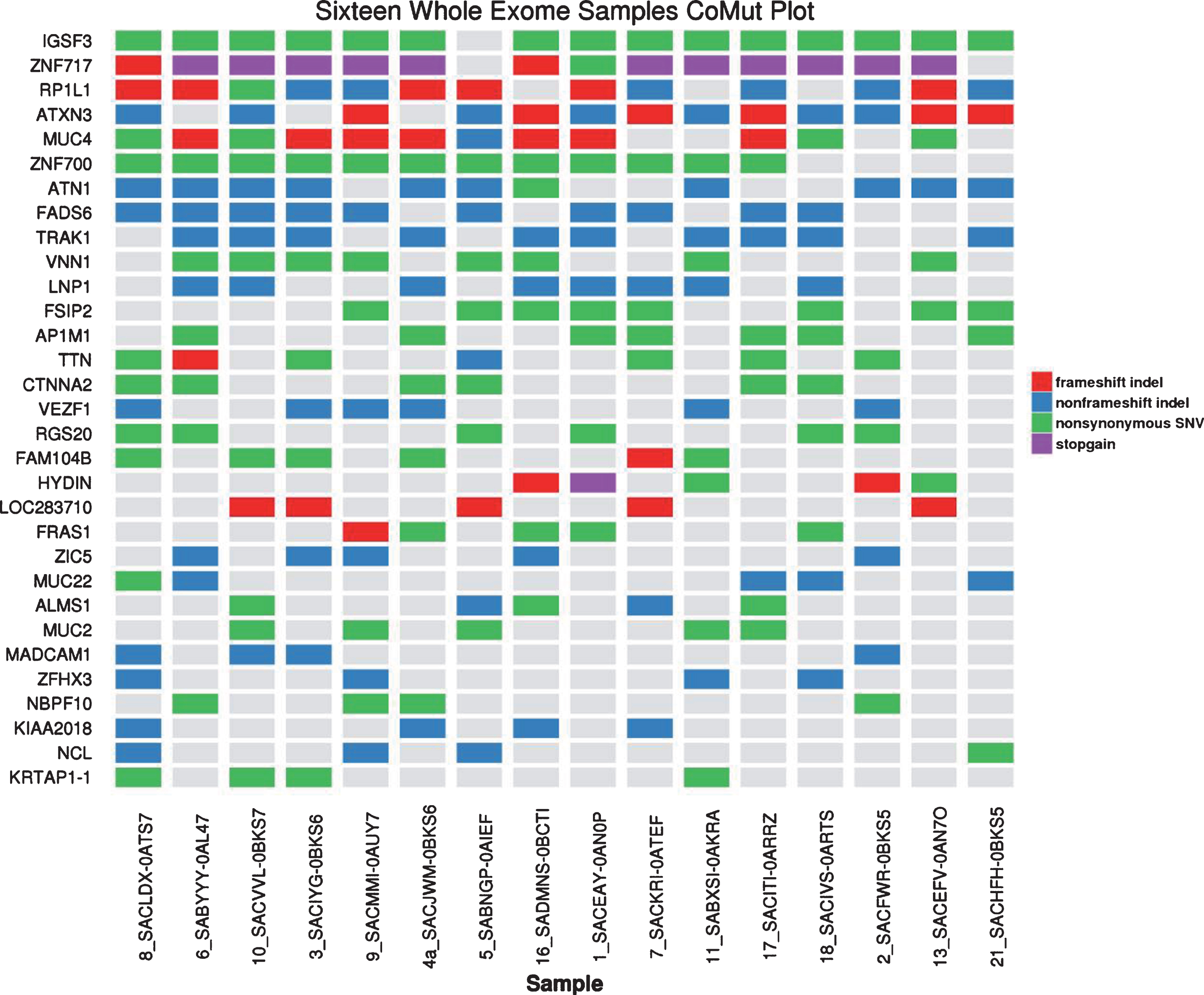
Table 2
Frequency of selected pRCC-related mutations among n = 16 out of 35 patient samples in whom adequate tumor tissue DNA was extracted for whole exome sequencing
| Gene | N (%) | Chr/exon | Function | AA Change |
| MET | 1 (6) | 7 / 18 | nsSNV | K1198I |
| FH | 0 | |||
| CDKN2A | 1 (6) | 9 /1 | Stopgain | P89_delG90 |
| PBRM1 | 3 (19) | 3 / 9,4,17 | nsSNV | Y738C |
| T113P | ||||
| I276V | ||||
| SETD2 | 2(12) | 3 /12,3 | Stopgain | E1991X |
| Frameshift deletion | D994fs | |||
| KDM6A | 2(12) | X / 15,20 | Frameshift deletion | L373fs I786fs |
| FAT1 | 2(12) | 4 / 2,14 | nsSNV | S3198F |
| Stopgain | Q283X | |||
| NF2 | 1 (6) | 22 / 5 | Deletion | P123_132del |
| CUL | 1 (6) | 2/1 | nsSNV | R17P |
nsSNV - non-synonymous single nucleotide variant.
Somatic copy number variation was difficult to obtain in these samples, as no matching normal tissues were collected. Initially, Conifer was used to derive CNV in these samples, which was based on the assumption that most of the cases do not share common CNVs. Only one case was identified to have MET amplification using this method. We then used case 8 and 13 as the baseline after visual inspection of their coverage at MET locus. Although these two cases have the lowest MET coverage, it is still difficult to conclude they are the best samples to be used as baseline. We describe this as virtual somatic copy number variants (Table 3). The results showed MET amplification in 6 cases (37.5%), and 3 cases with EGFR amplification and one case with EGFR deletion. We also observed 6 cases with CDKN2A amplification, which is surprising as only CDKN2A deletions were found in TCGA pRCC cohort [8].
Table 3
Virtual somatic copy number variants
| SAMPLE | MET | EGFR | ERRFI1 | CDKN2A |
| SACEAY_1 | dup | |||
| SACFWR_2 | ||||
| SACIYG_3 | ||||
| SACJWM_4a | del | dup | ||
| SABNGP_5 | dup | del | dup | |
| SABYYY_6 | dup | dup | ||
| SACKRI_7 | dup | |||
| SACMMI_9 | ||||
| SACVVL_10 | dup | dup | dup | |
| SABXSI_11 | dup | del | ||
| SADMNS_16 | dup | |||
| SACITI_17 | ||||
| SACIVS_18 | dup | |||
| SACHFH_21 | dup | dup |
DISCUSSION
Tivantinib - either alone or in combination with erlotinib induced no responses or exhibited no clinical activity in patients with advanced pRCC. The combination of tivantinib and erlotinib could not even replicate the previous experience of modest activity of single agent erlotinib. One of the possible explanations for the lack of activity of tivantinib in pRCC that was not appreciated at the time of study design is that tivantinib may not be an effective inhibitor of MET pathway after all, even though that was its originally postulated mechanism of action. Subsequent observations questioned the crucial role of MET inhibition in mediating pharmacologic activity of tivantinib and concluded that it is a cytotoxic agent with microtubule stabilizing effect [21, 22]. It is also important to remember that pRCC exhibits significant heterogeneity and is divided into type 1 and 2 based on different morphologic and underlying molecular characteristics [23]. How important is MET pathway in the pathophysiolgy of pRCC to significant degree depends on the specific histologic subset. In what is perhaps the most detailed assessment of MET aberrations to date along TCGA data, Albiges et al. assessed a series of 220 frozen tissues derived from patients with pRCC in the French RCC Network [24]. Gene expression was assessed in 98 of these specimens, and ultimately suggested high MET expression level across all patients with pRCC. However, assessment of copy number pointed to the importance of MET particularly in type I pRCC; specifically, 81% of patients with type I pRCC had copy number alternations, as compared to 46% in type II pRCC. MET alterations were also significantly more common in type 1 subset of pRCC in TCGA data [8]. In our study only 6% of patients were classified as type 1 pRCC versus 42% of type 2 and 52 % being indeterminate. Molecular characterization of our cohort was limited to 16 pts but we observed very low incidence of MET alterations with only 1 MET mutation and a relatively low number of copy number variants. One of the few reported clinical trials in pRCC utilizing MET inhibitor (specifically dual VEGFR2 and MET inhibitor) foretinib demonstrated clinical activity essentially restricted to rare patients with hereditary pRCC harboring germline MET mutations and did not reveal significant activity in more common patients with sporadic pRCC associated with other mechanisms of MET activation including MET amplification, duplication of chromosome 7 (location of MET gene) or other copy number aberrations [9]. Recently presented results of a clinical trial of selective MET inhibitor savolitinib in pRCC demonstrated RR of 18% in patients exhibiting MET- driven tumors and 0% RR in subset without MET alterations [10].
Although MET remains a potential therapeutic target of interest in pRCC, future clinical trials of MET inhibitors in this subset of RCC should consider more careful approach to patient selection. Central pathologic review with focus on patients with type 1 pRCC which have the highest rate of MET abnormalities within pRCC or even restriction to MET altered tumors may be considered. Selection of drug(s) with definitive ability to achieve robust inhibition of MET signaling and potential combinational approaches based on discovery of other important molecular drivers of pRCC may enhance the chances of improving outcomes in this important subset of kidney cancer. Recent emergence of PD1 inhibition as an effective approach for the treatment of ccRCC [25] poses important question regarding its potential applicability for non-clear cell variants. Clinical responses of nccRCC’s to PD-1 inhibitors in early clinical trials have been noted and interaction between PD-l and MET signaling pathways has also been recently reported [26]. In preclinical experiments MET induction markedly up-regulated the expression of PD-L1 [26] pointing out to the potential of exploring MET inhibitors and PD-1/PD-Ll targeted therapies for the treatments of patients with pRCC.
In summary tivantinib alone or in combination with erlotinib did not demonstrate any clinical activity in patients with metastatic pRCC. It appears that S1107 patient cohort had a high proportion of patients with molecular subtypes not driven by MET alterations and would not be expected to respond well to MET inhibitor therapy. Future clinical trials of MET inhibitors in pRCC while warranted will require more careful selection of drugs, patients and better understanding of interaction of MET with other pathways determining the pathophysiology of pRCC.
DISCLOSURE OF POTENTIAL CONFLICTS OF INTEREST DISCLOSURE STATEMENT
N. Agarwal is a consultant for Pfizer, Novartis, Merck, Genentech, Eisai, Exelixis, Clovis, EMD Serono. E. Plimack is a Consultant/Scientific Advisory Board member for AstraZeneca, Bristol-Myers Squibb, Eli Lilly and Company, Exelexis, Genentech, Horizon Pharma, Inovio, Novartis, Pfizer and Roche; reports receiving research grants from Acceleron, Agensys, AstraZeneca, Bristol-Myers Squibb, Merck, Peloton, Pfizer; and reports the following patents: Methods for Screening Muscle Invasive Bladder Cancer Patients for Neoadjuvant Chemotherapy Responsiveness, U.S. Patent Application No.: 14/588,503, filed 1/2/2015; Combination Of Immunomodulatory Agent With PD-1 Or PD-L1 Checkpoint Inhibitors In The Treatment Of Cancer. US Patent Application No: 15/226,474, filed on 7/1/15. No potential conflicts of interest were disclosed by the other authors.
AUTHORS’ CONTRIBUTIONS
Conception and design: P.Twardowski, P.Lara, C.Tangen, N.Vogelzang, I.Thompson.
Development of methodology: C.Tangen, M.Plets, X.Wu.
Acquisition of data: P.Twardowski, P.Lara, E.Plimack, N.Agarwal, N.Vogelzang, X.Wu, J. Wang, S. Tao.
Analysis and interpretation of data (e.g., statistical analysis, biostatistics, computational analysis): C.Tangen, M.Plets, X. Wu.
Writing, review, and/or revision of the manuscript: P. Twardowski, C. Tangen, X.Wu, M. Plets, E. Plimack, N. Agarwal, N. Vogelzang, J. Wang, S. Tao, I. Thompson, P. Lara Administrative, technical, or material support (i.e., reporting or organizing data, constructing databases): I.Thompson, C.Tangen, M.Plets.
Study supervision: P.Twardowski, P.Lara.
GRANT SUPPORT
The National Cancer Institute of the National Institutes of Health supported this research under grant awards CA180888; CA180819; CA180820, CA180818, CA189957, CA180835, CA189821, CA180835, CA189960, CA189954, CA189853, CA189829, CA180834, CA180801, CA180798, CA12644, CA22433, CA11083 and CA13612. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
SUPPLEMENTARY MATERIAL
[1] The supplementary table and figure are available in the electronic version of this article: http://dx.doi.org/10.3233/KCA-170018.
REFERENCES
[1] | Rini BI , Cambell SC , Escudier B . Renal cell carcinoma. Lancet (2009) ;373: :1119–32. |
[2] | Pai A , Brunson A , Brown M , et al. Evolving epidemiologic trends in nonclear cell renal cell cancer: An analysis of the California Cancer Registry. Urology (2013) ;82: (4):840–5. |
[3] | Twardowski PW , Mack PC , Lara PN Jr. . Papillary renal cell carcinoma: Current progress and future directions. Clin Genitourin Cancer (2014) ;12: (2):74–9. |
[4] | Birchmeier C , Birchmeier W , Gherardi E , et al. Met, metastasis, motility and more. Nat Rev Mol Cell Biol (2003) ;4: :915–25. |
[5] | Scagliotti GV , Novello S , von Pawel J . The emerging role of MET/HGF inhibitors in oncology. Cancer Treat Rev (2013) ;39: :793–801. |
[6] | Schmidt L , Duh FM , Chen F , Kishida T , Glenn G , Choyke P , Scherer SW , Zuang Z , Lubensky I , Dean M , et al. Germline and somatic mutations in the tyrosine kinase domain of the MET proto-oncogene in papillary renal cell carcinomas. Nat Genet (1997) ;16: :68–73. |
[7] | Bellon SF , et al. c-Met Inhibitors with novel binding mode show activity against several hereditary papillary renal cell carcinoma-related mutations. J Biol Chem (2008) ;283: (5):2675–83. |
[8] | The Cancer Genome Atlas Research Network. N Engl J Med (2016) ;374: :135–45. |
[9] | Choueri TK , Vaishampayan U , Rosenberg JE , Logan TF , Harzstark AL , Bukowski RM , Rini BI , Srinivas S , Stein MN , Adams LM , Ottesen LH , Laubscher KH , Sherman L , McDermott DF , Haas NB , Flaherty KT , Ross R , Eisenberg P , Meltzer PS , Merino MJ , Bottaro DP , Linehan WM , Srinivasan R . Phase II and biomarker study of the dual MET/VEGFR2 inhibitor foretenib in patients with papillary renal cell carcinoma. J Clin Oncol (2013) ;31: (2):181–6. |
[10] | Choueiri TK . A single-arm biomarker-based phase II trial of savolitinib in patients with advanced papillary renal cell cancer (PRCC). J Clin Oncol (2017) ;35: (suppl 6S;). |
[11] | Perera AD , Kleymenova EV , Walker CL . Requirement for the von Hippel-Lindau tumor suppressor gene for functional epidermal growth factor receptor blockade by monoclonal antibody C225 in renal cell carcinoma. Clin Cancer Res (2000) ;6: (4):1518–23. |
[12] | Gordon MS , Hussey M , Nagle RB , Lara PN Jr , Mack PC , Dutcher J , Samlowski W , Clark JI , Quinn DI , Pan CX , Crawford D . Phase II study of erlotinib in patients with locally advanced or metastatic papillary histology renal cell cancer: SWOG S0317. J Clin Oncol (2009) ;27: (34):5788–93. |
[13] | Munshi N , Jeay S , Li Y , et al. ARQ 197, a novel and selective inhibitor of the human c-Met receptor tyrosine kinase with antitumor activity. Mol Cancer Ther (2010) ;9: :1544–53. |
[14] | Yap TA , Frentzas S , Tunariu N , et al. Final results of pharmacokinetic (PK) and pharmacodynamic (PD) phase I trial of ARQ 197 incorporating dynamic contrast-enhanced (DCE) magnetic resonance imaging (MRI) studies investigating the anti-angiogenic activity of selective c-Met inhibition. J Clin Oncol (2009) ;27: :151s. |
[15] | Laux I , Goldman J , Just R , et al. Phase I dose escalation trial (ARQ 197-111) evaluating combination of selective c-MET inhibitor ARQ 197 and erlotinib. J Clin Oncol (2009) ;27: :158s. |
[16] | Scagliotti G , von Pawel J , Novello S , Ramlau R , Favaretto A , Barlesi F , Akerley W , Orlov S , Santoro A , Spigel D , Hirsh V , Shepherd FA , Sequist LV , Sandler A , Ross JS , Wang Q , von Roemeling R , Shuster D , Schwartz B . Phase III Multinational, Randomized, Double-Blind, Placebo-Controlled Study of Tivantinib (ARQ 197) Plus Erlotinib Versus Erlotinib Alone in Previously Treated Patients With Locally Advanced or Metastatic Nonsquamous Non-Small-Cell Lung Cancer. J Clin Oncol (2015) ;33: (24):2667. |
[17] | Therasse P , Arbuck SG , Eisenhauer EA , et al. New guidelines to evaluate the response to treatment in solid tumors. European Organization for Research and Treatment of Cancer, National Cancer Institute of the United States, National Cancer Institute of Canada. J Natl Cancer Inst (2000) ;92: :205–16. |
[18] | Lawrence MS , et al. Mutational heterogeneity in cancer and the search for new cancer genes. Nature (2013) ;499: (7457):214–18. |
[19] | Yonezawa S , et al. Expression profiles of MUC1, MUC2, and MUC4 mucins in human neoplasms and their relationship with biological behavior. Proteomics (2008) ;8: (16):3329–41. |
[20] | Rhind N , Russell P . Signaling Pathways that Regulate Cell Division. Cold Spring Harbor Perspectives in Biology (2012) ;4: (10). |
[21] | Basilico C , et al. Tivantinib (ARQ197) displays cytotoxic activity that is independent of its ability to bind MET. Clin Cancer Res (2013) ;19: (9):2381–92. |
[22] | Katayama R , et al. Cytotoxic activity of tivantinib (ARQ 197) is not due solely to c-MET inhibition. Cancer Res (2013) ;73: (10):3087–96. |
[23] | Lager DJ , Hustoin BJ , Timmerman TG , et al. Papillary renal tumors: Morphologic, cytochemial, and genotypic features. Cancer (1995) ;76: :669–73. |
[24] | Albiges L , Guegan J , Le Formal A , Verkarre V , Rioux-Leciercq N , Sibony M , Bernhard JC , Camparo P , Merabet Z , Molinie V , Allory Y , Orear C , Couve S , Gad S , Patard JJ , Escudier B . MET is potential target across all papillary renal cell carcinomas: Result from a large molecular study of pRCC with CGH array and matching gene expression array. Clin Cancer Res (2014) ;20: (13). |
[25] | Motzer RJ , Escudier B , McDermott DF , George S , Hammers HJ , Srinivas S , Tykodi SS , Sosman JA , Procopio G , Plimack ER , Castellano D , Choueiri TK , Gurney H , Donskov F , Bono P , Wagstaff J , Gauler TC , Ueda T , Tomita Y , Schutz FA , Kollmannsberger C , Larkin J , Ravaud A , Simon JS , Xu LA , Waxman IM , Sharma P ; CheckMate 025 Investigators. Nivolumab versus Everolimus in Advanced Renal-Cell Carcinoma. N Engl J Med (2015) . |
[26] | Balan M , Miery Teran E , Waaga-Gasser AM , Gasser M , Choueiri TK , Freeman G , Pal S . Novel roles of c-Met in the survival of renal cancer cells through the regulation of HO-1 and PD-L1 expression. J Biol Chem (2015) ;290: (13):8110–20. |


