
Prevalence of Concomitant Pathologies in Parkinson’s Disease: Implications for Prognosis, Diagnosis, and Insights into Common Pathogenic Mechanisms
Abstract
Pathologies characteristic of Alzheimer’s disease (i.e., hyperphosphorylated tau and amyloid-β (Aβ) plaques), cardiovascular disease, and limbic predominant TDP-43 encephalopathy (LATE) often co-exist in patients with Parkinson’s disease (PD), in addition to Lewy body pathology (α-synuclein). Numerous studies point to a putative synergistic relationship between hyperphosphorylation tau, Aβ, cardiovascular lesions, and TDP-43 with α-synuclein, which may alter the stereotypical pattern of pathological progression and accelerate cognitive decline. Here we discuss the prevalence and relationships between common concomitant pathologies observed in PD. In addition, we highlight shared genetic risk factors and developing biomarkers that may provide better diagnostic accuracy for patients with PD that have co-existing pathologies. The tremendous heterogeneity observed across the PD spectrum is most likely caused by the complex interplay between pathogenic, genetic, and environmental factors, and increasing our understanding of how these relate to idiopathic PD will drive research into finding accurate diagnostic tools and disease modifying therapies.
Parkinson’s disease (PD) is neuropathologically characterized by deposits of α-synuclein (α-syn) as Lewy bodies (LB) in the neuronal somata and as Lewy neurites (LN) in the axons and dendrites. There is evidence that in PD pathology originates in the enteric nervous system and propagates to the brainstem and follows a stereotypical pattern of progression throughout the brain. Limbic structures are affected next, and finally in end stages of the disease, there is widespread distribution of α-syn deposits throughout the cerebral cortex, where LB pathology would represent the neuropathological correlate of clinical diagnoses of Parkinson’s disease dementia (PDD) or dementia with Lewy bodies (DLB) [1]. However, the site of origin of α-syn deposition is of continuous debate as alternative hypotheses propose α-syn originates in the central nervous system rather than the enteric nervous system or independently in the olfactory bulb and progresses to interconnected brain regions [2, 3]. More recently neuropathological evidence has been published representing caudo-rostral and amygdala-based progression, representing the body-first and brain-first hypothesis with differing clinical trajectories and genetically distinct progression patterns [4, 5]. Similar to other neurodegenerative pathologies LB pathology rarely exists in isolation and already in 2008, it could be shown that 53.3% of neurodegenerative disease cases have lesions associated with more than one neurodegenerative condition [6]. This is particularly evident in PD as pathology associated with Alzheimer’s disease (AD), argyrophilic grain disease, vascular pathology, and to a lesser extent Creutzfeldt Jacob disease, and progressive supranuclear palsy, were observed at autopsy as additional pathologies [6]. Moreover, a recent large autopsy study utilizing 1,647 postmortem brains, found up to seven concurrent pathologies, resulting in 161 pathological combinations [7]. Another study focusing on concomitant pathology in Parkinsonian disorders demonstrated 38% of neuropathologically confirmed PD cases had additional pathologies associated with AD, progressive supranuclear palsy, argyrophilic grain disease, cerebral white matter rarefaction, and cerebral amyloid angiopathy (CAA) [8].
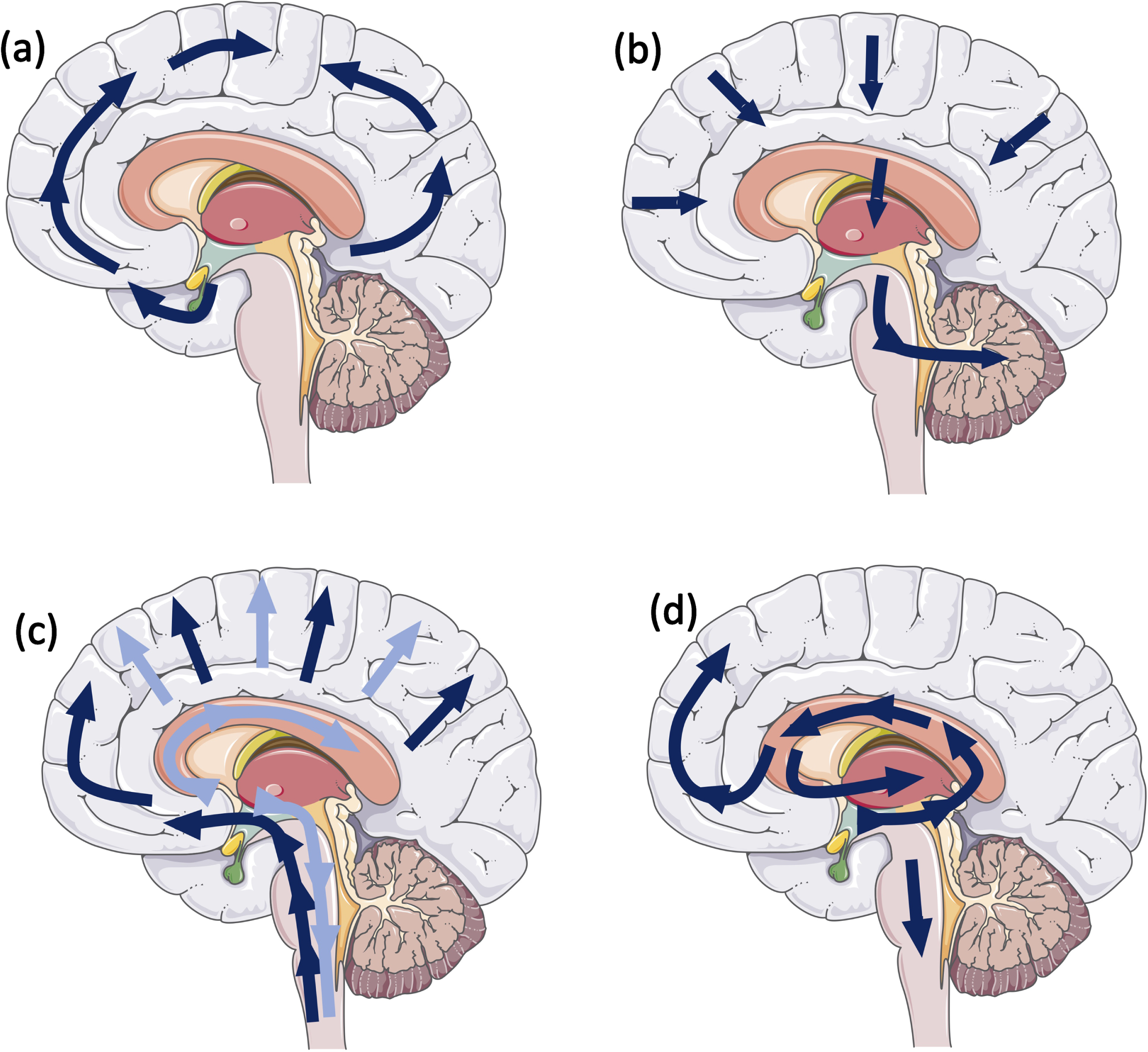
Different hallmark lesions of age associated neurodegenerative diseases (i.e., LB/LN, amyloid-β (Aβ), tau, and TDP-43) are initially deposited in different specific regions in the brain and progress though the brain in a stereotypical manner (Fig. 1) [1, 9–12]. There is growing evidence that a synergism exists between pathological proteins, which contributes to an accelerated disease trajectory observed in these cases. Since the observation was made that multiple pathologies can alter the prognosis, investigations into the potential mechanisms underlying this could be crucial in designing disease modifying therapies. This review will highlight the most common concomitant pathologies observed in PD cases, putative interactions between α-syn and tau, Aβ, cerebrovascular pathology, and TDP-43, and the clinical consequences in idiopathic and familial PD.
Fig. 1
Schematic illustrating stereotypical progression for (a) tau pathology: Neurofibrillary Braak stages (9); (b) amyloid-β pathology: Thal phases (10); (c) α-syn: Braak stages (1), dark blue arrows, Lewy pathology consensus criteria, light blue arrows (12); and (d) Limbic predominant age-related TDP-43 (11).
AD-RELATED PATHOLOGY
Tau
Both tau and Aβ are present at postmortem examination in PD patients, with a study from 2019 classifying 0% PD cases with a neurofibrillary tangle Braak stage of 0, 33.4% of cases with Braak stage I, 33.3% of cases with Braak stage II, and 33.3% of cases with Braak stage III [9, 13]. Neuropathological and experimental studies have suggested a direct synergistic interaction between AD pathology and α-syn (Fig. 2). In vitro studies using confocal microscopy and FRET based techniques found tau co-localizes and interacts with α-syn aggregates, changing the pattern of α-syn aggregation, reducing the size and increasing the number of aggregates [14]. Furthermore, α-syn has been highlighted as a candidate capable of polymerizing tau and initiating fibrilization. Once tau fibrilization is initiated, tau and α-syn synergistically interact to promote the polymerization of each other into amyloid fibrils [15, 16]. In a mouse model of synucleinopathy (TgA53T), tau is required for synaptic and memory deficits, and removal of endogenous mouse tau expression ameliorated cognitive and synaptic dysfunction [17]. However, in contrast to this, several studies have shown cognitive impairment in tau knock out mouse lines [18, 19]. Therefore, as it seems an excess reduction in tau protein results in motor and cognitive impairment, treatments that may restore tau function rather than targeting tau expression directly may be a safer therapeutic strategy. The in vivo relevance of a putative relationship between tau and α-syn was grounded further in animal studies. Tau inclusions are observed in 50% of mice expressing Ala53Thr mutant human α-syn, with the development of α-syn positive inclusions that led to motor dysfunction [15]. Supporting evidence demonstrated hyperphosphorylated tau deposits accumulate in transgenic mice overexpressing human α-syn under the PDGF promoter, which increases in an age-dependent manner [20, 21]. Transgenic animal studies have also demonstrated increased accumulation of tau aggregates following simultaneous inoculation of α-syn mouse pre-formed fibrils and AD lysate-derived tauseeds [22].
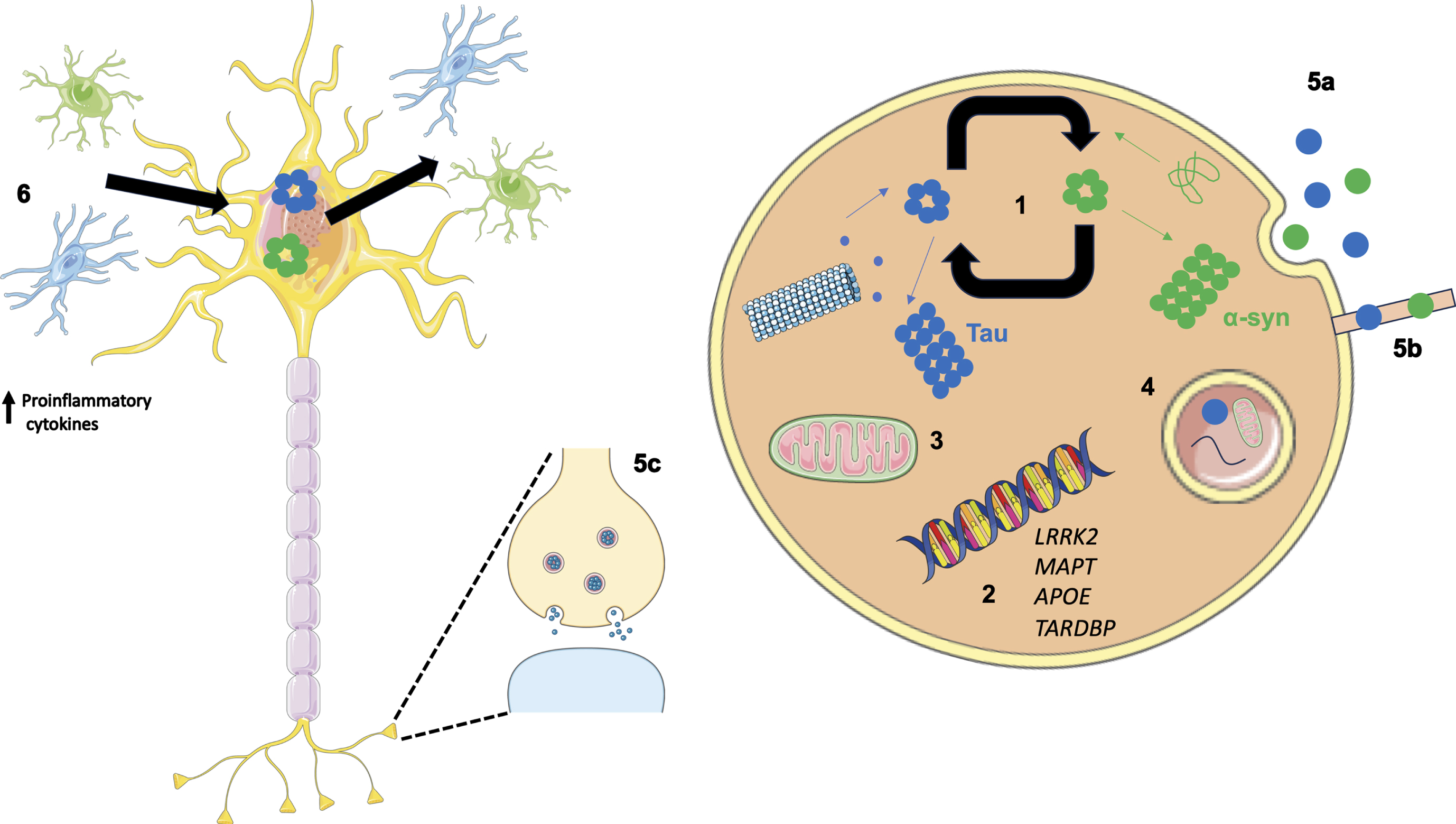
Fig. 2
Overview of potential mechanisms illustrating how co-pathologies may arise and interact. (1) Tau and α-syn monomers assemble into oligomers followed by larger aggregates. Intracellular tau and α-syn have been shown to interact and promote the fibrilization of each other. (2) Genetics can play a role in the relationship between tau and α-syn as both protein aggregates have been observed in LRRK2 carriers. In addition, genome wide association studies have revealed the MAPT locus is associated with increased risk in Alzheimer’s disease (AD) and Parkinson’s disease (PD). APOE ɛ4 is a well-known risk factor for AD; however, when APOE ɛ4 is expressed in α-syn mice motor dysfunction and general neurodegeneration is observed. TARDBP mutations are associated with motor neuron disease; however, the presence of a TDP-43 mutation (p.N267S substitution) has been found in a patient clinically diagnosed with PD. (3) Aβ and α-syn exist in different subcellular compartments; however, in pathological states both have been detected in mitochondria, lysosomes, and autophagosomes. (4) Failing cellular mechanisms can also contribute to the increased accumulation of pathologies observed in neurodegenerative diseases such as PD, as disrupted autophagy has been shown to impact α-syn, Aβ, and tau metabolism and clearance from the brain. (5) α-syn and tau are thought to propagate in a manner reminiscent of prions increasing the likelihood of aggregating in the same cell. Methods associated with this include exocytosis (5a), formation of tunnelling nanotubes (5b) and via synapses (5c). (6) Oligomeric species of tau and α-syn are capable of eliciting and inflammatory response from astrocytes and microglia resulting in an increase in proinflammatory cytokines. This in turn has a feed-forward affect where the upregulated immune system can lead to further protein aggregation and neuronal damage.
Uncovering the pathomechanisms that could potentially drive the accelerated decline observed in patients with considerable levels of α-syn pathology and high AD neuropathologic change would greatly benefit the design of disease modifying therapeutics. Indeed, several groups have conducted studies to spatially localize intracellular proteins α-syn and tau in pathological protein aggregates. Arima and colleagues reported tau can be incorporated into filaments in brainstem LBs [23]. Further to this, evidence from electron microscopy experiments demonstrated that α-syn selectively labelled 9- to 13-nm-thick straight filaments, which are analogous to LB filaments and AT8 antibody for hyperphosphorylated tau recognized twisted tubules with 80–100 nm-interval constrictions which correspond to the twisted tubules seen in AD [24]. Further studies in the brainstem structures indicate that, in particular, phosphorylated tau antibodies, preferentially labelled LBs compared to non-phosphorylated antibodies [25]. Colom-Cadena and colleagues also reported co-localizations between tau and α-syn extended to limbic regions [26]. Although α-syn is primarily located at the axonal terminal, during axonal transport it is also present at low levels in the axonal processes. On the occasion of aberrant dissociation of tau from the microtubule in disease states, this may give rise to tau-α-syn interactions and subsequent increase in aggregated forms of both proteins [20]. Increased tau phosphorylation at Ser396 has been reported in synapse-enriched fractions of PD brains in addition to phosphorylation α-syn Ser129. This suggests synapses are a site for putative protein interactions [27]. Further to this, in vitro NMR experiments have demonstrated monomeric α-syn and tau variants Tau23 and K19 synergistically promoted amyloid fibrilization, whereas S129D (a mimic of C-terminal phosphorylation of α-syn phosphorylated at Ser129) significantly enhanced the activity of α-syn in facilitating Tau23 and K19 aggregation [28]. Taken together these studies provide convincing evidence for the mutual promotion and aggregation of tau and α-syn to further advance and aggravate neurodegenerative processes. On the other hand, some studies provide evidence against an interaction between the proteins. Benussi and colleagues reported the P301L tau mutation prevented the interaction between tau and α-syn leading to an increase in unbound α-syn [29]. However this may lead to an increased burden of α-syn as a result of self-polymerization [30]. Furthermore, Esposito and colleagues demonstrated α-syn and its disease-related mutants interact differently with tau as in cell culture the interaction between α-syn and tau is decreased by the A30P mutation unlike the A53T and E46K mutations [31]. Finally Morris and colleagues demonstrated that reduction of tau does not protect mice against motor deficits and pathological alterations in the 6-OHDA mouse model or mice expressing human wildtype α-syn (hSYN) [32]. These studies implicate the tau-α-syn interaction is complicated and further study is required to elucidate the crosstalk between the proteins.
We have shown that the stereotypical patterns of pathology can be altered in Lewy body disease (LBD) cases with concomitant AD pathology [33], potentially sparing the hippocampus in a small number of cases [34]. The mechanisms by which tau and α-syn propagate to different brain regions are under debate, with increasing evidence suggesting they progress from neuron-to-neuron in a manner reminiscent of prions [35–37]. Evidence from case studies involving the transplant of fetal graft tissue into PD patients demonstrated tau and α-syn proteins at much denser concentrations compared to surrounding host tissue [38]. The trauma of the transplantation itself could leave the graft vulnerable to pathogenic insults or create a suitable environment to stimulate intracellular progression after initial seeding. Mechanisms associated with prion like spread include exocytosis, formation of tunnelling nanotubes, and via synapses (Fig. 2).
Aβ
Aβ plaques are not as common in PD; one study found 55.6% showing any Aβ plaques (22.2% Thal phase 2, 22.2%, Thal phase 3, and 11.2% Thal phase 4) and none showed neuritic plaques [10, 13]. Regional prevalence of Aβ pathologies differ in PD cases compared to PDD. Temporal cortex is significantly more affected in PDD and frontal cortex, caudate nucleus, and putamen trending towards significance [39]. Aβ burden as such was significantly increased in parietal, and occipital cortices, striatum and nucleus accumbens in PDD compared to PD [39]. Associations between Aβ and α-syn have been observed in human autopsy tissue. A neuropathological study by Lashley and colleagues found a significant correlation between Aβ plaque load and LB density in postmortem tissue from 40 PD cases and 20 controls [40]. This was further supported by Swirski and colleagues as an association was established between soluble and insoluble Aβ with insoluble levels of α-syn phosphorylated at Serine 129 [41]. Additionally, transgenic mice expressing Aβ and α-syn develop severe cognitive and motor deficits compared to single transgenic animals [42]. At a cellular level exposure of SH-SY5Y cells transfected with wild-type human SNCA cDNA to aggregated Aβ42 significantly increased the phosphorylation of α-syn at the Serine 129 position [41]. While α-syn fibrils are capable of catalyzing the heterogeneous nucleation of Aβ42 aggregates [43]. As the majority of Aβ is visualized as extracellular plaques mechanisms of the putative interaction between Aβ and α-syn is an interesting point for debate. A study conducted by Friedrich and colleagues in 2010 demonstrated internalized Aβ contributes to the formation of extracellular plaques [44]. Aβ becomes sorted to multivesicular bodies and detrimental to cell survival. Ultimately cells die and intracellular structures including intracellular amyloid species are released into the extracellular space [44]. Interestingly mice expressing neuronal Aβ and α-syn develop severe cognitive and motor deficits and showed prominent age-dependent degeneration of cholinergic neurons and presynaptic terminals compared to single transgenic animals [42]. In healthy neurons, Aβ and α-syn do not exist in the same subcellular compartment therefore limiting the potential for direct interaction. However, in pathological states both have been detected in mitochondria, lysosomes and autophagosomes (Fig. 2) [45, 46]. These data provide supporting evidence of a putative synergistic relationship between the proteins.
CEREBRAL AMYLOID ANGIOPATHY
In addition to extracellular plaques, Aβ can be deposited in the cerebral vessels including arteries, arterioles, meninges, capillaries and very rarely veins, a condition which is termed CAA. Although most commonly in AD (20–100%) [47–50], CAA has been observed in LBDs including PD [51, 52]. CAA exists in 2 forms: type 1, involvement of capillaries, with or without arterial involvement, and type 2, where arteries are affected without capillary involvement [53]. Similar to the other AD-related pathologies CAA in PD cases is less common compared to other LBDs [51], and severity scores are lower in PD cases. Postmortem studies have indicated that type 1 CAA, in particular, is associated with the presence of dementia in LBDs which could in part account for PDD and DLB having an increased presence of type 1 CAA compared to PD without dementia [54]. The prevalence of CAA is highest in the occipital lobe [55], and a neuroimaging study has also indicated this region is susceptible to cerebral microbleeds in PD cases that have developed dementia [56], suggesting that CAA could contribute to the vascular impact of cognitive impairment in PD.
TDP-43
TDP-43 inclusions are the pathological hallmark of amyotrophic lateral sclerosis (ALS) and a subtype of fronto-temporal lobar degeneration (FTLD-TDP) that is tau negative with neurons and sometimes glia positive for ubiquitin (FTLD-U). However, TDP-43 positive inclusions have been identified in aging and age-associated neurodegenerative diseases, and this has been referred to as limbic-predominant TDP-43 encephalopathy (LATE) [11]. Although these inclusions are phosphorylated and considered pathological, they do not follow the stereotypical pattern of deposition of TDP-43 observed in ALS and FTLD-TDP [57]. In LATE, TDP-43 first appears in the amygdala then progresses to the limbic areas, followed by the neocortex (Fig. 1) [58, 59]. The prevalence of LATE in PD is up to 7%, while in PDD 19% of cases are affected [60]. Within LBD cases there was a relationship between LATE and more severe symptomatology suggesting TDP-43 may have co-morbid effects in the neurogenerative pathways of LBDs. Although, the precise mechanisms behind this are not yet known as TDP-43 and α-syn are not co-localized in LBs; however, they were occasionally colocalized within dystrophic neurites [60]. Nonetheless, in vivo and in vitro experiments suggest the existence of a synergistic relationship between the pathological protein aggregates. When concomitantly over expressed in mice, TDP-43 and mutant α-syn synergistically induced dopaminergic neurodegeneration [61]. Wild type mice injected with α-syn fibrils resulted in dot-like inclusions of TDP-43 [62]. In addition, SH-SY5Y cells double infected with α-syn and TDP-43 lacking nuclear localization and incubated with synthetic α-syn fibrils resulted in inclusions positive for phosphorylated TDP-43 [63]. Interestingly, TDP-43 and α-syn have been found to be co-localized in cases with a G51D SNCA mutation [64] and in glial cytoplasmic inclusions in multiple system atrophy, another synucleinopathy [65].
INDIRECT INTERACTIONS
Inflammation
Neurodegenerative diseases are multi-factorial disorders with numerous pathological mechanisms contributing to the clinical phenotype. Neuroinflammation has been associated with several neurodegenerative diseases such as PD, AD, and FTLD with growing evidence suggesting the innate and adaptive immune system can contribute to cognitive decline in dementia [66–68].
Oligomeric species of proteins associated with neurodegeneration are capable of activating an inflammatory response as oligomeric α-syn has been shown to be an endogenous agonist for Toll-like receptor 2, which activates an inflammatory response in microglia [69]. Oligomeric tau has been shown to co-localize with astrocytes, microglia, and HMGB1, a pro-inflammatory cytokine [70]. Whereas soluble Aβ oligomers caused an increase in pro-inflammatory cytokines via a sensitized response of Toll-like receptor 4 over a period of time [71]. Ultimately activation of the immune response may initiate a feed-forward cycle. Upregulated inflammatory responses may lead to neuronal damage and protein accumulation, and thus more neuroinflammation (Fig. 2).
Triggering receptor expressed on myeloid cells-2 (TREM2) is one of the most highly expressed receptors in microglia. It suppresses cellular activation and inhibits cytokine production in response to both Toll-like receptor 2 (TLR2) and Toll-like receptor 4 (TLR4) stimulations [72]. In addition, it has been found to inhibit microglia mediated pro-inflammatory responses induced by apoptotic cells [73]. Defects in the TREM2 gene have been implicated in several neurodegenerative diseases, for example the TREM2 p.R47H substitution is a risk factor for both PD and AD [74, 75].
Impaired protein degradation
Failing cellular mechanisms can also contribute to the increased accumulation of pathologies observed in neurodegenerative diseases such as PD. Disrupted autophagy has been shown to impact α-syn, Aβ, and tau metabolism and clearance from the brain (Fig. 2). For example, knock out mice with depletion of Atg7 (an autophagy gene encoding E1-like enzyme in the two ubiquitin-like conjugation systems that are essential for the autophagosome biogenesis) leads to presynaptic accumulation of α-syn [76] and inclusions positive for hyperphosphorylated tau [77]. While knockdown of Atg7 results in an accumulation of Aβ in the Golgi [78]. Conversely abnormal protein accumulation has also been shown to cause disruptions in autophagy, for example α-syn compromises autophagy via Rab1a inhibition [79]. In addition, as microtubule stability and cytoskeletal elements are required for the formation and maturation of autophagic vacuoles [80], dissociation of tau from the microtubules and subsequent disruption of microtubule-dependent transport may result in further impairments autophagy and promotion of tau pathology. It stands to reason that if autophagy is impaired by the presence of one neurodegenerative pathology, this will affect the ability to carry out the fundamental task of degrading other aggregate prone misfolded proteins and will result in accumulation of numerous protein aggregates in the brain parenchyma.
CHALLENGES WHEN INVESTIGATING PROTEIN INTERACTIONS
While human post-mortem tissue provides the perfect cellular environment to study mixed pathologies in neurodegenerative diseases, attempting to understand disease mechanisms particularly early events in end-stage patients is difficult. Therefore, a lot of work in this field has employed animal and cellular models to investigate specific mechanisms and pathways. However, there are caveats with these experiments; firstly age-associated neurodegenerative disease are largely human-specific diseases and naturally animals do not develop the full neuropathological and clinical phenotype. Therefore, transgenic animals have been generated; however, while recapitulating specific neuropathological features of the human disease, transgenic animals cannot recapitulate the full spectrum of pathology seen in human idiopathic disease. Another means of inducing pathology in mice is via intracerebral injection of recombinant pre-formed fibrils and while some studies were able to show successful seeding and aggregation of tau pathology [81–83], others have failed to replicate this [84]. Interestingly in the same study the authors were able to demonstrate the formation of abundant tau inclusions in anatomically connected brain regions in non-transgenic mice when inoculating with AD derived tau fibrils, demonstrating different conformational features between synthetic and AD-derived brain seeds. Additionally recombinant tau fibrils have been shown to differ structurally from brain derived fibrils [85] and are therefore thought to have less translational impact compared to patient derived fibrils. Furthermore, as co-factors are necessary to form and stabilize tau fibrils [86, 87] and as cell-free assays are by design devoid of co-factors the results from these experiments should be interpreted with caution.
CEREBROVASCULAR DISEASE
In addition to proteinopathies, cerebrovascular pathologies (CVP) including lacunes, CAA, white matter lesions, old and recent ischemic infarcts and hemorrhages can present in patients with PD [88, 89]. In a large retrospective pathological study comprising 617 PD and 535 control cases, increased prevalence of CVP was observed in PD cases (44%) compared to age-matched controls (32.8%). However, in PD this included mainly mild to moderately severe lesions (26.8% and 10.6% vs. 20.9% and 6.5%, respectively), and only 6.8% had severe pathology compared to 5.4% in controls [90]. The relationship between motor impairment, vascular burden, and cognition in PD has been explored, and whole brain white matter lesion volume was associated with dementia in PD, freezing of gait and attention deficits [91]. Numerous neuroimaging studies have investigated the link between cognitive decline and CVP in PD, reporting mixed results. Some studies indicate CVP has a negative effect on cognition [92–96], while others have not been able to replicate this [97, 98]. Therefore, the clinical contribution of CVP to cognitive impairment in PD remains controversial. Nevertheless, the mechanisms behind a possible association have been investigated further: in a mouse model of α-synucleinopathy, Elabi and colleagues describe compromised blood-brain barrier integrity at early stages of disease, which is associated with dynamic alterations in vessel density and pathological activation of pericytes reflecting microvascular changes [99]. At 8 months, the density of microvessels is increased in transgenic animals compared to controls, then significantly decreased at 13 months. This could signify initial compensatory angiogenesis. Furthermore, in the human brain, using ultrasonographic examinations of extracranial vessels measuring intima-media thickness, the presence of atherosclerotic plaques, and intracranial vessels was observed. This suggests that comorbid atherosclerosis and subclinical impairment to vessels may contribute to mortality in PD [100].
In addition to studying the contribution of vascular lesions to the pathogenesis and clinical outcome of PD, the influence of vascular risk factors has been investigated. Hypertension, dyslipidemia, diabetes, and obesity are well-established risk factors for cognitive impairment and dementia in older adults. In a clinical study comprising 367 older adults with early PD, hypertension exerted a detrimental effect on memory and verbal fluency suggesting management of blood pressure and cardiovascular health may have implications in reducing cognitive decline [101]. In addition to baseline blood pressure, orthostatic hypertension has also been associated with increased dementia risk in PD [102]. While in the Parkinson Progression Markers Initiative study conducted by Chahine and colleagues a combined vascular risk score, comprising self-reported hypertension, diabetes, body mass index, and measured blood pressure was associated with an annual rate of change in global cognition [103]. Furthermore, in addition to influencing cognitive decline, the presence of at least two vascular risk factors was associated with gait impairment [104]. Diabetes mellitus (DM) has also been linked to cognitive changes in PD, and may induce a more aggressive phenotype as patients with DM demonstrated a faster motor progression and cognitive decline [105]. This is not surprising given evidence that suggests both PD and DM share common molecular pathways [106, 107].
CLINICAL AND BEHAVIORAL OBSERVATIONS
Limitations of studies investigating isolated pathologies are that they may not reflect the complex cellular environment and pathology loads evident in neurodegenerative diseases. To address this, Clinton and colleagues generated a mouse model that expressed Aβ, tau, and α-syn by crossing 3xTg-AD with mice the A53T mutation in α-syn. The subsequent DLB-AD mice developed all three pathological lesions at a significantly increased load compared to the single transgenic animals and demonstrated an accelerated cognitive decline [108]. In a large autopsy study comprising 670 cases spanning multiple neurodegenerative diseases, 63% of cases exhibited at least one additional pathology, which increased the odds of transitioning from mild cognitive impairment to mild dementia by 20-fold [109]. This is particularly evident in PD, as summated pathology scores of tau, Aβ, and α-syn were the best predictor of cognitive decline rather than one single pathological lesion [110, 111]. There is strong evidence that the burden of Aβ is a contributor to conversion to dementia in patients with PD, with total Aβ plaque burden being an independent predictor or shorter latency to dementia from onset of motor symptoms [112–116]. However, in a large multi-center cohort of patients diagnosed with synucleinopathies, both the progression of tau pathology and quantitative tau burden were the strongest predictors of a decreased interval between the onset of motor symptoms, the onset of dementia, and overall survival time [117]. In addition, shorter duration of parkinsonism prior to dementia is associated with higher CERAD scores [115]. Together these data suggest AD related pathologies are tightly linked to the accelerated clinical disease progression experienced by patients with PD. However, there have been mixed results when examining the relationship between concomitant LB pathology and AD clinical phenotype. Stern and colleagues did not find a relationship between the presence or absence of LB pathology and any clinical features in a cohort of fifty-one clinically and neuropathologically diagnosed AD cases [118]. In addition, Holtzer and colleagues found no relationship between quantitative indexes of LB abnormalities and clinical outcome [119]. To fully understand how the relationship between multiple pathologies contribute to the clinical picture in patients with dementia, it is important to understand which brain regions are affected by multiple pathologies; for example in PD, the hippocampus can be affected by numerous co-existing protein aggregates such as hyperphosphorylated tau, Aβ, α-syn, and TDP-43 (Fig. 3). Furthermore, identifying which pathologies are important in driving cognitive decline particularly in the early stages of disease or before the development of neuropathologically defined neurodegenerative diseases will inform drug discovery into disease modifying therapeutics. In a group of cognitively normal controls 63% of cases reported mixed pathologies, with tau and vascular lesions associated with poorer memory scores, tau with poorer general cognition and amyloid neuritic plaques with higher depression scores [120].
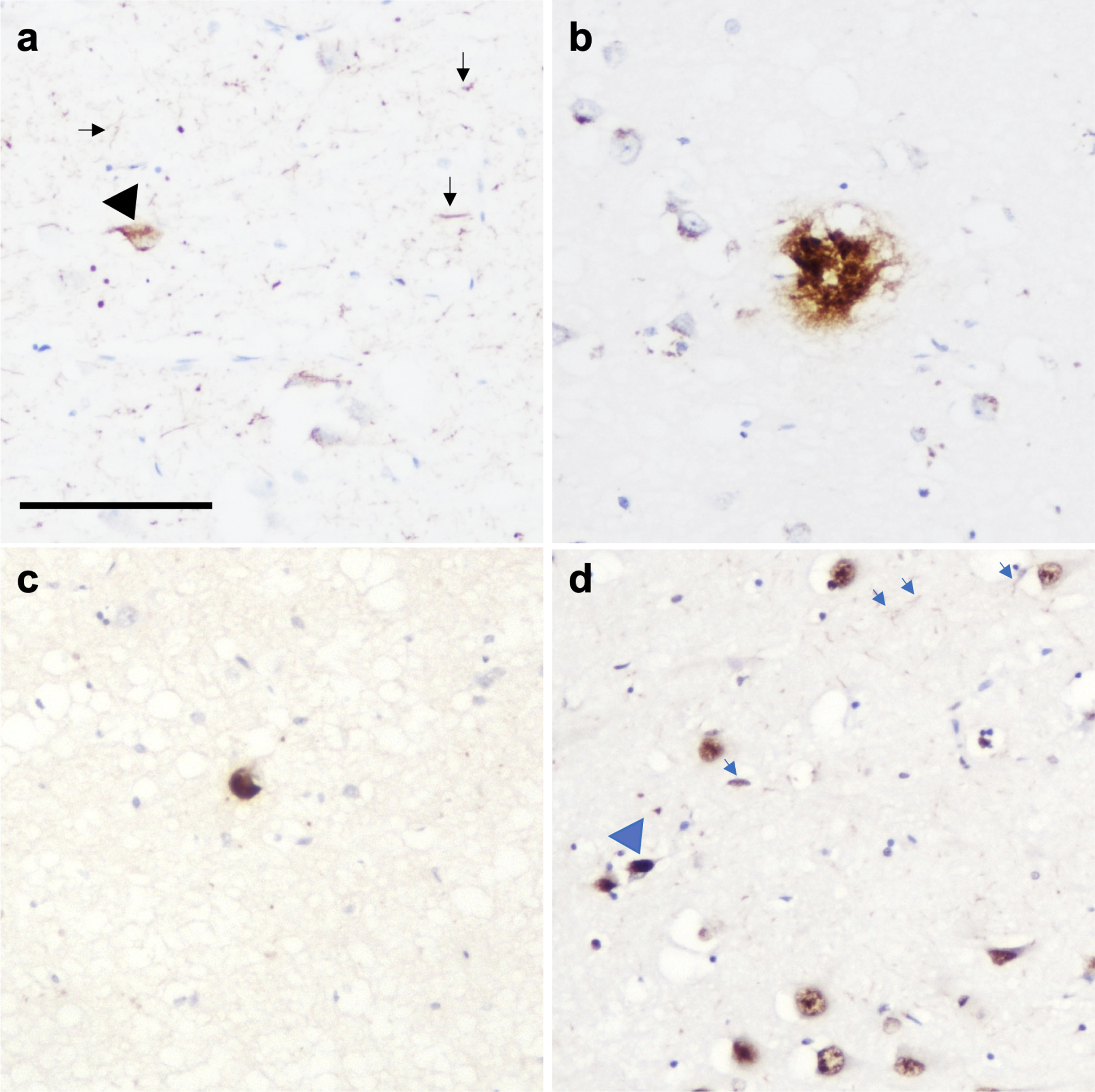
Fig. 3
Photomicrograph demonstrating the presence of multiple pathologies in CA1 region of the hippocampus in a PD case. (a) Neurofibrillary tangles (black arrowhead) and neuropil threads (black arrows), (b) amyloid-β plaque, (c) Lewy body, and (d) TDP-43 neuronal intranuclear inclusion (blue arrowhead) and dystrophic neurites (blue arrows). Scale bar represents 100μm.
EFFECTS OF PATHOLOGICAL LOAD
The amount of pathological load has to be considered since control cases lacking neurological deficits can exhibit low levels of pathology, which are mainly restricted to smaller amounts of pathological load and are generally considered to be age associated. This includes mainly tau and Aβ; however, incidental LBs have been found in a subset of cases, in particular neocortical LBs in PD cases without dementia [121]. TDP-43 pathology has also been observed in control cases [122–124]. It seems unlikely that the presence of a small amount of concomitant pathology will elicit a significant decline in cognition when restricted to specific areas; therefore, when assessing the presence of multiple pathologies in the context of neurodegenerative diseases we must pay close attention to the quantitative pathological load of each protein within each region. This has been demonstrated in studies where the increased burden of α-syn in the parahippocampal gyrus and cingulate cortex allowed to distinguish PDD from PD [125, 126]. Similar results are seen with AD-related pathology as in a longitudinal population-based study of aging and dementia, quantitative measures of both tau and Aβ in the neocortex and hippocampus were strongly associated with the presence of dementia, whereas using semi-quantitative values this association was not observed [127]. It seems there may also be a similar relationship with concomitant pathologies in PD as both imaging and fluid biomarker studies demonstrated Aβ at baseline was not able to distinguish PD patients from normal controls. However, longitudinal data suggested Aβ was associated with a more rapid cognitive decline in patients therefore acting as a future predictor of increased amyloid burden rather than a current marker of cognitive decline when pathological burden becomes high enough to be detected [128]. Tau neuroimaging studies in LBD have reported variable findings as some studies report an increase in tau PET binding in PDD compared to PD without cognitive impairment [129, 130]. Additionally other studies that have found an increased tau PET signal in DLB compared to control participants [131–133]. However, a conflicting study conducted by Mak and colleagues were unable to replicate these findings, reporting [18F]-AV1451 binding is not elevated in DLB relative to normal aging [134]. Interestingly, it has been noted that the amount of [18F]AV-1451 binding in DLB and PDD is substantially lower than that observed in AD [131, 135, 136]. This is in line with our neuropathological study which quantified tau pathology in cases that fulfilled neuropathological criteria for both AD and DLB, i.e., Braak stage VI, Thal phase 5, and neocortical LB disease. Even though all cases were Braak stage VI, those cases that present clinically with AD had significantly more tau pathology in cortical, subcortical, and brain stem regions compared to DLB, which was not observed using semi-quantitative staging criteria [34]. In the future implementation of quantitative methodologies will help disentangle the contribution of multiple concomitant pathologies to clinical and pathological phenotypes.
GENETIC CONSIDERATIONS
While the majority of PD cases are idiopathic, we have gained tremendous insights into disease pathogenesis from genetic mutations that cause PD and others that increase the risk of PD. The SNCA gene was the first recognized to cause autosomal dominant PD and subsequently there have been several missense mutations identified including A30P, E46K, H50Q, G51D, A53T, A53E, A53V, and A30G [137]. Several other genes have been convincingly associated with PD risk such as LRRK2, VPS35, PRKN, PINK1, GBA, and DJ-1. Interestingly, in mice overexpressing A30P, phosphorylated tau also develops in parallel with aggregates α-syn [138]. Furthermore in LRRK2 carriers, while α-syn pathology can been seen in 63.6% of individuals, tau pathology can be found in 100% of carriers, and is abundant in 91% of carriers in a small cohort [139].
Significant efforts have been focused on investigating the contribution of common variants to PD risk and age-at-onset. Several of these mutations, most commonly associated with other neurodegenerative diseases, have been observed in patients with a clinical diagnosis of PD such as APOE and MAPT. It is well known that APOE ɛ4 carriers have an increased risk of AD; however, two recent studies indicate that APOE4 can directly regulate levels of α-syn. Mice with different APOE genotypes were either genetically altered to express mutant α-syn or inoculated with α-syn preformed fibrils. The animals that expressed the ɛ4 allele exhibited the most severe α-syn pathology [140]. In a parallel study by Zhao and colleagues an adeno-associated virus gene delivery of α-syn was performed in human APOE-targeted replacement mice expressing APOE ɛ2, APOE ɛ3, or APOE ɛ4. In addition to expressing abundant α-syn, the mice expressing APOE ɛ4 had the most motor dysfunction and general neurodegeneration [141].
Genome wide association studies (GWAS) of 1,713 patients with PD and 3,978 controls in a cohort consisting of individuals from European ancestry, demonstrated a strong association at the MAPT locus [142], and this agreed with a study by Edwards and colleagues that combined data from two previous GWAS [143], confirming the MAPT region is a major gene whose common variants are influencing risk of PD. However, this failed to be replicated in a Japanese cohort [144] highlighting a population specific heterogeneity in PD.
Mutations in the TARDBP gene have been shown to cause motor neuron disease/ALS; however, in addition to TDP-43 aggregations in the brains of patients with PD mutations, Rayaprolu and colleagues report the presence of a TDP-43 mutation (p.N267S substitution), in a patient with clinically diagnosed PD [145]. Subsequently three additional missense mutations, p. G294A, p.G295S, and p.S393L, in patients with typical and atypical parkinsonism have been identified [146].
Taken together these data suggest that common pathomechanisms are present across the neurodegenerative spectrum, and particularly in patients with PD (Fig. 2). Harmonizing genetic and pathological substrates in terms of mixed pathology and disease pathogenesis will accelerate our upstanding of PD and drive design of disease modifyingtherapies.
BIOMARKERS
To aid the development of disease modifying therapies, it is critical that we are able to classify patients correctly during enrolment into clinical trials. Numerous biomarker studies have focused on the detection of α-syn in patients during life including in biofluids and peripheral tissue [147–152]. However, the presence of mixed pathologies in PD can be exploited in the biomarker field to aid diagnosis, prognosis, and the enrolment of patients into clinical trials.
Exciting developments in the AD biomarker field has proven tau (particularly detection in biofluids) is an accurate and reliable biomarker and has the potential to identify PD patients that are more likely to experience an accelerated disease trajectory. In 2009, Compta and colleagues found high total tau and phosphorylated tau levels in cerebrospinal fluid (CSF) in PD cases were associated with cognitive impairment and worsening motor symptoms [153, 154]. However, longitudinal evaluation of CSF total and phosphorylated tau measured 6 and 12 months from baseline demonstrated no significant changes over this time period. This suggests CSF tau and phosphorylated tau does not mirror disease progression, in particular progressive striatonigral degeneration as evaluated by clinical motor ratings (MDS-UPDRS III) and DaTscan measures [155]. More recently advances in plasma biomarkers (which provides a more accessible and less invasive option) have indicated plasma ptau181 is significantly increased in PD compared to controls. However no association was observed between ptau181 and longitudinal cognitive performance, supporting findings in CSF studies [156]. Reduced Aβ42 is associated with reduced cognitive function in PD compared to healthy controls (with cut-off levels being lower than those for AD) [157–159], with lower CSF Aβ42 associated with higher rate of Mini-Mental State Examination and Montreal Cognitive Assessment score decline [160].
It seems diagnostic panels incorporating α−syn and concomitant pathology may provide the most accurate diagnostic accuracy, perhaps highlighting the underlying pathological burden. For example, in a study comprising 45 participants, a diagnostic panel incorporating plasma α-syn and ptau181 combined with age and sex showed good performance in discriminating de novo PD cases from healthy controls [161]. Furthermore, the combination of CSF oligomeric and total α-syn, and Aβ42/tau ratios increase the diagnostic accuracy of PD [157, 160]. In addition to fluid biomarkers, neuroimaging has been trialed in PD biomarker studies. In 2016, Gomperts and colleagues investigated AV-1451 PET in PDD and PD cases, indicating higher standardized uptake value ratios in the inferior temporal gyrus than in healthy controls [131]; however, this difference was not recapitulated in a subsequent study [162]. Further work is required to elucidate the pathological signature of tau ligands in LBDs, particularly including a more quantitative approach to assessing pathological protein loads (see section ‘Effects of pathological load’).
RACIAL AND ETHNIC VARIATION IN PD
To date, racial and ethnic minorities are underrepresented in PD research. This was highlighted by GWAS studies where the MAPT locus was strongly associated with PD in a European population but not in a Japanese population [144]. Examination of diverse cohorts will allow us to investigate the enormous heterogeneity observed in neurodegenerative disease including PD. In a recent study investigating health-related quality of life in PD patients, differences were observed between racial and ethnic groups. After adjusting for co-factors, non-White patients scored lower than White counterparts, with further mediation analysis demonstrating lower cognition partly contributing to this association [163]. In a seminal study by Barnes and colleagues, Black decedents with AD dementia were more likely to have mixed pathologies compared to matched White decedents with AD [164]. A more recent neuropathological study investigating mixed LBD and AD pathologies across different ethnicities demonstrated prevalence of combined LBD and AD pathologies in 18% of non-Hispanic White individuals, 11% of Black individuals, and 35% of Hispanic individuals [165]. It is well documented that racial and ethnic groups are under-represented in brain donation programs for research into neurodegenerative diseases. Studies involving African Americans have identified religious beliefs [166], the effect of the removal process on physical appearance [167], and the lack of understanding regarding the rationale for brain donation in dementia research [167, 168] as contributing factors to lower participation rates in brain tissue donation initiatives. A study conducted by Boise and colleagues investigated this further conducting a survey of research volunteers from four racial/ethnic groups for their willingness to assent to brain donation including African American, Caucasian, Asian, and Latino research volunteers. Positive predictors included older age, Latino ethnicity, understanding how the brain is used by researchers, and an understanding of the process that participants need to follow to ensure the tissue is donated. Negative predictors included African/African American race, belief that the body should remain whole at burial, and concerns that researchers may not be respectful of the tissue following donation [169]. While the research community should be respectful of different religious beliefs and cultures regarding opinions on brain tissue donation, there are aspects that may be amendable to increase participation in such programs. Taking opportunities to familiarize under-represented minority groups with the enormous impact post-mortem tissue has on the research into neurodegenerative diseases should be prioritized. In addition to this, researchers should be transparent with research practices involving the use of donated tissues. Future studies focused on PD should include a more representative population including diverse ethnicities and also social, economic, and cultural backgrounds, and strive to make this data accessible to researchers, to fully appreciate the disease heterogeneity across the PD spectrum.
CONCLUSION
Aging and the accumulation of age-associated pathologies is the biggest risk factor for developing dementia in later life. Common protein aggregations such as hyperphosphorylation tau, Aβ, α-syn, and TDP-43 are assumed to lead to one specific neurodegenerative disease; however, it is rare for pathologies exist in isolation. Multiple pathologies can be observed in the PD brain at post-mortem examination, and while this is less frequent and severe compared to PDD and indeed even less compared DLB, a growing body of evidence suggests concomitant pathologies, even at low levels, can contribute to cognitive impairment in PD patients. It is assumed concomitant pathologies have a synergistic relationship that drives cognitive decline and research is ongoing to resolve the pathomechanisms underpinning this to inform the design of disease modifying therapies. Biomarker research (in particular fluid biomarkers) are demonstrating tremendous promise in identifying PD patients during life and will aid the stratification of patients for clinical trials. Finally, to fully appreciate the prevalence of concomitant pathologies and identify disease pathways in PD, it is crucial to include diverse populations of patients in research studies and clinical trials.
ACKNOWLEDGMENTS
This paper presents independent research funded and supported by the NIHR Newcastle Biomedical Research Centre (BRC). The views expressed are those of the authors and not necessarily those of the NIHR or the Department of Health and Social Care. Figures 1 and 2 were partly generated using Servier Medical Art, provided by Servier, licensed under a Creative Commons Attribution 3.0 unported license.
FUNDING
LW is funded by an Alzheimer’s Research UK fellowship (ARUK-RF2020A-010). Brain tissue provided for the images in this manuscript was provided by Newcastle Brain Tissue Resource, which is funded in part by a grant from the UK Medical Research Council (G0400074), by NIHR Newcastle Biomedical Research Centre awarded to the Newcastle upon Tyne NHS Foundation Trust and Newcastle University, and by a grant from the Alzheimer’s Society and Alzheimer’s Research UK as part of the Brains for Dementia Research initiative. The NIHR Newcastle Biomedical Research Centre (BRC) is a partnership between Newcastle Hospitals NHS Foundation Trust, Newcastle University, and Cumbria, Northumberland and Tyne and Wear NHS Foundation Trust and is funded by the National Institute for Health and Care Research (NIHR).
CONFLICT OF INTEREST
The authors have no conflict of interest to report.
REFERENCES
[1] | Braak H , Del Tredici K , Rub U , de Vos RA , Jansen Steur EN , Braak E ((2003) ) Staging of brain pathology related to sporadic Parkinson’s disease, Neurobiol Aging 24: , 197–211. |
[2] | Beach TG , Adler CH , Lue L , Sue LI , Bachalakuri J , Henry-Watson J , Sasse J , Boyer S , Shirohi S , Brooks R , Eschbacher J , White CL , 3rd, Akiyama H , Caviness J , Shill HA , Connor DJ , Sabbagh MN , Walker DG ((2009) ) Unified staging system for Lewy body disorders: Correlation with nigrostriatal degeneration, cognitive impairment and motor dysfunction. Acta Neuropathol 117: , 613–634. |
[3] | Rey NL , Petit GH , Bousset L , Melki R , Brundin P ((2013) ) Transfer of human α-synuclein from the olfactory bulb to interconnected brain regions in mice. Acta Neuropathol 126: , 555–573. |
[4] | Borghammer P , Horsager J , Andersen K , Van Den Berge N , Raunio A , Murayama S , Parkkinen L , Myllykangas L ((2021) ) Neuropathological evidence of body-first vs. brain-first Lewy body disease. Neurobiol Dis 161: , 105557. |
[5] | Raunio A , Kaivola K , Tuimala J , Kero M , Oinas M , Polvikoski T , Paetau A , Tienari PJ , Myllykangas L ((2019) ) Lewy-related pathology exhibits two anatomically and genetically distinct progression patterns: A population-based study of Finns aged 85. Acta Neuropathol 138: , 771–782. |
[6] | Kovacs GG , Alafuzoff I , Al-Sarraj S , Arzberger T , Bogdanovic N , Capellari S , Ferrer I , Gelpi E , Kovari V , Kretzschmar H , Nagy Z , Parchi P , Seilhean D , Soininen H , Troakes C , Budka H ((2008) ) Mixed brain pathologies in dementia: The BrainNet Europe consortium experience. Dement Geriatr Cogn Disord 26: , 343–350. |
[7] | Robinson JL , Xie SX , Baer DR , Suh E , Van Deerlin VM , Loh NJ , Irwin D , McMillan CT , Wolk D , Chen-Plotkin A , Weintraub D , Schuck T , Lee VM , Trojanowski JQ , Lee EB ((2023) ) Pathological combinations in neurodegenerative disease are heterogeneous and disease-associated. Brain 146: , 2557–2569. |
[8] | Dugger BN , Adler CH , Shill HA , Caviness J , Jacobson S , Driver-Dunckley E , Beach TG ((2014) ) Concomitant pathologies among a spectrum of parkinsonian disorders. Parkinsonism Relat Disord 20: , 525–529. |
[9] | Braak H , Braak E ((1991) ) Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol 82: , 239–259. |
[10] | Thal DR , Rub U , Orantes M , Braak H ((2002) ) Phases of A beta-deposition in the human brain and its relevance for the development of AD. Neurology 58: , 1791–1800. |
[11] | Nelson PT , Dickson DW , Trojanowski JQ , Jack CR , Boyle PA , Arfanakis K , Rademakers R , Alafuzoff I , Attems J , Brayne C ((2019) ) Limbic-predominant age-related TDP-43 encephalopathy (LATE): Consensus working group report. Brain 142: , 1503–1527. |
[12] | Attems J , Toledo JB , Walker L , Gelpi E , Gentleman S , Halliday G , Hortobagyi T , Jellinger K , Kovacs GG , Lee EB , Love S , McAleese KE , Nelson PT , Neumann M , Parkkinen L , Polvikoski T , Sikorska B , Smith C , Grinberg LT , Thal DR , Trojanowski JQ , McKeith IG ((2021) ) Neuropathological consensus criteria for the evaluation of Lewy pathology in post-mortem brains: A multi-centre study. Acta Neuropathol 141: , 159–172. |
[13] | Walker L , Stefanis L , Attems J ((2019) ) Clinical and neuropathological differences between Parkinson’s disease, Parkinson’s disease dementia and dementia with Lewy bodies - current issues and future directions. J Neurochem 150: , 467–474. |
[14] | Badiola N , de Oliveira RM , Herrera F , Guardia-Laguarta C , Goncalves SA , Pera M , Suarez-Calvet M , Clarimon J , Outeiro TF , Lleo A ((2011) ) Tau enhances alpha-synuclein aggregation and toxicity in cellular models of synucleinopathy, PLoS One 6: , e26609. |
[15] | Giasson BI , Forman MS , Higuchi M , Golbe LI , Graves CL , Kotzbauer PT , Trojanowski JQ , Lee VM ((2003) ) Initiation and synergistic fibrillization of tau and alpha-synuclein. Science 300: , 636–640. |
[16] | Waxman EA , Giasson BI ((2011) ) Induction of intracellular tau aggregation is promoted by alpha-synuclein seeds and provides novel insights into the hyperphosphorylation of tau. J Neurosci 31: , 7604–7618. |
[17] | Singh B , Covelo A , Martell-Martinez H , Nanclares C , Sherman MA , Okematti E , Meints J , Teravskis PJ , Gallardo C , Savonenko AV , Benneyworth MA , Lesne SE , Liao D , Araque A , Lee MK ((2019) ) Tau is required for progressive synaptic and memory deficits in a transgenic mouse model of alpha-synucleinopathy. Acta Neuropathol 138: , 551–574. |
[18] | Lei P , Ayton S , Moon S , Zhang Q , Volitakis I , Finkelstein DI , Bush AI ((2014) ) Motor and cognitive deficits in aged tau knockout mice in two background strains. Mol Neurodegener 9: , 29. |
[19] | Ikegami S , Harada A , Hirokawa N ((2000) ) Muscle weakness, hyperactivity, and impairment in fear conditioning in tau-deficient mice. Neurosci Lett 279: , 129–132. |
[20] | Lee VM , Giasson BI , Trojanowski JQ ((2004) ) More than just two peas in a pod: Common amyloidogenic properties of tau and alpha-synuclein in neurodegenerative diseases. Trends Neurosci 27: , 129–134. |
[21] | Haggerty T , Credle J , Rodriguez O , Wills J , Oaks AW , Masliah E , Sidhu A ((2011) ) Hyperphosphorylated Tau in an alpha-synuclein-overexpressing transgenic model of Parkinson’s disease. Eur J Neurosci 33: , 1598–1610. |
[22] | Bassil F , Meymand ES , Brown HJ , Xu H , Cox TO , Pattabhiraman S , Maghames CM , Wu Q , Zhang B , Trojanowski JQ , Lee VM ((2021) ) α-Synuclein modulates tau spreading in mouse brains, J Exp Med 218: , e20192193. |
[23] | Arima K , Hirai S , Sunohara N , Aoto K , Izumiyama Y , Uéda K , Ikeda K , Kawai M ((1999) ) Cellular co-localization of phosphorylated tau- and NACP/alpha-synuclein-epitopes in Lewy bodies in sporadic Parkinson’s disease and in dementia with Lewy bodies. Brain Res 843: , 53–61. |
[24] | Arima K , Mizutani T , Alim MA , Tonozuka-Uehara H , Izumiyama Y , Hirai S , Uéda K ((2000) ) NACP/α-synuclein and tau constitute two distinctive subsets of filaments in the same neuronal inclusions in brains from a family of parkinsonism and dementia with Lewy bodies: Double-immunolabeling fluorescence and electron microscopic studies. Acta Neuropathol 100: , 115–121. |
[25] | Ishizawa T , Mattila P , Davies P , Wang D , Dickson DW ((2003) ) Colocalization of tau and alpha-synuclein epitopes in Lewy bodies. J Neuropathol Exp Neurol 62: , 389–397. |
[26] | Colom-Cadena M , Gelpi E , Charif S , Belbin O , Blesa R , Marti MJ , Clarimon J , Lleo A ((2013) ) Confluence of alpha-synuclein, tau, and beta-amyloid pathologies in dementia with Lewy bodies. J Neuropathol Exp Neurol 72: , 1203–1212. |
[27] | Muntane G , Dalfo E , Martinez A , Ferrer I ((2008) ) Phosphorylation of tau and alpha-synuclein in synaptic-enriched fractions of the frontal cortex in Alzheimer’s disease, and in Parkinson’s disease and related alpha-synucleinopathies. Neuroscience 152: , 913–923. |
[28] | Lu J , Zhang S , Ma X , Jia C , Liu Z , Huang C , Liu C , Li D ((2020) ) Structural basis of the interplay between α-synuclein and tau in regulating pathological amyloid aggregation: The interaction between α-synuclein and tau. J Biol Chem 295: , 7470–7480. |
[29] | Benussi L , Ghidoni R , Paterlini A , Nicosia F , Alberici AC , Signorini S , Barbiero L , Binetti G ((2005) ) Interaction between tau and alpha-synuclein proteins is impaired in the presence of P301L tau mutation. Exp Cell Res 308: , 78–84. |
[30] | Giasson BI , Uryu K , Trojanowski JQ , Lee VM-Y ((1999) ) Mutant and wild type human α-synucleins assemble into elongated filaments with distinct morphologies, J Biol Chem 274: , 7619–7622. |
[31] | Esposito A , Dohm CP , Kermer P , Bähr M , Wouters FS ((2007) ) α-Synuclein and its disease-related mutants interact differentially with the microtubule protein tau and associate with the actin cytoskeleton. Neurobiol Dis 26: , 521–531. |
[32] | Morris M , Koyama A , Masliah E , Mucke L ((2011) ) Tau reduction does not prevent motor deficits in two mouse models of Parkinson’s disease, PLoS One 6: , e29257. |
[33] | Toledo JB , Gopal P , Raible K , Irwin DJ , Brettschneider J , Sedor S , Waits K , Boluda S , Grossman M , Van Deerlin VM , Lee EB , Arnold SE , Duda JE , Hurtig H , Lee VM , Adler CH , Beach TG , Trojanowski JQ ((2016) ) Pathological α-synuclein distribution in subjects with coincident Alzheimer’s and Lewy body pathology. Acta Neuropathol 131: , 393–409. |
[34] | Walker L , McAleese KE , Thomas AJ , Johnson M , Martin-Ruiz C , Parker C , Colloby SJ , Jellinger K , Attems J ((2015) ) Neuropathologically mixed Alzheimer’s and Lewy body disease: Burden of pathological protein aggregates differs between clinical phenotypes. Acta Neuropathol 129: , 729–748. |
[35] | Kordower JH , Chu Y , Hauser RA , Freeman TB , Olanow CW ((2008) ) Lewy body-like pathology in long-term embryonic nigral transplants in Parkinson’s disease. Nat Med 14: , 504–506. |
[36] | Li JY , Englund E , Holton JL , Soulet D , Hagell P , Lees AJ , Lashley T , Quinn NP , Rehncrona S , Bjorklund A , Widner H , Revesz T , Lindvall O , Brundin P ((2008) ) Lewy bodies in grafted neurons in subjects with Parkinson’s disease suggest host-to-graft disease propagation. Nat Med 14: , 501–503. |
[37] | Frost B , Jacks RL , Diamond MI ((2009) ) Propagation of tau misfolding from the outside to the inside of a cell. J Biol Chem 284: , 12845–12852. |
[38] | Ornelas AS , Adler CH , Serrano GE , Curry JR , Shill HA , Kopyov O , Beach TG ((2020) ) Co-Existence of tau and α-synuclein pathology in fetal graft tissue at autopsy: A case report. Parkinsonism Relat Disord 71: , 36–39. |
[39] | Hepp DH , Vergoossen DL , Huisman E , Lemstra AW , Berendse HW , Rozemuller AJ , Foncke EM , van de Berg WD ((2016) ) Distribution and load of amyloid-beta pathology in Parkinson disease and dementia with Lewy bodies. J Neuropathol Exp Neurol 75: , 936–945. |
[40] | Lashley T , Holton JL , Gray E , Kirkham K , O’Sullivan SS , Hilbig A , Wood NW , Lees AJ , Revesz T ((2008) ) Cortical alpha-synuclein load is associated with amyloid-beta plaque burden in a subset of Parkinson’s disease patients. Acta Neuropathol 115: , 417–425. |
[41] | Swirski M , Miners JS , de Silva R , Lashley T , Ling H , Holton J , Revesz T , Love S ((2014) ) Evaluating the relationship between amyloid-beta and alpha-synuclein phosphorylated at Ser129 in dementia with Lewy bodies and Parkinson’s disease. Alzheimers Res Ther 6: , 77. |
[42] | Masliah E , Rockenstein E , Veinbergs I , Sagara Y , Mallory M , Hashimoto M , Mucke L ((2001) ) beta-amyloid peptides enhance alpha-synuclein accumulation and neuronal deficits in a transgenic mouse model linking Alzheimer’s disease and Parkinson’s disease. Proc Natl Acad Sci U S A 98: , 12245–12250. |
[43] | Chia S , Flagmeier P , Habchi J , Lattanzi V , Linse S , Dobson CM , Knowles TP , Vendruscolo M ((2017) ) Monomeric and fibrillar α-synuclein exert opposite effects on the catalytic cycle that promotes the proliferation of Aβ42 aggregates. Proc Natl Acad Sci U S A 114: , 8005–8010. |
[44] | Friedrich RP , Tepper K , Rönicke R , Soom M , Westermann M , Reymann K , Kaether C , Fändrich M ((2010) ) Mechanism of amyloid plaque formation suggests an intracellular basis of Abeta pathogenicity. Proc Natl Acad Sci U S A 107: , 1942–1947. |
[45] | Hansson Petersen CA , Alikhani N , Behbahani H , Wiehager B , Pavlov PF , Alafuzoff I , Leinonen V , Ito A , Winblad B , Glaser E , Ankarcrona M ((2008) ) The amyloid beta-peptide is imported into mitochondria via the TOM import machinery and localized to mitochondrial cristae. Proc Natl Acad Sci U S A 105: , 13145–13150. |
[46] | Chinta SJ , Mallajosyula JK , Rane A , Andersen JK ((2010) ) Mitochondrial alpha-synuclein accumulation impairs complex I function in dopaminergic neurons and results in increased mitophagy. }, Neurosci Lett 486: , 235–239. |
[47] | Geinisman RV , Oksova EE ((1988) ) Morphologic diagnosis of vascular and senile dementia (the significance of Congophilic angiopathy). Zh Nevropatol Psikhiatr Im S S Korsakova 88: , 35–39. |
[48] | Joachim CL , Morris JH , Selkoe DJ ((1988) ) Clinically diagnosed Alzheimer’s disease: Autopsy results in 150 cases. Ann Neurol 24: , 50–56. |
[49] | Matthews FE , Brayne C , Lowe J , McKeith I , Wharton SB , Ince P ((2009) ) Epidemiological pathology of dementia: Attributable-risks at death in the Medical Research Council Cognitive Function and Ageing Study, PLoS Med 6: , e1000180. |
[50] | Parker JC , Jr. , Philpot J ((1985) ) Postmortem evaluation of Alzheimer’s disease. South Med J 78: , 1411–1413. |
[51] | Jellinger KA ((2021) ) Significance of cerebral amyloid angiopathy and other co-morbidities in Lewy body diseases. J Neural Transm 128: , 687–699. |
[52] | Jellinger KA , Attems J ((2008) ) Cerebral amyloid angiopathy in Lewy body disease. J Neural Transm (Vienna) 115: , 473–482. |
[53] | Thal DR , Ghebremedhin E , Rub U , Yamaguchi H , Del Tredici K , Braak H ((2002) ) Two types of sporadic cerebral amyloid angiopathy. J Neuropathol Exp Neurol 61: , 282–293. |
[54] | Attems J , Jellinger KA , Lintner F ((2005) ) Alzheimer’s disease pathology influences severity and topographical distribution of cerebral amyloid angiopathy. Acta Neuropathol 110: , 222–231. |
[55] | Attems J ((2005) ) Sporadic cerebral amyloid angiopathy: Pathology, clinical implications, and possible pathomechanisms. Acta Neuropathol 110: , 345–359. |
[56] | Daida K , Tanaka R , Yamashiro K , Ogawa T , Oyama G , Nishioka K , Shimo Y , Umemura A , Hattori N ((2018) ) The presence of cerebral microbleeds is associated with cognitive impairment in Parkinson’s disease. J Neurol Sci 393: , 39–44. |
[57] | Mackenzie IR , Neumann M ((2017) ) Reappraisal of TDP-43 pathology in FTLD-U subtypes. Acta Neuropathol 134: , 79–96. |
[58] | Josephs KA , Murray ME , Whitwell JL , Tosakulwong N , Weigand SD , Petrucelli L , Liesinger AM , Petersen RC , Parisi JE , Dickson DW ((2016) ) Updated TDP-43 in Alzheimer’s disease staging scheme. Acta Neuropathol 131: , 571–585. |
[59] | Abner EL , Kryscio RJ , Schmitt FA , Santacruz KS , Jicha GA , Lin Y , Neltner JM , Smith CD , Van Eldik LJ , Nelson PT ((2011) ) “End-stage” neurofibrillary tangle pathology in preclinical Alzheimer’s disease: Fact or fiction? J Alzheimers Dis 25: , 445–453. |
[60] | Nakashima-Yasuda H , Uryu K , Robinson J , Xie SX , Hurtig H , Duda JE , Arnold SE , Siderowf A , Grossman M , Leverenz JB , Woltjer R , Lopez OL , Hamilton R , Tsuang DW , Galasko D , Masliah E , Kaye J , Clark CM , Montine TJ , Lee VM , Trojanowski JQ ((2007) ) Co-morbidity of TDP-43 proteinopathy in Lewy body related diseases. Acta Neuropathol 114: , 221–229. |
[61] | Tian T , Huang C , Tong J , Yang M , Zhou H , Xia X-G ((2011) ) TDP-43 potentiates alpha-synuclein toxicity to dopaminergic neurons in transgenic mice. Int J Biol Sci 7: , 234. |
[62] | Hasegawa M , Nonaka T , Masuda-Suzukake M ((2017) ) Prion-like mechanisms and potential therapeutic targets in neurodegenerative disorders. Pharmacol Ther 172: , 22–33. |
[63] | Nonaka T , Masuda-Suzukake M , Hasegawa M ((2018) ) Molecular mechanisms of the co-deposition of multiple pathological proteins in neurodegenerative diseases. Neuropathology 38: , 64–71. |
[64] | Kiely AP , Ling H , Asi YT , Kara E , Proukakis C , Schapira AH , Morris HR , Roberts HC , Lubbe S , Limousin P , Lewis PA , Lees AJ , Quinn N , Hardy J , Love S , Revesz T , Houlden H , Holton JL ((2015) ) Distinct clinical and neuropathological features of G51D SNCA mutation cases compared with SNCA duplication and H50Q mutation. Mol Neurodegener 10: , 41. |
[65] | Koga S , Lin WL , Walton RL , Ross OA , Dickson DW ((2018) ) TDP-43 pathology in multiple system atrophy: Colocalization of TDP-43 and α-synuclein in glial cytoplasmic inclusions. Neuropathol Appl Neurobiol 44: , 707–721. |
[66] | Heneka MT , Carson MJ , El Khoury J , Landreth GE , Brosseron F , Feinstein DL , Jacobs AH , Wyss-Coray T , Vitorica J , Ransohoff RM , Herrup K , Frautschy SA , Finsen B , Brown GC , Verkhratsky A , Yamanaka K , Koistinaho J , Latz E , Halle A , Petzold GC , Town T , Morgan D , Shinohara ML , Perry VH , Holmes C , Bazan NG , Brooks DJ , Hunot S , Joseph B , Deigendesch N , Garaschuk O , Boddeke E , Dinarello CA , Breitner JC , Cole GM , Golenbock DT , Kummer MP ((2015) ) Neuroinflammation in Alzheimer’s disease. Lancet Neurol 14: , 388–405. |
[67] | Amin J , Erskine D , Donaghy PC , Surendranathan A , Swann P , Kunicki AP , Boche D , Holmes C , McKeith IG , O’Brien JT , Teeling JL , Thomas AJ ((2022) ) Inflammation in dementia with Lewy bodies. Neurobiol Dis 168: , 105698. |
[68] | Bright F , Werry EL , Dobson-Stone C , Piguet O , Ittner LM , Halliday GM , Hodges JR , Kiernan MC , Loy CT , Kassiou M , Kril JJ ((2019) ) Neuroinflammation in frontotemporal dementia. Nat Rev Neurol 15: , 540–555. |
[69] | Kim C , Ho DH , Suk JE , You S , Michael S , Kang J , Joong Lee S , Masliah E , Hwang D , Lee HJ , Lee SJ ((2013) ) Neuron-released oligomeric α-synuclein is an endogenous agonist of TLR2 for paracrine activation of microglia. Nat Commun 4: , 1562. |
[70] | Nilson AN , English KC , Gerson JE , Barton Whittle T , Nicolas Crain C , Xue J , Sengupta U , Castillo-Carranza DL , Zhang W , Gupta P , Kayed R ((2017) ) Tau oligomers associate with inflammation in the brain and retina of tauopathy mice and in neurodegenerative diseases. J Alzheimers Dis 55: , 1083–1099. |
[71] | Hughes C , Choi ML , Yi JH , Kim SC , Drews A , George-Hyslop PS , Bryant C , Gandhi S , Cho K , Klenerman D ((2020) ) Beta amyloid aggregates induce sensitised TLR4 signalling causing long-term potentiation deficit and rat neuronal cell death. Commun Biol 3: , 79. |
[72] | Turnbull IR , Gilfillan S , Cella M , Aoshi T , Miller M , Piccio L , Hernandez M , Colonna M ((2006) ) Cutting edge: TREM-2 attenuates macrophage activation. J Immunol 177: , 3520–3524. |
[73] | Hsieh CL , Koike M , Spusta SC , Niemi EC , Yenari M , Nakamura MC , Seaman WE ((2009) ) A role for TREM2 ligands in the phagocytosis of apoptotic neuronal cells by microglia. J Neurochem 109: , 1144–1156. |
[74] | Rayaprolu S , Mullen B , Baker M , Lynch T , Finger E , Seeley WW , Hatanpaa KJ , Lomen-Hoerth C , Kertesz A , Bigio EH , Lippa C , Josephs KA , Knopman DS , White CL , 3rd, Caselli R , Mackenzie IR , Miller BL , Boczarska-Jedynak M , Opala G , Krygowska-Wajs A , Barcikowska M , Younkin SG , Petersen RC , Ertekin-Taner N , Uitti RJ , Meschia JF , Boylan KB , Boeve BF , Graff-Radford NR , Wszolek ZK , Dickson DW , Rademakers R , Ross OA ((2013) ) TREM2 in neurodegeneration: Evidence for association of the p.R47H variant with frontotemporal dementia and Parkinson’s disease. Mol Neurodegener 8: , 19. |
[75] | Guerreiro R , Wojtas A , Bras J , Carrasquillo M , Rogaeva E , Majounie E , Cruchaga C , Sassi C , Kauwe JS , Younkin S , Hazrati L , Collinge J , Pocock J , Lashley T , Williams J , Lambert JC , Amouyel P , Goate A , Rademakers R , Morgan K , Powell J , St George-Hyslop P , Singleton A , Hardy J ((2013) ) TREM2 variants in Alzheimer’s disease. N Engl J Med 368: , 117–127. |
[76] | Friedman LG , Lachenmayer ML , Wang J , He L , Poulose SM , Komatsu M , Holstein GR , Yue Z ((2012) ) Disrupted autophagy leads to dopaminergic axon and dendrite degeneration and promotes presynaptic accumulation of α-synuclein and LRRK2 in the brain. J Neurosci 32: , 7585–7593. |
[77] | Inoue K , Rispoli J , Kaphzan H , Klann E , Chen EI , Kim J , Komatsu M , Abeliovich A ((2012) ) Macroautophagy deficiency mediates age-dependent neurodegeneration through a phospho-tau pathway. Mol Neurodegener 7: , 48. |
[78] | Nilsson P , Sekiguchi M , Akagi T , Izumi S , Komori T , Hui K , Sörgjerd K , Tanaka M , Saito T , Iwata N , Saido TC ((2015) ) Autophagy-related protein 7 deficiency in amyloid β (Aβ) precursor protein transgenic mice decreases Aβ in the multivesicular bodies and induces Aβ accumulation in the Golgi. Am J Pathol 185: , 305–313. |
[79] | Winslow AR , Chen CW , Corrochano S , Acevedo-Arozena A , Gordon DE , Peden AA , Lichtenberg M , Menzies FM , Ravikumar B , Imarisio S , Brown S , O’Kane CJ , Rubinsztein DC ((2010) ) α-Synuclein impairs macroautophagy: Implications for Parkinson’s disease. J Cell Biol 190: , 1023–1037. |
[80] | Aplin A , Jasionowski T , Tuttle DL , Lenk SE , Dunn WA Jr ((1992) ) Cytoskeletal elements are required for the formation and maturation of autophagic vacuoles. J Cell Physiol 152: , 458–466. |
[81] | Iba M , Guo JL , McBride JD , Zhang B , Trojanowski JQ , Lee VM ((2013) ) Synthetic tau fibrils mediate transmission of neurofibrillary tangles in a transgenic mouse model of Alzheimer’s-like tauopathy. J Neurosci 33: , 1024–1037. |
[82] | Peeraer E , Bottelbergs A , Van Kolen K , Stancu IC , Vasconcelos B , Mahieu M , Duytschaever H , Ver Donck L , Torremans A , Sluydts E , Van Acker N , Kemp JA , Mercken M , Brunden KR , Trojanowski JQ , Dewachter I , Lee VM , Moechars D ((2015) ) Intracerebral injection of preformed synthetic tau fibrils initiates widespread tauopathy and neuronal loss in the brains of tau transgenic mice. Neurobiol Dis 73: , 83–95. |
[83] | Gibbons GS , Banks RA , Kim B , Xu H , Changolkar L , Leight SN , Riddle DM , Li C , Gathagan RJ , Brown HJ , Zhang B , Trojanowski JQ , Lee VM ((2017) ) GFP-mutant human tau transgenic mice develop tauopathy following CNS injections of Alzheimer’s brain-derived pathological tau or synthetic mutant human tau fibrils. J Neurosci 37: , 11485–11494. |
[84] | Guo JL , Narasimhan S , Changolkar L , He Z , Stieber A , Zhang B , Gathagan RJ , Iba M , McBride JD , Trojanowski JQ , Lee VM ((2016) ) Unique pathological tau conformers from Alzheimer’s brains transmit tau pathology in nontransgenic mice. J Exp Med 213: , 2635–2654. |
[85] | Zhang W , Falcon B , Murzin AG , Fan J , Crowther RA , Goedert M , Scheres SH ((2019) ) Heparin-induced tau filaments are polymorphic and differ from those in Alzheimer’s and Pick’s diseases, Elife 8: , e43584. |
[86] | Goedert M , Jakes R , Spillantini M , Hasegawa M , Smith M , Crowther R ((1996) ) Assembly of microtubule-associated protein tau into Alzheimer-like filaments induced by sulphated glycosaminoglycans. Nature 383: , 550–553. |
[87] | Kampers T , Friedhoff P , Biernat J , Mandelkow EM , Mandelkow E ((1996) ) RNA stimulates aggregation of microtubule-associated protein tau into Alzheimer-like paired helical filaments. FEBS Lett 399: , 344–349. |
[88] | Hughes AJ , Daniel SE , Kilford L , Lees AJ ((1992) ) Accuracy of clinical diagnosis of idiopathic Parkinson’s disease: A clinico-pathological study of 100 cases. J Neurol Neurosurg Psychiatry 55: , 181–184. |
[89] | Mastaglia FL , Johnsen RD , Kakulas BA ((2002) ) Prevalence of stroke in Parkinson’s disease: A postmortem study. Mov Disord 17: , 772–774. |
[90] | Jellinger KA ((2003) ) Prevalence of cerebrovascular lesions in Parkinson’s disease. A postmortem study. Acta Neuropathol 105: , 415–419. |
[91] | Stojkovic T , Stefanova E , Soldatovic I , Markovic V , Stankovic I , Petrovic I , Agosta F , Galantucci S , Filippi M , Kostic V ((2018) ) Exploring the relationship between motor impairment, vascular burden and cognition in Parkinson’s disease. J Neurol 265: , 1320–1327. |
[92] | Beyer MK , Aarsland D , Greve OJ , Larsen JP ((2006) ) Visual rating of white matter hyperintensities in Parkinson’s disease. Mov Disord 21: , 223–229. |
[93] | Meyer JS , Huang J , Chowdhury MH ((2007) ) MRI confirms mild cognitive impairments prodromal for Alzheimer’s, vascular and Parkinson-Lewy body dementias. J Neurol Sci 257: , 97–104. |
[94] | Santangelo G , Vitale C , Trojano L , De Gaspari D , Bilo L , Antonini A , Barone P ((2010) ) Differential neuropsychological profiles in Parkinsonian patients with or without vascular lesions. Mov Disord 25: , 50–56. |
[95] | Sıawek J , Roszmann A , Robowski P , Dubaniewicz M , Sitek EJ , Honczarenko K , Gorzkowska A , Budrewicz S , Mak M , Gołab-Janowska M , Koziorowska-Gawron E , Droździk M , Kurzawski M , Bandurski T , Białecka M ((2012) ) The impact of MRI white matter hyperintensities on dementia in Parkinson’s disease in relation to the homocysteine level and other vascular risk factors. Neurodegener Dis 12: , 1–12. |
[96] | Kandiah N , Mak E , Ng A , Huang S , Au WL , Sitoh YY , Tan LCS ((2013) ) Cerebral white matter hyperintensity in Parkinson’s disease: A major risk factor for mild cognitive impairment. Parkinsonism Relat Disord 19: , 680–683. |
[97] | Dalaker TO , Larsen JP , Dwyer MG , Aarsland D , Beyer MK , Alves G , Bronnick K , Tysnes O-B , Zivadinov R ((2009) ) White matter hyperintensities do not impact cognitive function in patients with newly diagnosed Parkinson’s disease. Neuroimage 47: , 2083–2089. |
[98] | González-Redondo R , Toledo J , Clavero P , Lamet I , García-García D , García-Eulate R , Martínez-Lage P , Rodríguez-Oroz MC ((2012) ) The impact of silent vascular brain burden in cognitive impairment in Parkinson’s disease. Eur J Neurol 19: , 1100–1107. |
[99] | Elabi O , Gaceb A , Carlsson R , Padel T , Soylu-Kucharz R , Cortijo I , Li W , Li J-Y , Paul G ((2021) ) Human α-synuclein overexpression in a mouse model of Parkinson’s disease leads to vascular pathology, blood brain barrier leakage and pericyte activation. Sci Rep 11: , 1120. |
[100] | Rektor I , Goldemund D , Bednarík P , Sheardová K , Michálková Z , Telecká S , Dufek M , Rektorová I ((2012) ) Impairment of brain vessels may contribute to mortality in patients with Parkinson’s disease. Mov Disord 27: , 1169–1172. |
[101] | Doiron M , Langlois M , Dupré N , Simard M ((2018) ) The influence of vascular risk factors on cognitive function in early Parkinson’s disease. Int J Geriatr Psychiatry 33: , 288–297. |
[102] | Anang JB , Gagnon JF , Bertrand JA , Romenets SR , Latreille V , Panisset M , Montplaisir J , Postuma RB ((2014) ) Predictors of dementia in Parkinson disease: A prospective cohort study. Neurology 83: , 1253–1260. |
[103] | Chahine LM , Dos Santos C , Fullard M , Scordia C , Weintraub D , Erus G , Rosenthal L , Davatzikos C , McMillan CT ((2019) ) Modifiable vascular risk factors, white matter disease and cognition in early Parkinson’s disease, Eur J Neurol 26: , 246–e218. |
[104] | Malek N , Lawton MA , Swallow DMA , Grosset KA , Marrinan SL , Bajaj N , Barker RA , Burn DJ , Hardy J , Morris HR , Williams NM , Wood N , Ben-Shlomo Y , Grosset DG , Consortium obot PC ((2016) ) Vascular disease and vascular risk factors in relation to motor features and cognition in early Parkinson’s disease. Mov Disord 31: , 1518–1526. |
[105] | Pagano G , Polychronis S , Wilson H , Giordano B , Ferrara N , Niccolini F , Politis M ((2018) ) Diabetes mellitus and Parkinson disease, Neurology 90: , e1654–e1662. |
[106] | Chegão A , Guarda M , Alexandre BM , Shvachiy L , Temido-Ferreira M , Marques-Morgado I , Fernandes Gomes B , Matthiesen R , Lopes LV , Florindo PR , Gomes RA , Gomes-Alves P , Coelho JE , Outeiro TF , Vicente Miranda H ((2022) ) Glycation modulates glutamatergic signaling and exacerbates Parkinson’s disease-like phenotypes. NPJ Parkinsons Dis 8: , 51. |
[107] | Cheong JLY , de Pablo-Fernandez E , Foltynie T , Noyce AJ ((2020) ) The association between type 2 diabetes mellitus and Parkinson’s disease. J Parkinsons Dis 10: , 775–789. |
[108] | Clinton LK , Blurton-Jones M , Myczek K , Trojanowski JQ , LaFerla FM ((2010) ) Synergistic Interactions between Abeta, tau, and alpha-synuclein: Acceleration of neuropathology and cognitive decline. J Neurosci 30: , 7281–7289. |
[109] | McAleese KE , Colloby SJ , Thomas AJ , Al-Sarraj S , Ansorge O , Neal J , Roncaroli F , Love S , Francis PT , Attems J ((2021) ) Concomitant neurodegenerative pathologies contribute to the transition from mild cognitive impairment to dementia, Alzheimers Dement 17: , 1121–1133. |
[110] | Howlett DR , Whitfield D , Johnson M , Attems J , O’Brien JT , Aarsland D , Lai MK , Lee JH , Chen C , Ballard C , Hortobagyi T , Francis PT ((2015) ) Regional multiple pathology scores are associated with cognitive decline in Lewy body dementias. Brain Pathol 25: , 401–408. |
[111] | Compta Y , Parkkinen L , O’Sullivan SS , Vandrovcova J , Holton JL , Collins C , Lashley T , Kallis C , Williams DR , de Silva R , Lees AJ , Revesz T ((2011) ) Lewy- and Alzheimer-type pathologies in Parkinson’s disease dementia: Which is more important? Brain 134: , 1493–1505. |
[112] | Ruffmann C , Calboli FC , Bravi I , Gveric D , Curry LK , de Smith A , Pavlou S , Buxton JL , Blakemore AI , Takousis P , Molloy S , Piccini P , Dexter DT , Roncaroli F , Gentleman SM , Middleton LT ((2016) ) Cortical Lewy bodies and Abeta burden are associated with prevalence and timing of dementia in Lewy body diseases. Neuropathol Appl Neurobiol 42: , 436–450. |
[113] | Colom-Cadena M , Grau-Rivera O , Planellas L , Cerquera C , Morenas E , Helgueta S , Munoz L , Kulisevsky J , Marti MJ , Tolosa E , Clarimon J , Lleo A , Gelpi E ((2017) ) Regional overlap of pathologies in Lewy body disorders. J Neuropathol Exp Neurol 76: , 216–224. |
[114] | Sabbagh MN , Adler CH , Lahti TJ , Connor DJ , Vedders L , Peterson LK , Caviness JN , Shill HA , Sue LI , Ziabreva I , Perry E , Ballard CG , Aarsland D , Walker DG , Beach TG ((2009) ) Parkinson disease with dementia: Comparing patients with and without Alzheimer pathology. Alzheimer Dis Assoc Disord 23: , 295–297. |
[115] | Ballard C , Ziabreva I , Perry R , Larsen JP , O’Brien J , McKeith I , Perry E , Aarsland D ((2006) ) Differences in neuropathologic characteristics across the Lewy body dementia spectrum, Neurology 67: , 1931–1934. |
[116] | Myers PS , O’Donnell JL , Jackson JJ , Lessov-Schlaggar CN , Miller RL , Foster ER , Cruchaga C , Benitez BA , Kotzbauer PT , Perlmutter JS , Campbell MC ((2022) ) Proteinopathy and longitudinal cognitive decline in Parkinson disease, Neurology 99: , e66–e76. |
[117] | Irwin DJ , Grossman M , Weintraub D , Hurtig HI , Duda JE , Xie SX , Lee EB , Van Deerlin VM , Lopez OL , Kofler JK , Nelson PT , Jicha GA , Woltjer R , Quinn JF , Kaye J , Leverenz JB , Tsuang D , Longfellow K , Yearout D , Kukull W , Keene CD , Montine TJ , Zabetian CP , Trojanowski JQ ((2017) ) Neuropathological and genetic correlates of survival and dementia onset in synucleinopathies: A retrospective analysis. Lancet Neurol 16: , 55–65. |
[118] | Stern Y , Jacobs D , Goldman J , Gomez-Tortosa E , Hyman BT , Liu Y , Troncoso J , Marder K , Tang MX , Brandt J , Albert M ((2001) ) An investigation of clinical correlates of Lewy bodies in autopsy-proven Alzheimer disease. Arch Neurol 58: , 460–465. |
[119] | Holtzer R , Irizarry MC , Sanders J , Hyman BT , Wegesin DJ , Riba A , Brandt J , Albert M , Stern Y ((2006) ) Relation of quantitative indexes of concurrent alpha-synuclein abnormalities to clinical outcome in autopsy-proven Alzheimer disease. Arch Neurol 63: , 226–230. |
[120] | Wennberg AM , Whitwell JL , Tosakulwong N , Weigand SD , Murray ME , Machulda MM , Petrucelli L , Mielke MM , Jack CR Jr , Knopman DS , Parisi JE , Petersen RC , Dickson DW , Josephs KA ((2019) ) The influence of tau, amyloid, alpha-synuclein, TDP-43, and vascular pathology in clinically normal elderly individuals. Neurobiol Aging 77: , 26–36. |
[121] | Martin WRW , Younce JR , Campbell MC , Racette BA , Norris SA , Ushe M , Criswell S , Davis AA , Alfradique-Dunham I , Maiti B , Cairns NJ , Perrin RJ , Kotzbauer PT , Perlmutter JS ((2023) ) Neocortical Lewy body pathology parallels Parkinson’s dementia, but not always. Ann Neurol 93: , 184–195. |
[122] | Ince PG ((2001) ) Pathological correlates of late-onset dementia in a multicentre, community-based population in England and Wales. Neuropathology Group of the Medical Research Council Cognitive Function and Ageing Study (MRC CFAS). Lancet 357: , 169–175. |
[123] | Jellinger KA , Attems J ((2012) ) Neuropathology and general autopsy findings in nondemented aged subjects. Clin Neuropathol 31: , 87–98. |
[124] | McAleese KE , Walker L , Erskine D , Thomas AJ , McKeith IG , Attems J ((2017) ) TDP-43 pathology in Alzheimer’s disease, dementia with Lewy bodies and ageing. Brain Pathol 27: , 472–479. |
[125] | Harding AJ , Halliday GM ((2001) ) Cortical Lewy body pathology in the diagnosis of dementia. Acta Neuropathol 102: , 355–363. |
[126] | Kovari E , Gold G , Herrmann FR , Canuto A , Hof PR , Bouras C , Giannakopoulos P ((2003) ) Lewy body densities in the entorhinal and anterior cingulate cortex predict cognitive deficits in Parkinson’s disease. Acta Neuropathol 106: , 83–88. |
[127] | Robinson JL , Geser F , Corrada MM , Berlau DJ , Arnold SE , Lee VM , Kawas CH , Trojanowski JQ ((2011) ) Neocortical and hippocampal amyloid-beta and tau measures associate with dementia in the oldest-old. Brain 134: , 3708–3715. |
[128] | Gomperts SN , Locascio JJ , Rentz D , Santarlasci A , Marquie M , Johnson KA , Growdon JH ((2013) ) Amyloid is linked to cognitive decline in patients with Parkinson disease without dementia. Neurology 80: , 85–91. |
[129] | Buongiorno M , Antonelli F , Compta Y , Fernandez Y , Pavia J , Lomeña F , Ríos J , Ramírez I , García JR , Soler M , Cámara A , Fernández M , Basora M , Salazar F , Sanchez-Etayo G , Valldeoriola F , Barrio JR , Marti MJ ((2017) ) Cross-sectional and longitudinal cognitive correlates of FDDNP PET and CSF amyloid-β and tau in Parkinson’s disease. J Alzheimers Dis 55: , 1261–1272. |
[130] | Tang Y , Li L , Hu T , Jiao F , Han L , Li S , Xu Z , Fan Y , Sun Y , Liu F , Yen T-C , Zuo C , Wang J ((2023) ) 18F-florzolotau tau positron emission tomography imaging in Parkinson’s disease dementia, Mov Disord 38: , 147–152. |
[131] | Gomperts SN , Locascio JJ , Makaretz SJ , Schultz A , Caso C , Vasdev N , Sperling R , Growdon JH , Dickerson BC , Johnson K ((2016) ) Tau positron emission tomographic imaging in the Lewy body diseases. JAMA Neurol 73: , 1334–1341. |
[132] | Chen Q , Przybelski SA , Senjem ML , Schwarz CG , Lesnick TG , Botha H , Knopman DS , Graff-Radford J , Savica R , Jones DT , Fields JA , Jain MK , Graff-Radford NR , Ferman TJ , Kremers WK , Jack CR Jr , Petersen RC , Boeve BF , Lowe VJ , Kantarci K ((2022) ) Longitudinal tau positron emission tomography in dementia with Lewy bodies. Mov Disord 37: , 1256–1264. |
[133] | Kantarci K , Lowe VJ , Boeve BF , Senjem ML , Tosakulwong N , Lesnick TG , Spychalla AJ , Gunter JL , Fields JA , Graff-Radford J , Ferman TJ , Jones DT , Murray ME , Knopman DS , Jack CR Jr , Petersen RC ((2017) ) AV-1451 tau and beta-amyloid positron emission tomography imaging in dementia with Lewy bodies. Ann Neurol 81: , 58–67. |
[134] | Mak E , Nicastro N , Malpetti M , Savulich G , Surendranathan A , Holland N , Passamonti L , Jones PS , Carter SF , Su L , Hong YT , Fryer TD , Williams GB , Aigbirhio F , Rowe JB , O’Brien JT ((2021) ) Imaging tau burden in dementia with Lewy bodies using [(18)F]-AV1451 positron emission tomography. Neurobiol Aging 101: , 172–180. |
[135] | Johnson KA , Schultz A , Betensky RA , Becker JA , Sepulcre J , Rentz D , Mormino E , Chhatwal J , Amariglio R , Papp K , Marshall G , Albers M , Mauro S , Pepin L , Alverio J , Judge K , Philiossaint M , Shoup T , Yokell D , Dickerson B , Gomez-Isla T , Hyman B , Vasdev N , Sperling R ((2016) ) Tau positron emission tomographic imaging in aging and early Alzheimer disease. Ann Neurol 79: , 110–119. |
[136] | Ossenkoppele R , Schonhaut DR , Schöll M , Lockhart SN , Ayakta N , Baker SL , O’Neil JP , Janabi M , Lazaris A , Cantwell A , Vogel J , Santos M , Miller ZA , Bettcher BM , Vossel KA , Kramer JH , Gorno-Tempini ML , Miller BL , Jagust WJ , Rabinovici GD ((2016) ) Tau PET patterns mirror clinical and neuroanatomical variability in Alzheimer’s disease. Brain 139: , 1551–1567. |
[137] | Liu H , Koros C , Strohäker T , Schulte C , Bozi M , Varvaresos S , Ibáñez de Opakua A , Simitsi AM , Bougea A , Voumvourakis K , Maniati M , Papageorgiou SG , Hauser A-K , Becker S , Zweckstetter M , Stefanis L , Gasser T ((2021) ) A novel SNCA A30G mutation causes familial Parkinson’s disease. Mov Disord 36: , 1624–1633. |
[138] | Frasier M , Walzer M , McCarthy L , Magnuson D , Lee JM , Haas C , Kahle P , Wolozin B ((2005) ) Tau phosphorylation increases in symptomatic mice overexpressing A30P alpha-synuclein. Exp Neurol 192: , 274–287. |
[139] | Henderson MX , Sengupta M , Trojanowski JQ , Lee VMY ((2019) ) Alzheimer’s disease tau is a prominent pathology in LRRK2 Parkinson’s disease. Acta Neuropathol Commun 7: , 183. |
[140] | Davis AA , Inman CE , Wargel ZM , Dube U , Freeberg BM , Galluppi A , Haines JN , Dhavale DD , Miller R , Choudhury FA , Sullivan PM , Cruchaga C , Perlmutter JS , Ulrich JD , Benitez BA , Kotzbauer PT , Holtzman DM ((2020) ) APOE genotype regulates pathology and disease progression in synucleinopathy, Sci Transl Med 12: , eaay3069. |
[141] | Zhao N , Attrebi ON , Ren Y , Qiao W , Sonustun B , Martens YA , Meneses AD , Li F , Shue F , Zheng J , Van Ingelgom AJ , Davis MD , Kurti A , Knight JA , Linares C , Chen Y , Delenclos M , Liu CC , Fryer JD , Asmann YW , McLean PJ , Dickson DW , Ross OA , Bu G ((2020) ) APOE4 exacerbates α-synuclein pathology and related toxicity independent of amyloid, Sci Transl Med 12: , eaay1809. |
[142] | Simón-Sánchez J , Schulte C , Bras JM , Sharma M , Gibbs JR , Berg D , Paisan-Ruiz C , Lichtner P , Scholz SW , Hernandez DG , Krüger R , Federoff M , Klein C , Goate A , Perlmutter J , Bonin M , Nalls MA , Illig T , Gieger C , Houlden H , Steffens M , Okun MS , Racette BA , Cookson MR , Foote KD , Fernandez HH , Traynor BJ , Schreiber S , Arepalli S , Zonozi R , Gwinn K , van der Brug M , Lopez G , Chanock SJ , Schatzkin A , Park Y , Hollenbeck A , Gao J , Huang X , Wood NW , Lorenz D , Deuschl G , Chen H , Riess O , Hardy JA , Singleton AB , Gasser T ((2009) ) Genome-wide association study reveals genetic risk underlying Parkinson’s disease. Nat Genet 41: , 1308–1312. |
[143] | Edwards TL , Scott WK , Almonte C , Burt A , Powell EH , Beecham GW , Wang L , Züchner S , Konidari I , Wang G , Singer C , Nahab F , Scott B , Stajich JM , Pericak-Vance M , Haines J , Vance JM , Martin ER ((2010) ) Genome-wide association study confirms SNPs in SNCA and the MAPT region as common risk factors for Parkinson disease. Ann Hum Genet 74: , 97–109. |
[144] | Satake W , Nakabayashi Y , Mizuta I , Hirota Y , Ito C , Kubo M , Kawaguchi T , Tsunoda T , Watanabe M , Takeda A , Tomiyama H , Nakashima K , Hasegawa K , Obata F , Yoshikawa T , Kawakami H , Sakoda S , Yamamoto M , Hattori N , Murata M , Nakamura Y , Toda T ((2009) ) Genome-wide association study identifies common variants at four loci as genetic risk factors for Parkinson’s disease. Nat Genet 41: , 1303–1307. |
[145] | Rayaprolu S , Fujioka S , Traynor S , Soto-Ortolaza AI , Petrucelli L , Dickson DW , Rademakers R , Boylan KB , Graff-Radford NR , Uitti RJ , Wszolek ZK , Ross OA ((2013) ) TARDBP mutations in Parkinson’s disease. Parkinsonism Relat Disord 19: , 312–315. |
[146] | Tiloca C , Goldwurm S , Calcagno N , Verde F , Peverelli S , Calini D , Zecchinelli AL , Sangalli D , Ratti A , Pezzoli G , Silani V , Ticozzi N ((2022) ) TARDBP mutations in a cohort of Italian patients with Parkinson’s disease and atypical parkinsonisms. Front Aging Neurosci 14: , 1020948. |
[147] | Tokuda T , Qureshi MM , Ardah MT , Varghese S , Shehab SA , Kasai T , Ishigami N , Tamaoka A , Nakagawa M , El-Agnaf OM ((2010) ) Detection of elevated levels of α-synuclein oligomers in CSF from patients with Parkinson disease. Neurology 75: , 1766–1772. |
[148] | Mollenhauer B , Locascio JJ , Schulz-Schaeffer W , Sixel-Döring F , Trenkwalder C , Schlossmacher MG ((2011) ) α-Synuclein and tau concentrations in cerebrospinal fluid of patients presenting with parkinsonism: A cohort study. Lancet Neurol 10: , 230–240. |
[149] | Hansson O , Hall S , Ohrfelt A , Zetterberg H , Blennow K , Minthon L , Nägga K , Londos E , Varghese S , Majbour NK , Al-Hayani A , El-Agnaf OM ((2014) ) Levels of cerebrospinal fluid α-synuclein oligomers are increased in Parkinson’s disease with dementia and dementia with Lewy bodies compared to Alzheimer’s disease. Alzheimers Res Ther 6: , 25. |
[150] | El-Agnaf OM , Salem SA , Paleologou KE , Curran MD , Gibson MJ , Court JA , Schlossmacher MG , Allsop D ((2006) ) Detection of oligomeric forms of alpha-synuclein protein in human plasma as a potential biomarker for Parkinson’s disease. FASEB J 20: , 419–425. |
[151] | Beach TG , Adler CH , Dugger BN , Serrano G , Hidalgo J , Henry-Watson J , Shill HA , Sue LI , Sabbagh MN , Akiyama H ((2013) ) Submandibular gland biopsy for the diagnosis of Parkinson disease. J Neuropathol Exp Neurol 72: , 130–136. |
[152] | Donadio V , Incensi A , Leta V , Giannoccaro MP , Scaglione C , Martinelli P , Capellari S , Avoni P , Baruzzi A , Liguori R ((2014) ) Skin nerve α-synuclein deposits: A biomarker for idiopathic Parkinson disease. Neurology 82: , 1362–1369. |
[153] | Compta Y , Martí MJ , Ibarretxe-Bilbao N , Junqué C , Valldeoriola F , Muñoz E , Ezquerra M , Ríos J , Tolosa E ((2009) ) Cerebrospinal tau, phospho-tau, and beta-amyloid and neuropsychological functions in Parkinson’s disease. Mov Disord 24: , 2203–2210. |
[154] | Hall S , Surova Y , Öhrfelt A , Zetterberg H , Lindqvist D , Hansson O ((2015) ) CSF biomarkers and clinical progression of Parkinson disease. Neurology 84: , 57–63. |
[155] | Mollenhauer B , Caspell-Garcia CJ , Coffey CS , Taylor P , Shaw LM , Trojanowski JQ , Singleton A , Frasier M , Marek K , Galasko D , Parkinson’s Progression Marker Initiative ((2017) ) Longitudinal CSF biomarkers in patients with early Parkinson disease and healthy controls. Neurology 89: , 1959–1969. |
[156] | Batzu L , Rota S , Hye A , Heslegrave A , Trivedi D , Gibson LL , Farrell C , Zinzalias P , Rizos A , Zetterberg H , Chaudhuri KR , Aarsland D ((2022) ) Plasma p-tau181, neurofilament light chain and association with cognition in Parkinson’s disease. NPJ Parkinsons Dis 8: , 154. |
[157] | Stav AL , Aarsland D , Johansen KK , Hessen E , Auning E , Fladby T ((2015) ) Amyloid-β and α-synuclein cerebrospinal fluid biomarkers and cognition in early Parkinson’s disease. Parkinsonism Relat Disord 21: , 758–764. |
[158] | Delgado-Alvarado M , Gago B , Navalpotro-Gomez I , Jiménez-Urbieta H , Rodriguez-Oroz MC ((2016) ) Biomarkers for dementia and mild cognitive impairment in Parkinson’s disease. Mov Disord 31: , 861–881. |
[159] | Kang JH , Irwin DJ , Chen-Plotkin AS , Siderowf A , Caspell C , Coffey CS , Waligórska T , Taylor P , Pan S , Frasier M , Marek K , Kieburtz K , Jennings D , Simuni T , Tanner CM , Singleton A , Toga AW , Chowdhury S , Mollenhauer B , Trojanowski JQ , Shaw LM ((2013) ) Association of cerebrospinal fluid β-amyloid 1-42, T-tau, P-tau181, and α-synuclein levels with clinical features of drug-naive patients with early Parkinson disease. JAMA Neurol 70: , 1277–1287. |
[160] | Parnetti L , Farotti L , Eusebi P , Chiasserini D , De Carlo C , Giannandrea D , Salvadori N , Lisetti V , Tambasco N , Rossi A ((2014) ) Differential role of CSF alpha-synuclein species, tau, and Aβ42 in Parkinson’s Disease. Front Aging Neurosci 6: , 53. |
[161] | Ren J , Pan C , Wang Y , Xue C , Lin H , Xu J , Wang H , Zhang W , Xu P , Chen Y , Liu W ((2022) ) Plasma α-synuclein and phosphorylated tau 181 as a diagnostic biomarker panel for de novo Parkinson’s disease. J Neurochem 161: , 506–515. |
[162] | Hansen AK , Damholdt MF , Fedorova TD , Knudsen K , Parbo P , Ismail R , Østergaard K , Brooks DJ , Borghammer P ((2017) ) cortical tau in Parkinson’s disease using 18F-AV-1451 positron emission tomography, Mov Disord 32: , 922–927. |
[163] | Luca DGD , Luo S , Liu H , Cohn M , Davis TL , Ramirez-Zamora A , Rafferty M , Dahodwala N , Naito A , Neault M , Beck J , Marras C ((2023) ) Racial and ethnic differences in health-related quality of life for individuals with Parkinson disease across centers of excellence, Neurology 100: , e2170–e2181. |
[164] | Barnes LL , Leurgans S , Aggarwal NT , Shah RC , Arvanitakis Z , James BD , Buchman AS , Bennett DA , Schneider JA ((2015) ) Mixed pathology is more likely in black than white decedents with Alzheimer dementia. Neurology 85: , 528–534. |
[165] | Filshtein TJ , Dugger BN , Jin LW , Olichney JM , Farias ST , Carvajal-Carmona L , Lott P , Mungas D , Reed B , Beckett LA , DeCarli C ((2019) ) Neuropathological diagnoses of demented Hispanic, Black, and non-Hispanic white decedents seen at an Alzheimer’s disease center. J Alzheimers Dis 68: , 145–158. |
[166] | Connell CM , Avey H , Holmes SB ((1994) ) Attitudes about autopsy: Implications for educational interventions. Gerontologist 34: , 665–673. |
[167] | Bonner GJ , Darkwa OK , Gorelick PB ((2000) ) Autopsy recruitment program for African Americans. Alzheimer Dis Assoc Disord 14: , 202–208. |
[168] | Jefferson AL , Lambe S , Cook E , Pimontel M , Palmisano J , Chaisson C ((2011) ) Factors associated with African American and White elders’ participation in a brain donation program. Alzheimer Dis Assoc Disord 25: , 11–16. |
[169] | Boise L , Hinton L , Rosen HJ , Ruhl MC , Dodge H , Mattek N , Albert M , Denny A , Grill JD , Hughes T , Lingler JH , Morhardt D , Parfitt F , Peterson-Hazan S , Pop V , Rose T , Shah RC ((2017) ) Willingness to be a brain donor: A survey of research volunteers from 4 racial/ethnic groups. Alzheimer Dis Assoc Disord 31: , 135–140. |


