
The AAV-α-Synuclein Model of Parkinson’s Disease: An Update
Abstract
Targeted delivery of α-synuclein using AAV vectors has over the two decades since its introduction developed into a versatile tool for modeling different aspects of synucleinopathy, mimicking those seen in Parkinson’s disease and related Lewy body disorders. The viral vector approach to disease modeling is attractive in that the expression of α-synuclein, wild-type or mutated, can be confined to defined anatomical structures and targeted to selected cell populations using either cell-type specific promoter constructs or different natural or engineered AAV serotypes. AAV-α-synuclein was initially used to model progressive α-synuclein pathology in nigral dopamine neurons, and, like the standard 6-OHDA model, it has most commonly been applied unilaterally, using the non-injected side as a reference and control. In recent years, however, the AAV-α-synuclein model has become more widely used to induce Parkinson-like synuclein pathology in other relevant neuronal systems, such as the brainstem noradrenergic and serotonergic neurons, the vagal motor neurons, as well as in oligodendrocytes, the prime target relevant to the pathology seen in multiple system atrophy. The purpose of this review is to give an overview of the progress made in the use of the AAV-α-synuclein model over the last two decades and summarize the state-of-the art in the use of the AAV-α-synuclein model for disease modeling in rats and mice.
Plain Language Summary
Misfolding of the neuronal protein α-synuclein is central to the cellular processes that underlie the development of Parkinson’s disease and related disorders, such as dementia with Lewy bodies and multiple system atrophy. Targeted delivery of α-synuclein using adeno-associated virus, AAV, has become a standard tool to model the disease process in animals. This AAV-α-synuclein model of Parkinson’s disease was introduced two decades ago and over the ensuing decades it has become a widely used standard tool for experimental studies in animals. The usefulness of the AAV-α-synuclein model is largely due to its flexibility and versatility as an experimental tool. In this review the authors summarize the state-of-the art in this field and review the range of applications that has been developed using AAV-α-synuclein alone, in single hit models, or in combinations with other interacting risk factors, in double hit models.
INTRODUCTION
α-Synuclein related pathology, synucleinopathy, is at the core of the pathogenetic processes seen in patients with Parkinson’s disease (PD), dementia with Lewy bodies (DLB) and multiple system atrophy (MSA). Modeling of different forms of synucleinopathy in rodents has been developed using either transgenic technology, viral vector mediated gene transfer, or inoculation with pre-formed fibril (PFF) seeds.1–3 These models are largely complementary as experimental tools. The adeno-associated virus (AAV) model was developed 20 years ago as a tool to overexpress wild-type (WT) or mutant forms of α-synuclein (α-syn) in nigral dopamine (DA) neurons, initially in rats,4–6 and later also in mice and non-human primates.7–11 While the transgenic and PFF models are particularly useful in mice, the AAV-α-syn model has become a standard tool for studies in rats, and increasingly so also in mice.
Although the use of the AAV-α-syn model demands some basic skills in stereotaxic surgery and handling of AAV vectors, it is experimentally attractive in that it makes it possible to express wild-type (WT) or mutated α-syn locally in selected targets. It was initially developed to model progressive α-syn pathology in nigral DA neurons, and like the standard 6-OHDA model it has typically been applied unilaterally, using the non-injected side as a reference. In recent years, however, the AAV-α-syn model has become a more widely used tool to induce and model PD-like synuclein pathology in other targets. Not only in neurons, such as the brainstem noradrenergic and serotonergic neurons,12 but also in oligodendrocytes, the prime target relevant for the synucleinopathy seen in MSA.13 The use of different AAV serotypes, and different cell type-specific promoters to drive the α-syn transgene, offers experimental flexibility and interesting opportunities for wider application of the AAV-α-syn model(Table 1).
Table 1
Summary of AAV-α-synuclein based rodent models discussed in this review. In these models, vector driven synuclein pathology is aimed at different cellular targets with the goal to replicate different aspects of PD-like pathology using the AAV-α-synuclein vector alone (“Single hit models”) in A, or in combination with a second, interacting risk factor (“Double hit models”) in B
| A Single hit AAV-α-synuclein models | |||
| Anatomical target | Cellular target | Disease modeling | Representative references |
| Substantia nigra | Dopamine neurons | Pre symptomatic/advanced PD | 4, 5, 6, 17 |
| Locus coeruleus | Noradrenaline neurons | Prodromal non-motor symptoms | 52, 53 |
| Midbrain raphe nuclei | Serotonin neurons | Prodromal non-motor symptoms | 54, 55 |
| Basal forebrain nuclei | Cholinergic neurons | Acetylcholine dependent cognitive impairment | 54, 56 |
| Dorsal motor nucleus of Vagus | Cholinergic motor neurons | Mediator of spread of synuclein pathology | 57, 58, 60 |
| Striatum | Oligodendrocytes | Modeling pathology seen in Multiple System Atrophy | 62, 65, 70 |
| B Double hit AAV-α-synuclein models | |||
| Combination | Added risk factor | Impact | Representative references |
| AAV-α-synuclein + Rotenone | Mitochondrial dysfunction | Conversion of pre-symptomatic to symptomatic PD | 73, 74 |
| AAV-α-synuclein + impaired GBA1 | Lysosomal dysfunction | Conversion of pre-symptomatic to symptomatic PD | 79, 82 |
| AAV-α-synuclein + LRRK2 mutation | Increased α-syn aggregation | Conversion of pre-symptomatic to symptomatic PD | 92, 93 |
The purpose of this review is to summarize the progress made in the use of the AAV-α-syn model over the last two decades and discuss the relevance of the α-syn overexpression as a model of the pathology seen in PD and other synucleinopathies. In human PD increased expression of α-syn, as seen in patients carrying duplications or triplications of the α-syn gene, can be causative, and in sporadic PD elevated α-syn levels is likely to be a susceptibility or risk factor for the development of the disease. Although the AAV-α-syn overexpression model can be said to mimic the condition seen in patients carrying duplications or triplications of the α-syn gene, it should be noted that profound motor impairments in the AAV-α-syn model require expression levels in the order of 4–6 times above normal, which is well above the level of α-syn seen in duplication and triplication patients, which is estimated at 1.5- to 2-fold above normal.14,15 This difference may be explained by the time-course of the degenerative changes that develops over decades in humans and over weeks and months in the AAV-α-syn model.
INDUCTION OF PROGRESSIVE PD-LIKE PATHOLOGY IN NIGRAL DA NEURONS
The synucleinopathy induced by AAV-α-syn injected into the substantia nigra is remarkably selective for the DA-containing neurons. Thus, despite that AAV-derived α-syn is expressed broadly in both DA and non-DA neurons, the degenerative changes—the α-syn aggregates, dystrophic neurites, and cell death—develop only in the DA neurons.4,16,17 This vulnerability is in line with studies showing that α-syn toxicity in DA neurons is mediated by an interaction with cytosolic DA18–20 and mitochondrial oxidant stress.21 Increased cellular α-syn levels, as achieved in the AAV model, may thus interact with other interacting factors, such as cytosolic DA and DA-related oxidative stress, to initiate and drive a toxic process in those neuron systems which are prone to develop the classic signs of PD toxicity, i.e., Lewy bodies and Lewy neurites.21,22
The toxic impact of α-syn overexpression in nigral DA neurons is clearly dose dependent. As is the case in patients with duplication or triplication of the α-syn gene higher α-syn expression levels are associated with earlier disease onset and more severe and rapidly progressing disease.15,23 An attractive feature of the AAV model, therefore, is that the vector dose can be adjusted so as to obtain α-syn expression levels compatible with either advanced, symptomatic disease (seen at high vector titers) or early stage/pre-symptomatic disease (seen at moderate vector titers).
Modelling advanced stage PD using high α-syn expression levels
The development of the AAV-α-syn model was initially focused on its use in rats, administered unilaterally into the substantia nigra, and from the outset efforts were made to maximize the toxic impact of the vector in order to model the structural and functional impairments seen in of advanced PD, characterized by major DA neuron loss and manifest impairments in standard motor tests. This turned out to be possible by shifting from the use of the (then standard) AAV2/2 serotype to more efficient alternatives, such as AAV2/5,24 2/6,5 2/7,17 and 2/925,26 with optimized promoter and enhancer constructs, and used at high viral titers. The AAV-α-syn model is applicable also in mice (AAV2/2;27 AAV2/7;8 AAV2/9;25 AAV2/6;28 AAV1/2;9 AAV2/529), although the vector types and titers that work well in rats tend to be less potent in mice, with more limited TH+ cell loss and behavioral impairments. (See1,30 for comprehensive reviews of studies performed in rats and mice until 2021).
The vectors used in these studies express human WT or A53T mutant α-syn, driven by strong promoters, such as CMV, CBA/CMV hybrid, Synapsin-1, or CBAie-enhanced Synapsin-1, in some cases also including a WPRE enhancer element. Use of human α-syn has become a common standard in the field. The use of human α-syn has the advantage that the delivered α-syn can be visualized and quantified using antibodies specific for the human version. Comparisons made between the WT and A53T versions suggest that they are fairly similar in their potency to induce α-syn pathology in DA neurons (see, e.g., 4,8).
In the rat studies quoted above the AAV-α-syn vectors have been used in working titers spanning two orders of magnitude, in the range of 1012-1014 genome copies (gc)/ml. In one case,17 the selected working titer was as low as 3×1011 gc/ml. The in vivo efficiency of the AAV-α-syn vectors used varies a lot due to a number of factors, including the serotype, the promoters and enhancers, and the production and purification methods used. Vector titer is commonly expressed as gc/ml, determined by DNA dot blot or qPCR. This measure, however, is a poor predictor of the efficiency of transduction, i.e., the number of infectious particles. Despite efforts to optimize the vector constructs and standardize the vector production, therefore, the impact, i.e., the magnitude of cell loss and the extent of motor impairment, is quite variable, not only between different vector constructs and different laboratories, but also from batch to batch generated by the same procedure. This is, at least in part, due to the difficulty to standardize the purity and infectivity of the vector batches. There are methods for determination of the infectious titer, but they are seldom used, and since they are performed on cells in culture with the equivalent vector expressing a fluorescent reporter they provide only an indirect measure of the efficiency displayed by the α-syn expressing vector on DA neurons in vivo.
As a more realistic and relevant alternative, therefore, it is recommended to perform an in vivo pilot test to select the working titer for each new batch. In our laboratory we have selected the working titer for each new vector batch in a test where we inject the vector unilaterally into the substantia nigra at three different dilutions, spanning from around 1012 to around 1013 gc/ml, with 3 animals/dose. As illustrated in Fig. 1, we select the working dose based on the magnitude of TH+ cell loss in the substantia nigra and loss of TH+ innervation in striatum assessed at 3-4 weeks post-injection, as determined by TH immunostaining. Based on available data5,24,25,31 the selected dose should result in a TH+ cell loss of at least 25–30% at 3-4 weeks, combined with a similar loss of TH+ innervation in parts of the striatum (Fig. 1E) and significant impairment in paw use and amphetamine-induced rotation when tested at longer time points (Fig. 1B,C). In addition, we use h-α-syn immunostaining to secure that the vector derived human α-syn is expressed in the terminals of the nigrostriatal projection throughout the ipsilateral caudate-putamen, the dorso-lateral part of the head of the caudate-putamen in particular, the part most closely linked to the motor symptoms (Fig. 1D). At the selected high doses α-syn will be expressed on the contralateral side but, as illustrated in Fig. 1D, the nigral DA neurons will not be transduced to a level needed to induce a-Syn expression in nigro-striatal axons projecting to the contralateral striatum.
Fig. 1
Selection of vector dose for the pre-symptomatic and advanced, symptomatic versions of the intranigral AAV-α-synuclein model, induced by a single, unilateral injection into the substantia nigra (A). A high vector dose is required to induce neurodegenerative pathology associated with a significant impairment in paw use and amphetamine-induced turning, as determined in this case at 7 weeks post-injection (B,C). At this dose the vector-derived human α-synuclein is expressed at a high level throughout substantia nigra (SN), along the axons in the nigrostriatal pathway (NSP) and in the terminals in the caudate-putamen (CPu) (D), and it induces already at 3-4 weeks a loss of at least 25–30% of the TH+ nigral neurons, and a corresponding loss of TH+ innervation in the striatum (E). At the moderate, 3-fold lower dose, TH+ cell number and TH+ striatal innervation is unaffected, or at most marginally reduced (at most 20–25%) when assessed at 3 weeks post-injection (G). TH+ cell loss may develop at longer survival times, 12–24 weeks, but the animals remain essentially non-symptomatic at shorter survival times (B,C). Importantly, to match the requirements for a prodromal level of synuclein pathology the vector-derived α-synuclein should cover a large part of the substantia nigra and be expressed at high level in the striatal terminals, most importantly in the motor-related dorsolateral part of the head of the caudate-putamen (F). Dashed line in C represents the threshold rate, 3 turns/min, commonly used to signify a significant motor impairment in the amphetamine rotation test. (Own original data).
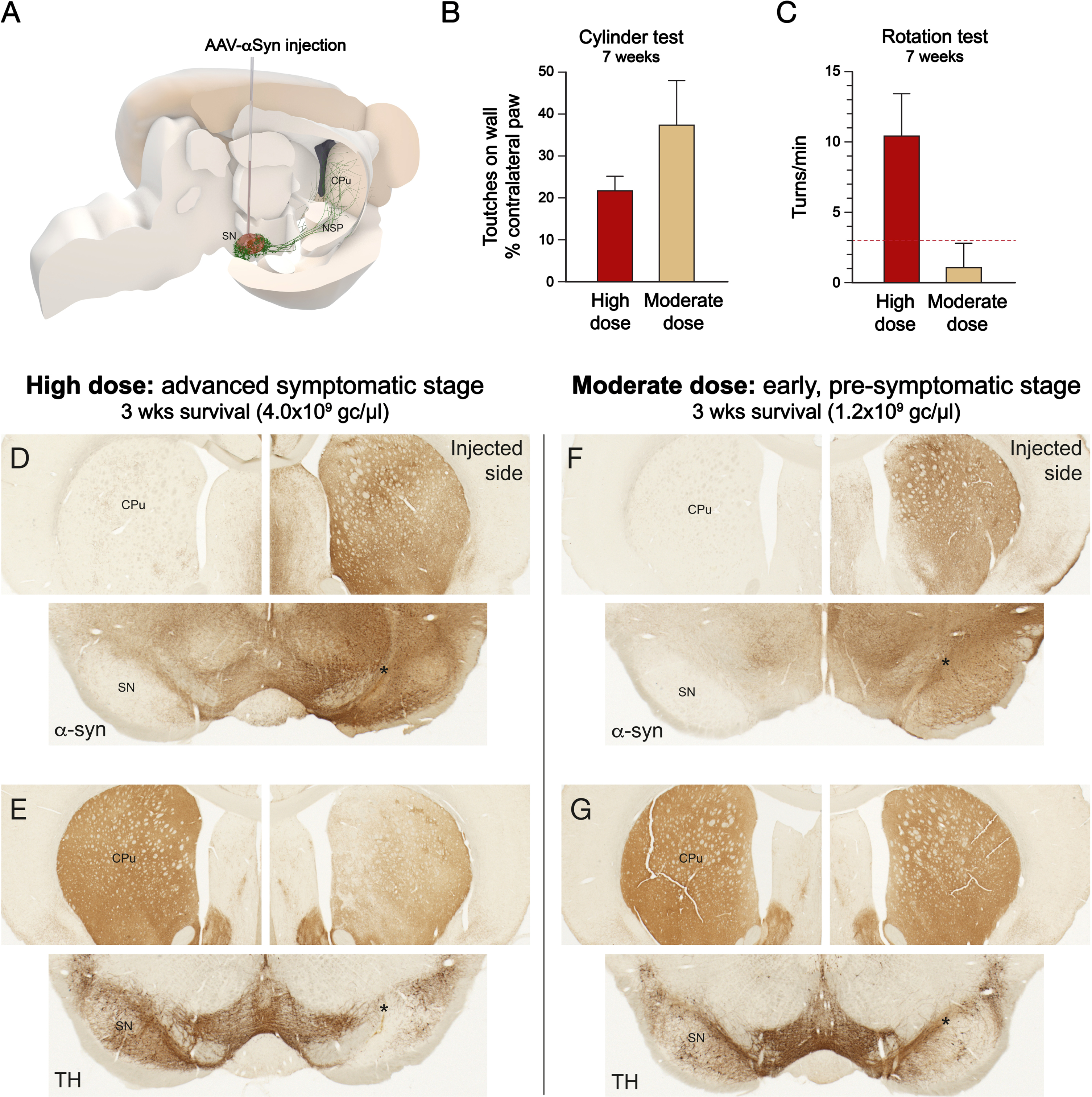
This test has a three-fold purpose: to make sure (i) that the infectivity of the selected titer is high enough to induce the level of DA neuron cell loss and axonopathy compatible with advanced stage PD, (ii) that the h-α-syn transgene is expressed in the vast majority of the nigral A9 neurons, and (iii) to avoid using the vector at excess titer, i.e., at a titer where toxicity unrelated to α-syn expression can kick-in. The use of very high titers of AAV-α-syn vectors carries the risk of overdosing, i.e., toxicity unrelated to α-syn expression. Control vectors expressing GFP is commonly used to control for this factor. At non-toxic levels this control vector will cause minimal or no damage, but there are examples in the literature where significant cell loss has been induced by the GFP control vector (see, e.g.,26,32). In such cases the results obtained with the matching α-syn vector may be difficult to interpret.
Time-course of degenerative changes seen at high vector titers
The impact of α-syn overexpression on DA neuron integrity and function is progressive. It hits the axons and terminals first and progresses over time to involve also the cell bodies. At high expression levels, in the range of 4-6-fold above the endogenous α-syn level,5,17,26,33 the retrograde progression of neurodegeneration resembles that described in human PD,34 although the degenerative changes seen in the AAV-α-syn model happen much faster than in the human disease. The time-course of changes, however, is spread out in time which makes it possible to distinguish stages that matches pre-symptomatic, early symptomatic, and advanced stages of the disease, allowing studies of stage-specific interventions and identification of targets for neuroprotective or disease-modifyinginterventions:
A pre-symptomatic stage, which spans over the first 2-3 weeks after vector injection, is characterized by an α-syn positive axonopathy that involves the appearance of axonal swellings that stain positively for phosphorylated form of α-synuclein (pSer129α-syn; p-syn), loss of TH+ terminals in striatum, downregulation of the DA synaptic machinery (TH, DAT and VMAT-2) and impaired synaptic DA release.35–37 The reduction in TH+ cell numbers seen at this early stage is in part due to downregulation of the TH enzyme.
These early changes are followed by an early symptomatic stage that evolves over the following 2-3 weeks, characterized by more extensive axonal pathology and TH+ terminal loss, partial loss of TH and VMAT-2 positive neurons and early signs of impairment in standard motor tests.
An advanced symptomatic stage is reached at about 6–8 weeks after vector injection. At this time-point the degenerative changes have reached a level when significant motor impairments and more substantial cell loss have developed. Part of the still surviving α-syn expressing nigral DA neurons remain in a dysfunctional state and the degenerative process may continue to progress during the following weeks.5,17,24,26
Amphetamine-induced rotation and forelimb use in the cylinder or stepping tests are commonly used to monitor motor impairment in unilaterally AAV-α-syn treated rats. Since none of the tests, used alone, can reliably identify well-lesioned animals in an experimental group it is recommendable to use a combination of at least two of them. Since these motor asymmetry tests are less informative in mice, tests of more general motor behavior, such as open field activity and rotarod and pole tests, are often included to monitor the impact of α-syn overexpression in mice with either unilateral or bilateral vector injections.
In contrast to the 6-OHDA lesion model where motor impairment is a direct consequence of the loss of DA neurons, the functional impairment in the AAV-α-syn model reflects a combination of cell death and dysfunction in still surviving dystrophic neurons. Thus, the magnitude of impairment seen in the standard cylinder and stepping tests is poorly correlated with the extent of DA neuron loss.5,24,26 Significant behavioral impairment can be seen with as little as 40–50% loss of TH+ cells in substantia nigra, while similar extent of impairment in the 6-OHDA model occurs only after more than 70–80% of the nigral neurons are lost.38,39 In the neurotoxin model the spared DA neurons remain functional and may even compensate, to some extent, for the lost neurons, while in the AAV-α-syn model spared neurons survive in a dystrophic state with p-syn+ cytoplasmic inclusions and swollen and distorted axons. Interestingly, these spared nigral neurons survive long-term in this pathological condition despite that the level of α-syn (judged by immunostaining) remains high. This suggests that the individual DA neurons may vary in their vulnerability to α-syn overexpression, and that some of the nigral A9 neurons, like the A10 neurons in the VTA, are able to resist α-syn toxicity, perhaps by more efficient handling and elimination of toxic α-syn species.
Modelling early-stage/pre-symptomatic PD using moderate and more physiological α-syn expression levels
Dose-response studies in rats24,32 and mice8 indicate that a moderate level of α-syn expression, compatible with a non-symptomatic, early-stage DA neuron pathology, as illustrated in Fig. 1F,G, is obtained with one-third to one-fourth of the genome copy (gc) titers used to generate the high dose advanced stage pathology described above.
The gc titer is a useful measure to standardize the doses used in different experiments as long as they are derived from the same batch. For each new vector batch, however, it is advisable to select the working titer based on an in vivo pilot test as described above. In this case the selected dose should transduce the entire A9 region of the substantia nigra and express the vector-derived h-α-syn at high level in the nigrostriatal terminals, as assessed by human-α-syn immunostaining, most importantly in the motor-related dorsolateral part of the head of the caudate-putamen. The TH+ cell number and the TH+ striatal innervation should be unaffected or at most marginally reduced (less than 20–25%) when assessed at 3-4 weeks post-injection. TH+ cell loss may develop at longer survival times, 12–24 weeks, but the animals remain essentially non-symptomatic.5,16 The early reduction in TH+ cell number is, in part at least, due to downregulation of TH in cells expressing pSer129-α-syn (p-syn). The appearance of p-syn+ granular cytoplasmic aggregates in the affected neurons is an additional feature of sub-threshold α-syn pathology that appears early and remains also at longer time-points.
The level of α-syn expression compatible with pre-symptomatic DA neuron pathology has been estimated to be 2-3-fold above the endogenous α-syn level.5 At this level of expression, which is in the range of those seen in patients carrying duplication or triplication mutations of the α-syn gene,14,15 α-syn may act as a susceptibility factor that can interact with other genetic or environmental risk factors to drive the development of a disease-causing degenerative process.21,22 The use of this sub-threshold AAV-α-syn model in such “double-hit” combinations (see Table 1) will be discussed further below.
Combination of AAV-α-synuclein with preformed fibril (PFF) seeds
The level of α-syn overexpression needed to induce nigral DA neuron cell death of a magnitude sufficient to induce significant motor impairments is well above what may occur in human PD. This raises the question whether the toxicity and pathology associated with such high α-syn expression levels is relevant as a model for the clinical condition. One way to circumvent this limitation is to combine AAV-mediated α-synuclein overexpression with preformed fibril (PFF) seeds, delivered into the substantia nigra either mixed in a single injection,40,41 or as two separate injections.16 PFFs are known to act as seeds for the recruitment of monomeric α-synuclein into toxic fibrillar aggregates, and the speed by which this happens is dependent on the level of α-synuclein present in the cell.42,43 The seeding is most efficient if PFFs and monomeric α-synuclein are from the same species.44
In the combined AAV-PFF (SynFib) model45 the AAV vector expressing human WT α-synuclein is used at the moderate dose level used to induce early-stage, non-symptomatic DA neuron pathology, as described above, and combined with a dose of human PFFs that, by itself, is not able to induce significant DA neuron loss at short survival times. Rats or mice receiving this combination exhibit enhanced and accelerated development of pathology with p-syn+ inclusions appearing in cell bodies, axons and dendrites, accompanied by a prominent inflammatory response that develops already within the first month.16,41,45 As illustrated in Fig. 2, the magnitude of these early changes far exceeds those seen in animals treated with the AAV-α-syn vector alone, both at the level of the cell bodies in the nigra (Fig. 2B-E) and at the level of the axonal in the striatum (Fig. 2G-I).
Fig. 2
In the SynFib model where the AAV-α-synuclein vector is used in combination with pre-formed fibrils (PFFs) the PFFs act as seeds to accelerate and amplify the formation of toxic α-synuclein aggregates. The vector is used at a moderate dose level to induce early-stage, non-symptomatic DA neuron pathology when the vector is used alone, as illustrated in panels D and E in Fig. 1, combined with a dose of human PFFs that, by itself, is not able to induce DA neuron loss at shorter survival times (B,G). The development of pS129-α-synuclein (p-syn) pathology and microglial activation (A,F) is enhanced and accelerated, both in nigra (D,E) and striatum (H,I) and is observed already at 4 weeks post-injection. (Own original data).
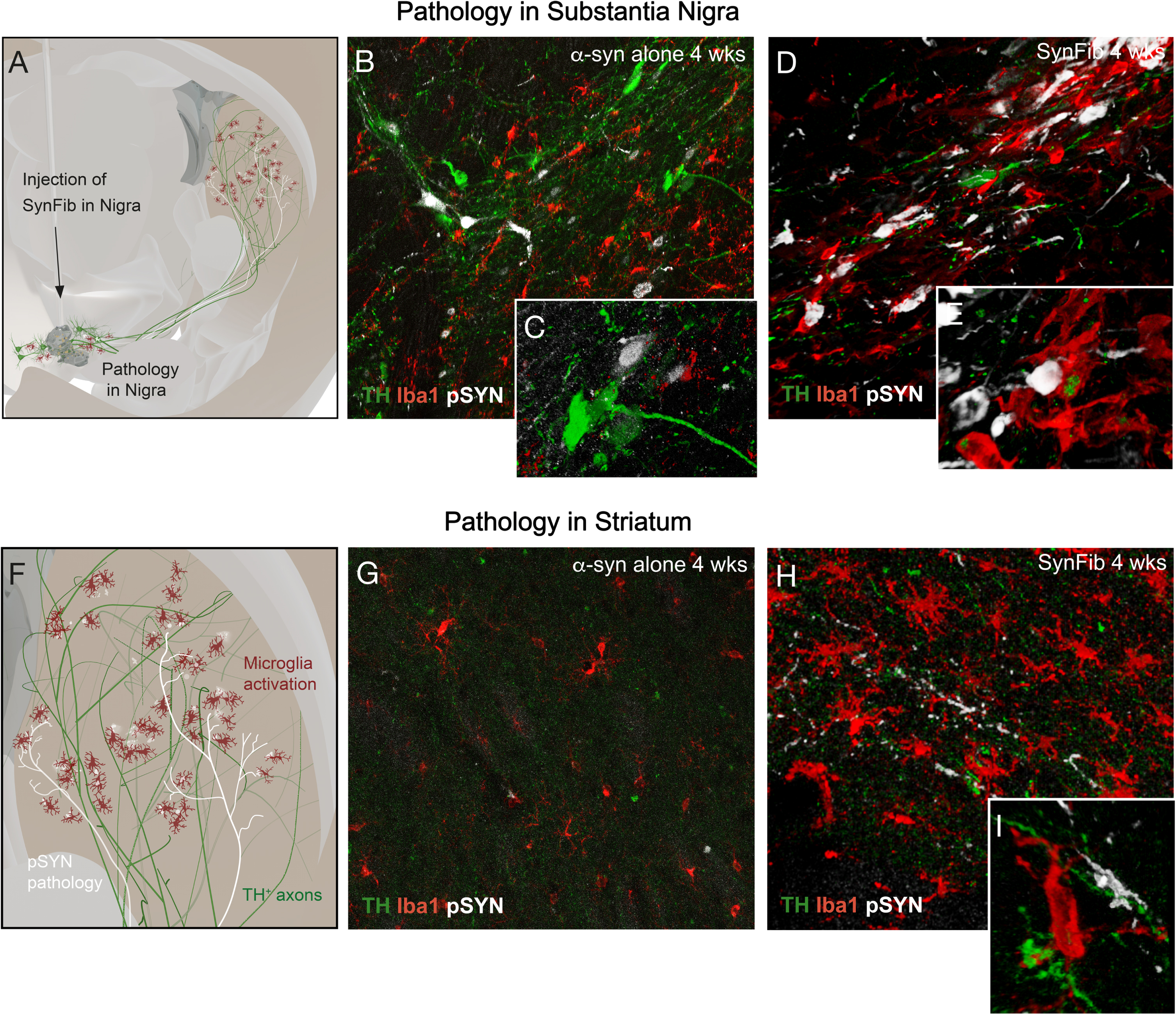
In the Hoban et al. study,41 motor impairment (assessed in the stepping and cylinder tests) and significant loss of nigral TH+ neurons were observed already at 4 weeks post-injection. Notably, at this early time-point most of the p-syn expressing nigral neurons stained negatively for TH, indicating that the reduction in the TH+ cell number, which amounted to about 50–60% at 4 weeks, reflected a combination of cell loss and TH downregulation in still surviving p-syn expressing neurons. At the longer time-point, 16 weeks, these p-syn positive/TH negative nigral neurons were gone, indicating a progressive loss of the p-syn expressing DA neurons over time. Thus, part of the affected DA neurons initially remains intact but in dysfunctional state linked to axonopathy and impaired DA neurotransmission, which is followed by a progressive loss of the p-syn expressing neurons that takes place over the subsequent weeks.
The SynFib model shares features with the 6-OHDA lesion model in that it is applied unilaterally in a single dose, and that the toxic impact is sufficient to induce significant impairments in both spontaneous and drug-induced motor behavior in a high percentage of the treated animals. In the studies conducted so far, the long-term nigral TH+ cell loss has been consistently above 50% provided that the injections have been correctly placed. Similar levels of nigral cell loss can be obtained in the AAV-α-syn only model but only at very high α-syn expression levels. As described in detail elsewhere45 the magnitude of cell loss obtained in the SynFib model, and the speed by which it happens, makes it possible to use simple behavioral tests to monitor the extent and progress of DA neuron pathology and degeneration over time. The early changes in the rotation and paw use tests seen at 4 weeks post-injection may be used as a predictor of the long-term loss of nigral TH+ neurons and the loss of TH+ innervation in the striatum. An amphetamine rotation score of >3 turns/min or, alternatively, a score of ≤35% contralateral touches, in % of total touches, in the cylinder test, recorded at 4 weeks, have been proposed to be used as a selection criterion to identify rats with more than 60% loss of nigral TH+ neurons recorded at longer timepoints.45
AAV-α-SYN INDUCED SYNUCLEINOPATHY IN NORADRENERGIC, SEROTONERGIC AND CHOLINERGIC NEURONS
The dorsal motor nucleus of the vagus (DMV), the locus coeruleus (LC) and the midbrain raphe nuclei (mRN) are among the first structures in the brain to be hit by synuclein pathology. DMV, in particular, develops α-syn inclusions early and is likely to play a role in the caudal-to-rostral spread of synuclein pathology.46 Dysfunction of the noradrenergic neurons in the LC and the serotonergic neurons in the mRN may contribute to the prodromal non-motor symptoms that precede the DA-related motoric impairments, such as anxiety, depression, anhedonia and disrupted sleep behavior.47,48 Like the nigral DA neurons, these neuronal systems have extensive and widely spread axonal networks and that may make them particularly vulnerable due to the excessive metabolic demand linked to the maintenance of activity in a highly branched axonal network, and similar to the nigral DA neurons these neurons exhibit the kind of autonomous peacemaking activity that is associated with calcium entry and high metabolic demands.22,49 The selective vulnerability of LC neurons may also be explained by their content of noradrenaline, a catecholamine that possesses latent toxic oxidative properties similar to those of DA.
AAV-mediated expression of α-synuclein offers a flexible tool to induce synucleinopathy selectively in these neuronal populations. Although the small size of these structures is challenging and demands precise stereotaxic surgery, AAV-mediated α-syn transfer offers interesting possibilities to target and adapt the vector construct to each neuron type selectively.
AAV-α-syn targeting the LC has been explored in mice in Wolfgang Oertel’s lab using an AAV1/2 vector expressing the A53T-α-synuclein mutant under the CBA promoter.50,51 The cellular changes replicate some of the features of α-syn pathology seen in the LC in PD patients: aggregation of phosphorylated α-syn with signs of proteasomal and lysosomal dysfunction, gradual development of signs of pathology including dystrophic axons and dendrites, a prominent microglial and astroglial response, and a progressive loss of the TH+ noradrenergic neurons. The progression of degenerative changes, in fact, is quite similar to that seen in AAV-α-syn transduced nigral DA neurons (see above), with cell loss starting at around 3 weeks and reaching about 60% at the longest timepoint studied, 9 weeks. Notably, the α-syn overexpression obtained with this vector, driven by the non-selective CBA promoter, was not confined to the noradrenergic neurons but included also neurons in adjacent structures. Nevertheless, it seems that α-syn pathology developed selectively in LC neurons, which is in line with the idea that the presence of noradrenaline in the cells confers vulnerability to α-syn induced toxicity. The impact of α-syn overexpression on neuronal firing properties was explored electrophysiologically,51 but since the vector injection was applied unilaterally the impact on LC-dependent behaviors was not pursued in this experiment.
AAV-α-syn targeting the midbrain raphe nuclei has been explored in two studies using either an AAV 2/6 vector expressing WT human α-syn driven by the cell-specific tryptophan hydroxylase promoter, performed in rats,52 or an AAV2/5 vector expressing human WT α-syn under the non-selective CBA promoter, performed in mice.53 The α-syn pathology obtained in these two studies was relatively mild, characterized by α-syn and p-syn positive inclusions and swollen and distorted α-syn+ axons and dendrites that developed over the first 4–6 weeks. The number of tryptophan hydroxylase positive cell bodies remained unchanged, up to 8 weeks in the mouse study and up to 20 weeks in the rat study. The density of SERT positive fibers in cortical and limbic forebrain regions was reduced, by about 50% at 8 weeks in the mouse study. It remained unchanged at up to 12 weeks in the rat study, followed by a loss of about 40% seen at 20 weeks post-injection. 5-HT tissue levels in cortical and limbic forebrain areas, however, remained unchanged in both cases. Anxiety-like behavior, as assessed in the elevated plus maze or the dark/light box test, was unaffected in both studies, while depressive-like behavior in the forced swim test was significantly enhanced in the mouse study but remained unaffected in the rat study. This difference may be due to the expression of α-syn in non-serotonergic neurons linked to the non-selectivity of the CBA-driven AAV vector used in the mouse study.
Considering the relatively moderate level of α-syn protein expression obtained in these studies it is not surprising that the resulting degenerative changes were quite modest. More severe pathology and serotonergic cell loss may be obtained using higher vector titers, or alternatively using a mixture of AAV-α-syn and pre-formed fibrils (PFFs), as in the SynFib model described above, allowing the added PFFs to act as seeds for the generation of toxic α-syn aggregates.
The cholinergic basal forebrain cholinergic neurons located in the medial septum /diagonal band area and the nucleus basalis of Meynert are affected later in the disease. The synucleinopathy that develops over time in these neurons is accompanied by degenerative changes, axonopathy and cell loss that correlate with the development of cognitive impairments.54,55 AAV-α-syn targeting the basal forebrain cholinergic neurons has been explored in rats in two studies, using either an AAV2/5 vector56 or an AAV2/6 vector expressing human WT α-synuclein driven by the synapsin-1 promoter.52 In both cases the vector was injected bilaterally and targeted on the medial septum/diagonal band area, in some of the animals in combination with an injection of the same vector in the ventral tegmental area (VTA)56 or an injection of the TPH-driven vector (see above) in the midbrain raphe nuclei.52 Following a series of behavioral tests, the animals were processed for histological analysis at 24 weeks post-injection. The pathological changes seen at this long-term timepoint were confined to an extensive axonal pathology characterized by axonal swellings and proteinase-K resistant α-syn aggregates, without any cholinergic cell loss. The number of ChAT-expressing septal/DB neurons was reduced by 40–50% due to downregulation of the enzyme in surviving neurons. The functional tests performed in the Wan et al. study52 did not show any impairments in either place learning in the water-maze test, anxiety-like behavior in the elevated plus maze, or depressive-like behavior in the forced swimtest.
Although limited and preliminary, these studies suggest that the basal forebrain cholinergic neurons are less vulnerable to elevated α-syn levels than nigral DA neurons and noradrenergic LC neurons, but somewhat similar to the more resistant DA neurons in the VTA.56 An experimentally useful model of basal forebrain cholinergic synucleinopathy will require a more efficient vector tool, or most likely, a double-hit approach, such as the combination of an AAV vector with synclein PFFs.
A method for AAV-α-syn targeting the dorsal motor nucleus of the vagus (DMV) has been developed by Donato Di Monte and colleagues,46 applied to either rats57,58 or mice.59,60 In this method an AAV-α-syn vector of the 2/6 serotype, expressing human WT α-syn and driven by the synapsin-1 promoter, is injected unilaterally into the vagal nerve using either a thin 60μm glass capillary fitted to a 5μl Hamilton syringe (in rats57) or a thin 36 gauge metal needle fitted to a 10μl NanoFil syringe (in mice59). The AAVs are transported from the injection site retrogradely to the cholinergic motor neurons located in the DMV, and anterogradely to the neurons in the sensory vagal ganglia, resulting within 1-2 weeks in widespread expression of human α-syn in the DMV neurons and their axonal and dendritic projections, as well as in the vagal afferents innervating the dorsal medulla oblongata, the solitary nucleus inparticular.
The h-α-syn protein was expressed in 30–40% of the DMV cholinergic neurons, exclusively on the injected side. Their number declined over time, by 30% at 3 months, 85% at 6 months and 95% at 12 months, indicating a progressive degenerative process induced by the elevated α-syn level.58 The h-α-syn was largely cytoplasmic, in the absence of h-α-syn+ inclusions or aggregates, but with signs of oxidative stress, indicated by elevated ROS levels. Interestingly, a further 1.5-fold increase in ROS levels, induced by systemic treatment with the ROS-generating agent paraquat, reduced the number of surviving h-α-syn expressing neurons by 25%. This was accompanied by an increase in the oxidized or nitrated form of α-syn, seen as granular inclusions in the affected neurons, and an increased aggregation of oligomeric forms of the protein.60 Thus, similar to what has been shown to occur in nigral DA neurons, the toxic impact of oxidative stress and h-α-syn overexpression act in synergy to induce degenerative changes in the DMV neurons.
The Di Monte lab has used the vagal model to study the role of DMV in the caudal-to-rostral spread of α-synuclein pathology.46,57,58 In support of this idea, they observe a gradual spread of vector-derived h-α-syn in axons distributed in areas of the pons and midbrain, including the coeruleus-subcoeruleus region and dorsal raphe nuclei, suggesting transfer of h-α-syn to afferents terminating on the transduced DMV neurons. This spread is dependent on the continuous supply of h-α-syn from the transduced DMV neurons, but independent of any seeding mechanism involving endogenous α-syn. It expands over the first 3 months, when it reaches its maximum, and declined during the following months. α-Syn toxicity is observed in the form of axonal pathology and partial loss of neurons in the LC, amounting to 15% at 1 year, accompanied by microglial activation that was evident already by 3 months.
Although experimentally valuable, the clinical relevance of the intra-vagal AAV-α-syn model is limited by the lack of α-syn accumulation or aggregates in neuronal cell bodies beyond the DMV, and the absence of h-α-syn spreading and pathology in the substantia nigra. This is in contrast to the gut-to-brain model where α-syn PFFs are injected into the gut, and where the spread to the brain is mediated by seeding and transfer of endogenous α-syn. In this case neuronal α-syn pathology, in the form of p-syn+ inclusions and overt nigral DA neuron loss is evident (see, e.g.,61).
AAV-α-SYN INDUCED SYNUCLEINOPATHY IN OLIGODENDROCYTES AS A MODEL OF MULTIPLE SYSTEM ATROPHY
Unlike PD and DLB, where the α-syn inclusions are confined to neurons, the inclusions formed in patients with MSA are predominantly located in the cytoplasm of oligodendroglia. These glial cytoplasmic inclusions (GCI) contain α-syn filaments that are structurally different from those in PD or DLB. The neuropathological features of MSA include demyelination, axonal damage and neurodegeneration combined with microglial activation, astrogliosis and T-cell infiltration.62 The accumulation of α-syn containing GCIs, which occurs early in the disease, seems to drive the neurodegenerative changes and is also well correlated with the extent of neuronal loss and demyelination.63,64 Based on pathology and symptoms one distinguishes two major types: a parkinsonian variant, MSA-P, with prominent nigrostriatal degeneration and parkinsonian features, and a cerebellar variant, MSA-C, with olivopontocerebellar atrophy and ataxia.
The viral vector models of MSA are based on the use of AAV vectors that allow expression of h-α-syn with high selectivity, and at high levels, in oligodendrocytes. The oligo selective AAV vector constructs developed so far are of two types. In one version selectivity is obtained by including a promoter sequence that drives the expression of human WT α-syn efficiently in oligodendrocytes, using either the promoter for myelin basic protein, MBP,65 or the promoter for myelin associated glycoprotein, MAG,66,67 In another version selectivity is obtained by using a synthetic capsid, called Olig001, that contains a chimeric mixture of elements from AAV1,2,6,8 and 9 that provides the vector with a high affinity for transduction of oligodendrocytes.62,68–70 In both cases the vectors are injected into the striatum in order to mimic the striato-nigral pathology of the parkinsonian variant, MSA-P.
In the study of Bassil et al.,65 the AAV1/2-MBP-hα-syn vector was injected bilaterally in rat striatum at a titer of 2.33×1013 gc/ml, and the rats were perfused for analysis at 3- and 6-months post-op. α-Syn was preferentially expressed in oligos (84%) and to a minor extent in neurons (11%), in part in an insoluble, proteinase-K resistant form. The α-syn expressing oligodendrocytes covered around 60% of the striatal volume and they occurred also outside striatum, in corpus callosum, motor cortex, globus pallidus and substantia nigra, suggesting spread of either vector derived α-syn, or the vector itself, along known anatomical pathways. As illustrated in Fig. 3A-F, the AAV1/2-MBP-hα-syn injected rats developed a progressive L-dopa-resistant motor deficit that was detectable at 2 months and became prominent at 6 months, accompanied by a progressive loss of neurons in both striatum (–23% at 6 months) and substantia nigra (–42% at 3 months and –67% at 6 months). Myelin damage and immune/inflammatory changes were not investigated in this study.
Fig. 3
Features of the MSA-like neurodegenerative changes induced by overexpression of human α-synuclein in oligodendrocytes, obtained by intrastriatal injection of an AAV1/2 vector driven by the promoter for myelin basic protein, MBP (A-F),65 or intrastriatal injection of a chimeric AAV-α-synuclein vector, Olig001, with high affinity for oligodendrocytes (G-J).70 In the Bassil et al. study,65 α-synuclein was selectively expressed in oligodendrocytes and covered a large part of the striatum (A,B). The rats developed motor impairments over time (C), associated with a progressive loss of TH+ and Nissl-stained neurons in the nigra (D,E) and NeuN+ neurons in the striatum (F). In the Marmion et al. study,70 the Olig001 vector was efficient in inducing widespread pS129-α-syn positive and proteinase K resistant cytoplasmic inclusions selectively in oligodendrocytes (A,B), accompanied by a dose-dependent loss of TH+ neurons in the nigra and NeuN+ neurons in the striatum, most pronounced at high titer (HT) of the vector (I,J). (Data compiled from65 and70).
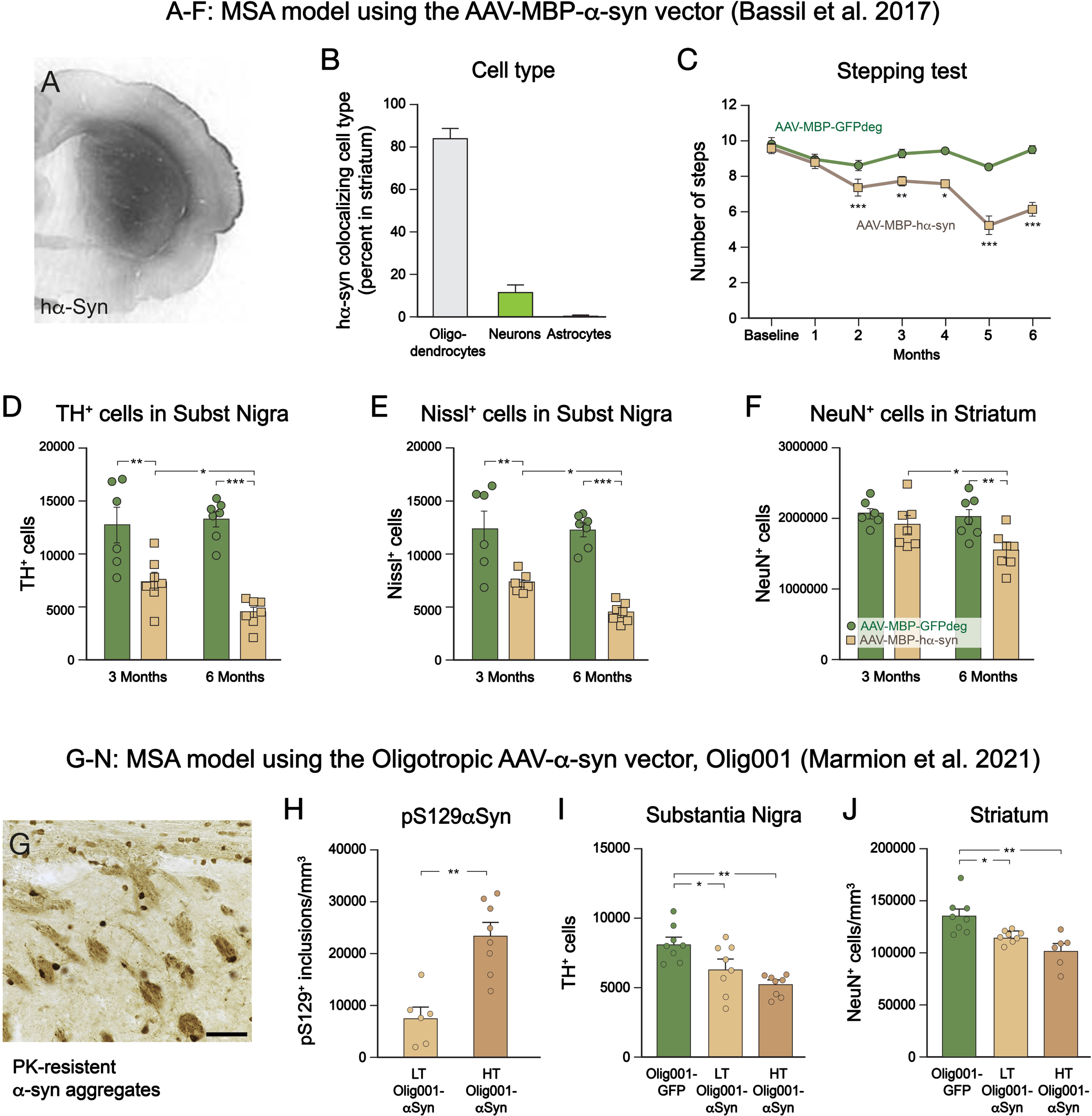
In the studies using the Olig001-hα-syn vector, the vector was delivered bilaterally in the striatum in rats at two different doses, 2.4×1012 gc/ml and 2.3×1012 gc/ml,70 and unilaterally in mice at a dose of 1.0×1013 gc/ml.62 The vector was highly efficient: over 95% of the transduced cells are oligos, with little or no expression in neurons or astrocytes.69
The pathology induced by intrastriatal delivery of the Olig001-hα-syn vector shares key features with human MSA of the parkinsonian type: wide-spread appearance of insoluble GCI-like α-syn+ inclusions in oligodendrocytes, myelin damage, and neuronal cell loss in both striatum and substantia nigra.70 (Fig. 3G-J). The distribution of pSer129-α-syn+ inclusions outside the striatum, in globus pallidus, thalamus and substantia nigra, suggests transfer of oligo-derived α-syn along anatomical pathways. These changes were dose-dependent and fully expressed only at the highest dose.
In the Williams et al. study in mice62 the wide-spread appearance of p-syn+ inclusions in oligodendrocytes, seen at 4 weeks in the striatum and corpus callosum, was accompanied by signs of demyelination, microglial activation and infiltration of CD4+ and CD8+ T-cells, closely associated with the area of demyelination and the p-syn expressing oligodendrocytes, similar to what have been observed in MSA patients. No neuronal cell loss was observed at this early time-point which is consistent with the idea that the accumulation of p-syn+ inclusions precede neuronal degeneration. Together with the associated T-cell response, these GCI-like inclusions may be driving the progression of pathology in this MSA model.
Compared to the transgenic mouse models where α-syn is constitutively expressed under the control of oligodendrocyte-specific promoters, these AAV vector models offer advantages in that the insult can be targeted to MSA-specific brain regions with α-syn expression levels high enough to induce more prominent neuropathology, neuronal cell death and motor deficits. And they are applicable not only in rats and mice, but also in non-human primates.65,69,70
USE OF AAV-α-SYN IN DOUBLE HIT MODELS
Increased cellular levels of α-syn, as obtained with AAV-α-syn vectors, can be toxic to neurons at high doses. At more physiological levels, however, α-syn can be viewed as a vulnerability factor that interacts with other risk factors to trigger the characteristic pathogenic process seen in symptomatic PD and DLB. As discussed above, this opens possibilities to use AAV-syn at pre-symptomatic moderate doses in combination with other insults to drive the kind of interactive pathogenic mechanisms seen in so-called double hit models. In this section we will discuss three such examples where AAV-induced α-syn overexpression is combined with either mitochondrial dysfunction (Rotenone treatment) or mutations in the GBA1 or LRRK2 genes, three major risk factors that interact with α-syn in the pathogenesis of the disease.21,71
AAV-α-syn combined with rotenone
Mitochondrial dysfunction, and deficits in complex I of the respiratory chain in particular, is an integral part of PD pathology. In human DA neurons elevated mitochondrial oxidative stress can trigger a toxic cascade leading to lysosomal dysfunction and α-syn accumulation, suggesting that oxidative stress resulting from mitochondrial impairment can increase the toxicity induced by elevated levels of α-syn.21,71 Thus, moderate overexpression levels of α-syn may become toxic if they are combined with complex I inhibition.
This double hit approach has been explored in rats by Eilís Dowd and her collaborators using intranigral AAV-α-syn in combination with the complex I inhibitor rotenone, given either systemically72 or locally in the substantia nigra73 or striatum.74 In their design rotenone was administered 3 months after AAV-α-syn, i.e., at a time-point when the vector derived α-syn has reached a stable level and the initial impact has subsided. Rotenone was administered either as a single intracerebral injection, or subcutaneously over 4 weeks using an osmotic minipump.
In all three cases the sequential administration of these two risk factors resulted in more pronounced pathology and motor impairments, at a level that was significantly greater than that seen after either insult alone. The most interesting results were obtained in the Naughton et al. study74 where rotenone was injected into the striatum in combination with intranigral AAV-α-syn given at a moderate, pre-symptomatic dose. Neither insult alone was sufficient to induce significant motor impairment as assessed in the stepping, corridor and whisker tests, while the rats with combined insults developed marked and significant impairments in all three tests, as well as a high, ipsilateral turning rate, 4–6 turns/min, in response to amphetamine. The motor impairment, observed on the side contralateral to the injections, appeared within 1 week after rotenone injection and remained stable for the following 4 weeks. TH immunohistochemistry, performed at this timepoint, showed an additive effect of the two insults on TH+ cell loss in the ipsilateral substantia nigra: around 30% in the AAV only and rotenone only groups, and around 60% in the combined group. As discussed above, the magnitude of cell loss obtained in the combined group, in combination with extensive axonopathy, is well within the range needed to induce significant motor impairment in animals given high doses of AAV-α-syn.
The results obtained in this study support the idea that α-syn, at the moderately elevated cellular levels seen in patients with sporadic PD, makes the affected nigral DA neurons vulnerable to a subsequent mitochondrial insult. The combination of the two insults, as used by Naughton et al.,74 thus provides an interesting example of a double hit AAV model where the development of pathology and motor impairment akin to symptomatic PD can be triggered in animals primed with moderate, pre-symptomatic doses of AAV-α-syn.
AAV-α-syn combined with impaired GBA1 function
Mutations in the GBA1 gene is a prevalent genetic risk factor for PD.75 GBA1 encodes for a lysosomal enzyme, glucocerebrosidase (GCas). GCas level and activity is reduced, not only in GBA patients but also in PD patients without GBA1 mutation.76,77 It is estimated that 10–30% of individuals with GBA1 mutations will develop PD by the age of 80, suggesting that GBA1 mutations alone is not sufficient to cause PD pathology, but that an additional factor such as increased expression of α-syn is required. This is the same in GBA1 knock-out mice or L444P knock-in mice where brain GCas level and activity is reduced by 50–80%.
Transgenic knock-out or knock-in mice show progressive accumulation of α-syn, but no overt α-syn pathology or DA neuron cell loss.78–80 In α-syn overexpressing transgenic mice, by contrast, expression of mutant GBA1 compromises lysosomal function and causes accumulation of toxic α-syn oligiomeres, loss of nigral DA neurons and motor impairments.80 Enhanced α-syn mediated toxicity, induced by partial depletion of GCas, are observed also in cellular α-syn overexpression models.78,81
These data show that reduced GCas activity alone is not sufficient to cause PD like pathology and that additional factors, such as increase in the cellular α-syn level, are required for overt PD-like pathology to develop. This suggests a possible double-hit model where AAV-vector induced α-syn, expressed at a pre-symptomatic level, is combined with reduced expression of GCas. Schapira and collaborators79 and Stefanis and collaborators82 have explored this possibility in studies where AAV-α-syn was delivered unilaterally using a moderate dose of the AAV vector, either in GCas deficient mice or in combination with an AAV vector expressing a GCas-downregulating microRNA. In both versions, overexpression of α-syn in the presence of reduced levels of GCas resulted in increased accumulation of α-syn in nigra and striatum, and a 30–50% loss of TH+ and Nissl-stained neurons in the ipsilateral substantia nigra. In the Polissidis et al. study82 this was accompanied by a 50% reduction in striatal DA levels and a significant impairment in paw use the cylinder test.
These findings point to a role of GCas as a regulator of α-syn toxicity, which is further supported by studies showing that increased expression of GBA1 prevents the development of α-syn pathology and cell death in the AAV-α-syn rat model.83,84 The concept underlying this combined double-hit AAV-α-syn/GCas model is compelling and deserves to be explored further, e.g., by extending the survival time beyond 2 months in order to give more time for the full impact to develop, and/or by using transgenic mice with more prominent reduction in GCas activity. The absence of p-syn positive inclusions and signs of lysosomal dysfunction in the Migdalska-Richards et al. study79 suggests that the impact of the combined AAV-α-syn/GCas insult in this case was rather mild. As discussed by the authors, this may be due to the relatively modest reduction in GCas activity, 30–40%, obtained in the transgenic mouse lines used in this study.
AAV-α-syn combined with mutations in the LRRK2 gene
Although mutations in the LRRK2 gene is a common genetic risk factor many mutation carriers will never develop PD, even at very old age. The lifetime penetrance of the most common mutation, G2019S, for example, varies widely among different populations, from 17 to 80%, implying that other contributing factors are needed to convert the LRRK2 induced prodromal state into symptomatic PD.85
Increased cellular levels of α-syn may be one such factor. In many, but not all, human cases increased LRRK2 activity is linked to increased α-syn aggregation and Lewy body formation, and studies in cellular and transgenic mouse models have shown that the G2019S-LRRK2 mutation increases α-syn aggregation and DA neuron degeneration caused by delivery of α-syn PFFs.86,87 Notably, in transgenic mice and rats the G2019S-LRRK2 mutation is by itself not sufficient to generate a neurodegenerative PD phenotype.88–90
These data suggest that LRRK2 and α-syn interact in the disease process, and that the toxicity of increased cellular levels of α-syn is amplified in the presence of increased LRRK2 activity. Conversely, increased cellular levels of α-syn achieved by moderate, pre-symptomatic doses of AAV-α-syn should be possible to use to convert a LRRK2 induced prodromal state88,91 into symptomatic PD.
This possibility has so far been investigated in two studies.92,93 In the Daher et al. study,92 G2019S-LRRK2 transgenic rats and WT controls received a unilateral injection of AAV1/2-α-syn into the substantia nigra, given at a moderate dose. In the WT mice, this resulted in a minor, 20%, TH+ cell loss with a low level of microglial activation and CD68+ myeloid cell infiltration, as assessed 2 and 4 weeks after injection. By contrast, both TH+ cell loss, microgliosis, and CD68+ myeloid cell infiltration was markedly increased in the AAV-α-syn treated G2019S-LRRK2 transgenics. This effect could be reversed by daily injections of a LRRK2 kinase inhibitor. Longer time-points, and changes in motor function, were not investigated in this study. The magnitude of DA neuron cell loss, however, around 40–50% at 4 weeks, should be sufficient to induce motor impairments at longer survival times. ∥In the Novello et al. study,93 young (3 months old) and middle aged (18 months old) G2019S-LRRK2 knock-in mice, and age-matched WT controls, received a bilateral intranigral injection of an AAV 2/9 vector overexpressing human mutant A53T α-syn. In this study AAV-α-syn was given at a dose that, by itself, caused a 50% loss of TH+ nigral neurons and a significant impairment in motor performance in the stepping and rotarod tests. In the young mice, the magnitude of cell loss, motor impairment and p-syn aggregation was similar in the LRRK2 mutant and WT mice. In the middle-aged mice, an increase TH+ cell loss (from 40 to 55%), and a 3-fold increase in the level of p-syn positive aggregates, was observed in the LRRK2 mutants relative to the WT controls, while motor impairment was similar in bothgroups.
Although preliminary and limited in scope, the results of these two studies suggest that the toxic impact of α-syn overexpression is enhanced in the presence of mutation-induced increase in LRRK2 activity. This is in line with findings in the α-syn-PFF model showing increased α-syn aggregation and PFF-induced DA neuron cell loss in G2019S-LRRK2 knock-in mice compared to WT controls,86,87 pointing to a role of LRRK2 as a regulator of α-syn homeostasis and aggregation, although the underlying mechanisms are unclear.94
PERSPECTIVE
Targeted delivery of α-syn using AAV vectors has over the two decades since its introduction developed into a useful and versatile tool for modeling different aspects of synucleinopathy seen in PD, DLB and MSA in rodents and non-human primates. The viral vector approach to disease modeling is attractive in that α-syn delivery can be confined to selected anatomical structures and the expression of α-syn, WT or mutated, can be targeted to selected cell populations using either cell-type specific promoter constructs or different natural or engineered AAV serotypes.
AAVα-syn delivered unilaterally to the substantia nigra, alone or together with α-syn-PFFs, shares attractive features with the standard 6-OHDA lesion model: a single unilateral stereotaxic intervention; pathology and cell loss developing over a short time span; and the possibility to monitor the degenerative changes using standard tests of motor behavior. For labs familiar with the 6-OHDA lesion model, the AAV-α-syn model is easy to adopt: the surgery and injection technique are the same, and the same functional tests are applicable in both models. The two methods are clearly complementary in that they replicate different aspects of the pathogenesis of human PD: ROS-dependent neurodegeneration in the 6-OHDA model, and progressive synuclein pathology, neuroinflammation, axonopathy and DA neuron loss in the AAV-α-syn model.
The progressive nature of the vector-induced synucleinopathy and the possibility to vary the strength of the toxic impact by the vector dose are interesting features of the AAV-α-syn model, making it ideally suited for studies aimed at disease modification and neuroprotection. An interesting possibility is to use the AAV-α-syn vector in combination with a second AAV vector, mixed in the same injection. This second vector can, for instance, be designed to modulate the expression of components of the autophagy-lysosomal pathway (ALP) with the goal to increase the resistance of the nigral DA neurons to α-syn aggregation and toxicity. Good examples of this mixed vector approach are studies aimed to modulate the expression of either GCas or the ALP-regulating transcription factor EB (TFEB). In the study of Rocha et al.,83 the AAV-α-syn vector was mixed with an AAV vector expressing GBA-1 to obtain protection against α-syn overexpression, and in a second study, Polissides et al.82 mixed the AAV-α-syn vector with a vector expressing a GBA-1 downregulating microRNA in order to exacerbate α-syn toxicity. Similarly, co-injecting the AAV-α-syn vector with an AAV vector expressing TFEB has been shown to efficiently prevent the build-up of α-syn aggregates and block α-syn toxicity and DA neuron death.95,96
In conclusion, the usefulness of the AAV-α-syn model is largely due to its flexibility and versatility as an experimental tool. The AAV vector can be targeted on selected structures and neuronal cell groups and used at different levels of toxicity; it can be applied unilaterally to allow the unaffected side to serve as a control; it can be designed to achieve selectivity for specific neuronal and glial cell types; and it can be used in both rodents and non-human primates. Further refinement of vector technology, such as the use of regulatable vectors97 and the development of vectors that can pass across the blood-brain barrier, and thus administered systemically,98 will add further to the utility of this method.
ACKNOWLEDGMENTS
The authors have no acknowledgements to report.
FUNDING
The authors have no funding to report.
CONFLICT OF INTEREST
The authors have no conflict of interest to report.
REFERENCES
1. | Volpicelli-Daley LA , Kirik D , Stoyka LE , et al. How can rAAV-alpha-synuclein and the fibril alpha-synuclein models advance our understanding of Parkinson’s disease? J Neurochem (2016) ; 139: (Suppl 1): 131–155. |
2. | Cenci MA and Bjorklund A. Animal models for preclinical Parkinson’s research: An update and critical appraisal. Prog Brain Res (2020) ; 252: : 27–59. |
3. | Carta AR , Boi L , Pisanu A , et al. Advances in modelling alpha-synuclein-induced Parkinson’s diseases in rodents: Virus-based models versus inoculation of exogenous preformed toxic species. J Neurosci Methods (2020) ; 338: : 108685. |
4. | Kirik D , Rosenblad C , Burger C , et al. Parkinson-like neurodegeneration induced by targeted overexpression of alpha-synuclein in the nigrostriatal system. J Neurosci (2002) ; 22: : 2780–2791. |
5. | Decressac M , Mattsson B , Lundblad M , et al. Progressive neurodegenerative and behavioural changes induced by AAV-mediated overexpression of alpha-synuclein in midbrain dopamine neurons. Neurobiol Dis (2012) ; 45: : 939–953. |
6. | Van der Perren A , Van den Haute C and Baekelandt V. Viral vector-based models of Parkinson’s disease. Curr Top Behav Neurosci (2015) ; 22: : 271–301. |
7. | St Martin JL , Klucken J , Outeiro TF , et al. Dopaminergic neuron loss and up-regulation of chaperone protein mRNA induced by targeted over-expression of alpha-synuclein in mouse substantia nigra. J Neurochem (2007) ; 100: : 1449–1457. |
8. | Oliveras-Salva M , Van der Perren A , Casadei N , et al. rAAV2/7 vector-mediated overexpression of alpha-synuclein in mouse substantia nigra induces protein aggregation and progressive dose-dependent neurodegeneration. Mol Neurodegener (2013) ; 8: : 44. |
9. | Ip CW , Klaus LC , Karikari AA , et al. AAV1/2-induced overexpression of A53T-alpha-synuclein in the substantia nigra results in degeneration of the nigrostriatal system with Lewy-like pathology and motor impairment: a new mouse model for Parkinson’s disease. Acta Neuropathol Commun (2017) ; 5: : 11. |
10. | Eslamboli A , Romero-Ramos M , Burger C , et al. Long-term consequences of human alpha-synuclein overexpression in the primate ventral midbrain. Brain (2007) ; 130: : 799–815. |
11. | Koprich JB , Johnston TH , Reyes G , et al. Towards a non-human primate model of alpha-synucleinopathy for development of therapeutics for Parkinson’s disease: optimization of AAV1/2 delivery parameters to drive sustained expression of alpha synuclein and dopaminergic degeneration in macaque. PLoS One (2016) ; 11: : e0167235. |
12. | Jellinger KA . Pathology of Parkinson’s disease. Changes other than the nigrostriatal pathway. Mol Chem Neuropathol (1991) ; 14: : 153–197. |
13. | Wenning GK , Stefanova N , Jellinger KA , et al. Multiple system atrophy: a primary oligodendrogliopathy. Ann Neurol (2008) ; 64: : 239–246. |
14. | Singleton AB , Farrer M , Johnson J , et al. alpha-Synuclein locus triplication causes Parkinson’s disease. Science (2003) ; 302: : 841. |
15. | Dachsel JC , Lincoln SJ , Gonzalez J , et al. The ups and downs of alpha-synuclein mRNA expression. Mov Disord (2007) ; 22: : 293–295. |
16. | Thakur P , Breger LS , Lundblad M , et al. Modeling Parkinson’s disease pathology by combination of fibril seeds and alpha-synuclein overexpression in the rat brain. Proc Natl Acad Sci U S A (2017) ; 114: : E8284–E8293. |
17. | Van der Perren A , Toelen J , Casteels C , et al. Longitudinal follow-up and characterization of a robust rat model for Parkinson’s disease based on overexpression of alpha-synuclein with adeno-associated viral vectors. Neurobiol Aging (2015) ; 36: : 1543–1558. |
18. | Xu J , Kao SY , Lee FJ , et al. Dopamine-dependent neurotoxicity of alpha-synuclein: a mechanism for selective neurodegeneration in Parkinson disease. Nat Med (2002) ; 8: : 600–606. |
19. | Conway KA , Rochet JC , Bieganski RM , et al. Kinetic stabilization of the alpha-synuclein protofibril by a dopamine-alpha-synuclein adduct. Science (2001) ; 294: : 1346–1349. |
20. | Mosharov EV , Larsen KE , Kanter E , et al. Interplay between cytosolic dopamine, calcium, and alpha-synuclein causes selective death of substantia nigra neurons. Neuron (2009) ; 62: : 218–229. |
21. | Burbulla LF , Song P , Mazzulli JR , et al. Dopamine oxidation mediates mitochondrial and lysosomal dysfunction in Parkinson’s disease. Science (2017) ; 357: : 1255–1261. |
22. | Geibl FF , Henrich MT , Xie Z , et al. alpha-Synuclein pathology disrupts mitochondrial function in dopaminergic and cholinergic neurons at-risk in Parkinson’s disease. bioRxiv (2023) ; https://doi.org/10.1101/2023.12.11.571045. Posted December 11, 2023. |
23. | Eriksen JL , Przedborski S and Petrucelli L. Gene dosage and pathogenesis of Parkinson’s disease. Trends Mol Med (2005) ; 11: : 91–96. |
24. | Gombash SE , Manfredsson FP , Kemp CJ , et al. Morphological and behavioral impact of AAV2/5-mediated overexpression of human wildtype alpha-synuclein in the rat nigrostriatal system. PLoS One (2013) ; 8: : e81426. |
25. | Bourdenx M , Dovero S , Engeln M , et al. Lack of additive role of ageing in nigrostriatal neurodegeneration triggered by alpha-synuclein overexpression. Acta Neuropathol Commun (2015) ; 3: : 46. |
26. | Gubinelli F , Sarauskyte L , Venuti C , et al. Characterisation of functional deficits induced by AAV overexpression of alpha-synuclein in rats. Curr Res Neurobiol (2023) ; 4: : 100065. |
27. | Cao S , Theodore S and Standaert DG. Fcgamma receptors are required for NF-kappaB signaling, microglial activation and dopaminergic neurodegeneration in an AAV-synuclein mouse model of Parkinson’s disease. Mol Neurodegener (2010) ; 5: : 42. |
28. | Ulusoy A , Bjorklund T , Buck K , et al. Dysregulated dopamine storage increases the vulnerability to alpha-synuclein in nigral neurons. Neurobiol Dis (2012) ; 47: : 367–377. |
29. | Alarcon-Aris D , Pavia-Collado R , Miquel-Rio L , et al. Anti-alpha-synuclein ASO delivered to monoamine neurons prevents alpha-synuclein accumulation in a Parkinson’s disease-like mouse model and in monkeys. EBioMedicine (2020) ; 59: : 102944. |
30. | Huntington TE and Srinivasan R. Adeno-associated virus expression of alpha-synuclein as a tool to model Parkinson’s disease: current understanding and knowledge gaps. Aging Dis (2021) ; 12: : 1120–1137. |
31. | Sanchez-Guajardo V , Febbraro F , Kirik D , et al. Microglia acquire distinct activation profiles depending on the degree of alpha-synuclein neuropathology in a rAAV based model of Parkinson’s disease. PLoS One (2010) ; 5: : e8784. |
32. | Koprich JB , Johnston TH , Huot P , et al. Progressive neurodegeneration or endogenous compensation in an animal model of Parkinson’s disease produced by decreasing doses of alpha-synuclein. PLoS One (2011) ; 6: : e17698. |
33. | Faustini G , Longhena F , Varanita T , et al. Synapsin III deficiency hampers alpha-synuclein aggregation, striatal synaptic damage and nigral cell loss in an AAV-based mouse model of Parkinson’s disease. Acta Neuropathol (2018) ; 136: : 621–639. |
34. | Burke RE and O’Malley K. Axon degeneration in Parkinson’s disease. Exp Neurol (2013) ; 246: : 72–83. |
35. | Lundblad M , Decressac M , Mattsson B , et al. Impaired neurotransmission caused by overexpression of alpha-synuclein in nigral dopamine neurons. Proc Natl Acad Sci U S A (2012) ; 109: : 3213–3219. |
36. | Chung CY , Koprich JB , Siddiqi H , et al. Dynamic changes in presynaptic and axonal transport proteins combined with striatal neuroinflammation precede dopaminergic neuronal loss in a rat model of AAV alpha-synucleinopathy. J Neurosci (2009) ; 29: : 3365–3373. |
37. | Phan JA , Stokholm K , Zareba-Paslawska J , et al. Early synaptic dysfunction induced by alpha-synuclein in a rat model of Parkinson’s disease. Sci Rep (2017) ; 7: : 6363. |
38. | Kirik D , Rosenblad C and Bjorklund A. Characterization of behavioral and neurodegenerative changes following partial lesions of the nigrostriatal dopamine system induced by intrastriatal 6-hydroxydopamine in the rat. Exp Neurol (1998) ; 152: : 259–277. |
39. | Decressac M , Mattsson B and Bjorklund A. Comparison of the behavioural and histological characteristics of the 6-OHDA and alpha-synuclein rat models of Parkinson’s disease. Exp Neurol (2012) ; 235: : 306–315. 20120225. |
40. | Peelaerts W , Bousset L , Van der Perren A , et al. alpha-Synuclein strains cause distinct synucleinopathies after local and systemic administration. Nature (2015) ; 522: : 340–344. |
41. | Hoban DB , Shrigley S , Mattsson B , et al. Impact of alpha-synuclein pathology on transplanted hESC-derived dopaminergic neurons in a humanized alpha-synuclein rat model of PD. Proc Natl Acad Sci U S A (2020) ; 117: : 15209–15220. |
42. | Volpicelli-Daley LA , Luk KC , Patel TP , et al. Exogenous alpha-synuclein fibrils induce Lewy body pathology leading to synaptic dysfunction and neuron death. Neuron (2011) ; 72: : 57–71. |
43. | Volpicelli-Daley LA , Luk KC and Lee VM. Addition of exogenous alpha-synuclein preformed fibrils to primary neuronal cultures to seed recruitment of endogenous alpha-synuclein to Lewy body and Lewy neurite-like aggregates. Nat Protoc (2014) ; 9: : 2135–2146. 20140814. |
44. | Luk KC , Covell DJ , Kehm VM , et al. Molecular and biological compatibility with host alpha-synuclein influences fibril pathogenicity. Cell Rep (2016) ; 16: : 3373–3387. |
45. | Bjorklund A , Nilsson F , Mattsson B , et al. A combined alpha-synuclein/fibril (SynFib) model of Parkinson-like synucleinopathy targeting the nigrostriatal dopamine system. J Parkinsons Dis (2022) ; 12: : 2307–2320. |
46. | Pinto-Costa R , Harbachova E , La Vitola P , et al. Overexpression-induced alpha-synuclein brain spreading. Neurotherapeutics (2023) ; 20: : 83–96. |
47. | Maillet A , Krack P , Lhommee E , et al. The prominent role of serotonergic degeneration in apathy, anxiety and depression in de novo Parkinson’s disease. Brain (2016) ; 139: : 2486–2502. 20160817. |
48. | Ray Chaudhuri K , Leta V , Bannister K , et al. The noradrenergic subtype of Parkinson disease: from animal models to clinical practice. Nat Rev Neurol (2023) ; 19: : 333–345. |
49. | Surmeier DJ , Obeso JA and Halliday GM. Selective neuronal vulnerability in Parkinson disease. Nat Rev Neurosci (2017) ; 18: : 101–113. |
50. | Henrich MT , Geibl FF , Lee B , et al. A53T-alpha-synuclein overexpression in murine locus coeruleus induces Parkinson’s disease-like pathology in neurons and glia. Acta Neuropathol Commun (2018) ; 6: : 39. |
51. | Matschke LA , Komadowski MA , Stohr A , et al. Enhanced firing of locus coeruleus neurons and SK channel dysfunction are conserved in distinct models of prodromal Parkinson’s disease. Sci Rep (2022) ; 12: : 3180. |
52. | Wan OW , Shin E , Mattsson B , et al. alpha-Synuclein induced toxicity in brain stem serotonin neurons mediated by an AAV vector driven by the tryptophan hydroxylase promoter. Sci Rep (2016) ; 6: : 26285. |
53. | Miquel-Rio L , Alarcon-Aris D , Torres-Lopez M , et al. Human alpha-synuclein overexpression in mouse serotonin neurons triggers a depressive-like phenotype. Rescue by oligonucleotide therapy. Transl Psychiatry (2022) ; 12: : 79. |
54. | Hall H , Reyes S , Landeck N , et al. Hippocampal Lewy pathology and cholinergic dysfunction are associated with dementia in Parkinson’s disease. Brain (2014) ; 137: : 2493–2508. |
55. | Liu AKL , Chau TW , Lim EJ , et al. Hippocampal CA2 Lewy pathology is associated with cholinergic degeneration in Parkinson’s disease with cognitive decline. Acta Neuropathol Commun (2019) ; 7: : 61. |
56. | Hall H , Jewett M , Landeck N , et al. Characterization of cognitive deficits in rats overexpressing human alpha-synuclein in the ventral tegmental area and medial septum using recombinant adeno-associated viral vectors. PLoS One (2013) ; 8: : e64844. |
57. | Ulusoy A , Rusconi R , Perez-Revuelta BI , et al. Caudo-rostral brain spreading of alpha-synuclein through vagal connections. EMBO Mol Med (2013) ; 5: : 1119–1127. |
58. | Rusconi R , Ulusoy A , Aboutalebi H , et al. Long-lasting pathological consequences of overexpression-induced alpha-synuclein spreading in the rat brain. Aging Cell (2018) ; 17: : 20180130. |
59. | Helwig M , Ulusoy A , Rollar A , et al. Neuronal hyperactivity-induced oxidant stress promotes in vivo alpha-synuclein brain spreading. Sci Adv (2022) ; 8: : eabn0356. |
60. | Musgrove RE , Helwig M , Bae EJ , et al. Oxidative stress in vagal neurons promotes parkinsonian pathology and intercellular alpha-synuclein transfer. J Clin Invest (2019) ; 129: : 3738–3753. |
61. | Kim S , Kwon SH , Kam TI , et al. Transneuronal propagation of pathologic alpha-synuclein from the gut to the brain models Parkinson’s disease. Neuron (2019) ; 103: : 627–641 e627. |
62. | Williams GP , Marmion DJ , Schonhoff AM , et al. T cell infiltration in both human multiple system atrophy and a novel mouse model of the disease. Acta Neuropathol (2020) ; 139: : 855–874. |
63. | Marmion DJ , Peelaerts W and Kordower JH. A historical review of multiple system atrophy with a critical appraisal of cellular and animal models. J Neural Transm (Vienna) (2021) ; 128: : 1507–1527. |
64. | Fanciulli A , Stankovic I , Krismer F , et al. Multiple system atrophy. Int Rev Neurobiol (2019) ; 149: : 137–192. |
65. | Bassil F , Guerin PA , Dutheil N , et al. Viral-mediated oligodendroglial alpha-synuclein expression models multiple system atrophy. Mov Disord (2017) ; 32: : 1230–1239. |
66. | von Jonquieres G , Frohlich D , Klugmann CB , et al. Recombinant human myelin-associated glycoprotein promoter drives selective AAV-mediated transgene expression in oligodendrocytes. Front Mol Neurosci (2016) ; 9: : 13. |
67. | Peelaerts W , Brito F , Van den Haute C , et al. Widespread, specific, and efficient transgene expression in oligodendrocytes after intracerebral and intracerebroventricular delivery of viral vectors in rodent brain. Hum Gene Ther (2021) ; 32: : 616–627. |
68. | Powell SK , Khan N , Parker CL , et al. Characterization of a novel adeno-associated viral vector with preferential oligodendrocyte tropism. Gene Ther (2016) ; 23: : 807–814. |
69. | Mandel RJ , Marmion DJ , Kirik D , et al. Novel oligodendroglial alpha synuclein viral vector models of multiple system atrophy: studies in rodents and nonhuman primates. Acta Neuropathol Commun (2017) ; 5: : 47. |
70. | Marmion DJ , Rutkowski AA , Chatterjee D , et al. Viral-based rodent and nonhuman primate models of multiple system atrophy: Fidelity to the human disease. Neurobiol Dis (2021) ; 148: : 105184. |
71. | Coukos R , Krainc D . Key genes and convergent pathogenic mechanisms in Parkinson disease. Nat Rev Neurosci (2024) ; 25: (6), 393–413. |
72. | Mulcahy P , O’Doherty A , Paucard A , et al. The behavioural and neuropathological impact of intranigral AAV-alpha-synuclein is exacerbated by systemic infusion of the Parkinson’s disease-associated pesticide, rotenone, in rats. Behav Brain Res (2013) ; 243: : 6–15. |
73. | Mulcahy P , O’Doherty A , Paucard A , et al. Development and characterisation of a novel rat model of Parkinson’s disease induced by sequential intranigral administration of AAV-alpha-synuclein and the pesticide, rotenone. Neuroscience (2012) ; 203: : 170–179. |
74. | Naughton C , O’Toole D , Kirik D , et al. Interaction between subclinical doses of the Parkinson’s disease associated gene, alpha-synuclein, and the pesticide, rotenone, precipitates motor dysfunction and nigrostriatal neurodegeneration in rats. Behav Brain Res (2017) ; 316: : 160–168. |
75. | O’Regan G , deSouza RM , Balestrino R , et al. Glucocerebrosidase mutations in Parkinson disease. J Parkinsons Dis (2017) ; 7: : 411–422. |
76. | Gegg ME , Burke D , Heales SJ , et al. Glucocerebrosidase deficiency in substantia nigra of Parkinson disease brains. Ann Neurol (2012) ; 72: : 455–463. |
77. | Murphy KE and Halliday GM. Glucocerebrosidase deficits in sporadic Parkinson disease. Autophagy (2014) ; 10: : 1350–1351. |
78. | Mazzulli JR , Xu YH , Sun Y , et al. Gaucher disease glucocerebrosidase and alpha-synuclein form a bidirectional pathogenic loop in synucleinopathies. Cell (2011) ; 146: : 37–52. |
79. | Migdalska-Richards A , Wegrzynowicz M , Rusconi R , et al. The L444P Gba1 mutation enhances alpha-synuclein induced loss of nigral dopaminergic neurons in mice. Brain (2017) ; 140: : 2706–2721. |
80. | Kim D , Hwang H , Choi S , et al. D409H GBA1 mutation accelerates the progression of pathology in A53T alpha-synuclein transgenic mouse model. Acta Neuropathol Commun (2018) ; 6: : 32. |
81. | Fishbein I , Kuo YM , Giasson BI , et al. Augmentation of phenotype in a transgenic Parkinson mouse heterozygous for a Gaucher mutation. Brain (2014) ; 137: : 3235–3247. |
82. | Polissidis A , Koronaiou E , Nikolopoulou G , et al. A double-hit in vivo model of GBA viral microRNA-mediated downregulation and human alpha-synuclein overexpression demonstrates nigrostriatal degeneration. Neurobiol Dis (2022) ; 163: : 105612. |
83. | Rocha EM , Smith GA , Park E , et al. Glucocerebrosidase gene therapy prevents alpha-synucleinopathy of midbrain dopamine neurons. Neurobiol Dis (2015) ; 82: : 495–503. |
84. | Sucunza D , Rico AJ , Roda E , et al. Glucocerebrosidase gene therapy induces alpha-synuclein clearance and neuroprotection of midbrain dopaminergic neurons in mice and macaques. Int J Mol Sci (2021) ; 22: : 4825. |
85. | Taymans JM , Fell M , Greenamyre T , et al. Perspective on the current state of the LRRK2 field. NPJ Parkinsons Dis (2023) ; 9: : 104. |
86. | Volpicelli-Daley LA , Abdelmotilib H , Liu Z , et al. G2019S-LRRK2 expression augments alpha-synuclein sequestration into inclusions in neurons. J Neurosci (2016) ; 36: : 7415–7427. |
87. | Bieri G , Brahic M , Bousset L , et al. LRRK2 modifies alpha-syn pathology and spread in mouse models and human neurons. Acta Neuropathol (2019) ; 137: : 961–980. |
88. | Lee JW , Tapias V , Di Maio R , et al. Behavioral, neurochemical, and pathologic alterations in bacterial artificial chromosome transgenic G2019S leucine-rich repeated kinase 2 rats. Neurobiol Aging (2015) ; 36: : 505–518. |
89. | Sloan M , Alegre-Abarrategui J , Potgieter D , et al. LRRK2 BAC transgenic rats develop progressive, L-DOPA-responsive motor impairment, and deficits in dopamine circuit function. Hum Mol Genet (2016) ; 25: : 951–963. |
90. | Domenicale C , Magnabosco S and Morari M. Modeling Parkinson’s disease in LRRK2 rodents. Neuronal Signal (2023) ; 7: : NS20220040. |
91. | Longo F , Mercatelli D , Novello S , et al. Age-dependent dopamine transporter dysfunction and Serine129 phospho-alpha-synuclein overload in G2019S LRRK2 mice. Acta Neuropathol Commun (2017) ; 5: : 22. |
92. | Daher JP , Abdelmotilib HA , Hu X , et al. Leucine-rich repeat kinase 2 (LRRK2) pharmacological inhibition abates alpha-synuclein gene-induced neurodegeneration. J Biol Chem (2015) ; 290: : 19433–19444. |
93. | Novello S , Arcuri L , Dovero S , et al. G2019S LRRK2 mutation facilitates alpha-synuclein neuropathology in aged mice. Neurobiol Dis (2018) ; 120: : 21–33. |
94. | Erb ML and Moore DJ. LRRK2 and the endolysosomal system in Parkinson’s disease. J Parkinsons Dis (2020) ; 10: : 1271–1291. |
95. | Decressac M , Mattsson B , Weikop P , et al. TFEB-mediated autophagy rescues midbrain dopamine neurons from alpha-synuclein toxicity. Proc Natl Acad Sci U S A (2013) ; 110: : E1817–E1826. |
96. | Arotcarena ML , Bourdenx M , Dutheil N , et al. Transcription factor EB overexpression prevents neurodegeneration in experimental synucleinopathies. JCI Insight (2019) ; 4: : e129719. |
97. | Torre-Muruzabal T , Devoght J , Van den Haute C , et al. Chronic nigral neuromodulation aggravates behavioral deficits and synaptic changes in an alpha-synuclein based rat model for Parkinson’s disease. Acta Neuropathol Commun (2019) ; 7: : 160. |
98. | Berard M , Martinez-Drudis L , Sheta R , et al. Non-invasive systemic viral delivery of human alpha-synuclein mimics selective and progressive neuropathology of Parkinson’s disease in rodent brains. Mol Neurodegener (2023) ; 18: : 91. |

