
Genomic Alterations and Outcomes with VEGF-Targeted Therapy in Patients with Clear Cell Renal Cell Carcinoma
Abstract
Background: Mutations in VHL, PBRM1, SETD2, BAP1, and KDM5C are common in clear cell renal cell carcinoma (ccRCC), and presence of certain mutations has been associated with outcomes in patients with non-metastatic disease. Limited information is available regarding the correlation between genomic alterations and outcomes in patients with metastatic disease, including response to VEGF-targeted therapy.
Objective: To explore correlations between mutational profiles and cancer-specific outcomes, including response to standard VEGF-targeted agents, in patients with metastatic cc RCC.
Methods: A retrospective review of 105 patients with metastatic ccRCC who had received systemic therapy and had targeted next-generation sequencing of tumors was conducted. Genomic alterations were correlated to outcomes, including overall survival and time to treatment failure to VEGF-targeted therapy.
Results: The most frequent mutations were detected in VHL (83%), PBRM1 (51%), SETD2 (35%), BAP1 (24%), KDM5C (16%), and TERT (14%). Time to treatment failure with VEGF-targeted therapy differed significantly by PBRM1 mutation status (p = 0.01, median 12.0 months for MT versus 6.9 months for WT) and BAP1 mutation status (p = 0.01, median 6.4 months for MT versus 11.0 months for WT). Shorter overall survival was associated with TERT mutations (p = 0.03, median 29.6 months for MT versus 52.6 months for WT) or BAP1 mutations (p = 0.02, median 28.7 months for MT versus not reached for WT).
Conclusions: Genomic alterations in ccRCC tumors have prognostic implications in patients with metastatic disease. BAP1 and TERT promoter mutations may be present in higher frequency than previously thought, and based on this data, deserve further study for their association with poor prognosis.
KEY MESSAGE
The most frequent mutations in metastatic clear cell renal cell carcinoma (ccRCC) are similar to those in localized ccRCC. Certain genetic alterations, such as BAP1, PBRM1 and TERT promoter mutations, may have prognostic implications in metastatic ccRCC and should be further investigated.
INTRODUCTION
Clear cell renal cell carcinoma (ccRCC) is the most common subtype of kidney cancer, estimated to account for about 80% of the nearly 62,700 cases diagnosed annually in the United States [1, 2]. The majority of patients are cured by surgical resection, but a considerable portion is either diagnosed with metastatic disease or develops recurrence after surgery. Once the disease is metastatic, it is usually incurable with limited overall survival (OS) [2].
Over the last decade, approved therapies for metastatic ccRCC have expanded considerably. The growing number of available agents includes vascular-endothelial growth factor (VEGF)-directed compounds, mammalian target of rapamycin (mTOR) inhibitors, and recently, checkpoint inhibitors [3–7]. Lack of predictive biomarkers, however, prevents rational sequencing strategies for these regimens. This stands in contrast to other malignancies such as non-small cell lung cancer or melanoma, where validated molecular markers predict response to approved targeted agents [8–10].
Large scale sequencing efforts have helped unveil the genomic landscape of ccRCC [11, 12]. In addition to confirming VHL alterations as linchpin events in the pathogenesis of ccRCC, they uncovered other recurrent genomic events, including alterations in the tumor suppressors PBRM1, SETD2, BAP1 and KDM5C. These analyses, however, were mostly carried out in patients with early stage disease. Similarly, prior reports associating some of these genomic alterations with outcome were limited to non-metastatic ccRCC [13–15].
For patients with metastatic disease, key questions remain unanswered, such as the incidence of genomic alterations and the association of alterations with clinical outcomes, including response to targeted therapies. In this study, we retrospectively analyzed patients with metastatic ccRCC treated with systemic therapy that had undergone targeted exome sequencing of cancer-related genes. The aim was to explore correlations between mutational profiles and cancer-specific outcomes, including response to standard VEGF-targeted agents.
METHODS
Patients and samples
Patients were identified from an institutional database of RCC patients who had previously undergone next-generation targeted exome sequencing of archival tumor tissue. Subjects with metastatic ccRCC who had received at least one line of systemic therapy at Memorial Sloan Kettering Cancer Center (MSKCC) were included in the analysis. Demographics, clinical characteristics and details on treatment effect were collected for each case via retrospective review of clinical records. This study was approved by the MSKCC Institutional Review Board, and all patients consented to a tissue procurement protocol.
Tissue acquisition and next-generation sequencing (NGS) analysis
Formalin-fixed, paraffin-embedded (FFPE) tissue specimens from either the primary or a metastatic site were reviewed by genitourinary pathologists to select areas of maximum tumor content for DNA extraction; blood samples were used for matched comparison of germline DNA. For NGS analysis, the MSKCC IMPACT™ (Integrated Mutation Profiling of Actionable Cancer Targets) assay was employed as previously described [16]. IMPACT is designed to detect SNVs, short indels, copy number aberrations and structural rearrangements. NGS was performed using Illumina HiSeq, with deep coverage across all exonic sequences, plus select introns deemed relevant for fusion events. Variants are annotated with Annovar, then further filtered using several criteria, including previous literature, and technical characteristics of the variant call such as depth of coverage. Initial samples (20%) had 341 genes investigated per an earlier version of the assay, while 410 genes were tested for all subsequent samples (full gene list in Supplemental Table 1, both versions include all commonly mutated genes in ccRCC). Five samples underwent whole genome sequencing.
Statistical analysis
The relationship between mutation status and overall survival (OS) and time to treatment failure (TTF) from treatment initiation of first therapy was evaluated using time to event methods. For OS, patients still alive at the time of analysis were censored at the last clinic date on which survival status could be confirmed. For TTF, an event was considered to be the time of treatment discontinuation, for any reason (progression or toxicity). Patients still on therapy at the time of analysis were censored at that point in time. Kaplan-Meier estimates for both endpoints were summarized by calculating the median with 95% confidence intervals and for OS 2 year estimates. Comparisons by each mutation were tested using the Log-Rank statistic. Our analyses are hypothesis generating and as such, we do not adjust for multiple comparisons. All analyses were performed in SAS v 9.4 and R v 3.1.1.
RESULTS
Patient characteristics
A total of 105 patients were identified; patient characteristics are summarized in Table 1. Median age was 57 years (range 33–81). MSKCC Risk Group was available for 89 patients starting first-line therapy (35% favorable, 52% intermediate and 14% poor risk). Thirty-six (34%) patients had received 1 line of therapy at time of analysis, 49 (47%) had received 2-3 lines, and 20 (19%) had received 4 or more lines.
Table 1
Patient characteristics (N = 105)
| Age at diagnosis (median, range) | 57 (33–81) |
| Sex | |
| F | 28 (26.7%) |
| M | 77 (73.3%) |
| Nephrectomy | |
| Yes | 95 (90.5%) |
| No | 10 (9.5%) |
| Type of Nephrectomy | |
| Localized Disease | 44 (46.3%) |
| Cytoreductive | 51 (53.7%) |
| Sarcomatoid Features | |
| Yes | 30 (28.6%) |
| No | 65 (61.9%) |
| NA | 10 (9.5%) |
| Grade | |
| G1 | 0 (0.0%) |
| G2 | 14 (13.3%) |
| G3 | 37 (35.2%) |
| G4 | 45 (42.9%) |
| NA | 9 (8.6%) |
| Stage at Time of Diagnosis | |
| I | 11 (10.5%) |
| II | 10 (9.5%) |
| III | 26 (24.8%) |
| IV | 57 (54.3%) |
| NA | 1 (1.0%) |
| MSKCC Risk Group | |
| Favorable | 31 (34.8%) |
| Intermediate | 46 (51.7%) |
| Poor | 12 (13.5%) |
| NA | 16 (18.0%) |
| Received a VEGF-Targeted Agent | |
| Yes | 95 (90.5%) |
| No | 10 (9.5%) |
| Therapies Prior to VEGF-Targeted Agent | |
| None | 94 (89.5%) |
| mTOR inhibitor | 6 (5.7%) |
| Interferon | 5 (4.8%) |
| First VEGF-Targeted Agent | |
| Sunitinib | 60 (57.2%) |
| Pazopanib | 31 (29.5%) |
| Bevacizumab | 1 (1.0%) |
| Axitinib | 0 (0.0%) |
| Sorafenib | 1 (1.0%) |
| Sunitinib/bevacizumaba | 2 (1.9%) |
aThis regimen was given as part of a clinical trial [32]. *VEGF, vascular endothelial growth factor; NA, not applicable; MSKCC, Memorial Sloan Kettering Cancer Center.
Mutation analysis
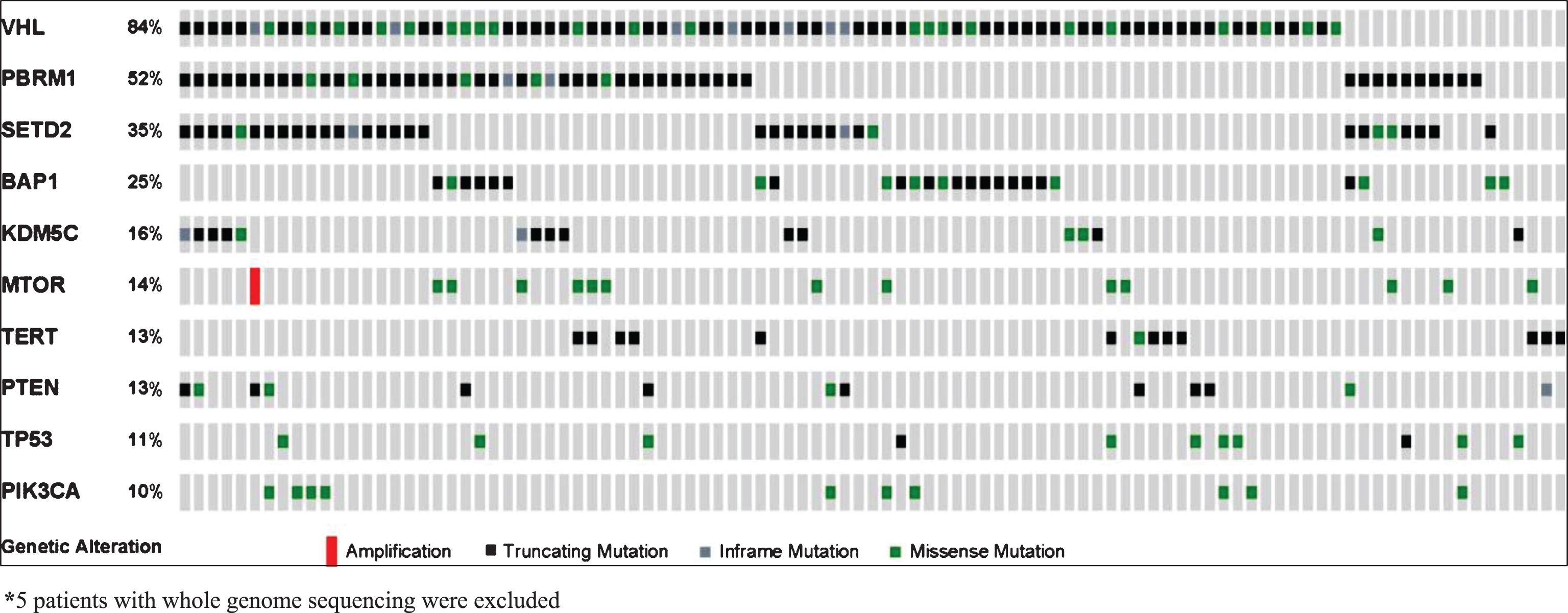
Mutation frequencies detected in our analysis of 105 tumors (66% primary tumors, 34% metastatic lesions) are outlined in Fig. 1. Mean depth of coverage across the cohort was 608x (range 147–1028). The majority of samples (90.5%) were collected prior to initiation of treatment with VEGF-targeted agents. The most frequent mutations were VHL (83%), PBRM1 (51%), SETD2 (35%), BAP1 (24%), KDM5C (16%), and TERT (14%). KDM5C and BAP1 mutations were mutually exclusive, while 15 of 17 tumors with KDM5C alterations also harbored mutations in PBRM1. Only one tumor had mutations in both BAP1 and TERT (Fig. 1). All but one of the TERT mutations were in the promoter region. When ranking mutation frequencies separately from primary tumors vs. metastatic sites, the top 4 mutated genes (VHL, PBRM1, SETD2, BAP1) remained the same (Supplemental Table 2).
Fig.1
Frequency of genetic alterations.
Mutation status and OS
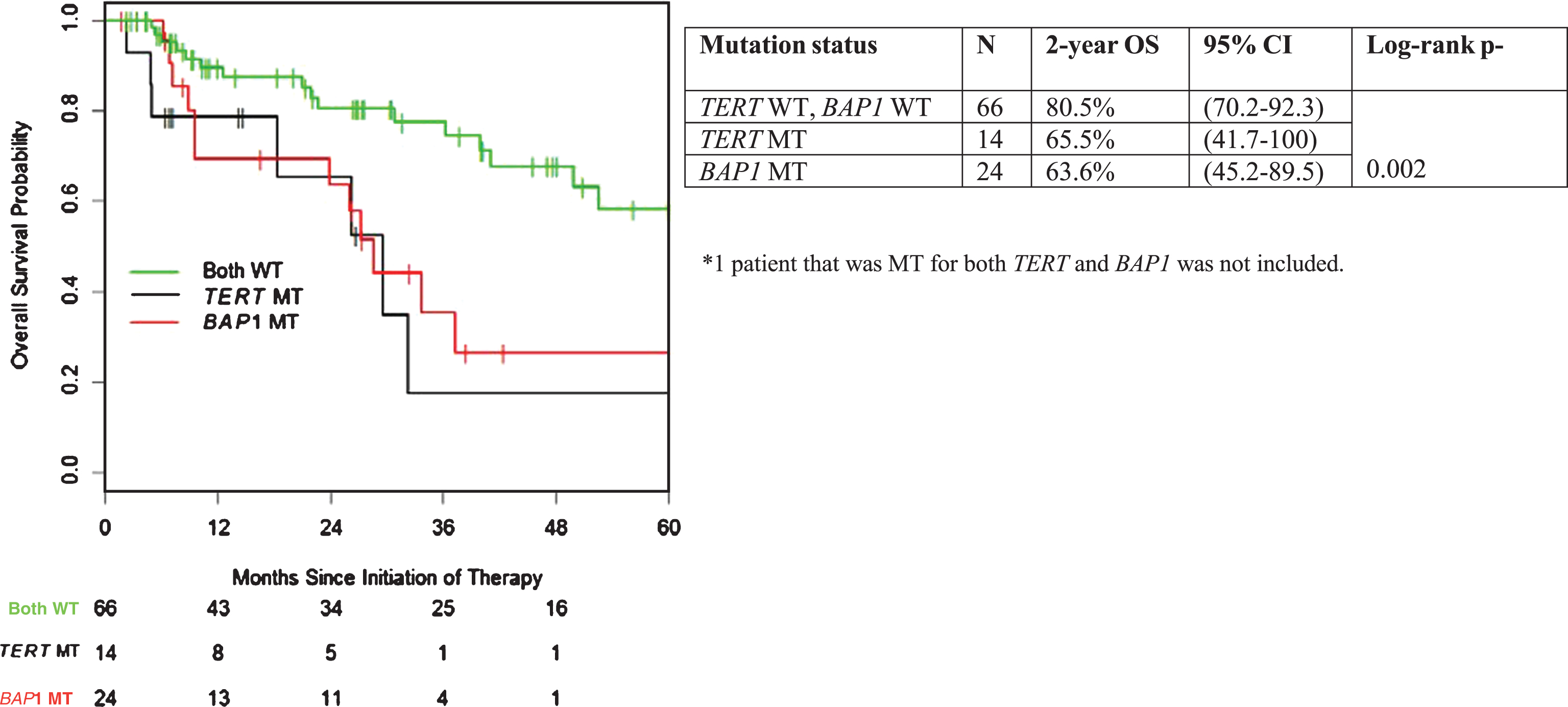
With a median follow-up of 26 months, median OS from the initiation of treatment for the entire cohort was 50 months (95% CI 36.3 – not reached). Correlation of mutation status in individual genes of interest with OS is summarized in Table 2. Patients with TERT mutant (MT) tumors had a significantly shorter OS compared to TERT wild-type (WT) tumors (median 29.6 vs. 52.6 months; p = 0.03). Similar adverse association with outcome was apparent for mutation status in BAP1 (p = 0.02, median 28.7 months vs. not reached) (Fig. 2). There were no statistically significant OS differences when comparing MT vs. WT for VHL, PBRM1, SETD2, and KDM5C (Table 2).
Table 2
Overall survival (OS) by mutation status
| Mutation | N | Median OS | 95% CI | Log-rank | 2 year | 95% CI |
| status | (months) | p-value | OS | |||
| VHL WT | 18 | 26.1 | (18.4-NA) | 0.13 | 62.0% | (37–100) |
| VHL MT | 87 | 49.9 | (37.3-NA) | 77.0% | (67–87) | |
| PBRM1 WT | 52 | 36.3 | (29.6-NA) | 0.12 | 72.8% | (61–88) |
| PBRM1 MT | 53 | NR | (49.9-NA) | 77.6% | (66–92) | |
| SETD2 WT | 68 | 40.0 | (29.6-NA) | 0.29 | 72.7% | (62–86) |
| SETD2 MT | 37 | NR | (37.3-NA) | 79.3% | (66–96) | |
| BAP1 WT | 80 | NR | (41.2-NA) | 0.02 | 78.0% | (68–89) |
| BAP1 MT | 25 | 28.7 | (23.9-NA) | 65.4% | (48–90) | |
| KDM5C WT | 88 | 40 | (32.3-NA) | 0.15 | 73.4% | (64–85) |
| KDM5C MT | 17 | NR | (41.2-NA) | 84.0% | (66–100) | |
| TERT WT | 90 | 52.6 | (40.0-NA) | 0.03 | 76.1% | (67–87) |
| TERT MT | 15 | 29.6 | (18.4-NA) | 68.6% | (46–100) |
*MT, mutant; WT, wild-type; NA, not applicable.
Fig.2
Overall survival stratified by presence of TERT promoter and BAP1 mutation.
Mutation status and TTF to VEGF-targeted therapy
Ninety-five patients had received VEGF-targeted agents; 20 were still on treatment at the time of this analysis and the remaining 75 had stopped therapy for progression (n = 59), toxicity (n = 14) or other reason (n = 2). TTF on first VEGF-targeted therapy by mutation status is summarized in Table 3. TTF with first VEGF-targeted therapy differed significantly by PBRM1 mutation status, where presence of acquired somatic mutation associated with more favorable TTF (p = 0.01; median 12.0 months vs. 6.9 months for WT). BAP1 mutation status correlated adversely with TTF in VEGF-targeted therapy treated patients (p = 0.01; median 6.4 months for MT vs. 11.0 months for WT). We found no significant correlation for patients harboring mutations in VHL, SETD2, TERT, and KDM5C.
Table 3
Time to failure (TTF) on first VEGF-targeted therapy by mutation status
| Mutation status | N | Median TTF | 95% CI | Log-rank |
| (months) | p-value | |||
| VHL WT | 16 | 6.5 | (5.8–NA) | 0.82 |
| VHL MT | 79 | 10.6 | (7.4–16.6) | |
| PBRM1 WT | 48 | 6.9 | (4.6–10.8) | 0.02 |
| PBRM1 MT | 47 | 12.0 | (10.0–21.6) | |
| SETD2 WT | 61 | 8.6 | (6.7–12.2) | 0.72 |
| SETD2 MT | 34 | 10.0 | (6.5–19.5) | |
| BAP1 WT | 73 | 11.0 | (8.4–19.3) | 0.01 |
| BAP1 MT | 22 | 6.4 | (3.9–16.6) | |
| KDM5C WT | 78 | 8.6 | (6.5–12.2) | 0.90 |
| KDM5C MT | 17 | 11.4 | (9.7–24.3) | |
| TERT WT | 82 | 10.6 | (7.1–12.3) | 0.25 |
| TERT MT | 13 | 6.9 | (2.3–NA) |
*MT, mutant; WT, wild-type; NA, not applicable.
DISCUSSION
With tumor genomic profiling increasingly being used in the diagnostic workup of RCC patients, knowledge of mutation spectrum has grown, but prior studies were largely limited to non-metastatic ccRCC [13–15]. This report represents an effort to correlate genomic profiling and outcomes with VEGF-targeted therapy in metastatic ccRCC patients.
In this cohort, recurrent alterations were detected across genes previously implicated in early stage disease, including VHL, PBRM1, SETD2, and BAP1, but at a higher frequency than in prior reports. For example, as compared to results from The Cancer Genome Atlas (TCGA), PBRM1 mutations were identified in 51 vs. 33%, SETD2 in 37 vs. 12%, and BAP1 in 24 vs. 10%; this finding supports their suggested significance in the pathogenesis of this disease, including development of a metastatic phenotype [12]. Furthermore, recurrent alterations were detected in TP53 (11%), and the TERT promoter region (13%), which were not reported as commonly altered genes in earlier non-metastatic datasets. These may represent late events in the evolution of metastatic disease.
The presence of BAP1 or TERT promoter mutations was associated with a significantly worse OS. These two mutations were mostly mutually exclusive, with only one tumor harboring mutations in both genes. BAP1 codes for a deubiquitinating enzyme involved in chromatin remodeling, and several studies in mostly localized RCC have found that loss of BAP1 is associated with poor prognosis [13, 14]. Although TERT mutations were not described in the original TCGA study of ccRCC, a recent study of mostly non-metastatic cases identified approximately 10% prevalence in ccRCC tumors, and also showed that there may be an association with aggressive disease [17]. TERT promoter mutations are the most common somatic noncoding mutations in cancer, and have also been associated with adverse outcomes in several malignancies, including melanoma, thyroid cancer, and bladder cancer [18–24]. An exploratory analysis was performed to investigate if acquired mutations in BAP1 or TERT were associated with MSKCC prognostic risk group or the presence of sarcomatoid features on histopathologic review. While there was no association with mutation status and risk group, there was a significantly higher rate of sarcomatoid features in MT vs. WT (p = 0.008). Due to sample size constraints, we were unable to investigate whether these are independent risk factors for adverse outcomes; this should be analyzed in larger cohorts. Patients whose tumors displayed sarcomatoid features had worse overall survival regardless of mutation status, but among those without sarcomatoid features, BAP1 or TERT MT had worse OS than WT (Supplemental Figure 1; Supplemental Table 3).
In this study, mutations in PBRM1 correlated with statistically longer TTF with VEGF-targeted therapy, with a trend towards superior OS (not reached for MT vs. 36.3 months for WT; p = 0.12). Similar findings were previously reported for metastatic patients treated on the RECORD-3 trial, with a trend towards superior outcome for patients with PBRM1 MT tumors, and in a recent analysis of ccRCC patients with extreme responses to VEGF-targeted therapy. In contrast to these findings in metastatic disease, early-stage studies have reported adverse association with cancer-specific outcome [14]. PBRM1 is a component of the SWI/SNF chromatin remodeling complex, and mutations of PBRM1 are an early event in ccRCC tumor development [25, 26]. Together, these findings may suggest that loss of PBRM1 is biologically relevant for invasiveness and metastatic spread; but once tumor cells metastasize, those with loss of PBRM1 may behave less aggressively than tumors which metastasize by other molecular means (e.g. mutations in BAP1 or TERT) [14, 25, 27]. Such observations are limited by sample size, and without a control arm, it is not possible to determine whether PBRM1 mutation status is a predictive or prognostic biomarker.
In the prior analysis of the RECORD-3 trial, there was a significant correlation between presence of KDM5C mutation and PFS in 111 patients receiving sunitinib (median PFS 8.3 for KDM5C WT vs. 20.6 months for MT; p = 0.03), but not for 109 everolimus-treated patients (median PFS 8.2 for KDM5C WT vs. 9.8 months for MT; p = 0.03). In the present analysis, TTF did not correlate significantly with KDM5C mutation status across 95 VEGF-targeted therapy treated patients (TTF 8.6 months for KDM5C WT vs. 11.4 months for MT; p = 0.9). The inability to confirm the previously suggested correlation may relate to sample size and a more heterogeneous patient population.
This study has several other limitations. As a single-institution experience of limited sample size, findings will need to be corroborated in larger independent datasets. In addition, TTF, which is subject to individual decision making of treating physicians, was used as a measure of therapeutic benefit and limits comparison to more rigid assessment approaches, such as Response Evaluation Criteria in Solid Tumors (RECIST) [28]. Archival samples were derived from both primary tumor and metastatic sites, some having been collected long before the initiation of targeted therapy. Finally, intratumor heterogeneity is a well-known phenomenon in RCC and limits our accuracy in assessing the true mutant allele frequency in each sample [25, 29]. Nevertheless, biology of metastatic sites should guide survival for most patients, and recent data shows a high correlation of mutation profiles in primary vs. metastatic sites [30].
The data presented here cannot inform the choice of agent for patients with metastatic ccRCC, although the ever expanding number of approved targeted therapies leaves prescribers with a clear need for predictive biomarkers in this space. Our findings can, however, help better inform our understanding of pathobiology and clinical course of this disease. Certain genetic alterations, such as BAP1 and TERT promoter mutations, may be present in higher frequency than previously thought and, based on this data, deserve further study for their prognostic implications. Ultimately, they may prove useful in refining our current standards of assessing risk status in metastatic disease, and could inform surveillance strategies and future trial design [31].
FUNDING
This work was funded in part through the Marie-Josée and Henry R. Kravis Center for Molecular Oncology and National Institutes of Health/National Cancer Institute Cancer Center Support (Grant P30 CA008748), Ruth L. Kirschstein National Research Service Award T32CA082088, Clinical and Translational Science Center at Weill Cornell Medical Center UL1TR00457 and the Randall MacDonald Kidney Cancer Research Fund. MIC is supported through a Kidney Cancer Association Young Investigator Award.
DISCLOSURES
MHV reports consulting fees (Pfizer, Eisai, Novartis, Exelixis, Calithera, Natera), research grants (BMS, Genentech/Roche) and travel funding (Novartis and Takeda). DRF reports consulting fees (Bayer and Seattle 75 Genetics) and research funding (Novartis). RJM reports consulting (Pfizer, Novartis, Exelixis, Eisai) and funding to MSKCC for clinical trial support (Pfizer, Genentech/Roche, Novartis, BMS, Eisai, and Exelixis). WL is employed by Helix, a for-profit company. CHL receives funding to MSKCC for clinical trial support (Pfizer and Eisai), and consulting (Exelixis). JJH reports honoraria (Chugai Pharmaceutical), consulting (Novartis, Chugai Pharmaceutical, Eisai) and research funding (Novartis, Pfizer, CGI). All the remaining authors have no disclosures.
ACKNOWLEDGMENTS
We gratefully acknowledge the members of the Molecular Diagnostics Service in MSKCC’s Department of Pathology.
Appendices
The supplementary table and figure are available in the electronic version of this article: http://dx.doi.org/10.3233/KCA-160003.
REFERENCES
[1] | Patard JJ , Leray E , Rioux-Leclercq N , et al. Prognostic value of histologic subtypes in renal cell carcinoma: A multicenter experience. J Clin Oncol (2005) ;23: :2763–71. |
[2] | Siegel RL , Miller KD , Jemal A . Cancer statistics, 2016. CA Cancer J Clin (2016) ;66: :7–30. |
[3] | Motzer RJ , Hutson TE , Tomczak P , et al. Sunitinib versus interferon alfa in metastatic renal-cell carcinoma. N Engl J Med (2007) ;356: :115–24. |
[4] | Hudes G , Carducci M , Tomczak P , et al. Temsirolimus, interferon alfa, or both for advanced renal-cell carcinoma. N Engl J Med (2007) ;356: :2271–81. |
[5] | Choueiri TK , Escudier B , Powles T , et al. Cabozantinib versus everolimus in advanced renal-cell carcinoma. New England Journal of Medicine (2015) ;373: :1814–23. |
[6] | Motzer RJ , Hutson TE , Glen H , et al. Lenvatinib, everolimus, and the combination in patients with metastatic renal cell carcinoma: A randomised, phase 2, open-label, multicentre trial. The Lancet Oncology 16: :1473–82. |
[7] | Motzer RJ , Escudier B , McDermott DF , et al. Nivolumab versus everolimus in advanced renal-cell carcinoma. N Engl J Med (2015) ;373: :1803–13. |
[8] | Chapman PB , Hauschild A , Robert C , et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. New England Journal of Medicine (2011) ;364: :2507–16. |
[9] | Shaw AT , Kim DW , Nakagawa K , et al. Crizotinib versus chemotherapy in advanced ALK-positive lung cancer. N Engl J Med (2013) ;368: :2385–94. |
[10] | Mok TS , Wu YL , Thongprasert S , et al. Gefitinib or carboplatin-paclitaxel in pulmonary adenocarcinoma. N Engl J Med (2009) ;361: :947–57. |
[11] | Sato Y , Yoshizato T , Shiraishi Y , et al. Integrated molecular analysis of clear-cell renal cell carcinoma. Nat Genet (2013) ;45: :860–7. |
[12] | Comprehensive molecular characterization of clear cell renal cell carcinoma. Nature (2013) ;499: :43–9. |
[13] | Hakimi AA , Ostrovnaya I , Reva B , et al. Adverse outcomes in clear cell renal cell carcinoma with mutations of 3p21 epigenetic regulators BAP1 and SETD A report by MSKCC and the KIRC TCGA research network. Clin Cancer Res (2013) ;19: :3259–67. |
[14] | Joseph RW , Kapur P , Serie DJ , et al. Clear cell renal cell carcinoma subtypes identified by BAP1 and PBRM1 expression. J Urol (2016) ;195: :180–7. |
[15] | Kapur P , Pena-Llopis S , Christie A , et al. Effects on survival of BAP1 and PBRM1 mutations in sporadic clear-cell renal-cell carcinoma: A retrospective analysis with independent validation. Lancet Oncol (2013) ;14: :159–67. |
[16] | Cheng DT , Mitchell TN , Zehir A , et al. Memorial sloan kettering-integrated mutation profiling of actionable cancer targets (MSK-IMPACT): A hybridization capture-based next-generation sequencing clinical assay for solid tumor molecular oncology. J Mol Diagn (2015) ;17: :251–64. |
[17] | Wang K , Liu T , Liu L , et al. TERT promoter mutations in renal cell carcinomas and upper tract urothelial carcinomas. Oncotarget (2014) ;5: :1829–36. |
[18] | Vinagre J , Almeida A , Populo H , et al. Frequency of TERT promoter mutations in human cancers. Nat Commun (2013) ;4: :2185. |
[19] | Bell RJ , Rube HT , Xavier-Magalhaes A , et al. Understanding TERT promoter mutations: A common path to immortality. Mol Cancer Res (2016) ;14: :315–23. |
[20] | Huang FW , Bielski CM , Rinne ML , et al. TERT promoter mutations and monoallelic activation of TERT in cancer. Oncogenesis (2015) ;4: :e176. |
[21] | Melo M , da Rocha AG , Vinagre J , et al. TERT promoter mutations are a major indicator of poor outcome in differentiated thyroid carcinomas. J Clin Endocrinol Metab (2014) ;99: :E754–65. |
[22] | Rachakonda PS , Hosen I , de Verdier PJ , et al. TERT promoter mutations in bladder cancer affect patient survival and disease recurrence through modification by a common polymorphism. Proc Natl Acad Sci U S A (2013) ;110: :17426–31. |
[23] | Qu Y , Dang S , Wu K , et al. TERT promoter mutations predict worse survival in laryngeal cancer patients. Int J Cancer (2014) ;135: :1008–10. |
[24] | Griewank KG , Murali R , Puig-Butille JA , et al. TERT promotermutation status as an independent prognostic factor in cutaneousmelanoma. J Natl Cancer Inst (2014) ;106. |
[25] | Gerlinger M , Rowan AJ , Horswell S , et al. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. New England Journal of Medicine (2012) ;366: :883–92. |
[26] | Varela I , Tarpey P , Raine K , et al. Exome sequencing identifiesfrequent mutation of the SWI/SNF complex gene PBRM1 in renalcarcinoma. Nature (2011) ;469: :539–42. |
[27] | Gerlinger M , Horswell S , Larkin J , et al. Genomic architecture andevolution of clear cell renal cell carcinomas defined bymultiregion sequencing. Nat Genet (2014) ;46: :225–33. |
[28] | Eisenhauer EA , Therasse P , Bogaerts J , et al. New response evaluation criteria in solid tumours: Revised RECIST guideline (version 1.1). Eur J Cancer (2009) ;45: :228–47. |
[29] | Sankin A , Hakimi AA , Mikkilineni N , et al. The impact of genetic heterogeneity on biomarker development in kidney cancer assessed by multiregional sampling. Cancer Med (2014) ;3: :1485–92. |
[30] | Guillermo de Velasco RM , Mahamed Ali S , Signoretti S , Mullane SA , Ross JS , Miller VA , Stephens PJ , Schrock AB , Young L , Pal SK , Choueiri TK . Genomic profiling of nephrectomy and metastatic sites in patients with advanced clear cell renal cell carcinoma (RCC) [abstract]. In Genitourinary Cancers Symposium (2017) . |
[31] | Rini BI , Dorff TB , Elson P , et al. Active surveillance inmetastatic renal-cell carcinoma: A prospective, phase 2 trial. The Lancet Oncology 17: :1317–24. |
[32] | Feldman DR , Baum MS , Ginsberg MS , et al. Phase I trial of bevacizumab plus escalated doses of sunitinib in patients with metastatic renal cell carcinoma. J Clin Oncol (2009) ;27: :1432–9. |


