
Are Preformed Fibrils a Model of Parkinson’s Disease?
Abstract
Pre-formed fibrils (PFFs) made from recombinant α-synuclein are broadly used throughout the field in cellular and animal models of Parkinson’s disease. However, their ability to successfully recapitulate disease biology is a controversial topic. In this article, two researchers debate this issue with Amanda Woerman taking the view that PFFs are a model of synucleinopathy but not Parkinson’s disease, while Kelvin Luk defends their use as an important tool in the field.
Plain Language Summary
Parkinson’s disease patients develop accumulations of misfolded proteins in the brain called Lewy bodies. An important protein found in Lewy bodies is α-synuclein, which aggregates into large fibrils that are thought to have toxic effects on neurons. To study the effect of α-synuclein fibrils, many scientists use pre-formed fibrils (PFFs), which are α-synuclein fibrils made in the lab under a variety of conditions. In this article, two researchers will debate the use of PFFs as a model of Parkinson’s disease. Amanda Woerman discusses the data suggesting why PFFs are an inadequate tool for understanding Parkinson’s disease. Kelvin Luk discusses the data supporting the use of PFFs as a Parkinson’s disease model.
PRE-FORMED FIBRILS ARE NOT A MODEL OF PARKINSON’S DISEASE
Parkinson’s disease (PD) is a neurodegenerative disorder that is neuropathologically defined by the presence of Lewy bodies (LB) and Lewy neurites in neurons. These inclusions contain a misfolded conformation of the protein α-synuclein and are known to be particularly toxic to dopaminergic neurons in the substantia nigra. While previous attempts to use fetal tissue grafts to replace the lost dopaminergic neurons were unsuccessful therapeutically, the finding that LBs spread from the host to the grafted tissue in patients surviving >10 years post-transplant led to the discovery that pathogenic α-synuclein can spread from cell-to-cell to cause disease.1 This key finding served as an important catalyst leading to extensive and ongoing research investigating the ability of α-synuclein to use the prion mechanism of disease to cause progressive degeneration in PD patients. To enable this work, pre-formed fibrils (PFFs) made from recombinant α-synuclein monomer emerged as a leading source of pathogenic or fibrillar protein for cellular and animal models of disease. Currently, either mouse or human α-synuclein fibrils (both wild-type and mutant) are made using a variety of buffer conditions, or human α-synuclein fibrils are ‘seeded’ by spiking in PD patient samples to induce amplification of the pathogenic conformation. These fibrils are then used in cellular and animal models of disease to investigate a wide range of questions related to PD pathogenesis, as well as in drug discovery programs around the world. However, as is detailed below, neither the spontaneously formed PFFs nor the currently available methods for generating amplified fibrils are capable of replicating disease-relevant α-synuclein conformations or structures, raising major concerns about the relevance of the data generated using PFFs in understanding PD pathogenesis.
To understand the importance of α-synuclein conformation in studying PD, we must draw on key lessons about protein misfolding diseases first learned in the prion field. An important early argument against Dr. Stanley Prusiner’s protein-only hypothesis was the need to explain how a single protein could cause a variety of distinct clinicopathological diseases, such as Creutzfeldt-Jakob disease (CJD) and fatal familial insomnia, in the absence of a nucleic acid. What emerged from this debate is the strain hypothesis, which argues that the conformation a protein misfolds into determines the disease an individual will develop.2 Because each protein conformation has unique biochemical and biophysical properties, these structures differ in the rate of fibril elongation and fragmentation, the length of pre-clinical and clinical stages of disease, and the regions of selective vulnerability in the brain that determine the clinical signs a patient exhibits. Several decades of research relied on these measures in animal models, along with fingerprints of biochemical stability, to differentiate between prion strains. However, recent advances in cryo-electron microscopy (cryo-EM) have finally enabled prion biologists to truly investigate the structure-function relationship underlying each prion strain.
An important example of this impact is seen in the recent cryo-EM structures of three laboratory strains derived from sheep scrapie— aRML and a22L from mice3, 4 and 263K from hamsters.5 The three conformations share an almost identical arrangement of the N-terminal residues of the prion protein but exhibit substantial structural variability in the C-terminal disulfide arches (Fig. 1). The profound impact of the structural rearrangement of a small portion of the overall conformation on the resulting disease measures (e.g., clinical signs, pathology distribution, host species susceptibility) underscores the ability of minor changes in protein conformation to exert substantial impacts on disease pathogenesis. The effect of prion strain has also greatly impacted therapeutic development for human prion diseases. Human prion strains do not replicate in cultured cells, which led to the use of lab strains in drug discovery screening assays. A prominent example of this was the use of the RML strain to identify the small molecule IND24, a 2-aminothiazole that inhibits prion replication through an unknown mechanism both in vitro and in vivo. IND24 doubles the lifespan of mice inoculated with RML prions, and is also effective against the ME7, 22L, and chronic wasting disease strains.6 However, it has no effect on mice inoculated with human CJD prions or two natural scrapie isolates, SSBP/1 and CH1641.6
Fig. 1
Small changes in protein structure underly distinct strain properties. Cryo-EM structures of rodent-adapted and lab cloned scrapie strains show a conserved N-terminal sequence in (A) aRML (PDB ID: 7TD6), (B) a22L (PDB ID: 8EFU), and (C) 263K (PDB ID: 7LNA) prion strains. Variation is largely restricted to the C-terminal disulfide arches, highlighted in periwinkle. aRML and a22L were isolated from mouse brain homogenates3, 4 and 263K was isolated from hamster brain homogenates.5
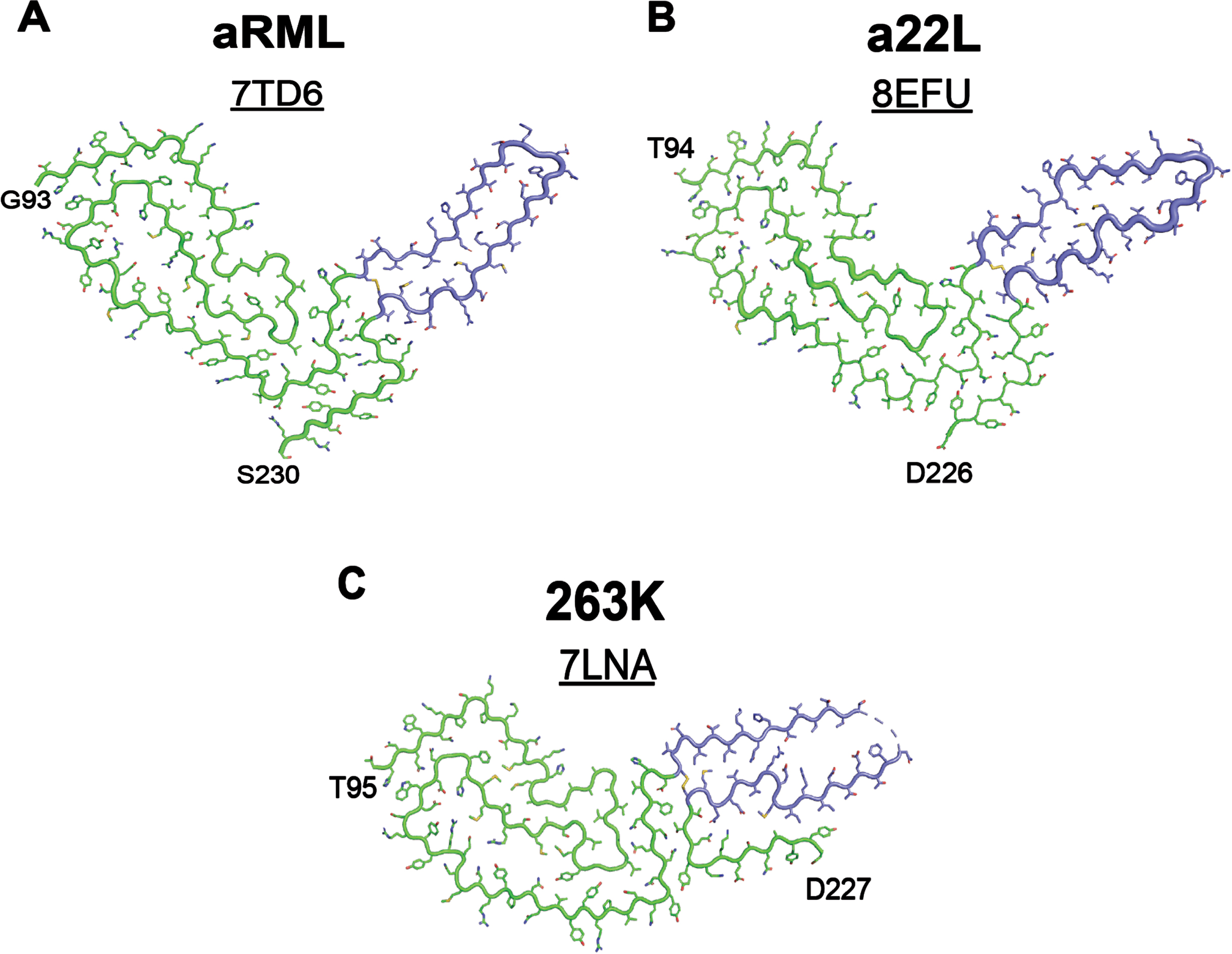
These fundamental findings demonstrate a critical facet of protein misfolding diseases: structure dictates dysfunction. The strain you choose to use in your experiment is the strain you are learning about. It remains unknown how much of the knowledge gained from studies using one strain will pertain or be relevant to other strains of the same protein. This reasoning resulted in an important shift away from the use of recombinant tau fibrils in research on human tauopathies. In 2019, cryo-EM studies showed that the conformations adopted by heparin-induced tau fibrils are distinct from those found in human patient samples.7 As a result, tau research has recently shifted to using disease-associated fibrils, rather than laboratory-derived conformations, to facilitate tauopathy-specific studies.
While there are important differences between prion diseases and other proteinopathies, including tauopathies and synucleinopathies, there are also notable similarities. As discussed earlier for prion diseases, synucleinopathies consist of multiple disorders, which structural, biological, and biochemical data indicate are caused by distinct α-synuclein strains. Moreover, cryo-EM data for each of these proteins, including the prion protein, tau, and α-synuclein, indicate that the structural changes that occur during misfolding result in a predominantly flat conformation with in-register templating that is likely driven by similar mechanisms. Moreover, several early studies using PFFs were important for establishing the protein-only hypothesis for the cell-to-cell spread of misfolded α-synuclein, as occurs in prion disease.
Consistent with the previously discussed research on prion diseases and tauopathies, the synucleinopathy literature is also rife with data pointing to distinct biological consequences arising from differences in α-synuclein fibril conformation. In addition to the cryo-EM structures of fibrils isolated from patients with LB diseases,8 multiple system atrophy (MSA),9 and juvenile-onset synucleinopathy,10 PFFs generated under different buffer conditions are repeatedly shown to induce distinct pathologies, clinical signs, and strain properties after inoculation into wild-type or transgenic mouse models (reviewed in11, 12). Notably, none of the PFF conformations reported to date show structural homology with the conformations resolved from human LB disease patient samples (Fig. 2). While there may be shared structural motifs, the cryo-EM data for aRML, a22L, and 263K emphasize the profound impact that small differences in protein conformation have on disease biology. As a result, it is unclear which datasets generated using PFFs are consistent with the underlying disease biology in PD. Differences in protein source and length, buffer conditions, and fibrillization conditions all influence the resulting PFF conformation, and the biological consequences of each structure have not been fully investigated (discussed in12). In an ideal world, we would be able to pair biological data with cryo-EM structures for every PFF conformation generated, which would enable the field to probe the structure-function relationship for each α-synuclein strain. However, in the absence of these data, it is not only unclear what data are disease-relevant, but it is also unclear how we can accurately interpret conflicting findings in the literature.
Fig. 2
PFFs do not adopt the same α-synuclein conformation that is present in Lewy bodies. A) The cryo-EM structure of α-synuclein fibrils isolated from patients with Lewy body (LB) diseases (PDB ID: 8A9L, published in8) differs substantially from the cryo-EM structures of fibrils generated either (B, C) by seeding monomeric α-synuclein with LB diseases or (D-F) by spontaneous fibrilization of monomeric protein. B, C) Using RT-QuIC methods to amplify α-synuclein in LB disease patients has not resulted in faithful replication of the Lewy fold seen in (A). B) PDB ID: 7OZG published in.16 C) PDB ID: 8FPT published in 15. D-F) Spontaneous fibrilization of monomeric α-synuclein does not recapitulate the Lewy fold, though some PFF structures show similarity to the amplified LB fibrils (compare B and E). D) PDB ID: 6CU7 published in37. E) PDB ID: 6RT0 published in.38 F) PDB ID: 6CU8 published in37.
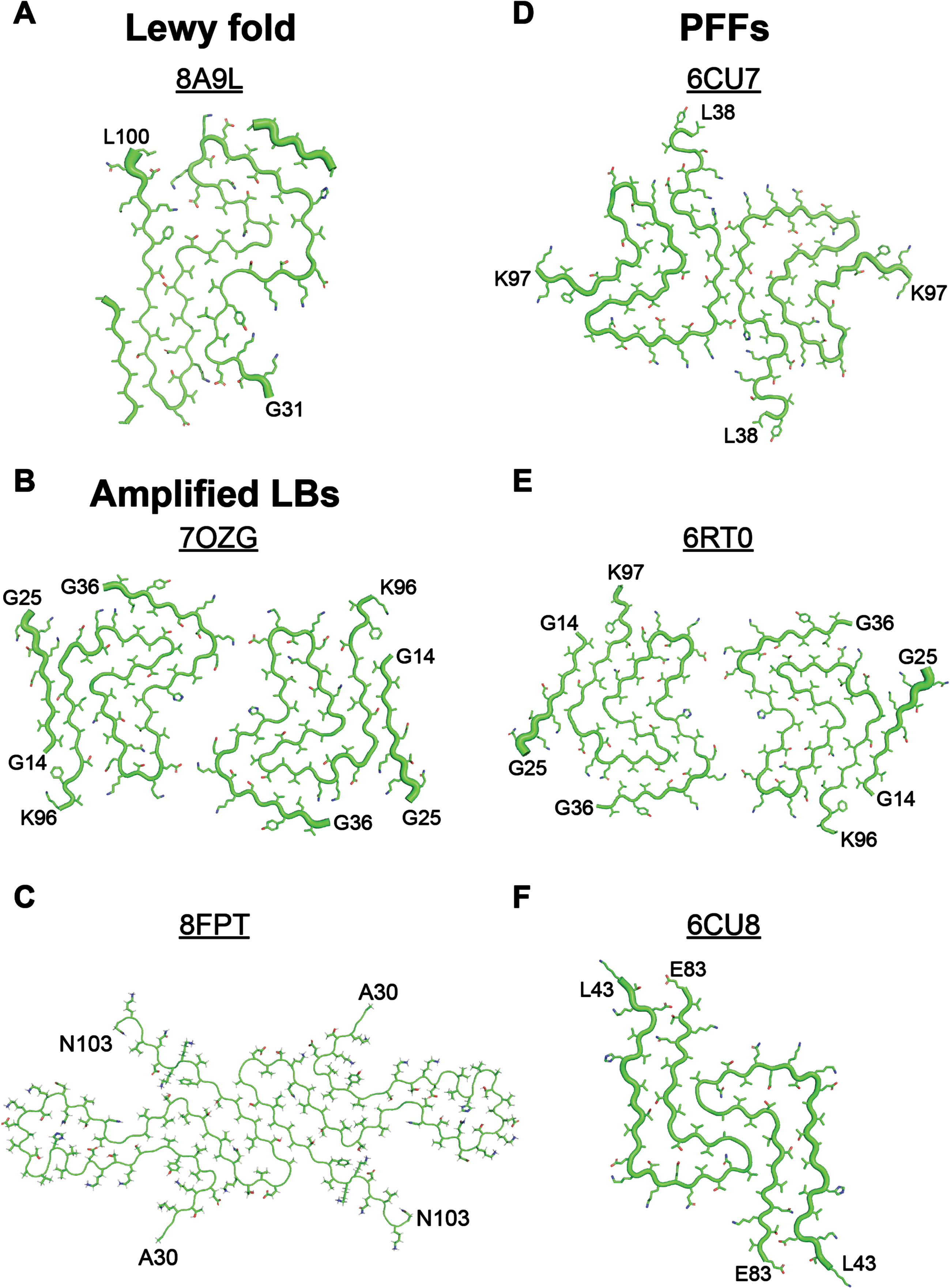
To overcome this limitation, many groups have moved to using PFFs generated by ‘seeding’ the fibrillization reaction with the pathogenic α-synuclein in PD patient samples. In these experiments, recombinant monomer is incubated with a small amount of patient sample that is spiked into the reaction to induce protein misfolding. This approach, often referred to as a seeding amplification assay, is based on the real-time quaking-induced conversion (RT-QuIC) assay developed to detect prions. It is important to note that RT-QuIC does not result in the faithful amplification of prion strains, and that the RT-QuIC product is not transmissible.13 Instead, cell-free amplification of prions is done using the protein misfolding cyclic amplification (PMCA) assay, which uses brain homogenate as the monomer source to facilitate prion replication. PMCA does faithfully amplify prion strains and generates an infectious product.14 This crucial difference may be because brain homogenates contain all necessary co-factors that contribute to protein misfolding, whereas the lack of brain homogenate in the RT-QuIC assay hinders replication of prion strains with high fidelity. Additionally, the lack of posttranslational modifications (PTMs; e.g., glycosylation, GPI anchoring) on recombinant protein used in the RT-QuIC assay may contribute to this result. There are similar concerns to consider when using RT-QuIC to amplify pathogenic α-synuclein. The reported Lewy fold contains a non-protein co-factor that acts to stabilize interactions between four positively charged lysines and one hydrophobic tyrosine. In the absence of a negatively charged co-factor to neutralize this interaction, the charge from the four lysines would repel one another, preventing α-synuclein from adopting the Lewy fold. Pathogenic α-synuclein also contains several PTMs, including phosphorylation and acetylation. Given the lack of co-factors and PTMs in the RT-QuIC reaction, it is fairly easy to understand why none of the reported protocols for amplifying LB α-synuclein15, 16 have been able to fully recapitulate the Lewy fold (Fig. 2). As a result, it remains unknown how predictive the amplified PFFs are of LB strain biology.
Consistent with this concern, it is important to recognize that PFFs and PD patient samples induce different effects in cellular and animal models of disease. For example, while PFFs are readily transmissible to transgenic mouse models, PD patient samples are not. Similarly, PD α-synuclein induces very little to no pathology when injected into mouse brains, but PFFs are frequently used for their ability to induce pathology within synaptic circuits in the brain. While PFFs can induce α-synuclein inclusions in brain regions affected by PD, it is unclear if PFFs use the same pathobiological mechanisms to disrupt neuronal function. These strain-specific differences are highlighted by our recent studies comparing wild-type PFF and MSA transmission to the TgM20+/– mouse model, where we showed that both strains induced neurological disease in the mice; however, the PFF-induced disease was faster and α-synuclein pathology was present in a larger number of brain regions compared to the MSA patient samples.17 By comparison, MSA is a more aggressive disease than PD, with an earlier age of onset and shorter clinical period. Additionally, MSA pathology is typically much more abundant in the brain compared to the Lewy pathology seen in PD patients. In considering these two pieces of information, one would expect that MSA transmission to an animal model would be more robust than PD transmission; however, the PFFs that are used to model PD show a greater proclivity to replicate in vivo than the MSA samples. Along with the structural differences discussed above, these biological differences indicate that PFFs are not accurately modeling PD α-synuclein biology.
While it is important to note that PFFs have been useful for several key discoveries, including the fact that misfolded α-synuclein fibrils alone are capable of inducing disease, caution is warranted in their use as a model of PD. While PFFs are undoubtedly a model of synucleinopathy, their ability to adequately recapitulate the LB strain, or any other strain isolated from a human patient sample, has not been shown. Until that goal is accomplished, PFFs should not be considered a model for PD. Moreover, their continued use in drug discovery programs is concerning, particularly in light of the known strain specificity of anti-prion therapeutics. Systems that more accurately model pathogenic α-synuclein in PD should replace PFFs to better support the development of effective therapeutics.
PRE-FORMED FIBRILS DO MODEL PARKINSON’S DISEASE
Accurate models of disease have the potential to transform the development of novel treatments for complex conditions, especially in the case of complex neurodegenerative disorders such as PD. But what constitutes a faithful model of PD? When attempting to answer this question, an even more difficult question is inevitably invoked: What is PD?
Based on current diagnostic criteria, sporadic PD presents as a syndrome consisting of a myriad of non-motor symptoms beyond the rest tremor, bradykinesia, instability, gait and rigidity which are classically associated with PD. These additional clinical features range from autonomic (e.g., constipation, urinary or erectile dysfunction, orthostatic hypotension) to sleep disturbances (e.g., REM-sleep behavior disorder) and cognitive impairment. Other common symptoms include depression, hallucinations, and hyposmia (loss of smell). There is general consensus regarding the breadth of symptoms associated with PD, yet individuals also show a high degree of heterogeneity in terms of which of these symptoms are present and when each of them emerge during the course of disease.
Indeed, this level of heterogeneity has fueled considerable debate as to whether PD represents a single (albeit complex) disease entity with multiple subtypes or a collection of distinct syndromes with different underlying mechanisms but overlapping phenotypes. This further raises the question of whether modeling PD entails a unifying approach or a pluralistic one. Despite this clinical heterogeneity, the large majority of individuals with sporadic PD (and most recognized forms of genetic PD) also share at least three common biological features.18 Firstly, PD is characterized by the degeneration of monoaminergic and cholinergic systems, most notably dopamine neurons in the nigrostriatal system. Secondly, neurodegeneration in PD is typically accompanied by neuroinflammation. Lastly, postmortem studies of PD brains and other tissues indicate that nearly all individuals with PD, with the exception of a small subset of patients with genetic PD, develop intracellular deposits of misfolded α-synuclein (a.k.a. Lewy bodies and Lewy neurites).19 The neuroanatomical distribution of Lewy pathology correlates with the nature of the observed clinical symptoms while the temporal sequence in which it appears also point to a strong relationship to specific phenotypes. The stereotypical patterns of Lewy pathology across postmortem brains further suggest that α-synuclein pathology spreads from affected neurons to their neighbors, reminiscent of the behavior of classical prions. Consistent with this hypothesis of disease and progression, more recent studies have demonstrated the presence of seeding-competent α-synuclein species in cerebrospinal fluid, plasma and various tissues obtained from individuals with PD.20
Combining these clinical and biological insights provide a working definition of sporadic PD. By extension then, a model of PD should aspire to recapitulate at least four main features: 1) intraneuronal deposition of aggregated α-synuclein, 2) progressive spread of Lewy-like pathologies, 3) impairment or neurodegeneration across multiple neurotransmitter systems, and 4) a neuroinflammatory response. Surprisingly, recombinant α-synuclein PFFs can elicit all these features when administered to animals (summarized in Fig. 3). For example, PFFs injected into the nigrostriatal system results in all four of the processes above, specifically progressive loss of nigral dopaminergic neurons and functional changes in associated networks, emergence of behavioral deficits, and alterations in the neuroimmune landscape.21 - 23 Modifications to the anatomical location of inoculation can elicit changes in other functional circuits leading to phenotypes that resemble those observed in individuals with PD, such as hyposmia,24 loss of REM-sleep muscle atonia,25 and cognitive deficits.26–28 Data from animals inoculated with PFFs at peripheral sites to elicit gastrointestinal29 and urogenital symptoms are also emerging.30 Thus, many of the key domains across the PD clinical spectrum, including those associated with early or prodromal disease, can be recapitulated when PFFs are inoculated into different functional circuits. In each of these experimental paradigms, the disease-relevant phenotype occurs in sequence with the amplification and spread of α-synuclein pathology, providing further insights into the relationship(s) between protein misfolding, transmission, and disease. When examined at the subcellular level, PFF-seeded inclusion pathology bears the morphological and biochemical signatures of Lewy bodies, composed of fibrillar hyperphosphorylated and poly-ubiquitinated α-synuclein mixed together with organelles such as lysosomes and mitochondria.31, 32
Fig. 3
α-synuclein PFFs induce multiple features of PD in vivo. Administration of recombinant α-synuclein PFFs to the CNS or peripheral organs recapitulates 4 characteristic features observed in human PD, including accumulation and spread of Lewy-like pathology, dysfunction or degeneration of selective neuronal circuits, and neuroinflammation. Illustration prepared using BioRender.
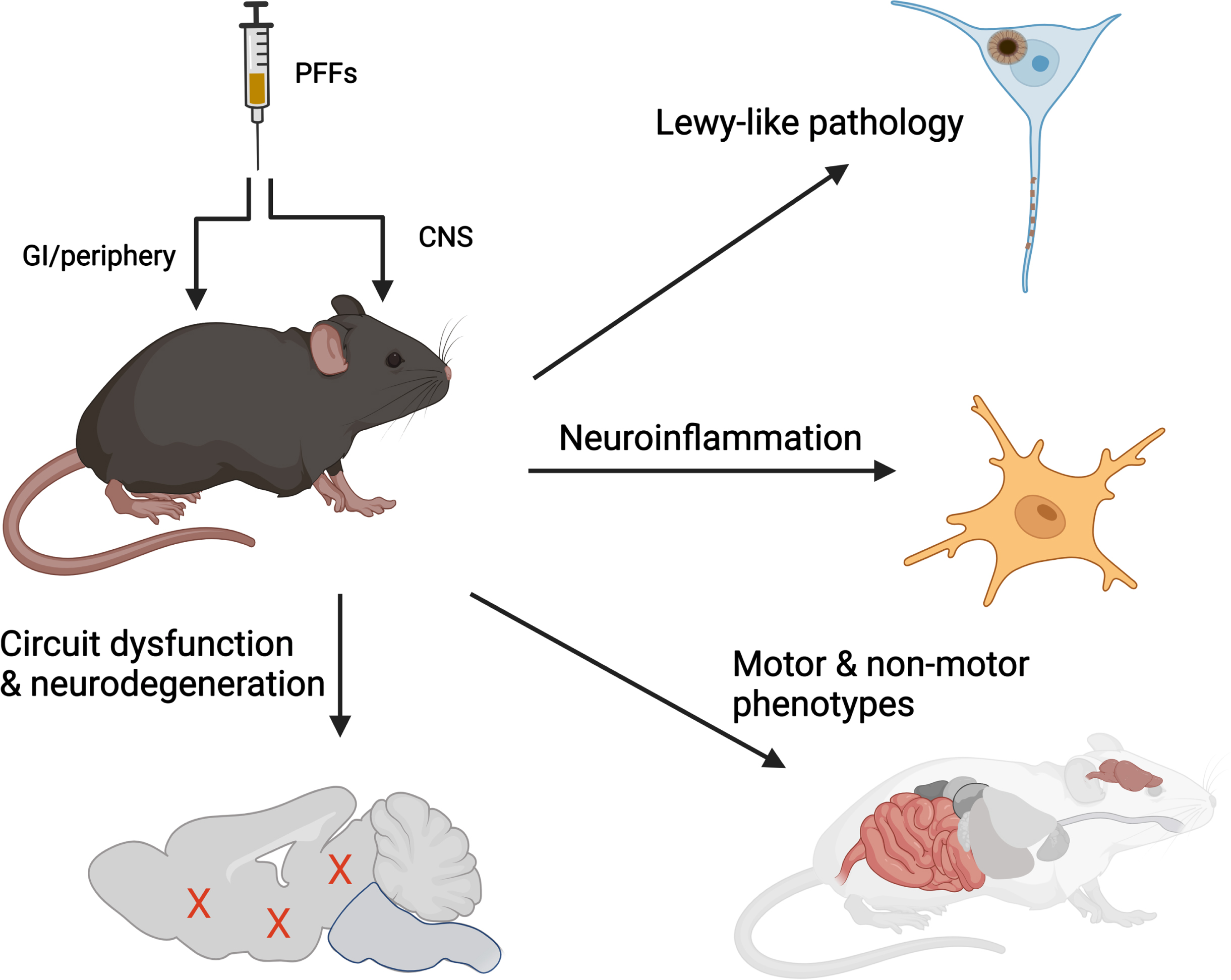
PFF-based models offer an additional technical advantage in that physiological α-synuclein expression is sufficient for supporting pathological seeding. This has permitted researchers to employ this approach across multiple vertebrate species including non-human primates. Furthermore, with no transgene requirement, the method can be applied across cells or animals with most genetic backgrounds, making it highly amenable for studying interactions with genetic risk factors (e.g., TMEM175) or co-pathologies (e.g., tau33 or amyloid-beta34). Interestingly, the disease phenotype responds faithfully (i.e., directionally similar to that observed in patients) in the listed examples. Since both clinical, genetic and experimental data thus far points to PD being the result of interactions between environment and/or pathogenic factors and the host, continuing to expand the types of host systems (i.e., cells or animal model) in which fibrils are injected into will likely yield impactful new biological insights.
As the statistician George Box noted: “All models are wrong, but some useful.” A little over a decade since its introduction, PFF-based seeding for PD evolved into a federation of related approaches that collectively mimic a clinically heterogeneous syndrome with decades-long disease duration and unclear etiology. This has provided important new insights with regards to how pathological spread is governed and the biological processes taking place downstream of α-synuclein misfolding. Arguably, this meets the basic criteria for being “useful”.
Nonetheless, this experimental paradigm is not without drawbacks and there remains room for improvement. From the technical perspective, although PFF inoculation into different anatomical targets can recapitulate several major PD symptoms, the list is far from complete, especially non-motor ones such as depression, anxiety and pain. In addition, most published studies have used recombinant α-synuclein PFFs as the seeding material owing to the relative ease that this material can be generated and characterized. Yet recent studies indicate that the core fibril structure of recombinant PFFs differs considerably from fibrils isolated from patients with Lewy-pathology or those with MSA, a synucleinopathy characterized by predominantly glial pathology, prominent autonomic involvement, and more rapid progression.8, 9 As these disease-specific “strains” also differ in their functional properties (e.g., the rate at which they can propagate in vitro or in vivo), it has been postulated that recombinant PFFs may be recapitulating an atypical form of PD or an altogether different entity.
Both of these concerns are justified but experimental strategies to address them are already started. For example, challenging previously untested inoculation sites will likely expand the spectrum of phenotypes that can be achieved. Though the concept of brain- vs. body-first PD subtypes is gaining traction,35 there remains a distinct possibility that pathology originates at multiple sites simultaneously, a hypothesis that can be tested experimentally. Regarding the issue of strains, in vitro amplification of patient-derived Lewy pathology and glial cytoplasmic inclusions has recently been described by multiple labs.15, 36 Deep characterization of these materials across commonly used in vitro and in vivo hosts will be essential in connecting how specific structural features lead to unique biological properties, and this should be a priority in the near and medium term. These studies may also help us better understand human PD where, despite their prominence, the toxic contributions of Lewy pathology and its spread have yet to be defined.
In conclusion, the PFF-based approaches have allowed users to specify the anatomical target and temporal window in which synucleinopathy is initiated. This has provided a versatile platform to model the family of diseases. Although the definition of PD has evolved since these tools were first introduced, PFF-based methods have already satisfied several of the key criteria for modeling this heterogeneous and progressive disorder.
ACKNOWLEDGMENTS
Figure 3 was made using BioRender.
FUNDING
A.L.W. is supported by grants from the NIH (R01NS121294 and accompanying supplement; R21NS127002), the Rainwater Charitable Foundation, and Colorado State University. K.C.L. is supported by grants from the NIH (R01NS088322, U19AG062418), and the Michael J. Fox Foundation.
CONFLICT OF INTEREST
A.L.W. is an Editorial Board Member of this journal but was not involved in the peer-review process of this article nor had access to any information regarding its peer-review. A.L.W. is a co-inventor on U.S. Patent Application PCT/US2023/072173, which is related to some of the research discussed. A.L.W. is a co-founder of Allagus Therapeutics. K.C.L. has received research support from GSK, Teva, Avid Radiopharmaceuticals, and WaveBreak Therapeutics, although this work is not discussed here.
REFERENCES
1. | Olanow CW and Prusiner SB . Is Parkinson’s disease a prion disorder? Proc Natl Acad Sci U S A (2009) ; 106: : 12571–12572. |
2. | Bessen RA and Marsh RF . Biochemical and physical properties of the prion protein from two strains of the transmissible mink encephalopathy agent. J Virol (1992) ; 66: : 2096–2101. |
3. | Hoyt F , Alam P , Artikis E , et al. Cryo-EM of prion strains from the same genotype of host identifies conformational determinants. PLoS Pathog (2022) ; 18: : e1010947. |
4. | Hoyt F , Standke HG , Artikis E , et al. Cryo-EM structure of anchorless RML prion reveals variations in shared motifs between distinct strains. Nat Commun (2022) ; 13: : 4005. |
5. | Kraus A , Hoyt F , Schwartz CL , et al. High-resolution structure and strain comparison of infectious mammalian prions. Mol Cell (2021) ; 81: : 4540–4551.e6. |
6. | Giles K , Olson SH and Prusiner SB . Developing therapeutics for PrP prion diseases. In: Prusiner SB (ed) Prion Diseases. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press, (2017) , pp. 579–597. |
7. | Zhang W , Falcon B , Murzin AG , et al. Heparin-induced tau filaments are polymorphic and differ from those in Alzheimer’s and Pick’s diseases. eLife (2019) ; 8: : e43584. |
8. | Yang Y , Shi Y , Schweighauser M , et al. Structures of alpha-synuclein filaments from human brains with Lewy pathology. Nature (2022) ; 610: : 791–795. |
9. | Schweighauser M , Shi Y , Tarutani A , et al. Structures of α-synuclein filaments from multiple system atrophy. Nature (2020) ; 585: : 464–469. |
10. | Yang Y , Garringer HJ , Shi Y , et al. New SNCA mutation and structures of α-synuclein filaments from juvenile-onset synucleinopathy. Acta Neuropathol (2023) ; 145: : 561–572. |
11. | Holec SAM and Woerman AL . Evidence of distinct α-synuclein strains underlying disease heterogeneity. Acta Neuropathol (2021) ; 142: : 73–86. |
12. | Holec SAM , Liu SL and Woerman AL . Consequences of variability in alpha-synuclein fibril structure on strain biology. Acta Neuropathol (2022) ; 143: : 311–330. |
13. | Groveman BR , Raymond GJ , Campbell KJ , et al. Role of the central lysine cluster and scrapie templating in the transmissibility of synthetic prion protein aggregates. PLoS Pathog (2017) ; 13: : e1006623. |
14. | Castilla J , Morales R , Saa P , et al. Cell-free propagation of prion strains. EMBO J (2008) ; 27: : 2557–2566. |
15. | Dhavale DD , Barclay AM , Borcik CG , et al. Structure of alpha-synuclein fibrils derived from human Lewy body dementia tissue. Nat Commun (2024) ; 15: : 2750. |
16. | Frieg B , Geraets JA , Strohäker T , et al. Quaternary structure of patient-homogenate amplified α-synuclein fibrils modulates seeding of endogenous α-synuclein. Commun Biol (2022) ; 5: : 1040. |
17. | Holec SAM , Lee J , Oehler A , et al. Multiple system atrophy prions transmit neurological disease to mice expressing wild-type human α-synuclein. Acta Neuropathol (2022) ; 144: : 677–690. |
18. | Höglinger GU , Adler CH , Berg D , et al. A biological classification of Parkinson’s disease: The SynNeurGe research diagnostic criteria. Lancet Neurol (2024) ; 23: : 191–204. |
19. | Schneider SA and Alcalay RN . Neuropathology of genetic synucleinopathies with parkinsonism: Review of the literature. Mov Disord (2017) ; 32: : 1504–1523. |
20. | Schmitz M , Candelise N , Canaslan S , et al. α-Synuclein conformers reveal link to clinical heterogeneity of α-synucleinopathies. Transl Neurodegener (2023) ; 12: : 12. |
21. | Luk KC , Kehm V , Carroll J , et al. Pathological alpha-synuclein transmission initiates Parkinson-like neurodegeneration in nontransgenic mice. Science (2012) ; 338: : 949–953. |
22. | Sossi V , Patterson JR , McCormick S , et al. Dopaminergic positron emission tomography imaging in the alpha-synuclein preformed fibril model reveals similarities to early Parkinson’s disease. Mov Disord (2022) ; 37: : 1739–1748. |
23. | Duffy MF , Collier TJ , Patterson JR , et al. Lewy body-like alpha-synuclein inclusions trigger reactive microgliosis prior to nigral degeneration. J Neuroinflammation (2018) ; 15: : 129. |
24. | Rey NL , George S , Steiner JA , et al. Spread of aggregates after olfactory bulb injection of alpha-synuclein fibrils is associated with early neuronal loss and is reduced long term. Acta Neuropathol (2018) ; 135: : 65–83. |
25. | Shen Y , Yu WB , Shen B , et al. Propagated alpha-synucleinopathy recapitulates REM sleep behaviour disorder followed by parkinsonian phenotypes in mice. Brain (2020) ; 143: : 3374–3392. |
26. | Goralski TM , Meyerdirk L , Breton L , et al. Spatial transcriptomics reveals molecular dysfunction associated with cortical Lewy pathology. Nat Commun (2024) ; 15: : 2642. |
27. | Dues DJ , Nguyen APT , Becker K , et al. Hippocampal subfield vulnerability to α-synuclein pathology precedes neurodegeneration and cognitive dysfunction. NPJ Parkinsons Dis (2023) ; 9: : 125. |
28. | Blumenstock S , Sun F , Klaus C , et al. Cortical circuit dysfunction in a mouse model of alpha-synucleinopathy in vivo. Brain Commun (2021) ; 3: : fcab273. |
29. | Kim S , Kwon S-H , Kam T-I , et al. Transneuronal propagation of pathologic α-synuclein from the gut to the brain models Parkinson’s disease. Neuron (2019) ; 103: : 627–641.e7. |
30. | Ding X , Zhou L , Jiang X , et al. Propagation of pathological α-synuclein from the urogenital tract to the brain initiates MSA-like syndrome. iScience (2020) ; 23: : 101166. |
31. | Mahul-Mellier AL , Burtscher J , Maharajan N , et al. The process of Lewy body formation, rather than simply α-synuclein fibrillization, is one of the major drivers of neurodegeneration. Proc Natl Acad Sci U S A (2020) ; 117: : 4971–4982. |
32. | Shahmoradian SH , Lewis AJ , Genoud C , et al. Lewy pathology in Parkinson’s disease consists of crowded organelles and lipid membranes. Nat Neurosci (2019) ; 22: : 1099–1109. |
33. | Vermilyea SC , Christensen A , Meints J , et al. Loss of tau expression attenuates neurodegeneration associated with α-synucleinopathy. Transl Neurodegener (2022) ; 11: : 34. |
34. | Bassil F , Brown HJ , Pattabhiraman S , et al. Amyloid-beta (Aβ) plaques promote seeding and spreading of alpha-synuclein and tau in a mouse model of Lewy body disorders with Aβ pathology. Neuron (2020) ; 105: : 260–275.e266. |
35. | Horsager J , Andersen KB , Knudsen K , et al. Brain-first versus body-first Parkinson’s disease: A multimodal imaging case-control study. Brain (2020) ; 143: : 3077–3088. |
36. | Uemura N , Marotta NP , Ara J , et al. α-Synuclein aggregates amplified from patient-derived Lewy bodies recapitulate Lewy body diseases in mice. Nat Commun (2023) ; 14: : 6892. |
37. | Li B , Ge P , Murray KA , et al. Cryo-EM of full-length α-synuclein reveals fibril polymorphs with a common structural kernel. Nat Commun (2018) ; 9: : 3609. |
38. | Guerrero-Ferreira R , Taylor NM , Arteni AA , et al. Two new polymorphic structures of human full-length alpha-synuclein fibrils solved by cryo-electron microscopy. Elife (2019) ; 8: : e48907. |


