
Tissue Factor and Its Cerebrospinal Fluid Protein Profiles in Parkinson’s Disease
Abstract
Background:
Prior investigations have elucidated pathophysiological interactions involving blood coagulation and neurodegenerative diseases. These interactions pertain to age-related effects and a mild platelet antiaggregant function of exogenous α-Synuclein.
Objective:
Our study sought to explore whether cerebrospinal fluid (CSF) levels of tissue factor (TF), the initiator of the extrinsic pathway of hemostasis, differ between controls (CON) compared to patients with Parkinson’s disease (PD) and dementia with Lewy bodies (DLB), considering that these conditions represent a spectrum of α-Synuclein pathology. We further investigated whether TF levels are associated with longitudinal progression in PD.
Methods:
We examined CSF levels of TF in 479 PD patients, 67 patients diagnosed with DLB, and 16 CON in order to evaluate potential continuum patterns among DLB, PD, and CON. Of the 479 PD patients, 96 carried a GBA1 variant (PD GBA1), while the 383 non-carriers were classified as PD wildtype (PD WT). We considered both longitudinal clinical data as well as CSF measurements of common neurodegenerative markers (amyloid-β 1-42, h-Tau, p-Tau, NfL, α-Synuclein). Kaplan-Meier survival and Cox regression analysis stratified by TF tertile levels was conducted.
Results:
Higher CSF levels of TF were associated with an older age at examination in PD and a significant later onset of postural instability in PD GBA1. TF levels were lower in male vs. female PD. DLB GBA1 exhibited the lowest TF levels, followed by PD GBA1, with CON showing the highest levels.
Conclusions:
TF as representative of blood hemostasis could be an interesting CSF candidate to further explore in PD and DLB.
Plain Language Summary
Parkinson’s disease is a common age-related condition, primarily affecting older individuals. However, it shows a wide range of symptoms and clinical courses, influenced by genetic mutations, neuroinflammatory processes and lifestyle factors. Research into the disease’s mechanisms is important for developing new therapies that could potentially slow its progression. Early diagnosis is also essential, as new disease-modifying therapies are most effective when started at an early stage of the disease. In this paper, we focus on a protein called tissue factor, which plays a role in both blood coagulation and neuroinflammation. Proteins involved in blood-coagulation also exhibit an increase in blood-concentration with higher age. Also, a subtle platelet antiaggregant function of exogenous α-Synuclein was found, a protein which aggregates in the brain of patients with Parkinson’s disease. Additionally, higher tissue factor levels were found in plaques of proteins (amyloid-β 1-42) found in Alzheimer’s disease. Thus, tissue factor could be a promising biomarker candidate for neurodegenerative diseases. We analyzed the concentration of this protein in the cerebrospinal fluid of 479 patients with Parkinson’s disease, 16 control participants, and 67 patients with dementia with Lewy bodies, a sub-type of Parkinson’s disease with exceptionally high levels of α-Synuclein in the brain. Our findings showed the lowest levels of tissue factor in patients with dementia with Lewy bodies, followed by those with typical Parkinson’s disease, and the highest levels in controls. Additionally, older patients had higher tissue factor levels than younger patients, and levels were lower in male patients compared to female patients. Thus, measuring tissue factor levels could help in diagnosing Parkinson’s disease. Further studies, especially with larger control groups, are needed to confirm these results. Additionally, exploring the connections between blood coagulation and Parkinson’s disease could improve our understanding of the disease’s mechanisms, potentially leading to new pharmaceutical developments.
INTRODUCTION
Parkinson’s disease (PD) stands as a paradigm for age-related disorders, as its incidence escalates with advancing age, highlighting age as a predominant risk factor for PD.1 Additionally, outcomes from community-based longitudinal cohorts consistently demonstrate that chronological age and an elevated age at disease onset independently forecast cognitive decline, deterioration in motor function and heightened mortality in individuals with PD.2–4
Neuroinflammation plays a crucial role in both the onset and progression of PD. In this context, the cytokines interleukin-1 beta (IL-1β), interleukin-6 (IL-6), monocyte chemoattractant protein 1 (MCP-1), and tumor necrosis factor alpha (TNF- α) are consistently highlighted in literature.5
It is firmly established that components of blood coagulation, particularly fibrinogen, factor V and factors VII-IX, exhibit an age-related increase. This phenomenon contributes to a procoagulatory condition associated with advanced age.6
Additionally, these proteins such as fibrinogen and prothrombin are involved in inflammatory processes, serving as acute-phase proteins.7 Tissue factor (TF), also known as coagulation factor III, plays a crucial role in initiating the extrinsic pathway of hemostasis. Thrombin, as illustrated in Fig. 1, acts as a downstream component in this cascade.8 Cytokines like IL-1, endotoxin and TNF can induce TF expression on monocytes. Conversely, chemokine production, including macrophage inflammatory protein-1 α (MIP-1α), has been found to be induced following TF injections.9 Injections of thrombin in a mouse model resulted in heightened microglial activation, subsequently leading to the degeneration of nigral dopaminergic neurons.10
Fig. 1
Tissue factor (TF), also known as coagulation factor III, stands at the beginning of the coagulation cascade. An injury of blood vessels leads to the association of a complex with FVIIa, which activates FX and FIX. The complex of FXa and FVa transforms prothrombin (FII) to thrombin (FIIa), which itself leads to an activation of platelets via protease-activated receptors (PARs) and fibrin-cross-linkage via FXIIIa, causing a clot in the end.8 α-Synuclein (α-Syn) might exceed a mild platelet antiaggregant function physiologically, mostly via inhibition of P-selectin expression on the surface of platelets and thus inhibiting thrombin-induced platelet activation.15 Created with BioRender.com
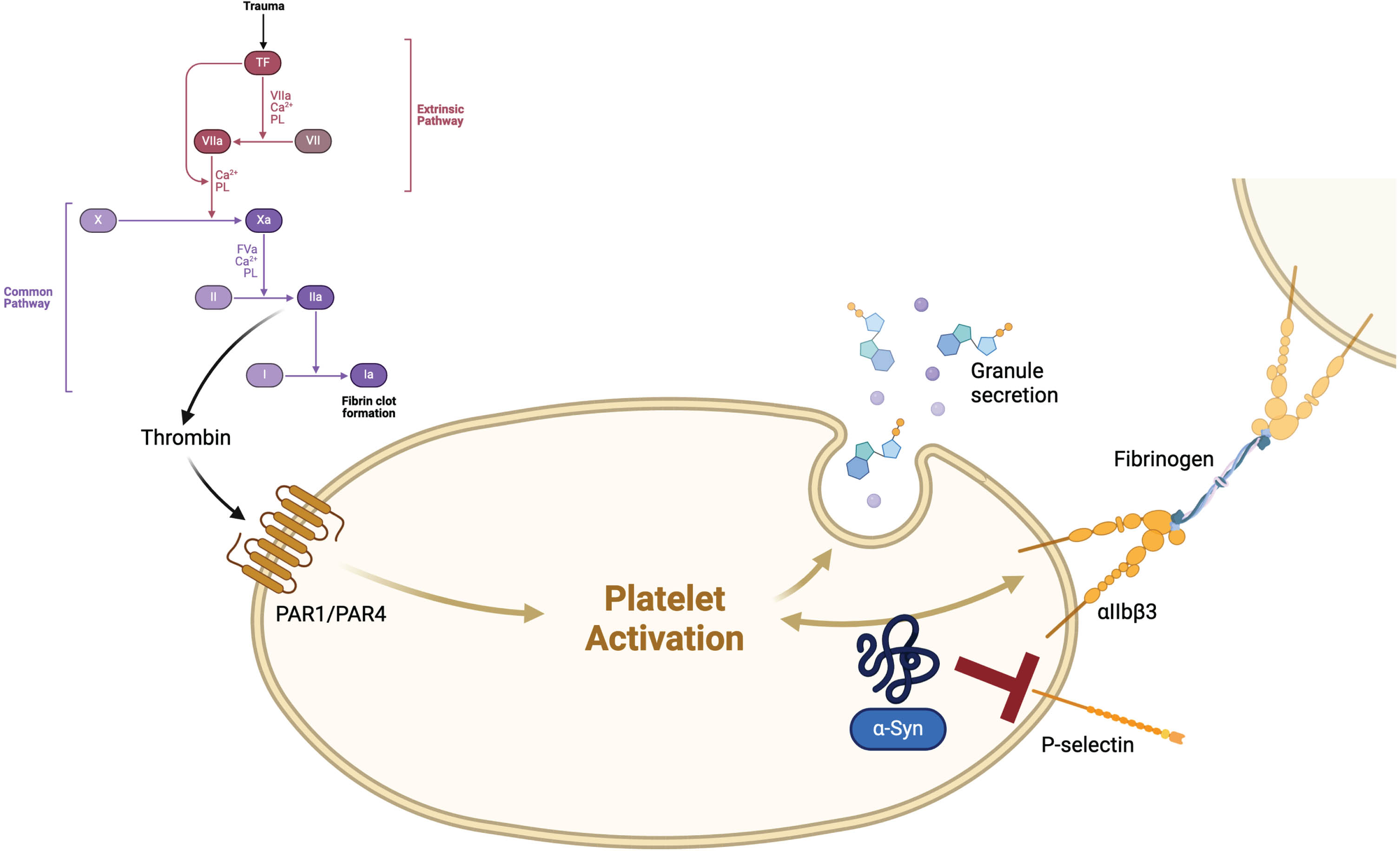
Therefore, TF and also thrombin might be promising links between coagulation, (neuro-) inflammation and neurodegeneration.
Growing evidence reveals other potential connections between blood coagulation and PD. Accumulation of α-Synuclein in the central and peripheral nervous system, referred to as Lewy bodies, is a pathophysiological hallmark in both PD and dementia with Lewy bodies (DLB).11 Within this framework, α-Synuclein aggregates serve as damage-associated molecular patterns (DAMPs), triggering microglial activation and cytokine production.12,13 Recently, a disease spectrum between PD, PD with dementia (PDD), and DLB based on α-Synuclein deposition was proposed, with the most pronounced Lewy body pathology in DLB patients,14 especially when exhibiting variants in GBA1. 11 In the context of blood coagulation, a subtle platelet antiaggregant function of exogenous α-Synuclein, which is also present in platelets, has been elucidated. This physiological function of α-Synuclein is primarily attributed to the inhibition of P-selectin expression on the platelet surface, thereby impeding thrombin-induced platelet activation.15 In animal models, P-selectin has been found to increase TF biosynthesis.16
Notably, heightened immunoreactivity for tissue factor antigen17 and an increased expression of thrombin have been observed within amyloid-β 1–42 plaques in individuals with Alzheimer’s disease (AD).18 Given that concurrent AD pathology, alongside additional vascular risk profiles, has been demonstrated to contribute to cognitive decline in PD patients,19 it is plausible that these findings may also apply, at least in part, to PD, PDD, and DLB.
Considering these pathophysiological connections, our objective was to investigate TF in PD and DLB through a two-pronged hypothesis:
1. TF levels might reflect disease stages and progression of PD. We analyzed cross-sectional and longitudinal clinical data in a first step. We also considered common neurodegenerative proteins (amyloid-β 1-42, h-Tau, p-Tau, NfL, and α-Synuclein), as they reflect different stages of neuronal damage and highlight additional AD pathology. This is particularly relevant because high levels of TF antigen have been found in senile plaques.
2. CSF TF levels might mirror the continuum of Lewy body pathology observed in patients with PD, PDD, and DLB, with the most pronounced α-Synuclein deposition in patients with DLB and GBA1 variants. Therefore, we stratified by GBA1 mutational status and included DLB as an additional cohort in all analyzes.
METHODS
Participants and clinical investigations
All 479 PD patients (referred to as PD total cohort) and 67 patients with DLB were recruited and examined between 2001 and 2022 from the ward and outpatient clinic for PD at the University of Tuebingen. All patients were examined by a movement disorder specialist. The diagnosis of PD was defined according to UK Brain Bank criteria.20 The following demographic and clinical data were obtained: age, sex, age at onset of parkinsonism. Additionally, we calculated the disease duration. We assessed the severity of motor symptoms using the Unified Parkinson’s Disease Rating scale part III (UPDRS III); from 2000–2008, and from 2009, the MDS-UPDRS were applied.21 The clinical diagnosis of DLB was established in accordance with the revised consensus criteria for DLB, as outlined in the fourth report of the DLB consortium.22 Cognitive function was tested using the Montreal Cognitive Assessment (MoCA)23 or the Mini-Mental Status Examination (MMSE).24 As the MoCA has only been available since 2009, all MMSE scores were converted to MoCA equivalent scores according to an algorithm published recently.25 MoCA cutoff ≤25 indicated cognitive impairment (point of maximum combined sensitivity and specificity).26 Postural instability was characterized using the UPDRS III score, whereby patients demonstrating three or more compensatory steps in the pull test were classified as having postural instability. Depressive symptoms were assessed using Beck’s Depression Inventory (BDI) II.27 The PD cohort, subjected to tissue factor measurements, was chosen based on the criterion of possessing a comprehensive clinical data set. Of the 479 PD patients, 297 had longitudinal data pertaining to the onset of postural instability, forming a smaller longitudinal sub-cohort.
All 16 control participants (CON) were spouses of patients with PD or patients with functional disorders in whom neurodegenerative diseases have been extensively excluded on the ward.
All CSF samples were obtained from the Neuro-Biobank of the University of Tuebingen, Germany (https://www.hih-tuebingen.de/en/about-us/core-facilities/biobank/). The biobank is supported by the local University, the Hertie Institute and the DZNE.
All participants gave written informed consent. The study was approved by the Ethics Committee of the Faculty of Medicine at the University of Tuebingen (Project-Nr. 458/2023BO2). All procedures are in accordance with the Declaration of Helsinki (1975).
Collection of CSF samples
Spinal tap was performed between 9:00 AM and 1:00 PM. Samples were directly taken from the bedside and centrifuged within 60 min after collection and frozen at –80°C within 90 min after collection. Samples with abnormal routine CSF diagnostics (white blood cell count >6 cells/μL; Immunoglobuline subtype G index >0.7) were excluded.
CSF measurements of amyloid-β 1-42, h-Tau, p181-Tau (p-Tau), neurofilament light protein (NfL) and α-Synuclein
CSF levels of amyloid-β 1-42, h-Tau and p-Tau were measured using ELISA kits from INNOTEST, Fujirebio GmbH, Germany. CSF levels of NFL were measured using the UmanDiagnostics NF- light® assay. Intra-assay coefficients of variation for each CSF parameter were below 15%. CSF levels of total α-Synuclein were assessed using an ELISA kit for human α-Synuclein (Roboscreen GmbH, Germany). Intra-assay imprecision of 4.4% was calculated from duplicate analyses and expressed as median of the range to average of the duplicates. Inter-assay imprecision of <10% was determined using two quality control CSF pool samples.
All measurements were performed by board-certified laboratory technicians who were blinded to the clinical data.
Bead-based detection of tissue factor in CSF
Tissue factor CSF levels (in pg/ml) were detected using a commercial 3-plex human magnetic Luminex assay (R&D Systems, Wiesbaden, Germany, cat no. LXSAHM-03, lot number L139408). Individual analyses were performed with 1:2 diluted samples according to the manufacturer’s protocol. For the multiplex assay, 50μL Luminex beads and 50μL standards, quality control or CSF samples were pipetted into the wells of a 96-well plate. The specific analytes are bound by the immobilized antibodies on the color-coded beads. Unbound sample was removed, and beads washed three times with wash buffer. 50μL biotinylated detection antibody was added to each well and incubated at 800 rpm for 1 h at room temperature. The beads were washed three times to remove unbound detection antibody. 50μL Streptavidin-PE conjugate was added to each well. After 30 min incubation at 800 rpm, unbound Streptavidin-PE was removed, and the beads washed three times. The beads were resuspended in 100μl of buffer and then analyzed using the FlexMap 3D® analyser and Luminex xPONENT® 4.2 software (Luminex, Austin, TX, USA). Median fluorescence intensity (MFI) values were back-calculated using a 5PL regression fit to the standard samples (Bio-Plex Manager, version 6) to determine tissue factor concentrations.
Genetic analysis
DNA was isolated from ethylenediaminetetraacetic acid blood by salting out method and stored at 4°C. Genetic screening for PD-associated mutations was done using the Neurochip. Known pathogenic point mutations in the genes GBA1, LRRK2, PRKN, PINK1, and DJ1 detected by Neurochip were confirmed by Sanger sequencing. Multiplex ligation-dependent probe amplification was used to detect deletions and duplications in the genes PRKN, PINK1 and DJ1.
We performed GBA1-subgroup classification for mutation severity according to established mutational risk reported for PD: low risk (“risk”, N = 48), mild risk (“mild”, N = 20), severe risk (“severe”, N = 28). Of note, some variants that have been reported as nonrelevant for Gaucher disease have been proven to increase the risk for PD and therefore have been included in our analysis, for example, p. E326K and p.T369M.
Statistics
Statistical analysis was performed using IBM-SPSS Statistics (IBM Corp. Released 2021. IBM SPSS Statistics for Macintosh, Version 28.0. Armonk, NY: IBM Corp.). Group comparisons were conducted among following groups: PD GBA1 (N = 96), comprising patients with variants in the GBA1 gene, PD wildtype (WT, N = 383) (patients without variants in GBA1), PD total cohort (PD GBA1 and PD WT, N = 479), CON (control participants, N = 16) and patients with dementia with Lewy bodies (DLB, N = 67).
The significance level was set at p≤0.007 for cross-sectional analysis (manual Bonferroni-correction due to comparison of 7 groups) (Table 1 and Supplementary Table 1), at p≤0.016 for comparison of TF levels between CON, PD, and DLB (Supplementary Table 2) and at p≤0.05 for longitudinal analysis (Supplementary Table 3).
Table 1
Demographic, clinical and CSF-biomarker data in patients with Parkinson’s disease (PD), dementia with Lewy Bodies (DLB), and controls (CON)
| CON | PD | DLB | |||||
| PD total | PD WT | PD GBA1 | DLB total | DLB WT | DLB GBA1 | ||
| age at examination | 65.6 (±9.5) | 65.4 (±9.9) | 65.9 (±9.9) | 63.4 (±9.4) | 72.5 (±6.5) | 73.6 (±6.4) | 69.4 (±6.0) |
| N = 16 | N = 479 | N = 383 | N = 96 | N = 67* | N = 49* | N = 18 | |
| age at onset | 58.1 (±10.5) | 58.8 (±10.5) | 55.1 (±10.2) | 69.3 (±7.3) | 70.6 (±7.3) | 66.1 (±6.2) | |
| N = 479 | N = 383 | N = 96 | N = 66 | N = 48 | N = 18 | ||
| disease duration | 7.3 (±5.2) | 7.1 (±5.0) | 8.3 (±5.6) | 3.2 (±2.1) | 3.1 (±2.2) | 3.4 (±1.8) | |
| N = 479 | N = 383 | N = 96 | N = 66 | N = 48 | N = 18 | ||
| UPDRS III | 1.3 (±2.6) | 26.1 (±11.5) | 26.0 (±11.5) | 26.6 (±11.7) | 26.8 (±9.2) | 25.5 (±7.9) | 28.0 (±10.4) |
| N = 7 | N = 446§§ | N = 354§§ | N = 92§§ | N = 21** | N = 10** | N = 11** | |
| MoCA | 27.7 (±2.2) | 25.3 (±3.9) | 25.4 (±3.6) | 24.8 (±4.8) | 15.1 (±4.3) | 14.6 (±4.0) | 16.0 (±4.7) |
| N = 11 | N = 403 | N = 315 | N = 88 | N = 43** | N = 28** | N = 15** | |
| BDI II | 4.5 (±6.2) | 9.1 (±7.2) | 8.9 (±7.1) | 10.2 (±7.3) | 10.5 (±6.5) | 12.0 (±6.0) | 3.0 |
| N = 10 | N = 348 | N = 275 | N = 73§ | N = 6 | N = 5 | N = 1 | |
| Amyloid-β 1–42 | 984.1 (±233.7) | 719.9 (±269.4) | 716.8 (±268.8) | 732.2 (±272.9) | 487.8 (±218.4) | 455.5 (±219.7) | 573.8 (±195.5) |
| (in pg/ml) | N = 14 | N = 457§§ | N = 364§§ | N = 93§§ | N = 66** | N = 48** | N = 18** |
| h-Tau | 270.6 (±79.6) | 249.7 (±130.5) | 250.0 (±129.6) | 248.8 (±134.7) | 320.9 (±222.7) | 356.7 (±242.0) | 225.4 (±120.3) |
| (in pg/ml) | N = 14 | N = 457 | N = 364 | N = 93 | N = 66 | N = 48 | N = 18 |
| p-Tau | 48.9 (±14.8) | 42.2 (±17.0) | 42.6 (±17.0) | 40.6 (±16.8) | 46.0 (±25.8) | 49.4 (±27.6) | 36.9 (±17.8) |
| (in pg/ml) | N = 14 | N = 446 | N = 355 | N = 91 | N = 62 | N = 45 | N = 17 |
| NfL | 776.5 (±423.2) | 969.6 (±797.6) | 983.9 (±840.5) | 914.6 (±605.6) | 1709.9 (±1711.2) | 1843.5 (±1927.3) | 1372.0 (±937.0) |
| (in pg/ml) | N = 10 | N = 437 | N = 347 | N = 90 | N = 60* | N = 43* | N = 17 |
| α-Synuclein | 652.0 (±237.9) | 614.2 (±298.0) | 630.9 (±306.3) | 545.2 (±250.9) | 515.5 (±302.0) | 531.9 (±313.5) | 471.9 (±273.2) |
| (in pg/ml) | N = 11 | N = 451 | N = 363 | N = 88 | N = 62 | N = 45 | N = 17 |
| tissue factor | 534.4 (±161.6) | 454.0 (±155.9) | 457.3 (±160.1) | 440.7 (±137.8) | 450.4 (±182.1) | 469.7 (±200.3) | 398.0 (±107.3) |
| (in pg/ml) | N = 16 | N = 479 | N = 383 | N = 96 | N = 67 | N = 49 | N = 18* |
Non-parametric Mann-Whitney-U test with significant p-values ≤0.007 (manual Bonferroni-correction) presented as following: § PD vs. CON; * DLB vs. CON; 0.001 < p≤0.007: §/*; p≤0.001: §§/**. WT, wildtype; GBA1, variant in the gene for glucocerebrosidase 1; UPDRS III, Unified Parkinson’s Disease Rating Scale part III; MoCA, Montreal Cognitive Assessment; BDI II, Beck’s Depression Inventory II; NfL, neurofilament light chain.
Cross-sectional analysis
Group comparisons for continuous variables of demographic, clinical and CSF data between DLB, PD, and CON as well as between GBA1 subgroups were conducted using either the non-parametric Mann-Whitney U test (Table 1; due to limited number of CON participants) or parametric analysis of covariance for all other analysis (ANCOVA), with covariates sex, age at examination and disease duration as appropriate (for comparison of TF levels between male and female and for GBA1-subgroup analysis).
Group comparisons of categorical variables (such as the incidence of cognitive impairment and postural instability; prevalence of intake of blood-thinning medication; prevalence of GBA1 variants) were conducted using the chi-squared test.
Pearson correlation analysis was employed to investigate the influence of TF levels and age at examination on demographic, clinical and biomarker data (Supplementary Table 1).
Analysis of TF levels
Comparison of CSF TF levels between PD, DLB, and CON was performed using non-parametric Kruskal-Wallis test, followed by post-hoc tests (Supplementary Table 2).
Longitudinal analysis
Kaplan-Meier curves, along with Cox regression analysis incorporating the factor “tertile group”, were employed for longitudinal analysis. For these analyses, we compared the lowest vs. mid vs. highest tertile of CSF TF levels (Supplementary Table 3).
RESULTS
Demographic and cross-sectional data
PD total cohort
Of the 479 PD patients, 313 were male (65.3%). The mean age at examination was 65.4 years (±9.9), the mean age at onset 58.1 years (±10.5), the mean disease duration 7.3 years (±5.2), the mean MoCA score 25.3 (±3.9), the mean UPDRS III score 26.1 (±11.5) and the mean BDI II score 9.1 (±7.2) (Table 1). There was no distinction between male and female PD patients concerning the age at examination (65.4 (±9.9) vs. 64.9 (±10.2), p = 0.102).
Within the PD patient cohort, there were 59 individuals (12.3%) using acetylsalicylic acid alone, 4 individuals (0.8%) using a combination of acetylsalicylic acid and clopidogrel, 2 individuals (0.4%) using a combination of acetylsalicylic acid and ticagrelor, 1 individual (0.2%) using clopidogrel alone and 1 individual (0.2%) using rivaroxaban (a direct factor X inhibitor). We observed a heightened prevalence of blood-thinning medication intake, including inhibitors of platelet aggregation or anticoagulants, in PD patients with the highest levels of TF (lowest tertile: 17/133 (12.8%), mid tertile: 16/139 (11.5%), highest tertile: 34/140 (24.3%), p = 0.006).
Higher TF levels in CSF were associated with a higher age at examination (r (correlation coefficient according to Pearson) = 0.292, p≤0.001), a higher age at onset (r = 0.221, p≤0.001) and higher CSF levels of amyloid-β 1–42 (r = 0.250, p≤0.001), h-Tau (r = 0.513, p≤0.001), p-Tau (r = 0.603, p≤0.001), NfL (r = 0.213, p≤0.001), and α-Synuclein (r = 0.538, p≤0.001).
A higher age at examination was associated with a higher age at onset, a longer disease duration, a lower MoCA score, a higher UPDRS III score and higher CSF levels of h-Tau, p-Tau, NfL, and α-Synuclein (Supplementary Table 1).
Male PD patients demonstrated lower CSF levels of TF compared to female patients (436.8 (±142.4) vs. 486.5 (±174.3), p = 0.003).
PD WT
383 out of 479 patients had no variant in the GBA1 gene, referred to as PD WT. 65.0% (249) of those patients were male. The mean age at examination was 65.9 years (±9.9), the mean age at onset 58.8 years (±10.5), the mean disease duration 7.1 years (±5.0), the mean MoCA score 25.4 (±3.6), the mean UPDRS III score 26.0 (±11.5) and the mean BDI II score 8.9 (±7.1) (Table 1).
There was no significant difference between male versus female PD WT regarding the age at examination (65.5 (±10.2) vs. 66.7 (±9.4), p = 0.268).
We observed an increased prevalence of blood-thinning medication intake in PD WT who exhibited the highest levels of TF (lowest tertile: 12/103 (11.7%), mid tertile: 12/110 (10.9%), highest tertile: 28/110 (25.5%), p = 0.004).
Higher TF levels in CSF were associated with a higher age at examination (r = 0.328, p≤0.001), a higher age at onset (r = 0.256, p≤0.001) and higher CSF levels of amyloid-β 1–42 (r = 0.233, p≤0.001), h-Tau (r = 0.548, p≤0.001), p-Tau (r = 0.613, p≤0.001), NfL (r = 0.207, p≤0.001), and α-Synuclein (r = 0.540, p≤0.001) (Supplementary Table 1).
Male PD patients demonstrated significantly lower CSF levels of TF compared to female patients (436.5 (±142.7) vs. 496.0 (±182.4), p≤0.001).
PD GBA1
Among the 479 patients with PD, 96 patients had a genetic variant in the gene for glucocerebrosidase 1 (referred to as PD GBA1). In PD GBA1, 64 out of 96 patients were male (66.7%). The mean age at examination was 63.4 years (±9.4), the mean age at onset 55.1 years (±10.2), the mean disease duration 8.3 years (±5.6), the mean MoCA score 24.8 (±4.8), the mean UPDRS III score 26.6 (±11.7) and the mean BDI II score 10.2 (±7.3) (Table 1).
There was no significant difference between male versus female PD GBA1 regarding the age at examination (62.4 (±9.7) vs. 65.4 (±8.7), p = 0.154).
There was no significant difference observed in terms of the use of blood-thinning medication (p = 0.507) or the prevalence of severe GBA1 variants (p = 0.078) among the CSF TF tertiles.
Higher TF levels in CSF were associated with higher CSF levels of amyloid-β 1–42 (r = 0.331, p≤0.001), h-Tau (r = 0.371, p≤0.001), p-Tau (r = 0.557, p≤0.001), and α-Synuclein (r = 0.520, p≤0.001) (Supplementary Table 1).
No significant difference was observed between male and female patients regarding CSF levels of TF (p = 0.931). Furthermore, no variance was identified in CSF TF levels based on the severity of GBA1 variants (risk: 438.3 (±142.6), mild: 476.5 (±109.6), severe: 419.5 (±146.6), p = 0.474).
DLB cohort
Out of 67 patients diagnosed with DLB, 46 were male, constituting a prevalence of 68.7%. The mean age at examination was 72.5 years (±6.5), which is notably higher than the age at examination in CON (p≤0.001) and PD (p≤0.001). The mean age at onset was 69.3 years (±7.3), the mean disease duration 3.2 years (±2.1), the mean MoCA score 15.1 (±4.3), the mean UPDRS III score 26.8 (±9.2) and the mean BDI II score 10.5 (±6.5). 18 patients had a variant in GBA1 (26.9%) (Table 1). 88.9% (16/18) of these patients were male.
Higher TF levels in CSF were associated with higher CSF levels of h-Tau (r = 0.689, p≤0.001), p-Tau (r = 0.812, p≤0.001), and α-Synuclein (r = 0.750, p≤0.001) (Supplementary Table 1).
No statistically significant differences were observed between males and females with DLB regarding the CSF TF levels (p = 0.766).
CON
Of the 16 control participants (CON), 7 were male (43.8%). The mean age at examination was 65.6 years (±9.5), the mean MoCA score 27.7 (±2.2), the mean UPDRS III score 1.3 (±2.6) and the mean BDI II score 4.5 (±6.2) (Table 1).
There were no significant differences concerning the age at examination between CON vs. PD total cohort (p = 0.935), CON vs. PD WT (p = 0.794) or CON vs. PD GBA1 (p = 0.549).
Higher TF levels in CSF were associated with higher CSF levels of h-Tau (r = 0.844, p≤0.001) and p-Tau (r = 0.786, p≤0.001) (Supplementary Table 1).
Group comparison of TF levels between CON, PD, and DLB
There was a significant difference between the three cohorts regarding CSF TF levels with the lowest levels in DLB GBA1 (398.0 pg/ml (±107.3)), followed by PD GBA1 (440.7 pg/ml (±137.8)) and CON (534.4 pg/ml (±161.6)) (p = 0.034). Post-hoc tests revealed a significant difference in TF levels between DLB GBA1 vs. CON (p = 0.012; PD GBA1 vs. CON: p = 0.030; PD GBA1 vs. DLB GBA1: p = 0.271) (Supplementary Table 2).
Longitudinal analysis
PD total cohort
The follow-up duration (in years) revealed no statistically significant differences among the three tertiles of CSF TF levels (p = 0.337).
Likewise, the incidence of cognitive impairment (p = 0.712) and postural instability (p = 0.976) did not demonstrate statistically significant variations across the three tertile groups throughout the longitudinal study.
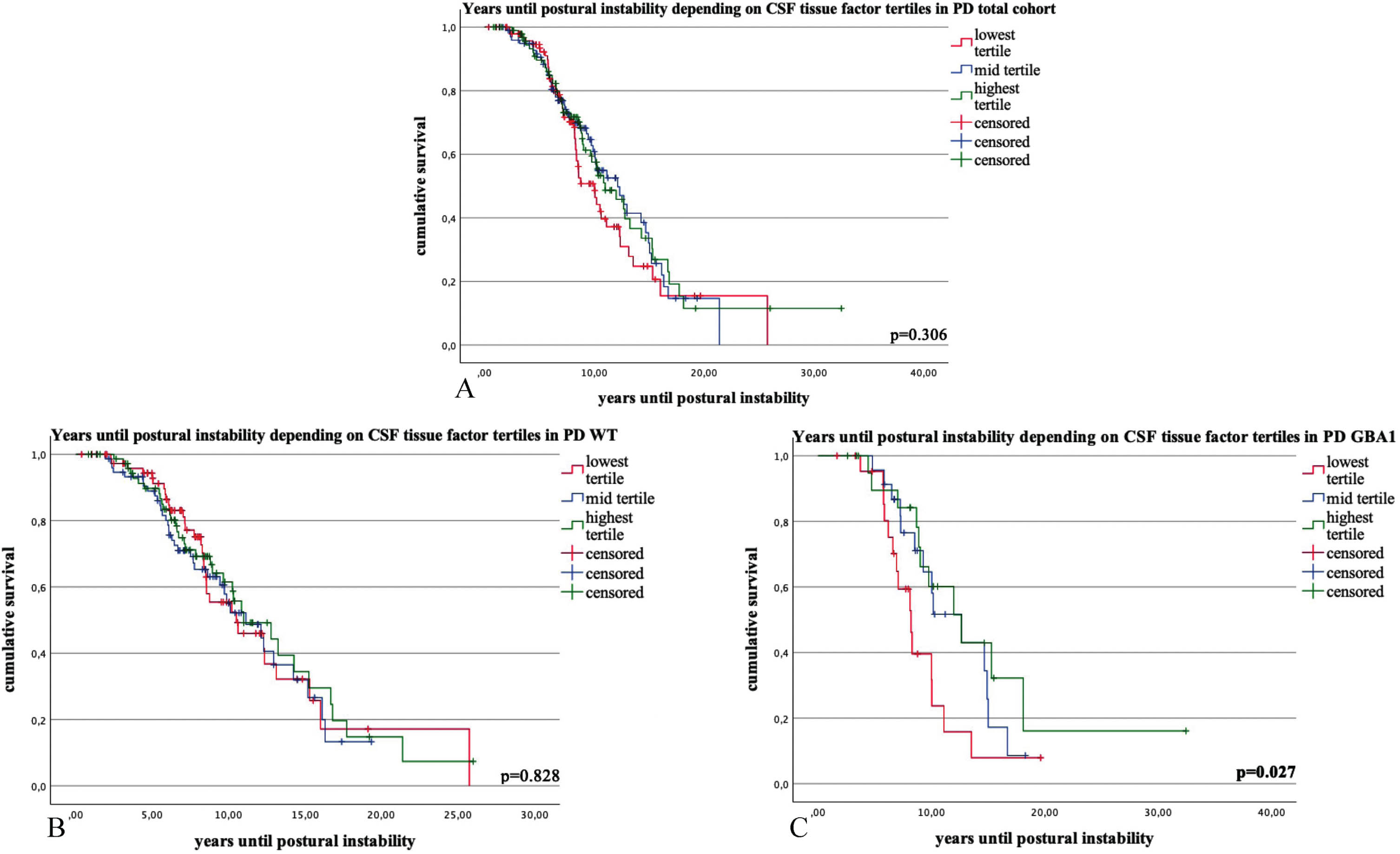
The time interval until 50% of patients reached postural instability did not exhibit any statistically significant differences based on the TF tertiles (p = 0.306) (Fig. 2 and Supplementary Tables 3 and 4).
Fig. 2
Kaplan-Meier survival curves and COX regression analysis for the time interval (in years) until 50% of the Parkinson’s disease (PD) patients reached the milestone postural instability (PI) in PD total cohort (1A), PD wildtype (1B) and PD GBA1 (1C), stratified by tertiles of CSF tissue factor (TF) levels. Categories: lowest tertile of tissue factor CSF levels; mid tertile of tissue factor levels; highest tertile of tissue factor levels. p-values ≤0.05 are highlighted in bold.
PD WT
Patients with the highest TF tertiles exhibited statistically significant longer follow-up times (lowest tertile: 7.9 (±4.3), N = 77; mid tertile: 8.1 (±3.9), N = 76; highest tertile: 8.2 (±4.7), N = 76; p = 0.047).
Throughout the follow-up times, there were no discernible differences in the incidence of postural instability (p = 0.769) and cognitive impairment (p = 0.130) among the three tertile groups. No statistically significant differences were observed in the time interval until 50% of patients reached postural instability based on the TF tertiles (p = 0.828) (Fig. 2 and Supplementary Tables 3 and 4).
PD GBA1
There were longer follow-up times in patients with the highest TF tertile (lowest tertile: 7.7 (±3.7), mid tertile: 9.9 (±3.8), highest tertile: 10.5 (±6.5), p = 0.033).
The time interval (in years) until 50% of patients reached postural instability differed significantly among the three tertiles of CSF TF levels. Specifically, in the lowest tertile, it was 9.2 years (95% confidence interval: 7.2–11.1, N = 23), in the mid tertile, it was 11.8 years (9.9–13.7, N = 23) and in the highest tertile, it was 14.9 years (9.8–20.1, N = 22) (p = 0.027). There was no significant difference in the age at examination (lowest tertile: 59.6 years (±9.5), mid tertile: 60.8 (±8.8), highest tertile: 64.6 (±10.7), p = 0.254).
The analysis yielded non-significant findings for the incidence of cognitive impairment (p = 0.261) and postural instability (p = 0.567) across the three tertile groups throughout the follow-up period (Fig. 2 and Supplementary Tables 3 and 4).
DISCUSSION
Our results support the hypothesis that tissue factor might serve as a novel biomarker for PD and other Lewy body diseases.
As a first step, we analyzed the association between TF levels and clinical data to determine if these levels reflect the progression of PD.
Higher TF levels in CSF were associated with a higher age at examination in PD, along with increased CSF levels of h-Tau, p-Tau, NfL, and α-Synuclein. Given that this pattern was similarly observed in the correlation analysis between the age at examination and the clinical and biomarker data, it is plausible to attribute these findings to age-related effects. Former studies also investigated on age-dependent changes of those CSF biomarkers showing an age-dependent increase in Tau,28 NfL,29 or α-Synuclein.30 However, an exception arises in the correlation between elevated TF levels and increased amyloid-β 1–42 in CSF. This deviation contrasts with the expected outcome linked to aging progression, wherein there is typically a decline in amyloid-β 1–42 levels, as partially evidenced by previous studies.31 Future studies should address the potential association between TF and amyloid-β 1–42 suggested by this finding, particularly considering the elevated deposition of TF antigen within senile plaques.17
Additionally, a significantly higher prevalence of blood-thinning medication intake was observed in PD patients with the highest tertile of TF levels. This finding may also be attributed to the older age at examination and the subsequently increased prevalence of neurovascular diseases. Previous studies demonstrated a reduction in circulating tissue factor following the administration of antiplatelet agents in individuals with peripheral arterial disease,32 especially with clopidogrel.33,34 This calls for further studies investigating interactions between CSF TF levels and blood-thinning medication.
Elevated CSF levels of TF were linked to a significantly delayed onset of postural instability in PD GBA1. Remarkably, patients with the highest TF levels exhibited a trend towards a higher age at examination, although this did not achieve statistical significance in the subgroup of patients with Kaplan-Meier data. Those patients with the highest TF levels also had longer follow-up times. It is essential to note that this observation could introduce potential bias, particularly if patients with a more protective disease course continued participation while others became dropouts.
Another limitation of our current study is the use of both the UPDRS III and, since 2009, the MDS-UPDRS III, which has a broader scale.
It is pertinent to acknowledge that the identification of age-related effects in our study serves not merely as a limitation but also as an opportunity. The quantification of TF protein levels could potentially serve as a surrogate marker, depicting (pathological) aging processes. This aligns with the hypothesis of accelerated aging in PD, dementia and other neurodegenerative conditions.
Our findings also revealed sex-specific differences, with male PD patients exhibiting lower CSF TF levels than their female counterparts. Investigations into sex-specific differences in blood coagulation and fibrinolysis have been conducted in various studies, including PD patients. Estrogen is recognized for its role in increasing coagulation proteins,35 which might serve as an explanation for sex-differences at least premenopausal, but not for our cohorts of mainly postmenopausal female patients. Other studies also showed sex-specific differences in coagulation proteins. In this line, elevated levels of Factor VIII were detected in the blood of female PD patients.36 Another study noted an age-related increase in TF pathway inhibitor and activated Factor VIIa, particularly in female participants.37 This calls for further investigations into those sex-specific differences of blood-coagulation factors especially in female PD patients, also considering postmenopausal supplementation of hormones like estrogen.
We found a significant difference between the three cohorts regarding CSF TF levels with the lowest levels in DLB GBA1, higher levels in PD GBA1 and the highest levels in CON.
This finding might mirror the continuum of Lewy body pathology observed in patients with PD and DLB,14 with the most pronounced Lewy body pathology in DLB patients, especially when exhibiting variants in GBA1.11 A proinflammatory condition preceding and/or induced by Lewy bodies might lead to the local deposition of TF, similar to the situation observed in senile plaques in AD, causing reduced levels of TF in the CSF of patients, analogous to the reduced CSF levels of α-Synuclein found in PD patients.
In pathological conditions characterized by Lewy body deposition, there may also be a local procoagulatory state, which contrasts with the proposed mild antiaggregant effects of α-Synuclein under normal physiological conditions.15 The fact that we only found significant results when comparing CON with PD and DLB patients exhibiting GBA1 variants might be explained by the higher α-Synuclein pathology in these groups compared to PD and DLB wild-type patients. Future studies should address this assumption.
The most significant limitation of our study is the small number of control participants. Therefore, our observations highlight the need for further investigations in larger cohorts.
First studies found effects of thrombin-inhibitors in models of PD. In this line, treatment with direct thrombin inhibitors has demonstrated neuroprotective effects, such as the amelioration of motor deficits and reduction of oxidative stress.18,38 This treatment resulted in increased Nurr1 expression and reduced thrombin accumulation in the substantia nigra, activating genes associated with tyrosine hydroxylase and vascular monoamine transporter. This led to elevated dopamine levels and improved motor function.39 A deficiency of PAR-1 and a treatment with a PAR-1 antagonist were associated with protective effects against 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)- induced toxicity.40 In this line, compensatory mechanisms in response to neurodegeneration, as previously discussed with the increased expression of PAR-1 in astrocytes of PD patients, merit particular consideration.41
Therefore, further investigations into the connections between blood coagulation and PD would be of interest.
ACKNOWLEDGMENTS
We acknowledge support from the Open Access Publication Fund of the University of Tuebingen.
FUNDING
This work was funded by the State Ministry of Baden-Württemberg for Economic Affairs, Labour and Tourism (“Predictive Diagnostic of immune-associated diseases for personalized medicine”, AZ 35-4223.10/8). The funder played no role in study design, data collection, analysis and interpretation of data, or the writing of this manuscript.
CONFLICT OF INTEREST
There are no conflicts of interest among the authors associated with the manuscript.
Dr. Kathrin Brockmann received Research Grants from the Michael J Fox Foundation for Parkinson’s Research (“LRRK2 Kinase Activity”, “Influence of Inflammatory Profiles on PD Phenotype and Progression”, “Prevent Dementia in GBA1-associated PD”), from the University of Tuebingen (“Endophenotyping of GBA1-PD”), from the German Society for Parkinson DPG, from the Health Forum Baden Wuerttemberg (“Predictive Diagnostic of immune-associated diseases for personalized medicine”, AZ 35-4223.10/8), from the Else Kröner Fresenius Stiftung (“ClinBrain”), and from the German Research Foundation DFG (“CORO-TREND”). She serves on advisory boards for F. Hoffmann-La Roche Ltd and VanqaBio. She received speaker honoraria from Abbvie, Lundbeck, UCB and Zambon.
Christian Deuschle has nothing to disclose.
Prof. Dr. Thomas Gasser serves on the editorial board of the Journal of Parkinson’s Disease, but was not involved in the peer-review process of this article nor had access to any information regarding its peer review. He holds a patent re: KASPP (LRRK2) gene, its production and use for the detection and treatment of neurodegenerative diseases. Prof. Gasser has received speaker’s honoraria from UCB Pharma, Novartis, Sanofi and MedUpdate. He has received consulting fees from Bayer AG, BlueRock Therapeutics and Biogen. He is Chairman of the Scientific Advisory Board of the “Joint Programming for Neurodegenerative Diseases” program, funded by the European Commission. He has received grant support from the German Research Foundation (DFG), the German Federal Ministry of Education and Research (BMBF), the Ministry for Science, Research and Art Baden-Württemberg (MWK), the European Commission, the Helmholtz Association and The Michael J. Fox Foundation.
Madeleine Fandrich has nothing to disclose.
Dr. Meike Jakobi has nothing to disclose.
Dr. Thomas O. Joos has nothing to disclose
Dr. Stefanie Lerche has nothing to disclose.
Dr. Benjamin Röben has received a research grant from the University of Tuebingen (Clinician Scientist; Project-Nr. 480-0-0).
Dr. Nicole Schneiderhan-Marra has received funding from the State Ministry of Baden-Württemberg for Economic Affairs, Labour and Tourism (“Predictive Diagnostic of immune-associated diseases for personalized medicine”, AZ 35-4223.10/8).
Claudia Schulte has nothing to disclose.
Dr. Isabel Wurster receives funding from The Michael J. Fox Foundation (MJFF) as an Edmond J. Safra Fellow in Movement Disorders.
Dr. Milan Zimmermann has received a research grant from the University of Tuebingen (Clinician Scientist; Project-Nr. 481-0-0).
Shahrzad Zimmermann has nothing to disclose.
DATA AVAILABILITY
The pseudonymized data of this study are available from the corresponding author upon reasonable request.
SUPPLEMENTARY MATERIAL
[1] The supplementary material is available in the electronic version of this article: https://dx.doi.org/10.3233/JPD-240115.
REFERENCES
[1] | Bower JH , Maraganore DM , McDonnell SK , et al. Incidence and distribution of parkinsonism in Olmsted County, Minnesota, 1976-1990. Neurology (1999) ; 52: : 1214–1220. |
[2] | Forsaa EB , Larsen JP , Wentzel-Larsen T , et al. What predicts mortality in Parkinson disease? a prospective population-based long-term study. Neurology (2010) ; 75: : 1270–1276. |
[3] | Buter TC , van den Hout A , Matthews FE , et al. Dementia and survival in Parkinson disease: a 12-year population study. Neurology (2008) ; 70: : 1017–1022. |
[4] | Alves G , Wentzel-Larsen T , Aarsland D , et al. Progression of motor impairment and disability in Parkinson disease: a population-based study. Neurology (2005) ; 65: : 1436–1441. |
[5] | Zimmermann M and Brockmann K. Blood and cerebrospinal fluid biomarkers of inflammation in Parkinson’s disease. J Parkinsons Dis (2022) ; 12: (s1): S183–S200. |
[6] | Franchini M Hemostasis and aging. Crit Rev Oncol Hematol (2006) ; 60: : 144–151. |
[7] | Jain S , Gautam V and Naseem S. Acute-phase proteins: As diagnostic tool. J Pharm Bioallied Sci (2011) ; 3: : 118–127. |
[8] | Mackman N . Role of tissue factor in hemostasis, thrombosis, and vascular development. Arterioscler Thromb Vasc Biol (2004) ; 24: : 1015–1022. |
[9] | Bokarewa MI , Morrissey JH and Tarkowski A. Tissue factor as a proinflammatory agent. Arthritis Res (2002) ; 4: : 190–195. |
[10] | Choi SH , Joe EH , Kim SU , et al. Thrombin-induced microglial activation produces degeneration of nigral dopaminergic neurons in vivo, J Neurosci (2003) ; 23: : 5877–5886. |
[11] | Lerche S , Machetanz G , Wurster I , et al. Dementia with lewy bodies: GBA1 mutations are associated with cerebrospinal fluid alpha-synuclein profile. Mov Disord (2019) ; 34: : 1069–1073. |
[12] | Lerche S , Zimmermann M , Roeben B , et al. Inflammatory CSF profiles and longitudinal development of cognitive decline in sporadic and GBA-associated PD. NPJ Parkinsons Dis (2023) ; 9: : 38. |
[13] | Lerche S , Zimmermann M , Wurster I , et al. CSF and serum levels of inflammatory markers in PD: sparse correlation, sex differences and association with neurodegenerative biomarkers. Front Neurol (2022) ; 13: : 834580. |
[14] | Mensikova K , Matej R , Colosimo C , et al. Lewy body disease or diseases with Lewy bodies? NPJ Parkinsons Dis (2022) ; 8: : 3. |
[15] | Acquasaliente L , Pontarollo G , Radu CM , et al. Exogenous human alpha-Synuclein acts in vitro as a mild platelet antiaggregant inhibiting alpha-thrombin-induced platelet activation, Sci Rep (2022) ; 12: : 9880. |
[16] | Furie B and Furie BC. P-selectin induction of tissue factor biosynthesis and expression. Haemostasis (1996) ; 26: (Suppl 1): 60–65. |
[17] | McComb RD , Miller KA and Carson SD. Tissue factor antigen in senile plaques of Alzheimer’s disease. Am J Pathol (1991) ; 139: : 491–494. |
[18] | Iannucci J , Renehan W and Grammas P. Thrombin, a mediator of coagulation, inflammation, and neurotoxicity at the neurovascular interface: implications for Alzheimer’s disease. Front Neurosci (2020) ; 14: : 762. |
[19] | Smith C , Malek N , Grosset K , et al. Neuropathology of dementia in patients with Parkinson’s disease: a systematic review of autopsy studies. J Neurol Neurosurg Psychiatry (2019) ; 90: : 1234–1243. |
[20] | Hughes AJ , Daniel SE , Kilford L , et al. Accuracy of clinical diagnosis of idiopathic Parkinson’s disease: a clinico-pathological study of 100 cases. J Neurol Neurosurg Psychiatry (1992) ; 55: : 181–184. |
[21] | Goetz CG , Tilley BC , Shaftman SR , et al. Movement disorder society-sponsored revision of the unified Parkinson’s disease rating scale (MDS-UPDRS): scale presentation and clinimetric testing results. Mov Disord (2008) ; 23: : 2129–2170. |
[22] | McKeith IG , Boeve BF , Dickson DW , et al. Diagnosis and management of dementia with Lewy bodies: Fourth consensus report of the DLB Consortium. Neurology (2017) ; 89: : 88–100. |
[23] | Nasreddine ZS , Phillips NA , Bedirian V , et al. The Montreal Cognitive Assessment, MoCA: a brief screening tool for mild cognitive impairment. J Am Geriatr Soc (2005) ; 53: : 695–699. |
[24] | Folstein MF , Folstein SE and McHugh PR. “Mini-mental state”. A practical method for grading the cognitive state of patients for the clinician. J Psychiatr Res (1975) ; 12: : 189–198. |
[25] | Bergeron D , Flynn K , Verret L , et al. Multicenter validation of an MMSE-MoCA conversion table. J Am Geriatr Soc (2017) ; 65: : 1067–1072. |
[26] | Hoops S , Nazem S , Siderowf AD , et al. Validity of the MoCA and MMSE in the detection of MCI and dementia in Parkinson disease. Neurology (2009) ; 73: : 1738–1745. |
[27] | Beck AT , Steer RA and Brown GK. Manual for the Beck Depression Inventory-II. San Antonio, TX: Psychological Corporation, (1996) . |
[28] | Glodzik-Sobanska L , Pirraglia E , Brys M , et al. The effects of normal aging and ApoE genotype on the levels of CSF biomarkers for Alzheimer’s disease. Neurobiol Aging (2009) ; 30: : 672–681. |
[29] | Vermunt L , Otte M , Verberk IMW , et al. Age- and disease-specific reference values for neurofilament light presented in an online interactive support interface. Ann Clin Transl Neurol (2022) ; 9: : 1832–1837. |
[30] | Winkel I , Ermann N , Zelwetro A , et al. Cerebrospinal fluid alpha synuclein concentrations in patients with positive AD biomarkers and extrapyramidal symptoms. J Neural Transm (Vienna) (2021) ; 128: : 817–825. |
[31] | Peskind ER , Li G , Shofer J , et al. Age and apolipoprotein E*4 allele effects on cerebrospinal fluid beta-amyloid 42 in adults with normal cognition. Arch Neurol (2006) ; 63: : 936–939. |
[32] | Rao AK , Vaidyula VR , Bagga S , et al. Effect of antiplatelet agents clopidogrel, aspirin, and cilostazol on circulating tissue factor procoagulant activity in patients with peripheral arterial disease. Thromb Haemost (2006) ; 96: : 738–743. |
[33] | Savi P , Bernat A , Dumas A , et al. Effect of aspirin and clopidogrel on platelet-dependent tissue factor expression in endothelial cells. Thromb Res (1994) ; 73: : 117–124. |
[34] | Stellbaum C , Willich T , Boltzen U , et al. Clopidogrel-mediated reduction of circulating tissue factor in patients with stable coronary artery disease. Eur J Haematol (2007) ; 78: : 347–352. |
[35] | Abou-Ismail MY , Citla Sridhar D and Nayak L. Estrogen and thrombosis: A bench to bedside review. Thromb Res (2020) ; 192: : 40–51. |
[36] | Sharma A , Muller J , Schuetze K , et al. Comprehensive profiling of blood coagulation and fibrinolysis marker reveals elevated plasmin-antiplasmin complexes in Parkinson’s disease. Biology (Basel) (2021) ; 10: : 716. |
[37] | Ariens RA , Coppola R , Potenza I , et al. The increase with age of the components of the tissue factor coagulation pathway is gender-dependent. Blood Coagul Fibrinolysis (1995) ; 6: : 433–437. |
[38] | Johnson SL , Iannucci J , Seeram NP , et al. Inhibiting thrombin improves motor function and decreases oxidative stress in the LRRK2 transgenic Drosophila melanogaster model of Parkinson’s disease. Biochem Biophys Res Commun (2020) ; 527: : 532–538. |
[39] | Kandil EA , Sayed RH , Ahmed LA , et al. Modulatory role of Nurr1 activation and thrombin inhibition in the neuroprotective effects of dabigatran etexilate in rotenone-induced Parkinson’s disease in rats. Mol Neurobiol (2018) ; 55: : 4078–4089. |
[40] | Hamill CE , Caudle WM , Richardson JR , et al. Exacerbation of dopaminergic terminal damage in a mouse model of Parkinson’s disease by the G-protein-coupled receptor protease-activated receptor 1. Mol Pharmacol (2007) ; 72: : 653–664. |
[41] | Ishida Y , Nagai A , Kobayashi S , et al. Upregulation of protease-activated receptor-1 in astrocytes in Parkinson disease: astrocyte-mediated neuroprotection through increased levels of glutathione peroxidase. J Neuropathol Exp Neurol (2006) ; 65: : 66–77. |


