
A Phase I, Randomized, SAD, MAD, and PK Study of Risvodetinib in Older Adults and Parkinson’s Disease
Abstract
Background:
Pre-clinical studies suggest that c-Abl activation may play an important role in the etiology of Parkinson’s disease, making c-Abl an important target to evaluate for potential disease-modification.
Objective:
To assess safety, tolerability, and pharmacokinetics of the c-Abl inhibitor risvodetinib (IkT-148009) in healthy subjects and participants with Parkinson’s disease.
Methods:
Part 1 (single ascending dose (SAD)) and Part 2 (7-day multiple ascending dose (MAD)) studies were in healthy volunteers. Participants were randomized 3 : 1 across 9 SAD doses and 3 MAD doses of risvodetinib (IkT-148009) or placebo. Part 3 was a MAD study conducted at two doses in 14 participants with mild-to-moderate PD (MAD-PD). Primary outcome measures were safety, tolerability and pharmacokinetics. Exploratory outcomes in PD participants included clinical measures of PD state, GI function, and cerebrospinal fluid (CSF) concentration.
Results:
108 patients were treated with no dropouts. The SAD tested doses ranging from 12.5 to 325 mg, while the MAD tested 25 to 200 mg and MAD-PD tested 50 to 100 mg in Parkinson’s participants. All active doses had a favorable safety profile with no clinically meaningful adverse events. Single dose pharmacokinetics were approximately linear between 12.5 mg and 200 mg for both Cmax and AUC0 - inf without distinction between healthy volunteers and participants with PD. Exposures at each dose were high relative to other drugs in the same kinase inhibitor class.
Conclusions:
Risvodetinib (IkT-148009) was well tolerated, had a favorable safety and pharmacology profile over 7-day dosing, did not induce serious adverse events and did not appear to induce deleterious side-effects in participants administered anti-PD medications.
INTRODUCTION
Parkinson’s disease (PD) is the second most prevalent neurodegenerative disorder [1], affecting up to 1.2 million persons in the United States, with 90,000 new cases annually [1– 3]. PD is a progressive disorder characterized clinically by bradykinesia, rigidity, resting tremor, and gait disturbances with postural instability [1, 3]. Pathologically, PD involves degeneration of dopaminergic (DA) and specific non-DA neurons coupled with the accumulation of alpha-synuclein aggregates [4– 6]. Recent studies in slowly progressive murine models of inherited or sporadic PD demonstrated that inhibition of the activated, non-receptor Abelson Tyrosine Kinases, termed c-Abl hereafter, can modify disease and substantially reduce alpha-synuclein pathology in the affected areas of the brain [7– 13]. The hallmarks of c-Abl activation have also been observed in post-mortem patient samples and in model studies of Multiple System Atrophy [14].
Marketed anti-cancer c-Abl inhibitors are poorly active in the central nervous system (CNS) due to their removal across the blood-brain barrier by efflux transporters such as P-glycoprotein (PGP). Risvodetinib (IkT-148009) is a selective inhibitor of c-Abl that was designed to have a reduced affinity for transporters like PGP. In a series of prophylactic and therapeutic animal models, once-daily oral administration of risvodetinib blocked disease initiation and progression in the MPTP acute toxicity model [13] and was therapeutically effective following once daily oral administration in models of inherited or sporadic Parkinson’s-like disease [13]. These observations prompted clinical evaluation of the safety, tolerability and clinical pharmacokinetics following single and multiple oral doses of risvodetinib in older healthy adults and participants with mild-to-moderate PD who remained on symptomatic therapies.
METHODS
Trial design
Clinical evaluation was performed as either a single ascending (SAD) or multiple ascending dose (7-day, MAD) escalation study in three parts. Part 1 (the SAD study) measured safety, tolerability and pharmacokinetics of single oral doses of risvodetinib succinate salt (hereafter referred to as risvodetinib) in healthy volunteers (see the Supplement-Protocols for details). Risvodetinib was administered as Type 0 gelatin capsule at nine doses ranging between 12.5 mg and 325 mg freebase. Part 2 (the MAD study) measured safety, tolerability, and pharmacokinetics of risvodetinib in older healthy adults at doses of 12.5 mg, 25 mg, and 200 mg. Part 3 (the MAD-PD study) measured doses of 50 and 100 mg of risvodetinib in participants with mild-to-moderate PD who remained on a stable regimen of symptomatic therapies. Voluntary lumbar puncture (LP) was used to measure the concentration of risvodetinib in cerebrospinal fluid (CSF) after six days of dosing in Parts 2 and 3. An independent Safety Review Committee (SRC) reviewed data prior to each dose escalation and for entry into Parts 2 and 3. The trial was conducted in accordance with the Principles of the Declaration of Helsinki and Good Clinical Practice Guidelines.
Setting and participants
Participants were recruited at a single Phase 1 clinical research unit for older healthy adults (Parts 1 and 2) or at two movement disorder specialty clinics (Part 3). Eligibility criteria included healthy adults and PD participants aged 45 to 70 with a Mini-Mental State Exam (MMSE) score ≥28 (≥26 for participants in Part 3), women who were either postmenopausal, sterile or men and women who agreed to practice two methods of highly effective birth control given the known risk of fatality to a developing embryo or fetus exposed to a c-Abl inhibitor [15, 16]. In Part 3, additional enrollment criteria included a diagnosis of PD consistent with the UK PD Society Brain Bank Criteria, evident bradykinesia, a clear motor response to dopamine replacement therapy, Hoehn & Yahr staging of 3 or less in the ON state, stable doses of all PD-related symptomatic therapy in the 4 weeks preceding screening and approval by a movement disorder specialist Enrollment Authorization Committee for suitability to participate.
Key exclusion criteria included clinically significant abnormal values for hematology, clinical chemistry or urinalysis, abnormal safety ECG or a QTcF interval ≥450 ms for male participants or ≥470 ms in female participants. An eGFR <60 mL/min and serum creatinine, amylase or lipase > upper limit of normal (ULN) were also exclusionary. For Part 3, additional key exclusion criteria included diagnosis of atypical parkinsonism, prior neurosurgery for PD or treatment with DUODOPA or infused apomorphine, concurrent use of neuroleptic medications, or clinically significant hallucinations, or major depression.
Randomizations and interventions
The properties of risvodetinib, including on-target and off-target inhibitory effects, inhibitory potential relative to commercial anti-cancer c-Abl inhibitors, as well as toxicity and histopathology studies from two species toxicology studies, including chronic toxicology studies, have been previously published [13 and accompanying supplemental materials].
In the SAD study, subjects were randomized 3 : 1 to escalating doses of 12.5 mg, 25 mg, 37.5 mg, 75 mg, 100 mg, 125 mg, 175 mg, 250 mg, or 325 mg risvodetinib freebase or placebo (N = 8) administered as a single Type 0 gelatin capsule. In the MAD studies, participants with PD were randomized to 50 mg or 100 mg risvodetinib or placebo as 2×25 mg or 4×25 mg Risvodetinib freebase gelatin capsules or placebo (N = 8), while the 200 mg dose was administered as 4×50 mg risvodetinib freebase gelatin capsules with no randomization (N = 6). Placebo was administered as hyprocellulose in a matching capsule. All dosing was done in the fed state.
Evaluations
Safety was evaluated throughout the study by monitoring adverse events and measures of vital signs with replicate measures of orthostatic blood pressure, ECGs with cardiodynamic Holter monitoring (in Part 1 only), chest x-ray, Columbia Suicide Severity Rating Scale (C-SSRS) and standard laboratory assessments. Tolerability was defined by percent of completers. Pharmacokinetic measures included area under the concentration-time curve from time 0 to 96 h (AUC0 - t) as well as AUC0 - inf, the maximum plasma concentration (Cmax), time to reach maximum concentration (Tmax), terminal half-life (t1/2), the maximum concentration at steady state (Cmax,ss) and the area under the concentration-time curve at steady-state (AUCss). Exploratory outcomes performed in PD participants included change from baseline to final visit in the Movement Disorder Society Unified Parkinson’s Disease Rating Scale (MDS-UPDRS) Parts III, II and I), Clinical Global Impression of Improvement (CGI-I), Patient Global Impression of Change (PGI-C) and Severity (PGI-S), Non-Motor Symptom Score (NMSS), Parkinson’s Disease Questionnaire (PDQ-39), Complete Satisfaction of Bowel Movement (CSBM) score, Upper GI Disorders Severity Index (PAGI-SYM) and CSF trough concentration of risvodetinib. For the MDS-UPDRS assessments, Part III baseline and Day 7 pre-dose assessment was attempted to be performed in the practically-defined OFF state, defined as 7– 12 h after withdrawal of anti-PD medications the evening before the assessment was to be performed. For Parts I and II, the Day 7 assessment was performed in the practically defined OFF state, but the baseline measure was performed while anti-Parkinson’s medications were still on board. All other assessments were performed with anti-Parkinson’s medications on board.
Statistical analyses
Data were summarized separately for Parts 1, 2, and 3, with placebo subjects pooled within each study part (see Supplement-Protocols and Statistics for details). All summaries (safety, tolerability, and pharmacokinetics (PK)) were grouped by dose defined as treated at each scheduled time point. For vital signs, the baseline value was defined as the last value observed prior to the first administration of study medication on Day 1. Change from baseline values were calculated at each time point and summarized using descriptive statistics. No adjustment was made for multiple comparisons in this study and no sample size was calculated. Phoenix WinNonlin™ version 8.3 or higher was used for the PK analyses.
RESULTS
Participants and adverse events
All participants provided IRB-approved written informed consent. Ninety-four older healthy adults and 14 people with PD participated in these studies, of which 72 older healthy adults participated in the SAD study and 22 participated in the MAD study at 12.5 mg, 25 mg, and 200 mg. Fourteen people participated in the MAD-PD study at 50 mg or 100 mg (Table 1).
Table 1
Baseline characteristics and demographics of older healthy adults or participants with PD administered single or 7-day daily doses of risvodetinib (IkT-148009)
| Category | Demographic | # Healthy Subjects(% of Total)N = 94 | # Participants with PD7-day, 1x/day (% of Total)N = 14 |
| Gender | Female | 36 (37.9) | 7 (50) |
| Male | 58 (61.1) | 7 (50) | |
| Age | Average | 57.9 | 62.6 |
| Median | 58.0 | 61.5 | |
| Range | 45, 68 | 57, 70 | |
| Ethnicity | Hispanic or Latino | 14 (14.9) | 3 (21.4) |
| Not Hispanic or Latino | 80 (85.1) | 11 (78.6) | |
| Race | Black or African American | 55 (58.5) | 2 (14.3) |
| White | 37 (39.4) | 9 (64.3) | |
| Other | 2 (2.1) | 0 (0) |
Treatment emergent adverse events were infrequent at all doses and dosing durations (Supplementary Table 1). In the SAD phase, there were 12 adverse events which were mild in severity and none were of clinical significance in the view of site investigators. In the MAD phase in healthy participants, 12 adverse events were recorded (3 among placebo-treated participants), all of which were of mild severity and none of clinical significance in the view of the site investigators. In participants with PD, 12 adverse events were recorded (3 in placebo-treated patients), all of which were mild in severity and none of clinical significance in the view of site investigators. Among all 36 recorded adverse events, 11 were deemed possibly or probably related to the active drug by the site investigators (Table 2).
Table 2
Treatment-related adverse events following single or multiple doses of risvodetinib (IkT-148009) in older healthy adults or participants with PD
| Category | Dose mg | Dose and Duration | # OccurrencesHealthy Subjects(N = 94) | # OccurrencesPD patients(N = 14) | Severity |
| Cardiovascular | 75 mg | Single Dose | 1 Palpitations1 | Mild | |
| Gastrointestinal | |||||
| 325 mg | Single Dose | 2 Diarrhea | Mild | ||
| 100 mg | 7-day, 1x/day | 1 Constipation2 | Mild | ||
| 200 mg | 7-day, 1x/day | 1 Elevated Lipase3 | Mild | ||
| Dermatological | |||||
| 50 mg | 7-day, 1x/day | 1 Dermatitis4 | Mild | ||
| Musculoskeletal | |||||
| 200 mg | 7-day, 1x/day | 5 Myalgias, joint pain, fatigue, edema3 | Mild |
1Appeared 2 weeks post-dose, no clinical basis found even after following by 3-day Holter monitoring; 2Appeared one day after last dosing day; 3Six AEs in a single subject of mild severity. Lipase elevation occurred on Day 6 of a 7-day dosing period and was normal pre-dose and on Days 5 and 7. 4Occured on first dosing day, was treated with a single dose of Benedryl and did not reoccur in the following 6 days.
Few laboratory abnormalities were noted at any dose or dose duration. The most frequent laboratory abnormality was an asymptomatic elevation in amylase and/or lipase (Supplementary Table 2). Eighteen of the participants, which included three participants on placebo, experienced sporadic elevations in either pancreatic enzyme and three participants displayed sporadic changes in both enzymes. In all but one case, amylase and/or lipase values returned to normal at the next timepoint without interruption of dosing, while the single case of progressive elevations in both enzymes returned to normal values following end of dosing. No participant was symptomatic or recorded an adverse event related to these sporadic changes in pancreatic enzyme values.
No participants displayed evidence of QTcF prolongation at any dose or dose duration, nor did any participant show signs of drug-related orthostatic changes in blood pressure. In participants with PD, risvodetinib did not exacerbate pre-existing orthostatic hypotension nor induced any abnormalities in heart rate, heart rhythm or blood pressure.
Functional assessments of motor and non-motor features of PD were performed for the 50 and 100 mg doses of risvodetinib or placebo (Table 3). No PD-related adverse events were recorded in the PD treatment or placebo treatment groups. The mean scores for MDS-UPDRS Part III increased by 6.2 (±9.50) points and 7.4 (±14.1) points, respectively, while the placebo treated participants experienced a 2.3 (±0.6) point decrease in mean value. Measurements over the 6-h window at baseline generally increased with time over pre-dose values, suggesting no acute treatment effect (Table 3).
Table 3
Assessment of motor and non-motor features of Parkinson’s disease in participants given risvodetinib (IkT-148009) once daily for 7-days
| Risvodetinib | Risvodetinib | Pooled Placebo | ||||
| 50 mg | 100 mg | Mean±SD | ||||
| Mean±SD | Mean±SD | (N = 3) | ||||
| (N = 6) | (N = 5) | |||||
| Parameter | Pre-dose | Day 7 Dose | Pre-dose | Day 7 Dose | Pre-dose | Day 7 Dose |
| MDS-UPDRS I1 | 8.2±2 | 5.8±3.3 | 5.4±3.5 | 4.8±3.6 | 9±4.6 | 5.7±4.2 |
| MDS-UPDRS II1 | 11.2±5 | 8.7±6.9 | 8.4±3.9 | 8.2±4.9 | 11.7±10.3 | 9.7±11.6 |
| MDS-UPDRS III2 | 28.2±9.6 | 34.3±10.7 | 41±18.3 | 48.4±20 | 40±18.4 | 37.7±18.7 |
| NMSS3 | 36.5±23.5 | 28.3±19.5 | 18.4±16.1 | 19.2±18.7 | 46.7±36.6 | 27.3±19.1 |
| PDQ-393,4 | 16.4±10.1 | 19.9±8.2 | 15.3±9.1 | 14.8±14.1 | 19±27 | 17.7±30.7 |
| PGI-S3 | 2.2±0.8 | 2.2±0.7 | 2.0±0.7 | 2.4±0.5 | 2±1 | 1.7±0.6 |
1MDS-UPDRS Parts I and II baseline measures were performed with anti-Parkinson’s medications on board. Day 7 measures were performed in the practically defined OFF state; 2MDS-UPDRS Part III baseline measure and Day 7 measures were performed in the practically defined OFF state; 3Baseline and Day 7 measures were performed with anti-Parkinson medications on board; 4PDQ-39 is reported using the weighting according to Reference 21.
Pharmacokinetics
Single dose PK were approximately linear between 12.5 mg and 175 mg, but absorption became non-linear at higher doses, particularly for Cmax (Fig. 1) and exposures were less than dose proportional (Table 4). Maximum concentrations were reached between 3-h and 7-h post-dose with an average terminal phase half-life ranging between 24 and 39 h (Table 4).
Fig. 1
Plasma pharmacokinetic Cmax and AUC0 - inf as a function of single oral dose for risvodetinib (IkT-148009). Risvodetinib (IkT-148009) was administered once and the plasma pharmacokinetics measured from 0 to 96 h in 9 cohorts of 8 subjects each (3 : 1 randomized to placebo). Risvodetinib was administered as a single Type 0 gelatin capsules which delivered the indicated dose as the freebase. A) Mean±standard deviation of Cmax (ng/mL) and B) Mean±standard deviation of AUC0 - inf (ng-h/mL).
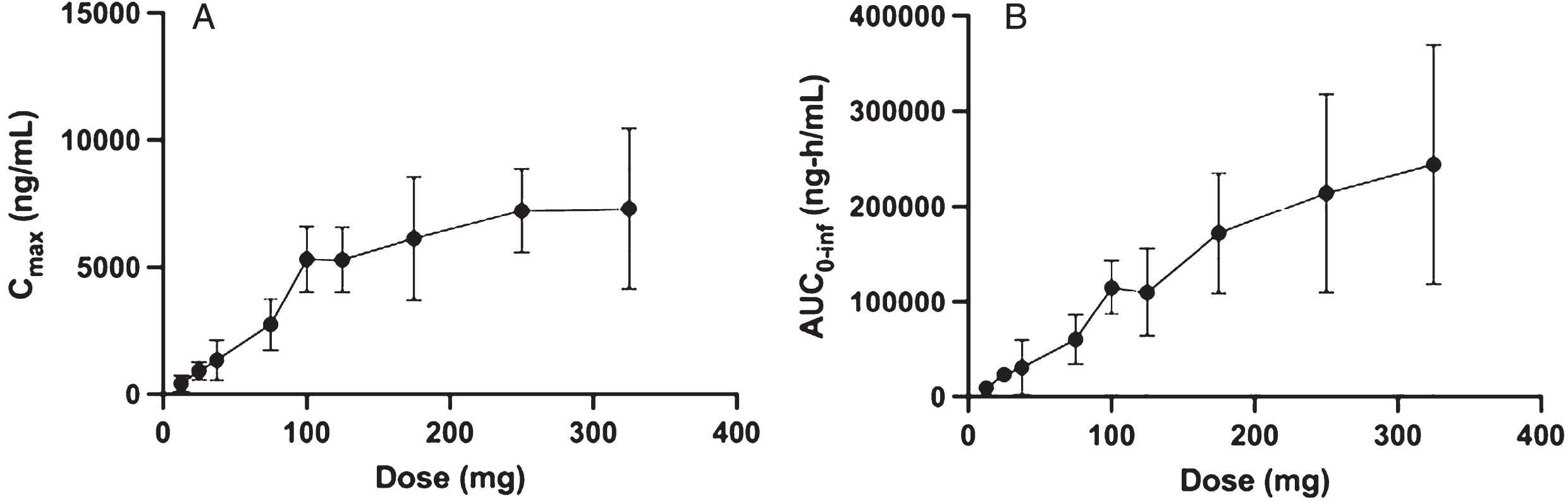
Table 4
Plasma pharmacokinetic mean and (standard deviation) values for single dose risvodetinib (IkT-148009) in older healthy adults (N = 6/dose)
| Dose | t1/2 | tlag | tmax | Cmax | AUC0 - t | AUC0 - ∞ | Cmax/D | AUC0 - ∞/D |
| (mg) | (h) | (h) | (h) | (ng/mL) | (h*ng/mL) | (h*ng/mL) | (ng/mL/mg) | (h*ng/mL/mg) |
| 12.5 | 25.0 (7.56) | 1.09 (1.48) | 7.33 (2.73) | 447 (333) | 9160 (4310) | 9470 (4310) | 35.8 (26.6) | 758 (345) |
| 25.0 | 25.9 (3.04) | 0.614 (0.367) | 5.97 (3.37) | 945 (348) | 22700 (7630) | 23400 (7860) | 37.8 (13.9) | 936 (314) |
| 37.5 | 24.4 (8.36) | 0.642 (0.393) | 5.67 (3.20) | 1350 (794) | 30400 (29200) | 30900 (29100) | 36.0 (21.2) | 823 (776) |
| 75.0 | 28.5 (8.38) | 0.342 (0.143) | 5.00 (1.10) | 2750 (1020) | 58400 (26900) | 60100 (25600) | 36.6 (13.6) | 802 (342) |
| 100 | 32.0 (4.83) | 0.414 (0.350) | 4.33 (1.51) | 5320 (1280) | 114000 (28400) | 115000 (28500) | 53.2 (12.8) | 1150 (285) |
| 125 | 30.2 (14.0) | 0.306 (0.284) | 3.34 (0.971) | 5290 (1270) | 109000 (46300) | 110000 (46200) | 42.3 (10.1) | 879 (370) |
| 175 | 36.6 (5.75) | 0.0944 (0.146) | 5.67 (5.28) | 6130 (2410) | 170000 (62800) | 172000 (62900) | 35.0 (13.7) | 982 (359) |
| 250 | 38.7 (6.86) | 0.547 (0.507) | 5.00 (1.67) | 7220 (1640) | 213000 (103000) | 214000 (104000) | 28.9 (6.55) | 858 (417) |
| 325 | 32.7 (9.20) | 0.186 (0.250) | 5.65 (5.54) | 7310 (3160) | 243000 (125000) | 244000 (125000) | 22.5 (9.71) | 749 (385) |
Multiple dose PK between 12.5 mg and 200 mg were measured across 3 older healthy adult cohorts (12.5 mg, 25 mg, and 200 mg) and two cohorts of participants with PD (50 mg and 100 mg). Exposure on Day 1 (Table 4) in the MAD studies was comparable to the exposures observed following single doses in the SAD studies (Table 4).
Consistent with the observed terminal t1/2, at doses less than or equal to 50 mg, the exposures at steady-state accumulated approximately two-fold and the trough concentration was constant over 7 days (Table 5, Fig. 2). However, at 100 mg or 200 mg, there is little or no accumulation and the exposures on Day 7 are comparable to Day 1 (Table 5, Fig. 2). Steady state was reached within one day at higher doses and the interpatient variability increased with dose.
Fig. 2
Plasma pharmacokinetic Cmax, AUC and Cmin as a function of daily oral dose for 7-days for risvodetinib (IkT-148009). Risvodetinib (IkT-148009) was administered once daily for 7 days and the plasma pharmacokinetics measured on Day 1 and Day 7 in cohorts of 8 subjects each (3 : 1 randomized to placebo). A) Mean±standard deviation of Cmax (ng/mL) on Day 1 and Day 7 as a function of dose; B) Mean±standard deviation of AUC0 - inf for Day 1 and AUC0 - τ for Day 7 as a function of dose and C) Trough concentration measured as a function of time. Measurements at 50 and 100 mg were done in participants with PD, while all other doses were measured in healthy volunteers.
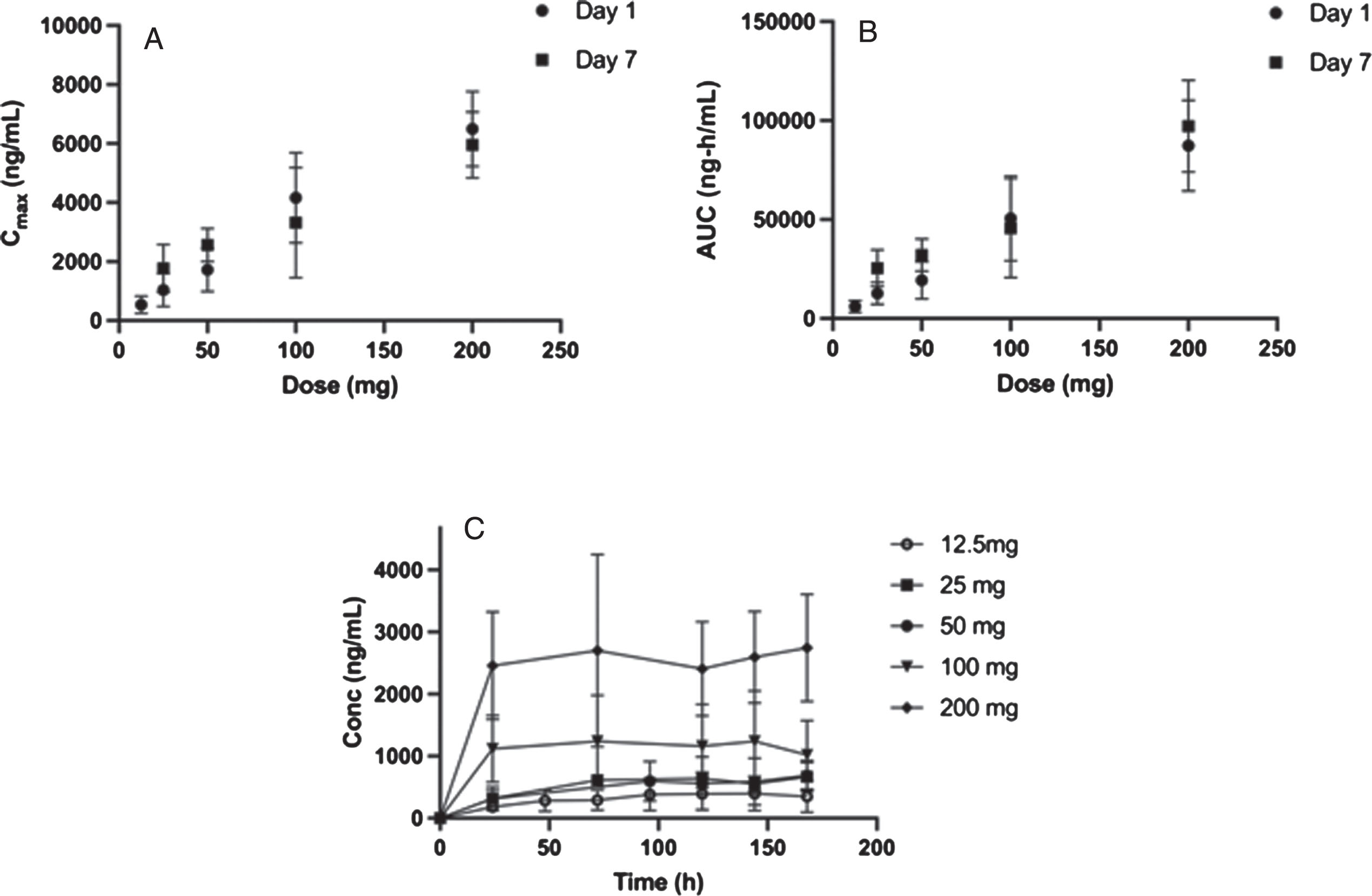
Table 5
Plasma pharmcokinetic mean and (standard deviation) values for 7-day dosing of risvodetinib (IkT-148009) to healthy volunteers and PD participants1
| DAY 1 | ||||||||
| Dose | t1/2 | tmax | Cmax | AUC0 - t | AUC0 - τ | AUC0 - ∞ | Cmax/D | AUC0 - ∞/D |
| (mg) | (h) | (h) | (ng/mL) | (h*ng/mL) | (h*ng/mL) | (h*ng/mL) | (ng/mL/mg) | (h*ng/mL/mg) |
| HV | ||||||||
| 12.5 | 13.1 (1.83) | 6.67 (3.01) | 538 (288) | 6700 (3150) | 6080 (3060) | 8830 (5300) | 43.0 (23.1) | 706 (424) |
| 25.0 | 15.3 (5.13) | 5.00 (1.10) | 1040 (557) | 12600 (5500) | 12700 (5510) | 19500 (7630) | 41.5 (22.3) | 780 (305) |
| 200 | 17.7 (4.15) | 4 (0) | 6493 (1271) | 87250 (22958) | 87250 (22958) | 151800 (52679) | 32.5 (6.4) | 759 (263) |
| PD2 | ||||||||
| 50.0 | 10.0 (2.59) | 4.66 (1.05) | 1720 (737) | 19300 (9370) | 19300 (9410) | 24500 (11800) | 34.3 (14.7) | 490 (237) |
| 100 | 11.3 (1.35) | 4.79 (1.82) | 4160 (1530) | 50200 (21300) | 50600 (21400) | 68700 (31000) | 41.6 (15.3) | 687 (310) |
| DAY 7 | ||||||||
| HV | ||||||||
| 25.0 | 27.9 (4.25) | 4.68 (1.63) | 1770 (807) | 46000 (15800) | 25400 (9250) | 70.9 (32.3) | 1020 (370) | |
| 200.0 | 23.3 (3.54) | 5 (1.67) | 5953 (1122) | 97190 (23147) | 29.8 (5.6) | 486 (116) | ||
| PD2 | ||||||||
| 50.0 | 24.8 (3.68) | 3.63 (1.43) | 2560 (564) | 51100 (17100) | 32100 (8190) | 51.2 (11.3) | 642 (164) | |
| 100 | 24.6 (4.73) | 4.41 (1.67) | 3320 (1870) | 76600 (43100) | 45700 (25000) | 33.2 (18.7) | 457 (250) | |
1Day 7 sample time errors occurred for the 12.5 mg dose and pharmacokinetic parameters were not determined. 2The 100 mg dose group was N = 5, all other groups N = 6.
Urinary excretion of intact risvodetinib (IkT-148009) was low from single and multiple doses, (between 0.2% and 0.4% of the administered dose). Metabolite and 14C-tracer studies underway suggest Rivodetinib is eliminated fecally (Werner et al., unpublished).
Eleven patients agreed to have LPs. Table 6 demonstrates that risvodetinib was detected pre-dose on the last dosing day of a 7-day dosing study, indicating that risvodetinib is getting into the CNS in older healthy adults and PD participants enrolled in this study who underwent LP.
Table 6
Mean and (standard deviation) trough concentration values for risvodetinib (IkT-148009) in cerebrospinal fluid following daily administration for 6 days1
| Dose (mg) | Concentration (ng/mL) |
| HV | |
| 25.0 | 4.51 (2.78) |
| PD Patients | |
| 50.0 | 3.07 (0.315) |
| 100 | 8.42 (ND) |
| 200 | NV |
1N = 6, 3, and 2 for the 25, 50, and 100 mg groups, respectively. NV = no voluntary LPs.
Calibration of the therapeutic dose
The proposed therapeutic dose range for risvodetinib in humans was calibrated by comparison of the steady-state plasma pharmacokinetics observed in animal models of therapeutic efficacy to the clinical pharmacokinetics in humans [13]. Doses of 50 and 100 mg/kg/day were used to establish therapeutic efficacy in models of inherited or sporadic PD [13]. Therapeutic doses in animal model studies that modified disease displayed steady-state Cmax and AUC0 - inf values that could be achieved in humans by once daily dosing between 50 and 200 mg (Supplementary Table 3), suggesting the possible therapeutic dosing range for treatment of PD.
DISCUSSION
Risvodetinib administered to 94 healthy subjects (ages 45– 68) and 14 participants with PD (ages 57– 70) gave rise to just 11 treatment-related adverse events, all were mild in severity, and none were dose or dose duration dependent. There were no deaths, no serious adverse events and no adverse events of clinical significance in the view of site investigators or Safety Monitoring Committee. The most frequent laboratory abnormality was a transient elevation in amylase and/or lipase, which is common to this drug class [17]. These observations are consistent with toxicology studies in rodent and non-human primates, respectively, where no histopathology in the pancreas was observed for daily dosing between 13 and 39 weeks duration [13]. In longer-term clinical studies, careful monitoring of these pancreatic enzyme values is warranted to detect the emergence of any risk from sporadic elevations in either enzyme, but given the similar exposures to risvodetinib in long-term toxicology studies and human clinical studies, it is not anticipated that pancreatic enzyme elevations will become adverse in humans.
Risvodetinib had a favorable side effect profile following single or multiple doses given once daily for up to 7 days. In contrast to the anti-cancer c-Abl inhibitors, risvodetinib did not give rise to hematological, musculoskeletal, gastrointestinal adverse events or QTcF prolongation over this period of time [15– 16]. Risvodetinib also did not induce orthostatic hypotension nor did it exacerbate PD clinical features. These are promising observations, but dosing to 7 days is insufficient for us to conclude that more serious adverse events will not emerge as the dosing duration is extended. As trial work continues, vigilance is warranted until a full safety data set is evaluated over 12 month and longer dosing durations.
C-Abl inhibitors passively cross the blood-brain barrier but are often substrates for efflux transporters that pump the inhibitors back into the systemic circulation. Thus, adequate brain exposure is often not achieved, which may account for the failure of one c-Abl inhibitor, nilotinib, to improve PD features in double blind clinical studies [18– 20]. Risvodetinib, on the other hand, is therapeutically effective in the CNS in model studies of PD [13] and was found in cerebrospinal fluid in humans at when plasma levels reached steady-state. LPs required voluntary agreement from trial participants and no participant was subjected to more than one LP during his or her participation, so this limited what could be learned from a single LP in a multi-dose study. A more thorough investigation of CSF pharmacokinetics is planned for the future.
The likely therapeutic dosing range was determined to be 50 to 200 mg given once daily. This was established by comparing the Cmax and AUC0 - inf observed in animal model studies where efficacy was observed to the pharmacokinetic exposures measured at different doses of risvodetinib in trial participants. With the assumption that the passage across the blood-brain barrier is approximately equal between test animal and human, the therapeutic dosing range should be between 50 and 200 mg. The safety and tolerability of risvodetinib at these doses, as well as preliminary efficacy, are being further explored in an ongoing Phase 2 clinical trial (NCT05424276).
The pharmacokinetics of risvodetinib are approximately linear up to 200 mg once daily but less than dose proportional at doses above 50 mg. The half-life of risvodetinib is approximately 24 h; however, it does not accumulate above 50 mg. Steady-state was reached by day 5 for doses ≤50 mg and reaches steady-state in one day at doses > 50 mg. The unusually high clinical exposures achieved were unexpected. A 75 mg dose of risvodetinib has the same exposure as a 500 mg dose of imatinib [15], a dose that is 25% above standard-of-care for imatinib in anti-cancer treatment [19]. Despite these high exposures, risvodetinib does not induce any clinically significant side effects when dosed for up to 7 days. This may be due to its selectivity for the non-receptor tyrosine kinases in the Abelson family. Many side effects associated with this class of inhibitor arise from their propensity to potently inhibit the receptor tyrosine kinases c-Kit and PDGFRα/β. The IC50 for risvodetinib is 30 to 100-fold lower for the receptor tyrosine kinases in the Abelson family relative to the intended targets c-Abl1 and c-Abl2/Arg in CNS applications [13]. This differential suggests that off-target effects within the Abelson tyrosine kinase family may be diminished for risvodetinib relative to other inhibitors within this drug class.
These studies were not designed to evaluate the potential of risvodetinib as a treatment for PD, but may provide some insights into the compatibility of risvodetinib with other medications administered to older adults with, or without, PD. At high steady-state exposures, risvodetinib did not induce adverse events in participants on a stable regimen of PD medications, suggesting that risvodetinib could be compatible with other common treatments in this patient population. This was true among participants who remained on their anti-PD medications as well as those who remained on other medications administered for co-morbid indications. The complete listing of concomitant medications administered to study participants (Supplementary Table 4) suggests risvodetinib was compatible with a wide array of medications a typical older adult or older adult with PD consumes.
Repeat dosing for 7-days in the MAD studies is an insufficient length to collect a fullsome picture of the safety or tolerability risks that could emerge from risvodetinib treatment. However, the typical hematological adverse events such as anemia, cytopenia, and leukopenia were not observed in any trial participant, nor was edema, cardiovascular or the frequent occurrence of GI disturbance. These are positive safety signals across 108 trial participants and contrast the experience of even single doses of the anti-cancer c-Abl inhibitors, such as imatinib or nilotinib [15, 16]. It is difficult to evaluate the meaning of observed changes in MDS-UPDRS Part III scores under treatment given the variability observed, the small group sizes (N = 6 at 50 mg; N = 5 at 100 mg). Scores within the 6-h window after first dose generally worsened over time, suggesting participants continued to turn OFF, which confounded interpretation of baseline values. We cannot exclude the possibility that risvodetinib may have contributed to this worsening effect, although given the variability in Part III scores, the short treatment duration and small sample size, this seems less likely. Despite this study limitation, this observation focuses our attention on key assessments to pay attention to as clinical development proceeds for risvodetinib. At the present level of knowledge, risvodetinib is relatively safe and well-tolerated even in the presence of a large number of concomitant medications, including anti-PD medications. The ongoing Phase 2 201 Trial (the201trial.com) with risvodetinib will evaluate the drug’s safety and tolerability and its potential to alter the course of PD in a multi-dose trial across 30 + centers in the United States (NCT05424276).
ACKNOWLEDGMENTS
The authors are grateful to the clinical staff and study participants in this study from the Hassman Research Institute, Quest Research and Velocity Clinical Research where these studies were performed.
FUNDING
This work was supported by Inhibikase Therapeutics, Inc.
CONFLICT OF INTEREST
M.H.W. is the Founder, Chief Executive and Largest Shareholder of Inhibikase Therapeutics, Inc. C.M., C.K. and J.P. are employees and shareholders of Inhibikase Therapeutics, Inc. C.W.O is Chief Executive of Clintrex Research Corporation which provides services for many pharmaceutical and biotech companies including Inhibikase, is a shareholder in Inhibikase and serves as an expert witness in the paraquat litigation.
DATA AVAILABILITY
The data supporting the findings of this study are available on request from the corresponding author. The data are not publicly available due to privacy or ethical restrictions as public release of individual patient data was not included in the informed consent. The authors will consider requests for additional data sharing on a per request basis.
SUPPLEMENTARY MATERIAL
[1] The supplementary material is available in the electronic version of this article: https://dx.doi.org/10.3233/JPD-230319.
REFERENCES
[1] | Bach JP , Ziegler U , Deuschl G , Dodel R , Doblhammer-Reiter G ((2011) ) Projected numbers of people with movement disorders in the years 2030 and 2050. Mov Disord 26: , 2286–2290. |
[2] | Pirooznia SK , Rosenthal LS , Dawson VL , Dawson TM ((2021) ) Parkinson disease: translating insights from molecular mechanisms to neuroprotection. Pharmacol Rev 73: , 33–97. |
[3] | Savitt JM , Dawson VL , Dawson TM ((2006) ) Diagnosis and treatment of Parkinson disease: molecules to medicine. J Clin Invest 116: , 1744–1754. |
[4] | Goedert M ((2001) ) Alpha-synuclein and neurodegenerative diseases. Nat Rev Neurosci 2: , 492–501. |
[5] | Goedert M , Spillantini MG , Del Tredici K , Braak H ((2013) ) 100 years of Lewy pathology. Nat Rev Neurol 9: , 13–24. |
[6] | Lee VM , Trojanowski JQ ((2006) ) Mechanisms of Parkinson’s disease linked to pathological alpha-synuclein: new targets for drug discovery. Neuron 52: , 33–38. |
[7] | Imam SZ , Zhou Q , Yamamoto A , Valente AJ , Ali SF , Bains M , Roters JL , Kahle PJ , Clark RA , Li S ((2011) ) Novel regulation of parkin function through c-Abl-mediated tyrosine phosphorylation: implications for Parkinson’s disease. J Neurosci 31: , 157–163. |
[8] | Brahmachari S , Ge P , Lee SH , Kim D , Karuppagounder SS , Kumar M , Mao X , Shin JH , Lee Y , Pletnikova O , Troncoso JC , Dawson VL , Dawson TM , Ko HS ((2016) ) Activation of tyrosine kinase c-Abl contributes to alpha-synuclein-induced neurodegeneration. J Clin Invest 126: , 2970–2988. |
[9] | Imam SZ , Trickler W , Kimura S , Binienda ZK , Paule MG , Slikker W Jr , Li S , Clark RA , Ali SF ((2013) ) Neuroprotective efficacy of a new brain-penetrating c-Abl inhibitor in a murine Parkinson’s disease model. PLoS One 31: , e65129. |
[10] | Ko HS , Lee Y , Shin JH , Karuppagounder SS , Gadad BS , Koleske AJ , Pletnikova O , Troncoso JC , Dawson VL , Dawson TM ((2010) ) Phosphorylation by the c-Abl protein tyrosine kinase inhibits parkin’s ubiquitination and protective function. Proc Natl Acad Sci U S A 107: , 16691–16696. |
[11] | Brahmachari S , Lee S , Kim S , Yuan C , Karuppagounder SS , Ge P , Shi R , Kim EJ , Liu A , Kim D , Quintin S , Jiang H , Kumar M , Yun SP , Kam TI , Mao X , Lee Y , Swing DA , Tessarollo L , Ko HS , Dawson VL , Dawson TM ((2019) ) Parkin interacting substrate zinc finger protein 746 is a pathological mediator in Parkinson’s disease. Brain 142: , 2380–2401. |
[12] | Werner MH and Olanow CW ((2022) ) Parkinson’s disease modification through Abl kinase inhibition: an opportunity. Mov Disord 37: , 6–15. |
[13] | Karuppagounder S , Wang H , Kelly T , Rush R , Nguyen R , Bisen S , Yamashita Y , Sloan N , Dang B , Sigmon A , Lee HW , Marino Lee S , Watkins L , Kim E , Brahmachari S , Kumar M , Werner MH , Dawson TM , Dawson VL ((2023) ) The c-Abl inhibitor IkT-148009 suppresses neurodegeneration in mouse models of heritable and sporadic Parkinson’s disease. Sci Transl Med 15: , eabp9352. |
[14] | Marmion DJ , Rutowski AA , Chatterjee D , Hiller BM , Werner MH , Bezard E , Kirik D , McCown T , Gray SJ , Kordower JH ((2021) ) Viral-based rodent and nonhuman primate models of multiple system atrophy: Fidelity to the human disease. Neurobiol Dis 148: , 105184. |
[15] | |
[16] | |
[17] | Pezzilli R , Corinaldesi R , Morselli-Labate AM ((2010) ) Tyrosine kinase inhibitors and acute pancreatitis. J Pancreas 11a: , 291–293. |
[18] | Pagan FL , Hebron ML , Wilmarth B , Torres-Yaghi Y , Lawler A , Mundel EE , Yusuf N , Starr NJ , Anjum M , Arellano J , Howard HH , Shi W , Mulki S , Kurd-Misto T , Matar S , Liu X , Ahn J , Moussa C ((2020) ) Nilotinib effects on safety, tolerability, and potential biomarkers in Parkinson disease: a phase 2 randomized clinical trial. JAMA Neurol 77: , 309–317. |
[19] | Pagan F , Wilmarth B , Torres-Yaghi Y , Hebron ML , Mulki S , Ferrante D , Matar S , Ahn J , Moussa C ((2021) ) Long-term safety and clinical effects of Nilotinib in Parkinson’s disease. Mov Disord 36: , 740–749. |
[20] | Simuni T , Fiske B , Merchant K , Coffey CS , Klingner E , Caspell-Garcia C , Lafontant DE , Matthews H , Wyse RK , Brundin P , Simon DK , Schwarzschild M , Weiner D , Adams J , Venuto C , Dawson TM , Baker L , Kostrzebski M , Ward T , Rafaloff G ((2021) ) Efficacy of nilotinib in patients with moderately advanced Parkinson disease: a randomized clinical trial. JAMA Neurol 78: , 312–320. |


