
Immunological Features of LRRK2 Function and Its Role in the Gut-Brain Axis Governing Parkinson’s Disease
Abstract
Emerging evidence implicates intestinal involvement in the onset and/or progression on the selective degeneration of dopaminergic neurons characterizing Parkinson’s disease (PD). On the one hand, there are studies supporting the Braak hypothesis that holds that pathologic α-synuclein, a hallmark of PD, is secreted by enteric nerves into intestinal tissue and finds its way to the central nervous system (CNS) via retrograde movement in the vagus nerve. On the other hand, there is data showing that cells bearing leucine-rich repeat kinase 2 (LRRK2), a signaling molecule with genetic variants associated with both PD and with inflammatory bowel disease, can be activated in intestinal tissue and contribute locally to intestinal inflammation, or peripherally to PD pathogenesis via cell trafficking to the CNS. Importantly, these gut-centered factors affecting PD development are not necessarily independent of one another: they may interact and enhance their respective pathologic functions. In this review, we discuss this possibility by analysis of studies conducted in recent years focusing on the ability of LRRK2 to shape immunologic responses and the role of α-synuclein in influencing this ability.
Emerging evidence implicates intestinal involvement in the onset and progression of Parkinson’s disease (PD). According to Braak et al. hypothesis, PD pathogenesis may originate in the gastrointestinal tract, with pathological α-synuclein, a hallmark of PD, secreted by enteric nervous system (ENS) and gaining access to the brain via the parasympathetic nervous system [1–3]. The leucine-rich repeat kinase 2 (LRRK2) gene, well recognized as one of the key genetic determinants of PD, is expressed in the enteric nervous system (ENS) [4] and can serve as a modulator of inflammation in the central nervous system (CNS) and the periphery [5, 6]. However, the impact of peripheral inflammation on PD initiation is insufficiently understood to inform disease interception, early diagnosis, or novel therapeutic strategies. This review will discuss the immunological features of LRRK2 function and the role that peripheral immune signaling may play in the regulation of neurodegeneration in LRRK2 as well as non-LRRK2-associated PD.
LRRK2 GENE PLEIOTROPY
The LRRK2 gene encodes a multi-domain protein that includes a domain with GTPase activity as well as a domain with serine/threonine kinase activity, the latter now known to have certain Rab GTPases as its main physiological substrate [7]. By phosphorylating such GTPases LRRK2 kinase activity can have protean effects on cell function including the regulation of intracellular vesicle trafficking, cell organelle maintenance, and processes associated with phagocytosis and autophagy (reviewed in [8]). On this basis it is not surprising that many of the PD-associated mutations in the LRRK2 gene are located in the GTPase and kinase domains and have been shown to affect the respective activities centered in these domains. In addition, LRRK2 kinase function may have important effects on cellular immune function altering fundamental capacities to mount inflammatory responses.
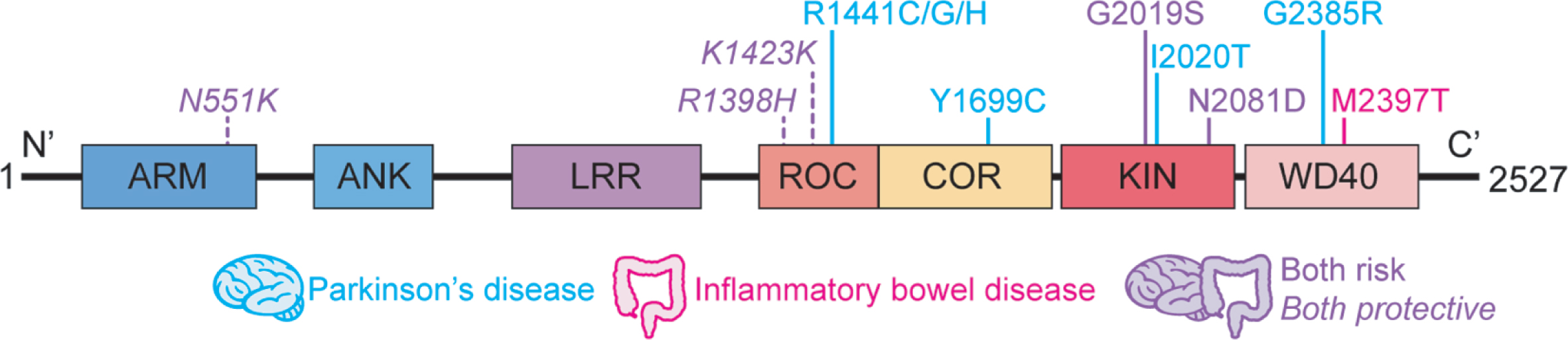
A series of genome-wide association studies has identified LRRK2 as a susceptibility locus for Crohn’s disease (CD) [9–11], a type of inflammatory bowel disease (IBD) characterized by chronic intestinal inflammation resulting from excessive immune response to commensal bacteria in genetically predisposed individuals. Follow-up exome sequencing studies have revealed that a functional LRRK2 variant, N2081D, conferred shared increased risk to develop CD and PD [12, 13], whereas LRRK2 R1398H and N551K, in strong linkage disequilibrium with each other, were associated with the protection against both diseases [12] (Fig. 1). The N2081D polymorphism is located in the same LRRK2 kinase domain, only 62 amino acids away from the major PD risk variant, G2019S, and both risk variants result in increased LRRK2 kinase activity with respect to Rab10-phosphorylation (though to a higher degree for G2019S) [12]. Along the same lines, the protective variants, located in a Ras of complex (RocCOR) domain harboring GTPase function, confers increased GTPase and Wnt activity [12, 14]. This pleiotropy, however, does not apply to all LRRK2 gene variants. Thus, the M2397T variant located in the WD40 domain of LRRK2 and purported to cause decreased LRRK2 expression, is linked to IBD but not to PD [15–17] (Fig. 1); in addition, this variant confers enhanced CD14 + cell TNF-α or IL-12 cytokine production in response to IFN-γ stimulation in CD patients, whereas these cells exhibit no increased TNF-α or IL-12 responses to the same stimulant in PD patients whether they bear G2019S and R1441C mutations or not [18]. In a similar vein, none of the other major PD-driving LRRK2 variants, including N1437H, R1441G/C/H, Y1699C, I2012T, I2020T, have been implicated in IBD development (Fig. 1). Overall, the picture that emerges is that whereas PD and IBD share a genetic basis related to LRRK2 variation, they also manifest unique LRRK2 polymorphisms as well. The genetic link is in accord with epidemiological studies from multiple national databases demonstrating an increased risk of PD development in patients with IBD [19–22], whereas other studies showed no or opposite effect [23, 24]. It was also suggested that early anti-inflammatory therapies for IBD may lead to reduced risk of developing PD [25].
Fig. 1
Schematic representation of LRRK2 domain structure. Respective locations of the amino acid substitutions previously linked to Parkinson’s disease and inflammatory bowel disease are shown. ARM, armadillo; ANK, ankyrin repeat region; LRR, leucine-rich repeat; ROC, Ras of complex proteins; COR, C terminal of ROC; KIN, mitogen-activated protein kinase; WD40, WD40 protein-protein interaction domain.
One way the genetic relation between IBD and PD can take part in a pathologic mechanism that is common to both of these diseases is through its possible effect on the relation of LRRK2 function to enhanced innate pro-inflammatory immune responses (Fig. 2). This possibility can be and, in fact, has been tested by studies evaluating the effect of LRRK2 on the output of pro-inflammatory cytokines by stimulated peripheral cells (monocytes/macrophages or dendritic cells) and equivalent cells in the CNS (microglia). However, with respect to the responses of peripheral cells bearing LRRK2 abnormalities the data so far obtained in such studies are equivocal.
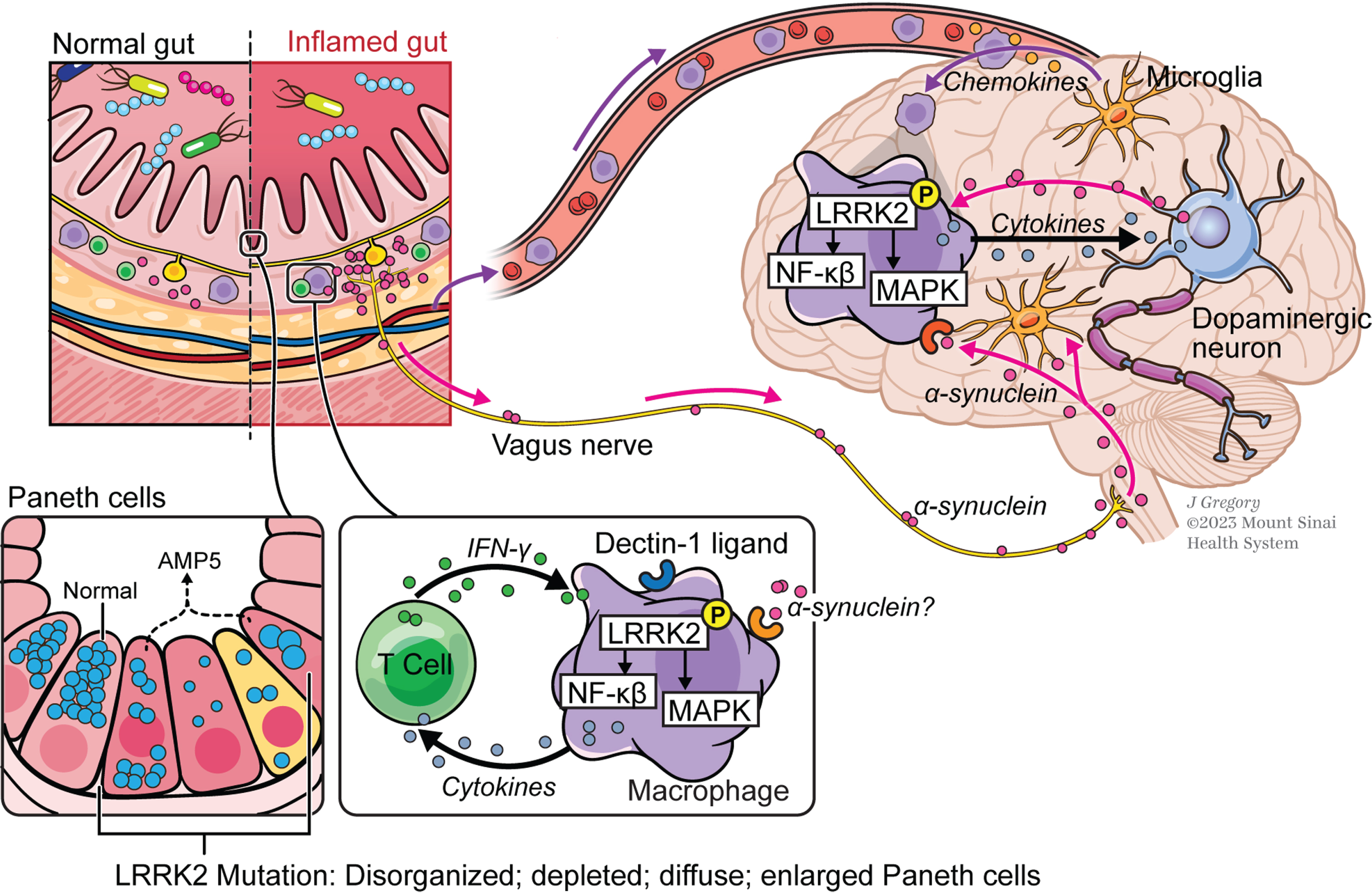
Fig. 2
The gut-brain axis in Parkinson’s disease (PD). In this concept of PD pathogenesis, macrophages in the gut lamina propria are induced by secreted pro-inflammatory cytokines in individuals carrying gain-of-function LRRK2 variants that express increased levels of activated LRRK2; stimulation of cells by α-synuclein from enteric nerves may also occur at this site but this is still unproven. Macrophages thus stimulated that gain entry into the circulation are attracted into brain tissue by chemokine-secreting microglia and in the brain tissue are induced to produce pathogenic inflammatory cytokines by as yet undefined stimuli, possibly including α-synuclein once again; these cytokines cause or aggravate neural degeneration. Also depicted are small intestinal Paneth cells whose function is regulated in part by LRRK2; thus, LRRK2 variants that affect this function and adversely regulate the composition of the gut microbiome may lead to PD by promoting gut inflammation.
LRRK2 AND PRO-INFLAMMATORY CYTOKINE RESPONSES OF CIRCULATING CELLS
A very recent study Ahmadi Rastegar et al. has shown that monocytes and macrophages derived from murine induced pluripotential stem cells (iPS cells) obtained from Wild Type (WT) and Lrrk2 knockout (KO) mice produce equivalent amounts of a broad range of cytokines and chemokines following stimulation with toll-like receptor 2 (TLR2), TLR3 and TLR7/8 ligands whereas the same type of cells from mice bearing the Lrrk2 G2019S mutation (and exhibiting increased LRRK2 kinase activity) produced strikingly increased amounts of a considerable number of these cytokines and chemokines [26]. However, two different LRRK2 kinase inhibitors failed to down-regulate the enhanced cytokine/chemokine responses.
Given the increased cytokine responses of cells bearing a gain-of-function LRRK2 mutation [26], one might have expected that cells lacking LRRK2 would exhibit decreased cytokine responses. This outcome is puzzling but nevertheless in accord with most previous studies of peripheral cells from Lrrk2 KO mice stimulated by TLR or Dectin-1 ligands. Thus, Wandu et al. reported that LPS-stimulated spleen cells from Lrrk2 KO mice exhibited about the same or only slightly more cytokine secretion than cells from normal mice [27]. Similarly, other studies found that macrophage cell lines or bone marrow-derived dendritic cells (BMDCs) from Lrrk2 wild-type (WT) and KO mice produced equal amounts of these cytokines upon TLR and Dectin-1 stimulation [28, 29]. To muddy the water further, Kubo et al. showed that lipopolysaccharide (LPS)-stimulated BMDCs from Lrrk2 KO mice or BMDCs from the latter mice with L. major infection produced very considerably more TNF-α and IL-6 than BMDCs from WT mice [30] and Liu et al. found that Dectin-1 ligand stimulated BMDMs from Lrrk2 KO mice produced increased amounts of IL-6 and IL-12p40 than cells from WT mice (although in this case responses to a range of TLR ligands were equivalent in cells from the two mouse sources) [17]. The only study in which cells from Lrrk2 KO mice did register an expected decreased cytokine response was in a study also conducted by Wandu et al. showing that BSA-stimulated spleen cells from BSA-immunized Lrrk2 KO mice produce less IFN-γ and IL-17 than cells from similarly-treated WT mice [27]. This study was in accord with the findings of Ikezu et al. showing that IFNγ-stimulated CD14+ monocytes from humans with CD bearing the M2397T LRRK2 polymorphism that, as mentioned, has been reported to result in reduced LRRK2 levels, exhibit increased TNF-α and IL-12 responses compared to similar cells from controls and that the increase is enhanced by a LRRK2 kinase inhibitor [18]. However, in this study the stimulated cells exhibited increased LRRK2 levels.
The finding by Ahmadi Rastegar et al. [26] showing that LRRK2 kinase inhibitors did not reverse the increased responses of cells originating from mice bearing LRRK2 with increased kinase function is also puzzling, but in this case is not supported by prior studies. Thus, Li et al. found that a LRRK2 inhibitor that inhibits LRRK2 GTP binding and kinase activity also inhibits TNF-α production by human B cell lymphoblasts [31]. This study is in agreement with those of Takagawa et al. [29] who showed that several (but not all) LRRK2 kinase inhibitors (including one of the inhibitors used by Ahmadi Rastegar et al. [26]) suppressed TNF-α production by Dectin-1 ligand stimulated BMDCs. These inhibitor studies are in accord with experiments conducted with murine microglia (see below).
Finally, the finding that cells bearing LRRK2 with increased kinase function due to a mutation exhibit increased cytokine secretion [26] does comport with a previous study showing that Dectin-1 ligand (ZymD)-stimulated BMDCs from mice bearing a WT-LRRK2 transgene exhibited increased TNF-α and IL-23 responses [29]. However, cells stimulated by either TLR2 ligands or TLR4 ligand did not exhibit increased responses. This might explain the finding that TLR4-ligand (LPS)-stimulated peritoneal macrophages from mice bearing a WT-LRRK2 transgene or a G2019S-LRRK2 transgene exhibited TNF-α and other cytokine production levels equivalent to that of similar macrophages from WT (non-transgenic) mice [32].
LRRK2 AND PRO-INFLAMMATORY CYTOKINE RESPONSES OF MICROGLIA
In contrast to the inconsistent findings relating to pro-inflammatory activity of LRRK2 in peripheral cells, studies of CNS macrophage-like cells, microglia, have yielded a seamless profile of LRRK2 as a significant pro-inflammatory factor. Thus, some ten years ago, Moehle et al. showed that TNF-α responses (and downstream iNOS responses) of isolated microglial cells stimulated by LPS were reduced by LRRK2 kinase inhibitors or LRRK2 down-regulation by shRNA [33]. Moreover, it was demonstrated that LRRK2 binds to and phosphorylate p53 and a microglial cell line transfected with phosphorylated p53 produces increased amounts of TNF-α, especially after LPS treatment [34]. Finally, Russo et al. showed that microglia obtained from knock-in G2019S mice produced increased amounts of IL-1β compared to cells from WT mice when stimulated with α-synuclein fibrils [35]. These more or less direct studies of LRRK2 pro-inflammatory activity are supported by studies by Kim et al. who showed that microglial cell lines in which LRRK2 is knocked down by specific shRNA transfection display greatly reduced IL-6 and TNF-α mRNA generation [36] and by Russo et al. who demonstrated that IL-1β and COX2 synthesis is decreased in a microglial cell line by LRRK2 inhibitors [35].
LRRK2 REGULATION OF NF-κB/MAPK RESPONSES
Studies of the relation of LRRK2 to NF-κB or MAPK activation, the processes upon which cytokine syntheses largely depend, are more consistent in showing that LRRK2 has a positive effect on pro-inflammatory responses than studies of cytokine output discussed above, regardless of whether the cell has a peripheral or CNS origin. The first such study addressing this question was conducted by Gardet et al. [37] who showed that HEK293 cells transfected with a WT LRRK2-expressing plasmid exhibited increased NF-κB activation compared to cells expressing control plasmid as assessed by a luciferase reporter [37]. A similar result was obtained in studies by Kim et al. [36] and both studies showed that transfection of LRRK2 plasmids with mutations that cause gain or loss of kinase activity had an effect similar to the WT plasmid [36, 37]. Kim et al. also found that phosphorylation of both p38 and JNK was also increased in the LRRK2-transfected HEK293 cells, but in this case LRRK2 with a mutation causing loss of kinase activity abrogated the LRRK2 effect on phosphorylation.
Other studies of LRRK2 effects on NF-κB activation conducted with peripheral cells or microglial cells rather than transfected (HEK) cells provided data verifying the enhancing effect of LRRK2. Thus, while Kubo et al. found that LPS-stimulated mouse BMDCs from Lrrk2 KO mice exhibited increased NF-κB (p65) expression (but not p-p38 or p-ERK expression) [30], Kim et al. showed that a microglial cell-line stimulated by several different TLR ligands in which LRRK2 was knocked down by shRNA exhibited decreased NF-κB activation by luciferase reported assay and decreased cytokine production. However, this was not associated with decreased NF-κB activation (already mentioned above) but was associated with increased binding of inhibitory p50, an NF-κB inhibitor, to its DNA binding site [36], leading to the conclusion that LRRK2 enhances NF-κB activation by somehow decreasing inhibitory p50 (or p50 homodimer) binding to the NF-κB promoter.
Subsequent studies expanded on this theme by showing that while LPS-stimulated primary microglia from LRRK2 KO mice exhibit no significant changes in their capacity to generate NF-κB components (including p65), they do exhibit increased expression of inhibitory phosphorylated p50; in addition, such inhibition correlated with the finding that α-synuclein fibril-stimulated microglial cells produce decreased amounts of IL-1β [35], indicating that, at least in microglial cells, deletion of LRRK2 and its associated inhibitory effect on NF-κB activation, leads to a decreased cytokine response.
Whereas these studies (and additional studies discussed below) suggest that LRRK2 has positive effect on NF-κB activation by preventing a negative effect (p-p50 generation), studies with LRRK2 inhibitors or with cells with increased LRRK2 expression or function go further by positing that LRRK2 has an unalloyed positive effect on the mechanisms of NF-κB/MAPK activation. Thus, Li et al. found that human B cells exposed to a LRRK2 GTPase and kinase inhibitor exhibited decreased pIκBα and pMAPK expression [31] and Takagawa et al. showed that ZymD-stimulated BMDCs from mice bearing a BAC-expressing LRRK2 transgene exhibited increased p-IκBα and p65 expression, as well as increased p-p38 and p-ERK expression [29]. Similarly, Hongge et al. demonstrated that IL-1β-stimulated human vascular endothelial cells (HUVACS) exhibited increased expression of nuclear p65, increased IκBα phosphorylation and increased NF-κB activation when transfected with an LRRK2-expressing plasmid, especially if the LRRK2 plasmid bears a G2019S mutation [38]. These studies, taken together, suggest that the enhancing effect of LRRK2 is upstream of NF-κB activation.
In reviewing the findings relating to LRRK2 regulation of cytokine responses, it becomes apparent that the inconsistency of the data resides mainly, if not exclusively, in studies of Lrrk2 KO peripheral cells, which exhibit either increased cytokine responses or responses no greater than WT peripheral cells; as already noted, this is contrary to the expectation that these cells would exhibit decreased responses that correspond to the decreased responses of cells exposed to LRRK2 kinase inhibitors. One explanation of this discrepancy is that Lrrk2 KO cells do not truly display (lack of) LRRK2 regulatory activity because mice with loss of Lrrk2 cannot survive without development of compensating mechanisms that mimic normal LRRK2 regulatory activity. In contrast, cells exposed to an LRRK2 inhibitor do display lack of LRRK2 regulatory activity because the cellular response to inhibitor is necessarily evaluated before compensation can be brought about. This explanation of the lack of decreased responses of Lrrk2 KO cells accords with the fact that studies with microglial cells uniformly suggest that LRRK2 kinase function has a positive pro-inflammatory effect because of the simple fact that studies of microglial cells have not been with cells from KO mice. Finally, it is important to note that the proposed compensation of LRRK2 function in KO cells may be limited to LRRK2 immunoregulatory function so that other LRRK2 functions in KO cells may be truly absent.
MECHANISMS OF LRRK2 EFFECTS ON NF-κB ACTIVATION
Accepting now the concept that LRRK2 has a positive effect on pro-inflammatory cytokine and chemokine synthesis, the question arises as to the mechanism of this effect. Russo et al. addressed this question initially with studies of LRRK2 KO cells (as described above) and later with cells exposed to an LRRK2 inhibitor, the latter providing more convincing data because of the aforementioned concerns about cells from LRRK2 KO mice [39]. In these studies, the authors again showed that lack of LRRK2 kinase function (due to presence of inhibitor) was associated with increased p-p50 expression, an NF-κB inhibitor. In further studies they attributed this to the fact that: 1) LRRK2 interacts with and phosphorylates PKA-RIIb (a PKA subunit that regulates PKA activity) and 2) LRRK2, in the presence of phosphorylated PKA-Rllb, activates phosphodiesterase E4 (PDE4), a factor that then degrades cAMP, thereby decreasing the concentration of a factor necessary for PKA catalytic function and ability to phosphorylate p50. The end result is that LRRK2, by its phosphorylation of PKA, facilitates NF-κB activation via decreased p50 phosphorylation and binding to the NF-κB promoter; consequently, increases in LRRK2 kinase function via mutation increases such inhibition and LRRK2 NF-κB-mediated pro-inflammatory function whereas decreases in LRRK2 kinase function via exposure to inhibitors has the opposite effect. Russo et al. emphasize the significance of the above findings by showing that stimulation of primary microglia from mice bearing a Lrrk2 G2019S knock-in mutation with α-synuclein fibrils causes increased Lrrk2 phosphorylation compared to microglia from WT mice and this is accompanied by greater IL-1β secretion by the knock-in cells.
This mechanistic explanation of LRRK2 enhancing function, while plausible, invites some skepticism because of its complexity and the lack of elucidation of certain key steps such as how LRRK2 regulates PDE4 and cAMP. In addition, it does not explain how LRRK2 enhances MAPK inflammatory function which is unrelated to p50. Finally, and perhaps most importantly, it is inconsistent with the fact that changes in LRRK2 function result in either positive or negative changes in the interactive function of the various adaptor proteins involved in NF-κB activation that are possible targets of LRRK2 kinase function. Thus, even if p50/p50 binding to NF-kB is, in fact, a feature of LRRK2 dysfunction, this could be a secondary effect to lack of primary NF-κB activation and p65/p50 formation that ordinarily blocks p50/p50 binding.
LRRK2 REGULATION OF THE INFLAMMASOME
The discussion of the effect of LRRK2 function on inflammation above has been mainly oriented to its positive effects on pro-inflammatory cytokine production arising from NF-κB/MAPK activation. Relatively recently, however, another LRRK2 mechanism of positive regulation has come into view, namely its role in the activation of the NLRC4 inflammasome. The key finding here was the observation by Liu et al., that NLRC4 activation by bacterial components such as flagellin or PrgJ expressed by Salmonella organisms is greatly reduced in the absence of LRRK2 (or in the presence of a LRRK2 inhibitor) and is increased in the presence of LRRK2 expressing the G2019S mutation [40]. Mechanistically, LRRK2 has this effect because it is the major kinase involved in the phosphorylation of NLRC4 at Ser533, an activation step necessary for NLRC4 interaction with ASC and caspase-1 to form the NLRC4 inflammasome capable of generating and secreting mature IL-1β and IL-18. Thus, in the absence of LRRK2, peritoneal macrophages mount curtailed IL-1β responses to Salmonella organisms and mice are more susceptible to Salmonella infection; in addition, LRRK2 G2019S transgenic mice mount more robust responses to Salmonella infection.
LRRK REGULATION OF NFAT TRANSLOCATION
The above discussion of the relation of LRRK2 to inflammation views LRRK2 function as having a positive influence on the various mechanisms that underlie inflammatory events, i.e., one that supports and/or enhances these mechanisms. There is, however, a body of work supporting the idea that LRRK2 has an inhibitory effect on pro-inflammatory mechanisms and this is, in fact, its dominant role.
Evidence of this inhibitory effect was first put forward by Liu et al. with data showing that LRRK2 is a necessary component of a suite of proteins, termed NRON, that together sequester NFAT1 in the cytoplasm of the cell and thus prevent translocation of NFAT1 into the nucleus where it has the potential to act as a cytokine transcription factor for a number of cytokines, especially IL-2 [17]. Among the data supporting this view of LRRK2 function were those from studies showing that over-expression of LRRK2 in LRRK2-transfected HEK293 cells suppresses NFAT activation in a luciferase assay and decreased nuclear expression of NFAT; in addition, such over-expression increased the concentration of NRON components and their association with NFAT. Finally, in functional studies, Dectin-1 ligand stimulated BMDMs from LRRK2 KO mice produced increased amounts of IL-12p40 and IL-6 than BMDMs from WT mice and, accordingly, Lrrk2 KO mice exhibited more severe DSS-induced colitis than WT mice. In this scenario, LRRK2 has a necessarily negative effect on cytokine formation and only when the cell manifests LRRK2 deficiency does it exhibit increased cytokine production. The authors therefore use this explanation of LRRK2 cytokine-related function to infer that an LRRK2 risk polymorphism that confers decreased LRRK2 survival and thus decreased LRRK2 concentration causes CD because it results in diminished NRON function and increased nuclear translocation of NFAT1 and NFAT-induced cytokine activity.
In critical evaluation of these findings, it is important to note that the authors found that only zymosan stimulation among a wide range of kinds of innate stimulation, including stimulation by various TLR ligands, led to increased cytokine responses in cells from Lrrk2 KO mice. This finding differed from that in several subsequent studies by the fact that in the latter studies increased responses were not found with any innate stimulus including zymosan. In any case, the fact that cells from Lrrk2 KO mice do not exhibit increased responses to most if not all innate stimuli does not comport with presumed increased NFAT translocation since the latter should exert a positive “piggy-back” effect that endows all innate stimuli with the ability to elicit an enhanced response. This logical inconsistency in the data is emphasized in a later study by Liu with Yan [41], in which it was shown that stimulation of cells with Lrrk2 deletion led to substantially decreased cytokine responses to stimulation by NOD1 and NOD2 ligands due to decreased down-stream RIPK2 phosphorylation; thus, the authors concluded that LRRK2 has a enhancing effect on cytokine production in spite presumed decreased NFAT translocation.
Subsequent studies of LRRK2 in relation to NFAT and its nuclear translocation have provided additional insight into the importance of this phenomenon. In one such study, NFAT translocation and IL-2 production were evaluated in a dendritic cell line expressing a luciferase NFAT reporter that enabled assay of NFAT activation [42]. The authors found that following stimulation of these indicator DCs with Aspergillis swollen coccidia (termed A-sw), i.e., fungal particles that activate cells via Dectin-1 and induce non-canonical autophagy, the cells express reduced amounts of LRRK2 presumably due to autophagic degradation of LRRK2. In addition, the indicator DCs stimulated in this way exhibited increased NFAT activation/translocation and IL-2 production compared to unstimulated cells and such activation/translocation was ablated by exposure to an autophagy inhibitor. In further studies, in this case of Lrrk2 KO cells stimulated with A-sw, the authors found that LRRK2 deficiency led to increased IL-2 secretion but not IL-12 and IL-23 secretion; this could also be attributed to NFAT activation/translocation because in a parallel study of cells in which LRRK2 was knocked down by shRNA, A-sw stimulation did lead to effects on NFAT. Whereas this study provides support for negative LRRK2 regulation of NFAT activation/translocation, its conclusions concerning the consequence of this LRRK2 effect is clouded by the fact that the studies were conducted with a stimulus that induces autophagy, a process that can itself cause increased cytokine secretion independent of LRRK2 [43]. More importantly, the negative regulation did not affect cytokines most implicated in inflammatory disease as implied by Liu et al. [17].
In the most recent and comprehensive study of the relation of LRRK2 to NFAT translocation, Panagiotakopoulos et al. investigated the function of iPSC-derived microglia and dopaminergic neurons subjected to IFN-γ stimulation based on the fact that interferon responses have been shown to contribute to brain aging and neurodegeneration [44]. They found that ionomycin-induced translocation was somewhat reduced in cells with an Lrrk2 mutation and increased in KO cells as compared to control cells; furthermore, translocation was reduced in both control cells and mutated cells by the presence of IFN-γ and this reduction was LRRK2-dependent since it was not seen in Lrrk2 KO cells. Perhaps the most important of several possible mechanisms explored in this study to explain this effect became evident from reports of LRRK2 effects on Ca+ mobilization from the endoplasmic reticulum (ER), the latter an indispensable initiator of NFAT translocation. Here they found that thapsigargen-induced release of Ca+ from the ER was impaired in mutated cells due to reduced ER storage of Ca+; in addition, they found that induced Ca+ release was diminished in control cells cultured with IFN-γ (presumably due to the effect of increased LRRK2 levels in IFN-γ-stimulated cells) and there was no reduction of Ca+ release in mutated cells possibly due to already depleted Ca+ stores. These findings thus led to the conclusion that LRRK2 does indeed cause decreased NFAT translocation, but this effect is mainly due to an unexplained effect on Ca+ storage in the ER.
In further studies, the LRRK2 regulation of NFAT translocation and NFAT activity was shown to be a possible mechanism of PD in that exposure of neuronal cells to an inhibitor of calcineurin-mediated NFAT activation or to IFN-γ was shown to result in neuronal structural abnormalities (such as neurite shortening) that was reversed with LRRK2 inhibitors. However, the effect of decreased NFAT translocation on cytokine generation was in this study equivocal. Thus, assessment of cytokine synthesis by human LPS-stimulated iPSC-derived microglia with an Lrrk2 mutation exhibited a mixed picture characterized by increased IL-1β/IL-12p70/MIP1β production and decreased IL-6/TNF-α/IL-8 production; and, perhaps more significantly, cells with LRRK2 deficiency exhibited only minor differences from control cells. On the basis of these data, the most reasonable conclusion is that whereas NFAT translocation may affect important neuronal or microglial cell signaling functions it has, at best, only a partial effect on cytokine production.
The observation that cells with decreased LRRK2 expression exhibit increased translocation of NFAT into the nucleus or that cells with increased LRRK2 kinase activity associated with LRRK2 mutations exhibit decreased translocation of NFAT made in the above studies leave one with little reason to doubt the validity of the phenomenon. The question remains: what is its significance with regard to LRRK2 effect on inflammation? If one assumes it implies that LRRK2 has mainly a negative effect on pro-inflammatory cytokine formation then it stands in opposition to the weight of evidence derived from the many studies reviewed above that point to the conclusion that LRRK2 enhances cytokine responses (knock-down studies) and such enhancement is augmented by both increases in LRRK2 levels (over-expression studies) and increased kinase function (mutational studies). Thus, even if LRRK2 has a negative effect on NFAT-mediated cytokine synthesis, the negative effect seems to be over-ridden by its positive effect on other pro-inflammatory mechanisms (such as NF-κB activation). This latter conclusion conforms to the fact that in contrast to the initial findings of Liu et al. [17], subsequent studies, especially that of Panagiotakopoulou et al. [44] showed that LPS-stimulated human microglial cells bearing the G2019S mutation that exhibit reduced NFAT translocation nevertheless produce increased amounts of important pro-inflammatory cytokines (IL-1β and IL-12p70) and LRRK2-deficient cells produce normal amounts of a wide range of cytokines.
RELATION OF LRRK2 ENHANCEMENT OF PRO-INFLAMMATORY FUNCTION TO PD (AND IBD)
Assessment of the kinase function of LRRK2 occurring in patients with PD bearing mutations in the kinase or GTP-ase domains of the molecule have revealed that the mutations cause increased kinase function [7]. On the other hand, polymorphisms associated with increased risk for development of PD or IBD (some shared between the two diseases as indicated above) occur at both coding and non-coding LRRK2 sites, not necessarily in kinase domains and have not always been assessed with respect to their effect on kinase function. Nevertheless, these genetic changes can reasonably be expected (at least in the majority of cases) to cause increased kinase function since they have been associated with increased LRRK2 expression and the latter can cause increased LRRK2 kinase function on the basis of a mass effect [29]. By the same logic, even cells bearing LRRK2 in the absence of a mutation or polymorphism could be expected to manifest increased LRRK2 kinase function when present in an inflammatory milieu because exposure to IFN-γ or to a TLR ligand (e.g., LPS) is known to increase LRRK2 levels. On the assumption that increased LRRK2 kinase activity occurring by one mechanism or another leads to enhanced pro-inflammatory activity, one can confidently assume that, for the most part, genetic abnormalities or inflammation itself leads to enhanced inflammatory activity on the basis of increased LRRK2 kinase function. Evidence that this is in fact true comes from extensive data, reviewed above, showing that cells expressing LRRK2 bearing mutations or, at least in one case an IBD-associated polymorphism, exhibit enhanced pro-inflammatory cytokine formation and NF-κB activity and that inhibitors of kinase activity abrogate such enhancement.
This chain of reasoning leads to the conclusion that abnormalities in LRRK2 function resulting in disease rely, at least in part, on the increased pro-inflammatory effect that is caused by such abnormalities. In the case of IBD this conclusion is obvious because the disease is widely understood as a poorly regulated inflammatory response to antigens/mitogens in the intestinal milieu. In the case of PD, however, this conclusion is not at all obvious and remains to be proven. What then are the main supports for the idea that the degenerative changes to dopaminergic neurons occurring in PD are in fact mainly or at least partly due to increased pro-inflammatory functions mediated by LRRK2 (Fig. 2)?
For the better part of two decades, studies have appeared showing that the kinase activity of LRRK2 is a necessary component of various aspects of neuronal function and markers of toxicity, but this by no means proves that this effect is necessarily channeled by LRRK2 pro-inflammatory immunological function. Likewise, ample evidence that neurodegeneration in patients or experimental animals is accompanied by increased cytokine levels that might be a result of increased LRRK2 kinase activity is not necessarily a mechanism of neurodegeneration. However, recent studies bearing on this issue are beginning to address this question. In one such study conducted by Kozina et al. [45] it was shown that mice bearing transgenes over-expressing mutant (R1441G) human LRRK2, but not wild-type human LRRK2, develop dopaminergic neuron loss upon systemic LPS administration and this was accompanied by increased levels of various inflammatory cytokines in the brain, notably cytokines previous associated with neurodegeneration, IFN-γ and IL-1α; in addition, there was evidence of microglial activation and expression of a gene activation pattern that had been induced by IFN-γ or TNF-α. However, while such LPS administration was accompanied by up-regulation of Lrrk2 expression in neuronal cells, no Lrrk2 expression was seen at any point in microglia. The source of the brain inflammatory cytokines was not brain infiltration by lymphocytes or myeloid cells since this was not seen upon careful search; rather the source was peripheral cells producing circulating cytokines which had crossed into the CNS. Thus, these studies suggest that LRRK2 mutations are associated with neurodegeneration because of their effect on the immune response at a site outside of the brain. More formal proof of this proposition came from subsequent studies by some of the same authors who showed first that Rag1–/– mice (lacking B/T cell expression) and reconstituted with R1441G/Lrrk2 BM cells develop neurodegeneration upon LPS administration, whereas double mutant Rag1–/–×R1441G/Lrrk2 chimeric mice reconstituted with WT BM cells do not, again indicating that the presence of the mutation in peripheral cells, not in the brain, is sufficient to cause neurodegeneration [46]. They then showed that R1441G/Lrrk2 transgenic mice express increased amounts of IL-6 upon LPS administration and neutralization of such IL-6 prevents neurodegeneration. Thus, in these studies LRRK2 is causing PD-like brain disease via peripheral cell elaboration of IL-6 alone. The authors called this finding a paradigm shift in our understanding of PD but important problems with their conclusion remain including the fact that these results imply that any inflammation causing chronic IL-6 hypersecretion should result in PD and this is manifestly untrue.
A very different picture of the role of LRRK2 in the causation of PD is based on recent studies by Xu et al. [5] that focus on the role of α-synuclein in PD. Here it was shown that α-synuclein fibrils (but not monomers) induce LRRK2 expression and downstream Rab10 phosphorylation in human and mouse monocyte-derived macrophages and that these processes depend on LRRK2 kinase activity as it is completely inhibited by a LRRK2 kinase inhibitor; however, such induction/phosphorylation is not seen in monocyte-derived microglial cells. The authors noted that accumulations of antigen-presenting cells can be observed in the human brain adjacent to α-synuclein inclusions; to determine the phenotype of such cells and their LRRK2 expression they isolated and characterized cells present in brain sections obtained from Lrrk2-transgenic mice (i.e., mice that facilitated LRRK2 phenotyping) and Lrrk2 KO mice following injection of α-synuclein. They found a much greater accumulation of monocytic cells in brain sections of the transgenic mice than in the KO mice and the LRRK2-positive cells in the transgenic mice consisted of cells bearing markers of monocytes derived from the circulation; cells bearing markers of microglia accumulated in the injected areas as well, but these cells were LRRK2-negative. In further studies the authors tied the monocyte migration into the brain implied by these studies to LRRK2 kinase activity by showing that LRRK2-positive monocytic cell accumulation in the brain after α-synuclein injection is enhanced in mice bearing Lrrk2 PD mutations and is inhibited by administration of a LRRK2-kinase inhibitor. In explanation of such activity the authors refer to their previous work showing that LRRK2, via its effect on Rab10 activation, facilitates endosomal loading of CCL5 by macropinosomes and thereby enhances CCL5 chemotactic function [47]. They therefore put forward a hypothesis holding that α-synuclein fibrils generated in the brain (or elsewhere) induce microglia or astrocytes to produce chemokines that attract circulating monocytes bearing chemokine receptors into the brain (Fig. 2). Although the authors do not make this point, increased expression of LRRK2 (and Rab10) presumably occurs outside of the brain to explain the fact that they require LRRK2-induced chemokine receptor expression to gain brain entry.
The view of LRRK2 function in PD inherent in the study of Xu et al. [5] seems to explain PD pathogenesis better than the view of such function put forward by the studies of Kozina et al. [45] because it incorporates one of the defining features of PD neurodegeneration, namely, the presence and function of α-synuclein in brain tissue. In this respect it comports with a previous study utilizing a mouse model in which α-synuclein is over-expressed that showed that α-synuclein mediates robust chemokine-mediated entry of peripheral monocytes into the brain and that such entry is necessary for degeneration of dopaminergic neurons [48]. Nevertheless, in focusing exclusively on the role of LRRK2 on chemotaxis it overlooks the possible pathogenic effect of LRRK2-bearing intra-cerebral cells on adjacent neurons after they enter the brain (Fig. 2). In addition, it provides little room for the possibility, emphasized by Kozina et al. [45] that cells can undergo LRRK2-mediated enhancement of pro-inflammatory function following encounter with α-synuclein or other stimuli in the periphery.
If indeed PD neurodegeneration depends on entry of LRRK2-positive macrophages into the brain as implied by the Xu et al. [5], it is reasonable to postulate that the cells bearing LRRK2 mutations or polymorphisms that cause increased LRRK2-mediated pro-inflammatory function described above play a key role in PD pathogenesis. One possible stimulus of such cells is α-synuclein itself, which in this case would confer on the latter the role of macrophage-cytokine inducer as well as microglial chemokine inducer. In studies supporting such α-synuclein-dependent pro-inflammatory function Russo et al. [35] showed that exposure of primary microglia (which in this study did express LRRK2) to α-synuclein causes LRRK2 phosphorylation to a greater extent than does LPS and induces activation of a suite of genes that overlaps with but is distinct from that induced by LPS. In addition, α-synuclein exposure induces microglial production of superoxide dismutase (SOD2) and IL-1β that is dependent on LRRK2 function since such production is reduced in LRRK2 KO microglia. Evidence that α-synuclein has a similar immunostimulatory effect on macrophages entering the brain comes from studies of a mouse model of neurodegeneration caused by injection of adenovirus-expressed α-synuclein in which it was demonstrated that the neurodegeneration is dependent on entry of circulating monocytes into brain tissue via chemokine receptor/ligand interactions [48]. Importantly, the infiltrating monocytes localized to sites of high α-synuclein expression and exhibited evidence of activation, MHC-class II expression. Thus, these studies complement those of Xu et al. [5] mentioned above in that they show that α-synuclein is not only necessary to enable chemotactic entry of macrophages into the brain, it is also necessary for the induction of macrophage-mediated neurodegeneration. Taken together, these data make the case that LRRK2-related pro-inflammatory function does, in fact, contribute to PD neurodegeneration, a concept previously supported by more general evidence of immune dysfunction in PD derived from experimental models, as well as clinical biomarker and imaging studies of human patients (reviewed by Tansey and colleagues [49, 50]).
LRRK2 ACTIVITY IN THE GUT AND ITS RELATION TO PD
Whereas there is substantial evidence, extensively described above, showing that LRRK2 is an important positive regulator of immune function and thus plays a role in an inflammatory response contributing to PD within the brain, there is a dearth of information of how LRRK2 immune function in the GI tract impacts PD. Studies conducted by Takagawa et al., building on the fact that PD and IBD (particularly CD) are associated with shared LRRK2 polymorphisms, offer some data relating to this issue [29]. These authors showed that cells bearing such shared polymorphisms express increased amounts of LRRK2 and exhibit increased Dectin-1-induced TNF-α responses, indicating that increased expression of LRRK2 alone leads to enhanced immune responses. In addition, they showed with studies of Lrrk2 transgenic mice that increased Lrrk2 expression causes increased NF-κB activation and increased experimental gut inflammation (more severe DSS-colitis) that is, in fact, ameliorated by a LRRK2 kinase inhibitor. These data thus implied that LRRK2 polymorphisms and resultant increased LRRK2 pro-inflammatory activity can be a genetic factor contributing to the inflammation of CD. More relevant to the present discussion, these data also suggested that individuals bearing CD/PD shared polymorphisms (Fig. 2) can be a “breeding ground” for activated LRRK2-bearing macrophages with the capacity to subsequently migrate to the brain and cause or exacerbate PD as suggested by the studies of Xu et al. [5] as well as by Kozina et al. [45, 46] as previously discussed, which may, at least in part, explain the higher than expected rate of IBD-PD comorbidity [19–22].
If, as implied above, the gut serves as a site for the enhanced development of cells bearing increased and/or activated LRRK2 in individuals with shared CD/PD risk polymorphisms, it is of interest to define the conditions in gastrointestinal tissues favoring such development. The inflammation of CD is one possible such condition since the inflamed mucosal tissue is replete with IFN-γ, a stimulator of LRRK2 expression. In this formulation, LRRK2 is both a driver of CD inflammation and is, in turn, activated by CD inflammation in a type of forward feedback. The concept that inflammation is a cause of increased/activated LRRK2-bearing intestinal cells also applies to PD inasmuch as PD is accompanied by an array of gastrointestinal tract symptoms that occur during a prodromal phase or during the disease course that could be interpreted as sub-clinical CD-like inflammation (reviewed in [51, 52]). This clinical evidence of gut inflammation in PD is accompanied by lab-based evidence showing that colonic biopsies from PD patients exhibit increased mRNA expression of pro-inflammatory cytokines, including TNF-α, IFN-γ, IL-6, and IL-1β [53], as well as elevated levels of IL-1α, 1β, CXCL8, and C-reactive protein in the stool [54]. Furthermore, several recent studies have reported that PD vs. control patients exhibit high levels of fecal calprotectin, a protein released into the gut lumen predominantly by neutrophils infiltrating the mucosa during an inflammatory response [55–57]. As in the case of CD, this evidence of gut inflammation in PD can be both a cause and effect of enhanced LRRK2 expression.
Another possible factor to consider in relation to LRRK2 activation in the gut of PD patients is the presence of increase amounts of α-synuclein at this site (see further discussion below). The significance of this arises from recent studies by Alam et al. showing that α-synuclein has a physiological role in mucosal immune responses in healthy individuals [58]. In these studies, it was shown first that α-synuclein is localized and secreted in enteric nerve tissue (ENT), is increased in gastrointestinal infection (such as Norovirus infection) and has a protective role in that α-synuclein is a potent chemotactic factor that draws neutrophils and other cells into the infected gut wall. In further work by the same authors, it was shown that α-synuclein induces maturation of macrophages and dendritic cells and thereby acts as an adjuvant to cause increased response to various immune stimuli as well as to α-synuclein itself. In part, such adjuvanticity was due to the capacity of α-synuclein to act as a TLR4 ligand and to thus initiate MyD88 and TRIF signaling. Finally, these authors marshaled data showing that α-synuclein emanating from nerve tissue is secreted into the peritoneal cavity and thus induces enhanced protective response to intra-peritoneal infection. These studies highlight α-synuclein as an innate immunological stimulus and thus opens the door to the possibility that this factor may be playing a role in the activation of LRRK2-bearing cells and LRRK2 itself in PD patients.
The above studies of α-synuclein complement previous studies of mucosal α-synuclein in PD that revolve around the Braak hypothesis (or Gut-First hypothesis) that advances the idea that PD neural degeneration is initiated in the gastrointestinal tract. This hypothesis draws upon the fact that α-synuclein concentrations are increased in the ENT of a substantial number of patients with PD and has been shown to be secreted by neural cells (microglia). Furthermore, there is evidence showing that misfolded α-synuclein accumulates first in the ENS and then travels caudad to the dopamine-producing neurons where it initiates definitive PD development (reviewed in Borghammer et al. [59]). Whether or not this hypothesis is true, the fact that α-synuclein is present in the mucosal tissues and has immunological function at this site suggests that this factor can be a contributor to enhanced activation of LRRK2 in the gut of PD patients. A final and related point is that the accumulation of α-synuclein in the ENT as well as the presence of LRRK2 in this tissue introduces the possibility that enteric nerve function is disrupted by α-synucleinopathy and this contributes to the GI symptoms of PD recounted above.
MUCOSAL IMMUNOLOGICAL MARKERS IN PD
Quite aside from studies of LRRK2 and LRRK2-bearing cells in PD pathogenesis discussed above are the many important studies of LRRK2 and related factors as mucosal immunological markers that are more accessible and perhaps earlier indications of the presence of PD.
Intestinal inflammatory biomarkers in PD
As noted above there are several studies documenting that many patients with PD display elevations of various cytokines, chemokines, and immune cell populations in mucosal tissues. Coupled with this there was the finding in one report that the fecal calprotectin levels in the stool, a reliable indication of the presence of gut inflammation widely used in the differential diagnostic and monitoring of IBD, was found in 43% of PD patients and in none of the controls. However, since in this study there was no correlation between fecal calprotectin and PD duration [55], these data suggested that fecal calprotectin is a marker of disease initiation rather than disease progression. Two other fecal markers of intestinal inflammation measuring in this case epithelial layer permeability and reduced expression of tight junction proteins, alpha-1-antitrypsin and zonulin, were significantly elevated in PD patients compared to age-matched controls in some [57] but not other reports [55]. Of note, in related studies, stool biomarkers of inflammation correlated with the presence of α-synuclein aggregates in a mouse model of PD [60] and in human PD samples [61], as well as PD onset and symptom severity [56]. These elevations in inflammatory markers, as alluded to above, could be an effect of LRRK2-related immune enhancement and α-synucleinopathy; in addition, as discussed below, it could reflect pro-inflammatory effects of an altered gut microbiota (reviewed in Chen & Lin [62]).
LRRK2 activity as a marker of PD in the gut
The possible role of the gut as an incubator of LRRK2-expressing cells with pathologic potential in PD, as discussed above, is supported by a study in which it was demonstrated that there is increased LRRK2 expression in colonic biopsies from patients with PD compared to age- and gender-matched control [63]. In this study, the colonic LRRK2 level in PD patients correlated with disease severity in both motor and cognitive function and the expression levels were higher in those carrying LRRK2 risk or pathogenic polymorphisms rather than wild type LRRK2 polymorphisms. Furthermore, PD patients in the prodromal phase had increased colonic LRRK2 expression compared to controls and, as the disease progressed, the expression of LRRK2 in colonic tissues increased, suggesting that colonic LRRK2 expression may serve as a surrogate gut-oriented marker of PD [63]. It should be noted, however, that in another study it was found that both LRRK2 expression and Rab10 phosphorylated at Thr73 kinase activity were significantly reduced in the colon of PD patients; this contrasted with findings in the CD gut, in which LRRK2 was upregulated in inflamed areas [64]. These results correlated with animal experiments wherein LRRK2 expression was significantly lower in the colons of aged α-synuclein transgenic mice bearing enteric α-synuclein deposits when compared to young mice not bearing such deposits and with studies of DSS-induced colitis in WT mice, in which the presence of induced inflammation was accompanied by increased LRRK2 levels [64]. These negative observations were obtained in studies of patients without evidence of gut inflammation and may reflect the possibility that gut-derived cells bearing LRRK2 may play a role in some but not all patients with PD and that the former consist of those bearing LRRK2 polymorphisms. Thus, the establishment of a correlation between prodromal disease progression and LRRK2 expression and kinase activity, as indicated by colonic LRRK2 activity, could be an early indicator of PD despite disease heterogeneity.
A totally different way that LRRK2 levels and activity may give rise to gut PD markers is related to LRRK2 activity in Paneth cells, i.e., secretory epithelial cells that reside at the bottom of small intestinal crypts that provide mucosal defense by secreting antimicrobial substances such as lysozyme and α-defensins into the intestinal lumen [65]. Previous reports have linked abnormal Paneth cell function (as shown in Fig. 2) to CD pathogenesis [66], presumably resulting from the fact that such cells affect the composition of the microbiome and/or affect mucosal barrier function. In addition, it has been reported that LRRK2 along with NOD2, a bacterial sensor with polymorphisms linked to both CD [67, 68] and PD [69], is involved in sorting of lysozyme in the Paneth cells [70]; thus, defects in either LRRK2 or NOD2 function result in lysosomal degradation of lysozyme rather than secretion of intact lysozyme. It has also been shown that LRRK2 M2397T, a CD susceptibility allele identified in European ancestry populations, is associated with an abnormal Paneth cell phenotype in a Japanese cohort [71]. As noted above, this LRRK2 polymorphism is not associated with increased risk of PD; however, other, as yet unidentified LRRK2 polymorphisms affecting Paneth cell function may have this property. As such, future studies are necessary to characterize transcriptomic and signaling signatures of Paneth cells in individuals with high risk for PD and PD patients with different genetic backgrounds to explore Paneth cell activity as an early biomarker, as well as therapeutic target, for PD development and progression.
α-synuclein in the gut as a marker in PD
In our current understanding of PD pathogenesis, the role of α-synuclein as an instigator of gut inflammation [72] and thus enhanced (and pathological) pro-inflammatory LRRK2 function suggested above is over-shadowed by its role as an effector of neuronal degeneration in brain tissue (Fig. 2). In this regard, there is substantial experimental evidence pointing towards the fact that in this respect LRRK2 is playing the supportive role in the effects of α-synuclein in that it accelerates the progression of α-synuclein-mediated neurodegeneration in the brain (reviewed in O’Hara et al. [73]). One aspect of such support is that LRRK2 is present within microglia (and perhaps within neurons as well) and that LRRK2 has been shown to retard intracellular clearance of α-synuclein by its effects on endocytic activity. Thus, LRRK2 within neural tissue promotes neuronal α-synuclein accumulation and the development and/or progression of PD in a manner that has little to do with its pro-inflammatory properties.
The above relation of α-synuclein to LRRK2 notwithstanding, the possibility that α-synuclein expression in the gut of PD patients also can serve as a marker of the presence of PD is still viable, albeit still unproven. Thus, whereas several studies have suggested that α-synuclein and Lewy bodies detected in the colonic biopsy samples are specific for PD [74, 75], others have suggested that it is a common finding not specific to PD [76, 77] (also reviewed in Fricova et al. [78]). Well-powered prospective studies with standardized sample location and mucosal depth, coupled with the use of multiple antibodies in patients with prodromal PD, IBD, and PD, stratified by LRRK2 mutation carrier status, might improve our understanding of the prognostic value of immunocytochemical detection of α-synuclein in the gut in patients with PD.
The role of the microbiome in PD pathogenesis
Given the possibility that gut inflammation may contribute to PD pathogenesis via its effect on LRRK2 expressing cells as discussed above, it is important to mention that the intestinal homeostasis (i.e., lack of inflammation) depends to some extent on proper interactions between the mucosal immune system and commensal bacteria. In particular, abnormalities in the gut microbiome (dysbiosis) have been thought to contribute to IBD inflammation by causing deficits in bacterial subpopulations that induce inflammation-suppressing regulatory cells (reviewed in Glassner et al. [79] and Qiu et al. [65]). Intestinal dysbiosis contributing to gut inflammation has likewise been suspected to be present in patients with PD, inasmuch as the latter frequently display reductions in beneficial intestinal components such as short chain fatty acids that are produced by intestinal microbiota [56, 80, 81]. This finding correlates with several studies centered on preclinical PD models. Here it was shown that gut microbes can promote neuroinflammatory responses leading to increased motor disability and α-synuclein deposition in the brain. Moreover, germ-free α-synuclein-overexpressing mice were shown to present with less motor deficits, microglia activation, and α-synuclein pathology compared to control animals [82]. Remarkably, colonization of α-synuclein-overexpressing mice with microbiota from PD patients enhanced physical impairments compared to microbiota transplants from healthy human donors, further suggesting that the gut microbiome represents a risk factor for PD. Lastly, reduced LRRK2 concentrations has been shown to increase the overall bacterial richness and alter bacterial community structure, while also alleviating experimental colitis development and progression [83]. In aggregate, these studies profiling the gut microbiome in Lrrk2-mutant mice and in response to LRRK2 inhibition therapy could help better understand the interplay between LRRK2 activity and the gut microbiome, thereby leading to the development of novel sensitive markers and/or treatment targets for PD, especially as fecal microbiota transplantation is currently being explored as a potential therapy for PD [84].
The impact of intestinal dysbiosis on clinical features of PD is still poorly understood. Emerging studies demonstrate that, compared with the tremor-dominant PD subtype, the diversity of Enterobacteriaceae bacteria was higher in PD patients with postural instability and gait difficulty, with disease severity positively correlating with these bacteria’s relative abundance [85]. Also, reports suggest the role of certain bacteria in the appearance of PD-associated symptoms. For example, low abundance of B. fragilis and Bifidobacterium has been associated with worsening of motivation/initiative and hallucinations/delusions, respectively, using the Movement Disorder Society-United Parkinson’s Disease Rating Scale (MDS-UPDRS) scoring system [86], while the Clostridium coccoides and Lactobacillus gasseri species have reportedly differentiated between early and advanced stages of the disease [87] (also reviewed in Dutta et al. [88]). Whether LRRK2 polymorphisms contribute to the gut dysbiosis or modify the relationship between intestinal microbiome and PD manifestation remains to be determined.
CONCLUDING REMARKS
In this review we have focused on the capacity of LRRK2 to mediate pro-inflammatory activities and thus to contribute to neurodegeneration in the brain. In addition, in that LRRK2 pro-inflammatory function may depend on genetically determined factors predisposing PD patients to gut inflammation similar to those operative in CD, we have marshaled evidence of a gut-brain axis in PD (Fig. 2). Having developed these concepts, however, we do not seek to imply that LRRK2 only affects PD pathogenesis via its pro-inflammatory function. On the contrary, we are fully aware of the ample evidence that LRRK2 may also contribute to PD development via its various effects on vesicle transport, autophagy, cytoskeletal organization or mitochondrial function, any of which could interfere with normal neuronal function directly or indirectly by facilitating downstream α-synuclein aggregation.
To place the pro-inflammatory role of LRRK2 in PD into prospective it is reasonable to assume that this role may vary among individual patients or patient groups depending on differences in genetic factors and co-factors affecting LRRK2 pro-inflammatory function. At one end of the spectrum at which LRRK2 exerts a maximal enhancing pro-inflammatory effect, LRRK2 mutations/polymorphisms may cause neurodegeneration in the absence of α-synuclein aggregation and α-synucleinopathy; this could explain the existence of the appreciable percentage of patients bearing LRRK2 mutations that lack Lewy body pathology at autopsy [89]. This is not in conflict with, and could in fact explain, the clinical observation that PD associated with LRRK2 mutations is somewhat milder than idiopathic PD [90] since LRRK2-driven PD under these circumstances may induce neurodegeneration in the absence of other pathological mechanisms that, when operative, intensify disease. At the other end of the spectrum at which LRRK2 polymorphisms exert minimal enhancing pro-inflammatory effects, LRRK2 abnormalities may give rise to generally milder neurodegeneration; this would provide a second reason that LRRK2-driven PD is less severe than idiopathic PD. Importantly, the above proposed spectrum applies to gain-of-function LRRK2 genetic abnormalities that lead to neurodegeneration. However, as noted above, there are protective LRRK2 polymorphisms (located in the RocCOR domain of LRRK2) that have been shown to cause increased GTPase and Wnt activity [12, 14]. These polymorphisms may be protective because they cause decreased LRRK2 kinase activity or because they have positive effects that oppose neuron degeneration; however, this remains to be demonstrated.
With respect to the role of gut inflammation as an instigator of PD via its effect on intestinal cells bearing activated LRRK2 capable of migration to the brain as suggested here, it is important to emphasize that the GI symptoms and signs of inflammation associated with or even preceding PD need not be indicative of full-blown Crohn’s disease which in any case may be difficult to define as present since this disease is lacking a unique marker. The rather convincing evidence that a sub-group of PD patients have gut inflammation is sufficient basis of the gut activation/migration concept whether or not Crohn’s disease is present because it is safe to say that the inflammation is associated with the secretion of cytokines (such as IFN-γ) that cause LRRK2 activation.
In future studies of the gut-brain axis in PD it is critical to conduct a systematic comparative profiling of the LRRK2 expression and activity along the entire digestive tract and across the gastrointestinal wall, not only in animal models but also in human biopsy tissue samples from individuals with IBD, prodromal and established PD, and well-matched controls. Ideally, this would be coupled with comprehensive profiling of ENS, including enteric neurons and enteric glial cells using multi-omics approaches. In addition, as shown in recent analyses of colonic biopsies obtained during routine cancer screening such studies can be utilized to correlate burden of PD markers with neurological and gastrointestinal symptoms (reviewed in Corbillé et al. [91]). Among the more pressing goals of these biopsy-based studies is the need to determine if ENS pathology can be used to diagnose and monitor PD and if ENS is a valid therapeutic target in PD, as it is currently considered in certain gastrointestinal diseases [92]. Additionally, it is imperative to identify early signs of PD before disease onset, especially in IBD patients who are at a higher risk of PD. Finally, there is a need to determine whether calprotectin, alpha-1-antitrypsin and zonulin, or other intestinal inflammatory biomarkers (e.g., lactoferrin or lipocalin), can be used to stratify individuals at risk of PD, define PD subtypes and/or monitor the effect of interventions in PD.
ACKNOWLEDGMENTS
This work was supported by the Michael J Fox Foundation. I.P. is currently supported from the NINDS/NIH (P20NS123220).
CONFLICT OF INTEREST
The authors have no conflict of interest to report.
REFERENCES
[1] | Braak H , Del Tredici K , Rub U , de Vos RA , Jansen Steur EN , Braak E ((2003) ) Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol Aging 24: , 197–211. |
[2] | Braak H , Rub U , Gai WP , Del Tredici K ((2003) ) Idiopathic Parkinson’s disease: Possible routes by which vulnerable neuronal types may be subject to neuroinvasion by an unknown pathogen. J Neural Transm (Vienna) 110: , 517–536. |
[3] | Hawkes CH , Del Tredici K , Braak H ((2007) ) Parkinson’s disease: A dual-hit hypothesis. Neuropathol Appl Neurobiol 33: , 599–614. |
[4] | de Guilhem de Lataillade A , Caillaud M , Oullier T , Naveilhan P , Pellegrini C , Tolosa E , Neunlist M , Rolli-Derkinderen M , Gelpi E , Derkinderen P ((2023) ) LRRK2 expression in normal and pathologic human gut and in rodent enteric neural cell lines. J Neurochem 164: , 193–209. |
[5] | Xu E , Boddu R , Abdelmotilib HA , Sokratian A , Kelly K , Liu Z , Bryant N , Chandra S , Carlisle SM , Lefkowitz EJ , Harms AS , Benveniste EN , Yacoubian TA , Volpicelli-Daley LA , Standaert DG , West AB ((2022) ) Pathological alpha-synuclein recruits LRRK2 expressing pro-inflammatory monocytes to the brain. Mol Neurodegener 17: , 7. |
[6] | Lee H , James WS , Cowley SA ((2017) ) LRRK2 in peripheral and central nervous system innate immunity: Its link to Parkinson’s disease. Biochem Soc Trans 45: , 131–139. |
[7] | Steger M , Tonelli F , Ito G , Davies P , Trost M , Vetter M , Wachter S , Lorentzen E , Duddy G , Wilson S , Baptista MA , Fiske BK , Fell MJ , Morrow JA , Reith AD , Alessi DR , Mann M ((2016) ) Phosphoproteomics reveals that Parkinson’s disease kinase LRRK2 regulates a subset of Rab GTPases. Elife 5: , e12813/. |
[8] | Alessi DR , Sammler E ((2018) ) LRRK2 kinase in Parkinson’s disease. Science 360: , 36–37. |
[9] | de Lange KM , Moutsianas L , Lee JC , Lamb CA , Luo Y , Kennedy NA , Jostins L , Rice DL , Gutierrez-Achury J , Ji SG , Heap G , Nimmo ER , Edwards C , Henderson P , Mowat C , Sanderson J , Satsangi J , Simmons A , Wilson DC , Tremelling M , Hart A , Mathew CG , Newman WG , Parkes M , Lees CW , Uhlig H , Hawkey C , Prescott NJ , Ahmad T , Mansfield JC , Anderson CA , Barrett JC ((2017) ) Genome-wide association study implicates immune activation of multiple integrin genes in inflammatory bowel disease. Nat Genet 49: , 256–261. |
[10] | Barrett JC , Hansoul S , Nicolae DL , Cho JH , Duerr RH , Rioux JD , Brant SR , Silverberg MS , Taylor KD , Barmada MM , Bitton A , Dassopoulos T , Datta LW , Green T , Griffiths AM , Kistner EO , Murtha MT , Regueiro MD , Rotter JI , Schumm LP , Steinhart AH , Targan SR , Xavier RJ , Libioulle C , Sandor C , Lathrop M , Belaiche J , Dewit O , Gut I , Heath S , Laukens D , Mni M , Rutgeerts P , Van Gossum A , Zelenika D , Franchimont D , Hugot JP , de Vos M , Vermeire S , Louis E , Cardon LR , Anderson CA , Drummond H , Nimmo E , Ahmad T , Prescott NJ , Onnie CM , Fisher SA , Marchini J , Ghori J , Bumpstead S , Gwilliam R , Tremelling M , Deloukas P , Mansfield J , Jewell D , Satsangi J , Mathew CG , Parkes M , Georges M , Daly MJ ((2008) ) Genome-wide association defines more than 30 distinct susceptibility loci for Crohn’s disease. Nat Genet 40: , 955–962. |
[11] | Liu JZ , van Sommeren S , Huang H , Ng SC , Alberts R , Takahashi A , Ripke S , Lee JC , Jostins L , Shah T , Abedian S , Cheon JH , Cho J , Dayani NE , Franke L , Fuyuno Y , Hart A , Juyal RC , Juyal G , Kim WH , Morris AP , Poustchi H , Newman WG , Midha V , Orchard TR , Vahedi H , Sood A , Sung JY , Malekzadeh R , Westra HJ , Yamazaki K , Yang SK ; International Multiple Sclerosis Genetics Consortium; International IBD Genetics Consortium; Barrett JC , Alizadeh BZ , Parkes M , Bk T , Daly MJ , Kubo M , Anderson CA , Weersma RK ((2015) ) Association analyses identify 38 susceptibility loci for inflammatory bowel disease and highlight shared genetic risk across populations. Nat Genet 47: , 979–986. |
[12] | Hui KY , Fernandez-Hernandez H , Hu J , Schaffner A , Pankratz N , Hsu NY , Chuang LS , Carmi S , Villaverde N , Li X , Rivas M , Levine AP , Bao X , Labrias PR , Haritunians T , Ruane D , Gettler K , Chen E , Li D , Schiff ER , Pontikos N , Barzilai N , Brant SR , Bressman S , Cheifetz AS , Clark LN , Daly MJ , Desnick RJ , Duerr RH , Katz S , Lencz T , Myers RH , Ostrer H , Ozelius L , Payami H , Peter Y , Rioux JD , Segal AW , Scott WK , Silverberg MS , Vance JM , Ubarretxena-Belandia I , Foroud T , Atzmon G , Pe’er I , Ioannou Y , McGovern DPB , Yue Z , Schadt EE , Cho JH , Peter I ((2018) ) Functional variants in the LRRK2 gene confer shared effects on risk for Crohn’s disease and Parkinson’s disease. Sci Transl Med 10: , eaai7795. |
[13] | Rivas MA , Avila BE , Koskela J , Huang H , Stevens C , Pirinen M , Haritunians T , Neale BM , Kurki M , Ganna A , Graham D , Glaser B , Peter I , Atzmon G , Barzilai N , Levine AP , Schiff E , Pontikos N , Weisburd B , Lek M , Karczewski KJ , Bloom J , Minikel EV , Petersen BS , Beaugerie L , Seksik P , Cosnes J , Schreiber S , Bokemeyer B , Bethge J ; International IBD Genetics Consortium; NIDDK IBD Genetics Consortium; T2D-GENES Consortium; Heap G , Ahmad T , Plagnol V , Segal AW , Targan S , Turner D , Saavalainen P , Farkkila M , Kontula K , Palotie A , Brant SR , Duerr RH , Silverberg MS , Rioux JD , Weersma RK , Franke A , Jostins L , Anderson CA , Barrett JC , MacArthur DG , Jalas C , Sokol H , Xavier RJ , Pulver A , Cho JH , McGovern DPB , Daly MJ ((2018) ) Insights into the genetic epidemiology of Crohn’s and rare diseases in the Ashkenazi Jewish population. PLoS Genet 14: , e1007329. |
[14] | Nixon-Abell J , Berwick DC , Granno S , Spain VA , Blackstone C , Harvey K ((2016) ) Protective LRRK2 R1398H variant enhances GTPase and Wnt signaling activity. Front Mol Neurosci 9: , 18. |
[15] | Fava VM , Manry J , Cobat A , Orlova M , Van Thuc N , Ba NN , Thai VH , Abel L , Alcais A , Schurr E , Canadian Lrrk2 in Inflammation T ((2016) ) A missense LRRK2 variant is a risk factor for excessive inflammatoryresponses in leprosy. PLoS Negl Trop Dis 10: , e0004412. |
[16] | Zhang DF , Wang D , Li YY , Yao YG ((2020) ) Is there an antagonisticpleiotropic effect of a LRRK2 mutation on leprosy and Parkinson’sdisease? Proc Natl Acad Sci U S A 117: , 10122–10123. |
[17] | Liu Z , Lee J , Krummey S , Lu W , Cai H , Lenardo MJ ((2011) ) The kinase LRRK2 is a regulator of the transcription factor NFAT that modulates the severity of inflammatory bowel disease. Nat Immunol 12: , 1063–1070. |
[18] | Ikezu T , Koro L , Wolozin B , Farraye FA , Strongosky AJ , Wszolek ZK ((2020) ) Crohn’s and Parkinson’s disease-associated LRRK2 mutations alter type II interferon responses in human CD14(+) blood monocytes ex vivo. J Neuroimmune Pharmacol 15: , 794–800. |
[19] | Lin JC , Lin CS , Hsu CW , Lin CL , Kao CH ((2016) ) Association between Parkinson’s disease and inflammatory bowel disease: A nationwide Taiwanese retrospective cohort study. Inflamm Bowel Dis 22: , 1049–1055. |
[20] | Villumsen M , Aznar S , Pakkenberg B , Jess T , Brudek T ((2019) ) Inflammatory bowel disease increases the risk of Parkinson’s disease: A Danish nationwide cohort study 1977-2014. Gut 68: , 18–24. |
[21] | Weimers P , Halfvarson J , Sachs MC , Saunders-Pullman R , Ludvigsson JF , Peter I , Burisch J , Olen O ((2019) ) Inflammatory bowel disease and Parkinson’s disease: A nationwide Swedish cohort study. Inflamm Bowel Dis 25: , 111–123. |
[22] | Peter I , Dubinsky M , Bressman S , Park A , Lu C , Chen N , Wang A ((2018) ) Anti-tumor necrosis factor therapy and incidence of Parkinson disease among patients with inflammatory bowel disease. JAMA Neurol 75: , 939–946. |
[23] | Camacho-Soto A , Gross A , Searles Nielsen S , Dey N , Racette BA ((2018) ) Inflammatory bowel disease and risk of Parkinson’s disease in Medicare beneficiaries. Parkinsonism Relat Disord 50: , 23–28. |
[24] | Coates MD , Ba DM , Liu G , Dalessio S , Leslie DL , Huang X ((2022) ) Revisiting the association between inflammatory bowel disease and Parkinson’s disease. Inflamm Bowel Dis 28: , 850–854. |
[25] | Zhu Y , Yuan M , Liu Y , Yang F , Chen WZ , Xu ZZ , Xiang ZB , Xu RS ((2022) ) Association between inflammatory bowel diseases and Parkinson’s disease: Systematic review and meta-analysis. Neural Regen Res 17: , 344–353. |
[26] | Ahmadi Rastegar D , Hughes LP , Perera G , Keshiya S , Zhong S , Gao J , Halliday GM , Schule B , Dzamko N ((2022) ) Effect of LRRK2 protein and activity on stimulated cytokines in human monocytes and macrophages. NPJ Parkinsons Dis 8: , 34. |
[27] | Wandu WS , Tan C , Ogbeifun O , Vistica BP , Shi G , Hinshaw SJ , Xie C , Chen X , Klinman DM , Cai H , Gery I ((2015) ) Leucine-rich repeat kinase 2 (Lrrk2) deficiency diminishes the development of experimental autoimmune uveitis (EAU) and the adaptive immune response. PLoS One 10: , e0128906. |
[28] | Nazish I , Arber C , Piers TM , Warner TT , Hardy JA , Lewis PA , Pocock JM , Bandopadhyay R ((2021) ) Abrogation of LRRK2 dependent Rab10 phosphorylation with TLR4 activation and alterations in evoked cytokine release in immune cells. Neurochem Int 147: , 105070. |
[29] | Takagawa T , Kitani A , Fuss I , Levine B , Brant SR , Peter I , Tajima M , Nakamura S , Strober W ((2018) ) An increase in LRRK2 suppresses autophagy and enhances Dectin-1-induced immunity in a mouse model of colitis. Sci Transl Med 10: , eaan8162. |
[30] | Kubo M , Nagashima R , Kurihara M , Kawakami F , Maekawa T , Eshima K , Ohta E , Kato H , Obata F ((2020) ) Leucine-rich repeat kinase 2 controls inflammatory cytokines production through NF-kappaB phosphorylation and antigen presentation in bone marrow-derived dendritic cells. Int J Mol Sci 21: , 1890. |
[31] | Li T , Ning B , Kong L , Dai B , He X , Thomas JM , Sawa A , Ross CA , Smith WW ((2021) ) A LRRK2 GTP binding inhibitor, 68, reduces LPS-inducedsignaling events and TNF-alpha release in human lymphoblasts. Cells 10: , 480. |
[32] | Moehle MS , Daher JP , Hull TD , Boddu R , Abdelmotilib HA , Mobley J , Kannarkat GT , Tansey MG , West AB ((2015) ) The G2019S LRRK2 mutation increases myeloid cell chemotactic responses and enhances LRRK2 binding to actin-regulatory proteins. Hum Mol Genet 24: , 4250–4267. |
[33] | Moehle MS , Webber PJ , Tse T , Sukar N , Standaert DG , DeSilva TM , Cowell RM , West AB ((2012) ) LRRK2 inhibition attenuates microglial inflammatory responses. J Neurosci 32: , 1602–1611. |
[34] | Ho DH , Seol W , Eun JH , Son IH ((2017) ) Phosphorylation of p53 by LRRK2 induces microglial tumor necrosis factor alpha-mediated neurotoxicity. Biochem Biophys Res Commun 482: , 1088–1094. |
[35] | Russo I , Berti G , Plotegher N , Bernardo G , Filograna R , Bubacco L , Greggio E ((2015) ) Leucine-rich repeat kinase 2 positively regulates inflammation and down-regulates NF-kappaB p50 signaling in cultured microglia cells. J Neuroinflammation 12: , 230. |
[36] | Kim B , Yang MS , Choi D , Kim JH , Kim HS , Seol W , Choi S , Jou I , Kim EY , Joe EH ((2012) ) Impaired inflammatory responses in murine Lrrk2-knockdown brain microglia. PLoS One 7: , e34693. |
[37] | Gardet A , Benita Y , Li C , Sands BE , Ballester I , Stevens C , Korzenik JR , Rioux JD , Daly MJ , Xavier RJ , Podolsky DK ((2010) ) LRRK2 is involved in the IFN-gamma response and host response to pathogens. J Immunol 185: , 5577–5585. |
[38] | Hongge L , Kexin G , Xiaojie M , Nian X , Jinsha H ((2015) ) The role of LRRK2 in the regulation of monocyte adhesion to endothelial cells. J Mol Neurosci 55: , 233–239. |
[39] | Russo I , Di Benedetto G , Kaganovich A , Ding J , Mercatelli D , Morari M , Cookson MR , Bubacco L , Greggio E ((2018) ) Leucine-rich repeat kinase 2 controls protein kinase A activation state through phosphodiesterase 4. J Neuroinflammation 15: , 297. |
[40] | Liu W , Liu X , Li Y , Zhao J , Liu Z , Hu Z , Wang Y , Yao Y , Miller AW , Su B , Cookson MR , Li X , Kang Z ((2017) ) LRRK2 promotes the activation of NLRC4 inflammasome during Salmonella Typhimurium infection. J Exp Med 214: , 3051–3066. |
[41] | Yan R , Liu Z ((2017) ) LRRK2 enhances Nod1/2-mediated inflammatory cytokine production by promoting Rip2 phosphorylation. Protein Cell 8: , 55–66. |
[42] | Wong AYW , Oikonomou V , Paolicelli G , De Luca A , Pariano M , Fric J , Tay HS , Ricciardi-Castagnoli P , Zelante T ((2018) ) Leucine-rich repeat kinase 2 controls the Ca(2+)/nuclear factor of activated T cells/IL-2 pathway during aspergillus non-canonical autophagy in dendritic cells. Front Immunol 9: , 210. |
[43] | Gao P , Liu H , Huang H , Sun Y , Jia B , Hou B , Zhou X , Strober W , Zhang F ((2022) ) The Crohn disease-associated ATG16L1(T300A) polymorphism regulates inflammatory responses by modulating TLR- and NLR-mediated signaling. Autophagy 18: , 2561–2575. |
[44] | Panagiotakopoulou V , Ivanyuk D , De Cicco S , Haq W , Arsic A , Yu C , Messelodi D , Oldrati M , Schondorf DC , Perez MJ , Cassatella RP , Jakobi M , Schneiderhan-Marra N , Gasser T , Nikic-Spiegel I , Deleidi M ((2020) ) Interferon-gamma signaling synergizes with LRRK2 in neurons and microglia derived from human induced pluripotent stem cells. Nat Commun 11: , 5163. |
[45] | Kozina E , Sadasivan S , Jiao Y , Dou Y , Ma Z , Tan H , Kodali K , Shaw T , Peng J , Smeyne RJ ((2018) ) Mutant LRRK2 mediates peripheral and central immune responses leading to neurodegeneration in vivo. Brain 141: , 1753–1769. |
[46] | Kozina E , Byrne M , Smeyne RJ ((2022) ) Mutant LRRK2 in lymphocytes regulates neurodegeneration via IL-6 in an inflammatory model of Parkinson’s disease. NPJ Parkinsons Dis 8: , 24. |
[47] | Liu Z , Xu E , Zhao HT , Cole T , West AB ((2020) ) LRRK2 and Rab10 coordinate macropinocytosis to mediate immunological responses in phagocytes. EMBO J 39: , e104862. |
[48] | Harms AS , Thome AD , Yan Z , Schonhoff AM , Williams GP , Li X , Liu Y , Qin H , Benveniste EN , Standaert DG ((2018) ) Peripheral monocyte entry is required for alpha-Synuclein induced inflammation and Neurodegeneration in a model of Parkinson disease. Exp Neurol 300: , 179–187. |
[49] | Tansey MG , Wallings RL , Houser MC , Herrick MK , Keating CE , Joers V ((2022) ) Inflammation and immune dysfunction in Parkinson disease. Nat Rev Immunol 22: , 657–673. |
[50] | Herrick MK , Tansey MG ((2021) ) Is LRRK2 the missing link between inflammatory bowel disease and Parkinson’s disease? NPJ Parkinsons Dis 7: , 26. |
[51] | Warnecke T , Schafer KH , Claus I , Del Tredici K , Jost WH ((2022) ) Gastrointestinal involvement in Parkinson’s disease: Pathophysiology, diagnosis, and management. NPJ Parkinsons Dis 8: , 31. |
[52] | Schaeffer E , Kluge A , Bottner M , Zunke F , Cossais F , Berg D , Arnold P ((2020) ) Alpha synuclein connects the gut-brain axis in Parkinson’s disease patients - a view on clinical aspects, cellular pathology and analytical methodology. Front Cell Dev Biol 8: , 573696. |
[53] | Devos D , Lebouvier T , Lardeux B , Biraud M , Rouaud T , Pouclet H , Coron E , Bruley des Varannes S , Naveilhan P , Nguyen JM , Neunlist M , Derkinderen P ((2013) ) Colonic inflammation in Parkinson’s disease. Neurobiol Dis 50: , 42–48. |
[54] | Houser MC , Chang J , Factor SA , Molho ES , Zabetian CP , Hill-Burns EM , Payami H , Hertzberg VS , Tansey MG ((2018) ) Stool immune profilesevince gastrointestinal inflammation in Parkinson’s disease. Mov Disord 33: , 793–804. |
[55] | Mulak A , Koszewicz M , Panek-Jeziorna M , Koziorowska-Gawron E , Budrewicz S ((2019) ) Fecal calprotectin as a marker of the gut immune system activation is elevated in Parkinson’s disease. Front Neurosci 13: , 992. |
[56] | Aho VTE , Houser MC , Pereira PAB , Chang J , Rudi K , Paulin L , Hertzberg V , Auvinen P , Tansey MG , Scheperjans F ((2021) ) Relationships of gut microbiota, short-chain fatty acids, inflammation, and the gut barrier in Parkinson’s disease. Mol Neurodegener 16: , 6. |
[57] | Schwiertz A , Spiegel J , Dillmann U , Grundmann D , Burmann J , Fassbender K , Schafer KH , Unger MM ((2018) ) Fecal markers of intestinal inflammation and intestinal permeability are elevated in Parkinson’s disease. Parkinsonism Relat Disord 50: , 104–107. |
[58] | Stolzenberg E , Berry D , Yang D , Lee EY , Kroemer A , Kaufman S , Wong GCL , Oppenheim JJ , Sen S , Fishbein T , Bax A , Harris B , Barbut D , Zasloff MA ((2017) ) A role for neuronal alpha-synuclein in gastrointestinal immunity. J Innate Immun 9: , 456–463. |
[59] | Borghammer P , Horsager J , Andersen K , Van Den Berge N , Raunio A , Murayama S , Parkkinen L , Myllykangas L ((2021) ) Neuropathological evidence of body-first vs. brain-first Lewy body disease. Neurobiol Dis 161: , 105557. |
[60] | Kelly LP , Carvey PM , Keshavarzian A , Shannon KM , Shaikh M , Bakay RA , Kordower JH ((2014) ) Progression of intestinal permeability changes and alpha-synuclein expression in a mouse model of Parkinson’s disease. Mov Disord 29: , 999–1009. |
[61] | Forsyth CB , Shannon KM , Kordower JH , Voigt RM , Shaikh M , Jaglin JA , Estes JD , Dodiya HB , Keshavarzian A ((2011) ) Increased intestinal permeability correlates with sigmoid mucosa alpha-synuclein staining and endotoxin exposure markers in early Parkinson’s disease. PLoS One 6: , e28032. |
[62] | Chen SJ , Lin CH ((2022) ) Gut microenvironmental changes as a potential trigger in Parkinson’s disease through the gut-brain axis. J Biomed Sci 29: , 54. |
[63] | Liao PH , Chiang HL , Shun CT , Hang JF , Chiu HM , Wu MS , Lin CH ((2021) ) Colonic leucine-rich repeat kinase 2 expression is increased and associated with disease severity in patients with Parkinson’s disease. Front Aging Neurosci 13: , 819373. |
[64] | de Guilhem de Lataillade A , Verchere J , Oullier T , Prigent A , Durand T , Pellegrini C , Neunlist M , Baron T , Rolli-Derkinderen M , Derkinderen P ((2021) ) LRRK2 is reduced in Parkinson’s disease gut. Acta Neuropathol 142: , 601–603. |
[65] | Qiu P , Ishimoto T , Fu L , Zhang J , Zhang Z , Liu Y ((2022) ) The gut microbiota in inflammatory bowel disease. Front Cell Infect Microbiol 12: , 733992. |
[66] | Kaser A , Blumberg RS ((2008) ) Paneth cells and inflammation dance together in Crohn’s disease. Cell Res 18: , 1160–1162. |
[67] | Ogura Y , Bonen DK , Inohara N , Nicolae DL , Chen FF , Ramos R , Britton H , Moran T , Karaliuskas R , Duerr RH , Achkar JP , Brant SR , Bayless TM , Kirschner BS , Hanauer SB , Nunez G , Cho JH ((2001) ) A frameshift mutation in NOD2 associated with susceptibility to Crohn’s disease. Nature 411: , 603–606. |
[68] | Hugot JP , Chamaillard M , Zouali H , Lesage S , Cezard JP , Belaiche J , Almer S , Tysk C , O’Morain CA , Gassull M , Binder V , Finkel Y , Cortot A , Modigliani R , Laurent-Puig P , Gower-Rousseau C , Macry J , Colombel JF , Sahbatou M , Thomas G ((2001) ) Association of NOD2 leucine-rich repeat variants with susceptibility to Crohn’s disease. Nature 411: , 599–603. |
[69] | Ma Q , An X , Li Z , Zhang H , Huang W , Cai L , Hu P , Lin Q , Tzeng CM ((2013) ) P268S in NOD2 associates with susceptibility to Parkinson’s disease in Chinese population. Behav Brain Funct 9: , 19. |
[70] | Zhang Q , Pan Y , Yan R , Zeng B , Wang H , Zhang X , Li W , Wei H , Liu Z ((2015) ) Commensal bacteria direct selective cargo sorting to promote symbiosis. Nat Immunol 16: , 918–926. |
[71] | Liu TC , Naito T , Liu Z , VanDussen KL , Haritunians T , Li D , Endo K , Kawai Y , Nagasaki M , Kinouchi Y , McGovern DP , Shimosegawa T , Kakuta Y , Stappenbeck TS ((2017) ) LRRK2 but not ATG16L1 is associated with Paneth cell defect in Japanese Crohn’s disease patients. JCI Insight 2: , e91917. |
[72] | Alam MM , Yang D , Li XQ , Liu J , Back TC , Trivett A , Karim B , Barbut D , Zasloff M , Oppenheim JJ ((2022) ) Alpha synuclein, the culprit in Parkinson disease, is required for normal immune function. Cell Rep 38: , 110090. |
[73] | O’Hara DM , Pawar G , Kalia SK , Kalia LV ((2020) ) LRRK2 and alpha-synuclein: Distinct or synergistic players in Parkinson’s disease? Front Neurosci 14: , 577. |
[74] | Shannon KM , Keshavarzian A , Mutlu E , Dodiya HB , Daian D , Jaglin JA , Kordower JH ((2012) ) Alpha-synuclein in colonic submucosa in early untreated Parkinson’s disease. Mov Disord 27: , 709–715. |
[75] | Stokholm MG , Danielsen EH , Hamilton-Dutoit SJ , Borghammer P ((2016) ) Pathological alpha-synuclein in gastrointestinal tissues from prodromal Parkinson disease patients. Ann Neurol 79: , 940–949. |
[76] | Visanji NP , Marras C , Kern DS , Al Dakheel A , Gao A , Liu LW , Lang AE , Hazrati LN ((2015) ) Colonic mucosal a-synuclein lacks specificity as a biomarker for Parkinson disease. Neurology 84: , 609–616. |
[77] | Jotanovic J , Milin-Lazovic J , Alafuzoff I ((2022) ) Gastrointestinal biopsy obtained during cancer screening, a biological marker for alpha-synucleinopathy? J Neuropathol Exp Neurol 81: , 356–362. |
[78] | Fricova D , Harsanyiova J , Kralova Trancikova A ((2020) ) Alpha-synuclein in the gastrointestinal tract as a potential biomarker for early detection of Parkinson’s disease. Int J Mol Sci 21: , 8666. |
[79] | Glassner KL , Abraham BP , Quigley EMM ((2020) ) The microbiome and inflammatory bowel disease. J Allergy Clin Immunol 145: , 16–27. |
[80] | Tan AH , Chong CW , Lim SY , Yap IKS , Teh CSJ , Loke MF , Song SL , Tan JY , Ang BH , Tan YQ , Kho MT , Bowman J , Mahadeva S , Yong HS , Lang AE ((2021) ) Gut microbial ecosystem in Parkinson disease: New clinicobiological insights from multi-omics. Ann Neurol 89: , 546–559. |
[81] | Romano S , Savva GM , Bedarf JR , Charles IG , Hildebrand F , Narbad A ((2021) ) Meta-analysis of the Parkinson’s disease gut microbiome suggests alterations linked to intestinal inflammation. NPJ Parkinsons Dis 7: , 27. |
[82] | Sampson TR , Debelius JW , Thron T , Janssen S , Shastri GG , Ilhan ZE , Challis C , Schretter CE , Rocha S , Gradinaru V , Chesselet MF , Keshavarzian A , Shannon KM , Krajmalnik-Brown R , Wittung-Stafshede P , Knight R , Mazmanian SK ((2016) ) Gut microbiota regulate motor deficits and neuroinflammation in a model of Parkinson’s disease. Cell 167: , 1469–1480 e1412. |
[83] | Yan J , Yu W , Wang G , Lu C , Liu C , Jiang L , Jiang Z , Liang Z , Liu D ((2022) ) LRRK2 deficiency mitigates colitis progression by favoring resolution of inflammation and restoring homeostasis of gut microbiota. Genomics 114: , 110527. |
[84] | Xue LJ , Yang XZ , Tong Q , Shen P , Ma SJ , Wu SN , Zheng JL , Wang HG ((2020) ) Fecal microbiota transplantation therapy for Parkinson’s disease: A preliminary study. Medicine (Baltimore) 99: , e22035. |
[85] | Scheperjans F , Aho V , Pereira PA , Koskinen K , Paulin L , Pekkonen E , Haapaniemi E , Kaakkola S , Eerola-Rautio J , Pohja M , Kinnunen E , Murros K , Auvinen P ((2015) ) Gut microbiota are related to Parkinson’s disease and clinical phenotype. Mov Disord 30: , 350–358. |
[86] | Minato T , Maeda T , Fujisawa Y , Tsuji H , Nomoto K , Ohno K , Hirayama M ((2017) ) Progression of Parkinson’s disease is associated with gut dysbiosis: Two-year follow-up study. PLoS One 12: , e0187307. |
[87] | Hasegawa S , Goto S , Tsuji H , Okuno T , Asahara T , Nomoto K , Shibata A , Fujisawa Y , Minato T , Okamoto A , Ohno K , Hirayama M ((2015) ) Intestinal dysbiosis and lowered serum lipopolysaccharide-binding protein in Parkinson’s disease. PLoS One 10: , e0142164. |
[88] | Dutta SK , Verma S , Jain V , Surapaneni BK , Vinayek R , Phillips L , Nair PP ((2019) ) Parkinson’s disease: The emerging role of gut dysbiosis, antibiotics, probiotics, and fecal microbiota transplantation. J Neurogastroenterol Motil 25: , 363–376. |
[89] | Schneider SA , Alcalay RN ((2017) ) Neuropathology of genetic synucleinopathies with parkinsonism: Review of the literature. Mov Disord 32: , 1504–1523. |
[90] | Menozzi E , Macnaughtan J , Schapira AHV ((2021) ) LRRK2 parkinsonism: Does the response to gut bacteria mitigate the neurological picture? Mov Disord 36: , 71–75. |
[91] | Corbille AG , Clairembault T , Coron E , Leclair-Visonneau L , Preterre C , Neunlist M , Derkinderen P ((2016) ) What a gastrointestinal biopsy can tell us about Parkinson’s disease? Neurogastroenterol Motil 28: , 966–974. |
[92] | Fleming MA , 2nd, Ehsan L , Moore SR , Levin DE ((2020) ) The enteric nervous system and its emerging role as a therapeutic target. Gastroenterol Res Pract 2020: , 8024171. |


