
Autopallidotomy: From Colloquial Term to Scientific Theory
Abstract
Levodopa-induced dyskinesia (LID), a frequent complication of Parkinson’s disease (PD), occurs in ∼30% of patients after five years’ treatment with levodopa. In atypical parkinsonism, LID occurs less frequently than in PD. Lower frequency of LID in atypical parkinsonism has traditionally been attributed to lower amounts of levodopa used by these patients; however, recent studies have shown lower frequency of LID in atypical parkinsonism compared with PD when adjusting for levodopa dose. The mechanism of LID is complex but requires pulsatile levodopa stimulation, progressive presynaptic dopaminergic degeneration, and a relatively intact postsynaptic dopaminergic system. The globus pallidus internus (GPi), the main inhibitory nucleus of the basal ganglia, may play a major role in the development and treatment of LID. Surgical lesioning of the posteroventral GPi is directly antidyskinetic; animal models showing GPi-associated striatal neurons are directly responsible for the development of LID. However, other cortical areas, particularly the primary sensory and motor cortices may also play a role in LID. In some cases of atypical parkinsonism, particularly progressive supranuclear palsy and corticobasal degeneration, severe degeneration of the GPi, a so-called “autopallidotomy,” may explain the absence of LID in these patients. In other atypical parkinsonisms, such as PD dementia and dementia with Lewy bodies, the lower incidence of LID may partly be attributed to more striatal degeneration but likely also relates to the degeneration of the motor cortex and resultant network dysfunction. Overall, atypical parkinsonism serves as a natural model that may ultimately reveal more effective therapies for LID.
Parkinson’s disease (PD) is a progressive neurodegenerative disorder characterized by motor symptoms, which robustly respond to treatment with levodopa. Motor complications such as levodopa-induced dyskinesia (LID), freezing of gait, and motor fluctuations evolve over time and are characteristic of more advanced PD with limited treatment options. LID, defined as involuntary, purposeless, predominantly choreiform movements, are feared complications of advanced PD and occur in up to 30% of patients after 5 years of treatment [1]. Higher risks of dyskinesias are associated with a younger age of PD onset and a higher daily dose of levodopa, leading to the erroneous assumption that early exposure to levodopa results in dyskinesias and, ultimately, avoidance of levodopa treatment until a later stage of PD. However, more recent data suggest the development of LID correlates with progressive presynaptic nigrostriatal degeneration with fairly spared postsynaptic nigrostriatal system in conjunction with pulsatile levodopa stimulation in the form of carbidopa-levodopa (as opposed to physiologic tonic stimulation) rather than prolonged exposure to levodopa per se [1, 2].
Treatment of LID primarily consists of reducing the levodopa dose or by adding the NMDA-receptor antagonist amantadine. Advanced PD therapies, including continuous subcutaneous apomorphine infusion and continuous intestinal infusion of levodopa, are less likely to correlate with dyskinesia due to greater physiologic tonic stimulation of dopamine receptors, despite the high doses of levodopa required in the continuous intestinal infusion [2]. Surgical treatments, including lesioning of the globus pallidus internus (GPi) through direct radiofrequency ablation and, more recently, through focused ultrasound as well as deep brain stimulation (DBS) targeting the GPi, ventrothalamic nuclei (VIM), and subthalamic nucleus (STN), have become treatment mainstays for motor symptoms of advanced PD, some of which have shown benefit for LID [3].
Although dyskinesia is common in PD, it is thought to occur less frequently in in other forms on the PD spectrum including PD dementia (PDD) and dementia with Lewy bodies (DLB). In one population-based cohort of parkinsonism, dyskinesias reportedly occurred in approximately 30% of PD patients compared with 12.6% in those with PDD or DLB despite adjusting the dosage of levodopa, suggesting factors other than purely lower doses of levodopa in PDD and DLB correlate with the decreased risk of dyskinesia compared with PD [1, 4]. LID is incredibly rare in progressive supranuclear palsy (PSP) or corticobasal degeneration (CBD), even at high doses of levodopa [5]. LID, primarily orofacial in nature, has been reported to occur into up to 27% of patients with multiple system atrophy (MSA), particularly those with higher doses of levodopa and longer disease duration, similar to risk factors for LID in PD [6, 7]. The reasons for the reported differences in dyskinesia frequency between different types of parkinsonism are likely multiple and include levodopa dosage, age, treatment duration, and length of follow-up; however, selective neuroanatomic degenerative differences and etiologies may also play a significant role.
THE ROLE OF THE GLOBUS PALLIDUS IN LID
The globus pallidus internus (GPi) is the major inhibitory output nucleus in the basal ganglia and is thought to be the major nucleus involved in generating and treating LID. Recent optogenetic studies in the 6-OHDA parkinsonian mouse model utilizing targeted recombination in active population (TRAP) have identified a subpopulation of medium spiny neurons in the striatum involved in the direct pathway responsible for LID development [8]. These investigators found that stimulation of these striatal neurons induces dyskinesia in the absence of levodopa while inhibition of this subpopulation of neurons prevents the development of LID [8]. Similarly, inhibition of non-TRAPed striatal neurons was insufficient to ameliorate LID [8]. On the other hand, this study did not assess the indirect pathway, so contributions of the indirect pathway to LID cannot be excluded by this study.
In the late 90 s, unilateral posteroventral medial pallidotomy (of GPi), initially pioneered by Svennilson and Leksell in the 1960 s, showed great promise for the treatment of LID, with several studies showing > 75% reduction in dyskinesias, primarily contralateral to the lesion [9, 10]. This effect remained 2 to 5 years after surgical lesioning [9, 11]. Surgical thalamotomy has proven highly efficacious for tremor; LID improvement is seen with lesioning of the ventralis oralis anterior and posterior nuclei (but not ventralis intermedius nucleus) but overall is not as antidyskinetic as pallidotomy [12]. DBS has identified new targets for ameliorating PD symptoms, namely the STN; however, the GPi has remained an important target, especially for patients with significant dyskinesias [13]. In fact, GPi stimulation appears to allow for increasing doses of levodopa without development of LID [14]. GPi stimulation induces greater direct antidyskinetic effects but less efficacy in treatment of PD symptoms compared with STN DBS [13, 15]. On the other hand, stimulation of the STNdoes appears to have no direct antidyskinetic effect, and stimulation may lead to increased dyskinesias in some cases [13, 15]. While LID decreases over time in STN DBS, this is attributed to lower levodopa doses required to treat PD symptoms following STN DBS. More recently, MR-guided, focused ultrasound ablation of the posteroventral GPi for PD patients with LID has begun clinical trials. Preliminary results show promise in a small number of patients, but these will need to be confirmed with larger trials [16].
CONTRIBUTIONS OF REGIONS OUTSIDE THE BASAL GANGLIA TO LID
While neuronal populations in the basal ganglia, particularly the GPi, striatum, and STN, are key contributors to LID, other cortical areas, primarily the primary motor and sensory cortex and network dysfunction in these regions, may also play a role in generating LID [2]. Changes in synchrony and firing patterns of neurons involved in LID are more important in LID generation than changes in the rate of firing [2]. Human and rat studies have shown maladaptive neuroplasticity, decreased long-term potentiation, and interrupted cortical oscillations in the primary motor and sensory motor cortices in LID [17–19]. Consistent with these studies, more recent data in the 6-OHDA mouse model found TRAPed neurons in the primary motor and sensory cortices also activating during LID [8]. However, activating these areas in the absence of levodopa was insufficient to cause dyskinesia, unlike specific medium spiny TRAPed neurons in the striatum [8]. Functional imaging studies have shown desynchrony in the inferior frontal cortex and pre-supplementary motor area in dyskinetic patients suggesting uncoordinated inhibitory control over motor circuits possibly contributing to LID [20]. Overall, LID contributions to structures outside the basal ganglia remain to be fully understood and likely play a supplementary rather than a direct causal role.
DYSKINESIA IN ATYPICAL PARKINSONISM AND THE PATHOPHYSIOLOGIC AUTOPALLIDOTOMY
As previously discussed, LID is significantly less frequent in atypical parkinsonism and some advanced stages of PD, such as PD dementia [4]. This has traditionally been attributed to these patients receiving lower doses of levodopa due to perceived lack of efficacy. However, studies have shown the degree of LID is less in these patients when adjusting for levodopa dose, and patients with PSP or CBD do not develop dyskinesia even when treated with high-dose levodopa [4, 5]. The lack of dyskinesia in patients with PSP despite high doses of levodopa is believed due to the widespread neurodegeneration of the GPi, which has been colloquially known as “autopallidotomy” [5]. Unlike PD, which spares the globus pallidus and striatum, the globus pallidus and striatum in PSP and CBD undergo severe degeneration, which likely limits their ability to generate dyskinesia despite the presence of levodopa [21]. Autopallidotomy may also play a role in clinical features of atypical parkinsonism such as postural instability or freezing of gait, which have been reported as complications of bilateral surgical pallidotomy [22]. Further, the motor cortex is also commonly involved in PSP but spared in PD, potentially contributing to less frequent LID in PSP. MSA, on the other hand, has more variable degeneration of the globus pallidus, which may explain the intermediate frequency of dyskinesias in MSA [1, 7, 21]. Interestingly, MSA patients who develop dyskinesias tend to have similar features as PD patients who develop dyskinesias (more presynaptic degeneration, longer duration of disease and higher levodopa dose) so MSA patients with LID may be less likely to have significant pathologic involvement of the GPi [7]. As such, atypical parkinsonism, particularly PSP, serves as a pathophysiologic model of the pallidotomy to treat LID, mirroring surgical and neurostimulation success over the last 50 years. An additional complementary mechanism to explain reduced LID in PSP and CBD may depend on the degree of striatal fluctuation in dopamine concentrations in response to levodopa dose. In PD, patients with LID have higher striatal concentrations of dopamine in response to equivalent doses of levodopa when compared with PD patients without LID, suggesting changes in striatal dopamine concentration may also contribute to development of LID [23]. However, striatal dopamine response to levodopa in PSP and CBD has not been evaluated so this mechanism requires further confirmatory testing.
Fig. 1
In the figure on the left, a lower Braak Stage is associated with less diffuse Lewy body accumulation and less aging changes; therefore autopallidotomy does not occur, whereas in more diffuse and higher Braak stage there are increased Lewy bodies and age-related changes leading to autopallidotomy.
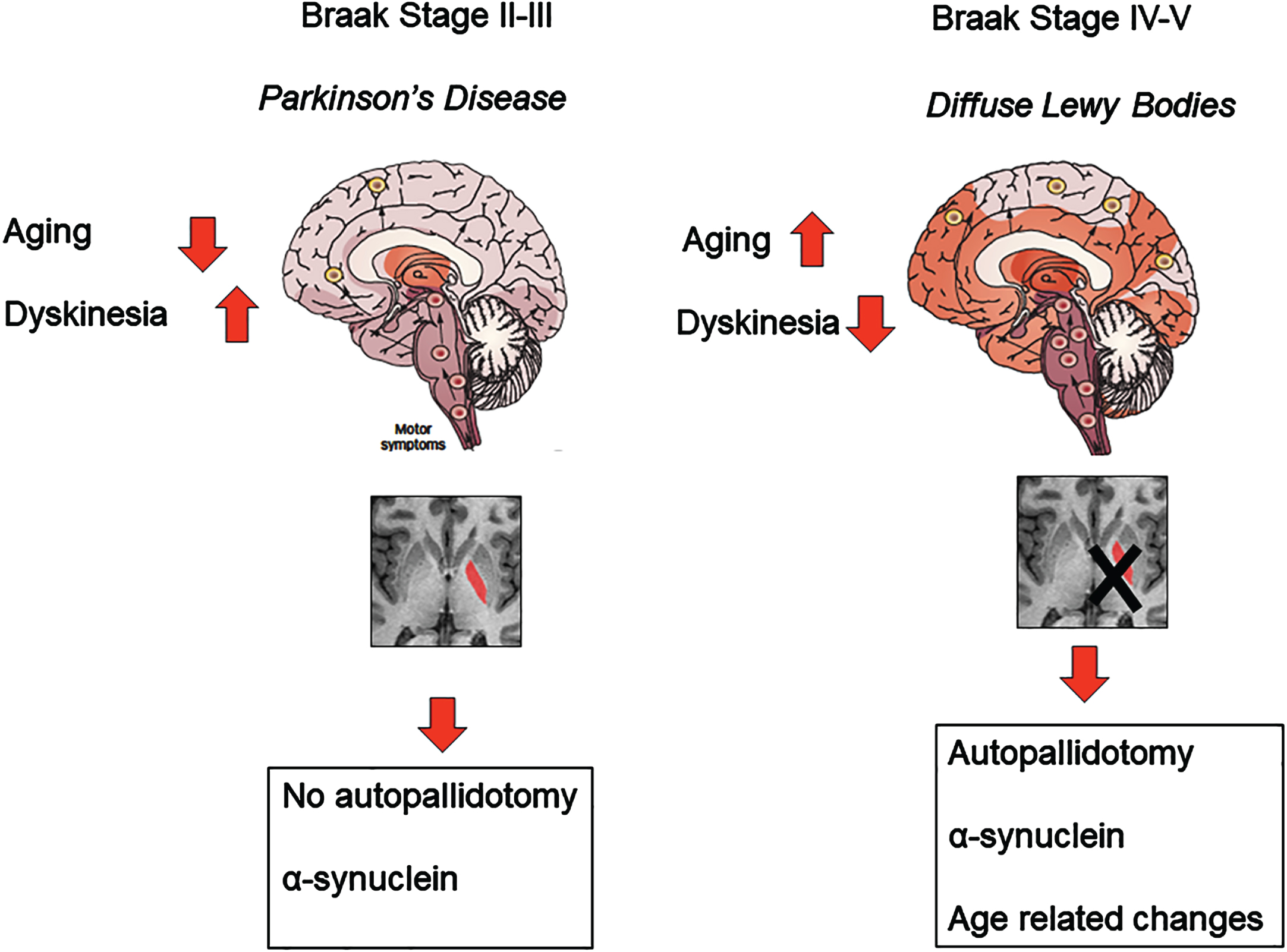
While the autopallidotomy largely explains the lack of LID in PSP and CBD and is potentially a component of less frequent dyskinesias in MSA, the explanation for less frequent dyskinesias in PDD and DLB is likely more complex, and this phenomenon has been poorly studied [4]. Meta-analysis of volumetric MRI studies in DLB patients has shown decreased volume of globus pallidus in DLB patients, suggesting the possibility of a mild form of autopallidotomy, but this has not been born out on autopsy [24]. One pathologic study of PDD vs. DLB patients found involvement of the globus pallidus of 1 of 11 total patients, suggesting the need for an alternate explanation for the lower LID frequency in PDD and DLB [25]. It is likely that lower doses of levodopa utilized in PDD and DLB out of concern for side effects play a role in fewer LID seen in DLB/PDD. While there was no difference in levodopa dose between dyskinetic and non-dyskinetic patients with PDD and DLB, the number of dyskinetic DLB and PDD patients were small, which may have explained lack of significant differences in levodopa dose in this study [4]. Pathologic studies comparing PD to PDD and DLB patients have shown significantly more alpha-synuclein pathology involving the striatum and more synuclein and tau pathology in the sensory and motor cortex in PDD and DLB [26]. As mouse models suggest, a specific subpopulation of neurons in the striatum is necessary to generate LID, but it is possible that more widespread neurodegeneration in the striatum may involve these neurons and lead to less LID [8]. On the other hand, more widespread, advanced cortical neurodegeneration may also contribute to decreased LID in PDD and DLB (Fig. 1). Overall, further work is needed to clarify the mechanisms of LID in patients with PDD and DLB.
CONCLUSION
Levodopa-induced dyskinesias are feared complications of PD but occur relatively infrequently in atypical parkinsonism. Degeneration of the globus pallidus, also known as autopallidotomy, likely explains the lack of LID in PSP and CBD and, to a degree, in MSA. Widespread cortical degeneration resulting in network disruption likely contributes to less dyskinesia in PDD and DLB, although more extensive striatal degeneration in PDD and DLB may also lower the dyskinesia risk. Atypical parkinsonism can serve as a model for further understanding of LID, which may allow for the identification of more efficacious treatments; however, the true frequency of LID in atypical parkinsonism compared with PD requires larger scale studies.
ACKNOWLEDGMENTS
The authors thank Lea C. Dacy for formatting and editing support.
CONFLICT OF INTEREST
SJM has no conflict of interest to report. RS receives funding from the National Institute on Aging, the National Institute of Neurological Disorders and Stroke, the Parkinson’s Disease Foundation, Inc., and an unrestricted grant from ACADIA Pharmaceuticals, Inc.
REFERENCES
[1] | Turcano P , Mielke MM , Bower JH , Parisi JE , Cutsforth-Gregory JK , Ahlskog JE , Savica R ((2018) ) Levodopa-induced dyskinesia in Parkinson disease: A population-based cohort study. Neurology 91: , e2238–e2243. |
[2] | Espay AJ , Morgante F , Merola A , Fasano A , Marsili L , Fox SH , Bezard E , Picconi B , Calabresi P , Lang AE ((2018) ) Levodopa-induced dyskinesia in Parkinson disease: Current and evolving concepts. Ann Neurol 84: , 797–811. |
[3] | Martini ML , Mocco J , Panov F ((2019) ) Neurosurgical approaches to levodopa-induced dyskinesia. World Neurosurg 126: , 376–382. |
[4] | Turcano P , Stang CD , Bower JH , Ahlskog JE , Boeve BF , Mielke MM , Savica R ((2020) ) Levodopa-induced dyskinesia in dementia with Lewy bodies and Parkinson disease with dementia. Neurol Clin Pract 10: , 156–161. |
[5] | Lang AE ((2005) ) Treatment of progressive supranuclear palsy and corticobasal degeneration. Mov Disord 20 Suppl 12: , S83–91. |
[6] | Wenning GK , Tison F , Ben Shlomo Y , Daniel SE , Quinn NP ((1997) ) Multiple system atrophy: a review of 203 pathologically proven cases. Mov Disord 12: , 133–147. |
[7] | Ueno SI , Oyama G , Kanai K , Hatano T , Shimo Y , Hattori N ((2020) ) A retrospective imaging evaluation of presynaptic dopaminergic degeneration in multiple system atrophy with levodopa induced dyskinesia. Tremor Other Hyperkinet Mov (N Y) 10: , 6. |
[8] | Girasole AE , Lum MY , Nathaniel D , Bair-Marshall CJ , Guenthner CJ , Luo L , Kreitzer AC , Nelson AB ((2018) ) A subpopulation of striatal neurons mediates levodopa-induced dyskinesia. Neuron 97: , 787–795 e786. |
[9] | Vitek JL , Bakay RA , Freeman A , Evatt M , Green J , McDonald W , Haber M , Barnhart H , Wahlay N , Triche S , Mewes K , Chockkan V , Zhang JY , DeLong MR ((2003) ) Randomized trial of pallidotomy versus medical therapy for Parkinson’s disease. Ann Neurol 53: , 558–569. |
[10] | Samuel M , Caputo E , Brooks DJ , Schrag A , Scaravilli T , Branston NM , Rothwell JC , Marsden CD , Thomas DG , Lees AJ , Quinn NP ((1998) ) A study of medial pallidotomy for Parkinson’s disease: clinical outcome, MRI location and complications. Brain 121 (Pt 1): , 59–75. |
[11] | Fine J , Duff J , Chen R , Chir B , Hutchison W , Lozano AM , Lang AE ((2000) ) Long-term follow-up of unilateral pallidotomy in advanced Parkinson’s disease. N Engl J Med 342: , 1708–1714. |
[12] | Narabayashi H , Yokochi F , Nakajima Y ((1984) ) Levodopa-induced dyskinesia and thalamotomy. J Neurol Neurosurg Psychiatry 47: , 831–839. |
[13] | Krause M , Fogel W , Heck A , Hacke W , Bonsanto M , Trenkwalder C , Tronnier V ((2001) ) Deep brain stimulation for the treatment of Parkinson’s disease: subthalamic nucleus versus globus pallidus internus. J Neurol Neurosurg Psychiatry 70: , 464–470. |
[14] | Oyama G , Foote KD , Jacobson CEt , Velez-Lago F , Go C , Limotai N , Zeilman PR , Romrell J , Wu SS , Neal D , Okun MS ((2012) ) GPi and STN deep brain stimulation can suppress dyskinesia in Parkinson’s disease. Parkinsonism Relat Disord 18: , 814–818. |
[15] | Krack P , Pollak P , Limousin P , Benazzouz A , Deuschl G , Benabid AL ((1999) ) From off-period dystonia to peak-dose chorea. The clinical spectrum of varying subthalamic nucleus activity. Brain 122 (Pt 6): , 1133–1146. |
[16] | Jung NY , Park CK , Kim M , Lee PH , Sohn YH , Chang JW ((2018) ) The efficacy and limits of magnetic resonance-guided focused ultrasound pallidotomy for Parkinson’s disease: a Phase I clinical trial. J Neurosurg 130: , 1853–1861. |
[17] | Alam M , Rumpel R , Jin X , von Wrangel C , Tschirner SK , Krauss JK , Grothe C , Ratzka A , Schwabe K ((2017) ) Altered somatosensory cortex neuronal activity in a rat model of Parkinson’s disease and levodopa-induced dyskinesias. Exp Neurol 294: , 19–31. |
[18] | Morgante F , Espay AJ , Gunraj C , Lang AE , Chen R ((2006) ) Motor cortex plasticity in Parkinson’s disease and levodopa-induced dyskinesias. Brain 129: , 1059–1069. |
[19] | Halje P , Tamte M , Richter U , Mohammed M , Cenci MA , Petersson P ((2012) ) Levodopa-induced dyskinesia is strongly associated with resonant cortical oscillations. J Neurosci 32: , 16541–16551. |
[20] | Gan C , Wang M , Si Q , Yuan Y , Zhi Y , Wang L , Ma K , Zhang K ((2020) ) Altered interhemispheric synchrony in Parkinson’s disease patients with levodopa-induced dyskinesias. NPJ Parkinsons Dis 6: , 14. |
[21] | Dickson DW ((2012) ) Parkinson’s disease and parkinsonism: neuropathology. Cold Spring Harb Perspect Med 2: , a009258. |
[22] | Horisawa S , Fukui A , Takeda N , Kawamata T , Taira T ((2021) ) Safety and efficacy of unilateral and bilateral pallidotomy for primary dystonia. Ann Clin Transl Neurol 8: , 857–865. |
[23] | de la Fuente-Fernandez R , Sossi V , Huang Z , Furtado S , Lu JQ , Calne DB , Ruth TJ , Stoessl AJ ((2004) ) Levodopa-induced changes in synaptic dopamine levels increase with progression of Parkinson’s disease: implications for dyskinesias. Brain 127: , 2747–2754. |
[24] | Zhong J , Pan P , Dai Z , Shi H ((2014) ) Voxelwise meta-analysis of gray matter abnormalities in dementia with Lewy bodies. Eur J Radiol 83: , 1870–1874. |
[25] | Jellinger KA , Attems J ((2006) ) Does striatal pathology distinguish Parkinson disease with dementia and dementia with Lewy bodies? Acta Neuropathol 112: , 253–260. |
[26] | Tsuboi Y , Uchikado H , Dickson DW ((2007) ) Neuropathology of Parkinson’s disease dementia and dementia with Lewy bodies with reference to striatal pathology. Parkinsonism Relat Disord 13 Suppl 3: , S221–224. |


